

Abstract
Background
Resistance to HER2-targeted therapies, including the monoclonal antibody trastuzumab and tyrosine kinase inhibitor lapatinib, frequently occurs and currently represents a significant clinical challenge in the management of HER2-positive breast cancer. We previously showed that the trastuzumab-resistant SKBR3-pool2 and BT474-HR20 sublines were refractory to lapatinib in vitro as compared to the parental SKBR3 and BT474 cells, respectively. The in vivo efficacy of lapatinib against trastuzumab-resistant breast cancer remained unclear.
Results
In tumor xenograft models, both SKBR3-pool2- and BT474-HR20-derived tumors retained their resistance phenotype to trastuzumab; however, those tumors responded differently to the treatment with lapatinib. While lapatinib markedly suppressed growth of SKBR3-pool2-derived tumors, it slightly attenuated BT474-HR20 tumor growth. Immunohistochemistry analyses revealed that lapatinib neither affected the expression of HER3, nor altered the levels of phosphorylated HER3 and FOXO3a in vivo. Interestingly, lapatinib treatment significantly increased the levels of phosphorylated Akt and upregulated the expression of insulin receptor substrate-1 (IRS1) in the tumors-derived from BT474-HR20, but not SKBR3-pool2 cells.
Conclusions
Our data indicated that SKBR3-pool2-derived tumors were highly sensitive to lapatinib treatment, whereas BT474-HR20 tumors exhibited resistance to lapatinib. It seemed that the inefficacy of lapatinib against BT474-HR20 tumors in vivo was attributed to lapatinib-induced upregulation of IRS1 and activation of Akt. Thus, the tumor xenograft models-derived from SKBR3-pool2 and BT474-HR20 cells serve as an excellent in vivo system to test the efficacy of other HER2-targeted therapies and novel agents to overcome trastuzumab resistance against HER2-positive breast cancer.
Keywords: Therapeutic resistance, HER2-targeted therapy, In vivo tumor models, Breast cancer
Introduction
Amplification/overexpression of HER2 (also known as erbB2/neu) is significantly associated with poor prognosis in breast cancer (BC) patients [1, 2]. HER2-targeted therapies, including trastuzumab (Herceptin) and lapatinib (Tykerb) are commonly used in the clinic and have dramatically improved the survival of BC patients with HER2-overexpressing (HER2-positive) tumors [3, 4]. It is well-known that trastuzumab is a humanized anti-HER2 monoclonal antibody (mAb) binding to the extracellular domain of HER2, blocks its signaling. Lapatinib, as a small molecule tyrosine kinase inhibitor (TKI), mainly inhibits the kinase activity of HER2 via targeting its intracellular domain. Although trastuzumab and lapatinib are effective for metastatic BC patients with HER2-positive tumors [5–8], both primary (de novo) and acquired resistance to the HER2-targeted therapies frequently occur and currently represent a significant clinical challenge for successful treatment of HER2-positive BC [9]. To date, we lack validated biomarkers to predict the response of HER2-positive BC to these therapies [4, 10]. There is an unmet need to identify novel approaches/agents to overcome the resistance with an aim to improve the survival of HER2-positive BC patients, especially those with advanced/metastatic diseases.
Several mechanisms of resistance to HER2-targeted therapy have been proposed in the treatment of HER2-positive BC [3, 9]. Among them, activation of compensatory signaling pathways-triggered by alternative receptor tyrosine kinases (RTKs), including HER3 and the insulin-like growth factor-1 (IGF-1R), plays an important role in attenuating the efficacy of an HER2-targeted therapy [11–13]. Whether HER2-positive BC cells utilize similar or different mechanisms to create resistance to trastuzumab and lapatinib remains an open question in our understanding of the molecular basis of HER2-targeted therapy resistance. Some studies have shown that activation of the signaling pathways initiated by other HER family members, such as EGFR and HER3, or non-HER receptors, including AXL, limits the response of HER2-positive BC cells to both trastuzumab and lapatinib [14–18]. Other reports suggest that the two agents may not share common mechanisms of resistance. While the PI-3K/Akt signaling pathway has been implicated as a major determinant of trastuzumab resistance [19], its role in lapatinib resistance remains elusive. Loss of PTEN-triggered PI-3K/Akt signaling has been shown to result in lapatinib resistance, which can be overcome by NVP-BEZ235, a dual inhibitor of PI-3K/mTOR [20]. Yet, other studies indicate that activation of the PI-3K/Akt signaling confers resistance to trastuzumab, but not lapatinib [21, 22], and lapatinib potently suppresses tumor growth of HER2-positive BC in a PTEN-independent manner [23].
The PI-3K/Akt signaling is one of the major downstream signaling pathways-initiated by HER3 and IGF-1R, and critically contributes to trastuzumab resistance [24–26]. We investigated the relationship of HER3 and IGF-1R in HER2-positive BC cells with acquired resistance to trastuzumab. Our data showed that both HER3 and IGF-1R directly interacted with HER2 in the trastuzumab-resistant BC sublines SKBR3-pool2 and BT474-HR20, which were derived from the parental BC cell lines SKBR3 and BT474, respectively [27]. In the resistant cells, HER2, HER3, and IGF-1R formed a heterotrimeric complex, which was responsible for enhanced activation of multiple downstream pathways, including the PI-3K/Akt signaling and Src kinase [27]. We next wondered whether the trastuzumab-resistant sublines would retain their resistant phenotypes to lapatinib. Our data showed that SKBR3-pool2 and BT474-HR20 cells as compared to their sensitive counterparts were relatively refractory to lapatinib treatment under our cell culture conditions [28]. Further examination revealed that HER3 and IGF-1R initiated distinct signaling pathways altering the responses of SKBR3-pool2 and BT474-HR20 cells to lapatinib in vitro [28]. In the current studies, we took advantage of the tumor xenograft models established with SKBR3-pool2 or BT474-HR20 cells in nude mice to determine the in vivo antitumor activity of lapatinib against trastuzumab-resistant BC.
