

Significance
RNA viruses adapt rapidly, sometimes switching hosts when given the opportunity. Many rely on arthropod vectors for transmission. Therefore, vector host switches expand evolutionary pathways for viruses, e.g., via spillover from the ancestral host, or via amplification of communities encountered posthost switch. We show that two closely related honey bee deformed wing virus (DWV) strains, transmitted by ectoparasitic varroa mites, show different patterns of spread. DWV-A was present in varroa’s ancestral host in East Asia and spread worldwide postswitch. By contrast, DWV-B was most likely absent in the original host and was acquired later, replacing the original strain in many regions. DWV illustrates the dynamic nature of viral adaptation and competition, as well as the risks posed by globalization.
Keywords: varroa, single-stranded RNA viruses, apis, phylogeography
Abstract
Novel transmission routes can allow infectious diseases to spread, often with devastating consequences. Ectoparasitic varroa mites vector a diversity of RNA viruses, having switched hosts from the eastern to western honey bees (Apis cerana to Apis mellifera). They provide an opportunity to explore how novel transmission routes shape disease epidemiology. As the principal driver of the spread of deformed wing viruses (mainly DWV-A and DWV-B), varroa infestation has also driven global honey bee health declines. The more virulent DWV-B strain has been replacing the original DWV-A strain in many regions over the past two decades. Yet, how these viruses originated and spread remains poorly understood. Here, we use a phylogeographic analysis based on whole-genome data to reconstruct the origins and demography of DWV spread. We found that, rather than reemerging in western honey bees after varroa switched hosts, as suggested by previous work, DWV-A most likely originated in East Asia and spread in the mid-20th century. It also showed a massive population size expansion following the varroa host switch. By contrast, DWV-B was most likely acquired more recently from a source outside East Asia and appears absent from the original varroa host. These results highlight the dynamic nature of viral adaptation, whereby a vector’s host switch can give rise to competing and increasingly virulent disease pandemics. The evolutionary novelty and rapid global spread of these host–virus interactions, together with observed spillover into other species, illustrate how increasing globalization poses urgent threats to biodiversity and food security.
Increased globalization, trade, and habitat fragmentation bring previously isolated species into contact, which risks increased transmission of diseases and parasites. These new connections caused global pandemics affecting populations of humans, animals, and plants. Many of these outbreaks have been caused by RNA viruses, and studies have highlighted them as primary etiological agents of human disease (1–4). The high adaptive potential of RNA viruses is linked to their high mutation rates, which are often an order of magnitude higher than those of DNA viruses (5) and their host range is frequently limited by their capacity to reach novel host populations (5, 6). Yet, our ability to predict specific viral host shifts and their consequences remains poor.
Many viruses solve the challenges of transmissibility by using vectors to connect potential hosts. As a result, changes in vector ecology, such as range expansions or host shifts, can drive viral pandemics. One model of viral spread involves anthropogenic changes in vector range, allowing viruses to spread to new geographical areas. For example, climate change has led to the poleward expansion of mosquito ranges bringing dengue to new human populations (7). Alternatively, viruses can acquire a novel host, driving their spread. This has happened in the case of the Chikungunya virus, originally found in arboreal mosquitoes feeding on primates in the sub-Saharan Africa, but has switched to urban-dwelling mosquitoes and spread through the human population to other continents (8). While human viruses have been studied extensively, these ecological and evolutionary dynamics are not limited to our species. Globalization has also caused pandemics among nonhuman animals (e.g., rabies) and plants (many diseases of crops) (9, 10). Yet, our ability to follow the dynamics of these pandemics is typically limited by a poor understanding of viral ecology in the original host, if the host is known at all.
Exceptionally, given their importance in world agriculture, there are excellent historical records on the spread of honey bee (genus Apis) parasites and viruses (11, 12). With their rapid generation times and large population sizes, arthropods provide an excellent opportunity to examine how vector host switches affect viral evolution. The eastern honey bee (Apis cerana) and the western honey bee (Apis mellifera) are sister species that diverged ~7 Mya (13). Eastern honey bees are found throughout Asia, while western honey bees naturally range from Europe to Africa but are nowadays distributed all over the world except Antarctica. Western honey bees have larger colony sizes and produce more honey, making them the mainstay of the beekeeping industry worldwide. Western honey bees existed in allopatry from other congenerics, isolating them from many honey bee diseases and parasites. However, that changed in the 19th century when beekeepers took western honey bees to Asia where they encountered eastern honey bees and Varroa mites, which are obligate parasites of honey bee brood. By the mid-20th century, Varroa destructor switched to its novel honey bee host (A. mellifera), followed by Varroa jacobsoni in the early 21st century (11, 14, 15). V. destructor then spread worldwide, transmitting a wide variety of bee viruses and causing global population collapses of western honey bees (12).
