

ABSTRACT
Type VI secretion systems (T6SSs) deliver cytotoxic effector proteins into target bacteria and eukaryotic host cells. Antibacterial effectors are invariably encoded with cognate immunity proteins that protect the producing cell from self-intoxication. Here, we identify transposon insertions that disrupt the tli immunity gene of Enterobacter cloacae and induce autopermeabilization through unopposed activity of the Tle phospholipase effector. This hyperpermeability phenotype is T6SS dependent, indicating that the mutants are intoxicated by Tle delivered from neighboring sibling cells rather than by internally produced phospholipase. Unexpectedly, an in-frame deletion of tli does not induce hyperpermeability because Δtli null mutants fail to deploy active Tle. Instead, the most striking phenotypes are associated with disruption of the tli lipoprotein signal sequence, which prevents immunity protein localization to the periplasm. Immunoblotting reveals that most hyperpermeable mutants still produce Tli, presumably from alternative translation initiation codons downstream of the signal sequence. These observations suggest that cytosolic Tli is required for the activation and/or export of Tle. We show that Tle growth inhibition activity remains Tli dependent when phospholipase delivery into target bacteria is ensured through fusion to the VgrG β-spike protein. Together, these findings indicate that Tli has distinct functions, depending on its subcellular localization. Periplasmic Tli acts as a canonical immunity factor to neutralize incoming effector proteins, while a cytosolic pool of Tli is required to activate the phospholipase domain of Tle prior to T6SS-dependent export.
IMPORTANCE Gram-negative bacteria use type VI secretion systems deliver toxic effector proteins directly into neighboring competitors. Secreting cells also produce specific immunity proteins that neutralize effector activities to prevent autointoxication. Here, we show the Tli immunity protein of Enterobacter cloacae has two distinct functions, depending on its subcellular localization. Periplasmic Tli acts as a canonical immunity factor to block Tle lipase effector activity, while cytoplasmic Tli is required to activate the lipase prior to export. These results indicate Tle interacts transiently with its cognate immunity protein to promote effector protein folding and/or packaging into the secretion apparatus.
KEYWORDS: contact-dependent growth inhibition, kin discrimination, self/nonself recognition, toxin immunity proteins
INTRODUCTION
Research over the past 20 years shows that bacteria commonly deliver growth-inhibitory toxins directly into neighboring competitors (1). This phenomenon was first described as contact-dependent growth inhibition (CDI) in Escherichia coli isolate EC93 (2). CDI is mediated by a specialized type V secretion system (T5SS) composed of CdiB and CdiA proteins. CdiB is an Omp85 family β-barrel protein responsible for transport of the CdiA effector protein to the cell surface (3). Upon binding a receptor on a neighboring bacterium, CdiA delivers its C-terminal toxin domain into the target cell to inhibit growth. CDI+ strains protect themselves from autoinhibition by producing immunity proteins that specifically neutralize the toxin domain. Shortly after the discovery of T5SS-mediated CDI, two reports demonstrated that type VI secretion systems (T6SSs) are also potent mediators of interbacterial competition (4, 5). T6SSs are dynamic multiprotein machines related in structure and function to the contractile tails of Myoviridae bacteriophages. The T6SS duty cycle begins with the assembly of a phage-like baseplate complex around trimeric VgrG, which is homologous to the β-tailspike protein of coliphage T4 (6). The baseplate then docks onto the cytosolic face of the transenvelope complex, which forms a secretion conduit across the cell envelope (7). The latter assembly nucleates the polymerization of a contractile sheath surrounding a central tube of Hcp proteins. Toxic effector proteins are typically linked to the VgrG β-spike (8, 9) and can also be packaged within the lumen of the Hcp tube (10). Once the sheath has grown to span the width of the cell, it contracts to expel the VgrG-capped tube through the transenvelope complex. The ejected projectile perforates neighboring bacteria and releases its toxic payload to inhibit target cell growth. As with toxic CdiA proteins, antibacterial T6SS effectors are encoded with cognate immunity proteins that protect the cell from intoxication by its isogenic siblings (1, 11). In this manner, T5SSs and T6SSs confer a competitive fitness advantage to bacteria, and their effector-immunity protein pairs play an important role in self/nonself discrimination.