Results
Trastuzumab-resistant breast cancer sublines maintain their resistance phenotypes in vivo
We previously reported that trastuzumab-resistant BT474-HR20 cells formed tumors in nude mice with a much shorter latency than parental BT474 cells [29]. Moreover, BT474-HR20-derived tumors were significantly less sensitive to trastuzumab treatment [29]. To determine if all trastuzumab-resistant BC cells would display aggressive characteristics and retain resistant phenotypes to trastuzumab in vivo, we performed similar experiments with the tumor xenograft models established from the trastuzumab-resistant SKBR3-pool2 cells in comparison with that established from the parental SKBR3 cells. We found that the tumors derived from SKBR3-pool2 cells grew significantly faster than that derived from SKBR3 cells (Fig. 1A). Treatment with trastuzumab had no significant effect on the growth of SKBR3-pool2 tumors at all-time points examined (Fig. 1B). The experiment was terminated at day 45 post cell injection. Our data indicated that although BT474-HR20 and SKBR3-pool2 were originally obtained via in vitro cell culture studies, they both maintained their trastuzumab resistance phenotypes in vivo. The aggressiveness of BT474-HR20 and SKBR3-pool2 cells to form tumors in nude mice enabled them to serve as excellent in vivo models to examine the efficacy of other therapeutic agents against trastuzumab-resistant BC.
Fig. 1.

SKBR3-pool2 cells as compared to the parental SBKR3 cells grew tumors in nude mice with a significantly faster rate and retained trastuzumab resistance phenotype in vivo. A SKBR3 or SKBR3-pool2 cells (5 × 106) were injected s.c into the flanks of 5-week-old female nude mice (n = 8/group). Mice were observed three times and tumor formation was measured twice a week. Tumor volume was calculated and expressed as cubic millimeters (mean ± SD). B When tumor volumes reached ~ 80mm3, the mice bearing tumors-derived from SKBR3-pool2 cells were treated with either control (PBS) or trastuzumab (20 mg/kg) twice a week for seven times (n = 5/group). Tumor volume was expressed as cubic millimeters (mean ± SD). Treatment with trastuzumab had no significant effect on SKBR3-pool2 tumor growth at all-time points examined. Statistical analyses were performed with two-sided student’s t tests
SKBR3-pool2 and BT474-HR20-derived tumors show distinct sensitivity to lapatinib in in vivo xenograft models
To test whether lapatinib would exert its antitumor effect on trastuzumab-resistant BC in vivo, we again used SKBR3-pool2 or BT474-HR20 cells to establish tumor xenograft models in nude mice. The tumor-bearing mice were treated with either vehicle control (DMSO) or lapatinib via intraperitoneal injection. At the end of the experiments, all tumors were photographed and weighted. Treatment with lapatinib profoundly suppressed SKBR3-pool2 tumor growth (Fig. 2A). The tumors obtained from lapatinib-treated mice were much smaller and significantly lighter than that obtained from controls (Fig. 2B & C). In contrast, the tumors-derived from BT474-HR20 cells were less sensitive to lapatinib treatment. Lapatinib slightly, but not significantly inhibited BT474-HR20 tumor growth (Fig. 3A). The size of the tumors obtained from lapatinib-treated mice was similar to those obtained from controls (Fig. 3B). There was no significant difference in the tumor weight between the two groups (Fig. 3C). Collectively, these data demonstrated that both SKBR3-pool2 and BT474-HR20 maintained their trastuzumab resistance phenotypes in vivo ( Fig. 1 and [29]), and they also were refractory to lapatinib treatment in in vitro cell culture experiments [28]. However, lapatinib exhibited different antitumor activity in xenograft models established from SKBR3-pool2 or BT474-HR20 cells. While lapatinib markedly inhibited SKBR3-pool2 tumor growth, it had little effect on BT474-HR20 tumor growth. Our studies suggest that some, but not all trastuzumab-resistant BCs respond well to lapatinib in in vivo tumor xenograft models.
Fig. 2.