The well-documented history of the varroa host switches makes this an excellent system to examine how novel transmission routes affect viral disease emergence, as well as providing insights into the evolution of viral diseases (16). Here, we focused on understanding (1) how host bee species affect the virome of varroa and (2) reconstructing the geographic spread of the two most common and important viruses vectored by varroa—deformed wing virus (DWV) strains A and B that diverged about 200 y ago (17). A previous phylogeographic investigation of DWV-A found that the virus most likely originated in Europe and has increased in frequency after the introduction of varroa mites (18), though the inference was based on less than 10% of the viral genome. The origins of DWV-B remain poorly understood to date, though its spread in honey bees appears to have happened in recent decades (19). While these two strains are similar in sequence and biology, they differ significantly in how they interact with both bees and mites (20, 21). For example, DWV-B, but not DWV-A, replicates in Varroa (22, 23), and is also more virulent to honey bees (24). This study aimed at using full-genome sequences to examine whether these viruses had similar or different phylogeographic histories, evolutionary origins, and population dynamics.
Materials and Methods
Data analysis pipelines and R scripts are available at: https://github.com/nonnohasegawa/varroa-transcriptomics.
Mite Collection, Sequencing, and Bioinformatic Analysis.
Varroa mites were collected from 1989 to 2019 from 43 countries and islands spanning six continents (SI Appendix, Tables S1 and S2, Dataset S1, and Fig. 1A). Mite RNA was extracted from single adult female mites using TRIzol and prepared for sequencing using the previously described methodology (25). Sequencing was performed on an Illumina NovaSeq S2 at 150 cycles with paired-end reads. Raw sequence data were first mapped to V. destructor reference genome Vdes_3.0 [GCF_002443255.1] (26) using Bowtie2 (27). The unmapped reads from the previous step were mapped to a selection of viruses that are known to be associated with varroa mites (21). Using VariantBam (28), duplicates were removed and downsampled to a maximum coverage to 200. A pileup format was created using SAMtools (29), then variants were called using VarScan (30). Indels, which are phylogenetically uninformative, were removed using vcftools (31). Then, viral sequences of each sample were extracted using BCFtools (29). In addition, we quantified viral abundances using Kallisto (32) on raw reads mapped to reference viral genomes using the set of viral genomes from ref. 21.
Fig. 1.
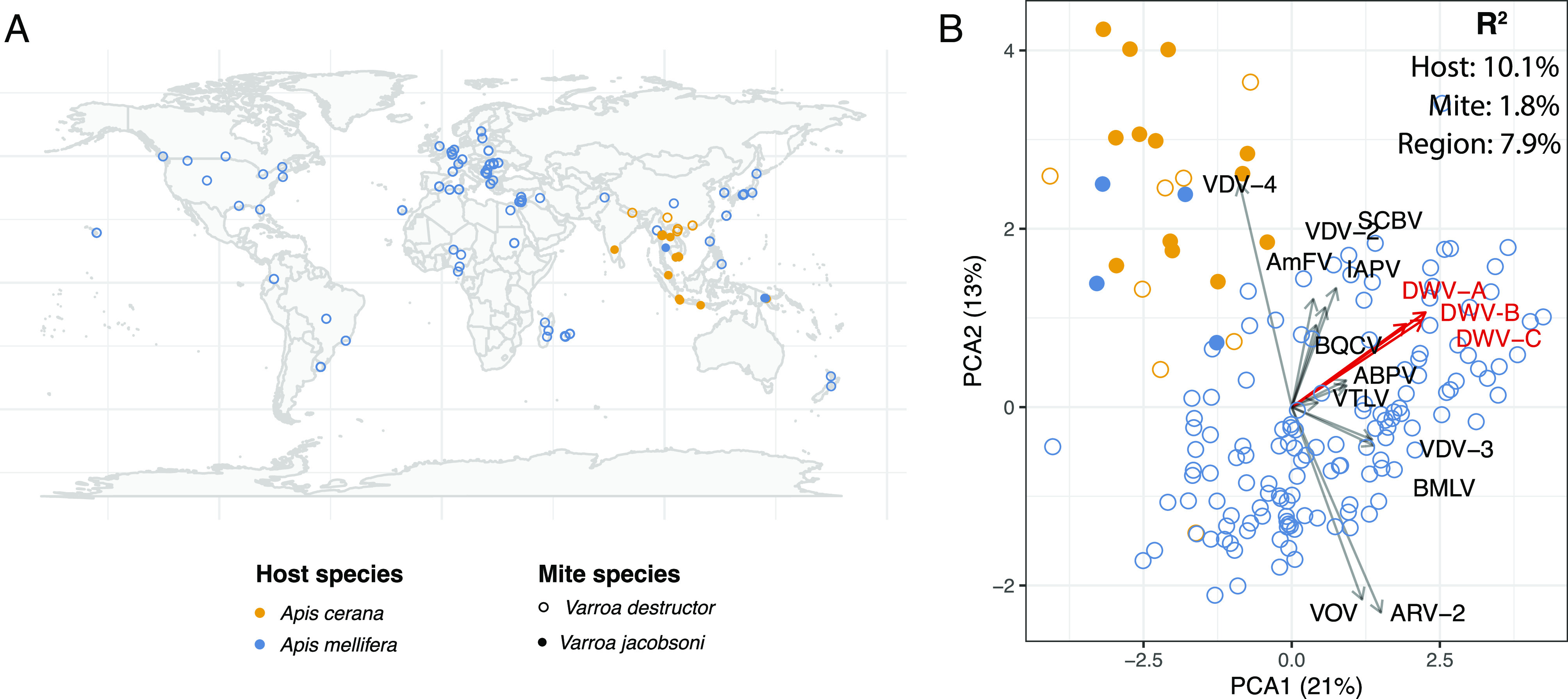
Varroa viromes are most strongly affected by the host bee species. (A) Map of 166 samples used in the virome analysis. (B) The Principal Component Analysis (PCA) plot of varroa virome composition. Arrows represent loadings of each virus onto the principal component axis (i.e., how correlated its titer is with each of the two PCA axes). Loadings show that mites from eastern honeybee have generally lower titers of deformed wing viruses (DWV-A, DWV-B, and DWV-C), as well as most other viruses, except VDV-4, which was more abundant. Note that short-read data will not distinguish between master and recombinant viral types, making loadings of closely related recombining strains, such as the DWV variants, more similar to each other than they are in reality. These community-level data show that the viral landscape inside the mite is most likely shaped by the host bee, either by direct uptake of viruses by mites from bees, or via tripartite coevolutionary relationships. This relationship is quantified by R2 values from an ANOVA of viral abundance distance matrices, suggesting that much of the mite virome were acquired from the host, relative to the effects of the mite itself. However, all effects were significant (P < 0.001), indicating that the virome is shaped by all the three factors. Abbreviations: AmFV–Apis mellifera filamentous virus; ABPV–acute bee paralysis virus; ARV–Apis rhabdovirus; BMLV–Bombyx mori latent virus; BQCV–black queen cell virus; IAPV–Israeli acute paralysis virus; SCBV–sacbrood virus; VTLV–Varroa Tymo-like virus; VOV–Varroa orthomyxovirus.