Given that T5SS- and T6SS-mediated competition relies on cell-to-cell contact, the systems are often only deployed under conditions that favor productive effector delivery. Several regulatory strategies have been described for T6SSs, which are controlled at all levels of gene expression. T6SS loci are commonly subject to H-NS-mediated transcriptional silencing (12–16), and many require specific bacterial enhancer proteins to initiate transcription in conjunction with σ54 (17, 18). Environmental signals also influence T6SS transcription through quorum sensing and two-component regulatory signaling pathways (16, 19, 20). In Pseudomonas aeruginosa, the stability and translation of T6SS transcripts are modulated by small noncoding RNAs (16). Finally, assembly of the secretion apparatus is subject to posttranslational control by a serine/threonine kinase and phosphatase (21). There are fewer studies on the regulation of antibacterial T5SSs. The originally identified cdiBAI locus from E. coli EC93 is expressed constitutively (2, 22), but systems from other species are strictly regulated. The CDI system of Burkholderia thailandensis E264 is activated through a quorum sensing pathway (23), and the loci of Dickeya dadantii EC16 and 3937 are expressed only when these bacteria are grown on plant hosts (24–26). The latter observations suggest that CDI promotes host colonization and pathogenesis, although the T5SS effector domains deployed by these phytopathogens are clearly antibacterial (26, 27).
We previously reported that the CDI system of Enterobacter cloacae ATCC 13047 (ECL) is not active under laboratory conditions, although it is functional when expressed from a heterologous promoter (27). The wild-type cdiBAIECL operon appears to be transcribed from a single promoter with possible operator sites for PurR and Fnr. These predictions suggest that cdi transcription may be repressed when ECL is grown in standard culture media. In an attempt to identify transcriptional regulators, we generated an ECL strain that harbors the gusA reporter gene under the control of the cdi promoter and then screened it for transposon mutants that exhibit increased β-glucuronidase activity. Surprisingly, the recovered mutations have no effect on cdi transcription and instead act to increase cell permeability to the chromogenic β-glucuronidase substrate. We identified multiple insertions in tli, which encodes a previously described immunity protein that neutralizes the T6SS phospholipase effector Tle (28). Thus, cell envelope integrity is disrupted by unrestrained phospholipase activity in these mutants. Increased permeability is T6SS dependent, indicating that tli mutants are intoxicated by Tle delivered from neighboring siblings rather than internally produced phospholipase. Notably, we find that Δtli mutants are not hyperpermeable because they fail to deploy active Tle. Taken together, our findings indicate that Tli has distinct functions, depending on its subcellular localization. In addition to its established role as a periplasmic immunity factor, we propose that a cytosolic pool of Tli is required to activate the Tle phospholipase prior to T6SS-dependent export.
RESULTS
Disruption of tli increases cell permeability.
We initially sought to identify regulators of the cdiBAI gene cluster in Enterobacter cloacae ATCC 13047 (ECL) using a β-glucuronidase (gusA) reporter to monitor expression. We fused the E. coli gusA gene to the cdiB (ECL_04452) promoter region and placed the construct at the ECL glmS locus using Tn7-mediated transposition. The resulting cdi-gusA reporter strain was subjected to transposon Tn5-mediated mutagenesis, and >25,000 insertion mutants were screened on agar medium supplemented with the chromogenic β-glucuronidase substrate X-Gluc (5-bromo-4-chloro-1H-indol-3-yl-β-d-glucopyranosiduronic acid). Nine different transposon insertion sites were identified from 17 mutants that formed pigmented colonies on indicator medium. One insertion is located within tle (ECL_01553), and the other eight disrupt the downstream tli cistron (ECL_01554) (Fig. 1A). The tle gene encodes a toxic phospholipase effector deployed by the type VI secretion system 1 (T6SS-1) of ECL, and tli encodes an ankyrin repeat immunity protein that neutralizes Tle activity (29). We transferred the transposon mutations into the original parental reporter strain using Red-mediated recombineering and found that all of the reconstructed mutants, except the tle-1106 strain, recapitulate the original phenotype on indicator medium (Fig. 1B). Quantification of β-glucuronidase activity in cell lysates from a subset of mutants revealed that each has the same specific activity as the parental reporter strain (Fig. 1C). These data indicate that cdi transcription is unaffected by the tli mutations. Moreover, immunoblot analyses showed that none of the mutants exhibits increased production of CdiB (Fig. 1D) or CdiA (Fig. 1E). Given these results, we hypothesized that the loss of Tli immunity function leads to unopposed Tle phospholipase activity, which in turn degrades cell membranes to increase permeability to X-Gluc. Consistent with this model, parental reporter cells grown on indicator medium become pigmented after treatment with SDS solution (Fig. 2A). Moreover, complementation with plasmid-borne tli suppresses pigment conversion (Fig. 2B), but it has little effect on specific β-glucuronidase activities (Fig. 2C). Together, these results indicate that diminished immunity function leads to increased cell permeability.
