

Significance
High plasma levels of histidine-rich protein II (HRPII), a secreted malaria protein, are associated with severe malaria, but the mechanism of HRPII virulence is not fully established. We observed that secreted HRPII in parasite culture and plasma forms large heme-laden nanoparticles. The virulence of HRPII is tied to its delivery of hundreds of hemes to endothelial cells, leading to iron toxicity and production of reactive oxygen species, and subsequently activating the NLRP3 inflammasome. This cascade disrupts endothelial barrier integrity. These data define the link between HRPII and cerebral edema, a hallmark of cerebral malaria. Disruption of this process by heme sequestration or inflammasome inhibition mitigated the effects of HRPII and may guide further studies into clinical therapies for severe malaria.
Keywords: blood–brain barrier, malaria pathogenesis, heme oxygenase
Abstract
Histidine-rich protein II (HRPII) is secreted by Plasmodium falciparum during the blood stage of malaria infection. High plasma levels of HRPII are associated with cerebral malaria, a severe and highly fatal complication of malaria. HRPII has been shown to induce vascular leakage, the hallmark of cerebral malaria, in blood–brain barrier (BBB) and animal models. We have discovered an important mechanism for BBB disruption that is driven by unique features of HRPII. By characterizing serum from infected patients and HRPII produced by P. falciparum parasites in culture, we found that HRPII exists in large multimeric particles of 14 polypeptides that are richly laden with up to 700 hemes per particle. Heme loading of HRPII is required for efficient binding and internalization via caveolin-mediated endocytosis in hCMEC/D3 cerebral microvascular endothelial cells. Upon acidification of endolysosomes, two-thirds of the hemes are released from acid-labile binding sites and metabolized by heme oxygenase 1, generating ferric iron and reactive oxygen species. Subsequent activation of the NLRP3 inflammasome and IL-1β secretion resulted in endothelial leakage. Inhibition of these pathways with heme sequestration, iron chelation, or anti-inflammatory drugs protected the integrity of the BBB culture model from HRPII:heme. Increased cerebral vascular permeability was seen after injection of young mice with heme-loaded HRPII (HRPII:heme) but not with heme-depleted HRPII. We propose that during severe malaria infection, HRPII:heme nanoparticles in the bloodstream deliver an overwhelming iron load to endothelial cells to cause vascular inflammation and edema. Disrupting this process is an opportunity for targeted adjunctive therapies to reduce the morbidity and mortality of cerebral malaria.
Death of children under age five from severe malaria infection remains a significant problem (1). Malaria is caused by several Plasmodium species, and Plasmodium falciparum is responsible for nearly all fatalities due to malaria. A severe form of P. falciparum infection is cerebral malaria which is clinically marked by the presence of coma. Children fortunate enough to survive cerebral malaria can go on to develop long-term neurocognitive deficits (2, 3). The mainstays of cerebral malaria treatment are antimalarial agents to treat P. falciparum infection and supportive care for clinical manifestations. A significant fraction of patients succumb to disease despite these interventions.
A distinguishing feature of P. falciparum infection is the release of the parasite protein, histidine-rich protein II (HRPII), into the host bloodstream (4, 5). Plasma concentrations of HRPII in malaria infection can range from 100 fM to 100 nM, and severe malaria is associated with HRPII concentrations > 50 pM (6). First identified in 1986 by Wellems and Howard (7), HRPII is a highly unusual protein mostly comprised of histidine and alanine repeats (7). The protein is synthesized at high levels in blood-stage P. falciparum with a Plasmodium export element (PEXEL) motif that is cleaved by the aspartyl protease plasmepsin V in the endoplasmic reticulum (8, 9). The processed protein is exported to the host erythrocyte cytoplasmic compartment and released into host circulation. It is widely used in rapid diagnostic tests as a diagnostic marker of P. falciparum infection and to differentiate from infection by other Plasmodium species with high sensitivity (10).
The effects of HRPII on the host during infection are multifaceted. Clinically, higher levels of HRPII on presentation have been associated with greater likelihood of progression to severe malaria disease (11–14). In vitro, HRPII was shown to inhibit factor Xa and thrombin through binding of heparin and other glycosaminoglycans, potentially contributing to the hypercoagulable state observed in malaria infection (15) and the anti-inflammatory signaling of antithrombin (16). Direct effects of HRPII bound to polyphosphate on vascular endothelium have been shown using permeability assays with endothelial cell line EA.hy926 and a mouse intestinal permeability model (16).
With specific regard to cerebral malaria, high plasma levels of HRPII are associated with progression to cerebral malaria in children with P. falciparum infection (12, 13). Postmortem brain tissue IHC staining has revealed deposition of HRPII in tissues and vasculature (17). Vasogenic edema is a hallmark of cerebral malaria, and higher plasma levels of HRPII are associated with cerebral edema on MRI in pediatric and adult patients (18). Using a cerebral endothelial cell tissue model with hCMEC/D3 cells, Pal et al. previously showed that HRPII was sufficient and necessary for disruption of blood–brain barrier (BBB) integrity (19). Transgenic expression of HRPII in an HRPII-negative strain of P. falciparum allowed parasites to active hCMEC/D3 cells and disrupted barrier integrity. Using a mouse model, Pal et al. also showed that purified HRPII caused cerebral endothelial leakage (20). In both models, treatment with HRPII resulted in inflammasome activation and IL-1β release. Regulation of the BBB by IL-1β occurs through modulation of tight junction proteins (21, 22), and in cerebral malaria, angiotensin II and β-catenin signaling pathways are important (23). HRPII has also been shown to cause neuronal injury in brain cortical organoids via TLR1/2 activation (24).
Here, we explored the mechanism by which HRPII induces IL-1β secretion and increases vascular permeability during cerebral malaria. We have further characterized the properties of HRPII from infected patients and have found that substantial amounts of heme are bound to parasite-produced HRPII nanoparticles and that heme iron delivery to endothelial cells is key to triggering inflammation and causing endothelial barrier leak.
Results
HRPII from Patient Serum and Parasite Culture Exists as Large Heme-Bound Nanoparticles.
We purified secreted HRPII from conditioned media of P. falciparum cultured in human red blood cells (RBCs) by nickel affinity chromatography. The parasite-produced HRPII formed large particles, approximately 10 nm in diameter, that were observable via negative-stain electron microscopy (Fig. 1A). Excess serum from two patients diagnosed with severe P. falciparum infection with cerebral malaria was provided by our local clinical laboratory. When visualized by negative-stain EM, particles resembling purified HRPII were observed in serum from the patients but not from an uninfected person (Fig. 1A). By blue-native PAGE, HRPII from patient serum migrated as a large complex comparable to that of purified HRPII from parasite culture (Fig. 1B).
Fig. 1.

HRPII secreted in the serum of P. falciparum–infected patients and from cultured parasites is a heme-laden nanoparticle. (A) Negative-stain EM images of purified HRPII from cultured parasites (HRPII) and serum from a healthy control and 2 P. falciparum–infected patients; bar = 100 nm. (B) Western blot for HRPII of purified HRPII and serum samples from A separated by blue-native PAGE. Detection was done using anti-HRPII mAb. (C) Fluorescence detection of heme using Alexa Fluor 405–conjugated hemopexin (HPX405) of blot from B. (D) Size-exculsion chromatography plot of log MW (kD) vs. retention time of apoHRPII (white square), urea-denatured apoHRPII (yellow square), apo recombinant HRPII (rHRPII, blue circle), and apo rHRPII mutant (rHRPIImt, red square). (E) Quantification of protein-bound heme by the pyridine hemochromagen assay. Comparison of HRPII, apoHRPII, recombinant HRPII (rHRPII), heme-saturated rHRPII (rHRPII:heme), rHRPIImt, and heme-saturated rHRIImt (rHRPIImt:heme) is shown. N = 6, from two independent experiments with technical triplicates.
