

Abstract
Repeated exposure to opioids causes tolerance, which limits their analgesic utility and contributes to overdose and abuse liability. However, the molecular mechanisms underpinning tolerance are not well understood. Here, we used a forward genetic screen in C. elegans for unbiased identification of genes regulating opioid tolerance which revealed a role for PTR-25/Ptchd1. We found that PTR-25/Ptchd1 controls μ-opioid receptor (MOR) trafficking and that these effects were mediated by the ability of PTR-25/Ptchd1 to control membrane cholesterol content. Electrophysiological studies showed that loss of Ptchd1 in mice reduced opioid-induced desensitization of neurons in several brain regions and the peripheral nervous system. Mice and C. elegans lacking Ptchd1/PTR-25 display similarly augmented responses to opioids. Ptchd1 knockout mice fail to develop analgesic tolerance and have greatly diminished somatic withdrawal. Thus, we propose that Ptchd1 plays an evolutionarily conserved role in protecting MOR against overstimulation.
Introduction
GPCRs comprise the largest family of cellular receptors that mount responses to a staggering range of stimuli in all eukaryotes. Studies over the past two decades have shown that GPCR signaling involves transducers, adaptors and scaffolding proteins that control receptor trafficking and function1–3. However, the molecular and cellular mechanisms underlying several key GPCR properties remain poorly understood. One such dark area is tolerance, a behavioral phenomenon that ensues from the cellular process of GPCR desensitization. The ability to scale down responses following excessive stimulation is a fundamental feature of many GPCRs and is thought to be an adaptive physiological mechanism that prevents signaling overflow. Yet, desensitization poses substantial limitations for pharmacologically targeting GPCRs and developing effective therapeutics. A quintessential example of this problem is opioid tolerance, a profound loss of analgesic and euphoric efficacy upon chronic drug exposure4–6. Tolerance limits analgesic utility, drives escalation of use and leads to overdose.
The clinically significant effects of opioids are mediated by their target GPCR, the μ-opioid receptor (MOR)7,8. Previous studies have shown that alterations in MOR signaling and trafficking impact the development of tolerance in a cell autonomous manner9,10. A critical role in this process has been attributed to a GPCR adaptor, β-arrestin11,12, although its involvement in opioid tolerance has been contested13. In addition, a few disparate molecules have been implicated in opioid tolerance14–16. While there has been significant progress in delineating the mechanisms of opioid tolerance and desensitization9,17, the molecular landscape of players and processes that GPCRs utilize to diminish their responses upon persistent stimulation remains incompletely defined.
Here, we designed and conducted an in vivo forward genetic screen in C. elegans for regulators of opioid tolerance, culminating in the identification of an evolutionarily conserved, poorly understood PTR-25/Ptchd1 system as a key enabler of opioid tolerance. Delineating its mechanism of action revealed that Ptchd1 controls desensitization of opioid responses by regulating MOR trafficking in a cholesterol-dependent manner.
Results
Genetic behavioral screen for modulators of opioid tolerance
We recently engineered an in vivo whole animal model for studying opioid-induced behavior and signaling by transgenically expressing mammalian MOR (tgMOR) in the nervous system of C. elegans18. Exposure of tgMOR animals to opioids suppresses their locomotion, which is used as a behavioral readout of opioid action (Fig. 1a; Extended Data Fig. 1a). We leveraged this model to design a behavioral assay for opioid tolerance (Fig. 1b). When tgMOR worms were repeatedly exposed to the opioid fentanyl, the paralysis-inducing effect of the drug was markedly attenuated but restored by increasing the fentanyl concentration, the hallmark of behavioral tolerance (Fig. 1c). Dose response studies further confirmed induction of tolerance (Fig. 1f, Extended Data Fig.1a–c), evident from the increase in the half maximal effective concentration (EC50) (Fig 1h). Similar behavioral tolerance was observed with morphine (Extended Data Fig. 1g, h).
Figure 1. Forward genetic screen in C. elegans identifies PTR-25/Ptchd1 as a regulator of opioid tolerance.
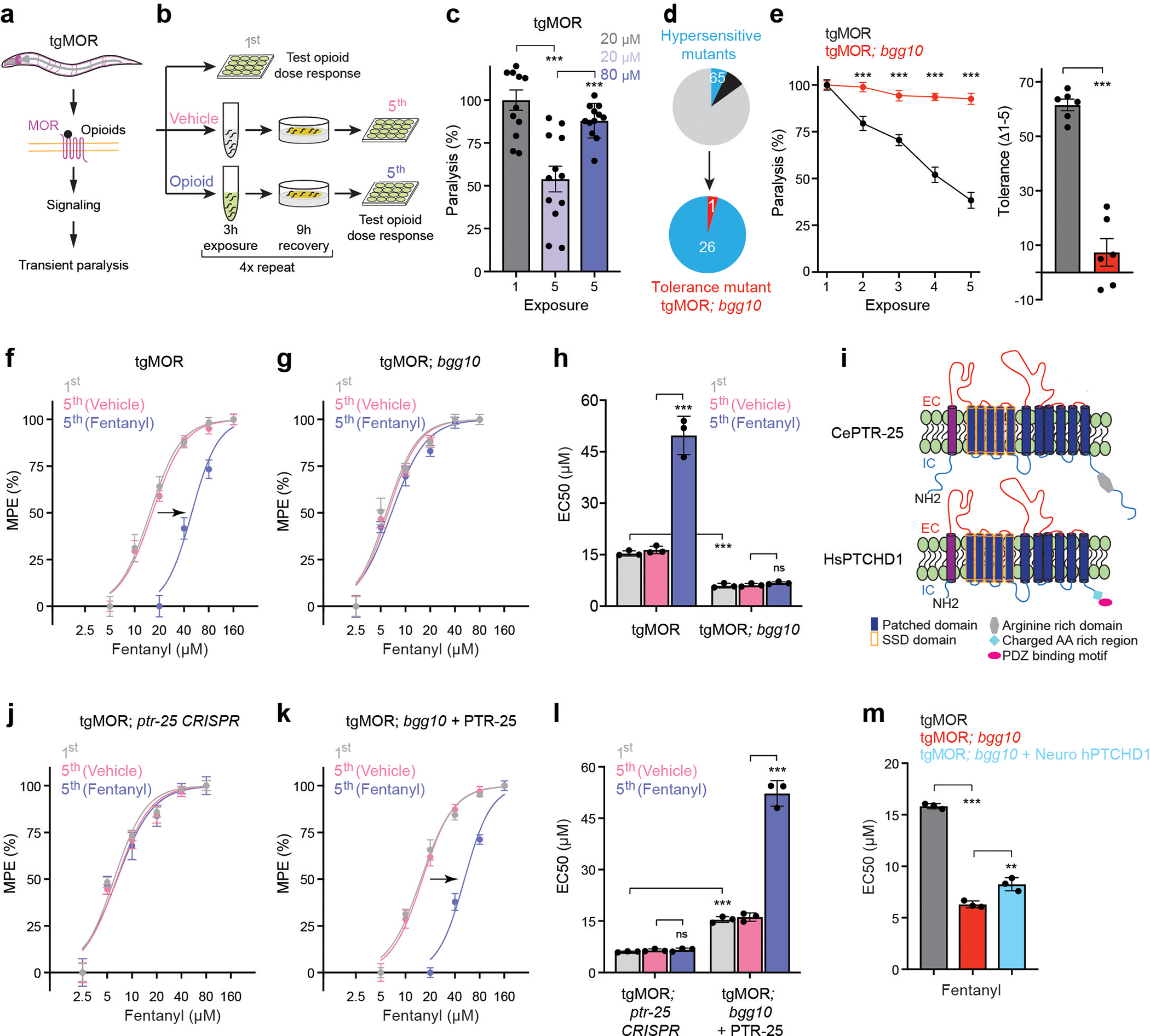
a, Schematic of C. elegans tgMOR opioid model.
b, Opioid tolerance assay using tgMOR C. elegans. Opioid dose-response analyzed before (1st) and after (5th) repeated exposures to vehicle or opioid.
c, Quantitation of fentanyl induced paralysis before (1st) and after (5th) repeated exposures. n=12 wells (4–5 animals/well) for each condition. Data derived from n=3 independent experiments.
d, Summary of results from two-stage forward genetic screen for tgMOR tolerance mutants from hypersensitive mutant collection.
e, Quantitation of fentanyl (10 μM) induced paralysis with repeated drug exposure (left). n=24 wells (4–5 animals/well). Change in fentanyl response calculated as difference between 1st and 5th exposure. n=6 independent experiments with 4 wells/experiment.
f, Dose-response curves showing that tgMOR animals develop tolerance upon repeated (5x) exposure to fentanyl (10 μM). Paralysis was measured and quantified as maximal possible effect (MPE) based on time to paralysis.
g, Dose-response curves analyzing tolerance in tgMOR; bgg10 animals after repeated (5x) exposure to fentanyl (10 μM).
h, Quantitation of fentanyl responsiveness for indicated genotypes (EC50 values for f and g).
i, Topology of C. elegans PTR-25 and human PTCHD1.
j, Dose-response curves analyzing tolerance in tgMOR; ptr-25 CRISPR animals after repeated (5x) exposure to fentanyl (10 μM).
k, Dose-response curves analyzing tolerance in tgMOR; bgg10 animals expressing transgenic PTR-25 after repeated (5x) exposure to fentanyl (10 μM).
l, Quantitation of fentanyl responsiveness for indicated genotypes (EC50 values for j and k).
m, Quantitation of fentanyl responsiveness for indicated genotypes (EC50 values).
For panels c, h, l, m, statistical comparisons were performed by one-way ANOVA with Bonferroni’s post hoc test. Data in pane1 e (left) was analyzed using two-way ANOVA followed by post hoc Bonferroni’s test and in panel e (right) using a two-tailed unpaired Student’s t-test. In panels c and e, data points and histograms show means with error bars representing SEM. In panels f-h and j-m, data points and histograms are means with error bars representing SD. For f-h and j-m, n = 3 independent experiments with each experiment derived from 4 wells (4–5 animals/well) per dose for all genotypes. **p<0.01, ***p<0.001
To further validate the tgMOR platform for tolerance, we evaluated the behavior of an opioid hypersensitive mutant, tgMOR; rsbp-1. Knockout of R7BP, the mammalian ortholog of RSBP-1, in mice produces opioid hypersensitivity and eliminates tolerance14. Indeed, tgMOR; rsbp-1 mutants failed to diminish responsiveness upon repeated exposure to fentanyl (Extended Data Fig. 2a, b). Thus, a known tolerance regulator in mice is required for opioid tolerance in C. elegans. To test whether opioid tolerance is selective or generally observed in mutants with increased opioid sensitivity, we evaluated tgMOR; frpr-13 mutants for tolerance. FRPR-13 is a recently discovered conserved inhibitor of MOR signaling18. In contrast to tgMOR; rsbp-1 animals, tgMOR; frpr-13 mutants had normal tolerance despite increased sensitivity to fentanyl (Extended Data Fig. 2c, d). Thus, opioid tolerance can be faithfully modeled in C. elegans and is governed by conserved genetic mechanisms.
Having established the validity of the tgMOR platform for studying opioid tolerance, we implemented a two-stage genetic behavioral screen to identify mutants with impaired tolerance (Fig. 1d). The first stage in this forward genetic approach isolated mutants with altered responses to opioids18. In the second stage presented here, we screened a collection of hypersensitive tgMOR mutants for altered opioid tolerance based on prior evidence that tgMOR hypersensitive mutants affect MOR signaling18 and observations from rodents that loss of tolerance is accompanied by increased opioid responsiveness12,14. In total, we screened 27 hypersensitive tgMOR mutants and identified only one strain, tgMOR; bgg10, with impaired opioid tolerance (Fig. 1d). In fact, when compared to the parental tgMOR strain, tgMOR; bgg-10 mutants developed no observable tolerance to repeated fentanyl administration across 5 testing sessions (Fig. 1e). Dose-response studies showed a prominent right-ward shift in potency in tgMOR worms following chronic fentanyl treatment (Fig. 1f) and no shift in tgMOR; bgg10 mutants (Fig. 1g). This was quantitatively evaluated by showing that EC50 increases in tgMOR animals but not tgMOR; bgg10 mutants (Fig. 1h). Collectively, these results demonstrate that tgMOR; bgg10 mutants are devoid of opioid tolerance.
Our analysis further confirmed that tgMOR; bgg10 mutants were indeed hypersensitive and responded to opioids with faster paralysis (Fig. 1h, Extended Data Fig. 3). Further, bgg10 mutants were similar to non-transgenic, wild-type animals in failing to respond to opioids in the absence of tgMOR (Extended Data Fig. 3), indicating observed effects were specifically mediated by changes in MOR responses. Thus, deploying a two-stage unbiased, forward genetic screen led to the identification of a rare mutant with impaired behavioral tolerance to opioids.
PTR-25/Ptchd1 regulates opioid tolerance in C. elegans
Next, we sought to identify the genetic lesion in tgMOR; bgg10 that causes the opioid tolerance phenotype. To do so, we mapped mutations in tgMOR; bgg10 using whole genome sequencing, opioid phenotypic selection, and computational analysis of genome sequence data19. We found that tgMOR; bgg10 mutants harbored a predicted premature stop codon, Q1032stop (3094C>T) that is likely to generate a loss-of-function mutation in an unannotated gene F43D9.1 (Extended Data Fig. 4a–c). F43D9.1 encodes a protein with a Patched (Ptch) family domain and twelve-transmembrane topology. These are canonical features of the Ptch protein family that includes Patched, Dispatched, and numerous Patched-like proteins (Extended Data Fig. 4d)20. In C. elegans, F43D9.1 is part of a large, evolutionarily expanded group of Patched related family (PTR) proteins (Extended Data Fig. 4d), prompting us to name this protein PTR-25. Our analysis indicates that Ptchd1 and Ptchd4 are the closest mammalian orthologs to PTR-25 (Fig. 1i; Extended Data Fig. 4d). Like PTR-25, Ptchd1 and Ptchd4 contain a Ptch domain and a sterol sensing domain (SSD). Only Ptchd1 is expressed in the mammalian nervous system and implicated in neuropsychiatric conditions21–23, but relatively little is known about its function or mechanism of action.
To confirm that the ptr-25 mutation affected opioid responses, we began by using CRISPR/Cas9 to edit the same premature stop codon, Q1032stop (3094C>T), present in tgMOR; bgg10 mutants into the parental tgMOR strain. This premature stop is predicted to truncate PTR-25 resulting in loss of the C-terminal intracellular portion of the receptor (Extended Data Fig. 4c). We found that tgMOR; ptr-25 CRISPR animals exhibited no opioid tolerance (Fig. 1j, l) and showed opioid hypersensitivity (Extended Data Fig. 5a). As a second validation, we performed transgenic rescue experiments. A Mos single copy insertion (MosSCI) was used to express PTR-25 with its endogenous promoter in tgMOR; bgg10 mutants. This completely restored behavioral tolerance to fentanyl (Fig. 1k, l) and opioid sensitivity (Extended Data Fig.5b). Transgenic MosSCI expression of PTR-25 using a pan-neuronal promoter also rescued opioid hypersensitivity of tgMOR; ptr-25 (bgg11) animals (Extended Data Fig. 5c). Collectively, these results indicate that PTR-25 is the causal gene responsible for loss of opioid tolerance and hypersensitivity in tgMOR; bgg10 mutants.
Finally, we tested whether Ptchd1 affects MOR-mediated opioid responses by transgenically expressing human PTCHD1 in the nervous system of tgMOR; bgg10 animals. We observed a significant rescue of hypersensitivity to fentanyl in these animals (Fig. 1m; Extended Data Fig. 5d, e). In summary, our results provide in vivo genetic evidence that PTR-25 and Ptchd1 are functional orthologs that regulate opioid tolerance and responsiveness.
Ptchd1 loss in mice enhances opioid efficacy and eliminates tolerance
To test the role of Ptchd1 in regulating opioid responses in mammals and to probe its translational relevance, we evaluated mice with a disruption in Ptchd1 (Ptchd1 KO). Previously, Ptchd1 KO mice were reported to be hyperactive22. When tested in the open field, Ptchd1 KO mice indeed showed increased locomotion upon introduction into a novel environment (Extended Data Fig. 6a,b). However, Ptchd1 KO animals gradually reduced their activity to levels seen in wild-type (WT) littermates (Extended Data Fig. 6c). We also found that hyperactivity of Ptchd1 KO mice was reversed by treatment with an opioid receptor antagonist, naltrexone (Extended Data Fig. 6d) suggesting that elevated activity in Ptchd1 KO mice is related to increased endogenous MOR signaling rather than developmental issues.
To initially evaluate behavioral responses of Pthcd1 KO mice to opioid drugs we used the conditioned place preference (CPP) assay. This test evaluates the reinforcing properties of opioids by measuring relative amount of time spent in a drug-paired chamber as compared to an unpaired chamber. Indeed, at the no-drug baseline, both Ptchd1 KO mice and their WT littermates behaved equally (Fig. 2a). When injected with morphine systemically, Ptchd1 KO mice exhibited substantially higher preference for the drug-paired chamber indicating they have an augmented response to the rewarding effects of morphine (Fig. 2a).
Figure 2. Knockout of Ptchd1 in mice enhances opioid efficacy and eliminates tolerance.
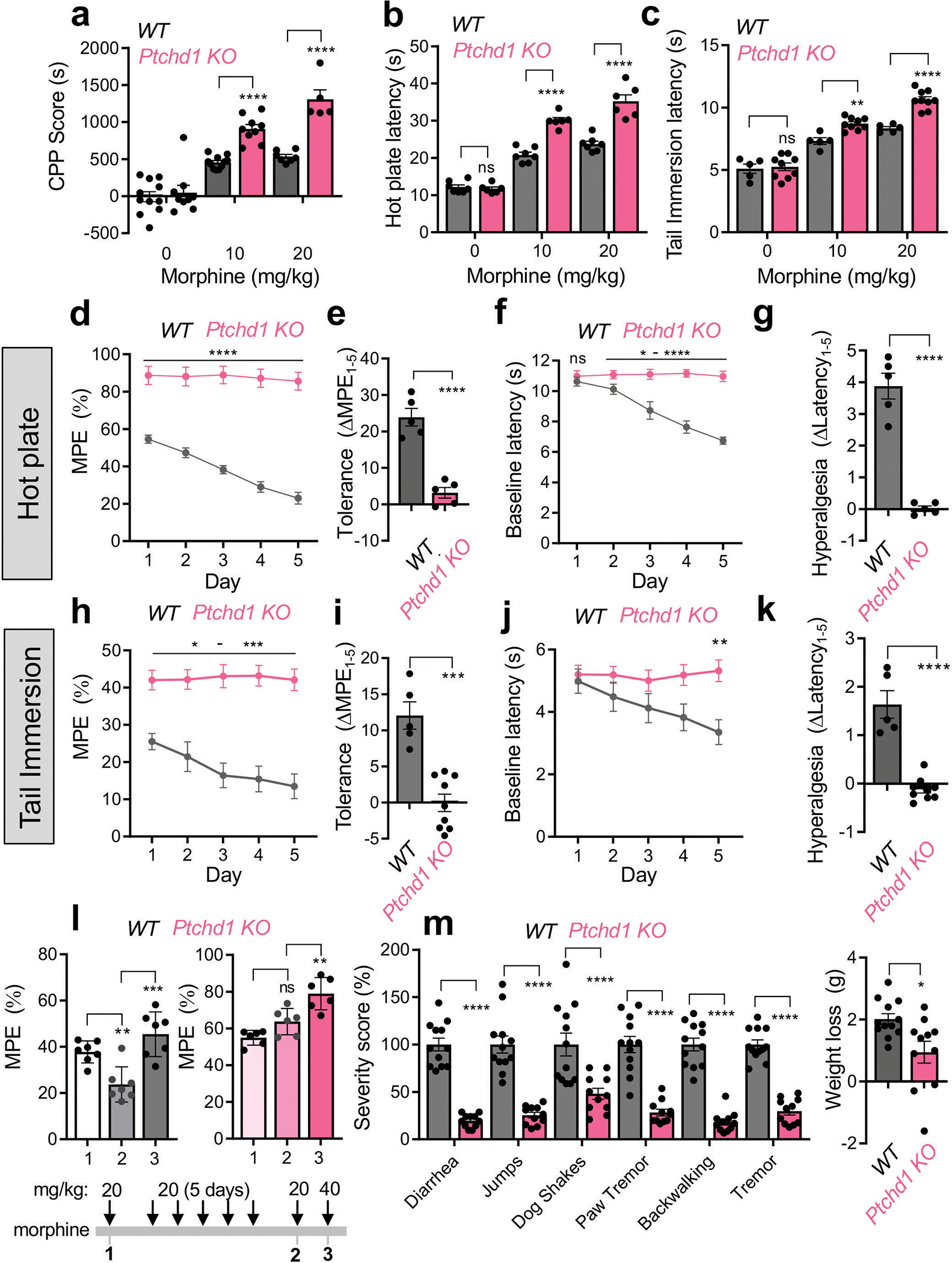
a, Evaluation of morphine reward by conditioned place preference test. For the 0 mg/kg and 10 mg/kg dose, n=11 animals per genotype; for the 20 mg/kg dose, n=7 WT and n=5 Ptchd1 KO mice. b, Evaluation of analgesia by hot plate assay. n=7 WT and n=6 Ptchd1 KO mice.
c, Evaluation of analgesia by tail immersion assay. n=5 WT and n=9 Ptchd1 KO mice.
d, Evaluation of analgesic tolerance by hot plate assay. n=5 male mice per genotype
e, Quantification of analgesic efficacy reduction as a difference in the MPE between session 1 and 5 from panel d.
f, Evaluation of opioid induced hyperalgesia by hot plate assay.
g, Quantification of opioid induced hyperalgesia as a difference in the baseline latencies between session 1 and 5 from panel f.
h, Evaluation of analgesic tolerance by tail immersion assay. n=5 WT and n=9 Ptchd1 KO mice.
i, Quantification of analgesic efficacy reduction as a difference in the MPE between session 1 and 5 from panel h.
j, Evaluation of opioid induced hyperalgesia by tail immersion assay. Baseline response latencies of mice receiving repeated morphine injections (in panel h).
k, Quantification of opioid induced hyperalgesia as a difference in the baseline latencies between session 1 and 5 from panel j.
l, Dose-response profile of analgesic tolerance. Mice were evaluated by tail immersion assay before and after repeated injections of morphine (20 mg/kg). n=7 WT and n=6 Ptchd1 KO mice.
m, Evaluation of somatic withdrawal following precipitated withdrawal from chronic morphine administration. n=12 WT and n=11 Ptchd1 KO mice.
