

ABSTRACT
DNA interstrand cross-links, such as those formed by psoralen-UVA irradiation, are highly toxic lesions in both humans and bacteria, with a single lesion being lethal in Escherichia coli. Despite the lack of effective repair, human cancers and bacteria can develop resistance to cross-linking treatments, although the mechanisms of resistance remain poorly defined. Here, we subjected E. coli to repeated psoralen-UVA exposure to isolate three independently derived strains that were >10,000-fold more resistant to this treatment than the parental strain. Analysis of these strains identified gain-of-function mutations in the transcriptional regulator AcrR and the alpha subunit of RNA polymerase that together could account for the resistance of these strains. Resistance conferred by the AcrR mutation is mediated at least in part through the regulation of the AcrAB-TolC efflux pump. Resistance via mutations in the alpha subunit of RNA polymerase occurs through a still-uncharacterized mechanism that has an additive effect with mutations in AcrR. Both acrR and rpoA mutations reduced cross-link formation in vivo. We discuss potential mechanisms in relation to the ability to repair and survive interstrand DNA cross-links.
IMPORTANCE Psoralen DNA interstrand cross-links are highly toxic lesions with antimicrobial and anticancer properties. Despite the lack of effective mechanisms for repair, cells can become resistant to cross-linking agents through mechanisms that remain poorly defined. We derived resistant mutants and identified that two gain-of-function mutations in AcrR and the alpha subunit of RNA polymerase confer high levels of resistance to E. coli treated with psoralen-UVA. Resistance conferred by AcrR mutations occurs through regulation of the AcrAB-TolC efflux pump, has an additive effect with RNA polymerase mutations, acts by reducing the formation of cross-links in vivo, and reveals a novel mechanism by which these environmentally and clinically important agents are processed by the cell.
KEYWORDS: DNA interstrand cross-links, psoralen-UVA, DNA repair, resistance
INTRODUCTION
The clinically important drugs psoralen, cisplatin, nitrogen mustard, and mitomycin vary widely in their origins. Some are naturally occurring: psoralens are defensive toxins produced by a variety of plants such as figs and celery (1), and mitomycins are antibiotics produced by bacteria of the genus Streptomyces (2). In contrast, nitrogen mustards were originally synthesized in the 1920s and 1930s for use in chemical warfare (3). Despite their differences, these drugs have a common mechanism of toxicity due to their ability to create particularly destructive lesions in DNA called interstrand cross-links (4, 5). Interstrand cross-links are generated when molecules such as psoralen intercalate between complementary strands of DNA and form covalent bonds with both strands (4, 6–8). In the case of psoralen and its derivatives, bond formation additionally requires the absorption of photons from the UVA wavelength (7, 9). Interstrand cross-links prevent the separation of the DNA strands, making essential cellular processes like transcription and replication impossible. Eventually, such interference is lethal to the cell (10). Furthermore, DNA interstrand cross-links are recalcitrant to known mechanisms of repair since no complementary strand is available for repair synthesis to occur (10).
The formidable lethality of interstrand cross-links makes cross-linking drugs potent treatments for conditions in which the destruction of rapidly dividing or metabolically active cells is required, such as in the treatment of cancers (11, 12). Cross-linking drugs are often first- and second-line chemotherapeutics, including cisplatin, which is curative for many testicular cancers (13–16). Despite the potency of these drugs, cells, including those in cancerous tumors, can develop resistance to these therapies (17–19). Much remains unknown about how resistance to cross-linking agents arises (19–21). Resistance may result from changes in the membrane permeation of the cross-linking drug or from the active efflux of the drug out of the cell (22), as seen in multidrug-resistant cancers (23, 24). Others have identified various gene products that seem to interact with interstrand cross-links and hypothesized that cells can remove interstrand cross-links from DNA and repair the genomic damage caused by these lesions (25–27).
Several DNA repair mutants that render cells hypersensitive to cross-linking agents have been isolated (25, 27–30). Notably, in humans, mutations in 22 genes result in the hereditary genetic disorder Fanconi anemia. Cells from patients exhibit hypersensitivity to cross-linking agents and accumulate chromosome breaks following treatment with cross-linking agents (31, 32), suggesting defects in the repair of these lesions. As in Escherichia coli, many of the cross-link-hypersensitive Fanconi anemia genes render cells hypersensitive to other types of DNA damage, including monoadducts formed by the same cross-linking agents (33, 34). Based on these hypersensitivities, a number of complex models have been proposed in which either base or nucleotide excision repair acts sequentially before and after translesion synthesis or recombination to effect repair (29, 35–37). However, in vivo evidence of intermediates predicted by these models is generally lacking. Complicating the interpretation of DNA repair mutant hypersensitivity is that all cross-linking drugs also induce other forms of DNA damage, including monoadduct intermediates (6, 38). Mutants proposed to be involved in cross-link repair are also hypersensitive to monoadducts, which may account for the sensitivity of these mutants to cross-linking drugs. By comparing psoralen derivatives that form only monoadducts to those that form both monoadducts and cross-links, Cole et al. found that the hypersensitivity of most mutants could be attributed to defects in monoadduct repair alone (39). Additionally, when the cross-links in DNA were quantified, the authors found that between one and two cross-links were sufficient to render E. coli inviable (9, 39). Using other cross-linking agents, others have arrived at similar conclusions (40). In human cells, studies measuring cross-link repair are limited but similarly suggest that the repair capacity is minimal, with lethality occurring at between 200 and 900 lesions per cell (41–44). In all of these studies, the estimates depend on extrapolation since lethality occurs at doses below the direct detection limit for cross-links.
These studies make it clear that the capacity to repair interstrand cross-links is limited and raise the possibility that effective repair mechanisms do not exist in the cell. This implies that alternative mechanisms are responsible for the development of resistance to cross-linking drugs like psoralen, which is the focus of this study. To investigate the mechanisms by which cross-link resistance develops, an iterative selection scheme was utilized to generate three independently derived strains of E. coli that are highly resistant to psoralen and UVA (PUVA) treatment. The genomes of these strains were then sequenced, and the mutations involved in psoralen resistance were characterized.
RESULTS
Generation of strains resistant to psoralen plus UVA irradiation.
To select for mutations that confer resistance to psoralen-UVA treatment, three independent cultures derived from a single colony were grown, spread onto plates, and exposed to various doses of UVA irradiation in the presence of 20 μg/mL 8-methoxypsoralen. The cells were then collected from the plate at the dose where survival was first noticeably reduced (see Fig. S1 in the supplemental material) and were used to inoculate cultures for the next successive round. Following six (for isolate 1) or seven (for isolates 2 and 3) rounds of selection, each culture exhibited a high level of resistance to psoralen-UVA treatment. Independent colonies were isolated from each culture and designated resistant isolates 1, 2, and 3. To quantify the level of the psoralen-UVA resistance of each isolate, 10-fold serial dilutions of a culture grown overnight were spotted onto plates containing 20 μg/mL 8-methoxypsoralen and exposed to increasing doses of UVA. Following incubation overnight at 37°C, the surviving colonies were counted and compared to those on the unexposed plate to determine survival. As shown in Fig. 1, the survival of the three isolates increased more than 104-fold relative to the parent strain at high psoralen-UVA doses. The survival of cells exposed to UVA alone or psoralen alone remained unaffected at these doses (Fig. S2). These results indicate that E. coli cells contain within their genomes the ability to become resistant to treatment with psoralen-UVA, a cross-linking agent.
FIG 1.

Isolates acquired high-level resistance to psoralen-UVA treatment following repeated exposure and selection. The survival of resistant isolate 1 (green open triangles), resistant isolate 2 (green open squares), resistant isolate 3 (green open diamonds), and the SR108 parent (black filled squares) following irradiation with the indicated UVA doses in the presence of 20 μg/mL 8-methoxypsoralen is plotted. The survival of the parental strain was below the detectable limit at UVA doses of >15.6 kJ/m2. Plots represent the averages from at least three independent experiments. Error bars represent the standard errors of the means.
Identification of mutations in resistant strains.
To identify the mutations responsible for conferring psoralen-UVA resistance, the genome of each isolate was sequenced using high-throughput sequencing and compared to the SR108 parent genome. Although several mutations were identified in each strain (Table 1), three genes were found to be mutated across multiple isolates. All three resistant isolates contained mutations in acrR, a transcriptional regulator (45). Resistant isolates 2 and 3 had point mutations in rpoA, which encodes the alpha subunit of the bacterial RNA polymerase (46). Finally, resistant isolate 2 contained a mutation in rclA, encoding an oxidoreductase involved in resistance to reactive chlorine species (47, 48). Similarly, resistant isolate 3 contained an intergenic mutation between rclR, the local activator of rclA expression, and a putative oxidoreductase, ykgE (47). It should be noted that while both isolate 1 and isolate 3 had the same large deletions of a region near the terminus, this deletion has been observed previously in our SR108 parent strain, appears to be driven by recombination between IS5 insertion elements, and does not exhibit any noticeable phenotype in our SR108 background. We therefore elected to further characterize the gene products of the acrR, rpoA, and rclA mutants for their potential role in psoralen-UVA resistance.
TABLE 1.