Host, Mite Species, and Geographic Effects on the Virome.
For the analysis of virome composition, we first filtered out 8 failed libraries. Read counts are compositional data, which means that an increase in one virus abundance in a sequenced sample must lead to a proportional decrease in the abundances of other viruses. To address this issue, we performed an additive log-ratio transform on the count data (33), using the number of reads mapped to the varroa genome as a common denominator. This procedure makes the viral titers independent of one another, so that increasing the titer of one virus does not lead to a corresponding decrease in the others, given a fixed number of reads, i.e., the resulting ratios should be independent of each other. We then used the ratios to perform a principal component analysis and to quantitatively assess the effects of honey bee host species, mite species, and geographic region using analysis of matrix distances analysis using Euclidean distances (34). Statistical analyses were conducted on a total of 166 libraries using the R programming environment using the vegan package (35).
Phylogeographic Analysis.
Consensus sequences extracted from varroa mite transcriptomes were aligned. We excluded genomes with insufficient sequence coverage, less than 90% for DWV-A and 80% for DWV-B. To this dataset, we added complete viral genomes of DWV-A and DWV-B from NCBI and consensus sequences from a published dataset (36), excluding those identified as recombinants by other studies (Dataset S2). While recombinant viruses play increasingly important roles in honey bee pathology (37, 38), their sequences do not follow the tree-like topology and excluding them is generally desirable for clock-based and phylgeographic analyses. To improve alignment quality, all genomes were trimmed to remove the untranslated regions and the polyprotein was codon aligned. All viral genomes were screened for possible recombination events using RDP4 (39). Sequences were considered likely recombinants if most detection methods implemented in RDP4 showed significant evidence for recombination. Overall, we identified one DWV-A sequence (out of 56) and two DWV-B sequences (out of 32) that were likely to be recombinants and excluded them from further analysis that relied upon a tree-like sequence topology.
The phylogenetic history of DWV isolates was reconstructed jointly with their geographic movement using a discrete-trait continuous-time Markov chain phylogeographic model in BEAST v. 2.6 (40, 41). Viral sequences were assumed to evolve under a general time reversible substitution model with gamma-distributed rate heterogeneity across sites. To obtain a time-calibrated tree, we assumed a strict molecular clock with a LogNormal (mean = 0.001; SD = 2.5) prior to the clock rate. For the phylogeographic reconstruction, sampling locations were binned into seven discrete locations (Africa, Asia, Europe, the Mediterranean, North America, Oceania, and South America). Bayesian Stochastic Search Variable Selection was used to identify movement rates between locations significantly higher than zero (41). The geographic origin of each DWV strain was then reconstructed based on the (posterior) ancestral state/location probabilities computed at the node representing the MRCA of each strain. Inferred origins, therefore, take into account uncertainty both in the phylogenetic (tree) and reconstructed ancestral states. We provide the input BEAST XML files for further details on the phylogenetic analysis (Dataset S1).