For panels a, b, and c, statistical comparisons were performed by 2-way ANOVA and Šídák’s post hoc comparison. For panels d, f, h, and j the statistical comparisons were performed by 2-way ANOVA and Bonferroni’s post hoc comparison. For panels e, g, i, k, and m, statistical comparisons were performed by unpaired two tailed Student’s t-test. In panel l data was analyzed by one-way ANOVA with Tukey’s post hoc comparison. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Mean values with S.E.M. errors are shown.
Next, we tested nocifensive behavior of mice in pain paradigms. We found that baseline nociceptive thresholds of Ptchd1 KO animals were similar to WT littermates in both hot plate and tail immersion assays (Fig. 2b, c; Supplemental Data Videos 1, 2). However, morphine produced a greater analgesic response in Ptchd1 KO mice relative to WT controls in both paradigms when injected systemically (Fig. 2b, c; Extended Data Fig. 6e; Supplemental Data Videos 3, 4). Morphine also produced greater analgesia in Ptchd1 KO mice when injected directly into the brain (Extended Data Fig. 6f, g). Furthermore, Ptchd1 KO mice exhibited similarly enhanced analgesia in response to fentanyl (Extended Data Fig. 6h, i). These data indicate that Ptchd1 restricts the analgesic efficacy of opioids in mice.
We next assessed tolerance by subjecting mice to repeated daily opioid injections. When injected with morphine, WT mice displayed a substantial reduction in opioid analgesia in the hot plate assay with each subsequent morphine application (Fig. 2d, e). This was accompanied by the development of hyperalgesia as evidenced by progressive reduction in baseline latency (Fig. 2f, g). In contrast, this chronic treatment regimen in Ptchd1 KO mice failed to diminish responsiveness suggesting chronic tolerance does not occur (Fig. 2d, e). Ptchd1 KO mice also did not develop morphine induced hyperalgesia (Fig. 2f, g). We then doubled our chronic tolerance induction window and used fentanyl (Extended Data Fig. 6j, k). Again, we found that Ptchd1 KO mice did not develop any signs of tolerance even in this extended chronic paradigm with a stronger opioid analgesic. Ptchd1 KO mice also did not develop opioid induced hyperalgesia in this paradigm (Extended Data Fig. 6l, m).
We further evaluated Ptchd1 KO mice using an acute tolerance paradigm where morphine injection was repeated immediately upon waning of the initial analgesic response. While WT mice completely lost their morphine responsiveness to repeated doses, there was no sign of acute tolerance in Ptchd1 KO (Extended Data Fig 6n, o).
Our observations using the tail immersion assay, which principally relies upon spinal reflexes, largely paralleled our findings with the hot plate assay. Once again, we observed that Ptchd1 KO mice do not develop tolerance or hyperalgesia (Fig. 2h–k). Dose-escalation studies showed that increasing morphine restored the loss of efficacy in WT mice, and augmented analgesia in Ptchd1 KO animals confirming tolerance was abrogated (Fig. 2l). In fact, the responses of Ptchd1 KO mice were sometimes sensitized following opioid exposure (e.g. Extended Data Fig. 6n,o). We further probed the local autonomy of these effects by intrathecal injections of morphine directly into spinal cord. Again, we observed both the augmentation of opioid responsiveness (Extended Data Fig 6p) and no tolerance in Ptchd1 KO mice (Extended Data Fig 6q,r) with this site specific manipulation.
Finally, we evaluated how loss of Ptchd1 affects dependence following chronic opioid exposure. Strikingly, Ptchd1 KO mice had greatly reduced somatic withdrawal signs and showed significantly less body weight loss following chronic morphine treatment (Fig. 2m). Thus, reduced withdrawal response accompanies impaired tolerance in Ptchd1 KO animals. Taken as a whole, our observations demonstrate that Ptchd1 negatively regulates behavioral responses to opioids and development of tolerance in mice, similar to its ortholog PTR-25 in C. elegans.
Ptchd1 controls MOR-mediated regulation of neuronal activity
To determine how loss of Ptchd1 affects behavioral responses to opioids, we examined the relationship between Ptchd1 and MOR in mouse neural circuits using slice electrophysiology. To identify neurons expressing Ptchd1 and MOR, we crossed Ptchd1 KO mice expressing YFP from a targeted allele with knock-in MOR-mCherry reporter mice (Extended Data Fig. 7a). Consistent with previous reports 21,22, we found Ptchd1 expression in several regions of the central nervous system. This included prominent expression in areas associated with reward and opioid actions including nucleus accumbens (NAc), ventral tegmental area (VTA), thalamus and locus coeruleus (Fig. 3a, b; Extended Data Fig. 7b–g). Importantly, we found that Ptchd1 was co-expressed with MOR in many of these brain regions (Fig 3a, b; Extended Data Fig. 7b–g). Furthermore, we observed Ptchd1 expression in peripheral dorsal root ganglia (DRG) neurons where opioids can also act (Fig 3i, j). Co-expression was particularly prominent in the VTA and in the DRGs where the majority of MOR-expressing neurons expressed Ptchd1 (Fig. 3c,i). We further confirmed co-expression of endogenous MOR and Ptchd1 mRNA using in situ hybridization (Extended Data Fig. 8a). This pattern of Ptchd1 expression in mice is consistent with its broad presence across the human nervous system23,24.
Figure 3. Ptchd1 regulates opioid efficacy and response desensitization.
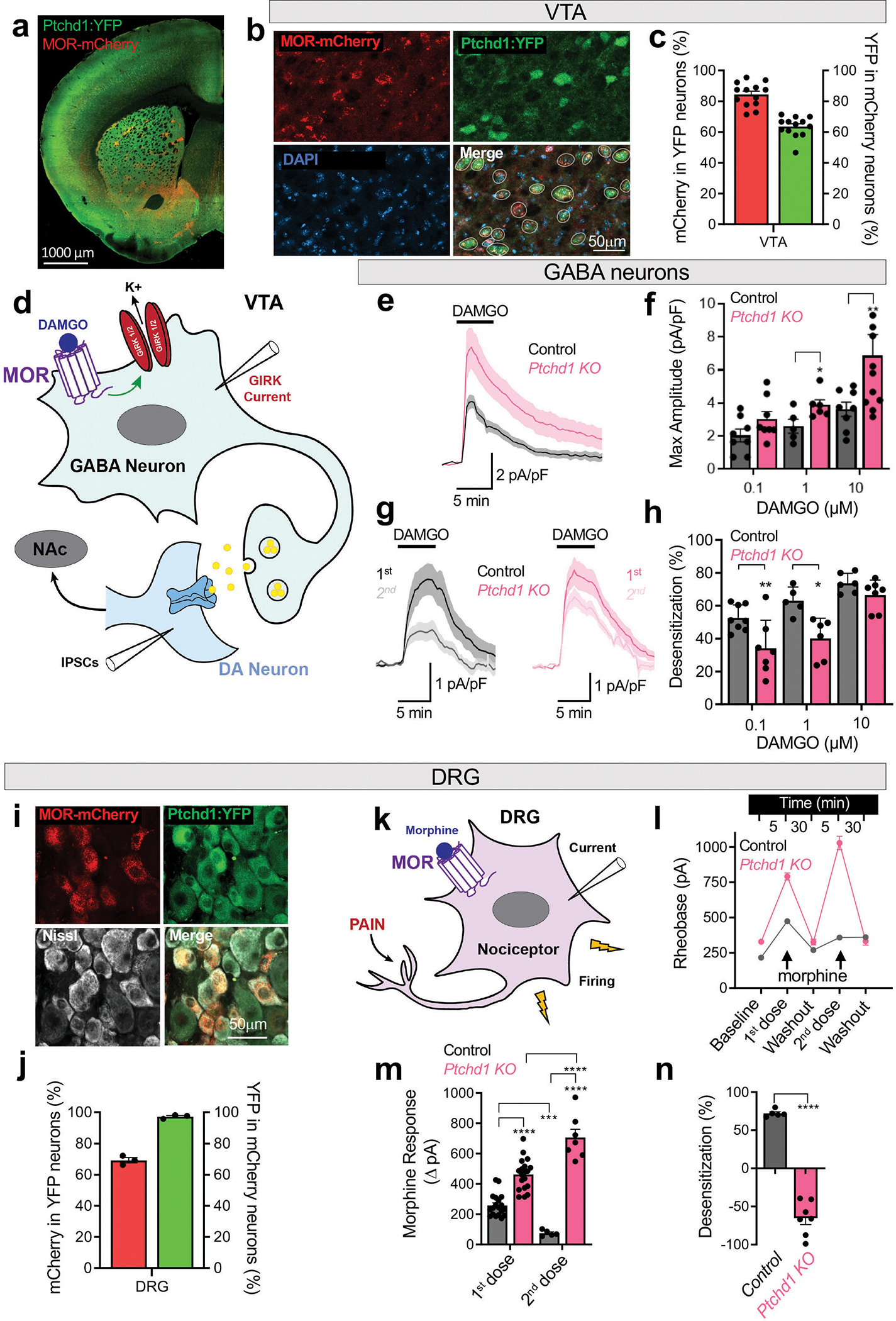
a, Distribution of MOR-mCherry and PTCHD1-YFP in a representative coronal section.
b, Co-expression of MOR and Ptchd1 in the ventral tegmental area (VTA).
c, Quantification of MOR and Ptchd1 coexpression in the VTA. 239 neurons, n=13 mice.
d, Circuit diagram of recording sites in the VTA.
e, Traces of averaged GIRK current evoked by 10μM DAMGO in GABA neurons. Amplitudes ranged from 19 pA to 333 pA across genotypes and conditions.
f, Quantification of maximal GIRK response amplitudes elicited by DAMGO in GABA neurons. In control n=8,5,8 cells from 4,4,5 mice; in Ptchd1 KO n=8,6,10 cells from 6,4,5 mice for 0.1, 1 and 10 μM DAMGO, respectively
g, Traces of averaged GIRK currents evoked by repeated application of 1 μM DAMGO.
h, Quantification of desensitization between paired first and second DAMGO-evoked GIRK currents. In control n=8,5,6 cells from 4,4,5 mice; in Ptchd1 KO n=7,6,7 cells from 6,4,5 mice for 0.1, 1 and 10 μM DAMGO, respectively
i, Co-expression of MOR-mCherry, PTCHD1-YFP and Nissl (white) in DRGs.
k, Diagram of a DRG nociceptor.
l, Quantification of rheobase at baseline and in response to repeated application of 10μM morphine in Ptchd1 KO and control DRG nociceptors. In control n=19 and 5 cells, 4 mice; in Ptchd1 KO n=19 and 7 cells, 4 mice for first and second treatment, respectively.
j, Quantification of MOR and Ptchd1 coexpression in DRG. 1126 neurons, n=3 mice.
m, Quantification of morphine responses as change in rheobase from baseline to 1st application of 10μM morphine, and washout to 2nd application of 10 μM morphine for Ptchd1 KO and control DRG nociceptors. Numbers are the same as in panel l.
n, Quantification of desensitization between first and second morphine responses in Ptchd1 KO and control DRG nociceptors. n=5 cells, 4 mice for control and n=7 cells, 4 mice in Ptchd1 KO.
For panels f and h and n, the statistical comparisons by unpaired two-tailed student’s t-test. For panel m, by a mixed-model ANOVA with Holm-Šídák post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Mean values with S.E.M. are shown.
We further examined brain region size, cell density and neuron size in Ptchd1 KO mice. Our results indicate that Ptchd1 elimination had no or minimal effects on the size of the VTA or NAc, and cell density in these brain regions (Extended Data Fig. 8b). Neuron size was also unaltered in the VTA and NAc, while a very small but significant increase was detected in the size of DRG neurons (Extended Data Fig. 8c). Overall, our results are consistent with previous morphological findings21,25.
To dissect the cellular mechanisms of Ptchd1 action, we first focused on the VTA neurons because the effects of opioids in this brain region are well understood. Here, MOR is predominantly expressed by the GABAergic interneurons where it activates G protein inwardly rectifying K+ (GIRK) channels reducing inhibitory output (Fig. 3d). Application of the MOR agonist DAMGO elicited GIRK-mediated inward currents, which were significantly augmented upon loss of Ptchd1 (Fig. 3e, f). Repeated application of DAMGO after washout resulted in smaller GIRK currents relative to the first exposure due to receptor desensitization (Fig. 3g, h), which is thought to be a cellular correlate of tolerance9,26. Importantly, less desensitization occurred in Ptchd1 KO neurons (Fig. 3g, h). Dose-response studies confirmed these observations and additionally revealed that loss of Ptchd1 preferentially augmented opioid efficacy, as the deletion of Ptchd1 had a larger effect on MOR-mediated GIRK regulation with increasing DAMGO concentration (Fig. 3f). In contrast, loss of desensitization was more prominent upon low level of MOR stimulation (Fig. 3h).
The increased MOR activation in GABAergic neurons in Ptchd1 KO would be expected to reduce their inhibitory influence over dopamine (DA)-releasing neurons. DA neurons in the VTA directly encode reward through changes in dopamine release, which is thought to be responsible for the reinforcing effects of opioids27 (Fig. 3d). Therefore, we next measured opioid modulation of GABAergic inputs into DA neurons by recording Inhibitory Post Synaptic Currents (IPSC). Application of DAMGO reduced IPSC frequency to a greater extent in the Ptchd1 KO compared to WT slice (Extended Data Fig. 9a). Challenging the same preparation with an escalating concentration of DAMGO did not augment the responses in WT neurons, likely due to MOR being fully desensitized (Extended Data Fig. 9b). In contrast, we observed increased inhibition in the Ptchd1 KO with increasing DAMGO concentrations, which is consistent with MOR-desensitization being impaired (Extended Data Fig. 9b).
To explore how general the role of Ptchd1 is in the brain, we next recorded from striatal medium spiny neurons (MSN) of the NAc (Extended Data Fig. 9c), a neuronal population relevant for opioid actions where Ptchd1 and MOR are co-expressed (Extended Data Fig. 7d, 8a). In these experiments, we used morphine to further probe agonist specificity of Ptchd1 effects. Similar to the VTA, we observed larger opioid responsiveness of MSNs lacking Ptchd1 (Extended Data Fig. 9d). Upon repeated morphine challenge, we observed pronounced desensitization in control neurons which did not occur in Ptchd1 KO neurons (Extended Data Fig. 9e, f).
Finally, we turned our attention to DRGs which co-express MOR and Ptchd1 (Fig. 3i, j; Extended Data Fig. 8a), and are a major peripheral site for MOR-mediated effects on analgesia and tolerance10,28. We relied on a genetic labeling strategy coupled with electrophysiological profiling to record specifically from the nociceptors that are positive for MOR and Ptchd1 expression (Fig. 3k). Similar to the central nervous system, we found that opioid-mediated suppression of excitability was significantly more pronounced in Ptchd1 KO nociceptors relative to controls (Fig. 3l, m). Strikingly, while control neurons substantially desensitized upon repeated application of morphine, we observed that excitability of Ptchd1 KO nociceptors was suppressed even more prominently upon morphine re-exposure (Fig. 3l, m). Thus, instead of desensitization, DRG nociceptors from Ptchd1 KO animals increase their sensitivity to morphine which parallels behavioral sensitization of Ptchd1 KO in the tail immersion paradigm (Fig 2h,i,l).
Based on these electrophysiological experiments, we conclude that Ptchd1 acts in the same neuronal populations as MOR across the nervous system to suppress opioid modulation of neuronal activity and to promote desensitization of MOR responses.
Ptchd1 controls MOR trafficking and surface abundance
We next set out to determine the molecular mechanism by which Ptchd1 influences MOR function. Given the increased efficacy of opioid responses at the behavioral and cellular levels in Ptchd1 KO animals, we examined whether Ptchd1 affects MOR expression and localization to impact its signaling strength. For these studies, we turned to cell-based assays in HEK293 cells reconstituted with MOR. When expressed alone, MOR was robustly localized on the plasma membrane (Fig. 4a–c). In contrast, introduction of Ptchd1 promoted intracellular retention of MOR and reduced its targeting to the plasma membrane (Fig. 4a–c; Extended Data Fig. 10a, b). To examine MOR localization dynamics, we employed a nanoluciferase complementation system (Fig. 4d). Again, over-expression of Ptchd1 decreased the surface abundance of MOR (Fig. 4e) without affecting overall MOR expression (Fig. 4f; Extended Data Fig. 10c). These results indicate that Ptchd1 is sufficient for regulating MOR trafficking under basal conditions.
Figure 4. Molecular mechanism of Ptchd1 action in MOR regulation.