In blood-stage parasites, HRPII is involved in heme detoxification by enhancing the crystallization of hemozoin (25, 26). Heme binding by HRPII has been described for recombinant HRPII (25–27). To determine whether circulating HRPII in our cerebral malaria patients was heme bound, we generated a heme probe (HPX405) by labeling a high heme-affinity protein, hemopexin (HPX), with a terminal amine-reactive Alexa Fluor™ 405 dye. Using HPX405, we observed that the corresponding HRPII bands in patient sera from patients one and two were highly heme bound (Fig. 1C). Additional less intense heme proteins were also detected in samples from uninfected and infected patients.
Native HRPII Is a Multimeric Nanoparticle Containing Large Amounts of Heme.
Noncovalent cofactors were removed from culture-derived HRPII by acid-acetone extraction to generate apoHRPII. The apoHRPII particles were approximately 400 kD when evaluated by size-exclusion chromatography (SEC) (Fig. 1D). An apo recombinant HRPII produced in ClearColi™ BL21 cells (apo rHRPII) also formed 400 kD particles by SEC. Mass estimation of apoHRPII by analytical ultracentrifugation (AUC) was 375 ± 16 kD, the equivalent of 14 HRPII polypeptides (SI Appendix, Fig. S1). An apo recombinant HRPII mutant with a 4 amino acid C terminus truncation (apo rHRPIImt) did not form large particles (Fig. 1D). By AUC, the apo rHRPIImt had a mass distribution of 25 ± 2.1 kD monomers and 80 ± 2.6 kD trimers (SI Appendix, Fig. S1). The culture-derived HRPII was bound to 48 ± 2 hemes per HRPII polypeptide (Fig. 1E) as quantified by the pyridine hemochromagen assay (28). By contrast, rHRPII was bound to 1 ± 1 heme per polypeptide. Incubation of rHRPII with excess heme followed by repurification showed saturation of heme binding at 49 ± 1 hemes per HRPII polypeptide, consistent with previously published titration results (25, 26). Similarly, the rHRPIImt showed saturation of heme binding at 45 ± 3 hemes per HPRII polypeptide.
Multimerization Is Required for Cell Binding, and Heme Loading Is Additionally Required for the Inflammatory Endothelial Response.
We used cultured hCMEC/D3 monolayers to study the effects of HRPII on cerebral vasculature (29). Using a modified enzyme-linked immunosorbent assay (ELISA), we found that parasite culture purified HRPII nanoparticles (denoted as HRPII:heme, >90% saturation) bound much more efficiently to hCMEC/D3 cells than apoHRPII, the heme-loaded nonparticle rHRPIImt (rHRPIImt:heme), and apoHRPIImt (Fig. 2A). For a quantifiable and real-time readout of the hCMEC/D3 inflammasome response to HRPII, we generated a Venus fluorescent protein-tagged IL-1β using CRISPR/Cas9 technology. This allowed detection of secreted IL-1β in the media. Our assay correlated well with traditional assays for endothelial activation and loss of endothelial barrier integrity by transendothelial electrical resistance (TEER) measurement and dextran permeability measurement and IL-1β release by ELISA (SI Appendix, Fig. S2). Treatment with HRPII:heme but not apoHRPII, rHRPIImt:heme, or aporHRPIImt led to a measurable increase in IL-1β-Venus fluorescence signal by 6 h posttreatment (Fig. 2B). IL-1β release was observed starting at HRPII:heme concentrations of 2 nM (SI Appendix, Fig. S3). We used 20 nM (540 ng of protein/mL) for subsequent experiments because this concentration yielded consistent peak levels of IL-1β. This is similar to reported levels of plasma HRPII (median 1,172 ng/mL) in patients with MRI-confirmed cerebral malaria, even though tissue culture models do not reflect the full complexity of serum–endothelium interactions.
Fig. 2.

HRPII oligomerization and heme loading are required for binding, caveolin-mediated internalization, and activation of hCMEC/D3 cells. (A and B) Comparison of 20 nM (by HPRII polypeptide) heme-saturated HRPII (HRPII:heme), apoHRPII, mutant HRPII with heme (rHRPIImt:heme), and heme-depleted mutant HRPII (apoHRPIImt). (A) Binding to hCMEC cell surface quantified by ELISA. (B) Quantification of IL-1β-Venus after 6 h of treatment. (C and D) Comparison of HRPII:heme to HRPII ± metallo-protoporphyrins (ZnPP and GaPP) and apoHRPII with free zinc. (C) Binding to hCMEC cell surface quantified by ELISA. (D) Secreted IL-1β-Venus was measured after 6 h of treatment and compared to treatment with heme alone (heme, 1 μM). (E) Secreted IL-1β-Venus after 6 h of treatment for HRPII:heme with 1 mM MβCD or caveolin 1 knockout line (CAV1 KO). (F) Comparison of HRPII:heme and apoHRPII binding to hCMEC cells vs. CAV1 KO quantified by ELISA. For all panels, N = 9 from three independent experiments and technical triplicates. ** indicates P < 0.01, *** indicates P < 0.001, **** indicates P < 0.0001, and ns indicates not significant by the t test for panels A, B, and F or ANOVA for panels C–E.
To investigate the role of heme, HRPII was loaded with different metallo-protoporphyrins. HRPII was able to bind equivalent amounts of gallium protoporphyrin IX (GaPP) and zinc protoporphyrin IX (ZnPP) compared to heme (SI Appendix, Table S3). HRPII loaded with GaPP retained the ability to bind to the cells (Fig. 2C) and be internalized (SI Appendix, Fig. S4) but was unable to induce IL-1β release (Fig. 2D). Interestingly, HRPII loaded with ZnPP did not bind hCMEC/D3 cells well and did not cause inflammasome activation. While heme, ZnPP, and GaPP are all metallo-protoporphyrins, Fe3+ and Ga3+ have more similar atomic radii and protein coordination preferences, so GaPP substitutions more closely mimic the hemoprotein compared to ZnPP substitution (30). The addition of zinc as zinc chloride as opposed to ZnPP also mediated apoHRPII binding to the cells, likely through a charge-mediated effect (Fig. 2C). Zinc cations are known to enhance binding of HRPII to negatively charged surface glycosaminoglycans (15, 16). There was no IL-1β release, however (Fig. 2D). Thus, binding is necessary but not sufficient for inflammasome activation, which also requires a heme cargo. High heme (60 μM) has been previously shown to induce cell death (31). However, in our assay, addition of 1 μM heme in the form of hemin (the equivalent amount of heme delivered by 20 nM HRPII:heme) had no effect on the cells as measured by IL-1β release (Fig. 2D).
IL-1β release by HRPII:heme was inhibited by the caveolae-dependent endocytosis inhibitor methyl-β-cyclodextrin (MβCD) and was blocked by CRISPR/Cas9 knockout of caveolin 1 (CAV1) in the hCMEC/D3-IL1β-Venus background (Fig. 2E). Interestingly, loss of CAV1 also resulted in decreased HRPII:heme binding to hCMEC/D3 (Fig. 2F). These data suggest the importance of caveolae-mediated internalization for inflammasome activation.
HRPII:Heme Is Sufficient to Cause Vascular Leakage in Mice.