Full list of mutations identified in psoralen-resistant isolates
| Resistant isolate | Mutationsa |
|---|---|
| 1 | ycdS(W555C), ycfQ (frameshift), flhC (frameshift), yghA/exbD (frameshift), yrbG (frameshift), ΔabgT ΔydeN, ykgD/ykgE (INS), acrR (INS) |
| 2 | rclA(A368G), acrR(L34Q), fadR (frameshift), purL(D162D), ygjR(M184T), yiiF/yiiE (frameshift), rpoA(E273G), lamB/malM (A→G) |
| 3 | dgt(L260*), gmhB(D127E), acrR (frameshift), ybeL/ybeQ (frameshift), ybfA (frameshift), rhsC(Y1276C), ssuC (frameshift), ychN(L28S), oppB (frameshift), cspI/ydfP (frameshift), torY (frameshift), wcaL (frameshift), yfeU(Y146Y), yfhM(P1609P), ygcE/ygcF (frameshift), ygiV/ygiW (T→A), qseB(Y41C), thrU/coaA (G→A), katG(D140E), wzxE (frameshift), aldB/yiaW (frameshift), yiaM (frameshift), rhsB (frameshift), rpoA(P323R), trkA(H51R), melR(L219P), yjiD (frameshift), ΔabgT -ΔydeN |
INS, insertion; /, intergenic.
Gain-of-function mutations in acrR and rpoA, but not rclA, confer resistance and can account for increased survival against psoralen and UVA irradiation.
To characterize these three candidate gene products, we focused on resistant isolate 2, which contained mutations in all three candidate genes, had the fewest total mutations, and exhibited levels of resistance as high as or higher than those of the other isolates (Fig. 1). Furthermore, despite numerous attempts, we have been unable to productively infect isolates 1 and 3 with phage P1 for transduction. To determine if any of these mutations contributed to psoralen-UVA resistance, each mutation was linked to a kanamycin resistance (Kanr) cassette and transduced from isolate 2 into BW25113, the parental strain used for the Keio collection of deletion mutants (49, 50). The resulting mutations were then examined using the same survival assay as the one used to test the resistant isolates, with BW25113 and resistant isolate 2 serving as negative and positive controls, respectively. As shown in Fig. 2A, both mutations acrR(L34Q) and rpoA(E273G) conferred moderate levels of resistance to psoralen-UVA in an otherwise wild-type background compared to their BW25113 parent. Neither the acrR(L34Q) nor the rpoA(E273G) mutant was as resistant as resistant isolate 2, arguing that each mutation could only partially account for the resistance phenotype of this isolate. In contrast, the sensitivity of strains containing the rclA(A368G) mutation did not change relative to the parental strain (Fig. 2A and Fig. S3), arguing that this mutation was not associated with the psoralen-UVA resistance phenotype.
FIG 2.
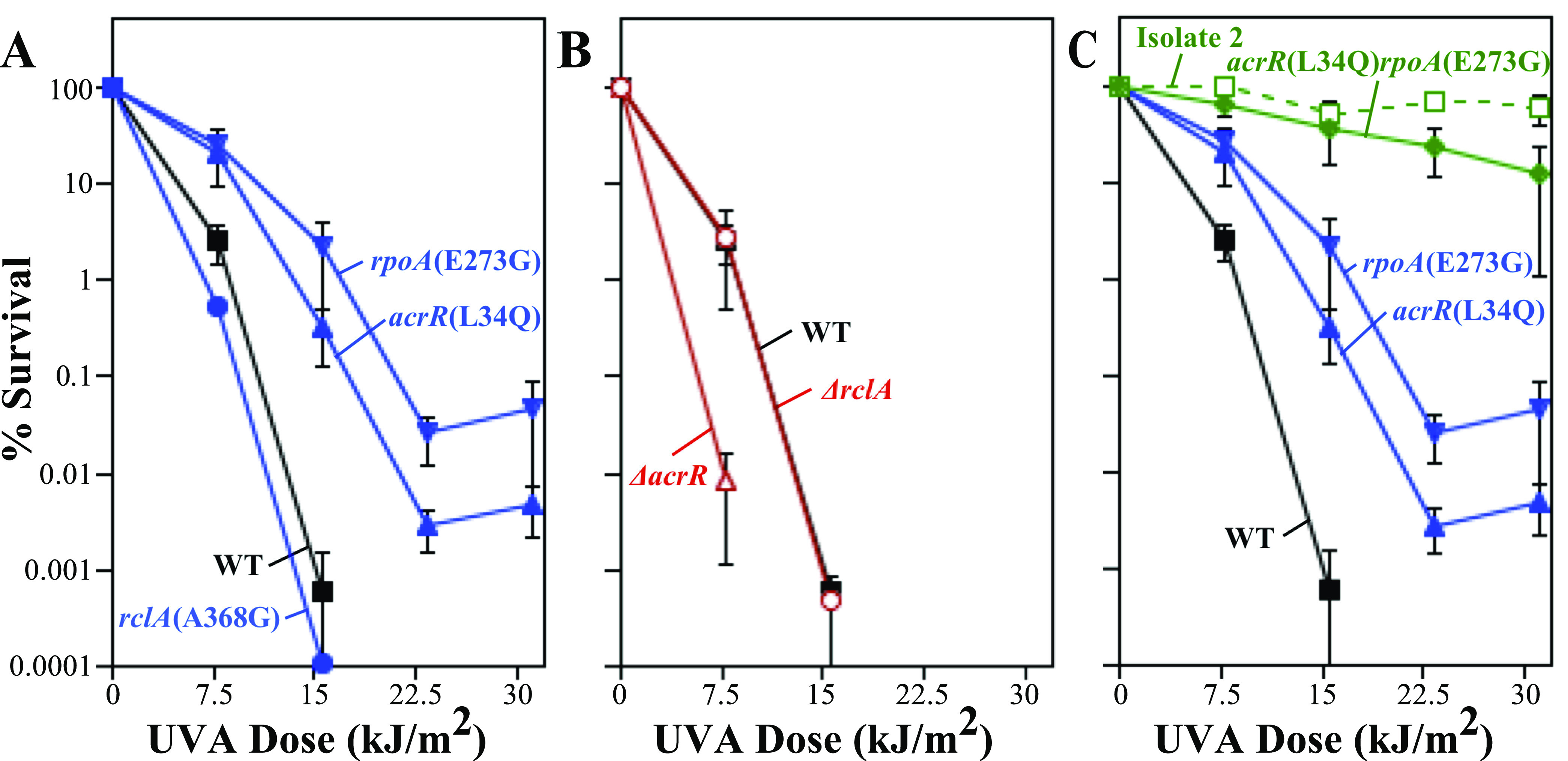
Gain-of-function mutations in acrR and rpoA, but not rclA, confer resistance to psoralen-UVA treatment. The survival of the acrR(L34Q) (blue triangles), rpoA(E273G) (blue inverted triangles), and rclA(A368G) (blue circles) mutants and the BW25113 parent (black squares) (A); the ΔacrR (red open triangles) and ΔrclA (red open circles) mutants (B); and the acrR(L34Q) mutant (blue triangles), the rpoA(E273G) mutant (blue inverted triangles), the acrR(L34Q) rpoA(E273G) double mutant (green diamonds), and resistant isolate 2 (green open squares) (C) in the presence of 20 μg/mL 8-methoxypsoralen at the indicated UVA doses is plotted as described in the legend of Fig. 1. The survival of the ΔacrR and ΔrclA mutants was below the detectable limit at UVA doses of >7.5 and >15.6 kJ/m2, respectively. Plots represent the averages from at least three independent experiments. Resistant isolate 2 was derived from the SR108 background (indicated by the dotted line), while all other strains are isogenic mutants of BW25113. Error bars represent the standard errors of the means. WT, wild type.
To determine whether the resistance-conferring mutations represent a loss or a gain of protein function, we examined the resistance of strains deleted for these genes. If deleting the gene confers resistance similar to that of the point mutation, the mutation likely results in a loss of the protein’s function. rpoA is an essential gene (49), implying that the resistance mutations arising in this gene product are gain-of-function mutations. However, the strains with deletions in acrR and rclA are viable. As shown in Fig. 2B, the deletion of acrR renders cells hypersensitive to psoralen-UVA irradiation, in contrast to the point mutation, arguing that acrR(L34Q) represents a gain-of-function mutation. The sensitivity of a mutant deleted for rclA was similar to those of both the rclA(A368G) point mutant and the parental strain. Furthermore, mutants deleted for the genes rclR and ykgE, which border an intergenic insertion in resistant isolate 3, also showed wild-type sensitivity to psoralen-UVA treatment (Fig. S4). These results are consistent with the interpretation that the rclA locus is not involved in the resistance phenotype.
The point mutations in rpoA and acrR could be acting in the same pathway to confer resistance, or they may function through separate mechanisms. If the mutations operate in different pathways to confer resistance, one might expect that cells containing both mutations would be more resistant than either single mutant alone. If the mutations operate in a single pathway, one might expect that the resistance of the double mutant cells would be similar to that of the single mutants. To examine this possibility, we constructed an acrR(L34Q) rpoAE(E273G) double mutant. As shown in Fig. 2C, the double mutant was more resistant than either single mutant and exhibited resistance comparable to that of isolate 2. We interpret these results to imply that the mechanisms by which mutations in acrR and rpoA confer resistance to psoralen-UVA are distinct and likely account for the resistance observed in isolate 2.
Neither acrR(L34Q) nor rpoA(E273G) alters the growth rate of cells.
