

Abstract
Five tungstopterin-containing oxidoreductases were characterized from the hyperthermophile Pyrococcus furiosus. Each enzyme catalyzes the reversible conversion of one or more aldehydes to the corresponding carboxylic acid, but they have different specificities. The physiological functions of only two of these enzymes are known: one, termed GAPOR, is a glycolytic enzyme that oxidizes glyceraldehyde-3-phosphate, while the other, termed AOR, oxidizes multiple aldehydes generated during peptide fermentation. Two of the enzymes have known structures (AOR and FOR). Herein, we focus on WOR5, the fifth tungstopterin enzyme to be discovered in P. furiosus. Expression of WOR5 was previously shown to be increased during cold shock (growth at 72 °C), although the physiological substrate is not known. To gain insight into WOR5 function, we sought to determine both its structure and identify its intracellular substrate. Crystallization experiments were performed with a concentrated cytoplasmic extract of P. furiosus grown at 72 °C and the structure of WOR5 was deduced from the crystals that were obtained. In contrast to a previous report, WOR5 is heterodimeric containing an additional polyferredoxin-like subunit with four [4Fe–4S] clusters. The active site structure of WOR5 is substantially different from that of AOR and FOR and the significant electron density observed adjacent to the tungsten cofactor of WOR5 was modeled as an aliphatic sulfonate. Biochemical assays and product analysis confirmed that WOR5 is an aliphatic sulfonate ferredoxin oxidoreductase (ASOR). A catalytic mechanism for ASOR is proposed based on the structural information and the potential role of ASOR in the cold-shock response is discussed.
Keywords: Tungstoenzyme, Taurine, Aliphatic sulfonates, Cold response protein, WOR5, Crystal structure, Pyrococcus furiosus
Graphical abstract
Introduction
The hyperthermophile Pyrococcus furiosus is a strictly anaerobic microorganism that grows optimally at 100 °C using carbohydrates or peptides as carbon and energy sources. Of particular metabolic significance and importance to this work is the fact that P. furiosus does not contain any cytochromes and the growth of the organism is strictly dependent on the presence of tungsten [1]. Genomic and biochemical analyses have identified five tungsten-containing oxidoreductase enzymes that are members of the phylogenetically diverse WOR family [2]. Transcriptomic and biochemical data are available on all five of the WOR family enzymes in P. furiosus, although the physiological function for only two of these enzymes has been firmly established. Glyceraldehyde-3-phosphate (GAP) ferredoxin oxidoreductase (GAPOR) is a glycolytic enzyme that replaces the expected GAP dehydrogenase [3]. GAPOR is specific for GAP and oxidizes it to 3-phosphoglycerate (3PG)-generating reduced ferredoxin. This is used by the electron-bifurcating enzyme NADH-dependent ferredoxin NADP+ oxidoreductase I, NfnI, (formally known as ferredoxin:NADP+ oxidoreductase, FNOR), which is thought to be critical to maintaining the redox balance of the cell [4]. In contrast to GAPOR, aldehyde ferredoxin oxidoreductase (AOR) has a broad substrate specificity and uses both aliphatic and aromatic aldehydes that are derived from amino acids [5], consistent with a protective role for AOR from aldehyde damage to the cell during the fermentation of amino acids.
The third member of the WOR family in P. furiosus, formaldehyde ferrodoxin oxidoreductase (FOR), is most active with small chain aliphatic aldehydes with one to three carbon atoms, but its true substrate and function are not known [6]. The physiological functions of the other two tungstoenzymes, WOR4 and WOR5, also remain to be established. WOR4 has been implicated in sulfur metabolism, as its gene expression is threefold higher when P. furiosus is grown in the presence of elemental sulfur [7]. Of the P. furiosus WOR family members, only AOR [8] and FOR [9] have been structurally characterized.
In the case of WOR5, cold-shock treatment of P. furiosus was previously shown to result in an almost a fivefold increase in the expression of wor5 (PF1480) occurring for up to 5 h after the temperature drops from 95 to 72 °C [10]. WOR5 is the most recent enzyme to be characterized in vitro and has some novel features [11], oxidizing many of the same aliphatic aldehydes oxidized by AOR and FOR, and also longer chain aldehydes. In particular, the ability to oxidize hexanal was an order of magnitude faster [11] by comparison to any other aldehyde tested, indicative of a potentially larger, more hydrophobic active site. Purification of WOR5 from P. furiosus biomass revealed a single 67 kDa band on SDS-PAGE, while native PAGE revealed a 135 kDa band that the authors attributed to a homodimer [11]. This was an interesting finding, given that the WOR5 gene is found in a two-gene operon. Specifically, PF1479 and PF1480 code for a polyferredoxin-like protein and WOR5, respectively. Based on the predicted cysteine content, the polyferredoxin could contain as many as four [4Fe–4S] clusters. However, it was not clear if this protein was a subunit of WOR5, as it was not detected by Hagen and coworkers [11], or the polyferredoxin actually served as its physiological electron carrier. Polyferredoxin domains and subunits are part of a superfamily of electron transfer proteins that are commonly found in oxidoreductases [12] and provide critical electron transfer conduits for enzymes involved in central metabolic processes, including integral membrane complexes [13]. Given the small size and fact that many ferredoxins do not stain well during electrophoretic analyses, it is not surprising that the smaller subunit was missed in previous investigations. Indeed, a tungsten-containing member of the WOR family that was recently isolated from a thermophilic bacterium was shown to be a heterodimer consisting of an WOR-like subunit and a polyferredoxin-like subunit [14]. Despite the wide range of aldehyde substrates that WOR5 has been reported to oxidize in vitro, its physiological function, as well as that of the adjacent “polyferredoxin” encoded in the genome, remains an open question.
In this work, we undertook a unique crystallization strategy in an attempt to ascertain the physiological substrate of WOR5 using cytoplasmic extracts of a P. furiosus strain genetically engineered to overproduce affinity 9 × His-tagged WOR5 and its predicted polyferredoxin-like subunit. To capture crystals of intracellular metabolite-bound WOR5, overexpressed WOR5 enzyme was crystallized from the cytoplasmic fraction of a concentrated cell-free extract. Anomalous scattering was observed from the resulting crystals and anomalous difference maps were consistent with a sulfur-containing molecule being bound to the tungsto-bispyranopterin. In addition, the putative polyferredoxin protein encoded by the gene adjacent to that encoding WOR5 is present with the larger catalytic WOR-like subunit showing that WOR5 is heterodimeric. Chemical soaking experiments and biochemical assays were performed to further address the true substrate of WOR5. We provide evidence for a novel function for WOR5 in the metabolism of sulfonated compounds that are abundant in the marine environment inhabited by P. furiosus.
Results
Crystallization and purification of WOR5
Initial attempts to obtain diffraction quality crystals of WOR5 using enzyme purified from P. furiosus biomass failed. With the knowledge that undefined components of the cellular milieu can often stabilize enzymes, we performed an uncontrolled experiment. Specifically, a concentrated cytoplasmic extract from temperature-induced (cold-shock) wild-type P. furiosus cells was screened under strictly anaerobic conditions using the capillary batch method. In addition to the identification of crystallization conditions, if a metabolite from the cytoplasmic extract was captured in the active site, then we would also gain insight into enzyme function. Identification of diffraction quality crystals took 18 months using sparse matrix crystal screening of concentrated extract from wild-type P. furiosus cells grown at 80 °C. Interestingly, when concentrated cytoplasmic extract was obtained from the WOR5 overexpression strain (also grown at 80 °C), reproduction of the crystallization conditions resulted in the appearance of WOR5 crystals in less than 1 month. The diffraction quality of the crystals obtained from the cytoplasmic extract varied from 2.0 to 3.5 Å (Table 1). The crystallization conditions could also be reproduced with purified, his-tagged enzyme and, although optimizations of pH and precipitant were tried, no improvement in diffraction quality was observed. Both crystals produced from cytoplasmic extracts or purified protein incubated with taurine formed thin brown plate crystals with no apparent changes in crystal morphology when bound with ligand. An overexpression strain containing 9x-his-tagged WOR5 with an S-layer protein promoter was used for optimizations and crystal screening using taurine. On average, isolation of WOR5 from the overexpression strain resulted in 5 mg of pure WOR5 from 10 g of wet cell paste (Table S1). These data also represent a substantial (~ tenfold) improvement in yield by comparison to the previously reported isolation of approximately 5 mg WOR5 per 100 g of wet cell paste reported by Bevers et al. [11].