We also reconstructed the demographic history of each DWV strain using a Bayesian Skyline analysis in BEAST 2 (42). To account for phylogenetic uncertainty, phylogenetic trees were jointly reconstructed along with demographic parameters using the same substitution and clock models as above. Historical changes in effective population sizes (Ne) were reconstructed under a Skyline coalescent tree prior 10 coalescent time intervals, each with a Jeffrey’s prior on Ne.
Results
Each library generated an average of 9,216,119 reads (SD 3,057,629), of which an average of 57.7% (SD 12.8%) mapped to viruses (ranging between 21.7 to 93.3%). We extracted 83 viral genome sequences, including 54 from our study, adding a further 29 reference sequences for phylogenetic analysis (Datasets S1 and S2).
Host Effects on the Virome.
Our dataset consisted of RNA-seq data that captured viruses from two species of mites collected from either original or novel bee hosts (A. cerana and A. mellifera). We used these data to determine the relative importance of host and vector species for the composition of the varroa virome, which showed that the host bee species played a greater role than the species of the mite (Fig. 1). Viromes of V. destructor mites which were collected from A. mellifera host had a higher viral load, notably of DWVs (Fig. 1B). On the contrary, viromes of varroa mites collected from A. cerana hosts exhibited higher association with VDV-4 and not to DWVs. We had far fewer V. jacobsoni samples, but they appeared to show a qualitatively similar trend (Fig. 1B).
Origins of DWVs.
We found that DWV-A and DWV-B diverged from each other 308 y ago (95% HPD 266 to 353 y), predating the varroa host switches. There are other DWV strains, the most common being DWV-C. While we did not encounter enough of this strain to conduct phylogeographic analysis, we could compute its divergence from the other two strains, which was even more ancient, 979 y ago (HPD 836 to 1128). We found strong evidence that DWV-A originated in East Asia (Fig. 2A), and that its population expanded rapidly after the varroa host switch to western honey bees. The expansion occurred around 1970 according to our analysis, consistent with spillover onto a novel host (posterior probability = 0.97). By contrast, while the most ancestral DWV-B samples originate from the Middle East, this origin was less supported (posterior probability = 0.48), and no evidence of an increase in global post-varroa host switch population size could be detected. However, when focusing only on Europe and North America, there was a signal of increased population size around 1970 (Fig. 2B). An East Asian origin for DWV-B is not likely (posterior probability = 0.12).
Fig. 2.
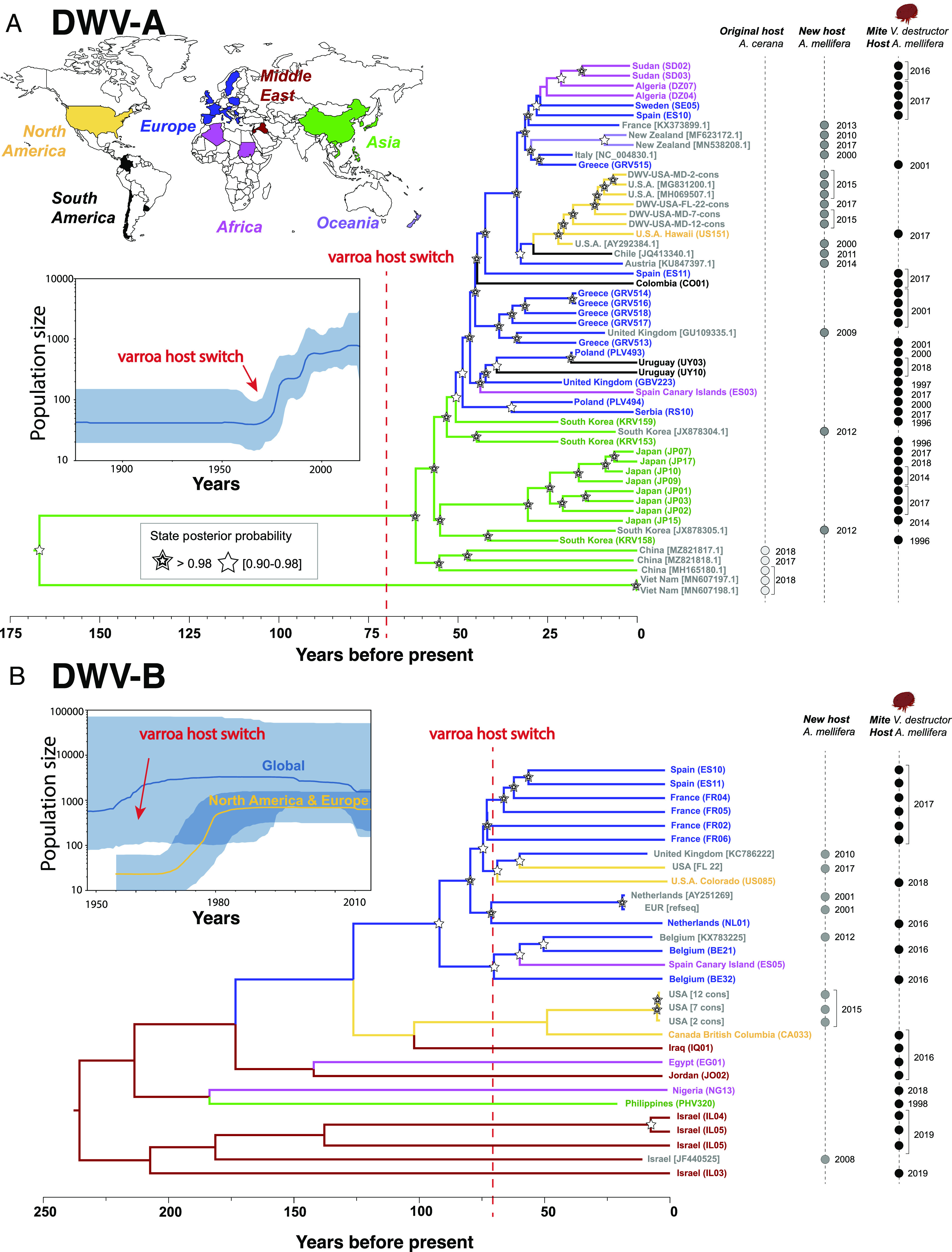
Divergence and phylogeography of deformed wing viruses based on sequences from mites and bees. Locations where samples were collected are color coded. Sample identifiers refer to samples sequenced in our study (Dataset S1) or via their NCBI identifiers (Dataset S2). (A) Divergences between the three main DWV strains predate the host switch by V. destructor by hundreds of years. Phylogeographic reconstruction indicates that DWV-A originated in Asia (posterior probability 0.97). (B) By contrast, the provenance of DWV-B remains unclear with the strongest evidence for a Middle Eastern origin (posterior probability 0.48). Bayesian skyline reconstruction shows a sharp increase in DWV-A populations in the mid-20th century concurrent with varroa’s host switch and subsequent