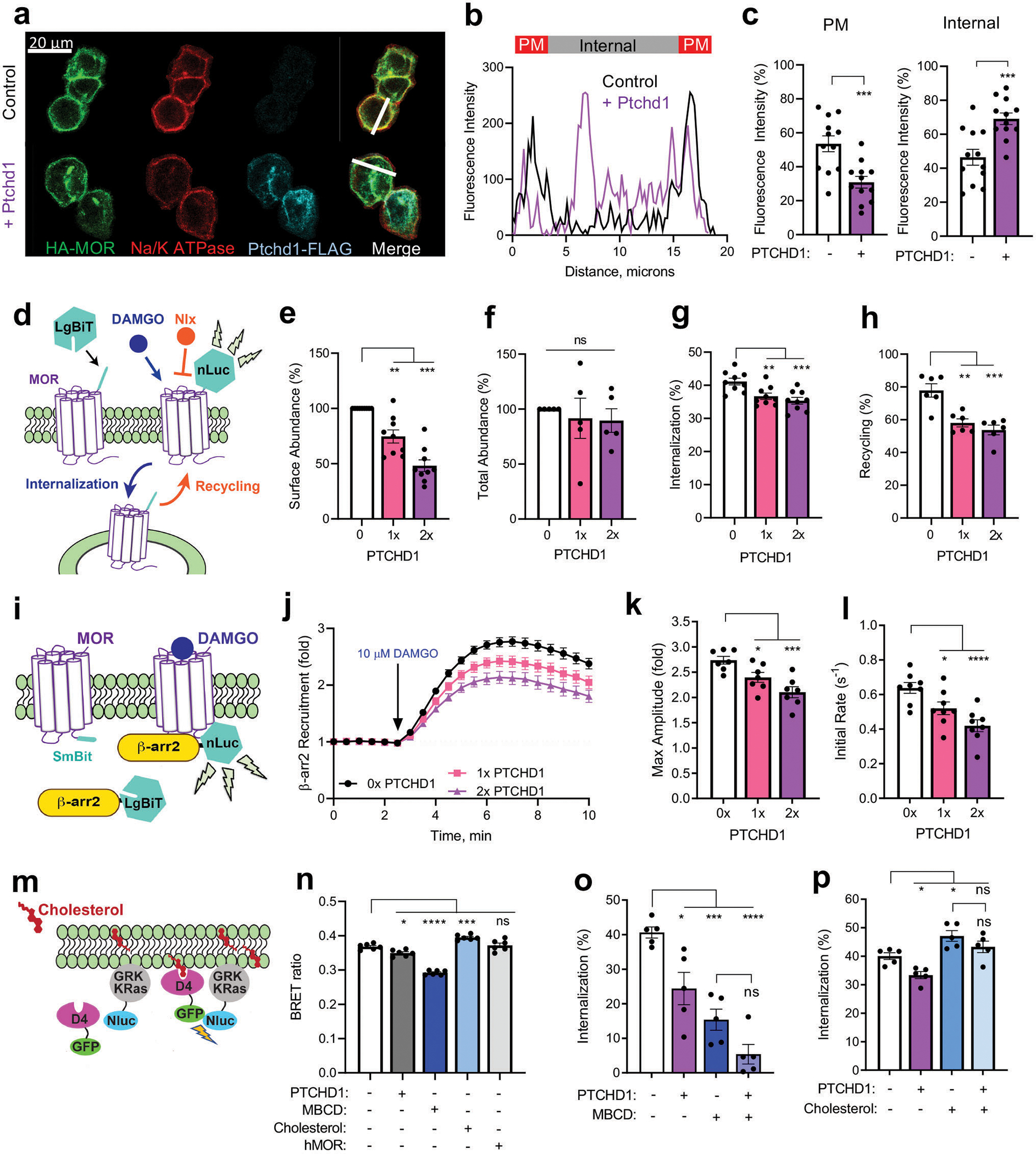
a, Co-expression of HA-MOR and PTCHD1-FLAG in HEK293 cells.
b, MOR fluorescence intensity distribution across white line in a. One representative cell is shown for each condition.
c, Quantification of subcellular MOR distribution from 12 cells for each genotype.
d, Experimental design of assays determining cell surface abundance, internalization and recycling of MOR.
e, Ptchd1 effect on MOR surface abundance from n=9 biologically independent experiments. Varying levels (1x, 2x) of Ptchd1 cDNA were transfected.
f, Ptchd1 effect on total MOR levels from n=5 biologically independent experiments.
g, Ptchd1 effect on MOR internalization. Loss of nLuc signal 17.5 min following addition of 10 μM DAMGO determined in n=9 biologically independent experiments.
h, Ptchd1 effect on MOR recycling. Restoration of nLuc signal 60 min after treatment with 100 μM naloxone determined in n=6 biologically independent experiments and normalized to surface abundance.
i, Experimental design of β-arrestin recruitment assay.
j, Time course of Ptchd1 effect on recruitment of β-arr2 to MOR determined in n=7 biologically independent experiments.
k, The effect of Ptchd1 on the maximum fold-change of β-arr2 recruitment from data in j.
l, The initial rate of β-arr2 recruitment to MOR from data in j. Data are from n=5 biologically independent experiments.
m, Schematic illustration of assay design for determining cholesterol content in the plasma membrane.
n, The effect of Ptchd1 and other manipulations in the cholesterol BRET assay. Data are from n=6 biologically independent experiments.
o, The effect of Ptchd1 on MOR internalization following cholesterol depletion by 4 mM MBCD (methyl-β-cyclodextrin). Data are from n=5 biologically independent experiments.
p, Ptchd1 effects of on DAMGO-mediated MOR internalization following treatment with 200 μg/ml cholesterol. Data are from n=5 biologically independent experiments.
For panel c statistical comparisons were performed by an unpaired two tailed Student’s t-test. In panels e, f, g, h, k and l the results were analyzed by two-way ANOVA with Dunett’s post-hoc test. In panels m-p significance was determined using a one-way ANOVA with Tukey’s post-hoc test. All data are presented as mean ± S.E.M. ns, p>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Next, we studied how Ptchd1 affects trafficking of MOR following activation by opioids. We found that co-expression of Ptchd1 significantly inhibited DAMGO-induced MOR internalization (Fig. 4g). Next, the MOR antagonist naloxone was added to examine the recovery of MOR back to the cell surface. We observed robust MOR recycling that was significantly inhibited by expressing Ptchd1 (Fig. 4h). Interestingly, overexpression of C. elegans PTR-25 similarly impaired MOR internalization triggered by DAMGO and produced an even stronger effect than human Ptchd1 (Extended Data Fig. 10d, e). This effect was specific to MOR, as Ptchd1 overexpression did not affect internalization of another GPCR, the β2 adrenergic receptor (Extended Data Fig. 10f, g). Thus, Ptchd1 and PTR-25 are general inhibitors of MOR trafficking both to and from the plasma membrane.
Previous studies established β-arrestins as key players that control trafficking of many GPCRs including MOR29. Therefore, we next examined the effects of Ptchd1 on the association of MOR with β-arrestin2 (Fig. 4i). Consistent with Ptchd1 inhibiting MOR internalization, we found that Ptchd1 expression inhibited agonist induced β-arrestin2 recruitment (Fig. 4j–l). We did not observe this influence with V2 vasopressin receptor, another GPCR prominently regulated by β-arrestin2 (Extended Data Fig. 10h–k). Overall, these results support the model that Ptchd1 regulates MOR trafficking by affecting its association with regulatory adaptors.
Cholesterol mediates effects of Ptchd1 on MOR
Prior studies have shown that several Patched family members regulate cholesterol transport30,31. Therefore, we tested whether Ptchd1 regulates cholesterol levels to affect MOR trafficking. Using a molecular biosensor approach (Fig. 4m), we found that overexpression of Ptchd1 significantly decreased the cholesterol content in the plasma membrane (Fig. 4n). This effect was similar to the action of the well-known cholesterol-depleting drug methyl-β-cyclodextrin (MBCD) and was the opposite from direct cholesterol supplementation (Fig. 4n). We tested another multi-pass transmembrane protein, MOR, as a control and did not observe effects on membrane cholesterol levels (Fig. 4n).
We next determined whether Ptchd1 modulation of cholesterol affects MOR trafficking. Consistent with prior observations32, depleting cholesterol with MBCD inhibited MOR internalization (Fig. 4o) whereas cholesterol addition had the opposite effect (Fig. 4p). Accordingly, increasing or reducing cholesterol levels had opposing effects on β-arrestin recruitment to MOR (Extended Data Fig. 10 l–o). Importantly, both depletion and enrichment of cholesterol enhanced and occluded, respectively, the effects of Ptchd1 overexpression on MOR internalization (Fig. 4o, p).
To examine an endogenous neuronal setting, we turned to acute cultures of adult nociceptors. First, we incubated genetic control DRG neurons with cholesterol, examining the impact on opioid-mediated suppression of excitability (Fig. 5a). We found that cholesterol treatment increased opioid inhibition of DRG firing (Fig. 5a, b). Remarkably, after the first application of morphine and subsequent washout, a repeated challenge with morphine resulted in the same degree of excitability decrease (Fig. 5a–c,). Thus, cholesterol supplementation prevents response desensitization to morphine. To test the role of Ptchd1, we performed similar experiments using DRGs from Ptchd1 KO animals. We observed that enhanced excitability of Ptchd1 KO cells was reversed by treatment with MBCD (Fig. 5d, e). Furthermore, cholesterol depletion with MBCD restored response desensitization of Ptchd1 KO neurons to repeated morphine application (Fig. 5d–f). We found that MBCD treatment similarly rescued both augmented opioid responses and their desensitization in the VTA GABAergic interneurons using slice preparation (Fig. 5. g–j). Thus, the cholesterol-related mechanism for Ptchd1 effects on MOR is broadly observed across different types of neurons in both the peripheral and central nervous system.
Figure 5. Role of cholesterol in mediating the effects of Ptchd1 on MOR desensitization in an endogenous neuronal setting.
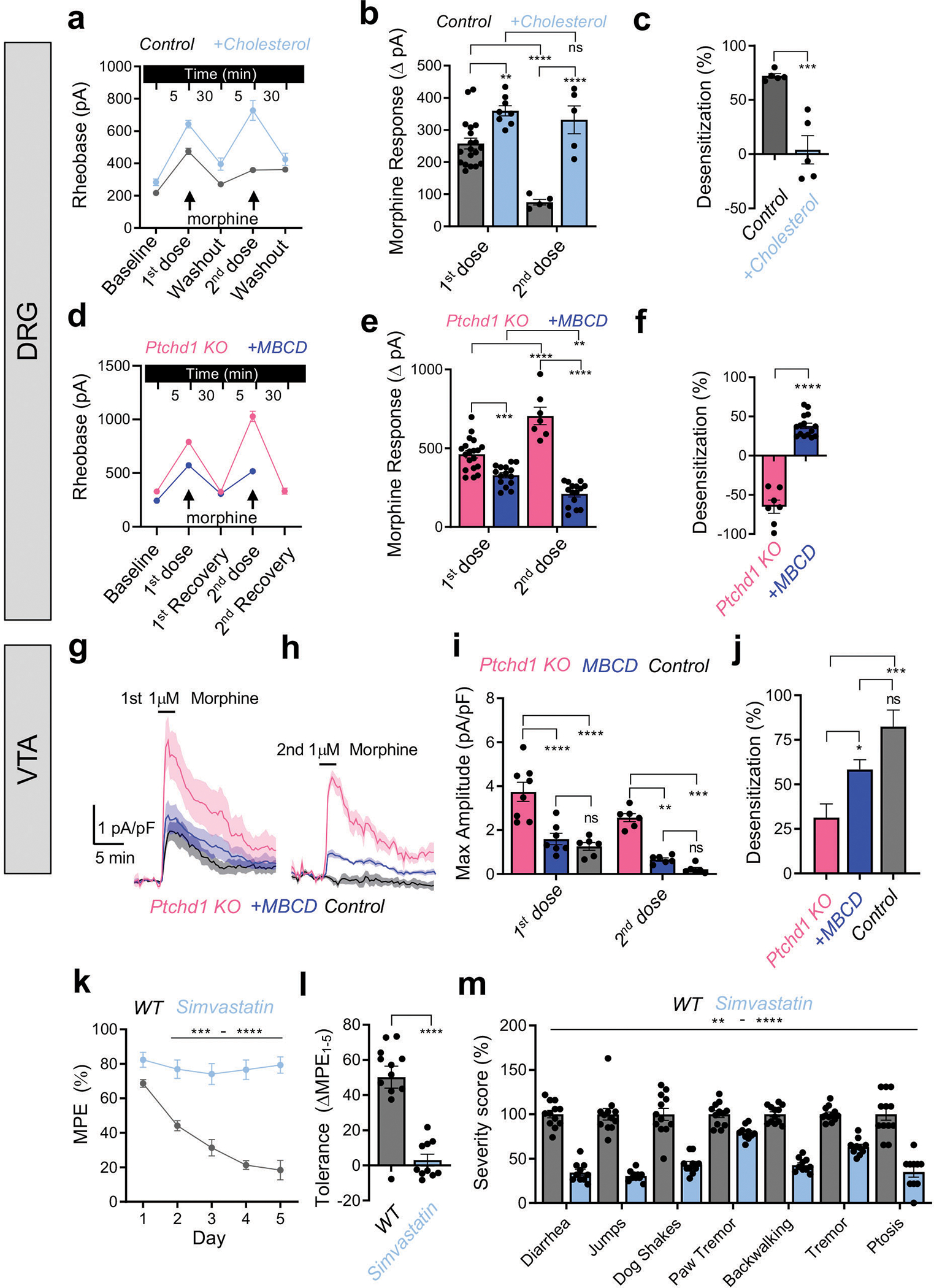
a, Changes in rheobase in control (Ptchd1 heterozygous) DRG induced by 200 μg/mL cholesterol. Control n=19 and 5 cells, 4 mice; Cholesterol n=8 and 5 cells, 3 mice first and second treatment, respectively.
b, Quantification of morphine responses as change in rheobase from panel a, same numbers.
c, Quantification of desensitization between first and second morphine responses in panel b. Control n=5 cells, 4 mice, cholesterol n=5 cells, 3 mice.
d, Changes in rheobase in Ptchd1 KO DRG upon 4 mM MBCD treatment. Ptchd1 KO n=19 and 7 cells, 4 mice; MBCD n=15 and 15 cells, 3 mice for first and second treatment, respectively.
e, Quantification of morphine responses as change in rheobase from panel d, same numbers.
f, Quantification of desensitization between first and second morphine responses in panel e. Ptchd1 KO n=7 cells, 4 mice, MBCD n=15 cells, 3 mice.
g and h, Mean GIRK current density profiles evoked in VTA GABA neurons evoked by 1 μM morphine.
i, Peak GIRK current density elicited by morphine in panels g and h. Control n=6 cells, 4 mice; Ptchd1 KO n=7 cells, 4 mice; MBCD n=7 cells, 4 mice.
j. Quantification of desensitization between the first and second morphine responses. Same numbers as in panel i.
k. Evaluation of analgesic tolerance by hot plate assay in WT mice treated with simvastatin (10mg/kg) for 5 days.
l. Quantification of analgesia reduction as a difference in the MPE between session 1 and 5 from panel k.
m. Evaluation of somatic withdrawal signs following precipitated withdrawal from chronic morphine administration.
For panels b and e, significance by two-way ANOVA with Holm-Šídák post-hoc test. For panels c, f, and l, significance by unpaired two-tailed Student’s t-test. In panel i, comparisons by mixed-model ANOVA, with Tukey’s multiple comparisons test. For panel j, comparisons were by a one-way ANOVA with Tukey’s multiple comparisons test. For panel k, comparisons by two-way ANOVA and Šídák post-hoc test. For panel m, comparisons by two-way ANOVA and Bonferroni post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Mean values with S.E.M. errors are shown.
Finally, we probed the in vivo relevance of Ptchd1 modulation of cholesterol on behavioral responses to opioids. For these experiments, we treated mice with simvastatin, a drug that lowers systemic cholesterol content but increases the high-density lipoprotein fraction of cholesterol33,34 potentially making cholesterol more available to influence MOR in neuronal cell membranes. Consistent with this prediction, we found that short-term administration of simvastatin completely abrogated analgesic tolerance (Fig. 5k,l). Moreover, simvastatin substantially reduced signs of somatic withdrawal (Fig. 5m). Collectively, these findings with native neurons and in vivo support the model that Ptchd1 regulates MOR desensitization by affecting cholesterol availability.
Discussion
In this study, we identify a poorly understood Patched family receptor, PTR-25/Ptchd1, as a novel regulator of opioid tolerance using two animal models, C. elegans and mice. Our findings indicate that Ptchd1 controls desensitization of opioid responses by regulating MOR trafficking in a cholesterol-dependent manner.
Chronic stimulation of MOR and other GPCRs triggers desensitization which leads to behavioral tolerance. A canonical example of tolerance, with particularly prominent clinical implications, is loss of therapeutic efficacy with prolonged use of opioids5,6. Tolerance diminishes both the analgesic and euphoric effects of opioid drugs, necessitating dose escalation. This in turn contributes to problematic side effects, such as dependence and overdose fatalities. Understanding the mechanisms underlying opioid tolerance and GPCR desensitization has been a major research focus for decades. Several processes affect MOR desensitization and influence tolerance including receptor phosphorylation, trafficking and interaction with G proteins and β-arrestins9. Although several mechanisms and players that regulate opioid tolerance have been described14,15,35–38, the process is not well understood at the molecular level. Here, we applied unbiased forward genetics to the study of opioid tolerance for the first time. Our in vivo behavioral approach identified PTR-25/Ptchd1 as an ancient evolutionarily conserved regulator of opioid tolerance. These findings introduce a new molecular target for intervention to combat tolerance, but not reward-associated dependence, which remains the most significant limitation of opioid therapies.
Because opioid tolerance is a behavioral phenomenon, genetic screens for tolerance require an in vivo setting, such as the animal model we deployed. C. elegans features an expansive repertoire of GPCRs that utilize conserved signaling mechanisms and has unsurpassed utility for addressing challenging questions in biology39–42. Our study further leverages the power of C. elegans in addressing important biomedical questions, which now includes drug tolerance.
The genetic and molecular mechanisms governing MOR trafficking and how they relate to opioid tolerance in vivo have remained intensely studied yet incompletely understood and controversial areas of pharmacology. Here, we introduce Ptchd1/PTR-25 as a new player that regulates MOR trafficking to enable behavioral tolerance. Our findings support a model in which Ptchd1 regulates MOR trafficking by changing the membrane lipid environment via effects on cholesterol (Fig. 6). Our results indicate that Ptchd1 regulates MOR internalization and inhibits its interaction with β-arrestin. This suggests that Ptchd1 restricts MOR endocytosis allowing prolonged signaling from the cell surface possibly reducing MOR re-sensitizing internalization and recycling43,44. Thus, Ptchd1 might facilitate persistent MOR signaling from the plasma membrane that ultimately induces tolerance. Ptchd1 may also link behavioral tolerance and acute MOR desensitization, as our findings indicate that loss of Ptchd1 makes neurons more resistant to the desensitizing effects of opioids.
Figure 6. Proposed mechanism for Ptchd1 regulation of MOR signaling and opioid tolerance.
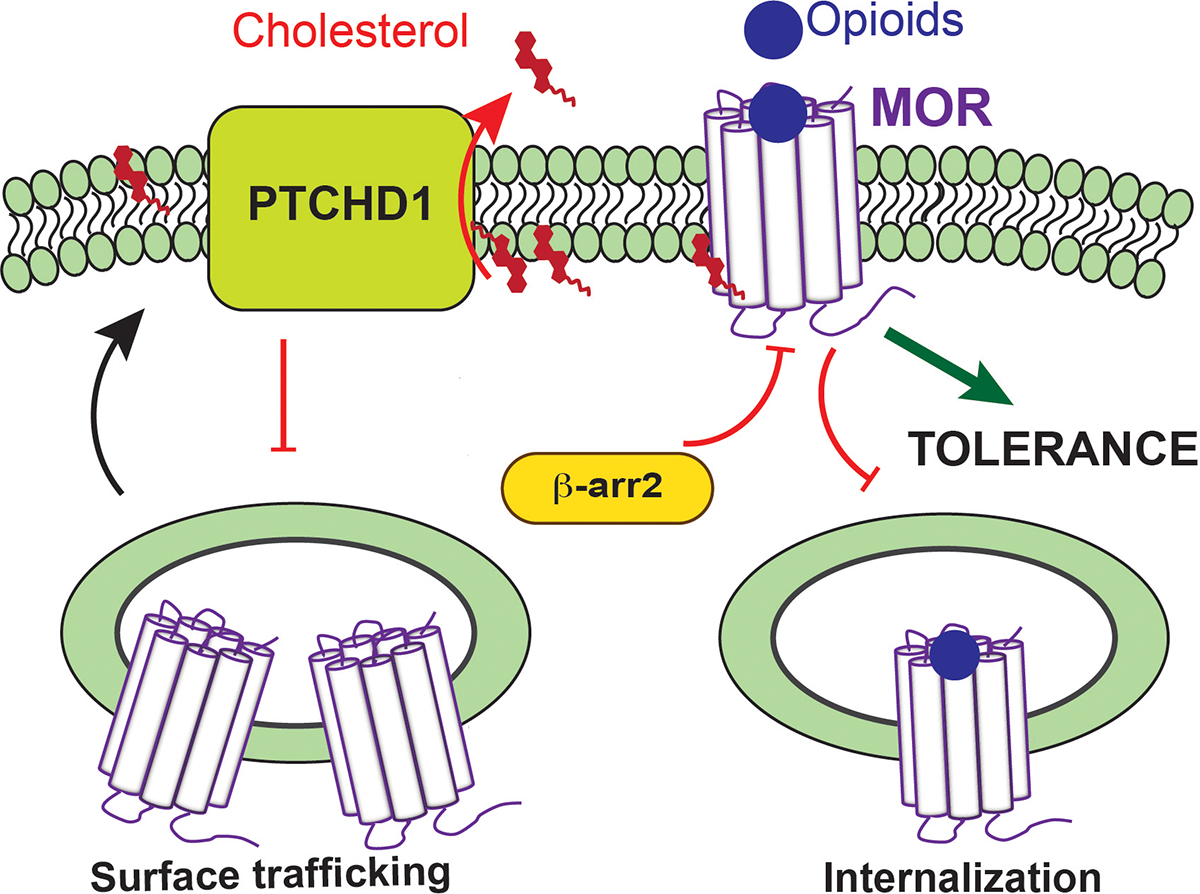
Ptchd1 acts to deplete membrane cholesterol which in turn affects MOR dynamics impeding its trafficking to the surface and internalization following activation by opioids. When trapped on the surface due to Ptchd1 action, MOR fails to undergo resensitizing internalization/recycling leading to prolonged signaling which triggers desensitization and tolerance. Red inhibitory bars and red arrows indicate negative regulatory functions and effects on reducing membrane cholesterol levels, respectively.