Mice were injected in the retro-orbital venous sinus with HRPII:heme, apoHRPII, or controls, and 4 kD dextran fluorescein extravasation into brain tissues was measured. Consistent with our tissue culture model, HRPII:heme treatment caused increased dextran leakage into mice brain tissue (Fig. 3A). This was also visualized using confocal microscopy of tissue sections. HRPII:heme treatment resulted in increased fluorescence around blood vessels (Fig. 3B). Recombinant HRPII-induced vascular leakage has been previously shown in mouse brain and intestines (16, 19, 20). An important distinction with our experiments is that vascular leakage was seen with 20 μg of HRPII:heme compared to 400 μg recombinant HRPII in mouse cerebral tissues in experiments by Pal et al (20) and 250 to 500 μg recombinant HRPII in mouse intestinal tissues in experiments by Dinarvand et al (16). As previously shown, recombinant HRPII is heme-deficient (Fig. 1E). Recombinant HRPII potency was increased by the addition of polyphosphate (16) which notably was present in Escherichia coli–produced HRPII but not parasite-produced HRPII (SI Appendix, Table S2). These experiments support our data that the delivery of heme by parasite-produced HRPII is a key driver of acute endothelial barrier disruption.
Fig. 3.

Heme loading was required for HRPII-mediated vascular leakage in mice. Mice were treated with 20 μg apoHRPII and HRPII:heme to assess cerebral vascular leakage. (A) Quantification of fluorescence in brain tissue homogenates (Ex 488, Em 520). N = 6; data are shown for two independent experiments with three mice per group. **** represents P < 0.0001, and ns represents not significant by ANOVA. (B) Fluorescent images of dextran fluorescein extravasation from representative images at 40× magnification.
Endosome Acidification Releases Labile Heme from Intracellular HRPII.
Based on our initial HRPII characterization and requirement for CAV1, we posited that endocytosis of HRPII:heme and acidification could allow for release of labile hemes from HRPII. Indeed, we found that treatment of cells with bafilomycin A, which inhibits endosome acidification, blocked HRPII:heme-induced IL-1β release (Fig. 4A). Decreased heme binding with low pH was previously shown with circular dichroism studies (25). We quantified the heme saturation of HRPII at different pHs and found that two-thirds of hemes were pH labile (Fig. 4B). At physiologic pH (7.4), HRPII was bound to 47 ± 2 hemes. After equilibration at pH 5.5 and repurification by desalting column, only 18 ± 2 hemes were bound per HPRII polypeptide. We generated HRPII loaded with different ratios of heme to polypeptide to expose cells to a gradient of HRPII and heme concentrations. We found that the ratio of heme to polypeptide was critical for inducing IL-1β release rather than absolute concentration of HRPII or heme (Fig. 4C). A threshold of about 30 hemes per HRPII polypeptide, corresponding to the number of pH-labile hemes, was required for activity. Interestingly, HRPII that is loaded with 20 GaPP per polypeptide under acidic conditions followed by 20 hemes at physiologic pH was able to activate cells but not HRPII loaded with 20 hemes under acidic conditions followed by 20 GaPP at physiologic pH (Fig. 4D). These data underscored the importance of release of acid-labile hemes for endothelial inflammasome activation.
Fig. 4.

Degradation of acid-labile heme released from HRPII:heme causes endothelial activation. (A) Quantification of secreted IL-1β-Venus after 6 h of treatment with 20 nM HRPII:heme and 100 nM bafilomycin A (BafA1) or dimethyl sulfoxide (DMSO) control. (B) Quantification of protein-bound heme after equilibration at different pH followed by protein repurification. Data for two independent experiments with technical triplicates are shown. (C) Quantification of secreted IL-1β-Venus after 6 h of treatment with 20 nM HRPII loaded with different ratios of heme (white bars) or 1 μM heme bound to different concentrations of HRPII (gray bars) to yield given ratio. (D) Quantification of secreted IL-1β-Venus after 6 h for cells treated with 20 nM HRPII loaded with 40 hemes per polypeptide (40 Heme), 20 hemes per polypeptide (20 Heme), 40 GaPP per polypeptide (40 GaPP), or mixed hemes and GaPP. HRPII was loaded with 20:1 GaPP followed by 20:1 heme (20 GaPP + 20 Heme) or in reverse with 20:1 heme first followed by 20:1 GaPP (20 Heme + 20 GaPP). (E) IL-1β-Venus secretion was compared between hCMEC/D3, heme oxygenase 1 knockout (HO1 KO) and heme transporter knockout (HRG1 KO) cells treated with 20 nM HRPII:heme. (F) ROS was quantified using CellROX deep red reagent of hCMEC/D3 cells treated with 20 nM HRPII:heme or 100 μM TBHP (ROS inducer) ± 10 mM N-acetylcysteine (NAC) and cells treated with 20 nM HRPII loaded with HRPII:GaPP. For panels A and C–F, N = 9 from three independent experiments and technical triplicates. * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0. 001, and ns indicates no significance.
Heme Oxygenase 1 and Heme Degradation Products Are Key to HRPII-Mediated Endothelial Activation.
Central to the sensing and handling of intracellular heme is heme oxygenase 1 (HO-1), a cytosolic enzyme that converts heme to biliverdin, carbon monoxide, and ferric iron. HO-1 usually protects cells from heme toxicity although proinflammatory effects due to iron toxicity have been reported (32, 33). We found that knockout of HO-1 decreased cell susceptibility to HRPII:heme (Fig. 4E). As shown above, heme is released from HRPII in the endosome but HO-1 resides in the cytoplasm. We found that knockout of HRG1, a heme transporter expressed on endolysosomes, also decreased cell susceptibility to HRPII:heme (Fig. 4E).
The ability of HRPII:heme but not HRPII:GaPP to activate endothelial cells in an HO-1-dependent manner suggested a mechanism. Unlike Fe3+, Ga3+ is redox inert. Iron species released from heme by HO-1 participate in Fenton reactions, generating reactive oxygen species (ROS) (34). Using an ROS dye, we found that HRPII:heme-treated cells had increased ROS at 6 h after treatment with HRPII:heme (Fig. 4F). Furthermore, pretreatment of the cells with the high concentrations of the antioxidant N-acetylcysteine blunted the amount of ROS generated and effects of HRPII:heme. These data suggest that HO-1 releases free iron which increases ROS, generating a proinflammatory response to HRPII:heme internalization.
Drug Inhibition of the NLRP3 Inflammasome Pathway and Heme-Mediated Inflammation Is Protective.
It was previously shown that heme-mediated endothelial inflammation can occur via the NLRP3 inflammasome (35). We generated a series of CRISPR knockouts in the hCMEC/D3-IL-1β-Venus cell line (Fig. 5A). Knockout of NLRP3 or caspase 1, but surprisingly not the apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), abrogated the IL-1β response to HRPII:heme. There was no obvious induction or condensation of ASC protein when cells were treated with HRPII:heme, unlike for TNF (Fig. 5B). We investigated other potential HRPII-sensing proteins through a series of CRISPR knockouts and found that RAGE, MD2, and TLRs-3/7/8/9 were not required for HRPII:heme endothelial activation (SI Appendix, Fig. S5).
Fig. 5.

Requirement of inflammasome components and potential anti-HRPII therapeutics. (A) CRISPR/Cas9-generated knockouts of NLRP3, caspase 1, IL-1 receptor, and ASC were treated with 20 nM HRPII:heme, and secreted IL-1β was measured at 6 h. (B) Immunofluorescent staining for ASC of untreated hCMEC/D3 cells compared to 6-h treatment with 100 pg TNF and 20 nM HRPII:heme. HCMEC/D3 cells were cotreated with 20 nM HRPII:heme and IL-1RA (100 ug/mL), MCC950 (20 nM), or VX-765 (1 μM). (C) Quantification of IL-1β-Venus secreted at 6 h. (D) Blue dextran flux across transwell cultured monolayers at 24 h. (E) HCMEC/D3 transwell monolayers were treated with 20 nM HRPII:heme and 1 μM hemopexin (HPX), 10 mM NAC, 20 μM deferoxamine (DFOA), or 1 μM ZnPP, and blue dextran flux was measured at 24 h. For panels A and C–E, N = 9 from three independent experiments and technical triplicates. * represents P < 0.05, ** represents P < 0.01, **** represents P < 0.0001, and ns represents not significant by ANOVA.