Table 1.
Data collection and refinement statistics
| Sample | Wor5 crystallized from crude extract | Taurine soak |
|---|---|---|
| Data collection | ||
| Beamline | APS 22-ID | APS 22-ID |
| Space group | P21212 | P21212 |
| Wavelength(s) (Å) | 1.74 | 0.98 |
| Resolution range (Å) | 50.0–2.10 | 50.0–1.97 |
| Outer shell (Å) | 2.18–2.10 | 2.04–1.97 |
| Unique observations | 130,184 | 157,481 |
| Completeness (%) | 99.3 (97.4)b | 87.3 (74.2) |
| Rsym (%)c | 5.5 (29.8) | 4.2 (22.2) |
| CC ½ | 99.4 (89.8) | 99.6 (91.5) |
| Redundancy | 6.3 (5.5) | 5.8 (5.4) |
| I/s | 15.5 (1.7) | 15.2 (2.0) |
| Phasing (Fe anomalous)b | ||
| Sites | 18(initial sites) | |
| BAYES-CC | 49.4 ± 15.3 | |
| Figure of merit | 0.49 | |
| Refinement | ||
| Unit cell (a, b, c) in Å | 123.0, 126.8, 141.2 | 125.2, 127.5, 140.7 |
| Protein atoms | 12,479 | 12,520 |
| Solvent atoms | 214 | 793 |
| Resolution limits (Å) | 47.1–2.1 | 38.1–1.97 |
| Rcryst (%) | 17.9 | 17.2 |
| Rfree (%) | 21.4 | 20.3 |
| rmsd bonds (Å) | 0.012 | 0.013 |
| rmsd angles (°) | 1.64 | 1.84 |
| Average B factor (Å2) | 32.5 | 22.0 |
Numbers in parentheses denote values for the outermost resolution shell
Phases were obtained from anomalous scattering of the iron and tungsten ions from data collected at 1.74 Å energy. The asymmetric unit contains 2 tungsten ions and 40 iron atoms
, where Ihkl is the intensity of an individual measurement of the reflection with indices hkl and < Ihkl > is the mean intensity of that reflection
Overall structure of WOR5
In both the crude-crystallized (crystallized from a cytoplasmic extract) and substrate-soaked WOR5 (purified WOR5 soaked with taurine) crystals (Table 1), the asymmetric unit of the WOR5 crystals (space group P21212) consists of two polyferredoxin-like subunits (WOR5-S for small subunit encoded by PF1479, calculated mass of 18.8 kDa) and two WOR-like subunits (WOR5-L for large subunit encoded by PF1480, calculated mass of 64.8 kDa). However, there is substantial evidence that the biological form of WOR5 is in fact a heterodimer that consists of the smaller WOR5-S subunit with four [4Fe–4S] clusters and the larger WOR5-L subunit containing the active site tungsto-bispyranopterin cofactor as well as another [4Fe–4S] cluster (Fig. 1A). In addition to the genetic organization, another line of support for this oligomeric arrangement comes from a PISA analysis of the crystal packing. Specifically, PISA analysis led to the identification of two distinct protein–protein interfaces between the polyferredoxin subunit and WOR-like subunit. However, only one interface is consistent with a viable electron transfer pathway. Specifically, this interface has a buried surface area of 2,079 Å2 and is shown in Fig. 1A. The relative orientation of the metal cofactors within this heterodimer are shown in Fig. 1B. Electron transfer in metalloproteins is well understood [15]; specifically, electron transfer in metalloproteins is optimized when the metal cofactors are between 10 and 12 angstroms apart [16]. Only one heterodimeric arrangement, that is shown in Fig. 1B, minimizes the distance between the [4Fe–4S] cluster in the large WOR-type subunit and the closest [4Fe–4S] cluster within the polyferredoxin subunit to a distance of approximately 11 Å (Fig. 1B). This electron transfer pathway is also consistent with the proposed ferredoxin binding site on FOR reported by Hu et al. [9]. Hagen and colleagues originally identified an open reading frame adjacent to that of WOR5 coding for a 19 kDa protein with 16 cysteines [11]; we have now identified this protein as the smaller subunit of heterodimeric WOR5. The other four WOR family members in P. furiosus, AOR, GAPOR, FOR and WOR4, are all single subunit proteins and, in contrast to WOR5, the genes encoding them do not have adjacent genes encoding polyferredoxin-like proteins.
Fig. 1.
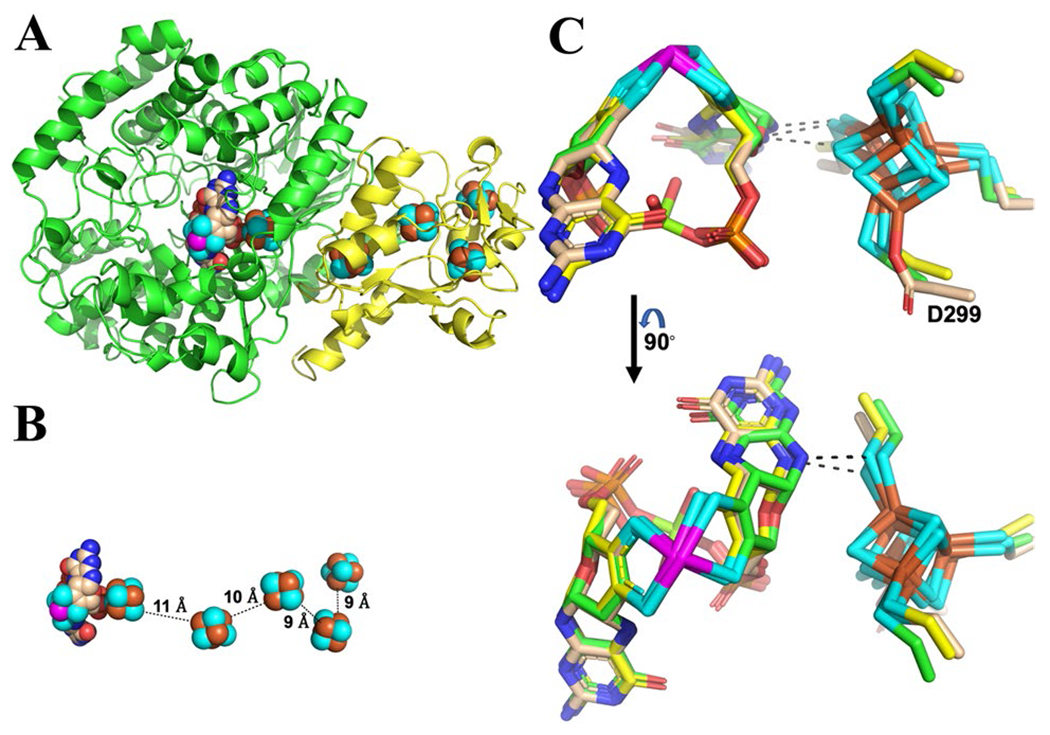
Cartoon representation showing the heterodimeric structure (Panel A) of WOR5, the relative spatial orientation of the cofactors (Panel B) shown as space filling spheres, and a stick representation showing a structural overlay (Panel C) of the tungsto-bispyranopterin and [4Fe–4S] cluster in the catalytic subunit of WOR5 (tan carbon atoms), AOR (yellow carbon atoms), and FOR (green carbon atoms). The buried surface area for the dimer interface between the large subunit (green cartoon) and the polyferredoxin subunit (yellow cartoon) is 2073 Å2. Nitrogen, oxygen, sulfur, phosphorus, magnesium, and tungsten are colored, blue, red, cyan, orange, lime green, and magenta, respectively. The aspartic acid (E299) ligand to [4Fe–4S] is identified and the dashed lines represent the closest contact distance (less then 3.3 Å) between the [4Fe–4S] cluster and the tungsto-bispyranopterin cofactor in all models
The catalytic domain of WOR5 shares several general features with those in AOR and FOR. In particular, the overall fold of the catalytic subunit is conserved, especially near the tungsto-bispyranopterin cofactor and nearby [4Fe–4S] cluster. Structural alignment of the catalytic domain of AOR and FOR with the catalytic domain of WOR5 results in an RMSD for the backbone atoms of 1.5 and 1.1 Å, respectively. However, the [4Fe–4S] cluster within the catalytic domain, immediately adjacent to the tungsto-bispyranopterin, is ligated by all cysteine residues in AOR and FOR. In contrast, the [4Fe–4S] cluster in the catalytic subunit of WOR5 has three cysteine ligands and one aspartic acid ligand (Fig. 1C). Based on previous studies with simple ferredoxins [17], the aspartic acid ligation should shift the midpoint reduction potential of the [4Fe–4S] cluster to a slightly more positive value by comparison to the fully cysteine coordinated cluster. The closest contact between the tungsten cofactor and the [4Fe–4S] cluster is similar in all three tungstoenzymes for which structures are available. Specifically, a sulfur atom from one of the cysteine ligands comes within 3.3 Å of the pterin (Fig. 1C, dashed line), consistent with a role for the [4Fe–4S] cluster in electron transfer to ferredoxin or, in the case of WOR5, to the polyferredoxin-like subunit.