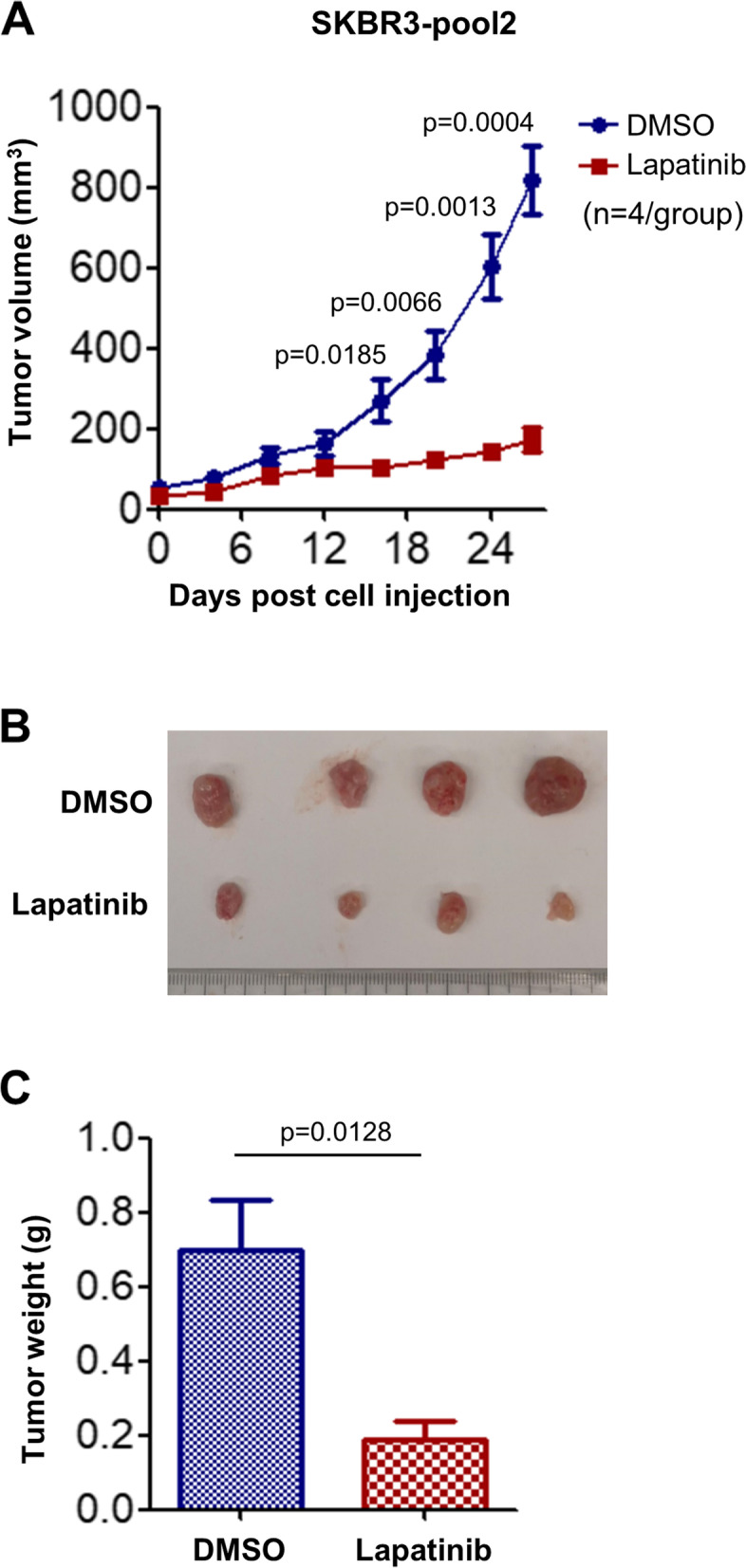
Lapatinib exhibited potent inhibitory effects on SKBR3-pool2 tumor growth in vivo. SKBR3-pool2 cells (8 × 106) were injected s.c into the flanks of 5-week-old female nude mice. When tumor volumes reached ~ 80 mm3, mice were treated with vehicle control (DMSO) or lapatinib (80 mg/kg, i.p.) every other day (n = 4/group). A Tumor growth curves were plotted using average tumor volume within each group at the indicated time points. Bars, SD. B & C at the end of the experiment, all mice were sacrificed. The tumors were dissected, imaged as indicated (B) and measured for weight (C). Two-sided student’s t tests were used for statistical analyses
Fig. 3.