We tested drug inhibitors that can potentially inhibit steps of HRPII:heme-mediated endothelial inflammation. Inhibition of inflammasome components with MCC950 (a selective NLRP3 inhibitor), VX765, (a selective caspase 1 inhibitor), and IL-1 receptor antagonist (IL1RA) also decreased IL-1β release at 6 h and leakage of blue dextran across the cell monolayer at 24 h (Fig. 5 C and D). More specific to HRPII:heme, sequestration of heme to “de-arm” HRPII with the high-affinity hemoprotein HPX (Kd < 1 pM) decreased endothelial barrier integrity disruption as measured by leakage of blue dextran across the cell monolayer at 24 h (Fig. 5E). Iron toxicity mitigation with N-acetylcysteine to reduce ROS, deferoxamine (DFOA) to chelate free iron, and ZnPP to inhibit HO-1 also blunted the effects of HRPII:heme (Fig. 5E).
Discussion
Pathogenicity of HRPII in promoting endothelial inflammation has been proposed to occur directly with purified protein (19, 20) and also indirectly through the inhibition of antithrombin (15, 20). Herein, we show that the direct effects of HRPII in promoting cerebral endothelial inflammation are due to the delivery of an overwhelming heme load to the cells and consequent toxicity of iron and ROS from heme breakdown products (Fig. 6). HO-1 is usually cytoprotective with low concentrations of carbon monoxide and biliverdin being anti-inflammatory (36, 37). However, at high concentrations, the products of heme breakdown by HO-1, notably carbon monoxide and iron, are toxic and proinflammatory (36, 38). Field studies have linked HO-1 polymorphisms resulting in increased HO-1 expression with more severe malaria (39, 40). In our model, HO-1 serves a proinflammatory role by generating excess iron from the heme-laden HRPII particles. Each HRPII polypeptide carries up to 50 hemes, so an HPRII particle composed of approximately 14 polypeptides has potential to carry 700 hemes, which appears to be more than the heme catabolic pathway can detoxify (Fig. 1). HRPII is likely loaded with heme in the host RBC where the labile heme concentration is approximately 20 mM (41) and in the bloodstream, which can have high heme levels due to hemolysis during severe P. falciparum infection (42). In acute severe illness, HRPII:heme circulates at high concentrations, promoting endothelial inflammation, and consequently, cerebral edema. Children with cerebral malaria continue to have elevated signs of endothelial activation up to 28 d after treatment which may be due in part to the persistence of HRPII which can remain detectable even weeks after treatment (43–45). To better understand the role of HRPII:heme in severe disease, clinical studies tracking disease severity and HRPII:heme measurements are needed.
Fig. 6.
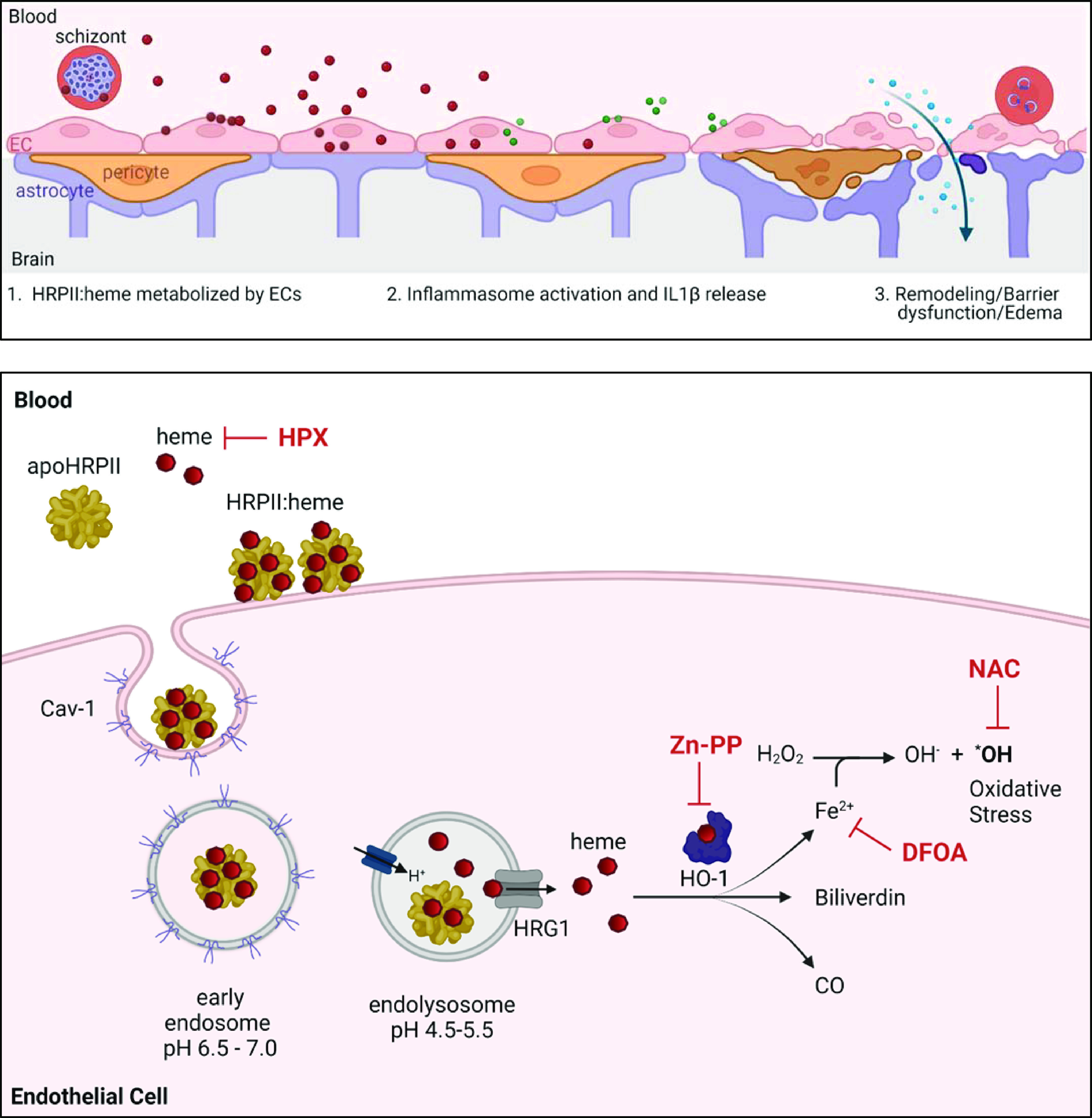
Model of heme-driven endothelial activation by HRPII:heme. (Top) Blood–brain barrier. HRPII particles (red) deliver heme load to endothelial cells (pink). Subsequent metabolization of heme and oxidative stress lead to inflammasome activation and IL-1β signaling cascade (green). Remodeling of tight junction proteins results in barrier dysfunction, increased flux, and edema (19). (Bottom) HRPII:heme is internalized via caveolin 1. Upon endosome acidification, acid-labile hemes are released from HRPII and reach the cytoplasm via heme transporter HRG1. Heme is metabolized by HO-1 to generate iron, CO, and biliverdin. Iron reactivity leads to oxidative stress. This process is blocked by heme sequestration with hemopexin (HPX), HO-1 inhibition by ZnPP, iron sequestration by deferoxamine (DFOA), and oxidative stress mitigation by N-acetylcysteine (NAC). Images were created using BioRender.com and a publication license was obtained.