Identification of taurine as a substrate for WOR5
Phases were obtained from the anomalous scattering of the tungsten and iron atoms in WOR5. However, at the iron edge (7112 eV), sulfur atoms also have a measurable anomalous signal that begins to appear at approximately 3–5 s (Supplemental Information, Figure S1). While some care must be taken when interpreting electron density around the tungsten atom due to “ripples” caused by series termination effects [18], it is clear that there is a significant anomalous signal near the tungsten cofactor as evidenced by comparable density around the [4Fe–4S] cluster and the sulfur atoms of nearby methionine residues (Figure S1). The anomalous signal (Df’’) for tungsten at this wavelength is over 15 electrons, while the Df’’ for iron is just over 4 electrons. These data indicate the presence of an additional, unknown, anomalous peak near the tungsten ion (Figure S1, black arrow). Given that these crystals were obtained from a cytoplasmic extract, a reasonable conclusion is that this signal is coming from an atom of an unknown metabolite present in the cytoplasm of this organism. More specifically, this atom is most likely due to a sulfur atom. Figure 2A, shows the Fo-Fc composite omit map contoured at 3 s for the diffraction data obtained from WOR5 crystals grown from crude extract. The best fit to the unknown density was (S)-1-hydroxy-1-butylsulfonate (HBS, Fig. 2A). It is important to point out that this is a complete guess based on a purely qualitative fit to the electron density with a molecule that also positions a sulfur atom to satisfy the anomalous signal. Interestingly, modeling the “R” enantiomer did result in an increase in both R and Rfree by 1%. No bonding restraints from the HBS molecule to the tungsten atom were used during refinement. Regardless, the HBS molecule satisfies the density well with one sulfonate oxygen atom coming within 2.5 Å of the tungsten ion and within 3.0 Å of the side chain of H469 and another sulfonate oxygen is within 2.7 Å of E328.
Fig. 2.
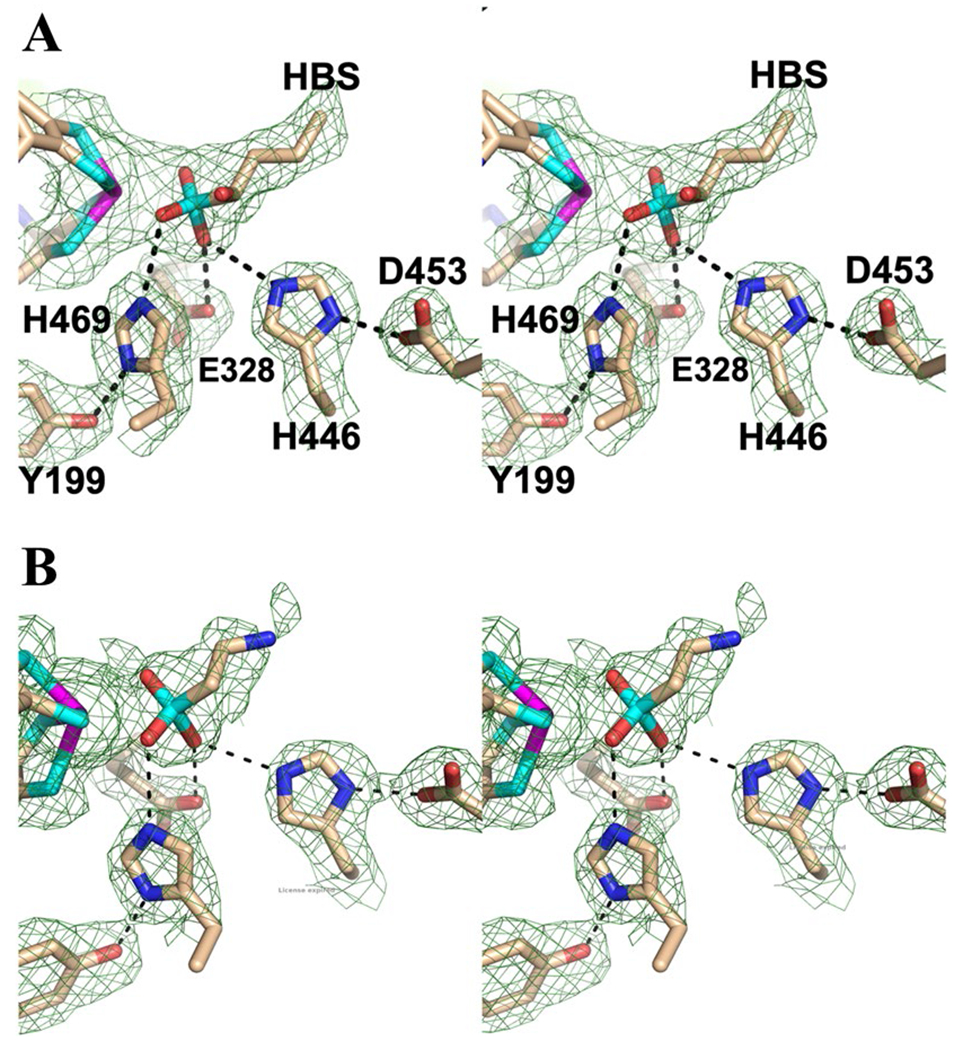
Wall-eyed stereo view showing the models (stick format) and Fo–Fc composite omit density (green cage) for WOR5 crystals obtained crude extract (Panel A) and following treatment with 5 mM taurine (Panel B). The composite omit maps are contoured at 3 s and were generated using the simulated annealing protocol. Carbon, nitrogen, oxygen, sulfur and tungsten atoms are colored tan, blue, red, cyan, and magenta, respectively. Dashed lines represent distances shorter than 3.3 Å
We therefore propose that the physiological substrate of WOR5 is an aliphatic sulfonate and investigated what compounds would be found in the physiological environment of P. furiosus. Interestingly, there are several sulfonated compounds, typically used as cellular osmolytes that are produced in high concentrations (> 1 mM) by marine organisms ranging from phytoplankton to mussels and clams [19]. Among the most prevalent molecules are 2-aminoethanesulfonate, which is also known as taurine, and 2,3-dihydroxylpropane-1-sulfonate (DHPS). In addition, taurine, together with the amino acids glycine and alanine, is common in marine metazoans and can represent up to 40% of the free amino acid pool in their release products [19]. To investigate whether an aliphatic sulfonate such as taurine might be a substrate for WOR5, crystals grown from purified WOR5 were allowed to soak overnight in mother liquor containing 5 mM taurine. As shown in Fig. 2B, distinct changes in the electron density were observed at the active site. Specifically, the electron density became smaller/shorter, consistent with a smaller molecule bound near the tungsten atom. Interestingly, while E328 and H469 are conserved in FOR and AOR, Y199, H446, and D453 are unique to WOR5. In addition to the specific differences mentioned above, the active site of WOR5 is much larger forming a 10 × 15 Å cleft that is almost 10 Å deep. The net result is an active site that is accessible to much larger substrates and considerably more solvent when compared to those of AOR and FOR (Supporting information, Figure S2) suggesting that the WOR5 reaction may be more complex than simply oxidizing an aldehyde to carboxylic acid. The larger active site of WOR5 is also consistent with its ability to oxidize the longer chain aldehyde, hexanal, which is unique among the P. furiosus WOR family [11].