Lapatinib slightly attenuated the growth of BT474-HR20-derived tumors in xenograft models. BT474-HR20 cells (8 × 106) were injected s.c into the flanks of 5-week-old female nude mice. When tumor volumes reached ~ 80 mm3, mice were treated with vehicle control (DMSO) or lapatinib (80 mg/kg, i.p.) every other day (n = 6/group). A Tumor growth curves were plotted using average tumor volume within each group at the indicated time points. Bars, SD. B & C at the end of the experiment, all mice were sacrificed. The tumors were dissected, imaged as indicated (B) and measured for weight (C). Two-sided student’s t tests were used for statistical analyses
Lapatinib differentially regulates the levels of phosphorylated Akt (p-Akt) and IRS1 expression in SKBR3-pool2- and BT474-HR20-derived tumors
We observed an enhanced activation of multiple signaling pathways, including PI-3K/Akt and Src kinase in SKBR3-pool2 and BT474-HR20 cells because of the trimeric complex formed by HER2, HER3, and IGF-1R [27]. Specific inhibition of either Akt or Src reversed the resistant phenotypes of both SKBR3-pool2 and BT474-HR20 cells [27]. This was consistent with other reports which indicated that PI-3K/Akt signaling and Src kinase played a pivotal role in the development of trastuzumab resistance [19, 30, 31]. Our studies also showed that HER3 induced activation of both PI-3K/Akt signaling and Src, whereas IGF-1R mainly activated Src kinase in SKBR3-pool2 and BT474-HR20 cells [28]. Further defining the underlying mechanisms of HER3-/IGF-1R-initiated signaling in trastuzumab resistance, we recently discovered elevated levels of phosphorylated FOXO3a (p-FOXO3a) in the trastuzumab-resistant cells due to transcriptional suppression of PPP3CB, which is a subunit of the serine/threonine-protein phosphatase 2B (PP2B) [32]. The increased p-FOXO3a disrupted a negative feedback inhibition loop formed by FOXO3a and several IGF2-/IRS1-targeting miRNAs (miR-193a-5p, miR-128-3p, and miR-30a-5p). This, thereby, increased the expression of both IGF2 and IRS1 [32]. Thus, we hypothesized that the distinct antitumor activity of lapatinib against SKBR3-pool2 and BT474-HR20 in vivo might be attributed to its differential effects on the aberrant activation of HER3 (phosphorylation) and/or the IGF2/IGF-1R/IRS1 signaling in the resistant tumors. To this end, we took advantage of the SKBR3-pool2 and BT474-HR20 tumors obtained from the animal experiments and performed immunohistochemistry (IHC) assays on HER3, p-HER3, p-FOXO3a, p-Akt, and IRS1. For both SKBR3-pool2 and BT474-HR20 tumors, we found no apparent difference in the levels of HER3, p-HER3, and p-FOXO3a between lapatinib-treated mice and controls (Fig. 4A & B), i.e. lapatinib treatment had little effect on HER3 protein expression and activation in vivo. In contrast, the levels of p-Akt and IRS1 were markedly increased in the BT474-HR20 tumors obtained from lapatinib-treated mice as compared to that from controls (Fig. 5A left). Since lapatinib treatment generally had no effect on Akt expression, we focused our studies on p-Akt. Quantification analyses of both p-Akt and IRS1 were significantly different between the two groups (Fig. 5A right). Taken together, our data suggest that lapatinib’s inefficacy against BT474-HR20 tumors in xenograft models is likely due to its capability to induce Akt activation (phosphorylation) and/or enhance IRS1 expression in vivo.
Fig. 4.

No significant changes of p-HER3, HER3, and p-FOXO3a were observed between DMSO- and lapatinib-treated tumors derived from SKBR3-pool2 or BT474-HR20 cells. The tumors derived from either SKBR3-pool2 (A) or BT474-HR20 (B) cells were formalin-fixed and paraffin-embedded (FFPE) and sectioned into five-micron-thick slides. The FFPE slides were analyzed with IHC staining assays for p-HER3 (Y1289), HER3, or p-FOXO3a (Ser253) following the procedures described in the materials and methods. Two individuals independently evaluated the IHC staining. The levels of p-HER3, HER3, and p-FOXO3a showed no apparent differences between control and lapatinib-treated groups
Fig. 5.

Lapatinib treatment markedly increased p-Akt levels and enhanced IRS1 expression in the tumors-derived from BT474-HR20, but not SKBR3-pool2 cells. The FFPE slides of BT474-HR20 (A) or SKBR3-pool2 (B) tumors were analyzed with IHC staining assays for p-Akt (Ser473) or IRS1. The IHC staining was evaluated by two individuals independently and quantified with ImageJ and ImageJ plugin IHC profiler. The average staining intensity of three random field of each slide was plotted and shown as bar graphs on the right. The data were statistically analyzed with two-sided student’s t tests. Bars, SD. ns, not significant. ** p < 0.01, *** p < 0.005
Discussion
Lapatinib is an orally bioavailable reversible TKI with dual targeting function against both EGFR and HER2 [33]. Its combination with capecitabine is indicated to treat patients with advanced/metastatic HER2-positive BC whose diseases have progressed after prior therapy with trastuzumab plus chemotherapy [33, 34]. Despite lapatinib’s success in HER2-positive BC patients being observed, both primary (de novo) and acquired resistances to lapatinib frequently occur in the clinical setting [35, 36]. Among those potential mechanisms of lapatinib resistance, reactivation of the downstream signaling pathways, especially the PI-3K/Akt signaling has been shown to play an important role in the development of resistance to lapatinib [34, 37]. In the current studies using tumor xenograft models established with the trastuzumab-resistant BC sublines, we observed a marked increase of p-Akt in BT474-HR20-derived tumors (Fig. 5A left), which were refractory to lapatinib treatment (Fig. 3A). In contrast, lapatinib exerted profound suppression on SKBR3-pool2 tumor growth (Fig. 2A), and there was no increase in p-Akt levels in the SKBR3-pool2 tumors of lapatinib-treated mice as compared to controls (Fig. 5B left). Collectively, our data suggest that enhanced activation of Akt (evidenced by increased p-Akt) likely contributes to the inefficacy of lapatinib against BT474-HR20 tumor growth in vivo.