One general mechanism that increases resistance to DNA damage is a reduction in the growth rate of cells. In general, this allows more time for repair to occur and reduces the frequency with which replication encounters DNA damage (51–53). To examine whether slower growth may be contributing to the resistance of these strains, we compared the growth rates of these mutants to those of their parents. The results showed that the growth rates of resistant isolates 2 and 3 were modestly slower than that of the SR108 parental strain from which they were derived (Fig. 3A). However, when the acrR(L34Q) or rpoAE(E273G) mutation was moved into an otherwise wild-type BW25113 background, no difference in the growth rate was observed for either the single or double mutant (Fig. 3B). These results suggest that the reduced growth rates of isolates 2 and 3 are likely caused by secondary mutations that accumulated in these strains. Notably, all growth rates remained relatively similar between strains, and no severe growth impairments were observed that could account for the extreme resistance observed in these strains.
FIG 3.

Growth rates remain similar between strains and are unlikely to account for the resistance conferred by acrR(L34Q) or rpoA(E273G). The absorbance (630 nm) values of SR108 parent (black filled squares), isolate 1 (green open inverted triangles), isolate 2 (green open squares), and isolate 3 (green open diamonds) cultures (A) and BW25113 parent (black filled squares), ΔacrR mutant (red open triangles), acrR(L34Q) mutant (blue triangles), rpoA(E273G) mutant (blue inverted triangles) and acrR(L34Q) rpoA(E273G) double mutant (green diamonds) cultures (B) grown at 37°C are plotted over time. The doubling times and ranges (in minutes) from duplicate experiments are indicated below each strain.
acrR(L34Q) and rpoA(E273G) affect the efflux capacity, but not the repair capacity, of the cell.
Reduced cross-link formation in the resistant isolates could result from an increased repair or efflux capacity of the cell. To address this, we examined survival following UVC irradiation, whose toxicity is associated with the formation of bulky DNA adducts that require removal by nucleotide excision repair. The UVC resistance of the three isolates as well as the acrR(L34Q) and rpoA(E273G) mutants remained statistically similar to that of the parental strain (Fig. 4A, B, and D, E). In contrast, the three isolates and the acrR(L34Q) and rpoA(E273G) mutants each conferred resistance to chloramphenicol, an aromatic molecule that affects translation but does not damage DNA directly (Fig. 4C and F). Chloramphenicol has a structure similar to that of psoralen and has been shown to be a substrate for the AcrAB efflux pump (54). Together, these observations are consistent with psoralen-UVA resistance being associated with an upregulation of the efflux capacity but not the DNA repair capacity.
FIG 4.
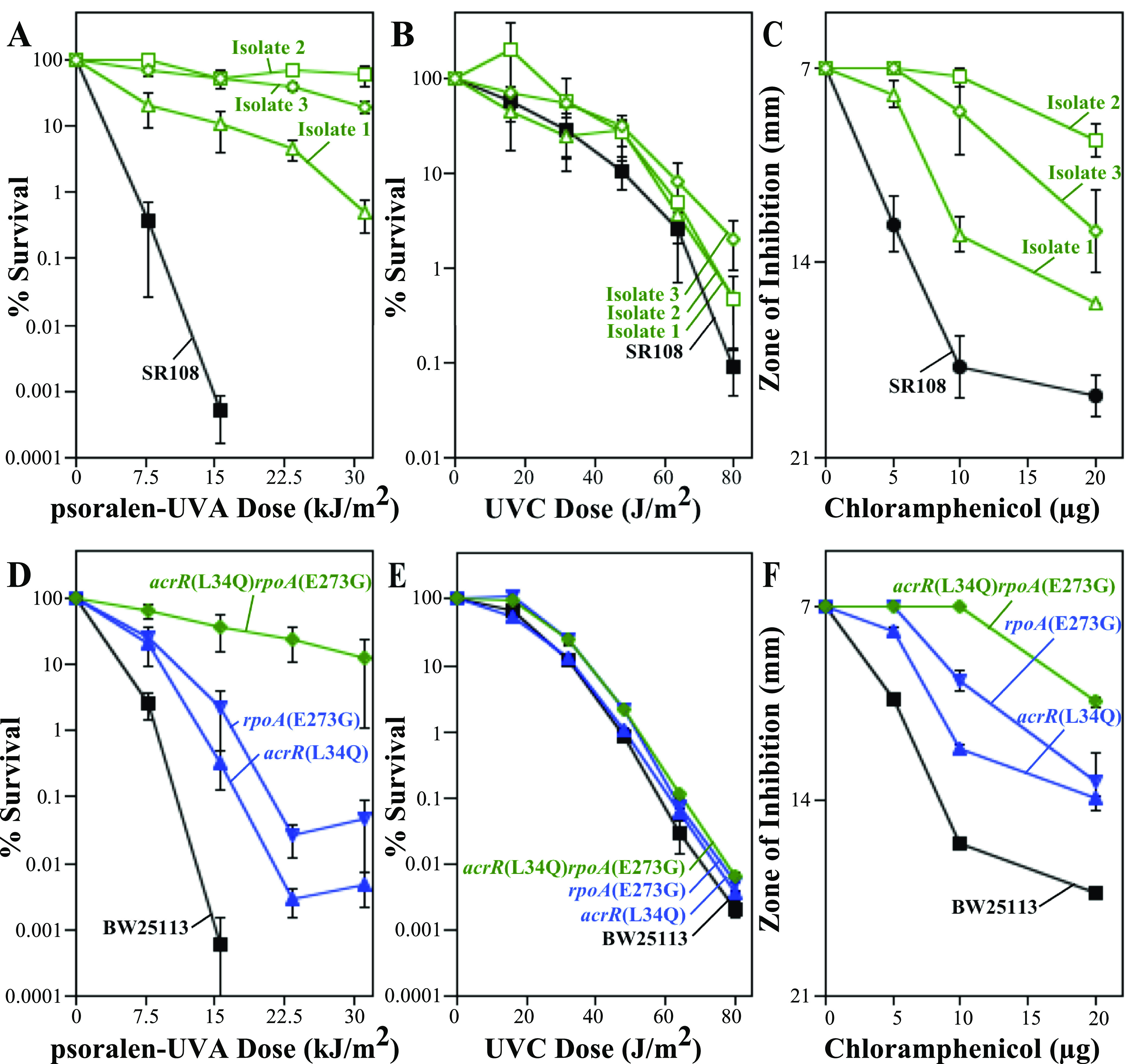
Isolates and mutants that exhibit increased resistance to psoralen-UVA also exhibit increased resistance to chloramphenicol but not UVC irradiation. (A) The survival of resistant isolate 1 (green open triangles), resistant isolate 2 (green open squares), resistant isolate 3 (green open diamonds), and the SR108 parent (black filled squares) in the presence of psoralen is replotted from Fig. 1 for comparison. (B) The survival of resistant isolate 1 (green open triangles), resistant isolate 2 (green open squares), resistant isolate 3 (green open diamonds), and the SR108 parent (black filled squares) following UVC irradiation at the indicated doses is plotted. (C) Diameters of the zones of inhibition around 7-mm paper discs treated with the indicated amounts of chloramphenicol are plotted for resistant isolate 1 (green open triangles), resistant isolate 2 (green open squares), resistant isolate 3 (green open diamonds), and the SR108 parent (black filled squares). (D) The survival of the acrR(L34Q) mutant (blue triangles), the rpoA(E273G) mutant (blue inverted triangles), the acrR(L34Q) rpoA(E273G) double mutant (green diamonds), and the BW25113 parent (black squares) is replotted from Fig. 2 for comparison. (E) The survival of the acrR(L34Q) mutant (blue triangles), the rpoA(E273G) mutant (blue inverted triangles), the acrR(L34Q) rpoA(E273G) double mutant (green diamonds), and the BW25113 parent (black squares) following UVC irradiation at the indicated doses is plotted. (F) The diameters of the zones of inhibition around 7-mm paper discs treated with the indicated amounts of chloramphenicol are plotted for the acrR(L34Q) mutant (blue triangles), the rpoA(E273G) mutant (blue inverted triangles), the acrR(L34Q) rpoA(E273G) double mutant (green diamonds), and the BW25113 parent (black squares). Plots represent the averages from at least two experiments. Error bars represent the standard errors of the means.
acrR(L34Q) and rpoA(E273G) reduce cross-link formation frequencies in vivo.
Another mechanism by which resistance could be conferred is by decreasing cross-link formation in cellular DNA. To examine this possibility, we utilized an alkali agarose gel and Southern blot analysis to monitor the accumulation of cross-links on plasmids growing in these strains. Cells containing the plasmid pBR322 were treated with psoralen and UVA before the total cellular DNA was purified, digested with a restriction enzyme to linearize the plasmid, and analyzed by denaturing alkali agarose gel electrophoresis. Interstrand cross-links covalently bind both DNA strands, preventing complete strand separation under denaturing conditions, and cross-linked plasmid forms migrate more slowly than the corresponding single-stranded fragments during electrophoresis (55, 56). In theory, the assay should produce gels containing only 2 bands, undamaged DNA and interstrand DNA cross-links. However, 1 to 2 cross-links per chromosome are lethal in E. coli (39, 40), and assays detecting these lesions in vivo require doses far beyond this, with long irradiation times. The long irradiation periods required allow cross-links formed in cultures to be further processed by enzymes that may unlink, excise, and exonucleolytically degrade the DNA surrounding adducts, resulting in a more diffuse signal that stretches from the cross-link down to the undamaged DNA. To quantify the overall levels of psoralen-UVA-induced adduct formation in cells, the loss of the undamaged single-strand DNA band remaining in the treated samples was compared to that in the unirradiated sample.