Previous studies showed considerably more effect with rHRPII than with apoHRPII in our assays. Results with rHRPII in our hands varied depending on the prep used. We have found that E. coli HRPII has varying amounts of polyphosphate (SI Appendix, Table S2), a mediator of endothelial activation (16). In the current experiments, we removed polyphosphate from E. coli–produced HRPII by acid–acetone extraction to generate apoHRPII. We also found that parasite-produced HRPII does not contain measurable polyphosphate but is instead loaded with heme. We found a striking correlation between the degree of heme saturation needed for activity and the heme-binding capacity at acidic pH (between 20 and 30 hemes per polypeptide). This suggests that the final 20 to 30 bound hemes are acid labile. Given that CAV1 and endosome acidification are required for HRPII-mediated effects, we suspect that upon endosome acidification, the HRPII histidines, which likely coordinate heme binding (27), are protonated, resulting in a quantity of heme dissociating from the particle. There are likely two distinct types of heme binding sites, rather than a mechanism such as μ-oxo heme dimer binding and dissociation, based on circular dichroism studies that showed no significant protein structure change below pH 6, reflecting dependence on histidine salt bridges (25). It is this labile heme that is then metabolized by HO-1, increasing intracellular reactive ferric iron, generating ROS, and triggering the inflammatory cascade. Noniron metalloporphyrins are heme mimetics but do not lead to ROS generation or trigger inflammation even when internalized.
Furthermore, pretreatment with DFOA, an iron chelator, decreased the effects of HRPII:heme. This could potentially explain a conundrum in the malaria field where iron supplementation in children (which boosts heme synthesis) has been linked to increased malaria-related mortality (46) and iron deficiency seems protective from severe malaria (47). In high iron and heme settings, HRPII:heme would be higher and consequently more pathogenic. In patients with iron deficiency, some ZnPP is produced instead of heme, and HRPII may alternatively be loaded with ZnPP (48, 49). Patients with circulating HRPII:ZnPP, based on our model, would have less HRPII-driven endothelial inflammation and potentially less severe disease.
As seen in some other inflammation models, the iron toxicity is due to ROS production and subsequent inflammasome activation (33). An unexpected result was that ASC, which facilitates NLRP3 inflammasome complex assembly, was not required for HRPII-mediated IL-1β release, and increased ASC was not seen by immunofluorescence (Fig. 5). Moreover, our hCMEC/D3 model did not require a separate priming signal for endothelial activation. Heme-driven Il-1β release via noncanonical pathways (40) has been described for other cell types in other inflammatory states such as sickle cell disease (50). Our data suggest that this may be the case for HRPII:heme as well, although additional key mediators such as other caspases and potassium channels have not been elucidated. Future studies of noncanonical heme and iron-driven inflammatory pathways may have implications beyond cerebral malaria.
Armed with the mechanism for HRPII-mediated BBB disruption, we examined potential avenues for clinically mitigating the cerebral effects of severe malaria. Disruption of cerebral vasculature relies on IL-1β. Anakinra, an FDA-approved IL-1 receptor antagonist, has been proposed as having beneficial anti-inflammatory effects in placental malaria (51) and may also have some utility in cerebral malaria. Inflammasome inhibitors are also being investigated to reduce cerebral edema associated with cerebral ischemia–reperfusion injury (52, 53). Blocking binding of HRPII to surface glycosaminoglycans such as caveolar glypicans by using antithrombin has also been suggested as a therapeutic avenue (16). While not specifically designed to treat cerebral malaria, these anti-inflammatory therapeutics may be useful adjuncts to treat the cerebral edema component of severe malaria, improving mortality and long-term neurological sequalae. Unfortunately, during acute malaria infection, broad anti-inflammatory agents may compromise the protective immune response. Using HRPII:heme specific strategies could provide a safer and more targeted intervention. As shown in our cell-based assay, HRPII could be disarmed using heme sequestration with HPX or haptoglobin. Iron chelation therapy has shown promise in some clinical trials (47, 54) but not all (55). These trials did not select for patients with normal to high iron status which may explain the inconsistency of the benefit of adjunctive chelation therapy. Being able to measure HRPII:heme levels could provide a means of determining patients who would benefit from chelation therapy or antioxidative agents.
Now that we have a better understanding of the role of heme and HRPII, it may be possible to develop a more useful prognostic marker for progression to severe malaria. HRPII is commonly used in rapid diagnostic tests. Plasma HRPII levels >1,700 ng/mL, based on pathology studies, have high sensitivity and specificity for cerebral malaria (56), although there is a subset of patients with elevated HRPII levels who do not develop cerebral malaria. Perhaps the HRPII heme saturation is different between those who develop cerebral malaria and those who have non-CM severe malaria. Recently, Watson et al. showed that elevated HRPII and thrombocytopenia (platelet count of ≤ 150,000/μL) has an estimated 74% sensitivity for severe malaria in African children (6). We posit that thrombocytopenia may reflect the degree of endothelial activation from infected RBCs and HRPII:heme. Activated endothelia produce vasoactive factors that promote clotting and consumption of platelets (57). This is exacerbated by direct antithrombin inhibition by HRPII (15). Thus, directly measuring HRPII:heme, such as with a heme-binding protein, or iron status with noninvasive excitation spectroscopy, may help triage which patients are more likely to develop cerebral malaria or benefit from HRPII:heme-targeted adjunctive therapies.
A large area of uncertainty is the emergence of PfHRP2-negative parasites which circumvent currently used HRPII-based rapid diagnostic tests and are of unclear clinical significance (1, 58, 59). HRP2/3-negative parasites have been reported in population surveillance studies from the Peruvian Amazon basin, South Asia, and Africa. Clinical cases of HRP2-negative severe malaria have not been reported alongside these studies. The clinical severity, treatment response, and long-term sequelae of HRP2-negative malaria infection are clinical areas in great need of study. Given the inflammatory effects of HRPII:heme on cerebral vasculature, it is possible that HRP2-negative malaria infections may present with lower rates of cerebral malaria as seen with non-falciparum malaria infections. However, consideration should also be given to the potential protective effects of HRPII sequestration of low levels of heme in acid-stable binding sites. Heme toxicity and HO-1 protective effects have been shown in mouse cerebral malaria models using Plasmodium berghei, which is HRPII negative (60, 61). The fate of the excess free heme during high hemolysis in the absence of HRPII may have implications for severity of disease. We postulate that the severity of disease depends on the balance between the heme scavenging by HRPII, lipoproteins, HPX, and albumin and the ability of different cell types to take up and metabolize heme. Cerebral endothelial cells may be more susceptible because of specific uptake of HRPII:heme and/or because of inability to process heme at the same rate as professional heme scavengers like macrophages.
In conclusion, we have shown the mechanism by which HRPII contributes to BBB disruption in cerebral malaria. The delivery of heme to endothelial cells and the generation of iron and ROS are key to IL-1β secretion and endothelial activation (Fig. 6). This results in IL-1β-driven modulation of junctional proteins leading to BBB disruption (19, 21, 22). While we have shown that HRPII from P. falciparum–infected patients forms large heme-bound particles, additional clinical studies comparing HRPII:heme levels and disease severity are needed to assess the contribution of iron toxicity and to determine which patients might best benefit from HRPII:heme-targeted therapy. In addition to adjunctive therapies for cerebral malaria, further studies into inhibition of heme-mediated endothelial inflammation have implications for treatment of other diseases such as sickle cell disease, intracranial hemorrhage, and sepsis.
Materials and Methods
Reagents.