Oxidation of taurine by WOR5
To address the hypothesis that WOR5 could desulfonate and oxidize aliphatic sulfonates represented by taurine, the purified enzyme (Figure S3, Table S1) was incubated with taurine and the artificial electron acceptor benzyl viologen (BV) in place of the putative natural acceptor P. furiosus ferredoxin. For taurine, and known substrates such as propionaldehyde and hexanal, BV reduction proceeded only in the presence of the substrate and WOR5. Based on a least-squares fit to the Michaelis–Menten equation, the Vmax value for taurine oxidation was 14.9 μmoles min−1 mg−1, confirming that WOR5 can utilize this compound as a substrate, with an apparent Kmapp value of 112 ± 4 μM. For comparison, the Vmax values with the aldehydes hexanal and propionaldehyde were 30.0 and 26.2 μmoles min−1 mg−1, with apparent Kmapp values of 230 ± 9 μM and 2,047 ± 67 μM, respectively. The Vmax and Kmapp values for hexanal are similar to those reported previously [11]. The higher affinity for taurine supports the idea that it is a physiological substrate, but its rate of oxidation is notably lower than that measured with the aldehydes. However, as shown in Fig. 3, taurine oxidation is not simply a one-step aldehyde to acid conversion. It involves two steps, an oxidative desulfonation reaction followed by the activation of a second water molecule and oxidation of the resulting aldehyde. This activity has not been reported for any other member of the WOR family and is so far unique to WOR5. For example, we found that purified AOR or FOR from P. furiosus did not catalyze the conversion of taurine to glycine. Specifically, no reduction of BV was observed with AOR or FOR under the same reaction conditions. Additional assays with DHPS (Vmax 14.5 μmoles min−1 mg−1) confirmed that the enzyme can utilize hydroxylated sulfonates analogous to the HBS modeled in the active site of the crystals obtained from the cytoplasmic extract. Moreover, in addition to taurine and DHPS, other sulfonated and phosphonated compounds, including hexyl-sulfonate, hexylphosphonate and aminomethyl-phosphonate, exhibited rates of BV reduction ranging from 1 to 3 μmoles min−1 mg−1.
Fig. 3.
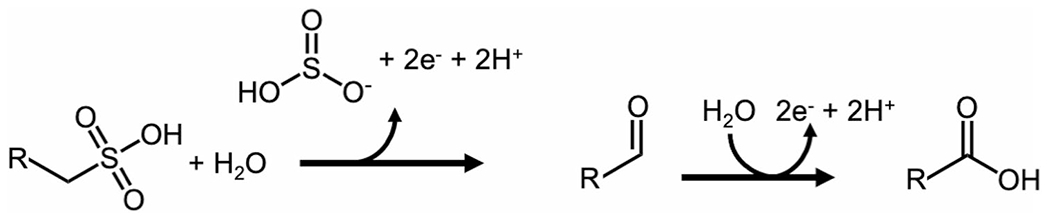
Proposed reaction catalyzed by ASOR. R represents an aliphatic side chain. See text for details
NMR analysis of taurine oxidation by WOR5
We utilized NMR to confirm the proposed oxidation products of taurine by WOR5 using the physiological electron acceptor, ferredoxin, rather than the artificial carrier BV. However this made the experiment particularly challenging because excess ferredoxin must be added to ensure efficient oxidation and accumulation of sufficient product so that it is detectable by NMR. Figure 4 shows the NMR spectra that were acquired from samples taken before and after reaction of taurine with WOR5. In the absence of WOR5, taurine exhibits a doublet of triplets at δH = 3.21 and 3.31 ppm. After reaction with WOR5 and ferredoxin, the taurine resonances almost completely disappear and are replaced by a singlet at δH = 3.54 ppm, a resonance frequency characteristic of glycine. A 1H–13C HSQC spectrum of the reaction product (Fig. 4B) enables correlation of the δH = 3.54 ppm to a δH = 44.13 ppm resonance, and a 1H–13C HMBC spectrum (Fig. 4C) allows correlation of the δH = 3.54 ppm to a δC = 175.2 ppm peak. That these chemical shifts arise from glycine was confirmed a) by comparison to the 1D 1H and 13C spectra obtained from a glycine standard (Supporting Information, Figure S4), and b), by comparison to the chemical shifts compiled in the Biological Magnetic Resonance Database.
Fig. 4.
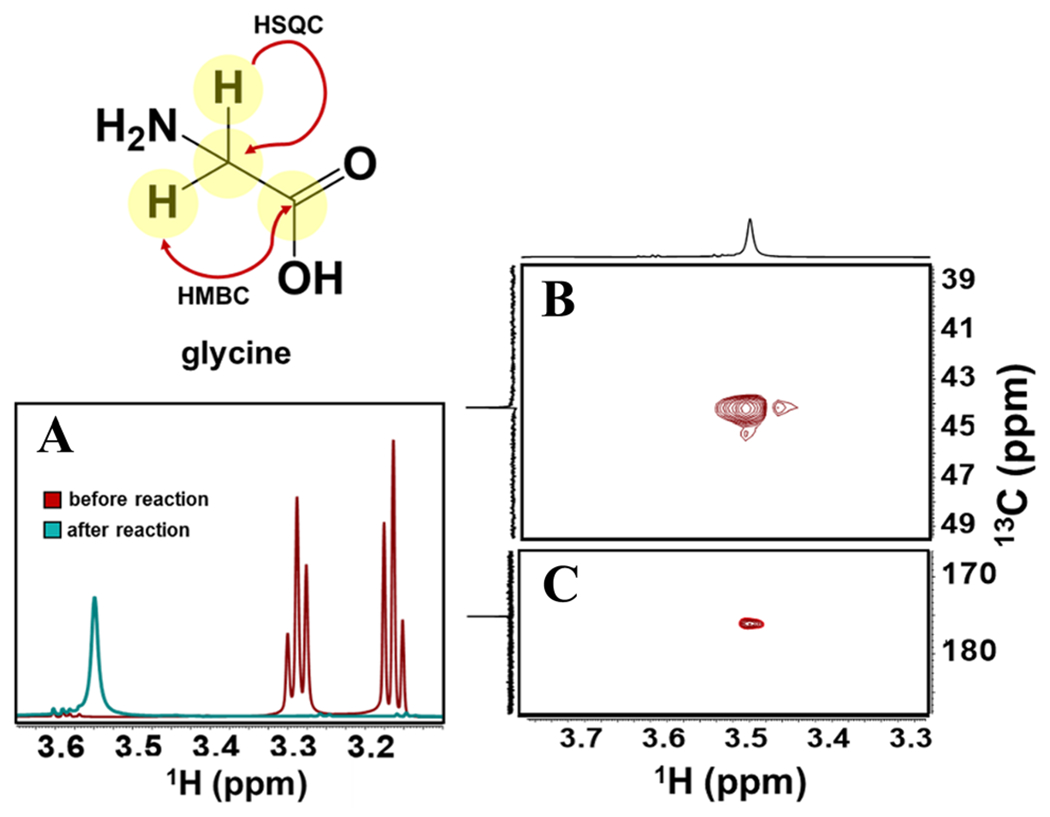
NMR Spectra monitoring the conversion of taurine to glycine. 1H NMR spectra (Panel A) were recorded of samples taken before and after the enzymatic reaction. In addition, 1H–13C HSQC (Panel B) and 1H–13C HMBC spectra of the post-turnover samples were also recorded.
Negative ion mass spectrometry of the WOR5-treated taurine samples further demonstrated the disappearance of taurine and formation of glycine over the course of the reaction (Fig. 5). Finally, we showed that taurine oxidation by WOR5 generated sulfate or sulfite. Specifically, using the barium sulfate turbidimetric assay (Supporting Information, Figure S5), the WOR5 reaction showed an increase in sulfate/sulfite compard to the negative control lacking enzyme. A mechanism for reaction of WOR5 with an aliphatic sulfonate involving desulfonation, liberation of sulfite and oxidation of the aldehyde, is discussed below.
Fig. 5.