The underlying mechanisms resulting in increased p-Akt in the BT474-HR20 tumors after lapatinib treatment remain unclear. Accumulating evidence indicates that compensatory induction of HER3 potently activates the PI-3 K/Akt signaling pathway, thereby leading to resistance to a wide variety of therapeutic agents [12, 38], including lapatinib [15]. However, we did not detect apparent alterations in either HER3 protein levels or its phosphorylation (p-HER3) in both BT474-HR20 and SKBR3-pool2 tumors upon lapatinib treatment (Fig. 4). This suggest that reactivation of Akt in lapatinib-treated BT474-HR20 tumors is likely, via a mechanism independent of HER3 upregulation and activation. We previously showed that increased p-FOXO3a and elevated expression of IGF2 and IRS1 were critical for the development of trastuzumab resistance in HER2-positive BC. This occurred via disruption of a negative feedback inhibition loop of FOXO3a and several IGF2-/IRS1-targeting miRNAs [32]. Here, our IHC analyses found little change in p-FOXO3a levels between control and lapatinib-treated BT474-HR20 and SKBR3-pool2 tumors (Fig. 4), whereas the expression of IRS1 was significantly increased upon lapatinib treatment only in BT474-HR20 tumors (Fig. 5). Interestingly, recent investigation showed that the expression of IRS4, a closely related member of the IRS family (consisting of IRS1, IRS2, and IRS4) activated the PI-3K/Akt signaling without significant activation of the upstream RTKs, and subsequently caused resistance to HER2-targeted therapy [39, 40]. Nonetheless, whether the elevated expression of IRS1 is responsible for the increased p-Akt levels in lapatinib-treated BT474-HR20 tumors remains unknown. It is also unclear if lapatinib treatment may upregulate IRS4 in the BT474-HR20 tumors. Alternatively, the increased p-Akt may be due to activation of the non-receptor tyrosine kinase Src [41], which functions as a key mechanism of trastuzumab resistance and indicates poor prognosis in HER2-positive/ER-negative BC [42]. Src activation has been shown to cause resistance to lapatinib in HER2-positive BC cells [43, 44]. Our studies of SKBR3-pool2 and BT474-HR20 cells revealed that IGF-1R-initiated Src activation did not influence the efficacy of lapatinib under in vitro cell culture conditions [28]. Currently, we are performing IHC analysis of Src and p-Src to examine if lapatinib treatment in vivo may alter Src expression and/or activation in the tumors derived from BT474-HR20 or SKBR3-pool2 cells.
In 2010, lapatinib was approved for use with an aromatase inhibitor (letrozole) in the treatment of post-menopausal women with metastatic BC co-expressing hormonal receptors (estrogen receptor (ER) and progesterone receptor (PR)) and HER2 [34, 36]. ER expression and/or activation have been shown to contribute to the development of different mechanisms of resistance to trastuzumab and lapatinib [45, 46]. BT474 cells are also ER-positive, a typical luminal B subtype of BC cells, whereas SKBR3 cells do not express ER/PR. Whether the expression of ER may play a role in the resistant phenotype of BT474-HR20 tumors to lapatinib treatment in vivo remains to be determined. It would be interesting and clinically relevant to test whether the combinations of lapatinib and letrozole may synergistically inhibit BT474-HR20 tumor growth in our xenograft models.
In summary, we demonstrate that trastuzumab-resistant BCs exhibit distinct sensitivity to the treatment of lapatinib in tumor xenograft models. Enhanced activation of Akt (evidenced by increased p-Akt) and the elevated expression of IRS1 seem to be associated with the resistance of BT474-HR20 tumors to lapatinib treatment in vivo. Our data support that SKBR3-pool2- and BT474-HR20-derived tumor xenograft models serve as an excellent in vivo system to test the antitumor activity of other HER2-targeted therapies, including lapatinib against trastuzumab-resistant BC. The model system may also be used to determine the potential of novel therapeutic agents to overcome trastuzumab resistance in HER2-positive BC.
Materials and methods
Reagents and antibodies
Trastuzumab (Herceptin) was purchased from Roche (Basel, Switzerland). Lapatinib (HY-50898) were purchased from MedChemExpress (Princeton, NJ, USA). Primary antibodies used for immunohistochemistry (IHC) assays were the following: Rabbit mAb against p-Akt (Ser473) (Cat. #4060, 1:100 dilution), rabbit mAb against p-HER3 (Y1289) (Cat. #4791, 1:100 dilution), and rabbit mAb against HER3 (cat# 12708, 1: 400 dilution) from Cell Signaling Technology (Beverly, MA, USA); Rabbit polyclonal Ab against p-FOXO3a (Ser253) (Cat. #PA5-36816, 1:50 dilution) and mouse mAb against IRS1 (Cat. #MA5-36222, 1:50 dilution) from Thermo Fisher Scientific Inc. (Waltham, MA, USA).
Cells and cell culture
Human BC cell lines SKBR3 and BT474 were obtained from the American Type Culture Collection (Manassas, VA). The trastuzumab-resistant sublines SKBR3-pool2 and BT474-HR20, derived from SKBR3 and BT474, respectively, were described previously [26, 27]. They were routinely maintained in the presence of 20μg/ml of trastuzumab. Cell line authentication was confirmed by Short Tandem Repeat (STR) analysis with PowerPlex® 18D System from Promega (Madison, WI, USA). All cell lines were free of mycoplasma contamination, determined by the MycoAlert™ Mycoplasma Detection Kit (Lonza Group Ltd. Basel, Switzerland) every six months. Cells were cultured with DMEM/F-12 (1:1) medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) in a 37°C humidified atmosphere containing 95% air and 5% CO2 and split twice a week.