global spread (95% posterior probability shown in blue shading). This suggests that the host switch was mechanistically linked to the demographic expansion of the virus. A similar pattern is seen in the clade of North American and European DWV-B. In contrast, the globally DWV-B population size is more stable, suggesting that transmission dynamics may vary regionally. Also, note the different time scales in the two panels, with most divergences in the DWV-B tree being much deeper than those in DWV-A. These data show that the two closely related viruses most likely have distinctly different geographic and evolutionary origins as well as dynamics of spread, while predominantly relying on the same host and vector.
Discussion
Host availability limits the spread of many RNA viruses, and changes in vector ecology can lead to novel transmission pathways. For example, climate-driven expansion of mosquito ranges leads to larger endemic ranges of many mosquito-borne diseases (7). Alternatively, acquiring a new host and/or vectors can allow a virus to spread globally, as in the case of the Chikungunya virus (8). Mirroring these human vector–borne viruses, we found evidence for both mechanisms in DWV. DWV-A spilled over into western honey bees in association with the varroa host switch. While the exact geographic origin and host of DWV-B remain unclear, several lines of evidence suggest that it was acquired by varroa after its host switch. First, it appears to be absent in eastern honey bees, whereas DWV-A is detected consistently (43–45). Second, its split from DWV-A predates the varroa pandemic by hundreds of years (Fig. 2). Third, epidemiological data suggest a spread that started over the past two decades, long after the varroa host switch that started in the mid-20th century (19, 36, 46). These lines of evidence point to the dynamic interplay between vector and disease ecology, and the potential for continuing viral emergence after a vector host switch.
Virome Analysis.
Taking advantage of viromes collected from different species of mites across different bee hosts, we could ask which factors play a greater role in shaping viral community composition. Most of the differentiation was associated with the bee host rather than the mite species (Fig. 1). These findings are interesting considering recent work suggesting that viruses interact with varroa gene expression in species-specific ways that affect viral titers (21). DWV-A titer was shown to be strongly correlated with changes in mite gene expression, whereas DWV-B titer was not. However, relatively few viruses replicate in varroa, and therefore must be taken up from the bee hosts. Thus, the biology of the host bee likely remains the primary driver of viral community composition.
An important caveat to the virome analysis is that short-read data cannot accurately distinguish recombinant genotypes which are increasing in frequency worldwide (37, 38). They were relatively rare in our phylogeographic dataset, much of which are historic, but are likely more prevalent in more modern collections. A larger database of recombinant sequences could allow for a more detailed study of how recombination affects DWV genomes, which is expected to play a major role in viral adaptation by generating sequence diversity and reducing genetic linkage (47). Additional insights could come from time series data tracking the frequency of recombinant lineages, which will provide additional insights into molecular mechanisms underlying viral competition.
Phylogeographic Analysis.