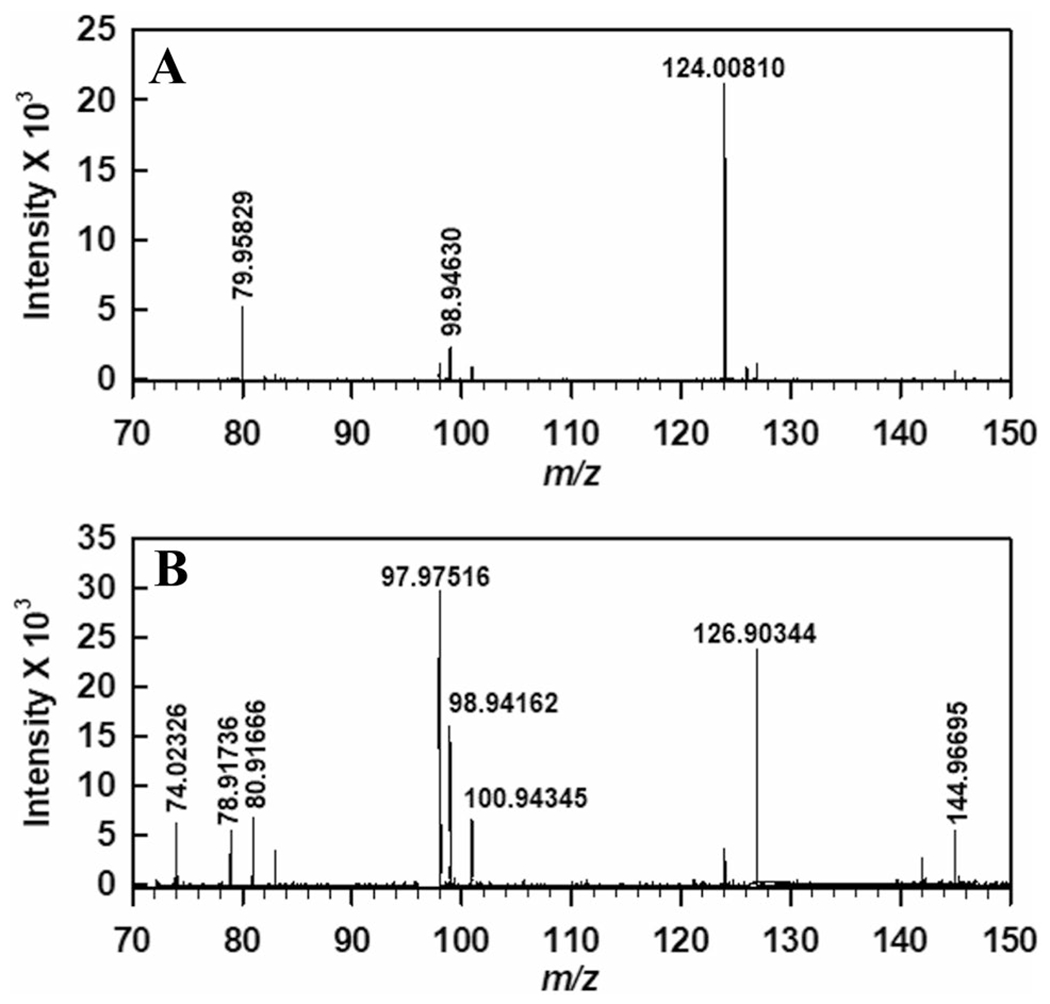
Negative ion mode mass spectra of sample taken A before and B after enzymatic reaction. The m/z 124 corresponds to taurine pre-turnover (Panel A). Following turnover, glycine is observed (m/z 74, Panel B). Sample preparation and experimental parameters are described in “Experimental procedures”
Discussion
This work presents the first demonstration of using crystallography to investigate the physiological substrate of an enzyme. In the case of WOR5, it was clear that an aliphatic hydroxysulfonate was captured in the active site during crystallization of the enzyme present in a cytoplasmic extract, and this provided essential clues that lead us to identify a new enzyme function. Specifically, we demonstrate that WOR5 is an aliphatic sulfonate ferredoxin oxidoreductase (ASOR). In addition, the structure reveals that ASOR is a heterodimeric enzyme with a WOR-like catalytic subunit and a second polyferredoxin-like subunit, as this crystallized from the concentrated cytoplasmic extract of P. furiosus. A significant clue to the physiological role of ASOR may be the observation that its expression is up-regulated as part of a “cold-shock” response of this organism, with mRNA levels increasing almost fivefold during both short (1–2 h) and long (4–5 h) cold (72 °C) adaptation experiments [10]. This is in contrast to three of the other four tungsten-containing enzymes of P. furiosus, AOR, FOR and GAPOR, which also oxidize aldehydes of various types, as the expression of the genes encoding them were not significantly affected by a drop in temperature. Due to the role of osmolytes, in particular, sugar glycerates, sulfonates and phosphonates [20], in the adaptation to lower growth temperatures, we propose that ASOR may be involved in the oxidation of various aliphatic sulfonates and also phosphonates, and that its true suite of physiological substrates has yet to be identified.
The proposed reaction scheme for the oxidation of aliphatic sulfonates catalyzed by ASOR is shown in Fig. 3. Such molecules are abundant in the marine environment inhabited by P. furiosus. Indeed, taurine itself is a key organic osmolyte across all cellular life, playing an important role in stabilizing protein structure in general as well as regulating cellular content in mammalian cells [21]. Recent work elucidated a glycyl radical enzyme which catalyzes C–S bond cleavage in isethionate, a taurine derivative, in commensal gut bacteria [22]. The proposed radical-mediated mechanism involves direct elimination of sulfonate, thereby generating acetaldehyde and sulfite from isethionate [23]. At the present time, there is no evidence for a radical species in any pyranopterin-dependent enzyme, including AOR or FOR. Regardless, the activity we have observed for ASOR is a significant finding that will require a multifaceted approach to identify its physiological substrate and function. The biochemistry and structural data reported here provide a foundation from which to build upon.
Overall structure and active site architecture
The enzymatic oxidation of aldehydes to carboxylic acids by pyranopterin-containing enzymes is well established [8]. Likewise, the mechanism of the pyranopterin-dependent oxidation of sulfite by sulfite oxidase has also been investigated in detail [24, 25]. However, the observation that a single enzyme can catalyze both a desulfonation reaction as well as the oxidation of an aldehyde and do so using a pyranopterin-based active site is unprecedented. As anticipated, there are notable differences in the active site architecture of sulfonate-oxidizing ASOR and aldehyde-oxidizing AOR and FOR, despite structural similarity in the overall fold of the catalytic subunit. The root mean square deviation (RMSD) for the alignment of the backbone carbon atoms in the catalytic subunit of ASOR with AOR and FOR is 1.5 Å and 1.1 Å, respectively. However, when these structural alignments are compared it becomes clear that there are several regions of the AOR and FOR that are extended, resulting in a considerably smaller active site cleft (Supporting Information, Figure S2) for both of these enzymes. In general, ASOR has a substantially larger active site cleft that can accommodate longer molecules. These observations are consistent with the report that ASOR can work on longer chain aldehydes with considerably higher turnover numbers. Domain 1 of ASOR also contains an alpha helix (residues 143–166) that extends out three turns further than the equivalent helix in FOR or AOR. This extension forms a majority of the interface between ASOR and its polyferredoxin-like redox partner (Figure S2).
ASOR and AOR share 58% sequence similarity within Domain 1 (Residues 1–210), 50% similarity in Domain 2 (Residues 211–417), and 52% similarity in Domain 3 (Residues 418–605). Domain 1 in ASOR, forming the characteristic “saddle” base for the tungsto-bispyranopterin, retains most of the same structural features seen in AOR [8]. The internal hydrogen bonding network of the tungsto-bispyranopterin contains two ordered water molecules, with slight asymmetry attributed to K77 (an arginine residue in AOR) hydrogen bonding to a phosphate oxygen. Similar to FOR, Domains 2 and 3 of ASOR lack the mononuclear iron binding site observed in AOR. Domains 2 and 3 are involved in substrate binding and catalysis, although there are no direct protein ligands to the tungsten ion. However, there are only three amino acids in the active site that are conserved across all five WOR-type enzymes in P. furiosus (Figure S2). These are Y327, E328, and H469 (ASOR numbering). Two of these amino acids, E328 and H469, have been proposed to be involved in proton-transfer reactions. The ASOR electron density indicates that the side chain of E328 would be within hydrogen bonding distance of a sulfonate oxygen at ~ 2.5 Å. The H469 N-ε nitrogen is also near another sulfonate oxygen at 2.8 Å (Fig. 6). However, in addition to these conserved active site residues, ASOR contains another group of polar residues that are not found in the other AOR-type enzymes. These residues form a hydrogen bonding network that is not observed in the AOR or FOR structures (Fig. 6). In the taurine-soaked crystals, the N-ε nitrogen of histidine 446 lies close to a substrate sulfonate oxygen, and the H446 N-δ nitrogen is within hydrogen bonding distance of a carboxylate oxygen from D453. Tyrosine 199 is within hydrogen bonding distance of the N-δ nitrogen of H469. The additional hydrogen bonding network in ASOR may explain the novel functionality.
Fig. 6.