When we compared the resistant isolates to their SR108 parent, all three resistant isolates retained significantly more un-cross-linked, linear, single-stranded DNA following psoralen-UVA treatment (Fig. 5A). In the SR108 parent, the fraction of undamaged plasmid remaining was significantly diminished following doses of both 35.1 kJ/m2 and 70.2 kJ/m2, whereas the resistant isolates could be seen to retain undamaged plasmid DNA at both the low and high doses used in these experiments. Thus, we interpret the observed persistence of the undamaged linear DNA in the resistant isolates to indicate that the formation of cross-links in DNA is reduced in the three isolates and that this likely accounts for their observed resistance.
FIG 5.
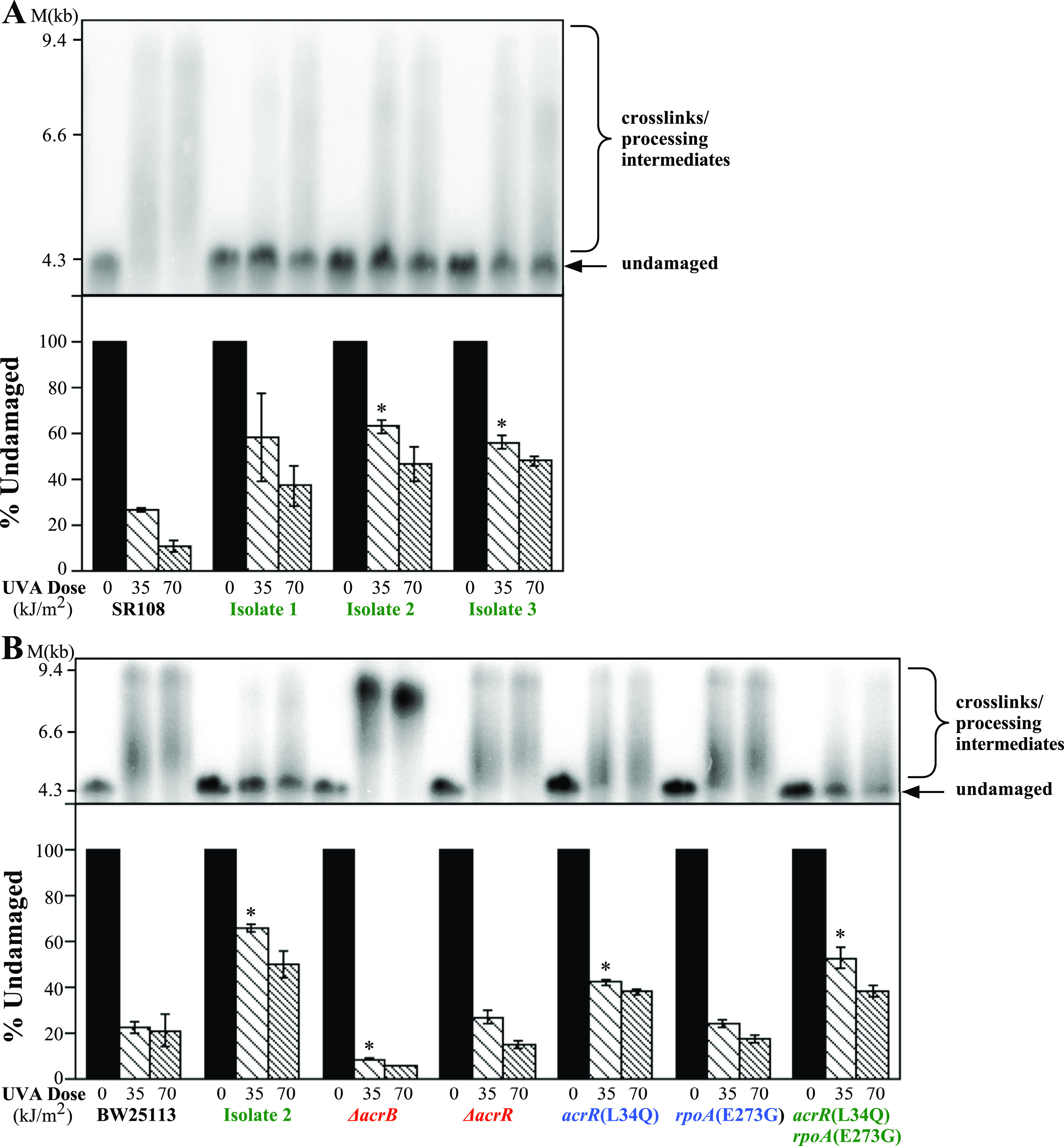
acrR(L34Q) rpoA(E273G) double mutants exhibit reduced formation of cross-links and other forms of DNA damage, similar to the PUVA-resistant isolates. (A) Resistant isolates do not accumulate PUVA-induced DNA damage. (Top) SR108, isolate 1, isolate 2, and isolate 3 cultures containing the plasmid pBR322 were UVA irradiated at the indicated doses in the presence of 20 μg/mL 8-methoxypsoralen before total genomic DNA was isolated, restricted with PvuII to linearize the plasmid, and examined by Southern analysis following alkali gel electrophoresis using 32P-labeled pBR322 as a probe. Undamaged DNA is indicated by the arrow. A shift from undamaged DNA to a higher-apparent-molecular-weight form indicates the presence of PUVA-induced DNA damage. (Bottom) The percentage of undamaged DNA remaining at each dose is plotted for each strain. (B) The acrR(L34Q) rpoA(E273G) mutants show reduced formation of PUVA-induced DNA damage. Cultures of the BW251113 parent and the acrR(L34Q), rpoA(E273G), and acrR(L34Q) rpoA(E273G) mutants containing the plasmid pBR322 were treated and analyzed as described above for panel A. (Top) Representative Southern blot; (bottom) percentage of undamaged DNA remaining at each dose. Plots represent averages from two experiments. Error bars represent the standard errors of the means. * indicates statistical significance relative to the corresponding wild-type strain (P < 0.05 by a t test).
To determine if the acrR(L34Q) and rpoA(E273G) mutations were responsible for the reduced cross-link formation, we next examined the formation of cross-links in strains containing the acrR(L34Q), rpoA(E273G), and both mutations. The acrR(L34Q) mutant partially reduced the formation of cross-links relative to its BW25113 parent, as measured by the loss of full-length undamaged DNA, although this effect appeared modest within the sensitivity of our assay. When the acrR(L34Q) rpoAE(E273G) double mutant was examined, a clear increase in the amount of undamaged DNA was observed (Fig. 5B). The amount of undamaged DNA in the double mutant was comparable to that seen in the original resistant isolate 2. These observations suggest that the two mutations act through separate mechanisms, are additive, and together can account for the reduced cross-link formation seen in the resistant isolate.
AcrR regulates the expression of the AcrAB-TolC efflux system. This suggests a mechanism in which the upregulation of the efflux system may increase psoralen export, thereby preventing its intercalation with DNA and cross-link formation. To examine this possibility, we examined how the deletion of the AcrB transporter component of the efflux system affected cell sensitivity to psoralen-UVA and cross-link formation. As shown in Fig. 5B, the deletion of acrB resulted in an increase in cross-link formation in DNA. In addition to the effect on cross-link formation, the inactivation of the efflux system severely hypersensitized the parental strain to psoralen-UVA treatment (Fig. 6). Furthermore, the deletion of acrB in isolate 2 completely abolished its resistance, rendering the cells as hypersensitive as the ΔacrB strain in an otherwise wild-type background (Fig. 6). The same was observed for the other two resistant isolates (Fig. S5). Taken together, these results indicate that the acrR(L34Q) mutation is likely effecting resistance through the regulation of the AcrAB-TolC complex and that AcrAB-TolC is capable of effluxing psoralen to reduce cross-link formation.
FIG 6.

The AcrAB-TolC efflux system is required for resistance to PUVA. (A) The survival of BW25113 (black filled squares), resistant isolate 2 (green open squares), an acrB deletion mutant (red open inverted triangles), and an acrB deletion mutant in the resistant isolate 2 background (red open diamonds) in the presence of psoralen at the indicated UVA doses is plotted. (B) The survival of BW25113 (black filled squares), resistant isolate 2 (green open squares), an acrB deletion mutant (red open inverted triangles), and an acrB deletion mutant in the resistant isolate 2 background (red open diamonds) in the presence of psoralen is replotted on a narrower UVA scale. Resistant isolate 2 and resistant isolate 2 ΔacrB mutant were derived from the SR108 background (indicated by the dotted line), while ΔacrB is an isogenic mutant of BW25113 (WT). Plots represent the averages from at least three independent experiments. Error bars represent the standard errors of the means.
To test this possibility directly, the acrAB operon was placed into an arabinose-inducible expression plasmid. The overexpression of AcrAB, by itself, in an otherwise wild-type background increased psoralen-UVA resistance, similar to the AcrR mutant allele (Fig. 7), demonstrating that resistance can be conferred directly by the upregulation of the efflux pump under AcrR control.
FIG 7.