Phylogeographic analysis suggests that DWV-A was already present in eastern honey bees and spilled over to western honey bees at the same time as the varroa host switch (Fig. 2). These findings differ significantly from earlier work that excluded an Asian origin and suggested that the virus was endemic to western honey bee populations before the arrival of varroa (18). The reason for these discrepancies is not clear, though our study used nearly complete viral genomes instead of fragments, and could pick up stronger signals. One of the signals detected in this study is the explosive growth of DWV-A associated with the origins of varroosis, which supports the link between mite host switch and viral spread (Fig. 2A). It is possible that other DWV strains existed in Europe prior to the arrival of the current East Asian DWV-A strain.
Our findings that varroa drove the spread of DWV-A need to be reconciled with other work that found DWV-A in regions where varroa has not yet been detected. For instance, in Australia (18), where subsequent work has shown DWV to be absent (Roberts et al. 48), or in advance of the invasion front in Hawaii (49). There are several possibilities for reconciling the absence of Varroa with the presence of DWV-A. It is possible that once DWV-A became established in western honey bees post-varroa host switch, it gained access to additional transmission pathways that allowed spread without varroa. Another possibility is that some of these results were caused by contamination; e.g., a subsequent in-depth study of Australian viruses, where libraries were prepared and sequenced in Australia, without the possibility of crosscontamination, failed to find DWV (48). To further illustrate this point, a cursory examination of sequenced negative controls in the NCBI SRA database (search terms negative control AND “A. mellifera”[orgn] in RNA data, 19 January 2023), yielded just two sequencing runs of library blanks (50), and both have DWV contamination present (accession numbers DRR333172 and DRR333191). We recommend that future studies aiming to ascertain the presence of DWV rule out contamination by sequencing negative controls. This is particularly important for studies aiming to detect viral spillover into non-Apis taxa, which are usually prepared and sequenced alongside honey bee samples.
In contrast to DWV-A, we found no evidence that DWV-B population history was affected by the Varroa host switch at a global level. It was first detected in the Netherlands in 2001 (51). The fact that it was detected decades after DWV-A suggests that it was either a virus novel to honey bees or was previously geographically restricted and not detected until later. Thus, it is possible that DWV-B was already present in some western honey bee populations before Varroa but the mites have offered it a new mode of transmission. Alternatively, it may have been acquired by honey bees from another host after the varroa host switch. While bearing in mind the caveat above regarding contamination, DWV-B is found in other taxa (52, 53), and some of them could be potential sources of infection. Yet, there are increases in DWV-B levels in honey bees, particularly in Europe and North America (19, 36, 54), and we see an increase in effective population size in that lineage of DWV-B that is contemporaneous with the spread of varroa in these regions (Fig. 2B). Future work should investigate the origins of DWV-B to ascertain how new viruses can enter the varroa–bee ecosystem. This may cause repeated viral emergence, particularly given the recent arrival of varroa in Australia, which is home to a diverse picornavirus community (48) that could be the source of future varroa-transmitted diseases.
One limitation of phylogeographic studies is that sampling biases can affect outcomes since reconstructions of ancestral states are sensitive to the number of samples from a particular geographic region (55). To minimize the potential impact of sampling bias we attempted to get as broad a range of specimens as possible. In the case of DWV-A, this approach yielded many sequences with good geographic coverage. The number of DWV-B sequences was considerably lower, making it impossible to infer its geographic origins with certainty. However, the reconstructed location of the most ancestral lineages in our dataset appears in the Middle East, and an East Asian origin is unlikely. Additional sampling of DWV-B could resolve this uncertainty as well as potentially provide more details into temporal changes in effective population size.
Our study illustrates the dynamic interaction between virus, vector, and host. Although the original host switch of V. destructor took place in the mid-20th century, (co-)evolutionary dynamics continue in this system with the acquisition of novel viruses and novel vectors (14, 15). Viral fitness depends on successfully exploiting both vectors and hosts, and how viruses evolve to do that remains an open question. For example, DWV-A appears to affect varroa gene expression more than DWV-B (21), suggesting that even relatively closely related viruses may have significantly different ways of interacting with their vector. The dynamism of the bee–varroa–virus system can make it an excellent model for viral emergence and evolution, as well as broadly applicable strategies for viral control.
Supplementary Material
Dataset S01 (GZ)
Dataset S02 (GZ)
Dataset S03 (XLSX)
Acknowledgments
We are grateful to all our generous material providers who supported this work by collecting fresh mites in their apiary or digging in their freezers: Lilia I. De Guzman and Mandy Frake (Brazil and Louisiana, U.S.A); Alison McAfee, Leonard Foster, Heather Higo (British Columbia, Canada); Alina Varaldi and the ROMAPIS beekeepers (Romania); Jevrosima Stevanovic (Serbia); Vivian Wepfer (Switzerland); Peter Rosenkranz (Germany); USDA APHIS (U.S.A); Henriette Rasolofoarivao (Madagascar); Tjeerd Blacquiere (Netherlands); Junichi Takahashi (Japan and New Zealand); and finally the Onna Village Office with the Honey Coral Project (Okinawa). We warmly thank Miyuki Suenaga for technical help with the RNA extraction and library preparation. Library sequencing was performed with the support of the OIST sequencing center. We thank the OIST Scientific Computing and Data Analysis Section for providing HPC to carry out data processing. A.S.M. was supported by a Future Fellowship from the Australian Research Council (FT160100178) and a Kakenhi Grant-in-Aid for Scientific Research from the JSPS (Japan Society for Promotion of Science) (18H02216). M.A.T.’s research was supported by a postdoctoral fellowship from the JSPS (P19723) and Kakenhi Grant-in-Aid (19F19723).