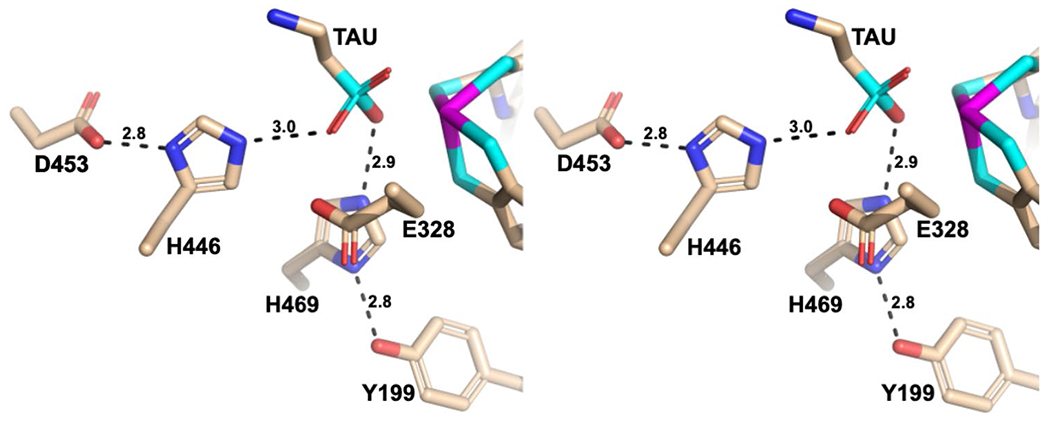
Wall-eyed stereo view highlighting proton-transfer networks that are unique to ASOR in the model for the taurine-soaked crystals. See text for details
Other than ASOR, the only other ligand-bound structure available for any WOR family enzyme in P. furiosus is the glutarate-bound structure of FOR (PDB ID: 1B4N). The carboxylate oxygens of glutarate in the FOR structure are 3.4 and 5.2 Å away. In our models, the proximity of the sulfonate oxygens to the tungsten site may suggest bidentate coordination, which could explain the higher affinity for taurine compared to aldehyde substrates. However, given the ambiguous nature of the electron density, it would be premature to make any specific assignments. Spectroscopic investigations are underway to further elucidate how sulfonated substrates may coordinate or interact with the tungsten metal center as well as the ASOR mechanism and the relevant oxidation states of the tungsten atom. Finally, it should be emphasized that the [4Fe–4S] cluster, adjacent to the tungsto-bispyranopterin cofactor, in the large subunit of ASOR is significantly different in that an aspartic acid is a ligand for one of the iron atoms. This will certainly influence the reduction potential of the [4Fe–4S] cluster and may have implications for the unique chemistry being catalyzed by ASOR.
The proposed mechanism for oxidative desulfonation by ASOR
Undoubtedly, the novel activity we observe for ASOR must be due to differences in the active site structure of this enzyme when compared to the structures and aldehyde oxidation reported for AOR and FOR. Based on all the available structural information as well as the biochemical and biophysical data discussed above, we propose the mechanism for ASOR shown in Fig. 7. The reaction that ASOR catalyzes consists of two sequential oxidation reactions where in each the W site undergoes reversible two electron reduction of the resting oxidized W(VI) state via the W(V) and W(IV) states, based on what has been proposed for the W site in FOR [9]. The W site in WOR5 was previously shown to generate an intermediate W(V) state upon addition of hexanal [11]. The overall reaction consists of the oxidative desulfonation of the aliphatic sulfonate, followed by the oxidation of the resulting aldehyde to a carboxylic acid (Fig. 7). The reduction potentials of these two reactions are approximately − 700 mV and − 510 mV [6, 26], respectively, more than sufficient to reduce P. furiosus ferredoxin (−385 mV) [17].
Fig. 7.
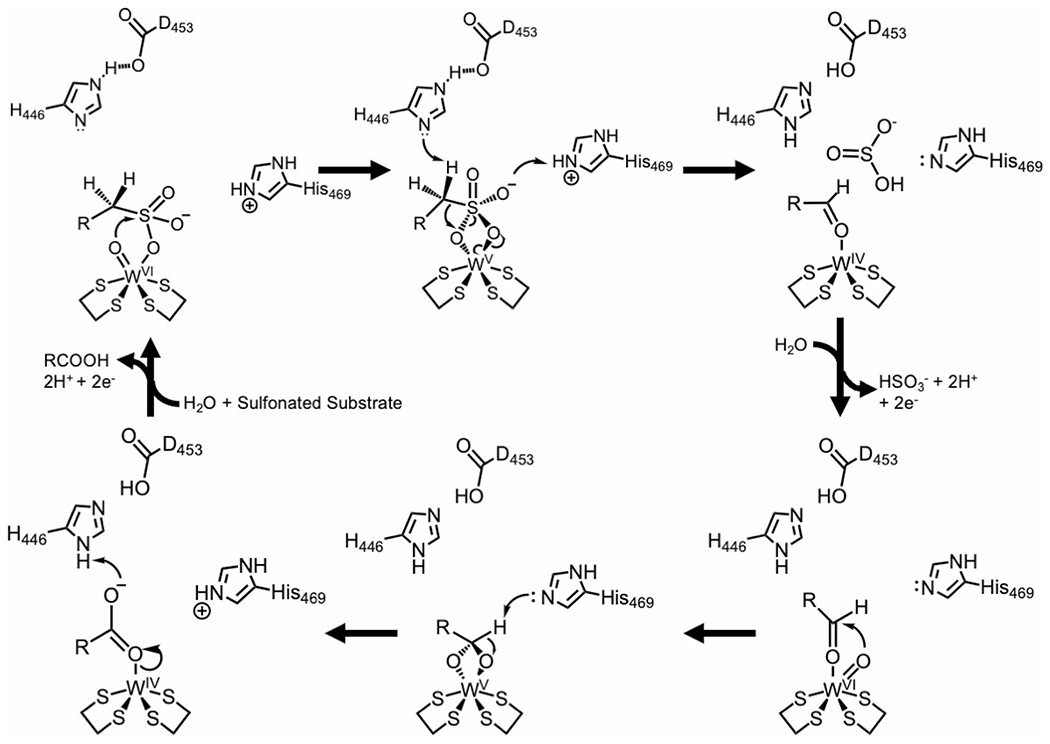
Proposed mechanism for the oxidation of aliphatic sulfonates by ASOR incorporating the roles of its unique active site residues. R represents an aliphatic side chain. The four sulfur atoms surrounding the W atom are provided by the dithioline groups of two pyranopterin molecules. See text for details
In trying to explain the mechanism of ASOR, a rational conclusion is that breaking the C–S bond must involve additional deprotonation and protonation steps compared to what is required for oxidation of the aldehyde to a carboxylic acid. This is supported by the presence of an additional Asp-His pair in the active site of ASOR (D453 and H446, Fig. 6), but not in the active sites of AOR or FOR. Moreover, D453 in ASOR is partially buried and also interacting with R264; therefore, this side chain is likely negatively charged. E328 and H469 are structurally conserved in all three of these WOR enzymes (AOR, FOR and ASOR) and are therefore most likely involved in the oxidation of water for subsequent conversion of the aldehyde to the corresponding acid. However, there is one notable difference. In ASOR, H469 is hydrogen bonded to Y199, potentially modulating the microscopic pKa for the H469 side chain lower. We ran the free software PROPKA [27] and found the general trend of the predicted pKa values, based on the protein environment, to be consistent with these predictions.
Considering these factors, QM/MM calculations[28], as well as what has been proposed previously for the mechanism of aldehyde oxidation at the tungsto-bispyranopterin site [29], our mechanism begins with the aliphate sulfonate binding via the sulfonate near the W=O center (Fig. 7). The oxidative reactions are facilitated by an additional proton-transfer network in ASOR that is not present in AOR or FOR, specifically D453 and H446 (Fig. 6). Upon substrate binding, the tungsten-bound oxygen acts as a nucleophile and attacks the sulfur atom of the sulfonate, forming a single bond with the sulfur atom (Fig. 7, top left). The intermediate is further stabilized by the protonated form of H469. H446 acts as a general base to help facilitate breaking the S–C bond and formation of the C–O bond (Fig. 7, top middle). Loss of sulfite is also made more favorable through protonation (Fig. 7, top right). E328 is conserved in all enzymes and has been proposed to facilitate the oxidation/activation of water to yield the W=O state and we propose that this is also the case for WOR5 (Fig. 7, bottom right). H469 is also a conserved amino acid and has been proposed to facilitate deprotonation as the tungsten-bound oxygen attacks the carbonyl carbon of the aldehyde (Fig. 7, bottom middle). Protonation of the carboxylic acid, which is glycine in the case of taurine, facilitates the release of product from the tungsten center. Oxidation of another water molecule, again facilitated by the strictly conserved residue E328, returns the active site to the resting state. Future work will address substrate diversity to include phosphonated compounds and probe the proposed reaction mechanism through spectroscopy and site specific mutagenesis.