Upregulation of the AcrAB efflux pump confers resistance to psoralen-UVA, similar to acrR(L34Q). (A) The expression of AcrAB functionally complements an acrB mutant. Ten-microliter drops of 10-fold serial dilutions are shown for the ΔacrB mutant containing the pBAD33 vector and the ΔacrB mutant containing pBAD33-AcrAB following UVA irradiation at the indicated doses in the presence of 20 μg/mL 8-methoxypsoralen. (B) The survival of BW25113 containing the pBAD33 vector (black open squares), BW25113 containing pBAD33-AcrAB (black filled squares), and the acrR(L34Q) mutant (blue filled triangles) in the presence of 20 μg/mL 8-methoxypsoralen at the indicated UVA doses is plotted. Plots represent the averages from two experiments. Error bars represent the standard errors of the means.
DISCUSSION
Here, we demonstrate that gain-of-function mutations in rpoA and acrR can confer high-level resistance to psoralen-UVA treatment. Considering that psoralen-UVA cytotoxicity arises from the formation of interstrand cross-links (40, 57), we initially expected that mutations conferring resistance might upregulate or induce mechanisms for repairing these lesions. However, although high-level resistance was observed, both mutations appeared to operate through mechanisms that reduce or prevent the formation of DNA cross-links, rather than increase repair. This would be consistent with the results of a previous study by Cole et al., in which the researchers failed to observe significant contributions from the known repair enzymes and found that as few as 1 to 2 cross-links per genome were lethal to E. coli (39). Those observations led the researchers to conclude that mechanisms for the effective repair of cross-links may not exist and that resistance therefore would likely involve mechanisms that primarily reduce or prevent cross-link formation, as reported here.
The results reported here identify psoralen as a novel substrate of the AcrAB-TolC efflux pump. This is based on the observations that mutations in the efflux regulator acrR were present in all three psoralen-resistant strains. The deletion of AcrB, which inactivates the efflux system, hypersensitized cells to psoralen and completely reversed the resistance phenotype of the resistant isolates (Fig. 6; see also Fig. S5 in the supplemental material). Additionally, mutations that upregulate or downregulate the acrR regulator correlate with the ability of psoralen to form cross-links in cells, as does the presence or absence of the transporter (Fig. 6 and 7). Notably, the AcrAB-TolC efflux pump is a tripartite efflux system that was originally identified for its ability to transport acridine dyes (58–60), which are structurally similar to the three-ringed psoralen molecule (Fig. S6) (9, 61). AcrAB-TolC has since been shown to pump a broad variety of substrates, with many sharing the carbon ring structures associated with multidrug resistance in Gram-negative bacteria (62, 63).
The observation that point mutations in acrR confer resistance to psoralen, whereas its deletion renders the strains hypersensitive, argues that regulation by this protein is more complex than previously appreciated. AcrR belongs to the TetR family of transcriptional regulators and has generally been thought to function as a repressor of the efflux genes acrA and acrB (45). If AcrR functioned as a simple repressor, one would expect that the deletion of acrR would upregulate expression and increase resistance. However, the opposite was observed, implying that regulation by AcrR may involve both activation and repression under various conditions. Based on the correlation of the absence of the efflux pump with increased cross-link formation (Fig. 5), we infer that the acrR point mutations in the resistant isolates upregulate the expression of the pump. Other regulators in the TetR family have similarly been reported to be able to function as both activators and repressors (64–66). One early study found that changes in acrAB operon expression occurred even if acrR was deleted and proposed that acrR is a secondary modulator of AcrAB expression (45).
How the various mutations in acrR affect protein function is unclear. Based on the reported structure of AcrR (67, 68), the point mutation in isolate 2 alters a single amino acid in the DNA binding domain. Isolates 1 and 3 contain an IS5 insertion and a frameshift, respectively, at the end of the DNA binding domain that effectively truncate AcrR after this region (Fig. S7A). Folding predictions suggest that the DNA binding domain could remain intact in all three mutants (Fig. S7B and C) (69), which we speculate may account for the gain-of-function phenotypes observed for these alleles.
It also remains unclear how the point mutations in rpoA confer resistance to cross-links. Both mutations map to the C-terminal domain of the RNA polymerase alpha subunit, which is known to interact with a variety of transcriptional regulators (70–75). In many cases, these regulators are associated with resistance to other cytotoxic agents (76–78). Thus, the rpoA mutations could confer resistance through the regulation of any number of operons. However, the finding that rpoA(E273G) conferred resistance to chloramphenicol suggests that resistance may involve the regulation of factors that may suppress membrane permeability or other importers or exporters that can accommodate psoralen. Finally, although rpoA(E273G) appears additive with acrR(L34Q), we cannot rule out the possibility that this occurs exclusively through the additional upregulation of the AcrAB-TolC efflux pump. Several alternative mechanisms for how the rpoA alleles affect interstrand cross-linking tolerance remain possible. In addition to its regulatory roles, RNA polymerase can function as an impediment to other cellular processes or as a sensor that recruits repair enzymes to specific lesions (79–81). It remains possible that mutations affecting these activities could alter cellular tolerance to cross-linking agents.
The resistant mutants isolated here are consistent with previous work that implied that the prevention, rather than the repair, of cross-links is the primary mechanism of survival for bacteria challenged with chemicals that form these lesions (39). These results demonstrate that this prevention in E. coli is dependent on the active efflux of the drug, a mechanism that has been shown to be responsible for multidrug resistance in both bacteria and human cancers (23, 24, 62, 63). These results also suggest that the regulation of efflux by acrR is more complicated than previously believed. Additionally, while it is suspected that the mutations in rpoA likely confer resistance by modulating the RNA polymerase’s interactions with a variety of transcriptional regulators, the actual mechanism remains uncharacterized. It will therefore be of interest to understand how the resistance observed in this study is regulated.
MATERIALS AND METHODS
Bacterial strains.
SR108, a thyA36 deoC2 derivative of W3110 (82), was used as the parent for the selection of psoralen-resistant strains. Thymine auxotrophy was used to confirm that the selected populations were derived from the parental population and were not contaminants. To characterize candidate mutations for their contribution to psoralen-UVA resistance, mutations were placed into the BW25113 background, which is the parental strain used for the Keio collection (49). Mutations present in psoralen-resistant strains were first linked to Kanr cassettes approximately 25 kb away by P1 transduction of ybaT::Kan for acrR(L34Q) and chiA::Kan for rpoA(E273G) from strains JW0475 and JW3300, respectively, selecting for resistance to kanamycin and psoralen-UVA irradiation (49). For rclA(A368G), the Kanr cassette was recombineered into ecpD using primers 5′-CAGCGGCCTCTCATCGTGGGCGGCGGTGACGCAGACAGGAGAAGAGAATGATTCCGGGGATCCGTCGACC-3′ and 5′-CCAGCATACAGACCGCTGTCAGCAGGGCCTTAGTTAATGTTACGCCACGTTGTAGGCTGGAGCTGCTTCG-3′ (Eurofins Genomics, Louisville, KY, USA) to amplify the Kanr cassette from JW0318 and transformed into electrocompetent arabinose-induced isolate 2 cells containing plasmid pKD46 as described previously (50). P1 transduction was then performed to cotransduce each target mutation and linked mini-Kan cassette into BW25113. In the case of acrR(L34Q) and rpoA(E273G), the linked genes were confirmed by psoralen-UVA resistance in the cotransductants. The presence of rclA(A368G) was confirmed by sequencing. Strains CL5333 to CL5336 were constructed by transforming pBAD33 or pBAD33-AcrAB plasmids into electrocompetent BW25113 or JW0451 cells (83). A complete list of the strains used in this study is shown in Table 2.
TABLE 2.
Strains used in this studya
| Strain | Relevant genotype or description | Reference or construction |
|---|---|---|
| SR108 | thyA deoC IN(rrnD-rrnE) | 81 |
| BW25113 | lacIq rrnB T14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 | 50 |
| CL3844 | Resistant isolate 1 | Mutagenized with psoralen and UVA |
| CL3845 | Resistant isolate 2 | Mutagenized with psoralen and UVA |
| CL3846 | Resistant isolate 3 | Mutagenized with psoralen and UVA |
| JW0453 | acrR::FRT–mini-Kan | 49 |
| JW5040 | rclA::FRT–mini-Kan | 49 |
| JW0298 | rclR::FRT–mini-Kan | 49 |
| JW5041 | ykgE::FRT–mini-Kan | 49 |
| JW0451 | acrB::FRT–mini-Kan | 49 |
| CL4426 | Resistant isolate 1 acrB::FRT–mini-Kan | P1 transduction of acrB::FRT–mini-Kan from JW0451 into resistant isolate 1 |
| CL4427 | Resistant isolate 2 acrB::FRT–mini-Kan | P1 transduction of acrB::FRT–mini-Kan from JW0451 into resistant isolate 2 |
| CL4428 | Resistant isolate 3 acrB::FRT–mini-Kan | P1 transduction of acrB::FRT–mini-Kan from JW0451 into resistant isolate 3 |
| JW0475 | ybaT::FRT–mini-Kan | 49 |
| JW3300 | chiA::FRT–mini-Kan | 49 |
| JW0284 | ecpD::FRT–mini-Kan | 49 |
| CL5227 | Resistant isolate 2 ybaT::FRT–mini-Kan | P1 transduction of ybaT::FRT–mini-Kan from JW0475 into resistant isolate 2 |
| CL5228 | Resistant isolate 2 chiA::FRT–mini-Kan | P1 transduction of chiA::FRT–mini-Kan from JW0475 into resistant isolate 2 |
| CL5229 | Resistant isolate 2 ecpD::FRT–mini-Kan | Recombineering to replace ecpD in resistant isolate 2 with FRT–mini-Kan |
| CL5230 | acrR(L34Q) | P1 cotransduction of acrR(L34Q) and ybaT::FRT–mini-Kan from CL5227 into BW25113 |
| CL5231 | rpoA(E273G) | P1 cotransduction of rpoA(E273G) and chiA::FRT–mini-Kan from CL5228 into BW25113 |
| CL5232 | acrR(L34Q) rpoA(E273G) | P1 cotransduction of rpoA(E273G) and chiA::FRT–mini-Kan from CL5231 into CL5234 |
| CL5233 | rclA(A368G) | P1 cotransduction of ecpD::FRT–mini-Kan and rclA(A368G) from CL5229 into BW25113 |
| CL5235 | Resistant isolate 2 chiA::FRT | Removal of the mini-Kan cassette from CL5227 via pCP20 expression of FLP recombinase |
| CL5333 | pBAD33 | Transformation of pBAD33b into BW25113 |
| CL5334 | pBAD33-AcrAB | Transformation of pBAD33-AcrABb into BW25113 |
| CL5335 | acrB::FRT–mini-Kan/pBAD33 | Transformation of pBAD33b into JW0451 |
| CL5336 | acrB::FRT–mini-Kan/pBAD33-AcrAB | Transformation of pBAD33-AcrABb into JW0451 |
FRT, FLP recombination target.