HPX and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (USA). Lipofectamine 3000 was purchased from Life Technology (Carlsbad, CA). Anti-ASC antibody and gallium (III) protoporphyrin IX were purchased from Santa Cruz (Dallas, TX). RPMI 1640 and Albumax were purchased from Gibco (USA). EGM-2 was purchased from Lonza (Morrisville, NC). Heat-inactivated fetal bovine serum (FBS) was purchased from HyClone (Logan, UT). MCC950, VX765, and ZnPP were purchased from Invitrogen (Waltham, MA). Opti-MEM, SYBR Green PCR master mix, IL1RA, blue dextran, and fluorescein dextran 4 kD were purchased from Thermo Fisher Scientific (Waltham, MA).
Protein Purification.
P. falciparum strain 3D7 parasites were cultured in RBCs (from the St Louis Children’s Hospital blood bank) at 2% hematocrit in RPMI 1640 supplemented with 0.25% (w/v) Albumax, 15 mg/L hypoxanthine, 110 mg/L sodium pyruvate, 2.52 g/L sodium bicarbonate, and 2 g/L glucose as previously described (62) except for increased HEPES at 11.9 g/L. HRPII was purified from spent media of parasite culture at 15 to 20% parasitemia. Albumin was depleted from the media using a HiTrap albumin depletion column (Cytiva Healthcare, Emeryville, CA). HRPII was then purified using nickel agarose beads (Gold Biotechnology, Olivette, MO), eluting with 50 mM HEPES pH 8, 200 mM NaCl, and 600 mM imidazole. The eluate was passed through a desalting column (Cytiva Healthcare) and further purified by SEC using Superdex 200 (Cytiva Healthcare). Samples were stored in Tris-buffered saline (TBS), pH 8, with 20% glycerol at −80 °C.
The coding sequence for a PEXEL-processed HRPII was cloned into pBAD/His and produced by heterologous expression in ClearColi® (RCTech, Tuscon, AZ). Induction was performed with 0.1% arabinose at 16 °C for 18 h. Recombinant HRPII was subsequently purified as previously described via nickel affinity chromatography and SEC (20). The protein samples were confirmed endotoxin-free by the LAL Assay (Lonza Bioscience) and HEK-Blue hTLR4 cell assay (InvivoGen, San Diego, CA). Protein was quantified by absorbance at 280 nm and Pauly’s stain as previously described (63).
Protein Size Determination.
Blue-native PAGE was performed using the NativePAGE kit (Invitrogen). SEC data were collected using an AKTA protein purification system (Cytiva Healthcare) with Superdex 10/300 column (Cytiva Healthcare) using gel filtration standards (Bio-Rad, Hercules, CA) and TBS at pH 7.4. AUC was performed on an Optima XL-A analytical ultracentrifuge using an An60Ti rotor (Beckman Coulter, Pasadena, CA) as described previously (64). For negative-stain electron microscopy, the sample was applied to a copper grid, mixed with 2% uranyl acetate solution, air dried, and examined with a JEOL 1200EX 120 keV microscope (Jeol USA, Peabody, MA).
Cofactor Binding.
Protein heme binding was determined using the pyridine hemochromagen assay as previously described (28). For heme saturation, purified protein was incubated with 100-fold excess heme in TBS, pH 7.5, and then repurified over a desalting column (Cytiva Healthcare). Acid–acetone extraction was used for heme extraction from parasite-produced HRPII and for heme and polyphosphate extraction from recombinant HRPII as previously described (65). ZnPP binding was quantified using fluorescence measurements on a multimodal plate reader (PerkinElmer Envision, Waltham, MA) (66).
Polyphosphate Quantification.
Polyphosphate bound to protein was measured by polyphosphate kinase assay as previously described (Ohtomo 2008).
Patient Samples.
Excess serum samples from patients diagnosed with P. falciparum infection were obtained from the Barnes Jewish Institute of Health clinical laboratory. Samples were deidentified, and an IRB exemption was granted for this study.
Western Blot.
Following blue-native PAGE as above, acrylamide gels were equilibrated in Tris–glycine buffer with 1% sodium dodecyl sulfate. Proteins were transferred by electrophoresis to the polyvinylidene fluoride membrane (Millipore). The PVDF membrane was washed with Tris–glycine and 10% (v/v) methanol to remove traces of Coomassie blue. Membranes were blocked with StartingBlock blocking buffer (Thermo Fisher) and then probed with 1:500 mouse 2G12 anti-HRPII antibody (gift from Diane Taylor, University of Hawaii). Membranes were washed with PBS + 0.1% Tween 20 and then incubated with goat anti-mouse IRDye 800CW 1:10,000 (Licor). Membranes were then washed with PBS + 0.1% Tween 20 and imaged using a Licor Odyssey platform.
Heme Blot.
Human HPX (Fisher) was derivatized with Alexa Fluor 405 succinimidyl ester (Thermo Fisher) in sodium bicarbonate buffer, pH 8.3, and purified by gravity-fed size-exclusion column containing BioGel™ P-30 resin (Thermo Fisher). For heme blotting, samples were separated by blue-native PAGE and transferred to PVDF as described above. Following western blot, membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Fisher). Membranes were blocked with PBS + 3% BSA. Membranes were then probed with HPX–Alexa Fluor 405 (HPX405) at a dilution of 1:200, washed with PBS, and imaged using the ChemiDoc MP Imaging System (Bio-Rad).
Mammalian Cell Culture and Cloning.
hCMEC/D3 cells were maintained in EGM-2 base media supplemented with 5% FBS as previously described (29). For experiments, cells were between passages 15 and 25. For monolayer experiments, cells were transitioned to phenol red-free EBM-2 (Invitrogen) supplemented with 5% FBS. CRISPR/Cas9 knockout constructs were generated by transfection of recombinant TrueCut Cas9 (Invitrogen) and synthesized guide RNAs (GenScript, Piscataway, NJ) as previously described (67). Cells were transitioned to Opti-MEM and transfected at 50 to 80% confluence using Lipofectamine 3000. Clones were obtained by limited dilution, and cells were screened using the GeneArt Genomic Cleavage Detection Kit (Invitrogen). Genetic modifications were confirmed by genomic DNA sequencing (Azenta). Primer and guide RNA sequences are listed in SI Appendix, Table S1.
Immunohistochemistry (IHC).
hCMEC/D3 cells were cultured on collagen-treated coverslips. Following treatment with 20 nM HRPII or controls, coverslips were fixed in ice-cold methanol followed by blocking with TBS + 1% FBS. Cells were then incubated with primary antibodies at 1:200 in blocking buffer, washed with PBS, incubated with secondary Alexa Fluor–conjugated antibodies, and washed again in PBS. Coverslips were affixed to glass slides with ProLong Gold antifade with DAPI (Thermo Fisher) and imaged by confocal microscopy (Carl Zeiss). For mouse albumin IHC, brain tissues were blocked with StartingBlock blocking buffer followed by incubation with 1:300 dilution of anti-mouse serum albumin antibody conjugated to fluorescein isothiocyanate (Alpha Diagnostic International). Tissues were washed with TBS, and then, coverslips were mounted with Vectashield (Vector Laboratories, Newark, CA). Tissue sections were imaged by confocal microscopy on an AxioObserver Z1 (Carl Zeiss, Oberkochen, Germany).
Cell-Binding ELISA.
hCMEC/D3 cells were cultured in 96-well collagen-treated black microplates and incubated with 20 nM HRPII or controls. Cell monolayers were washed with PBS and fixed to microplates with 8% paraformaldehyde. Cells were washed with PBS and blocked with ELISA blocking buffer (Abcam, Cambridge UK). Cells were incubated with 1:300 dilution of anti-HRPII antibody at 4 °C overnight, washed with PBS, then incubated with secondary Alexa Fluor–conjugated antibodies, and washed again with PBS. Total fluorescence was measured using an Envision multimodal plate reader (Perkin Elmer, Waltham, MA).
Endothelial Activation Assays.