Author contributions
M.A.T. and A.S.M. designed research; N.H., M.A.T., and A.S.M. performed research; M.A.T., N.A., M.S.a.-H., K.A., A.B., K.C., H.D., U.H.D., N.E., M.A.A.e.-N., O.E., J.D.E., N.J.H., B.L., I.M., G.N., D.P., J.M.K.R., P.D.l.R., M.A.S., and Z.S. contributed new reagents/analytic tools; N.H., M.A.T., D.A.R., and A.S.M. analyzed data; and N.H., M.A.T., and A.S.M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Although PNAS asks authors to adhere to United Nations naming conventions for maps (https://www.un.org/geospatial/mapsgeo), our policy is to publish maps as provided by the authors.
Data, Materials, and Software Availability
Raw sequence data are available under DDBJ/NCBI BioProject PRJDB14940 (56).
Supporting Information
References
- 1.Morens D. M., Folkers G. K., Fauci A. S., The challenge of emerging and re-emerging infectious diseases. Nature 430, 242–249 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weitz J. S., et al. , Phage-bacteria infection networks. Trends Microbiol. 21, 82–91 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Binder S., Levitt A. M., Sacks J. J., Hughes J. M., Emerging infectious diseases: Public health issues for the 21st century. Science 284, 1311–1313 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Woolhouse M. E. J., Gowtage-Sequeria S., Host range and emerging and reemerging pathogens. Emerg. Infect. Dis. 11, 1842–1847 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes E. C., The Evolution and Emergence of RNA Viruses (Oxford University Press, 2009). [Google Scholar]
- 6.Woolhouse M. E. J., Adair K., Brierley L., RNA viruses: A case study of the biology of emerging infectious diseases. Microbiol. Spectr. 1, 10.1128/microbiolspec.OH-0001-2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulkarni M. A., Duguay C., Ost K., Charting the evidence for climate change impacts on the global spread of malaria and dengue and adaptive responses: A scoping review of reviews. Global. Health 18, 1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver S. C., Lecuit M., Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 372, 1231–1239 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Jones R. A. C., Disease pandemics and major epidemics arising from new encounters between indigenous viruses and introduced crops. Viruses 12, 1388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher C. R., Streicker D. G., Schnell M. J., The spread and evolution of rabies virus: Conquering new frontiers. Nat. Rev. Microbiol. 16, 241–255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Traynor K. S., et al. , Varroa destructor: A complex parasite, crippling honey bees worldwide. Trends Parasitol. 36, 592–606 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Beaurepaire A., et al. , Diversity and global distribution of viruses of the Western honey bee, Apis mellifera. Insects 11, 239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C., et al. , Population genomics provide insights into the evolution and adaptation of the Eastern honey bee (Apis cerana). Mol. Biol. Evol. 35, 2260–2271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Techer M. A., Roberts J. M. K., Cartwright R. A., Mikheyev A. S., The first steps toward a global pandemic: Reconstructing the demographic history of parasite host switches in its native range. Mol. Ecol. 31, 1358–1374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts J. M. K., Anderson D. L., Tay W. T., Multiple host shifts by the emerging honeybee parasite, Varroa jacobsoni. Mol. Ecol. 24, 2379–2391 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Eliash N., Mikheyev A., Varroa mite evolution: A neglected aspect of worldwide bee collapses? Curr. Opin. Insect Sci. 39, 21–26 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Mordecai G. J., Wilfert L., Martin S. J., Jones I. M., Schroeder D. C., Diversity in a honey bee pathogen: First report of a third master variant of the Deformed Wing Virus quasispecies. ISME J. 10, 1264–1273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilfert L., et al. , Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science 351, 594–597 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Paxton R. J., et al. , Epidemiology of a major honey bee pathogen, deformed wing virus: Potential worldwide replacement of genotype A by genotype B. Int. J. Parasitol. Parasites Wildl. 18, 157–171 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norton A. M., et al. , Adaptation to vector-based transmission in a honeybee virus. J. Anim. Ecol. 90, 2254–2267 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Eliash N., Suenaga M., Mikheyev A. S., Vector-virus interaction affects viral loads and co-occurrence. BMC Biol. 20, 284 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gisder S., Genersch E., Direct evidence for infection of Varroa destructor Mites with the bee-pathogenic deformed wing virus variant B–but not variant A–via fluorescence-in situ-hybridization analysis. J. Virol. 95, e01786-20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posada-Florez F., et al. , Deformed wing virus type A, a major honey bee pathogen, is vectored by the mite Varroa destructor in a non-propagative manner. Sci. Rep. 9, 12445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McMahon D. P., et al. , Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R. Soc. 283, 20160811 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasegawa N., Techer M., Mikheyev A. S., A toolkit for studying Varroa genomics and transcriptomics: Preservation, extraction, and sequencing library preparation. BMC Genomics 22, 54 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Techer M. A., et al. , Divergent evolutionary trajectories following speciation in two ectoparasitic honey bee mites. Commun. Biol. 2, 357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wala J., Zhang C.