FIG 1.
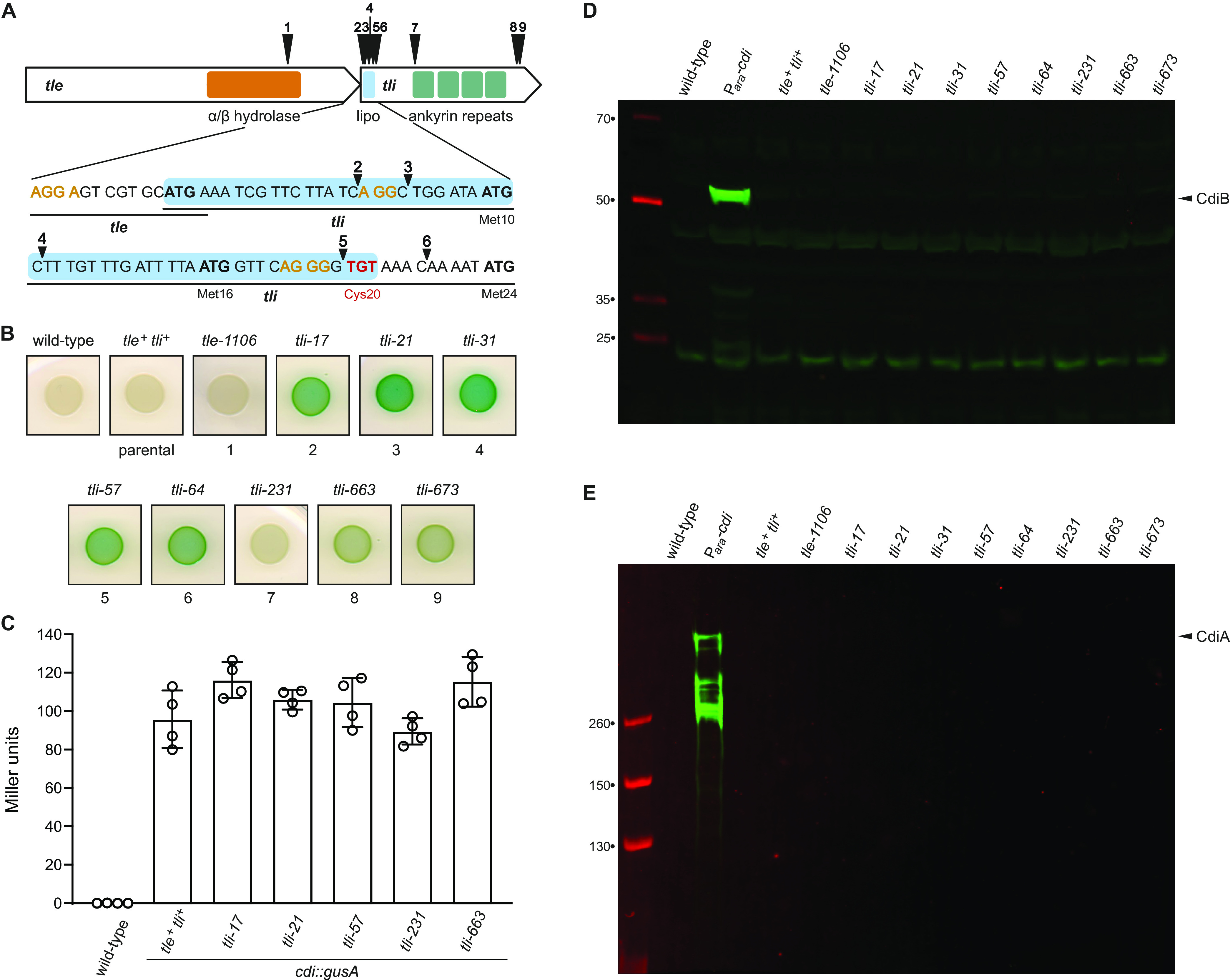
Identification of Tn5 insertions in the tli immunity gene. (A) Schematic of Tn5 insertion sites in tle and tli. Regions encoding the predicted α/β-hydrolase domain of Tle and the Tli lipoprotein signal peptide (lipo) and ankyrin repeats are depicted. Alternative translation initiation codons (Met10, Met16, and Met24) are indicated, and possible ribosome-binding sites are shown in orange font. The codon for the lipidated Cys20 residue is shown in red font. (B) Wild-type ECL, the parental cdi-gusA reporter strain (tle+ tli+), and tli mutants were grown on LB agar supplemented with X-Gluc. (C) Quantification of β-glucuronidase activity in selected tli mutants. Activities were quantified as Miller units as described in Materials and Methods. Data for technical replicates from two independent experiments are shown with the average ± standard deviation. (D and E) Tn5 insertions do not upregulate CdiB or CdiA production. Protein samples from wild-type ECL, the parental tle+ tli+ reporter strain, and tli mutants were subjected to immunoblot analysis using polyclonal antisera to CdiB (D) and CdiA (E). The Para-cdi sample is from an ECL strain that contains an arabinose-inducible promoter inserted upstream of the native cdiBAI gene cluster.
FIG 2.

Disruption of tli increases cell permeability. (A) ECL is impermeable to chromogenic X-Gluc substrate. Wild-type and cdi-gusA reporter cells were grown on LB agar supplemented with X-Gluc. Where indicated, SDS solution (1% [wt/vol]) was applied to the edge of the colonies prior to imaging. (B) The