IL-1β secretion assays were performed by plating hCMEC/D3-IL-1β-Venus cells to 96-well plates in phenol red-free EBM-2 supplemented with 5% FBS and grown to confluence >80%. After treatment under described conditions, 100 μL of the supernatant was transferred to black 96-well plates, and fluorescence was measured with excitation at 515 nm and emission measured at 530 nm using a Cytation three multimode plate reader with Gen5 software (Biotek, Winooski, VT). For dextran permeability assays, hCMEC/D3 were cultured on 24-well transwell 5-μm pore inserts (Corning, Corning, NY) coated with collagen in phenol red-free EBM-2 supplemented with 5% FBS and 0.5 ng/mL bFGF until confluent. Components of interest (protein and chemical inhibitors) were added to the apical compartment with blue dextran (4 kD) at 1 μM. Flux across the monolayer was measured by sampling 200 μL of media from the basolateral compartment and measuring absorbance 620 nm using an Envision multimodal plate reader (Perkin Elmer). TEER experiments were performed as previously described using hCMEC/D3 cells (68). Monolayers were treated by addition of protein to the apical compartment. Resistance recordings were measured via a chopstick electrode with an EVOM voltmeter (World Precision Instruments).
ROS Quantification.
hCMEC/D3 cells were seeded at 5,000 cells/100 μL/well in black clear bottom 96-well plates. Cells were treated with 20 nM HRPII:heme for 6 h or 100 μM TBHP for 30 min at 37 °C without or without pretreatment with 10 mM N-acetylcysteine for 1 h.
qRT-PCR.
First, total RNA was isolated from cells using the RNEasy kit (Qiagen) with DNase treatment (Qiagen). RNA was converted to cDNA using the SuperScript III first-strand synthesis system (Thermo Fisher). Quantification was performed using SYBR Green PCR Master Mix (Applied Biosystems) on a QuantStudio three Real-Time PCR System (Thermo Fisher).
Mouse Treatment and Fluorescein Quantification.
Seven-week-old C57B6/J mice were obtained from Jackson Laboratories. Mice received retro-orbital injections of protein. After 24 h, dextran fluorescein (4 kD) was injected into the contralateral retro-orbital sinus. One hour later, mice were anesthetized with ketamine/xylazine/acepromazine cocktail. Mice were perfused with ice-cold PBS followed by ice-cold 4% paraformaldehyde in PBS. Brains were collected and fixed in 4% PFA overnight followed by wash with PBS and storage in 0.1% sodium azide. The left hemispheres were homogenized for total fluorescence quantification. The right hemispheres were frozen in Tissue-Tek O.T.C., sectioned by Cryostat, mounted onto glass slides, and covered with Vectashield-mounted coverslips. Slides were imaged by confocal microscopy with excitation at 488 nm (fluorescein), 50 ms exposure (Carl Zeiss).
Statistical Analysis.
Statistical analyses were performed with GraphPad Prism (GraphPad Software Inc, San Diego, CA).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by the NIH grants R01 AI126909 to D.E.G. and T32 AI106688 to S.T.N. We thank Dr. Wandy Beatty [Molecular Microbiology Imaging Facility, Washington University School of Medicine (WUSM)] for electron microscopy and immunofluorescence assistance, the WUSM biochemistry core for analytical ultracentrifugation experiments, the Barnes Jewish Hospital clinical laboratories for patient serum samples, and the Saint Louis Children’s Hospital blood bank for human red blood cells. We especially thank Dr. Sergei Djuranovic (WUSM), Dr. L. David Sibley (WUSM), Dr. Jeffrey P. Henderson (WUSM), Dr. S. Celeste Morley (WUSM), and Vibert Putra (WUSM) for helpful suggestions.
Author contributions
S.T.N., M.D.C., R.S.K., I.R., and D.E.G. designed research; S.T.N., D.D., D.W., M.D.C., Q.W., and I.R. performed research; S.T.N., R.S.K., I.R., and D.E.G. contributed new reagents/analytic tools; S.T.N., D.D., D.W., M.D.C., Q.W., I.R., and D.E.G. analyzed data; and S.T.N., I.R., and D.E.G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: C.C.J., Indiana University School of Medicine; and D.J.S., Johns Hopkins University.
Contributor Information
Ilaria Russo, Email: i.russo@keele.ac.uk.
Daniel E. Goldberg, Email: dgoldberg@wustl.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.World Health Organization, World Malaria Report 2021 (World Health Organization, 2021) (June 12, 2022). [Google Scholar]
- 2.Bangirana P., et al. , Severe malarial anemia is associated with long-term neurocognitive impairment. Clin. Infect. Dis. 59, 336–344 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Idro R., Jenkins N. E., Newton C. R., Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 4, 827–840 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Howard R. J., et al. , Secretion of a malarial histidine-rich protein (Pf HRP II) from plasmodium falciparum-infected erythrocytes. J. Cell Biol. 103, 1269–1277 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parra M. E., Evans C. B., Taylor D. W., Identification of plasmodium falciparum histidine-rich protein 2 in the plasma of humans with malaria. J. Clin. Microbiol. 29, 1629–1634 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watson J. A., et al. , Improving the diagnosis of severe malaria in African children using platelet counts and plasma PfHRP2 concentrations. Sci. Transl. Med. 14, eabn5040 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wellems T. E., Howard R. J., Homologous genes encode two distinct histidine-rich proteins in a cloned isolate of plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 83, 6065–6069 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang H. H., et al. , N-terminal processing of proteins exported by malaria parasites. Mol. Biochem. Parasitol. 160, 107–115 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo I., et al. , Plasmepsin V licenses plasmodium proteins for export into the host erythrocyte. Nature 463, 632–636 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatton M. L., et al. , Pan-plasmodium band sensitivity for plasmodium falciparum detection in combination malaria rapid diagnostic tests and implications for clinical management. Malar. J. 14, 115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendriksen I. C. E., et al. , Defining falciparum-malaria-attributable severe febrile illness in moderate-to-high transmission settings on the basis of plasma PfHRP2 concentration. J. Infect. Dis. 207, 351–361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hendriksen I. C. E., et al. , Diagnosing severe falciparum malaria in parasitaemic african children: A prospective evaluation of plasma PfHRP2 measurement. PLoS Med. 9, e1001297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox L. L., et al. , Histidine-rich protein 2 plasma levels predict progression to cerebral malaria in malawian children with plasmodium falciparum infection. J. Infect. Dis. 208, 500–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahu P. K., et al. , Determinants of brain swelling in pediatric and adult cerebral malaria. JCI Insight 6, e145823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ndonwi M., et al. , Inhibition of antithrombin by plasmodium falciparum histidine-rich protein II. Blood 117, 6347–6354 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinarvand P., Yang L., Biswas I., Giri H., Rezaie A. R., Plasmodium falciparum histidine rich protein HRPII inhibits the anti-inflammatory function of antithrombin. J. Thromb. Haemost. 18, 1473–1483 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armah H., et al. , High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Ann. Trop. Med. Parasitol. 99, 629–647 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Sahu P. K., et al. , Determinants of brain swelling in pediatric and adult cerebral malaria. JCI Insight 6, e145823 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal P., et al. , Plasmodium falciparum histidine-rich protein II compromises brain endothelial barriers and may promote cerebral malaria pathogenesis. mBio 7, e00617–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pal P., et al. , Plasmodium falciparum histidine-rich protein II causes vascular leakage and exacerbates experimental cerebral malaria in mice. PLoS One 12, e0177142 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Argaw A. T., et al. , IL-1β regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 177, 5574–5584 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Israelov H., et al. , Caspase-1 has a critical role in blood-brain barrier injury and its inhibition contributes to multifaceted repair. J. Neuroinflammation 17, 267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallego-Delgado J., et al. , Angiotensin receptors and β-catenin regulate brain endothelial integrity in malaria. J. Clin. Invest. 