Interestingly, we found that Ptchd1 KO mice also exhibited less severe somatic withdrawal signs despite their augmented opioid sensitivity. Although how this occurs remains unknown, we think that it may reflect a selective role of Ptchd1 in processes triggered by chronic opioid exposure. These could be distinct from the reactions that MOR utilizes to produce immediate analgesia and euphoria, as observed in several other genetic mouse models where opioid effects on various behaviors are also dissociated 12,18,45. Alternatively, this could be explained by different molecular landscapes for signaling adaptations to chronic opioid administration versus changes in signaling sensitivity of a receptor upon acute opioid stimulation.
On a cellular level, our results suggest that Ptchd1 regulates MOR trafficking via effects on cholesterol. Cholesterol can affect GPCRs by direct binding and indirectly by regulating receptor dynamics in the lipid environment46,47. Structural studies have shown that MOR possesses a cholesterol binding pocket48. Cholesterol has also been shown to regulate trafficking and signaling of MOR 32,49. We have shown that Ptchd1 reduces the cholesterol content in the plasma membrane, and that its effects on MOR trafficking depend on cholesterol availability. We further demonstrated the relevance of Ptchd1 cholesterol-dependent mechanisms in opioid regulation of native neurons and behavioral tolerance, which substantially extends previous observations on the role of cholesterol in opioid effects50,51. Thus, we propose that Ptchd1 reduces cholesterol availability for MOR, thereby altering its trafficking dynamics and interaction with adaptor proteins. It remains to be determined whether cholesterol-regulated trafficking is the only mode by which Ptchd1 affects MOR, or if additional mechanisms further shape the interplay between the two to control the effects of opioids.
In addition to affecting receptor desensitization and tolerance, we report that Ptchd1 strongly influences the levels of MOR on the cell surface. Overexpression of Ptchd1 leads to intracellular sequestration of MOR. In contrast, loss of Ptchd1 substantially increases the efficacy of opioid signaling in neurons, presumably due to increased surface content of MOR. Thus, it appears that Ptchd1 also impedes forward targeting of MOR to the plasma membrane thereby serving as a general inhibitor of MOR trafficking.
Importantly, our results could also have broader implications for GPCR biology. Ptchd1 belongs to the extensive and enigmatic family of Ptch proteins. The best studied member of this family, Ptch1, is a component of the Sonic hedgehog pathway that operates exclusively in the primary cilium52,53. There are intriguing parallels between our findings and the established role of Ptch1 as an inhibitor of Smoothened receptor trafficking into the cilium52,54. Interestingly, Ptch1 has been shown to control cholesterol availability and recent structural studies suggest that Ptch1 affects cholesterol to modulate Smoothened signaling54. Considering that both Smoothened and MOR are GPCRs, it is plausible that Ptch family proteins may serve as general regulators of GPCR trafficking. While Ptch1 actions are restricted to the cilium, other Ptch family members may regulate receptor trafficking in other compartments and cell-types. Determining how different Ptch proteins affect the enormous repertoire of over 800 GPCRs in metazoans now becomes an expansive, exciting area for future investigation.
Methods
C. elegans strains
C. elegans strains were maintained using standard methods and cultivated at 20°C. The following strains were used: N2 (wild isolate, Bristol, UK), CB4856 polymorphic Hawaiian strain (wild isolate, Hawaii, USA), tgMOR (bggIs10), tgMOR; rsbp-1 (vs163), frpr-13 (bgg78), tgMOR; pha-1 (e2123), ptr-25 (bgg10), tgMOR; ptr-25 (bgg10), tgMOR; ptr-25 CRISPR (bgg85), tgMOR; ttTi5605mos1, tgMOR; ptr-25 (bgg10); ttTi5605mosI, tgMOR; ptr-25 (bgg10); bggSi44 [Pptr-25PTR-25], tgMOR; ptr-25 (bgg10); bggSi45 [Prab-3PTR-25] and tgMOR; ptr-25 (bgg10); bggSi46 [Prab-3hPTCHD1].
C. elegans genetics
To identify mutations in tgMOR; bgg10, we employed SNP-based mapping coupled with Whole Genome Sequencing (WGS). TgMOR; bgg10 hermaphrodites were crossed with CB4856 males to generate F1 progeny (introducing pre-determined SNPs). At least 20 F3 progeny (independent recombinants) were randomly selected and tested for opioid hypersensitivity. All phenotypically verified recombinants were cultivated to starvation and genomic DNA was extracted. WGS was done on purified genomic DNA using MiSeq DNA Sequencing System (Illumina, USA) with pair-end 250 nucleotide reads. WGS data was analyzed using a custom CloudMap pipeline on the Galaxy public server. The candidate mutation in ptr-25 (which results in a premature stop codon) identified in tgMOR; bgg10 mutants by WGS was validated by co-CRISPR gene editing using tgMOR; pha-1 (e2123) as described previously18. TgMOR; ptr-25 CRISPR animals were isolated using temperature-sensitive selection for pha-1 (25°C) and confirmed by PCR genotyping and sequencing. To generate tgMOR; ptr-25 CRISPR animals (tgMOR; ptr-25 CRISPR (bgg85)), the exact premature stop codon (Q1032stop; 3094C>T) present in ptr-25 (bgg10) was CRISPR edited into the ptr-25 locus of parental tgMOR animals. The CRISPR repair template included a silent mutation to prevent Cas9 re-cutting and to facilitate PCR-based genotyping of tgMOR; bgg85 mutants using the HpyCH4IV restriction enzyme (Supplementary Table 3).
Transgenic rescues were performed using Mos1-mediated single copy insertion (MosSCI). PTR-25 expressing MosSCI plasmids were constructed using MosSCI backbone vectors (neomycin-resistance cassette). Gibson Assembly® (NEB, USA) was used to generate Pptr-25PTR-25 (pBG-332) which included ptr-25 cDNA, ptr-25 promoter (2000bp) and ptr-25 3’UTR (2000bp) assembled into a MosSCI vector (pBG-264). Gateway® Cloning (Invitrogen, USA) was used to generate plasmids for pan-neuronal expression: Prab-3PTR-25 (pBG-GY862) and Prab-3hPTCHD1 (pBG-GY1050). C. elegans ptr-25 cDNA or codon-optimized human PTCHD1 cDNA (NM_173495.3) were cloned into a PCR8 vector which was recombined with a MosSCI vector (pBG-GY805). All final constructs were verified by sequencing, and all strains containing integrated MosSCI transgenes were confirmed by PCR genotyping and sequencing.
Specific details about all transgenes, injection conditions, and CRISPR constructs used for this study are provided in Supplemental Tables 1–3.
C. elegans opioid assays and tolerance screen
Multi-Worm Tracker (MWT) was used to record all opioid-induced behaviors as described previously55. For each experiment, 4–5 young adult hermaphrodites were placed in individual wells containing 15 μL assay buffer (M9 + 0.01% Tween20). Five-minute baseline locomotion in liquid was recorded, 15 μL 2x opioid drug was added to desired concentration, and recording was resumed for 180 minutes. Mean speed was calculated every 5 minutes for each well using custom-written scripts. For each genotype, drug dose and time point, data was collected from 12 or more wells obtained from at least 3 independent experiments.
To test opioid tolerance (illustrated in Fig 1B), synchronized young adults were divided into 3 groups of animals. The first group was evaluated with a full dose-response to opioids (annotated as 1st). The second group was exposed 4 times to opioids (10 μM fentanyl or 300 μM morphine; 3h exposure and 9h recovery in absence of drug) and tested with a full opioid dose-response (annotated as 5th, fentanyl or morphine). The third group of animals underwent the same protocol as the second group except using the vehicle (M9 + 0.01% Tween20) for repeated exposures (annotated as 5th, vehicle).
The screen for opioid tolerance contained two steps (illustrated in Fig 1D). The first step was a large-scale unbiased forward genetic screen for tgMOR mutants with altered opioid responses1. The second step was an unbiased screen for tolerance to repeated opioid exposure using twenty-seven tgMOR opioid hypersensitive mutants. Of the tgMOR hypersensitive mutants screened, only tgMOR; bgg10 mutants showed impaired tolerance. Statistical analyses and numbers for all experiments are specified in figure legends. Data for all experiments was obtained from a minimum of three independent experiments.
For dose responses, maximum possible effect (MPE) was calculated based on MWT data. As shown in Supplemental Figure S1 (a-f), different doses of fentanyl alter the onset times to maximum paralysis, while the amplitude and duration of effect are only modestly different between doses. Therefore, we use the onset time of maximum paralysis as the parameter to calculate the maximum possible effect and opioid dose-response curves.
For tgMOR animals, doses tested are 5 μM, 10 μM, 20 μM, 40 μM, 80 μM and 160 μM. All doses evoke maximum paralysis and subsequent recovery within a 180-minute maximum timeframe. We define time to paralysis for 5 μM as the minimum dose effect (180 min - time to paralysis 5 μM), which upon normalization is 0% MPE. We define time to paralysis for 160 μM as the maximum dose effect (180-time (min)160 μM), which upon normalization is 100% MPE. The time point for maximum dose effect was evaluated manually.
For hypersensitive tgMOR mutants (tgMOR; ptr-25 and tgMOR; frpr-13), doses tested are 2.5 μM, 5 μM, 10 μM, 20 μM, 40 μM and 80 μM. All doses evoke maximum paralysis and subsequent recovery within 180min. We define time to paralysis for 2.5 μM as the minimum dose effect (180 min - time to paralysis 2.5 μM), which upon normalization is 0% MPE. We define time to maximum paralysis for 80 μM as the maximum dose effect (180 min – time to paralysis 80 μM) which upon normalization is 100% MPE.
For tolerance assays with tgMOR animals or hypersensitive tgMOR mutants, fentanyl doses tested (after 4 exposures to a set dose) are 10 μM, 20 μM, 40 μM, 80 μM and 160 μM. We define time to paralysis for 10 μM as the minimum dose effect (180 min - time to paralysis 10 μM), which upon normalization is 0% MPE. We define time to paralysis for 160μM as the maximum dose effect (180 min – time to paralysis 160 μM), which upon normalization is 100% MPE.
Graphpad Prism software version 9.0.1 was used to generate dose-response curves by performing global nonlinear regression analysis and fitting to the log (agonist) vs. normalized response - variable slope model. Dose response curves are shown as best-fit curves, which are used to generate the corresponding EC50.
Mouse strains and behavior
This study was carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals, as drafted, updated and published by National Research Council and as approved by the Institutional Animal Care and Use Committee at The Scripps Research Institute. In all experiments, mice used were 3–6 months old and group housed in a normal 12:12 light cycle at 72°F with 40–50% humidity and fed ad libitum. The Ptchd1-YFP knock in/knock out mice (B6; 129-Ptchd1tmGfng/J, JAX stock #028986) express YFP from the endogenous Ptchd1 locus22. We refer to this strain as Ptchd1 KO. Because Ptchd1 is an X-linked gene, behavioral studies were performed with littermates derived from crossing hemizygous males and heterozygous females producing littermate males that are either knockout or wild-type and only heterozygous females. In order to strictly control behavioral studies with littermate comparisons, it was only possible to use males. For electrophysiological studies, the targeted Ptchd1 allele needed to be present for the cell type identification, thus all control animals were heterozygous or hemizygous for Ptchd1 allele. The MOR-mCherry reporter mice (B6;129S2-Oprm1tm4Kff/J, JAX stock # 029013) have the μ-opioid receptor (Oprm1) gene fused with the monomeric red fluorescent protein mCherry resulting in the expression of chimeric fluorescent protein56. For cell type identification in electrophysiological and histological studies, the Ptchd1 KO strain was crossed with MOR-mCherry mice. The Oprm1 allele was always kept heterozygous, maintaining one wild-type copy of Oprm1.
Conditioned Place Preference
Animals were initially placed into a three-chamber box (black and white designated chambers with connecting tunnel, built in house) and tracked (ANY-Maze, Stoelting Co., Wood Dale, IL USA) for 30 minutes. All animals had scored less than 60% of total time in all chambers and were deemed unbiased. Animals were then drug paired, using unbiased method, to either the white or black chamber for morphine stimulus (7.5, 10, 20 mg/kg, s.c.) and the other chamber paired to saline control. Animals were trained for 6 consecutive days, alternating in paired chambers. Once training was completed, mice were placed into the tunnel and given 30 minutes to explore. Conditioned place preference is graphed as Place Preference Score (time spent in morphine paired chamber - time spend in saline paired chamber); mean ± SEM.
Hotplate
Animals were tested for analgesic effects of morphine or fentanyl using hotplate set to 52°C. Mice were first tested with saline (s.c.) to establish baseline responses and then injected with either morphine (10, and 20 mg/kg, s.c.) or fentanyl (0.1 mg/kg, s.c.), then individually placed onto hotplate and observed for physical signs of heat sensitivity in 10 minute intervals. Latencies to the first nociceptive reaction including reflectory paw lifting, flinching or licking were determined at which point the mice were quickly removed from the hot plate. Any mice that showed no signs of discomfort after 50 second cut off were removed. Animals were tested every 30 minutes for 2 hours. Time (seconds) spent on hotplate is graphed as %MPE = [(test latency – baseline latency) / (cut off time latency – baseline latency) × 100], mean ± SEM.
To examine chronic tolerance, animals were injected initially with saline and tested for latency to react to establish baseline. Animals, were then injected with 20 mg/kg morphine or 0.15 mg/kg fentanyl and tested 30 minutes after for reaction latency for 5 (morphine) of 10 (fentanyl) consecutive days. Initial weight and daily weight during testing were recorded. Reaction time (seconds) was graphed as %MPE = [(test latency – baseline latency) / (cut off time latency – baseline latency) × 100].
To examine acute tolerance, mice following determining the baseline latencies, were injected with the first dose of morphine (20 mg/kg; s.c.) and latency was recorded 30 minutes later. Animals were allowed to recover for 90 minutes at which point their baselines were measured again followed by repeating the injection with the second dose of morphine (20mg/kg; s.c.) and testing on the hotplate 30 minutes after second injection.
In some experiments morphine was delivered intracerebroventricularly (ICV) using previously established protocols57. In these experiments, animals were first tested with saline injected ICV 24 hours prior to morphine to establish normal baselines. On testing day, animals were administered morphine (3.5 nmol) and tested in 10-minute increments on a hotplate set to 52°C for 1 hour.
Withdrawal
Animals were injected with saline to establish baseline and were tested for reaction time on hot plate prior to dose responses (Dose response 1 included 10, 20 and 40 mg/kg morphine; s.c. Dose response 2 contained 20, 40, 60 mg/kg morphine; s.c.) All animals were given doses and tested on consecutive days. Animals were injected with 20mg/kg morphine s.c. for 5 days between dose responses. Initial weight and daily weight during testing were recorded. Reaction time (seconds) was graphed as %MPE = [(test latency – baseline latency) / (cut off time latency – baseline latency) × 100], mean ± SEM. Data set was analyzed with two-Way ANOVA Bonferroni post-hoc. Naïve animals were injected once daily with increasing dose of 20, 40, 60, and 80 mg/kg morphine (s.c.) and weight monitored daily. On the day 5, animals were injected 100 mg/kg morphine s.c. Withdraw was precipitated 3 hours later by injecting 1m/kg naloxone (s.c.). Withdraw was scored immediately for 30 minutes by number of jumps, dog shakes, paw tremor, backwalking, and tremor. Scores are presented normalized to 100 for WT control.
Tail Immersion
Mice were restrained in a tube with airholes with 2/3 of entire tail immersed in a water bath heated to 50°C. Latency to remove tail was recorded. Animals were tested for baseline latency using saline and given 30 minutes for recovery before morphine injections (5, 10, 20, 40 and 60 mg/kg; s.c.). For intrathecal delivery either saline or 0.35 μg morphine in saline was injected. Latency for tail removal with was tested at 30 minutes after systemic morphine injections and 10 minutes after intrathecal delivery. To evaluate analgesic tolerance using tail immersion, mice were injected once daily either s.c. with 20 mg/kg morphine or intrathecally with 0.35 μg morphine followed by testing for five consecutive days.
Simvastatin study
Mice were acclimated to unheated hot plate for 3 consecutive days prior to start of study. Animals were then orally gavaged with either vehicle (distilled water) or simvastatin (10 mg/kg in distilled water) for 5 consecutive days. On the six day and on, the animals first received oral gavage followed by evaluation of baseline pain responses on hot plate 30 minutes later. Morphine (20 mg/kg) was then administered and animals re-tested 30 minutes after on the hotplate. This protocol was repeated for 5 consecutive days to evaluate analgesic tolerance. To evaluate withdrawal, mice continued to receive escalating doses of morphine (40, 60, 80 and 100 mg/kg, daily). On testing day of 100 mg/kg morphine, animals were injected with naloxone (1 mg/kg; i.p.) after 3 hours of morphine injection, placed into empty sterile house cages and recorded for 30 minutes.
Open Field and Naltrexone Study
Animals were injected with either saline or naltrexone solution in saline (4 mg/kg; i.p.), placed into open field arena (30 × 30 cm) and tracked for movement (Noldus EthoVision XT; Leesburg VA USA) for 60 minutes. Distance traveled was recorded. In parallel, naïve non-injected animals were also similarly evaluated by the open field test for 120 minutes.
Whole cell patch-clamp recordings
Male or female mice between 3–6 months of age were used for VTA and NAc slice electrophysiology. Due to X-linked nature of Ptchd1 gene and the need to rely on genetic labeling strategy to identify neurons expressing both MOR and Ptchd1 for recordings Ptchd1+/− female littermates were used as controls in lieu of WT. See genetic cross scheme in Extended Data Fig. 7 for the details on breeding scheme and controls. The mice were anesthetized with isoflurane, decapitated, and brains quickly removed and submerged in ice-cold NMDG cutting solution containing (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, 10 MgCl2, adjusted to pH 7.3–7.4 with HCl. VTA horizontal brain slices (250 μm) were cut on a vibratome (VT1200S, Leica), divided along the midline, incubated for 25 minutes at 34°C in NMDG cutting solution, then transferred and held in for up to 6 hours in 25°C recirculating oxygenated aCSF (containing in mM: 125 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 2 CaCl2, 10 dextrose, adjusted to pH 7.3–7.4 with HCl.) Slices were transferred to a submerged chamber and continuously perfused at 2 ml/min with oxygenated aCSF. Recordings were obtained with borosilicate glass pipettes (2–5 MΩ) filled with either Cs+2 internal (containing in mM: 110 CsMeSO3, 10 CsCl, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 HEPES, adjusted to pH 7.3–7.4 with CsOH) or filled with K+ internal (containing in mM: 6 NaCl, 4 NaOH, 130 K-gluconate, 11 EGTA, 1 CaCl2, 1 MgCl2, 10 HEPES, 2 Na2ATP, and 0.2 Na2GTP, adjusted to pH 7.3–7.4 with HCl, 285–295 mOsm).
For VTA slice morphine-evoked GIRK recordings, current was measured with K+ internal while clamping at −70 mV and monitoring input resistance with a periodic 10mV, 10ms hyperpolarizing step in 30 second intervals. Cells were excluded if input resistance varied >20% while recording. For each slice, 1 μM morphine was perfused twice with a 30 minute washout interval. In the MβCD rescue group, Ptchd1 KO slices were perfused with 2 mM MβCD for 10 minutes preceding bath application of 1μM morphine. Desensitization was calculated as the ratio of sample means between each application of morphine, and the standard error by Taylor expansion.