indicated ECL strains were complemented with plasmid-borne tli (pTli) or an empty vector and grown on LB agar supplemented with X-Gluc. (C) Complementation with tli does not affect cdi-gusA reporter expression. β-Glucuronidase activities were quantified as Miller units as described in Materials and Methods. Data for technical replicates from two independent experiments are shown with the average ± standard deviation.
Increased cell permeability is dependent on T6SS-1 activity.
Two scenarios could account for hyperpermeability: the tli mutants could be permeabilized by internally produced phospholipase, or they could be intoxicated by Tle delivered from neighboring sibling cells. These models are not exclusive, and the phenotype may reflect a combination of external and internal phospholipase activities. To eliminate the contribution from T6SS-delivered effector, we deleted tssM (Fig. 3A), which encodes a critical component of the transenvelope complex required for secretion. The resulting tli ΔtssM cells do not secrete Hcp into culture supernatants (Fig. 3B), confirming that T6SS-1 is inactivated. The tli ΔtssM strains also exhibit less pigmentation when grown on X-Gluc indicator medium (Fig. 3C, top row). To ensure that the ΔtssM mutation does not abrogate tle expression, we showed that complementation with plasmid-borne tssM restores Hcp secretion and hyperpermeability phenotypes to each strain (Fig. 3B and C, bottom row). These data indicate that increased cell permeability is largely due to T6SS-1-dependent exchange of phospholipase toxin between sibling cells.
FIG 3.