126, 4016–4029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harbuzariu A., et al. , Neuregulin-1/ErbB4 signaling modulates plasmodium falciparum HRP2-induced damage to brain cortical organoids. iScience 25, 104407 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynn A., Chandra S., Malhotra P., Chauhan V. S., Heme binding and polymerization by plasmodium falciparum histidine rich protein II: Influence of pH on activity and conformation. FEBS Lett. 459, 267–271 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Sullivan D. J., Gluzman L. Y., Goldberg D. E., Plasmodium hemozoin formation mediated by histidine-rich proteins. Science 271, 219–222 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Choi C. Y. H., Cerda J. F., Chu H.-A., Babcock G. T., Marletta M. A., Spectroscopic characterization of the heme-binding sites in Plasmodium falciparum histidine-rich protein 2†. Biochemistry 38, 16916–16924 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Barr I., Guo F., Pyridine hemochromagen assay for determining the concentration of heme in purified protein solutions. Bio. Protoc. 5, e1594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weksler B., Romero I. A., Couraud P.-O., The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 10, 16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolff N., Histidine pKa shifts and changes of tautomeric states induced by the binding of gallium-protoporphyrin IX in the hemophore HasASM. Protein Sci. 11, 757–765 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu M., et al. , Neuregulin-1 attenuates experimental cerebral malaria (ECM) pathogenesis by regulating ErbB4/AKT/STAT3 signaling. J. Neuroinflammation 15, 104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu J.-J., et al. , Heme oxygenase 1: A novel oncogene in multiple gynecological cancers. Int. J. Biol. Sci. 17, 2252–2261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menon A. V., et al. , Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 139, 936–941 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nitti M., et al. , Heme oxygenase 1 in the nervous system: Does it favor neuronal cell survival or induce neurodegeneration? Int. J. Mol. Sci. 19, E2260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erdei J., et al. , Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid. Med. Cell Longev. 2018, 4310816 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gozzelino R., Jeney V., Soares M. P., Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Takahashi T., et al. , Heme oxygenase-1 is an essential cytoprotective component in oxidative tissue injury induced by hemorrhagic shock. J. Clin. Biochem. Nutr. 44, 28–40 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim H. S., Loughran P. A., Rao J., Billiar T. R., Zuckerbraun B. S., Carbon monoxide activates NF-κB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G146–G152 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Kuesap J., Na-Bangchang K., The effect of ABO blood groups, hemoglobinopathy, and heme oxygenase-1 polymorphisms on malaria susceptibility and severity. Korean J. Parasitol. 56, 167–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendonça V. R. R., et al. , Association between the haptoglobin and heme oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infect. Immun. 80, 1445–1454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aich A., Freundlich M., Vekilov P. G., The free heme concentration in healthy human erythrocytes. Blood Cells Mol. Dis. 55, 402–409 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeo T. W., et al. , Relationship of cell-free hemoglobin to impaired endothelial nitric oxide bioavailability and perfusion in severe falciparum malaria. J. Infect. Dis. 200, 1522–1529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moxon C. A., et al. , Persistent endothelial activation and inflammation after plasmodium falciparum Infection in Malawian children. J. Infect. Dis. 209, 610–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biswas S., Tomar D., Rao D. N., Investigation of the kinetics of histidine-rich protein 2 and of the antibody responses to this antigen, in a group of malaria patients from India. Ann. Trop. Med. Parasitol. 99, 553–562 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Swarthout T. D., Counihan H., Senga R. K. K., van den Broek I., Paracheck-Pf® accuracy and recently treated plasmodium falciparum infections: Is there a risk of over-diagnosis? Malar J. 6, 58 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sazawal S., et al. , Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: Community-based, randomised, placebo-controlled trial. Lancet 367, 133–143 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Gwamaka M., et al. , Iron deficiency protects against severe plasmodium falciparum malaria and death in young children. Clin. Infect. Dis. 54, 1137–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmermann M. B., et al. , Serum transferrin receptor and zinc protoporphyrin as indicators of iron status in African children. Am. J. Clin. Nutr. 81, 615–623 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Hiremath G. S., Sullivan D. J., Tripathi A. K., Black R. E., Sazawal S., Effect of plasmodium falciparum parasitemia on erythrocyte zinc protoporphyrin. Clin. Chem. 52, 778–779 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Bolívar B. E., et al. , Noncanonical roles of caspase-4 and caspase-5 in heme-driven IL-1β release and cell death. J. Immunol. 206, 1878–1889 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reis A. S., et al. , Inflammasome activation and IL-1 signaling during placental malaria induce poor pregnancy outcomes. Sci. Adv. 6, eaax6346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franke M., et al. , The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 92, 221–231 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Feng Y.-S., et al. , Inhibition of NLRP3 inflammasome: A prospective target for the treatment of ischemic stroke. Front. Cell. Neurosci. 14, 155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gordeuk V., et al. , Effect of iron chelation therapy on recovery from deep coma in children with cerebral malaria. N. Engl. J. Med. 327, 1473–1477 (1992). [DOI] [PubMed] [Google Scholar]
- 55.Mohanty D., Ghosh K., Pathare A. V., Karnad D., Deferiprone (L1) as an adjuvant therapy for plasmodium falciparum malaria. Indian J. Med. Res. 115, 17–21 (2002). [PubMed] [Google Scholar]
- 56.Seydel K. B., et al. , Plasma concentrations of parasite histidine-rich protein 2 distinguish between retinopathy-positive and retinopathy-negative cerebral malaria in malawian children. J. Infect. Dis. 206, 309–318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang M., Hao H., Leeper N. J., Zhu L., Thrombotic regulation from the endothelial cell perspectives. Arterioscler. Thromb. Vasc. Biol. 38, e90-e95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gatton M. L., et al. , Implications of parasites lacking plasmodium falciparum histidine-rich protein 2 on malaria morbidity and control when rapid diagnostic tests are used for diagnosis. J. Infect. Dis. 215, 1156–1166 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Jejaw Zeleke A., et al. , Plasmodium falciparum histidine-rich protein 2 and 3 genes deletion in global settings (2010–2021): A systematic review and meta-analysis. Malar. J. 21, 26 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M., et al. , Heme mediated STAT3 activation in severe malaria. PLoS One 7, e34280 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pamplona A., et al. , Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 13, 703–710 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Istvan E. S., et al. , Esterase mutation is a mechanism of resistance to antimalarial compounds. Nat. Commun. 8, 14240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sahal D., et al. , Specific and instantaneous one-step chemodetection of histidine-rich proteins by Pauly’s stain. Analyt. Biochem. 308, 405–408 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Singh S. P., Kukshal V., De Bona P., Antony E., Galletto R., The mitochondrial single-stranded DNA binding protein from S. cerevisiae, Rim1, does not form stable homo-tetramers and binds DNA as a dimer of dimers. Nucleic Acids Res. 46, 7193–7205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fronticelli C., Bucci E., Acetone extraction of heme from myoglobin and hemoglobin at acid pH. Biochim. Biophys. Acta 78, 530–531 (1963). [DOI] [PubMed] [Google Scholar]
- 66.Leonard J. J., Yonetani T., Callis J. B., Fluorescence study of hybrid hemoglobins containing free base and zinc protoporphyrin IX. Biochemistry 13, 1460–1464 (1974). [DOI] [PubMed] [Google Scholar]
- 67.Liang X., et al. , Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 208, 44–53 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Daniels B. P., et al. , Immortalized human cerebral microvascular endothelial cells maintain the properties of primary cells in an in vitro model of immune migration across the blood brain barrier. J. Neurosci. Methods 212, 173–179 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.