For recordings from DRG neurons, acutely dissociated culture was prepared as previously described58. Briefly, DRG’s were dissected from 2–3 month old mice in HBSS and enzymatically dissociated with Collagenase A and Dispase II (Sigma-Aldrich) for 20–30 minutes at 37°C. DRG were centrifuged at 200x g for 5 minutes, washed with DMEM supplemented with 10% FBS, glutamate, sodium pyruvate, and penicillin/streptomycin and triturated in Neurobasal A medium supplemented with 10% fetal bovine serum, 1% penicillin, 1% streptomycin, 1% GlutaMax, 2% B27, 25 ng/L NGF, and 2 ng/L GDNF. After plating on laminin-coated coverslips, cells were incubated at 37°C overnight. Experiments were performed the day after dissection.
Nociceptors were identified both by genetic labeling of MOR, and previously described physiologic classification protocols59. To identify hyperpolarization-activated current and kinetics of resulting transient currents, membrane potential was stepped from −60 mV to −110 mV for 500 ms in 10 mV increments (Protocol 1). Inward currents were identified by with two protocols, first preconditioning membrane potential at −100 mV for 500 ms, then stepping from −60 mV to 40 mV in 10 mV increments for either 200 ms (Protocol 2) or 2 ms (Protocol 3). Protocol 2 was used to identify the activation threshold of A-current peaks and the A-current decay constant at 40 mv. Protocol 3 was used to determined the inactivation decay constant of the first inward current response. Membrane capacitance (Cm) and series resistance (Rs) were determined by a hyperpolarizing 10 mV pulse of 10ms duration initially and at a 30 second interval throughout the recording period. Cells were recorded only if initial series resistance ≤20 MΩ and excluded if Rs varied >20% while recording. For whole-cell voltage-clamp recordings, neurons were held at Vm = −70 mV. For spontaneous mIPSC recordings, K+ internal was used and tetrodotoxin (TTX, 1μM) was added to high-K+ perfusion ACSF (containing in mM: 118 NaCl, 10 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 2 CaCl2, 10 dextrose, adjusted to pH 7.3–7.4 with HCl). Current-voltage relationships were generated using a voltage-step protocol, from −100 mV to 80 mV in 10 mV increments. Signals were filtered at 10 kHz and sampled at 20 kHz using Patchmaster software (HEKA Instruments) with an EPC10 amplifier (HEKA Instruments) and transferred with an ITC-16 ADC (HEKA Instruments) for offline analysis. Liquid-junction potentials were not corrected.
Cell Culture
HEK239T/17 cells (ATCC; CRL-11268) were maintained in culture medium (DMEM supplemented with 10% v/v fetal bovine serum, MEM non-essential amino acids, and 1 mM sodium pyruvate). On the day prior to transfection, cells were split to 8 × 105 cells on 35 mm plates in culture medium supplemented with 1 μg/mL Matrigel and incubated at 37°C for 24 hours. Cells were then transfected using Lipofectamine LTX Reagent with PLUS Reagent (Invitrogen, A12621). All transfected DNA’s were balanced to 5 μg using pcDNA3.1 and added dropwise per manufacturer’s instructions. Cells were incubated for 24 hours at 37°C prior to experiments. For polyethylenimine (PEI) transfections, cells were plated at 1.6 × 106 cells per mL on 35 mm cell culture plates in culture media supplemented with 1 μg / mL Matrigel. Cells were incubated for 3–4 h at 37°C, 5% CO2. All transfected DNA’s were balanced to 5 μg using pcDNA3.1. and transfected using the polyethylenimine (PEI) transfection reagent as described60.
Imaging and Analysis
Imaging of brain sections was performed on a Zeiss LSM 880 confocal microscope using a 20x objective or the Zeiss LSM 980 microscope using a 10x objective. Imaging of immunocytochemistry cells was performed on a Leica SP8 MP using 25x water-immersion objective. Co-localization analysis was performed with ImageJ (https://imagej.nih.gov/ij/) and 12 cells for each condition (with and without PTCHD1). Lines were drawn across cells, avoiding the nucleus, and the fluorescence values from the plasma membrane (confirmed by sodium-potassium ATPase antibody) and intracellular membranes were quantified and reported as a percentage of total fluorescence intensity values along the line. Co-localization analysis using the entire cell was done using the JACoP plugin in Image J and the Pearson’s Correlation Coefficient was reported.
Immunohistochemistry
Mice were perfused with ice-cold PBS followed by 4% paraformaldehyde in PBS. Brains were removed and fixed at 4° C for 12–72 hours. 75 micron sections were taken and washed in PBS for 15 minutes. Sections were incubated in blocking solution (5% normal goat serum, 2% BSA, 0.2% Triton X-100, 1x PBS) for 1 hour, then incubated with rabbit anti-EGFP antibody (Invitrogen A11122) overnight at 4° C at 1:1000 dilution. Sections were washed with PBS for 1 hour, then incubated in Alexa Fluor 488 goat anti-rabbit secondary antibody for 4 hours at room temperature. Sections were washed in PBS for 1 hour then mounted onto glass slides using Fluoromount with DAPI.
Immunocytochemistry
HEK293T/17 cells were grown on coverslips coated in poly-d-lysine and transfected with 0.21 μg (1x) of HA-tagged MOR and 0.42 μg (2x) of codon-optimized Ptchd1-FLAG. The next day cells were fixed in 4% paraformaldehyde in PBS for 20 minutes and then permeabilized in 0.5% Triton X-100 in PBS for 5 minutes. Cells were blocked with 5% milk in 0.1% Triton X-100 in PBS for 1 hour at room temperature. The following primary antibodies were diluted in 1% milk in 0.1% Triton X-100 in PBS: anti-HA (Roche) dilution 1:500, anti-sodium potassium ATPase (Invitrogen MA5–32184) dilution 1:1000, and anti-FLAG M2 (Sigma-Aldrich, F1804) dilution 1:1000. Cells were incubated with primary antibody for 2 hours at RT, washed with 0.1% Triton X-100 in PBS for 15 min, then incubated with the following secondary antibodies diluted in 1% milk in 0.1% Triton X-100 in PBS: AlexaFluor Goat anti-rat 488, AlexaFluor donkey anti-mouse 647, and AlexaFluor Donkey anti-rabbit 546 for 1 hour. All secondary antibodies were used 1:500 dilution. Cells were then washed in 0.1% Triton X-100 in PBS for 15 minutes, washed in PBS for 15 minutes, then mounted onto slides with Fluoromount containing DAPI.
In Situ Hybridization
RNA in situ hybridization was performed using RNAscope V2 Assay (Advanced Cell Diagnostics) for Akt (Mm-Ptchd1-C1; Cat No. 489651), Oprm1 (Mm-Oprm1-C3; Cat No. 315841), and Rbfox3 (NeuN; Mm-Rbfox3-C4; Cat No. 313311) probes. Protocol was performed per manufacturer’s instructions. Wild-type mice were perfused with 4% paraformaldehyde (PFA) and the brains were left overnight in PFA. Brains were stored in cryoprotectant at −20°C until sliced in a vibratome (Leica VT1000S) at 40 μm. Dorsal root ganglion (DRG) extraction was performed in rapidly euthanized wild-type mice and placed into 4% PFA for 48 hrs and moved to cryoprotectant until use. DRGs were embedded in Tissue-Tek OCT (Sakura Cat: 4583) and sliced on a microtome at 40 μm. All slices were washed 10x in PBS every 5 minutes to remove OCT and remove detriment. Brain slices were mounted on Super Frost Plus slides (Fisher Scientific; Cat No. 12–550-15). Slides were postfixed in 4% PFA in 1x PBS for 1 hour at 4°C. The slides were dehydrated in 50, 70, and 100% ethanol, and subsequently dried at room temperature for 5 minutes; an ImmEdge Hydrophobic Barrier PAPpen (Vector Laboratories; Cat No. H-4000) was used to draw on the slides around the brain sections. The slides were then treated with Protease III solution at room temperature for 30 minutes, followed by washing in 1x PBS. Probes were then applied and incubated at 40°C for 2 hours in a humidified incubator. Slides were incubated with amplifier probes (AMP1, 40°C for 30 minutes; AMP3, 40°C for 30 minutes; AMP4, 40 °C for 15 minutes) and then incubated with fluorescently labeled probes Opal 520 nm, Opal 570 nm and Opal 690 nm for Ptchd1, Oprm1 and NeuN respectively. After washing, slides were cover slipped using ProLong Diamond Antifade Mountant (Thermo Fisher; P36961). Images were taken within 48 hours of staining on a Zeiss LSM980 confocal microscope. Confocal images were acquired with a 20× objective.
Surface Abundance Assays
In the following assays the Nano-Glo® HiBiT Extracellular Detection System (Promega, N2420) and the PHERAstar FSX plate reader (BMG Labtech) were used. For all assays, 0.21 μg (1x) HiBit-tagged human mu opioid receptor (HiBiT-hMOR) or HiBit-tagged β2 adrenergic receptor (HiBit-ADRB2) and 0, 0.21 (1x), or 0.42 μg (2x) of codon-optimized PTCHD1-FLAG or PTR-25-FLAG in pcDNA3.1 vector were transfected into HEK293T/17 cells. The day after transfection, cells were re-plated at 1.5 × 105 cells per well of a 96-well plate (in DMEM without phenol red, supplemented with 10% v/v fetal bovine serum, MEM non-essential amino acids, and 1 mM sodium pyruvate) and allowed to adhere for 3 hours. Media was then aspirated and 50 μl of BRET buffer (0.1 % (w/v) glucose, 1x phosphate-buffered saline, and 0.5 mM MgCl2) containing 1x NanoGlo® reagent and 1x LgBiT protein were added to each well. Plates were loaded into plate reader set at 37° C and luminescence was measured every 150 s for 15 minutes to allow for signal equilibration as described in manual. The final luminescence measurement was recorded as protein surface abundance. To measure total abundance, 200 μg/mL digitonin was added to BRET buffer containing 1x NanoGlo® reagent and 1x LgBiT protein to allow for cell lysis, after which luminescence was measured as described above. To measure internalization, surface abundance was first measured as described, then 10 μM DAMGO was added and luminescence was measured every 150 seconds for 20 minutes. The final time point was recorded as surface abundance after internalization and compared with the surface abundance prior to addition of DAMGO.
For the cholesterol depletion or enrichment, cells were incubated with 4 mM methylbetacyclodextrin (MBCD) (Acros Organics, 377110050) or 200 μg/mL water-soluble cholesterol (Sigma, C4951) in DMEM without phenol red supplemented with MEM non-essential amino acids, 1 mM sodium pyruvate, and 25 mM HEPES) for 1 hour prior to beginning the experiment. Final results were normalized to expression level.
To measure recycling, cells were incubated with 10 μM DAMGO for 20 minutes, after which BRET buffer containing 1x NanoGlo® reagent and 1x LgBiT protein and 10 μM DAMGO was added and luminescence was measured every 150 seconds for 15 minutes. The final time point was recorded as surface abundance after internalization. Then 100 μM naloxone was added and luminescence was measured every 150 seconds for 1 hour. The final time point was recorded as surface abundance after recycling and compared with the surface abundance after internalization.
Analysis was performed on 5–9 independent experiments with three technical replicates each.
Western Blotting
HEK293T/17 cells were transfected with 0.21 μg (1x) of HiBiT-hMOR and 0.21 μg (1x) of PTCHD1-FLAG. Cells were harvested in PBS, centrifuged at 6000 g for 1 min, and resuspended in 1% Triton X-100 lysis buffer (1 M Tris-HCl pH 7.4, 4M NaCl, 10% w/v Triton X-100, 1x Complete protease inhibitor cocktail). Cells were then sonicated for 10s on ice at 30% power, incubated on ice for 30 min, and centrifuged 20,000 g for 10 min at 4° C. 2x Laemmli sample buffer was added and 12 μl of each sample were loaded onto two gels. Gels were run at 200V for 30 min. Western blot was then run for each gel for 2 hours at 30V.
For the HiBiT western, the Nano-Glo® HiBiT Blotting System was used. The western blot PVDF membrane was incubated overnight at 4° C in PBS /Tween-20. After replacing PBS/Tween-20 with 1x Nano-Glo® Blotting Buffer, LgBit protein was added and incubated with rocking for 2 hours at room temperature. Nano-Glo® Luciferase Assay Substrate was added to Blotting buffer with LgBit and mixed well by rocking. Following incubation for 5 min at room temperature the membranes were imaged with chemiluminescence imager (KwikQuant).
For FLAG and GAPDH detection, PVDF membrane was incubated in 5% milk in PBS/Tween-20 for 1 hour at room temperature. Anti-FLAG M2 (Sigma-Aldrich, F1804), dilution 1:1000 or anti-GAPDH (EMD-Millipore, MAB374) dilution 1:5000 antibodies were diluted into 1% milk in PBS/Tween-20 and incubated at 4° overnight. After washing for 30 minutes with PBS-Tween20, the secondary antibodies were applied in 1% milk in PBS/Tween20 and incubated at room temperature for 1 hour. Following final washes for 30 minutes with PBS/Tween20 and rinse with water, KwikQuant western detection kit reagents were added and the blots were imaged with a KwikQuant imager.
Cholesterol Sensor BRET assay
HEK293T/17 cells were transfected with 1.26 μg (6x) of DNA encoding the D4 cholesterol sensing domain of Perfringolysin O with YDA mutations to enhance cytosolic plasma membrane cholesterol binding as described61. The resulting cholesterol sensor was placed in a pcDNA3.1 vector with an EGFP appended to the C-terminus. Cells were also transfected with 0.21 μg (1x) GRK3cterm-NanoLuc-KRas. The next day, cells were washed with 1 mL of BRET buffer then lifted from plates using 1 mL BRET buffer (0.1 % (w/v) glucose, 1x phosphate-buffered saline, and 0.5 mM MgCl2), centrifuged for 5 min at 500 × g, and then resuspended in 1.5 mL of BRET buffer. 25 μl of each cell suspension was added in triplicate to wells of the 96 well plate. Twenty five μl of 2x Nano-Glo® luciferase was injected into each well and BRET signal was then measured in cells with and without PTCHD1-FLAG and hMOR, as well as in cells treated for 1 hour prior to harvesting with 4 mM MBCD or 200 μg/mL water-soluble cholesterol. BRET ratio (535 nm / 475 nm) was reported. Quantitative analysis was performed on six independent experiments with three technical replicates each.
NanoBiT® β-arr2 Recruitment Assay
Assays were performed as described (Wang 2019) using the Promega NanoBiT® protein:protein interaction assay system with the following modifications: Cells were plated at 1.6 × 106 cells on a 35 mm cell culture dish and transfected using the polyethylenimine (PEI) transfection reagent. The following cDNAs were transfected and balanced to five μg using empty vector: 0.42 μg (2x) MOR-SmBiT pcDNA3.1, 0.21 μg (1x) β-arrestin-2-LgBiT pcDNA3.1, 0.21 μg (1x) or 0.42 μg (2x) codon optimized PTCHD1-FLAG pcDNA3.1 or VPR2-SmBit pcDNA3.1. For treatment with 4 mM MβCD or 200 μg / mL cholesterol, overlaying cell media was removed and replaced with 50 μl of freshly-prepared reagent in BRET buffer (1x PBS, 0.5 mM MgCl2, 0.1% (w/v) D-glucose) and incubated at 37°C, 5% CO2 1 hour prior to start of the assay.
Statistics and Reproducibility
All statistical tests and sample sizes are specified in the Figure Legends and in specific sections of the Methods where appropriate. Data distribution was assumed to be normal but this was not formally tested. All C. elegans behavioral data was acquired using computationally automated methods which are unbiased. For mouse experiments, the experimenters were blinded to the genotype when subjective scoring criteria were used (somatic withdrawal), other tests were videotaped and relied on measurable parameters during which the experimenters were not blinded. No statistical method was used to predetermine sample size. No data were excluded from the analyses. No specific randomization methods were used. Animals were randomly assigned to experimental groups depending on genotype. Cells were allocated into experimental groups based on experimental condition (DNA transfection). Control cells transfected with empty vectors were used in all experiments utilizing cultured cells.
Extended Data
Extended Data Fig.1. TgMOR; bgg10 mutants show impaired tolerance to fentanyl and morphine.
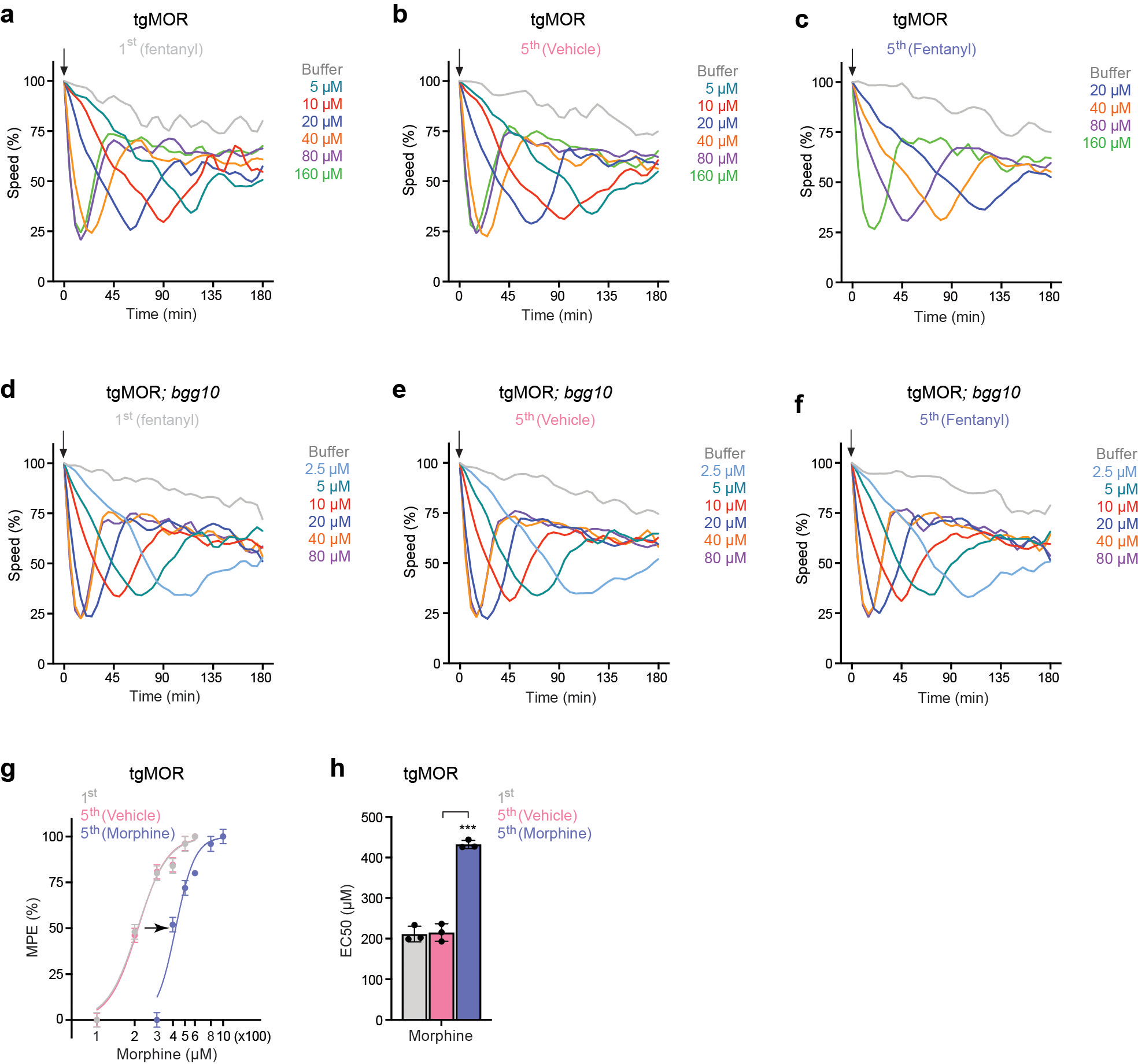
a, Time course showing animal speed after application of different concentrations of fentanyl (arrow) for tgMOR animals at 1st exposure (naïve animals). b, Time course of fentanyl doses for tgMOR after repeated (5x) exposure to negative control vehicle. c, Time course of fentanyl doses for tgMOR after repeated (5x) exposure to fentanyl (10μM). d, Time course of fentanyl doses for tgMOR; bgg10 mutants at 1st exposure (naïve animals). e, Time course of fentanyl doses for tgMOR; bgg10 after repeated (5x) exposure to vehicle. f, Time course of fentanyl doses for tgMOR; bgg10 after repeated (5x) exposure to fentanyl (10μM). g, Dose-response curves showing tgMOR animals develop tolerance after repeated (5x) exposure to morphine (300μM). h, Quantitation of opioid efficacy (EC50 values from g) shows decreased morphine efficacy and development of tolerance by tgMOR animals after repeated (5x) exposure to morphine. For a-h, automated MWT was used to acquire all data. Plotted is mean data for n=12 wells (4–5 animals/well) for all conditions and genotypes. Data for 12 wells were derived from 3 independent experiments. For g and h, data points and histograms are means. n=3 independent experiments with each experiment derived from 4 wells (4–5 animals/well) per dose. For g, error bars are ± SEM. For h, error bars are ± SD and data were analyzed using one-way ANOVA and Bonferroni’s post hoc test. ***p<0.001
Extended Data Fig 2. RSBP-1/R7BP and FRPR-13 show differential effects on opioid tolerance.