See reference 82.
Selection for psoralen-UVA resistance.
Aliquots (0.1 mL) of a fresh culture grown overnight in Luria-Bertani medium supplemented with 10 μg/mL thymine (LBthy) were spread onto LBthy agar plates supplemented with 20 μg/mL 8-methoxypsoralen and UVA irradiated using two 32-W UVA bulbs (peak emittance at 320 nm) at an incident dose of 5.05 J/m2/s for increasing exposure times. Following incubation overnight at 37°C, 100 μL of bacteria was scraped and collected from the plate irradiated with the lowest dose where cell lethality was evident (i.e., almost, but not quite, a lawn of bacteria), resuspended in 1 mL of LBthy medium, and used to inoculate a new 5-mL culture grown overnight before the selection process was repeated. A portion of the culture from each successive selection passage was frozen in LBthy medium supplemented with 20% glycerol and stored at −80°C for future characterization. This process was used to generate three independently derived psoralen-UVA-resistant strains, designated resistant isolate 1, resistant isolate 2, and resistant isolate 3. Resistant isolate 1 was isolated after six rounds of selection, whereas resistant isolates 2 and 3 were isolated after seven rounds of selection.
Genomic DNA purification.
Genomic DNA was purified from 0.75 mL of the culture by the addition of 0.75 mL of ice-cold 2× NET buffer (100 mmol/L NaCl, 10 mmol/L Tris [pH 8.0], 10 mmol/L EDTA) before cells were pelleted and resuspended in 140 μL of TE (10 mmol/L Tris [pH 8.0], 10 mmol/L EDTA) containing 1 mg/mL of lysozyme (Thermo Scientific, Waltham, MA, USA) and 0.2 mg/mL of RNase A (MP Biomedicals, Irvine, CA, USA). The samples were then treated with 10 μL each of 10 mg/mL of proteinase K (Thermo Scientific, Waltham, MA, USA) and 20% Sarkosyl (Thermo Scientific, Waltham, MA, USA) and incubated for 30 min at 37°C. Following incubation, samples were extracted with 4 volumes of a 1:1 mixture of phenol-chloroform. Finally, samples were dialyzed on 47-mm Whatman 0.05-μm-pore-size discs (Merck Millipore, Darmstadt, Germany) floating in 250-μL beakers of TE (10 mmol/L Tris [pH 8.0], 10 mmol/L EDTA).
Genome sequencing of resistant strains.
Purified genomic DNA from each strain was sequenced using seqWell (Beverly, MA, USA) library prep kits and 50-bp single-end Illumina (San Diego, CA, USA) NextSeq 2000 high-throughput DNA sequencing according to the manufacturers’ instructions. Sequence reads were then aligned and compared to the SR108 parent genome using Breseq (84) to identify mutations that arose in the resistant strains.
Psoralen-UVA survival.
Ten-microliter aliquots of 10-fold serial dilutions from cultures grown overnight were spotted onto LBthy plates containing 20 μg/mL 8-methoxypsoralen. The plates were then exposed to UVA irradiation at an incident dose of 6.5 J/m2/s for the indicated doses and incubated overnight at 37°C. The surviving colonies at each dose were then counted and compared to those on the nonexposed plates to calculate the percent survival.
For the overexpression of AcrAB from expression vectors, 5-mL LBthy subcultures were inoculated with 50 μL of cultures grown overnight containing the expression plasmid and grown in a 37°C shaking water bath to an optical density at 600 nm (OD600) of 0.4. l-Arabinose (1 mM) was added to the subcultures for the last 30 min of incubation before proceeding with the survival assay as described above.
UVC survival.
Ten-microliter aliquots of 10-fold serial dilutions from cultures grown overnight were spotted onto LBthy plates. The plates were then exposed to UVC irradiation at an incident dose of 0.8 J/m2/s for the indicated doses and incubated overnight at 37°C. The surviving colonies at each dose were then counted and compared to those on the nonexposed plates to calculate the percent survival.
Chloramphenicol resistance.
Ten microliters of 5, 10, or 20 mg/mL of chloramphenicol in ethanol (EtOH) was spotted onto 7-mm Whatman paper discs and allowed to dry for 1 h. Discs treated with only EtOH served as controls. One hundred fifty microliters of the cultures grown overnight were spread onto Davis medium supplemented with 0.4% glucose, 0.2% Casamino acids, and 10 μg/mL thymine (DGCthy) with a cotton swab, antibiotic discs were placed onto the surfaces of the plates, and the plates were incubated overnight at 37°C. The diameters of the zones of inhibition were measured using ImageJ software (85).
In vivo detection of DNA interstrand cross-links.
The detection of cross-linked DNA was performed as previously described (39, 56). Briefly, cultures containing the plasmid pBR322 were grown overnight in DGCthy medium supplemented with 50 μg/mL ampicillin at 37°C. A 0.1-mL aliquot of this culture was pelleted, resuspended in 10 mL DGCthy medium without ampicillin, and grown in a 37°C shaking water bath to an OD600 of 0.4. The cultures were treated with 20 μg/mL 8-methoxypsoralen for 10 min at 37°C and subsequently irradiated with the indicated doses of UVA light. Aliquots (0.75 mL) were collected and transferred to an equal volume of ice-cold 4× NET buffer, and the genomic DNA was purified as described above. The purified DNA was digested with PvuII (New England BioLabs, Ipswich, MA, USA), which linearizes pBR322, before samples were electrophoresed on a 0.75% alkaline agarose gel in a solution containing 30 mM NaOH and 1 mM EDTA at 1 V/cm for 16 h. The DNA in the gels was transferred to Hybond N+ nylon membranes (Cytiva, Marlborough, MA, USA), and the plasmid DNA was visualized by probing with 32P-labeled pBR322 prepared using a Prime-It RmT labeling kit (Agilent Technologies, Santa Clara, CA, USA) with >6,000 Ci/mmol [α-32P]dCTP (PerkinElmer, Waltham, MA, USA). Southern blots were visualized and quantitated using the Storm 840 phosphorimager and its associated ImageQuant analysis software (Cytiva, Marlborough, MA, USA).
Data availability.
The sequencing data for the parental and resistant isolates have been deposited in the NCBI Sequence Read Archive (www.ncbi.nlm.nih.gov/sra/) under BioProject accession number PRJNA952657.
ACKNOWLEDGMENTS
We thank Y. Wei and L. Yang for generously providing their AcrAB expression plasmids.
This study was supported by grant MCB1916625 from the National Science Foundation, grant R21ES034880 from the NIH National Institute of Environmental Health Sciences, and grant R16GM14554 from the NIH National Institute of General Medical Sciences.
Footnotes
Supplemental material is available online only.
Contributor Information
Travis K. Worley, Email: tworley@pdx.edu.
George O’Toole, Geisel School of Medicine at Dartmouth.