Conclusions
In this work, we report the structure of WOR5, now termed ASOR, as well as an unprecedented enzymatic activity. While the physiological activity and implications will require significantly more interrogation, our investigation demonstrates an exciting new structure and function for a tungsto-bispyranopterin enzyme that is consistent with the marine environment inhabited by P. furiosus. Admittedly, crystal screening of concentrated crude extract is, in fact, an uncontrolled experiment. However, given that the initial experiment was performed with cytoplasmic extract obtained from cells that were cold-shocked, as previously described [10], the density we have observed may have physiological relevance. Regardless, these data led us to identify a new substrate representing aliphatic sulphonates, which are abundant in the marine environment. SDS-PAGE of the crude extract from the overexpression strain does not provide any additional insight as to why ASOR crystallized as there are clearly numerous other proteins present (Supplemental Figure S6). However, the time to obtain crystals was greatly reduced using the crude extract from the overexpression strain, consistent with more AOSR being present. This work represents important implications for other difficult-to-crystalize enzymes, as we have shown that unknown metabolites or other stabilizing factors present in the crude extract may facilitate crystallization.
Experimental procedures
Preparation of a recombinant P. furiosus strain overexpressing genes pf1479 and pf1480
The genetically tractable P. furiosus strain COM1 was used for the overexpression of WOR5 [30]. An insertion cassette was assembled using overlapping PCR [31]. The upstream flanking region (UFR), downstream flanking region (DFR), and PF1479-PF1480 were amplified from COM1 genomic DNA, with primers designed to insert an N-terminal 9x-histidine tag on PF1480. The selection marker (pyrF-Pgdh-Pslp) was amplified using pGLW021 as the template [30]. The genomic DNA was prepared using the ZymoBead Genomic DNA Kit (Zymo Research). P. furiosus COM1 transformants were grown as previously described [30]. Genomic DNA was used for PCR screening, which was carried out by using GXL polymerase (Takara, ClonTech). The PCR screening was performed using a pair of primers outside the intergenic GR1 locus to confirm that the transformation cassette recombined into the correct locus. Once a colony was positively identified, the genomic DNA was further sequenced using Genewiz Sanger Sequencing service (South Plainfield, NJ).
Growth of P. furiosus strains
Growth of the wild-type P. furiosus strain and cold-shock induction of WOR5 were performed as previously described [32, 33]. Briefly, to construct the overexpression strain, the genetic system took advantage of the highly-expressed P. furiosus promoter (Pslp) for the gene encoding the S-layer protein. A 9x-Histidine tag was also introduced at the N-terminus of PF1480 to facilitate purification. The tag location was chosen to avoid the interface between genes encoding WOR5 and its potential polyferredoxin subunit. Previous work has shown that Pslp can lead to up-regulation of expression in some cases by greater than an order of magnitude [34]. Large scale growth of the WOR5 expression strain was carried out in a 28-L fermentor using maltose as the carbon source [35] at 80 °C for 14 h with constant flushing of N2/CO2. Cells (~ 15 g) were flash frozen in liquid nitrogen and stored at ࢤ 80 °C.
WOR5 purification from overexpression strain
WOR5 was purified anaerobically from a 40 g cell paste at 25 °C. Cells were resuspended in 100 mM sodium phosphate, pH 8.0, containing 150 mM KCl and 50 μg/ml DNase I and were lysed by osmotic shock. Following a 1-h incubation in an anaerobic chamber, cells were centrifuged at 30,000×g for 1 h. The supernatant was loaded on an Amintra-CoHis column (Expedeon) pre-equilibrated with the same buffer, but without DNase. Following a two-column volume buffer wash, the proteins were eluted using a stepwise gradient from 10 mM imidazole to 250 mM imidazole. Fractions containing WOR5 as determined by activity and SDS-PAGE were pooled and diluted 20-fold in 100 mM sodium phosphate, pH 8.0 before being loaded onto an anaerobic QHP column. Buffer A contained 100 mM sodium phosphate pH 8.0, and 1 mM cysteine, while Buffer B contained 100 mM sodium phosphate, pH 8.0, 1 M KCl and 1 mM cysteine. Following a three-column volume buffer wash, proteins were eluted using a linear gradient from 0 to 100% Buffer B. Following SDS-PAGE analysis, fractions containing WOR5 were again pooled, concentrated, and the protein concentration was measured using the Biuret assay.
Preparation of cytoplasmic extract for crystallization
Frozen cell paste (50 g) of the WOR5 overexpression strain was thawed in 150 mL of 50 mM Tris/HCl, pH 8.0, on a vacuum manifold while the gas phase was continuously exchanged under vacuum with oxygen-free argon. Cells were lysed using an anaerobic French Press system maintained under a slight argon pressure. The cell-free extract was centrifuged at 100,000×g for 1 h to remove insoluble material and the soluble fraction was concentrated using a gas concentrator and a 100 kDa cutoff membrane. The final protein concentration of the cytoplasmic extract was approximately 100 mg/mL, and this was again centrifuged at 100,000×g for 1 h to remove any insoluble material.
Crystallization of WOR5
Concentrated cytoplasmic extract (~ 100 mg/mL) was screened anaerobically in a soft-sided Coy™ glove box containing an atmosphere of 95% nitrogen and 5% hydrogen. The oxygen concentration was maintained below 1 ppm using a palladium catalyst that was regenerated on a bi-weekly basis. Crystal screening was carried out in 75 μL melting point capillaries by layering 20 μL of the concentrated crude extract on top of 30 μL of the crystallization condition. 480 unique conditions, from five sparse matrix crystallization kits, were screened. All crystallization experiments were carried out in an anaerobic chamber at room temperature (approximately 22–27 °C). Identification of crystalline material was facilitated by use of a polarizer. The identity of WOR5 crystals, when crystallized from concentrated crude extract, was confirmed by mass spectroscopy of crystals than had been resolubilized in 0.1 M HEPES pH 7.5. Optimization of the initial conditions resulted in a final crystallization solution consisting of 0.1 M HEPES pH 7.5, 2% (v/v) polyethylene glycol (PEG) 400, and 2.0 M ammonium sulfate. Cryo-protection of the crystals was achieved by incrementally (2.5%, w/v) increasing the concentration of PEG 400, along with and equal percentage (v/v) of glycerol, DMSO and polyethylene glycol to 10%. Although cumbersome, there is a significant advantage of the anaerobic capillary batch method for oxygen sensitive enzymes. Specifically, crystals can be grown in a sealed quartz capillary and therefore safely removed from the chamber, subjected to a quick test for diffraction, before being return to the anaerobic chamber.
Data collection, phasing, and refinement
Data was collected at 100 K on beamline 22ID, SER-CAT, Advanced Photon Source, Argonne National Laboratory using a 50-μm beam and an Eiger 16 M Detector. Phases were obtained by collecting data at two wavelengths, 12,782 eV and 7112 eV (the iron edge). The anomalous signal for tungsten at 12,651 eV is greater than ten electrons allowing two tungsten sites in the asymmetric unit to be readily identified. These positions were utilized by PHASER to further refine and search for the iron positions at 7112 eV and led to interpretable electron density maps. Subsequent rounds of model building and refinement were accomplished using COOT [36, 37] and PHENIX [38].