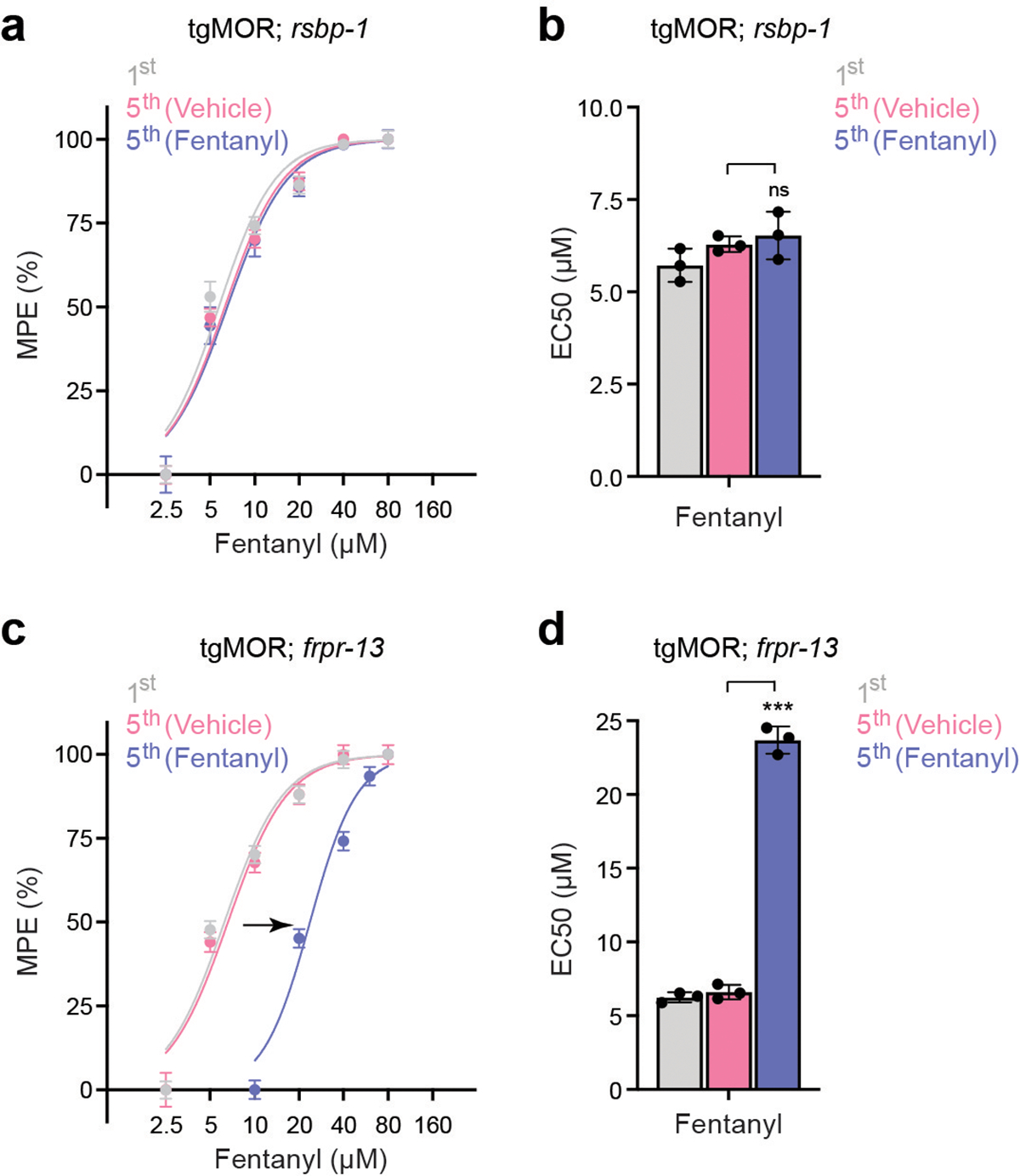
a, Dose-response curves showing tgMOR; rsbp-1 mutants do not develop tolerance after repeated (5x) exposure to fentanyl (10 μM). b, Quantitation of opioid efficacy (EC50 values from a) shows similar fentanyl efficacy and absence of tolerance in tgMOR; rsbp-1 mutants after repeated (5x) exposure to fentanyl. c, Dose-response curves showing tgMOR; frpr-13 mutants develop tolerance after repeated (5x) exposure to fentanyl (10 μM). d, Quantitation of opioid efficacy (EC50 values from c) shows decreased fentanyl efficacy and development of tolerance by tgMOR; frpr-13 mutants after repeated (5x) exposure to fentanyl. For a-d, data points and histograms are means ± SD. n=3 independent experiments with each experiment derived from 4 wells (4–5 animals/well) per dose for all genotypes. For b and d, data were analyzed using one-way ANOVA and Bonferroni’s post hoc test. ***p<0.001
Extended Data Fig. 3. Altered opioid responsiveness of TgMOR; bgg10.
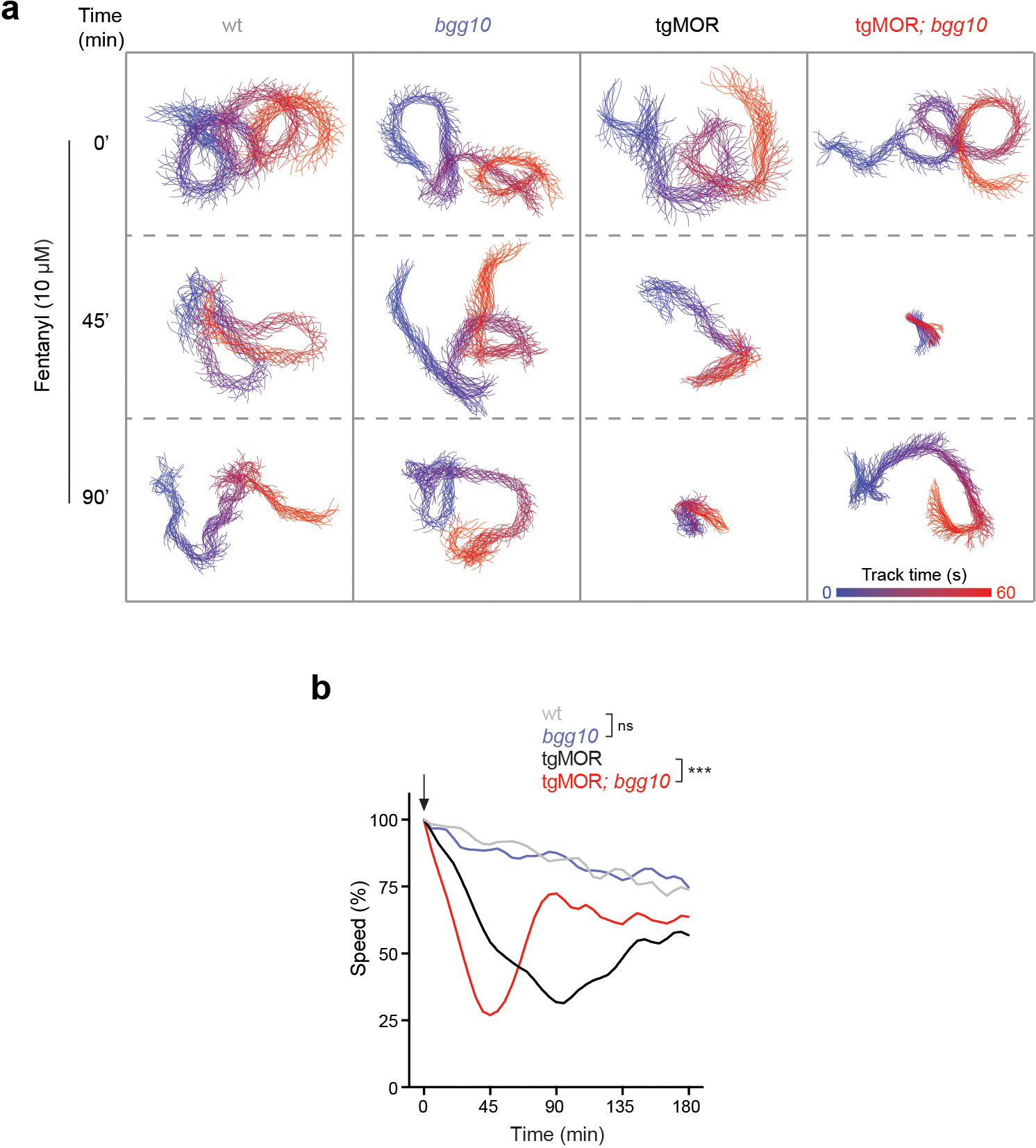
a, Representative traces of individual animal movement acquired using MWT after application of fentanyl (10μM) for indicated time and genotypes. b, Time course of fentanyl response (10μM, arrow) for indicated genotypes. Arrow denotes fentanyl application. Note only tgMOR and tgMOR; bgg10 animals respond to fentanyl. TgMOR; bgg10 mutants are hypersensitive reaching maximum paralysis after 45 minutes in fentanyl. For b, plotted is mean data for n=12 wells (4–5 animals/well) for all genotypes. Data for 12 wells were derived from three independent experiments. Data were analyzed using two-way ANOVA and Bonferroni’s post hoc test. ***p<0.001
Extended Data Fig. 4. Mutations in tgMOR; bgg10 occur in PTR-25/F43D9.1, a conserved member of the Patched protein family.
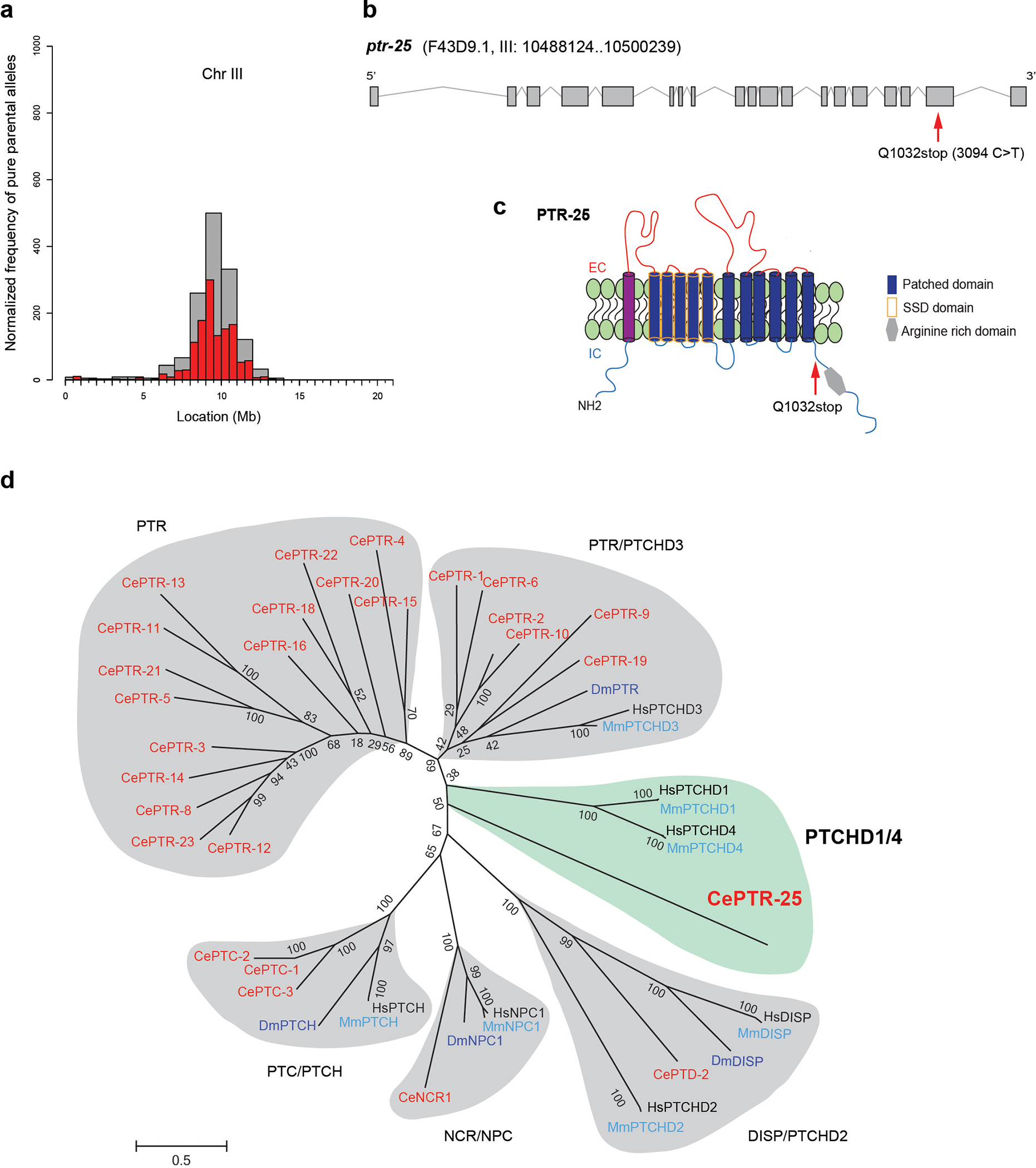
a, Plot showing analysis of whole genome sequence data with mapped region of chromosome III (red and grey bars) containing bgg10. b, Gene diagram showing Q1032stop nonsense mutation (red arrow) in ptr-25 (F43D9.1) contained in mapped region from tgMOR; bgg10 mutants. c, PTR-25 protein with Sterol Sensing Domain (SSD, yellow), Patched family domain (blue) and arginine rich domain (grey). bgg10 mutation, Q1032stop, highlighted (red arrow). d, Phylogenetic analysis of Patched family proteins including all major subfamilies: PTC/PTCH, NCR/NPC, DISP/PTCHD2, PTCHD1/4 (shaded in green) and the PTR group which is heavily expanded in C. elegans. Note PTR-25 has closest homology to mammalian PTCHD1/4. Protein sequences from human (black), mouse (light blue), fly (dark blue) and C. elegans (red) were aligned with Clustal W. Phylogenetic tree was constructed using maximum likelihood method with 1000 bootstrap replicates.
Extended Data Fig. 5. Validation of PTR-25/PTCHD1 as causal gene mutated in tgMOR; bgg10 animals that results in opioid hypersensitivity.
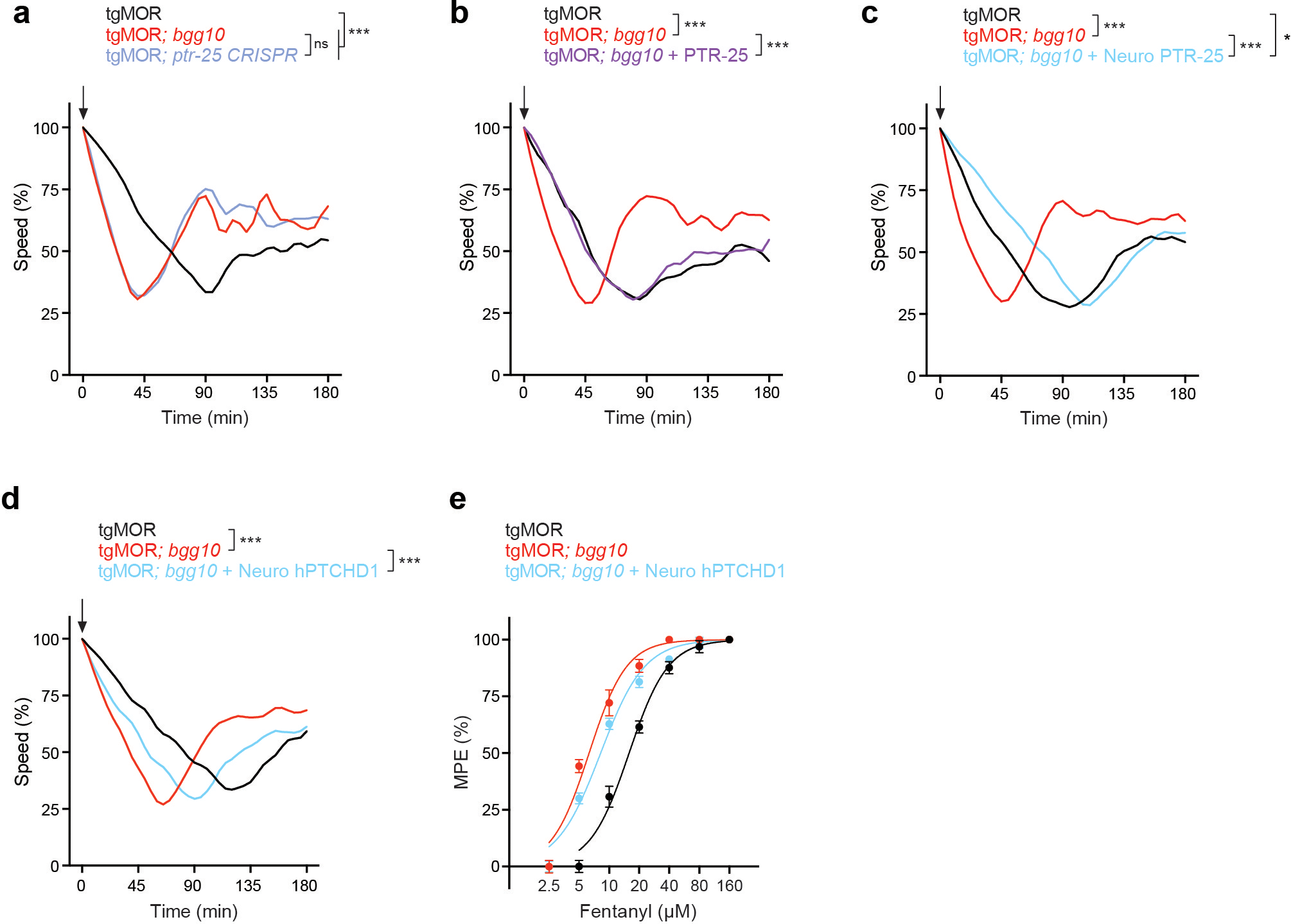
a, Time course of fentanyl application for indicated genotypes. Arrow indicates application of fentanyl (10μM). TgMOR; ptr-25 CRISPR animals phenocopied hypersensitivity in tgMOR; bgg10 mutants. b, Native rescue with PTR-25 restored opioid sensitivity of tgMOR; bgg10 animals. c, Pan-neuronal rescue (rab-3 promoter) with PTR-25 reversed opioid sensitivity of tgMOR; bgg10 mutants. d, Time course of fentanyl application (5μM, arrow) for indicated genotypes. Note transgenic expression of human PTCHD1 using the native ptr-25 promoter rescues opioid hypersensitivity of tgMOR; bgg10 animals. e, Dose-response curves with fentanyl showing transgenic human PTCHD1 decreased opioid sensitivity of tgMOR; bgg10 animals. For a-d, plotted is mean data for n=12 wells (4–5 animals/well) for all genotypes. For e, n=3 independent experiments with each experiment derived from 4 wells (4–5 animals/well) per dose for all genotypes. Data points are means ± SD. For a-d, data were analyzed by two-way ANOVA with Bonferroni’s post hoc test. ***p<0.001, * p<0.05
Extended Data Fig. 6. Behavioral characterization of opioid responsiveness of Ptchd1 KO mice.