REFERENCES
- 1.Pathak MA, Daniels F, Jr, Fitzpatrick TB. 1962. The presently known distribution of furocoumarins (psoralens) in plants. J Invest Dermatol 39:225–239. doi: 10.1038/jid.1962.106. [DOI] [PubMed] [Google Scholar]
- 2.Hata T, Hoshi T, Kanamori K, Matsumae A, Sano Y, Shima T, Sugawara R. 1956. Mitomycin, a new antibiotic from Streptomyces. I. J Antibiot (Tokyo) 9:141–146. [PubMed] [Google Scholar]
- 3.Boyland E. 1948. Biochemical reactions of chemical warfare agents. Nature 161:225–227. doi: 10.1038/161225a0. [DOI] [PubMed] [Google Scholar]
- 4.Brookes P, Lawley PD. 1960. The reaction of mustard gas with nucleic acids in vitro and in vivo. Biochem J 77:478–484. doi: 10.1042/bj0770478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iyer VN, Szybalski W. 1963. A molecular mechanism of mitomycin action: linking of complementary DNA strands. Proc Natl Acad Sci USA 50:355–362. doi: 10.1073/pnas.50.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brookes P, Lawley PD. 1961. The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem J 80:496–503. doi: 10.1042/bj0800496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole RS. 1970. Light-induced cross-linking of DNA in the presence of a furocoumarin (psoralen). Studies with phage lambda, Escherichia coli, and mouse leukemia cells. Biochim Biophys Acta 217:30–39. doi: 10.1016/0005-2787(70)90119-x. [DOI] [PubMed] [Google Scholar]
- 8.Hearst JE. 1981. Psoralen photochemistry. Annu Rev Biophys Bioeng 10:69–86. doi: 10.1146/annurev.bb.10.060181.000441. [DOI] [PubMed] [Google Scholar]
- 9.Dall’Acqua F, Marciani S, Ciavatta L, Rodighiero G. 1971. Formation of inter-strand cross-linkings in the photoreactions between furocoumarins and DNA. Z Naturforsch B 26:561–569. doi: 10.1515/znb-1971-0613. [DOI] [PubMed] [Google Scholar]
- 10.Lawley PD, Brookes P. 1968. Cytotoxicity of alkylating agents towards sensitive and resistant strains of Escherichia coli in relation to extent and mode of alkylation of cellular macromolecules and repair of alkylation lesions in deoxyribonucleic acids. Biochem J 109:433–447. doi: 10.1042/bj1090433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiraha Y, Sakai K, Teranaka T. 1958. Clinical trials of mitomycin C, a new antitumor antibiotic; preliminary report of results obtained in 82 consecutive cases in the field of general surgery. Antibiot Annu 6:533–540. [PubMed] [Google Scholar]
- 12.Adair FE, Bagg HJ. 1931. Experimental and clinical studies on the treatment of cancer by dichlorethylsulphide (mustard gas). Ann Surg 93:190–199. doi: 10.1097/00000658-193101000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogawa K, Hosokawa A, Ueda A, Saito S, Mihara H, Ando T, Kajiura S, Terada M, Tsukioka Y, Horikawa N, Kobayashi T, Note M, Sawasaki K, Fukuoka J, Sugiyama T. 2012. Irinotecan plus mitomycin C as second-line chemotherapy for advanced gastric cancer resistant to fluoropyrimidine and cisplatin: a retrospective study. Gastroenterol Res Pract 2012:640401. doi: 10.1155/2012/640401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saint A, Evesque L, Falk AT, Cavaglione G, Montagne L, Benezery K, Francois E. 2019. Mitomycin and 5-fluorouracil for second-line treatment of metastatic squamous cell carcinomas of the anal canal. Cancer Med 8:6853–6859. doi: 10.1002/cam4.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miki T, Mizutani Y, Akaza H, Ozono S, Tsukamoto T, Terachi T, Naito K, Nonomura N, Hara I, Yoshida O, Japan Blood Cell Transplantation Study Group for Testicular Germ Cell Tumor . 2007. Long-term results of first-line sequential high-dose carboplatin, etoposide and ifosfamide chemotherapy with peripheral blood stem cell support for patients with advanced testicular germ cell tumor. Int J Urol 14:54–59. doi: 10.1111/j.1442-2042.2006.01655.x. [DOI] [PubMed] [Google Scholar]
- 16.Liszewski W, Naym DG, Biskup E, Gniadecki R. 2017. Psoralen with ultraviolet A-induced apoptosis of cutaneous lymphoma cell lines is augmented by type I interferons via the JAK1-STAT1 pathway. Photodermatol Photoimmunol Photomed 33:164–171. doi: 10.1111/phpp.12302. [DOI] [PubMed] [Google Scholar]
- 17.Lengert AVH, Pereira LDNB, Cabral ERM, Gomes INF, de Jesus LM, Gonçalves MFS, da Rocha AO, Tassinari TA, da Silva LS, Laus AC, Vidal DO, Pinto MT, Reis RM, Lopes LF. 2022. Potential new therapeutic approaches for cisplatin-resistant testicular germ cell tumors. Front Biosci (Landmark Ed) 27:245. doi: 10.31083/j.fbl2708245. [DOI] [PubMed] [Google Scholar]
- 18.Johnson SW, Shen D, Pastan I, Gottesman MM, Hamilton TC. 1996. Cross-resistance, cisplatin accumulation, and platinum-DNA adduct formation and removal in cisplatin-sensitive and -resistant human hepatoma cell lines. Exp Cell Res 226:133–139. doi: 10.1006/excr.1996.0211. [DOI] [PubMed] [Google Scholar]
- 19.Jacobsen C, Honecker F. 2015. Cisplatin resistance in germ cell tumours: models and mechanisms. Andrology 3:111–121. doi: 10.1111/andr.299. [DOI] [PubMed] [Google Scholar]
- 20.Perez RP. 1998. Cellular and molecular determinants of cisplatin resistance. Eur J Cancer 34:1535–1542. doi: 10.1016/s0959-8049(98)00227-5. [DOI] [PubMed] [Google Scholar]
- 21.Shen D-W, Pouliot LM, Hall MD, Gottesman MM. 2012. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev 64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottesman MM. 2002. Mechanisms of cancer drug resistance. Annu Rev Med 53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 23.Gottesman MM, Fojo T, Bates SE. 2002. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 24.Gottesman MM, Pastan IH. 2015. The role of multidrug resistance efflux pumps in cancer: revisiting a JNCI publication exploring expression of the MDR1 (P-glycoprotein) gene. J Natl Cancer Inst 107:djv222. doi: 10.1093/jnci/djv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole RS. 1973. Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci USA 70:1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole RS. 1973. Repair of interstrand cross-links in DNA induced by psoralen plus light. Yale J Biol Med 46:492. [PMC free article] [PubMed] [Google Scholar]
- 27.Magaña-Schwencke N, Henriques JA, Chanet R, Moustacchi E. 1982. The fate of 8-methoxypsoralen photoinduced crosslinks in nuclear and mitochondrial yeast DNA: comparison of wild-type and repair-deficient strains. Proc Natl Acad Sci USA 79:1722–1726. doi: 10.1073/pnas.79.6.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinden RR, Cole RS. 1978. Repair of cross-linked DNA and survival of Escherichia coli treated with psoralen and light: effects of mutations influencing genetic recombination and DNA metabolism. J Bacteriol 136:538–547. doi: 10.1128/jb.136.2.538-547.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berardini M, Mackay W, Loechler EL. 1997. Evidence for a recombination-independent pathway for the repair of DNA interstrand cross-links based on a site-specific study with nitrogen mustard. Biochemistry 36:3506–3513. doi: 10.1021/bi962778w. [DOI] [PubMed] [Google Scholar]
- 30.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NGJ, Beverloo HB, Hoeijmakers JHJ, Kanaar R. 2004. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol 24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki MS, Tonomura A. 1973. A high susceptibility of Fanconi’s anemia to chromosome breakage by DNA cross-linking agents. Cancer Res 33:1829–1836. [PubMed] [Google Scholar]
- 32.Cohen MM, Fruchtman CE, Simpson SJ, Martin AO. 1982. The cytogenetic response of Fanconi’s anemia lymphoblastoid cell lines to various clastogens. Cytogenet Cell Genet 34:230–240. doi: 10.1159/000131810. [DOI] [PubMed] [Google Scholar]
- 33.Gueiderikh A, Rosselli F, Neto JBC. 2017. A never-ending story: the steadily growing family of the FA and FA-like genes. Genet Mol Biol 40:398–407. doi: 10.1590/1678-4685-GMB-2016-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duxin JP, Walter JC. 2015. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol 37:49–60. doi: 10.1016/j.ceb.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berardini M, Foster PL, Loechler EL. 1999. DNA polymerase II (polB) is involved in a new DNA repair pathway for DNA interstrand cross-links in Escherichia coli. J Bacteriol 181:2878–2882. doi: 10.1128/JB.181.9.2878-2882.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cole RS, Sinden RR. 1975. Repair of cross-linked DNA in Escherichia coli. Basic Life Sci 5B:487–495. doi: 10.1007/978-1-4684-2898-8_10. [DOI] [PubMed] [Google Scholar]
- 37.Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. 2008. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole RS. 1971. Psoralen monoadducts and interstrand cross-links in DNA. Biochim Biophys Acta 254:30–39. doi: 10.1016/0005-2787(71)90111-0. [DOI] [PubMed] [Google Scholar]
- 39.Cole JM, Acott JD, Courcelle CT, Courcelle J. 2018. Limited capacity or involvement of excision repair, double-strand breaks, or translesion synthesis for psoralen cross-link repair in Escherichia coli. Genetics 210:99–112. doi: 10.1534/genetics.118.301239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szybalski W, Iyer VN. 1964. Crosslinking of DNA by enzymatically or chemically activated mitomycins and porfiromycins, bifunctionally “alkylating” antibiotics. Fed Proc 23:946–957. [PubMed] [Google Scholar]
- 41.Crathorn AR, Roberts JJ. 1966. Mechanism of the cytotoxic action of alkylating agents in mammalian cells and evidence for the removal of alkylated groups from deoxyribonucleic acid. Nature 211:150–153. doi: 10.1038/211150a0. [DOI] [PubMed] [Google Scholar]
- 42.Ball CR, Roberts JJ. 1970. DNA repair after mustard gas alkylation by sensitive and resistant Yoshida sarcoma cells in vitro. Chem Biol Interact 2:321–329. doi: 10.1016/0009-2797(70)90054-2. [DOI] [PubMed] [Google Scholar]