Tumor xenograft model
Athymic nu/nu female mice were purchased from Charles River Laboratories Inc. (Wilmington, MA) and maintained according to the procedures and guidelines approved by the Institutional Animal Care and Use Committee (IACUC). SKBR3 or SKBR3-pool2 cells were suspended in 100μL of PBS, mixed (1:1) with Matrigel (BD Biosciences, Franklin Lakes, NJ) and inoculated subcutaneously into the right flank of female athymic mice to generate xenograft tumors. Tumor formation was measured with fine calipers twice a week. Tumor volume was calculated by the formula: Volume = (Length × Width2)/2, where length was the longest axis and width the measurement at a right angle to the length. When SKBR3-pool2 tumor volume reached ~ 80mm3, the tumor-bearing mice received intraperitoneal (i.p.) injections of PBS or trastuzumab (20 mg/kg). Tumor growth curves were plotted using average tumor volume and followed by statistical analysis as described previously [29, 32]. To examine the antitumor activity of lapatinib against trastuzumab-resistant BC, SKBR3-pool2 or BT474-HR20 cells were suspended in 100μL of PBS, mixed with Matrigel (BD Biosciences), and inoculated subcutaneously into the right flank of female athymic mice to generate xenograft tumors. When tumor sizes reached ~ 80 mm3, mice were randomly divided into 2 groups and treated with vehicle control (DMSO) or lapatinib (80mg/kg, i.p.). The treatment was administered every other day. Tumor growth was measured every 3 days. At the end of treatment, mice were sacrificed, and the tumors were dissected and measured for weight. All tumors were collected for further analysis.
Immunohistochemistry (IHC) assay
IHC assays were performed as we described previously [47–49]. In brief, the tumors were formalin-fixed and paraffin-embedded. Five-micron-thick sections were deparaffinized in xylene and rehydrated with a series of graded alcohols. The slides were then treated with 3% hydrogen peroxide in methanol for 15 min. To exhaust endogenous peroxidase activity, the antigens were retrieved in 0.01 M sodium cirate buffer (pH 6.0) using a microwave oven. The slides were blocked with a blocking sniper (Biocare Medical, Pacheco, CA), and then incubated with a primary Ab described in the figure legends at 4°C overnight. After washing with Tris Buffer Saline (pH 8.0), the slides were incubated with a MACH 1 HRP Polymer detection kit (Biocare Medical) according to the manufacturer’s instructions. The staining colors were developed with a DAB Chromogen Kit (Biocare Medical). Finally, all sections were counterstained in Mayer’s hematoxylin, nuclei blued in 1% ammonium hydroxide (v/v), dehydrated, and then mounted with permanent aqueous mounting medium (Bio-Rad). Two individuals independently evaluated the IHC slides.
Quantification of IHC assays
Quantification of IHC assays was conducted as we reported using ImageJ and ImageJ plugin IHC profiler [47]. Briefly, the IHC images were imported into the software and followed by color deconvolution with IHC profiler. The images were then inverted into 8-bit grayscale type under the “Edit” menu of ImageJ. The “Measure” function of ImageJ was used to examine the mean intensity of the IHC images. Three fields of each IHC slide were evaluated and followed by statistical analysis.
Statistical analysis
Experimental data were statistically analyzed using two-sided student’s t tests. Significance was set at the P < 0.05. All values are reported at the mean ± SD.
Acknowledgements
The authors are grateful to Ms. Susan Theodosiou (Stanley S. Scott Cancer Center, School of Medicine, LSU Health New Orleans) for her critical reading of the manuscript.
Abbreviations
- BC
Breast cancer
- RTK
Receptor tyrosine kinase
- TKI
Tyrosine kinase ihibitor
- EGFR
Epidermal growth factor receptor
- HER2
Human epidermal growth factor receptor-2
- HER3
Human epidermal growth factor receptor-3
- mAb
Monoclonal antibody
- IRS1
Insulin receptor substrate-1
- IGF-I
Insulin-like growth factor-I
- IGF-1R
IGF-I receptor
- PTEN
Phosphatase and tensin homolog
- PI-3 K
Phosphoinositide 3-kinase
- MAPK
Mitogen-activated protein kinase
- IHC
Immunohistochemistry
Authors’ contributions
H.Liu, B.Liu, and H.Lyu did Conceptualization; H.Liu, S.R., C.T., and H.Lyu did Investigation; B.Liu and H.Lyu Wrote the main manuscript draft; M.E.L., B.Liu did review & editing; Funding acquisition, B.Liu; Resources, H.Liu, and H.Lyu; Supervision, B.Liu. All authors have read and agreed to the final version of the manuscript.
Funding
This work was supported in part by a translational research award from METAvivor Research and Support Inc. and a start-up fund provided by the Stanley S. Scott Cancer Center of LSUHSC (BL).
Availability of data and materials
All data generated for this study are included within this article. The data used and analyzed in the current study are available from the corresponding authors on reasonable request.
Declarations
Ethics approval and consent to participate
Animal experiments were conducted after approval by the IACUC at Louisiana State University (LSU) Health Sciences Center New Orleans or Affiliated Cancer Hospital & Institute of Guangzhou Medical University. All animals were maintained in accordance with the IACUC guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Bolin Liu, Email: bliu2@lsuhsc.edu.
Hui Lyu, Email: hlyu@lsuhsc.edu.