Biochemical assays of WOR5, AOR and FOR
AOR and FOR were purified from wild-type P. furiosus cells as previously described [5, 6] and their published enzymatic activities verified using in vitro assays and the artificial electron acceptor benzyl viologen (BV). Unlike WOR5, neither AOR nor FOR showed any BV reduction in the presence of taurine. All assays were conducted anaerobically using 100 mM sodium phosphate, pH 8.0, 150 mM KCl, 100 nM enzyme, and 1 mM BV at 80 °C. Varying concentrations of substrate were injected using a gastight syringe to initiate the reaction. Specific activity was determined using a molar extinction coefficient for reduced BV at 600 nm of 7,400 M−1·cm−1.25
NMR analysis of WOR5 products
A 2.5-mL reaction mixture was flash frozen with liquid nitrogen and freeze-dried with the aid of a SpeedVac concentrator (Thermo SAVANT, SPD111V) overnight. The dry powder was dissolved in 300 μL of D2O (99.8%) containing 0.01% (w/v) 3-(trimethylsilyl) propane-1-sulfonate and sodium salt (DSS) for chemical shift referencing. The resultant solutions were centrifuged and then transferred to 4 mm NMR tubes (Norell). NMR spectra were acquired at 298 K on a Bruker AVANCE Neo 700 MHz spectrometer equipped with an HCN-Cryo-probe. One-dimensional 1H NMR spectra were acquired (64 scans) using the 1D-excitation sculpting for pre-saturation of the water peak, with a spectral width of 16 ppm, 32 K data points, 1.4 s acquisition time and 2.0 s relaxation delay. 1H–13C HSQC (heteronuclear single quantum correlation) spectra were acquired with a spectral width of 13 ppm, 2048 data points and 1.5 s relaxation delay in the direct-detected dimension (32 scans), and 256 data points over a spectral width of 165 ppm in the indirect-detected dimension. 1H–13C HMBC (heteronuclear multiple bond correlation) spectra were acquired with a spectral width of 13 ppm, 4096 data points and 1.0 s relaxation delay in the direct-detected dimension (32 scans), and 256 data points over a spectral width of 220 ppm in the indirect-detected dimension (32 scans). Adjustments to the baseline, peak phasing and chemical shift referencing were made using Bruker Topspin 3.6.2. To confirm product identity, the chemical shifts in the NMR spectra were compared to in-house standards and the Biological Magnetic Resonance Database for positive matches.
Negative ion mass spectrometry of samples
Samples were analyzed on an Agilent 6230B time-of-flight mass spectrometer operating in negative ion mode. Figure 5 contains the mass spectra of the samples taken before (T0) and after (Tf) the enzymatic reaction. In Fig. 5A, the presence of taurine is indicated by the 124 m/z peak. In Fig. 5B, the 124 m/z is not detectable; however, the emergence of glycine (74 m/z) is observed.
Turbidimetric determination of sulfate
30 μL of 4.2 M NaCl (10% v/v HCl) was added to the 300 μL T0 and Tf samples and vortexed for 1 min. After, 7 μL of 0.1 M Ba(NO3)2 was added to the mixtures, vortexed for 1 min, and settled for 5 min to observed the formation of precipitate (Supporting Information, Figure S4).
Supplementary Material
Acknowledgements
The authors wish to acknowledge the technical support of the Proteomics and Mass Spectrometry Facility at the University of Georgia for acquisition and interpretation of their mass spectrometry data. This work was supported in part by grants (GM124203 to WNL and GM136885 to MWWA) from the National Institutes of Health General Medical Sciences.
Footnotes
Conflict of interest
The authors declare no competing interests.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00775-022-01965-0.
Data availability
The accession number for the atomic coordinates reported in this paper are PDB ID 6X1O and 6X6U for the model obtained from crude extract and the taurine-soaked model of ASOR, respectively.
References
- 1.Schicho RN, Snowden LJ, Mukund S, Park JB, Adams MKW (1993) Arch Microbiol 159:380–385 [Google Scholar]
- 2.Schut GJ, Thorgersen MP, Poole FL 2nd, Haja DK, Putumbaka S, Adams MWW (2021) Proc Natl Acad Sci U S A. 10.1073/pnas.2109008118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mukund S, Adams MW (1995) J Biol Chem 270:8389–8392 [DOI] [PubMed] [Google Scholar]
- 4.Nguyen DMN, Schut GJ, Zadvornyy OA, Tokmina-Lukaszewska M, Poudel S, Lipscomb GL, Adams LA, Dinsmore JT, Nixon WJ, Boyd ES, Bothner B, Peters JW, Adams MWW (2017) J Biol Chem 292:14603–14616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mukund S, Adams MW (1991) J Biol Chem 266:14208–14216 [PubMed] [Google Scholar]
- 6.Roy R, Mukund S, Schut GJ, Dunn DM, Weiss R, Adams MW (1999) J Bacteriol 181:1171–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy R, Adams MW (2002) J Bacteriol 184:6952–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan MK, Mukund S, Kletzin A, Adams MW, Rees DC (1995) Science 267:1463–1469 [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Faham S, Roy R, Adams MW, Rees DC (1999) J Mol Biol 286:899–914 [DOI] [PubMed] [Google Scholar]
- 10.Weinberg MV, Schut GJ, Brehm S, Datta S, Adams MW (2005) J Bacteriol 187:336–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevers LE, Bol E, Hagedoorn PL, Hagen WR (2005) J Bacteriol 187:7056–7061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berks BC, Page MD, Richardson DJ, Reilly A, Cavill A, Outen F, Ferguson SJ (1995) Mol Microbiol 15:319–331 [DOI] [PubMed] [Google Scholar]
- 13.Jormakka M, Yokoyama K, Yano T, Tamakoshi M, Akimoto S, Shimamura T, Curmi P, Iwata S (2008) Nat Struct Mol Biol 15:730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott IM, Rubinstein GM, Poole FL 2nd, Lipscomb GL, Schut GJ, Williams-Rhaesa AM, Stevenson DM, Amador-Noguez D, Kelly RM, Adams MWW (2019) J Biol Chem 294:9995–10005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray HB, Winkler JR (2010) Biochim Biophys Acta 1797:1563–1572 [DOI] [PubMed] [Google Scholar]
- 16.Winkler JR, Gray HB (2014) Chem Rev 114:3369–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brereton PS, Duderstadt RE, Staples CR, Johnson MK, Adams MW (1999) Biochemistry 38:10594–10605 [DOI] [PubMed] [Google Scholar]
- 18.Schindelin H, Kisker C, Rees DC (1997) J Biol Inorg Chem 2:773–781 [Google Scholar]
- 19.Clifford EL, Hansell DA, Varela MM, Nieto-Cid M, Herndl GJ, Sintes E (2017) Limnol Oceanogr 62:2745–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esteves AM, Chandrayan SK, McTernan PM, Borges N, Adams MW, Santos H (2014) Appl Environ Microbiol 80:4226–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert IH (2004) Neurochem Res 29:27–63 [DOI] [PubMed] [Google Scholar]
- 22.Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, Schleheck D (2019) Proc Natl Acad Sci U S A 116:3171–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson CD, Irwin SM, Backman LRF, Le C, Wang JX, Vennelakanti V, Yang Z, Kulik HJ, Drennan CL, Balskus EP (2021) Cell Chem Biol 28(1333–1346):e1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Severen MC, Andrejic M, Li J, Starke K, Mata RA, Nordlander E, Ryde U (2014) J Biol Inorg Chem 19:1165–1179 [DOI] [PubMed] [Google Scholar]
- 25.Caldararu O, Feldt M, Cioloboc D, van Severen MC, Starke K, Mata RA, Nordlander E, Ryde U (2018) Sci Rep 8:4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arendsen AF, deVocht M, Bulsink YBM and Hagen WR (1996) J Biol Inorg Chem 1:292–296 [Google Scholar]
- 27.Bas DC, Rogers DM, Jensen JH (2008) Proteins 73:765–783 [DOI] [PubMed] [Google Scholar]
- 28.Liao RZ, Yu JG, Himo F (2011) J Inorg Biochem 105:927–936 [DOI] [PubMed] [Google Scholar]
- 29.Tong IT, Liao HH, Cameron DC (1991) Appl Environ Microbiol 57:3541–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipscomb GL, Stirrett K, Schut GJ, Yang F, Jenney FE Jr, Scott RA, Adams MW, Westpheling J (2011) Appl Environ Microbiol 77:2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bryksin AV, Matsumura I (2010) Biotechniques 48:463–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu CH, Ponir CA, Haja DK, Adams MWW (2018) Protein Eng Des Sel 31:337–344 [DOI] [PubMed] [Google Scholar]
- 33.Chandrayan SK, McTernan PM, Hopkins RC, Sun J, Jenney FE Jr, Adams MW (2012) J Biol Chem 287:3257–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandrayan SK, Wu CH, McTernan PM, Adams MW (2015) Protein Expr Purif 107:90–94 [DOI] [PubMed] [Google Scholar]
- 35.Verhagen MF, Menon AL, Schut GJ, Adams MW (2001) Methods Enzymol 330:25–30 [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K (2004) Acta Crystallogr D Biol Crystallogr 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Debreczeni JE (2012) Methods Mol Biol 841:143–159 [DOI] [PubMed] [Google Scholar]
- 38.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) Acta Crystallogr D Biol Crystallogr 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the atomic coordinates reported in this paper are PDB ID 6X1O and 6X6U for the model obtained from crude extract and the taurine-soaked model of ASOR, respectively.