- 43.Roberts JJ, Friedlos F, Scott D, Ormerod MG, Rawlings CJ. 1986. The unique sensitivity of Walker rat tumour cells to difunctional agents is associated with a failure to recover from inhibition of DNA synthesis and increased chromosome damage. Mutat Res 166:169–181. doi: 10.1016/0167-8817(86)90015-5. [DOI] [PubMed] [Google Scholar]
- 44.Knox RJ, Lydall DA, Friedlos F, Basham C, Rawlings CJ, Roberts JJ. 1991. The Walker 256 carcinoma: a cell type inherently sensitive only to those difunctional agents that can form DNA interstrand crosslinks. Mutat Res 255:227–240. doi: 10.1016/0921-8777(91)90026-l. [DOI] [PubMed] [Google Scholar]
- 45.Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE. 1996. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol Microbiol 19:101–112. doi: 10.1046/j.1365-2958.1996.357881.x. [DOI] [PubMed] [Google Scholar]
- 46.Jaskunas SR, Burgess RR, Nomura M. 1975. Identification of a gene for the alpha-subunit of RNA polymerase at the str-spc region of the Escherichia coli chromosome. Proc Natl Acad Sci USA 72:5036–5040. doi: 10.1073/pnas.72.12.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parker BW, Schwessinger EA, Jakob U, Gray MJ. 2013. The RclR protein is a reactive chlorine-specific transcription factor in Escherichia coli. J Biol Chem 288:32574–32584. doi: 10.1074/jbc.M113.503516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Derke RM, Barron AJ, Billiot CE, Chaple IF, Lapi SE, Broderick NA, Gray MJ. 2020. The Cu(II) reductase RclA protects Escherichia coli against the combination of hypochlorous acid and intracellular copper. mBio 11:e01905-20. doi: 10.1128/mBio.01905-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ganesan AK, Smith KC. 1968. Dark recovery processes in Escherichia coli irradiated with ultraviolet light. I. Effect of rec mutations on liquid holding recovery. J Bacteriol 96:365–373. doi: 10.1128/jb.96.2.365-373.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganesan AK, Smith KC. 1969. Dark recovery processes in Escherichia coli irradiated with ultraviolet light. II. Effect of uvr genes on liquid holding recovery. J Bacteriol 97:1129–1133. doi: 10.1128/jb.97.3.1129-1133.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma RC, Smith KC. 1985. A mechanism for rich-medium inhibition of the repair of daughter-strand gaps in the deoxyribonucleic acid of UV-irradiated Escherichia coli K12 uvrA. Mutat Res 146:177–183. doi: 10.1016/0167-8817(85)90008-2. [DOI] [PubMed] [Google Scholar]
- 54.George AM, Levy SB. 1983. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: involvement of a non-plasmid-determined efflux of tetracycline. J Bacteriol 155:531–540. doi: 10.1128/jb.155.2.531-540.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vos JMH. 1988. Analysis of psoralen monoadducts and interstrand crosslinks in defined genomic sequences, p 367–398. In Friedberg EC, Hanawalt PC (ed), DNA repair: a laboratory manual of research procedures, vol 3. Marcel Dekker, New York, NY. [Google Scholar]
- 56.Perera AV, Mendenhall JB, Courcelle CT, Courcelle J. 2016. Cho endonuclease functions during DNA interstrand cross-link repair in Escherichia coli. J Bacteriol 198:3099–3108. doi: 10.1128/JB.00509-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brookes P, Lawley PD. 1963. Effects of alkylating agents on T2 and T4 bacteriophages. Biochem J 89:138–144. doi: 10.1042/bj0890138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakamura H. 1965. Gene-controlled resistance to acriflavine and other basic dyes in Escherichia coli. J Bacteriol 90:8–14. doi: 10.1128/jb.90.1.8-14.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1993. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J Bacteriol 175:6299–6313. doi: 10.1128/jb.175.19.6299-6313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol 16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- 61.Lerman LS. 1963. The structure of the DNA-acridine complex. Proc Natl Acad Sci USA 49:94–102. doi: 10.1073/pnas.49.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okusu H, Ma D, Nikaido H. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol 178:306–308. doi: 10.1128/jb.178.1.306-308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X-Z, Poole K, Nikaido H. 2003. Contributions of MexAB-OprM and an EmrE homolog to intrinsic resistance of Pseudomonas aeruginosa to aminoglycosides and dyes. Antimicrob Agents Chemother 47:27–33. doi: 10.1128/AAC.47.1.27-33.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foster TJ, Ginnity F. 1985. Some mercurial resistance plasmids from different incompatibility groups specify merR regulatory functions that both repress and induce the mer operon of plasmid R100. J Bacteriol 162:773–776. doi: 10.1128/jb.162.2.773-776.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chattoraj P, Mohapatra SS, Rao JL, Biswas I. 2011. Regulation of transcription by SMU.1349, a TetR family regulator, in Streptococcus mutans. J Bacteriol 193:6605–6613. doi: 10.1128/JB.06122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson PJT, Shafer WM. 2015. The transcriptional repressor, MtrR, of the mtrCDE efflux pump operon of Neisseria gonorrhoeae can also serve as an activator of “off target” gene (glnE) expression. Antibiotics (Basel) 4:188–197. doi: 10.3390/antibiotics4020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li M, Gu R, Su C-C, Routh MD, Harris KC, Jewell ES, McDermott G, Yu EW. 2007. Crystal structure of the transcriptional regulator AcrR from Escherichia coli. J Mol Biol 374:591–603. doi: 10.1016/j.jmb.2007.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Routh MD, Su C-C, Zhang Q, Yu EW. 2009. Structures of AcrR and CmeR: insight into the mechanisms of transcriptional repression and multi-drug recognition in the TetR family of regulators. Biochim Biophys Acta 1794:844–851. doi: 10.1016/j.bbapap.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garrett S, Silhavy TJ. 1987. Isolation of mutations in the alpha operon of Escherichia coli that suppress the transcriptional defect conferred by a mutation in the porin regulatory gene envZ. J Bacteriol 169:1379–1385. doi: 10.1128/jb.169.4.1379-1385.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas MS, Glass RE. 1991. Escherichia coli rpoA mutation which impairs transcription of positively regulated systems. Mol Microbiol 5:2719–2725. doi: 10.1111/j.1365-2958.1991.tb01980.x. [DOI] [PubMed] [Google Scholar]
- 72.Gussin GN, Olson C, Igarashi K, Ishihama A. 1992. Activation defects caused by mutations in Escherichia coli rpoA are promoter specific. J Bacteriol 174:5156–5160. doi: 10.1128/jb.174.15.5156-5160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jafri S, Urbanowski ML, Stauffer GV. 1995. A mutation in the rpoA gene encoding the alpha subunit of RNA polymerase that affects metE-metR transcription in Escherichia coli. J Bacteriol 177:524–529. doi: 10.1128/jb.177.3.524-529.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Williams SM, Savery NJ, Busby SJ, Wing HJ. 1997. Transcription activation at class I FNR-dependent promoters: identification of the activating surface of FNR and the corresponding contact site in the C-terminal domain of the RNA polymerase alpha subunit. Nucleic Acids Res 25:4028–4034. doi: 10.1093/nar/25.20.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Camakaris H, Yang J, Fujii T, Pittard J. 2021. Activation by TyrR in Escherichia coli K-12 by interaction between TyrR and the α-subunit of RNA polymerase. J Bacteriol 203:e00252-21. doi: 10.1128/JB.00252-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caslake LF, Ashraf SI, Summers AO. 1997. Mutations in the alpha and sigma-70 subunits of RNA polymerase affect expression of the mer operon. J Bacteriol 179:1787–1795. doi: 10.1128/jb.179.5.1787-1795.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Webber MA, Whitehead RN, Mount M, Loman NJ, Pallen MJ, Piddock LJ. 2015. Parallel evolutionary pathways to antibiotic resistance selected by biocide exposure. J Antimicrob Chemother 70:2241–2248. doi: 10.1093/jac/dkv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Merchel Piovesan Pereira B, Wang X, Tagkopoulos I. 2021. Biocide-induced emergence of antibiotic resistance in Escherichia coli. Front Microbiol 12:640923. doi: 10.3389/fmicb.2021.640923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Selby CP, Sancar A. 1995. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J Biol Chem 270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- 80.Selby CP, Sancar A. 1993. Molecular mechanism of transcription-repair coupling. Science 260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 81.Trautinger BW, Lloyd RG. 2002. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J 21:6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mellon I, Hanawalt PC. 1989. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature 342:95–98. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 83.Rajapaksha P, Ojo I, Yang L, Pandeya A, Abeywansha T, Wei Y. 2021. Insight into the AcrAB-TolC complex assembly process learned from competition studies. Antibiotics (Basel) 10:830. doi: 10.3390/antibiotics10070830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, Meyer AG. 2014. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics 15:1039. doi: 10.1186/1471-2164-15-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S7. Download jb.00126-23-s0001.pdf, PDF file, 1.6 MB (1.6MB, pdf)
Data Availability Statement
The sequencing data for the parental and resistant isolates have been deposited in the NCBI Sequence Read Archive (www.ncbi.nlm.nih.gov/sra/) under BioProject accession number PRJNA952657.