References
- 1.Hurvitz SA, Hu Y, O’Brien N, Finn RS. Current approaches and future directions in the treatment of HER2-positive breast cancer. Cancer Treat Rev. 2013;39(3):219–229. doi: 10.1016/j.ctrv.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jelovac D, Emens LA. HER2-directed therapy for metastatic breast cancer. Oncol (Williston Park) 2013;27(3):166–75. [PubMed] [Google Scholar]
- 3.Pernas S, Tolaney SM. HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol. 2019;11:1758835919833519. doi: 10.1177/1758835919833519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ponde N, Brandao M, El-Hachem G, Werbrouck E, Piccart M. Treatment of advanced HER2-positive breast cancer: 2018 and beyond. Cancer Treat Rev. 2018;67:10–20. doi: 10.1016/j.ctrv.2018.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Incorvati JA, Shah S, Mu Y, Lu J. Targeted therapy for HER2 positive breast cancer. J Hematol Oncol. 2013;6:38. doi: 10.1186/1756-8722-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehta AI, Brufsky AM, Sampson JH. Therapeutic approaches for HER2-positive brain metastases: circumventing the blood-brain barrier. Cancer Treat Rev. 2013;39(3):261–269. doi: 10.1016/j.ctrv.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen DL, Kumler I, Palshof JA, Andersson M. Efficacy of HER2-targeted therapy in metastatic breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Breast. 2013;22(1):1–12. doi: 10.1016/j.breast.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Patil A, Sherbet GV. Therapeutic approach to the management of HER2-positive breast cancer metastatic to the brain. Cancer Lett. 2015;358(2):93–99. doi: 10.1016/j.canlet.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 9.Vernieri C, Milano M, Brambilla M, Mennitto A, Maggi C, Cona MS, Prisciandaro M, Fabbroni C, Celio L, Mariani G, Bianchi GV, Capri G, de Braud F. Resistance mechanisms to anti-HER2 therapies in HER2-positive breast cancer: current knowledge, new research directions and therapeutic perspectives. Crit Rev Oncol Hematol. 2019;139:53–66. doi: 10.1016/j.critrevonc.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Brandao M, Ponde NF, Poggio F, Kotecki N, Salis M, Lambertini M, de Azambuja E. Combination therapies for the treatment of HER2-positive breast cancer: current and future prospects. Expert Rev Anticancer Ther. 2018;18(7):629–649. doi: 10.1080/14737140.2018.1477596. [DOI] [PubMed] [Google Scholar]
- 11.Choong GM, Cullen GD, O’Sullivan CC. Evolving standards of care and new challenges in the management of HER2-positive breast cancer. CA Cancer J Clin. 2020;70(5):355–74. doi: 10.3322/caac.21634. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, Liu S, Lyu H, Riker AI, Zhang Y, Liu B. Development of effective therapeutics targeting HER3 for Cancer Treatment. Biol Proced Online. 2019;21:5. doi: 10.1186/s12575-019-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wynn CS, Tang SC. Anti-HER2 therapy in metastatic breast cancer: many choices and future directions. Cancer Metastasis Rev. 2022;41(1):193–209. doi: 10.1007/s10555-022-10021-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amin DN, Sergina N, Lim L, Goga A, Moasser MM. HER3 signalling is regulated through a multitude of redundant mechanisms in HER2-driven tumour cells. Biochem J. 2012;447(3):417–25. doi: 10.1042/BJ20120724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, Manning HC, Chang J, Arteaga CL. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108(12):5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia W, Petricoin EF, 3rd, Zhao S, Liu L, Osada T, Cheng Q, Wulfkuhle JD, Gwin WR, Yang X, Gallagher RI, Bacus S, Lyerly HK, Spector NL. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2 + breast cancer models. Breast Cancer Res. 2013;15(5):R85. doi: 10.1186/bcr3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69(17):6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 19.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 20.Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–30. doi: 10.1158/0008-5472.CAN-08-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, O’Donovan N, Slamon DJ. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9(6):1489–502. doi: 10.1158/1535-7163.MCT-09-1171. [DOI] [PubMed] [Google Scholar]
- 22.Hutchinson L. Targeted therapies: activated PI3K/AKT confers resistance to trastuzumab but not lapatinib. Nat Rev Clin Oncol. 2010;7(8):424. doi: 10.1038/nrclinonc.2010.113. [DOI] [PubMed] [Google Scholar]
- 23.Xia W, Husain I, Liu L, Bacus S, Saini S, Spohn J, Pry K, Westlund R, Stein SH, Spector NL. Lapatinib Antitumor activity is not dependent upon phosphatase and Tensin Homologue deleted on chromosome 10 in ErbB2-Overexpressing breast cancers. Cancer Res. 2007;67(3):1170–1175. doi: 10.1158/0008-5472.CAN-06-2101. [DOI] [PubMed] [Google Scholar]
- 24.Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, Sliwkowski MX. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2(2):127–137. doi: 10.1016/S1535-6108(02)00097-1. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (herceptin).[comment] J Natl Cancer Inst. 2001;93(24):1852–7. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 26.Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–28. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, Esteva FJ, Liu B. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-i receptor in breast cancer cells resistant to herceptin. Cancer Res. 2010;70(3):1204–1214. doi: 10.1158/0008-5472.CAN-09-3321. [DOI] [PubMed] [Google Scholar]