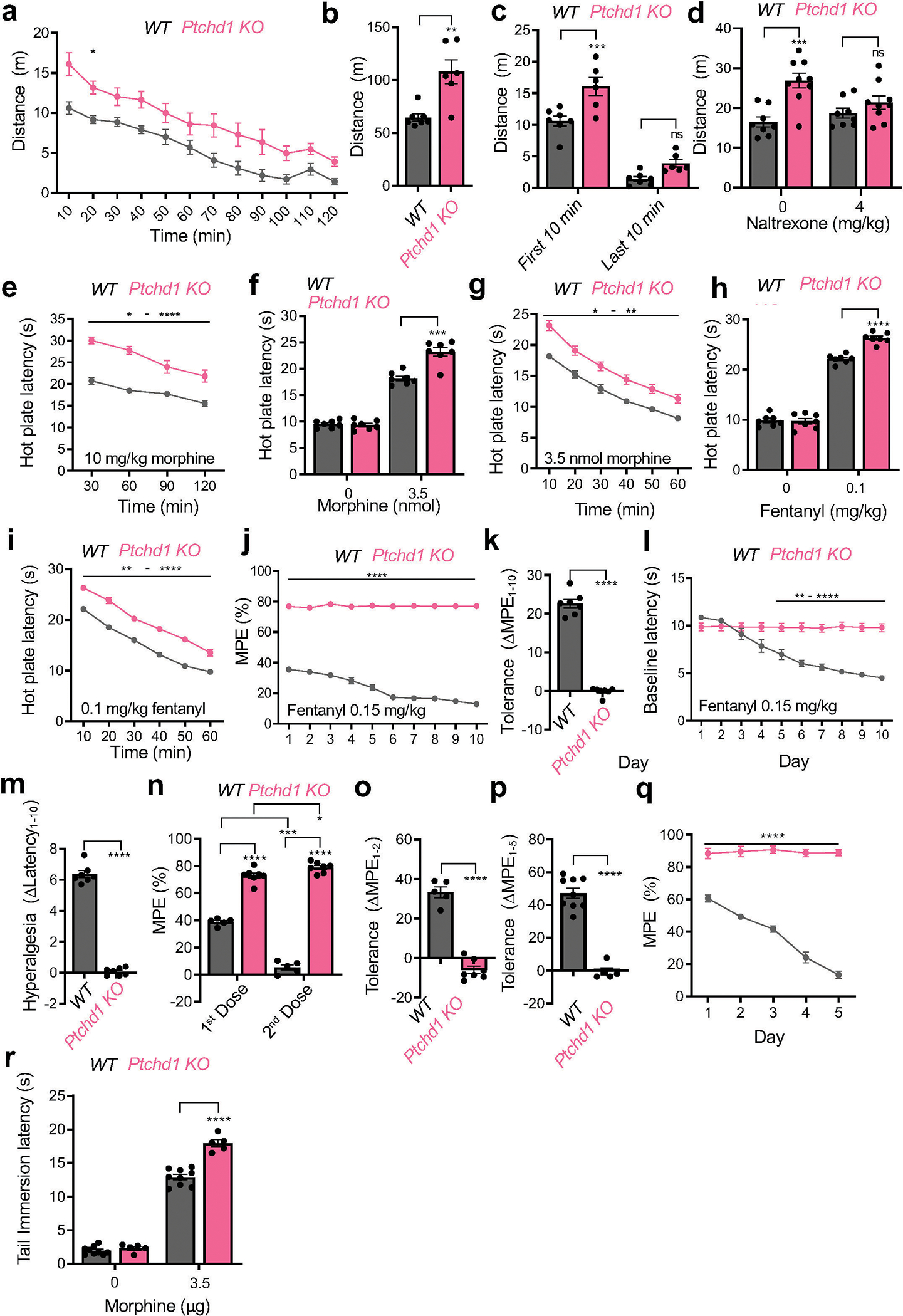
a, Evaluation of activity levels by tracking distance traveled in an open field. Data are from 7 WT and 6 Ptchd1 KO mice.
b, Evaluation of activity levels by calculating total distance traveled in an open field over 120 minutes (from panel a). Data are from 7 WT and 6 Ptchd1 KO mice.
c, Evaluation of activity levels by tracking distance traveled in an open field in first 10 minutes and last 10 minutes from panel a. Data are from 7 WT and 6 Ptchd1 KO mice.
d, Evaluation of activity levels by tracking distance traveled in an open field after naltrexone or saline injection. Data are from 7 WT and 6 Ptchd1 KO mice.
e, Analysis of time course of morphine analgesia by hot plate assay upon systemic morphine administration. Data are from 7 WT and 6 Ptchd1 KO mice.
f, Evaluation of morphine induced analgesia in hot plate assay upon intracerebroventricular (ICV) injection of morphine. Data are from 7 male mice per genotype.
g, Analysis of time course of morphine analgesia in the hot plate assay following ICV injection of morphine. Data are from 7 male mice per genotype.
h, Evaluation of analgesia by hot plate assay with systemic fentanyl administration. Data are from 7 male mice per genotype.
i, Analysis of time course of fentanyl analgesia by hot plate assay from the experiment in panel d.
j, Evaluation of chronic analgesic tolerance by hot plate assay. Data are from 7 male mice per genotype
k, Quantification of analgesic efficacy reduction as a difference in the MPE between session 1 and 10 from panel j. Data are from 7 male mice per genotype.
l, Evaluation of opioid induced hyperalgesia by hot plate assay. Baseline response latencies of mice receiving repeated fentanyl injections (in panel j) prior to drug administration.
m, Quantification of opioid induced hyperalgesia as a difference in the baseline latencies between session 1 and 10 from panel l. Data are from 7 male mice per genotype.
n, Evaluation of acute analgesic tolerance by hot plate assay. Mice received repeated morphine injections (20 mg/kg) 120 minutes apart. Data are from 5 WT and 7 Ptchd1 KO mice.
o, Quantification of analgesic efficacy reduction as a difference in the MPE between session 1 and 2 from panel n. Data are from 5 WT and 7 Ptchd1 KO mice.
p, Evaluation of analgesia by tail immersion assay after intrathecal administration of morphine. Data are from 9 WT and 5 Ptchd1 KO mice.
q, Evaluation of analgesic tolerance by tail immersion assay. Mice received repeated intrathecal morphine injections (3.5 μg) over 5 days. Data are from 9 WT and 5 Ptchd1 KO mice.
r, Quantification of analgesic efficacy reduction as a difference in the MPE between session 1 and 5 from panel q. Data are from 9 WT and 5 Ptchd1 KO mice.
For panel a, d, and q, statistical comparisons were performed by 2-way ANOVA and Šídák post-hoc test. For panel c, e, g, i, j, l, and p, statistical comparisons were performed by 2-way ANOVA and Bonferroni’s post hoc comparison. For panels f, h, and n, statistical comparisons were performed by multiple unpaired two tailed Student’s t-test with Bonferroni’s post hoc. For b, k, m, o, and r, statistical comparisons were performed by unpaired two tailed Student’s t-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Mean values with S.E.M. errors are shown. MPE, maximum possible effect.
Extended Data Fig. 7. Characterization of Ptchd1 – MOR co-expression across the brain.
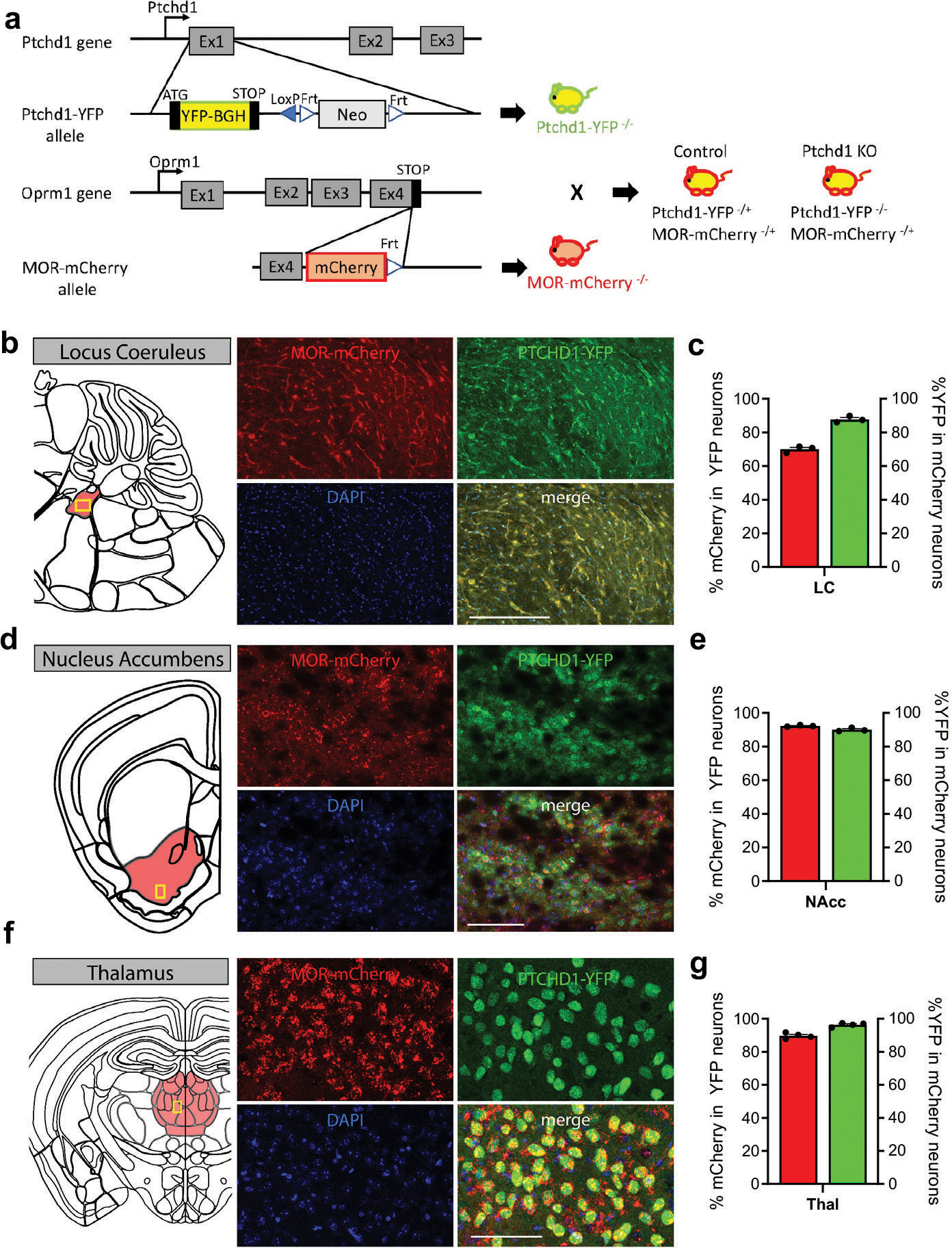
a, Schematic of Ptchd1:YFP and MOR-mCherry mouse genetic manipulations. In the Ptchd1-YFP mouse, exon 1 of the Ptchd1 gene has been replaced with YFP with a YFP-bovine growth hormone poly-A tail (BGH) cassette. In the MOR-mCherry mouse, mCherry was inserted just before the stop codon on exon 4. For electrophysiology and expression studies, the Ptchd1-YFP mouse was crossed with the MOR-mCherry mouse. All mice imaged were heterozygous for the MOR-mCherry allele and homozygous for the Ptchd1-YFP allele. For electrophysiology experiments, control mice were heterozygous the Ptchd1-YFP allele and the MOR-mCherry allele. Ptchd1 KO mice were homozygous for the Ptchd1-YFP allele and heterozygous for the MOR-mCherry allele. “-“ denotes genetically modified allele, “+” is the wild-type allele.
b-g, Co-expression of MOR and PTCHD1 in locus coeruleus (b-c), nucleus accumbens (d-e), and thalamus (f-g). Representative immunohistochemistry images of brain sections from Ptchd1−/− /
MOR-mCherry–/+ mice showing PTCHD1-YFP and MOR-mCherry co-expression. Scale bar is 250 μm in panel b and 100 μm in d and f. Quantification was performed by counting neurons with identified presence of indicated markers in each region from n=3 (panel c and e) and n=4 (panel g) mice per genotype. Mean values with S.E.M. errors are shown in all bar graphs.
Extended Figure 8. Analysis of MOR co-expression with Ptchd1 and morphological effects of Ptchd1 loss.
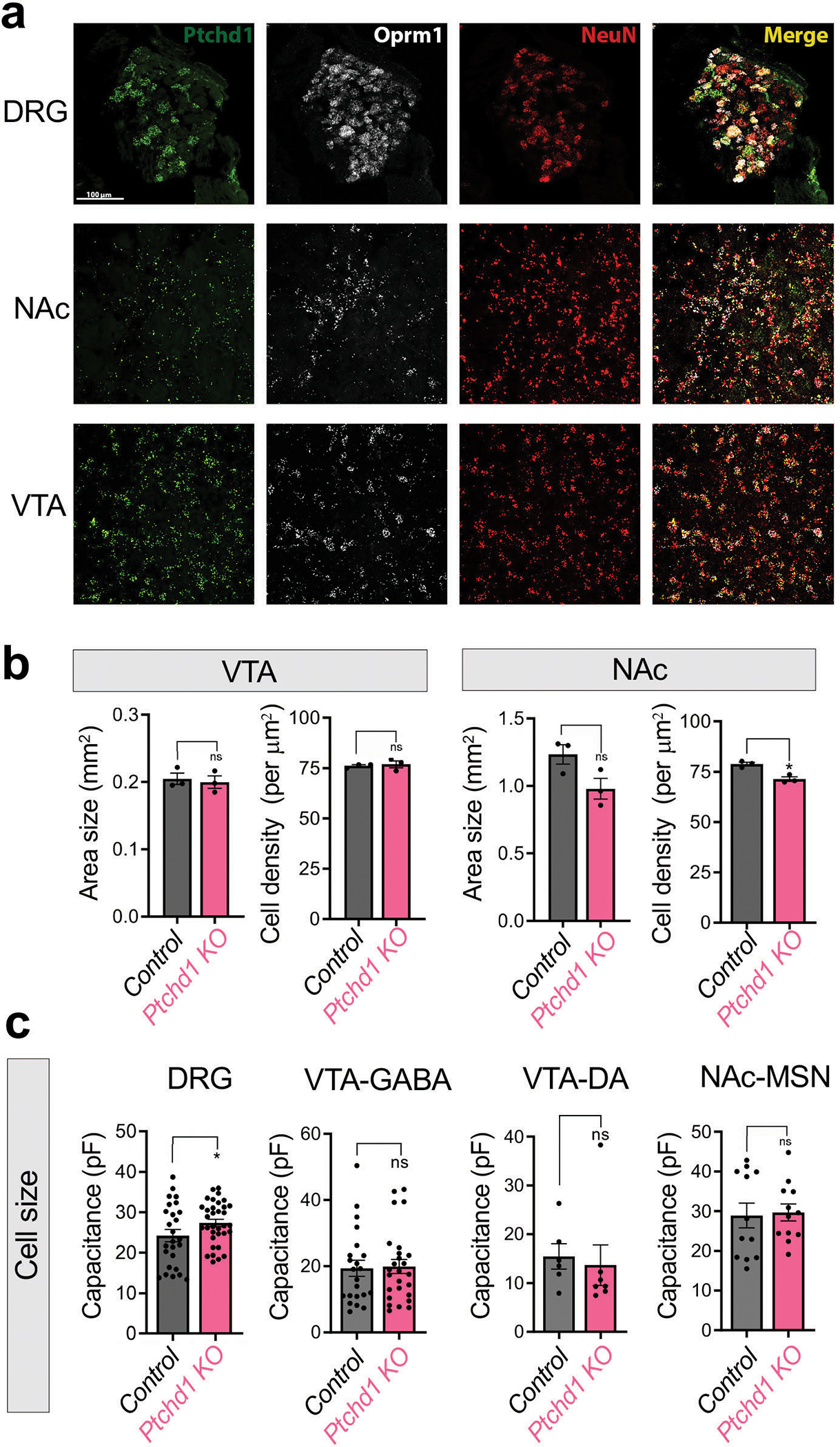
a, In situ hybridization in brain sections from WT animals. Slices were processed for the detection of the indicated probes in dorsal root ganglia (DRG), striatum (STR) and ventral tegmental area (VTA) using RNAScope. Neuronal marker NeuN was used to identify neurons.
b, Quantifications of regional size and cell density in VTA and NAc between Ptchd1 KO and control genotypes. VTA included 8,071 cells from n=3 in control and 7,812 cells from n=3 in Ptchd1 KO mice. NAc included 46,979 cells from n=3 in control and 41,111 cells from n=3 in Ptchd1 KO mice.
c, Quantification of membrane capacitance from recorded DRG nociceptors (control n=26 cells, 8 mice; Ptchd1 KO n=34 cells, 7 mice), VTA GABAergic interneurons (control n=22 cells, 8 mice; Ptchd1 KO n=25 cells, 12 mice), VTA dopamine projection neurons (control n=6 cells, 3 mice; Ptchd1 KO n=7 cells, 4 mice), and NAc medium spiny neurons (control n=12 cells, 4 mice; Ptchd1 KO n=12 cells, 5 mice) between Ptchd1 KO and control genotypes.
For panel b and c, the statistical comparisons were performed by unpaired two-tailed student’s t-test. *p<0.05, n.s. is p>0.05. Mean values with S.E.M. errors are shown.
Extended Data Fig. 9. Electrophysiological characterization of opioid responses in Ptchd1 KO neurons.
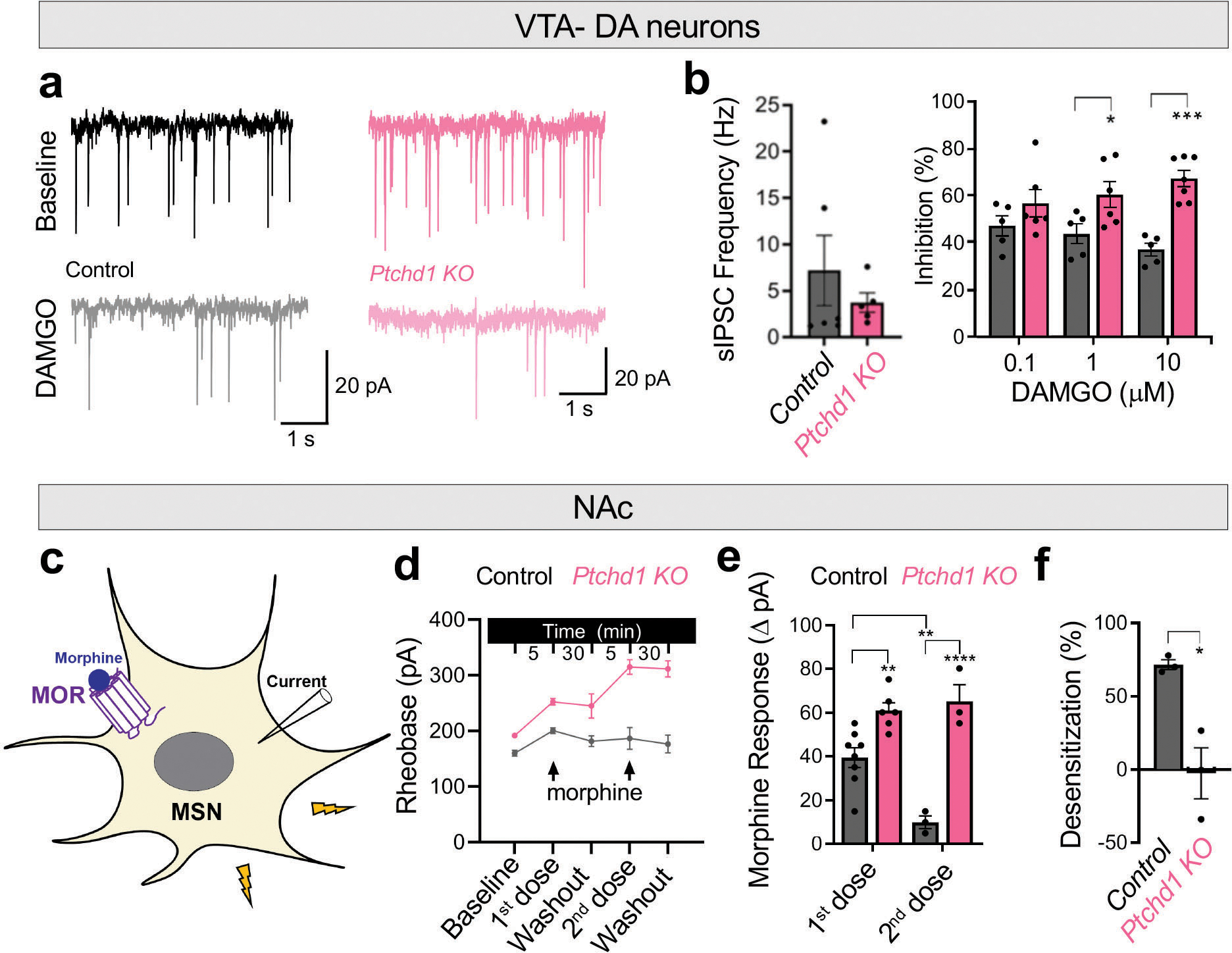
a, Representative whole-cell current recordings of Ptchd1 KO and control DA neurons illustrating inhibition of sIPSC frequency by application of 10 μM DAMGO.
b, Quantification of both baseline sIPSC frequencies and their inhibition in DA neurons upon sequential applications of increasing concentrations of DAMGO. Control n=6 cells, 3 mice; Ptchd1 KO n=7 cells, 4 mice
c, Schematic of recordings from medium spiny neurons (MSN) in the NAc. Activation of MOR on MSNs depresses their excitability.
d, Quantification of rheobase from baseline, through two applications of 10μM morphine and recovery. Control n=8 cells, 4 mice and 3 cells, 3 mice; Ptchd1 KO n=6 cells, 4 mice and 3 cells, 3 mice for the first and second application, respectively.
e, Quantification of 10μM morphine responses as the difference in rheobase.
f, Quantification of desensitization between first and second morphine responses. n=3 cells from 3 mice.