-Z., Meyerson M., Beroukhim R., VariantBam: Filtering and profiling of next-generational sequencing data using region-specific rules. Bioinformatics 32, 2029–2031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danecek P., et al. , Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koboldt D. C., et al. , VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danecek P., et al. , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bray N. L., Pimentel H., Melsted P., Pachter L., Erratum: Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 888 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Aitchison J., The statistical analysis of compositional data. J. R. Stat. Soc. 44, 139–160 (1982). [Google Scholar]
- 34.McArdle B. H., Anderson M. J., Fitting multivariate models to community data: A comment on distance–based redundancy analysis. Ecology 82, 290–297 (2021), 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2. [DOI] [Google Scholar]
- 35.Dixon P., VEGAN, A package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003). [Google Scholar]
- 36.Ryabov E. V., et al. , Recent spread of Varroa destructor virus-1, a honey bee pathogen, in the United States. Sci. Rep. 7, 17447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore J., et al. , Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. J. Gen. Virol. 92, 156–161 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Ryabov E. V., et al. , A virulent strain of deformed wing virus (DWV) of honeybees (Apis mellifera) prevails after Varroa destructor-mediated, or in vitro transmission. PLoS Pathog. 10, e1004230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin D. P., Murrell B., Golden M., Khoosal A., Muhire B., RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bouckaert R., et al. , BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemey P., Rambaut A., Drummond A. J., Suchard M. A., Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5, e1000520 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drummond A. J., Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Wang S., et al. , Occurrence of multiple honeybee viruses in the ectoparasitic mites Varroa spp. in Apis cerana colonies. J. Invertebr. Pathol. 166, 107225 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Deng Y., et al. , An investigation of honey bee virus prevalence in managed honey bees (Apis mellifera and Apis cerana) undergone colony losses: A case study in China. Res. Square (2020), 10.21203/rs.3.rs-48932/v1. [DOI]
- 45.Yañez O., et al. , Potential for virus transfer between the honey bees Apis mellifera and A. cerana. J. Apic. Res. 54, 179–191 (2015). [Google Scholar]
- 46.Grindrod I., Kevill J. L., Villalobos E. M., Schroeder D. C., Martin S. J., Ten years of deformed wing virus (DWV) in Hawaiian honey bees (Apis mellifera), the dominant DWV-A variant is potentially being replaced by variants with a DWV-B coding sequence. Viruses 13, 969 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pérez-Losada M., Arenas M., Galán J. C., Palero F., González-Candelas F., Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 30, 296–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roberts J. M. K., Anderson D. L., Durr P. A., Metagenomic analysis of Varroa-free Australian honey bees (Apis mellifera) shows a diverse Picornavirales virome. J. Gen. Virol. 99, 818–826 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Martin S. J., et al. , Global honey bee viral landscape altered by a parasitic mite. Science 336, 1304–1306 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Kowallik V., Mikheyev A. S., Honey bee larval and adult microbiome life stages are effectively decoupled with vertical transmission overcoming early life perturbations. MBio 12, e0296621 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ongus J. R., et al. , Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 85, 3747–3755 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Brettell L. E., Schroeder D. C., Martin S. J., RNAseq of deformed wing virus and other honey bee-associated viruses in eight insect taxa with or without Varroa infestation. Viruses 12, 1229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manley R., et al. , Knock-on community impacts of a novel vector: Spillover of emerging DWV-B from Varroa-infested honeybees to wild bumblebees. Ecol. Lett. 22, 1306–1315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kevill J. L., Stainton K. C., Schroeder D. C., Martin S. J., Deformed wing virus variant shift from 2010 to 2016 in managed and feral UK honey bee colonies. Arch. Virol. 166, 2693–2702 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Maio N., Wu C.-H., O’Reilly K. M., Wilson D., New routes to phylogeography: A bayesian structured coalescent approximation. PLoS Genet. 11, e1005421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hasegawa N., et al. , Evolutionarily diverse origins of honey bee deformed wing viruses, National Library of Medicine Bioproject PRJDB14940, https://www.ncbi.nlm.nih.gov/bioproject/923870. Accessed 31 May 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S01 (GZ)
Dataset S02 (GZ)
Dataset S03 (XLSX)
Data Availability Statement
Raw sequence data are available under DDBJ/NCBI BioProject PRJDB14940 (56).