- 28.Lyu H, Yang XH, Edgerton SM, Thor AD, Wu X, He Z, Liu B. The erbB3- and IGF-1 receptor-initiated signaling pathways exhibit distinct effects on lapatinib sensitivity against trastuzumab-resistant breast cancer cells. Oncotarget. 2016;7(3):2921–2935. doi: 10.18632/oncotarget.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, Liu B. The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer. 2013;12(1):134. doi: 10.1186/1476-4598-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muthuswamy SK. Trastuzumab resistance: all roads lead to SRC. Nat Med. 2011;17(4):416–418. doi: 10.1038/nm0411-416. [DOI] [PubMed] [Google Scholar]
- 31.Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, Sahin AA, Esteva FJ, Hortobagyi GN, Yu D. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17(4):461–469. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo L, Zhang Z, Qiu N, Ling L, Jia X, Song Y, Li H, Li J, Lyu H, Liu H, He Z, Liu B, Zheng G. Disruption of FOXO3a-miRNA feedback inhibition of IGF2/IGF-1R/IRS1 signaling confers herceptin resistance in HER2-positive breast cancer. Nat Commun. 2021;12(1):2699. doi: 10.1038/s41467-021-23052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlam I, Swain SM. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now. NPJ Breast Cancer. 2021;7(1):56. doi: 10.1038/s41523-021-00265-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins DM, Conlon NT, Kannan S, Verma CS, Eli LD, Lalani AS, Crown J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers. 2019;11(6):737. doi: 10.3390/cancers11060737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008;14(21):6730–6734. doi: 10.1158/1078-0432.CCR-08-0581. [DOI] [PubMed] [Google Scholar]
- 36.Xuhong JC, Qi XW, Zhang Y, Jiang J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am J Cancer Res. 2019;9(10):2103–19. [PMC free article] [PubMed] [Google Scholar]
- 37.D’Amato V, Raimondo L, Formisano L, Giuliano M, De Placido S, Rosa R, Bianco R. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev. 2015;41(10):877–883. doi: 10.1016/j.ctrv.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Haikala HM, Janne PA. Thirty years of HER3: from Basic Biology to therapeutic interventions. Clin Cancer Res. 2021;27(13):3528–3539. doi: 10.1158/1078-0432.CCR-20-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikink GJ, Hilkens J. Insulin receptor substrate 4 (IRS4) is a constitutive active oncogenic driver collaborating with HER2 and causing therapeutic resistance. MolCell Oncol. 2017;4(2):e1279722. doi: 10.1080/23723556.2017.1279722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikink GJ, Boer M, Bakker ER, Hilkens J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat Commun. 2016;7:13567. doi: 10.1038/ncomms13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rexer BN, Ham AJ, Rinehart C, Hill S, Granja-Ingram Nde M, Gonzalez-Angulo AM, Mills GB, Dave B, Chang JC, Liebler DC, Arteaga CL. Phosphoproteomic mass spectrometry profiling links src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 2011;30(40):4163–74. doi: 10.1038/onc.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peiro G, Ortiz-Martinez F, Gallardo A, Perez-Balaguer A, Sanchez-Paya J, Ponce JJ, Tibau A, Lopez-Vilaro L, Escuin D, Adrover E, Barnadas A, Lerma E. Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. Br J Cancer. 2014;111(4):689–695. doi: 10.1038/bjc.2014.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, Shou J, Bissell MJ, Osborne CK, Schiff R. beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011;13(4):R84. doi: 10.1186/bcr2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jegg AM, Ward TM, Iorns E, Hoe N, Zhou J, Liu X, Singh S, Landgraf R, Pegram MD. PI3K independent activation of mTORC1 as a target in lapatinib-resistant ERBB2 + breast cancer cells. Breast Cancer Res Treat. 2012;136(3):683–692. doi: 10.1007/s10549-012-2252-9. [DOI] [PubMed] [Google Scholar]
- 45.Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, Botero MF, Healy NA, Hilsenbeck SG, Phillips GL, Chamness GC, Rimawi MF, Osborne CK, Schiff R. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers–role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011;13(6):R121. doi: 10.1186/bcr3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, Yang SS, Yin PH, Chang T, Shi LX, Fang L, Fang GE. Activated estrogen receptor-mitogen-activated protein kinases cross talk confer acquired resistance to lapatinib. Thorac Cancer. 2015;6(6):695–703. doi: 10.1111/1759-7714.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S, Polsdofer EV, Zhou L, Ruan S, Lyu H, Hou D, Liu H, Thor AD, He Z, Liu B. Upregulation of endogenous TRAIL-elicited apoptosis is essential for metformin-mediated antitumor activity against TNBC and NSCLC. Mol Ther Oncolytics. 2021;21:303–14. doi: 10.1016/j.omto.2021.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyu H, Wang S, Huang J, Wang B, He Z, Liu B. Survivin-targeting mir-542-3p overcomes HER3 signaling-induced chemoresistance and enhances the antitumor activity of paclitaxel against HER2-overexpressing breast cancer. Cancer Lett. 2018;420:97–108. doi: 10.1016/j.canlet.2018.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu H, Lyu H, Jiang G, Chen D, Ruan S, Liu S, Zhou L, Yang M, Zeng S, He Z, Wang H, Li H, Zheng G, Liu B. ALKBH5-Mediated m6A demethylation of GLUT4 mRNA promotes glycolysis and resistance to HER2-Targeted therapy in breast Cancer. Cancer Res. 2022;82(21):3974–3986. doi: 10.1158/0008-5472.CAN-22-0800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated for this study are included within this article. The data used and analyzed in the current study are available from the corresponding authors on reasonable request.