For panel b, the statistical comparison was performed by 2-way ANOVA with Holm-Šídák post-hoc test. For panel e, the statistical comparison was performed by a mixed-model ANOVA with Holm-Šídák post-hoc test. For panel f, the statistical comparison was performed by an unpaired two-tailed student’s t-test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Mean values with S.E.M. errors are shown.
Extended Data Fig 10. Cell biological characterization of Ptchd1 actions.
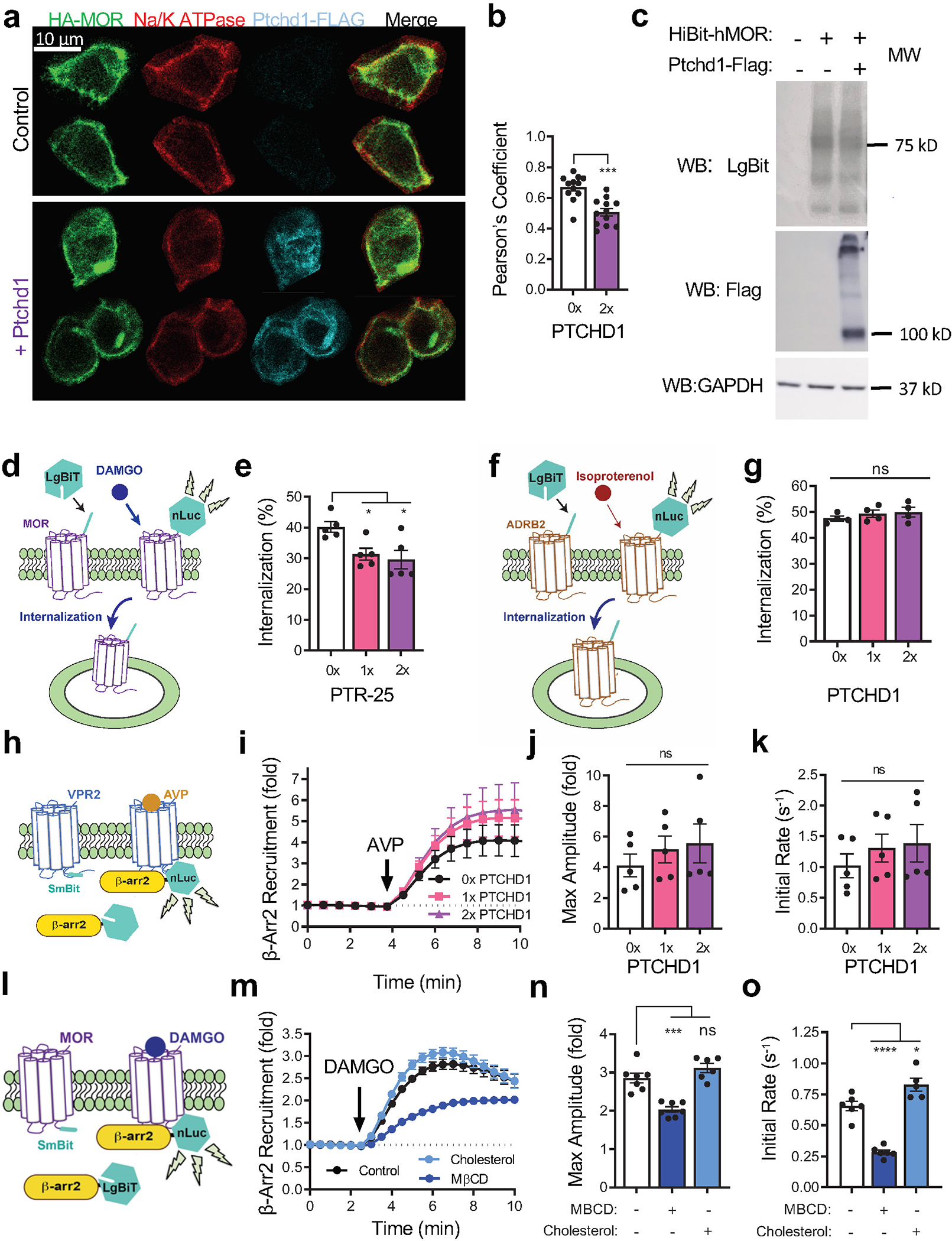
a, Representative immunofluorescence images examining co-localization of MOR with Na+/K+ ATPase. n=8 cells from each genotype.
b, Quantification of the Ptchd1 effect on co-localization based on Pearson’s correlation coefficient in confocal images of fixed cells. 0x and 2x refer to the amount of DNA transfected, where x = 0.21 μg. n=12 cells from each genotype. Significance determined using unpaired two tailed Student’s t-test. ***p<0.001
c, Western blot of HEK293T cells expressing HiBiT-MOR alone or with PTCHD1-flag.
d, Schematic of the assay design to study the effect of C. elegans protein PTR-25 on internalization of MOR. Addition of LgBit to cells expressing HiBit-MOR forms a nanoluciferase (nLuc) enzyme. DAMGO treatment causes internalization of the complex and quenching of the luminescence.
e, Quantification of DAMGO-induced internalization of HiBiT-MOR in HEK293T cells with or without PTR-25 co-expression. 0, 1x, and 2x refer to the amount of DNA transfected, where x = 0.21 μg. n=5 biologically independent experiments. Significance determined using one-way ANOVA and Dunnett’s post-hoc. *p<0.05
f, Schematic of assay design to study the effect of Ptchd1 on internalization of β2-adrenergic receptor (ADRB2). Addition of LgBit to cells expressing HiBit-ADR2B forms a nanoluciferase (nLuc) enzyme. Isoproterenol treatment causes internalization of the complex and quenching of the luminescence.
g, Quantification of isoproterenol induced internalization of HiBit-ADRB2 in HEK293T cells with or without PTCHD1 co-expression. 0x, 1x, and 2x refer to the amount of DNA transfected, where x = 0.21 μg. n=4 biologically independent experiments. Significance determined using one-way ANOVA and Dunnett’s post-hoc. n.s. is p>0.05
h, Experimental design of β-arrestin recruitment assay to study the effect of Ptchd1 on type 2 vasopressin receptor (VPR2). Addition of arginine vasopressin (AVP) induces recruitment of LgBiT-tagged β-arr2 to VPR2-SmBiT to form a functional nLuc enzyme.
i, Time course of Ptchd1 effect on recruitment of β-arr2 to VPR2 determined in n=5 biologically independent experiments.
j, Quantification of the effect of Ptchd1 on the maximum fold-change of β-arr2 recruitment from data in panel i. Significance determined using one-way ANOVA. n.s. is p>0.05
k, Quantification of the initial rate of β-arr2 recruitment to MOR from data in b. 0x, 1x, and 2x refer to the amount of DNA transfected, where x = 0.21 μg. n=5 biologically independent experiments. Significance determined using one-way ANOVA and Dunnett’s post-hoc. n.s.,p>0.05
l, Schematic of the assay design of β-arrestin recruitment assay. Addition of DAMGO induces recruitment of LgBiT-tagged β-arr2 to MOR-SmBiT to form a functional nLuc enzyme.
m, Modulation of DAMGO mediated- βarr2-LgBiT recruitment to MOR-SmBiT following incubation with 4 mM MBCD or 200 μg / mL cholesterol. n=7 biologically independent experiments for control and MβCD and n=6 biologically independent experiments for cholesterol.
n, Quantification of the maximum fold change and o, the initial recruitment rate from panel n. Significance was determined using a one-way ANOVA and Dunnett’s post-hoc. ns, p>0.05, *p<0.05, ***p<0.001, ****p<0.0001.
In all graphs mean values with S.E.M. errors are shown.
Supplementary Material
Acknowledgments
We wish to thank Ms. Natalia Martemyanova for producing and maintaining mice examined in this study and members of the Grill and Martemyanov labs for valuable input. This work was supported by an NIH grants DA040406 and DA048036 (to BG and KAM), DA036596 and EY028033 (to KAM), DA047771 (to HMS). Dr. Grill’s work is made possible in part by a generous gift in honor of Timothy Jackson. We thank the University of Kansas Genome Sequencing Core, which is supported by an NIH Center of Biomedical Research Excellence Grant (GM103638).
Footnotes
Competing Interests
B.G. and K.A.M have filed a patent on the utility of Ptchd1 and modulation of neuronal cholesterol for therapeutic purposes. They are also stockholders in Evodenovo Inc, which has financial interests in intellectual property associated with this study.
Code Availability
Multi-worm tracker is an automated behavioral tracking instrument that uses custom software. Additional information about custom software is available from corresponding authors (B.G.) upon reasonable request.
Data Availability
The data that support the findings of this study are published online as Source Data files for all display items. Additional information is available from the corresponding author upon reasonable request.
References
- 1.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ & Caron MG Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27, 107–144 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Abramow-Newerly M, Roy AA, Nunn C & Chidiac P RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal 18, 579–591 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Hilger D, Masureel M & Kobilka BK Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol 25, 4–12, doi: 10.1038/s41594-017-0011-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collett BJ Opioid tolerance: the clinical perspective. Br J Anaesth 81, 58–68, doi: 10.1093/bja/81.1.58 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Martyn JAJ, Mao J & Bittner EA Opioid Tolerance in Critical Illness. N Engl J Med 380, 365–378, doi: 10.1056/NEJMra1800222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colvin LA, Bull F & Hales TG Perioperative opioid analgesia-when is enough too much? A review of opioid-induced tolerance and hyperalgesia. Lancet 393, 1558–1568, doi: 10.1016/S0140-6736(19)30430-1 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Darcq E & Kieffer BL Opioid receptors: drivers to addiction? Nat Rev Neurosci 19, 499–514, doi: 10.1038/s41583-018-0028-x (2018). [DOI] [PubMed] [Google Scholar]
- 8.Matthes HW et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383, 819–823, doi: 10.1038/383819a0 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Williams JT et al. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65, 223–254, doi: 10.1124/pr.112.005942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corder G et al. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med 23, 164–173, doi: 10.1038/nm.4262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shenoy SK & Lefkowitz RJ beta-Arrestin-mediated receptor trafficking and signal transduction. Trends in pharmacological sciences 32, 521–533, doi: 10.1016/j.tips.2011.05.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ & Caron MG Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723, doi: 10.1038/35047086 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Gillis A et al. Critical Assessment of G Protein-Biased Agonism at the mu-Opioid Receptor. Trends in pharmacological sciences 41, 947–959, doi: 10.1016/j.tips.2020.09.009 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Terzi D, Cao Y, Agrimaki I, Martemyanov KA & Zachariou V R7BP modulates opiate analgesia and tolerance but not withdrawal. Neuropsychopharmacology 37, 1005–1012, doi: 10.1038/npp.2011.284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y et al. Blockade of PDGFR-beta activation eliminates morphine analgesic tolerance. Nat Med 18, 385–387, doi: 10.1038/nm.2633 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue M & Ueda H Protein kinase C-mediated acute tolerance to peripheral mu-opioid analgesia in the bradykinin-nociception test in mice. J Pharmacol Exp Ther 293, 662–669 (2000). [PubMed] [Google Scholar]
- 17.Zhou J et al. Molecular mechanisms of opioid tolerance: From opioid receptors to inflammatory mediators (Review). Exp Ther Med 22, 1004, doi: 10.3892/etm.2021.10437 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang D et al. Genetic behavioral screen identifies an orphan anti-opioid system. Science, doi: 10.1126/science.aau2078 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minevich G, Park DS, Blankenberg D, Poole RJ & Hobert O CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269, doi: 10.1534/genetics.112.144204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong Y, Gu LJ, Sun XG, Yang SH & Zhang XH Comprehensive analysis of patched domain-containing genes reveals a unique evolutionary pattern. Genet Mol Res 13, 7318–7331, doi: 10.4238/2014.February.13.11 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Tora D et al. Cellular Functions of the Autism Risk Factor PTCHD1 in Mice. J Neurosci 37, 11993–12005, doi: 10.1523/JNEUROSCI.1393-17.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wells MF, Wimmer RD, Schmitt LI, Feng G & Halassa MM Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature 532, 58–63, doi: 10.1038/nature17427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noor A et al. Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci Transl Med 2, 49ra68, doi: 10.1126/scitranslmed.3001267 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawrylycz MJ et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399, doi: 10.1038/nature11405 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ung DC et al. Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol Psychiatry 23, 1356–1367, doi: 10.1038/mp.2017.39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsui A, Jarvie BC, Robinson BG, Hentges ST & Williams JT Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron 82, 1346–1356, doi: 10.1016/j.neuron.2014.04.030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nestler EJ Is there a common molecular pathway for addiction? Nat Neurosci 8, 1445–1449 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Khomula EV, Araldi D, Bonet IJM & Levine JD Opioid-Induced Hyperalgesic Priming in Single Nociceptors. J Neurosci 41, 31–46, doi: 10.1523/JNEUROSCI.2160-20.2020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Claing A, Laporte SA, Caron MG & Lefkowitz RJ Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and *-arrestin proteins. Progress in Neurobiology 66, 61–79 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Li X, Saha P, Li J, Blobel G & Pfeffer SR Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc Natl Acad Sci U S A 113, 10079–10084, doi: 10.1073/pnas.1611956113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y et al. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 175, 1352–1364 e1314, doi: 10.1016/j.cell.2018.10.026 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu Y, Wang Y, Law PY, Chen HZ & Loh HH Cholesterol regulates micro-opioid receptor-induced beta-arrestin 2 translocation to membrane lipid rafts. Mol Pharmacol 80, 210–218, doi: 10.1124/mol.110.070870 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barter PJ, Brandrup-Wognsen G, Palmer MK & Nicholls SJ Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lipid Res 51, 1546–1553, doi: 10.1194/jlr.P002816 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McTaggart F & Jones P Effects of statins on high-density lipoproteins: a potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther 22, 321–338, doi: 10.1007/s10557-008-6113-z (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niu Z et al. Melanocortin 4 receptor antagonists attenuates morphine antinociceptive tolerance, astroglial activation and cytokines expression in the spinal cord of rat. Neurosci Lett 529, 112–117, doi: 10.1016/j.neulet.2012.09.034 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Lamberts JT, Rosenthal LD, Jutkiewicz EM & Traynor JR Role of the guanine nucleotide binding protein, Galphao, in the development of morphine tolerance and dependence. Psychopharmacology (Berl) 235, 71–82, doi: 10.1007/s00213-017-4742-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanna MD, Borgonetti V & Galeotti N mu Opioid Receptor-Triggered Notch-1 Activation Contributes to Morphine Tolerance: Role of Neuron-Glia Communication. Mol Neurobiol 57, 331–345, doi: 10.1007/s12035-019-01706-6 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Toda N, Kishioka S, Hatano Y & Toda H Modulation of opioid actions by nitric oxide signaling. Anesthesiology 110, 166–181, doi: 10.1097/ALN.0b013e31819146a9 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Vidal B et al. An atlas of Caenorhabditis elegans chemoreceptor expression. PLoS Biol 16, e2004218, doi: 10.1371/journal.pbio.2004218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beets I et al. Vasopressin/oxytocin-related signaling regulates gustatory associative learning in C. elegans. Science 338, 543–545, doi: 10.1126/science.1226860 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Fernandez RW et al. Cellular Expression and Functional Roles of All 26 Neurotransmitter GPCRs in the C. elegans Egg-Laying Circuit. J Neurosci 40, 7475–7488, doi: 10.1523/JNEUROSCI.1357-20.2020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flavell SW et al. Serotonin and the neuropeptide PDF initiate and extend opposing behavioral states in C. elegans. Cell 154, 1023–1035, doi: 10.1016/j.cell.2013.08.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quillinan N, Lau EK, Virk M, von Zastrow M & Williams JT Recovery from mu-opioid receptor desensitization after chronic treatment with morphine and methadone. J Neurosci 31, 4434–4443, doi: 10.1523/JNEUROSCI.4874-10.2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whistler JL, Chuang HH, Chu P, Jan LY & von Zastrow M Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron 23, 737–746, doi: 10.1016/s0896-6273(01)80032-5 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Zachariou V et al. Essential role for RGS9 in opiate action. 100, 13656–13661 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gimpl G, Burger K & Fahrenholz F Cholesterol as modulator of receptor function. Biochemistry 36, 10959–10974, doi: 10.1021/bi963138w (1997). [DOI] [PubMed] [Google Scholar]
- 47.Sejdiu BI & Tieleman DP Lipid-Protein Interactions Are a Unique Property and Defining Feature of G Protein-Coupled Receptors. Biophys J 118, 1887–1900, doi: 10.1016/j.bpj.2020.03.008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manglik A et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326, doi: 10.1038/nature10954 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng H et al. Palmitoylation and membrane cholesterol stabilize mu-opioid receptor homodimerization and G protein coupling. BMC Cell Biol 13, 6, doi: 10.1186/1471-2121-13-6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng H et al. Cholesterol level influences opioid signaling in cell models and analgesia in mice and humans. J Lipid Res 53, 1153–1162, doi: 10.1194/jlr.M024455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mansouri MT, Khodayar MJ, Tabatabaee A, Ghorbanzadeh B & Naghizadeh B Modulation of morphine antinociceptive tolerance and physical dependence by co-administration of simvastatin. Pharmacology, biochemistry, and behavior 137, 38–43, doi: 10.1016/j.pbb.2015.08.002 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Pedersen LB, Mogensen JB & Christensen ST Endocytic Control of Cellular Signaling at the Primary Cilium. Trends Biochem Sci 41, 784–797, doi: 10.1016/j.tibs.2016.06.002 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Qi X & Li X Mechanistic Insights into the Generation and Transduction of Hedgehog Signaling. Trends Biochem Sci 45, 397–410, doi: 10.1016/j.tibs.2020.01.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kowatsch C, Woolley RE, Kinnebrew M, Rohatgi R & Siebold C Structures of vertebrate Patched and Smoothened reveal intimate links between cholesterol and Hedgehog signalling. Curr Opin Struct Biol 57, 204–214, doi: 10.1016/j.sbi.2019.05.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 55.Giles AC et al. A complex containing the O-GlcNAc transferase OGT-1 and the ubiquitin ligase EEL-1 regulates GABA neuron function. J Biol Chem 294, 6843–6856, doi: 10.1074/jbc.RA119.007406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erbs E et al. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct 220, 677–702, doi: 10.1007/s00429-014-0717-9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haley TJ & McCormick WG Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br J Pharmacol Chemother 12, 12–15, doi: 10.1111/j.1476-5381.1957.tb01354.x (1957). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perner C & Sokol CL Protocol for dissection and culture of murine dorsal root ganglia neurons to study neuropeptide release. STAR Protoc 2, 100333, doi: 10.1016/j.xpro.2021.100333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petruska JC, Napaporn J, Johnson RD, Gu JG & Cooper BY Subclassified acutely dissociated cells of rat DRG: histochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol 84, 2365–2379, doi: 10.1152/jn.2000.84.5.2365 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Oner SS, Blumer JB & Lanier SM Group II activators of G-protein signaling: monitoring the interaction of Galpha with the G-protein regulatory motif in the intact cell. Methods Enzymol 522, 153–167, doi: 10.1016/B978-0-12-407865-9.00009-1 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Liu SL et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat Chem Biol 13, 268–274, doi: 10.1038/nchembio.2268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are published online as Source Data files for all display items. Additional information is available from the corresponding author upon reasonable request.