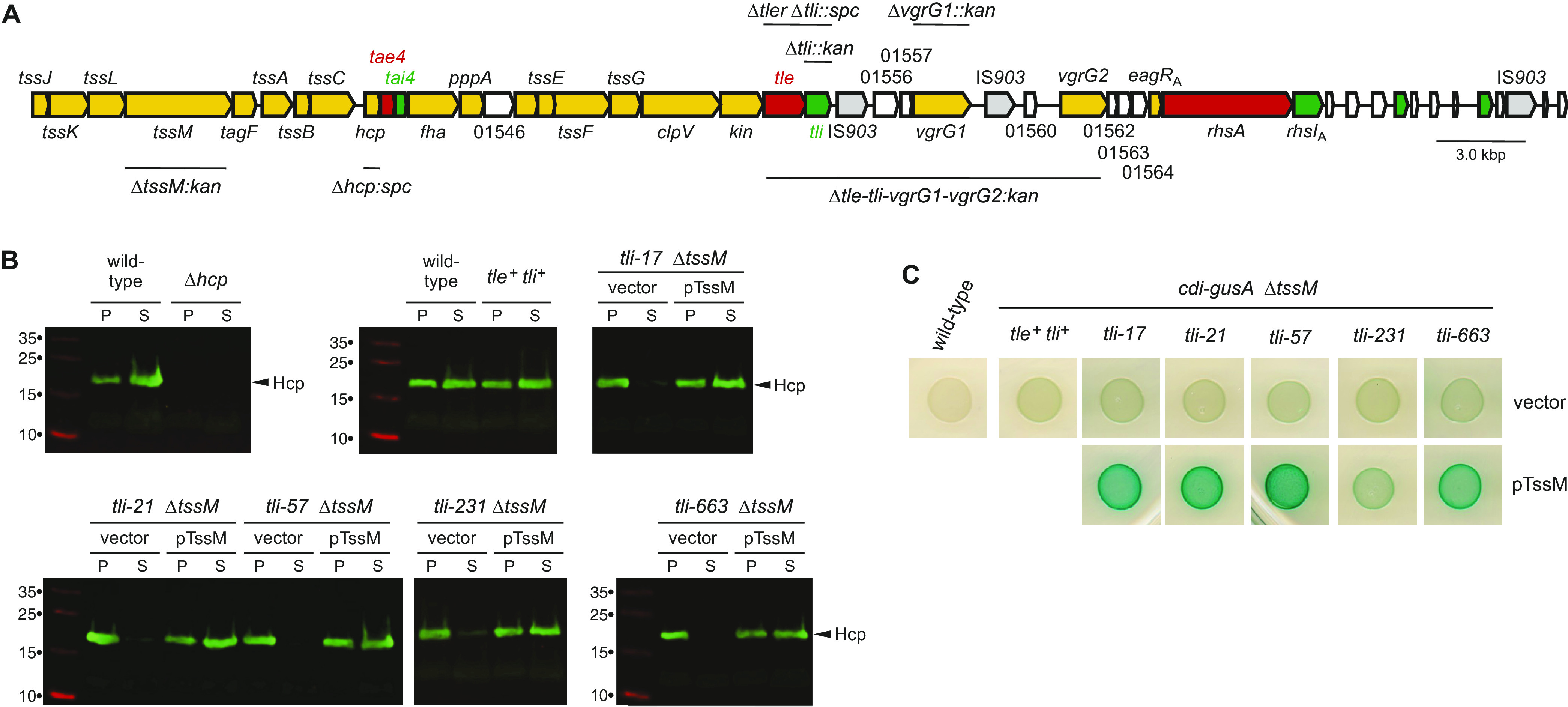
Hyperpermeability is T6SS-1 dependent. (A) Schematic of the ECL T6SS-1 locus. Core secretion system genes are colored light orange, and effector and immunity genes are shown in red and green, respectively. Genes of unknown function are not colored and are indicated by their ordered locus number. The extents of the gene deletions used in this study are indicated. (B) ECL ΔtssM strains do not secrete Hcp. The indicated ECL strains were grown in LB, and then cell pellet (P) and culture supernatant (S) fractions were prepared as described in Materials and Methods. Where indicated, the strains carried a plasmid-borne copy of tssM (pTssM) or an empty vector plasmid. Fractions were analyzed by SDS-PAGE and immunoblotting with polyclonal antisera to Hcp. (C) ECL tli ΔtssM mutants do not exhibit increased cell permeability. The indicated ECL strains were grown on LB agar supplemented with X-Gluc. Where indicated, the strains carried pTssM or an empty vector plasmid.
Although tli is disrupted in each mutant, most retain the capacity to produce a truncated form of the immunity protein. Insertions in the lipoprotein signal sequence (tli-17, tli-21, tli-31, tli-57, and tli-64) should allow cytosolic Tli to be synthesized using Met10, Met16, or Met24 as alternative translation initiation codons (Fig. 1A). Furthermore, the tli-663 and tli-673 alleles are predicted to produce secreted Tli lipoproteins that carry transposon-encoded residues fused at Asp221 and Lys224, respectively (Fig. 1A). Immunoblotting revealed that the tli-17, tli-21, tli-57, and tli-64 mutants accumulate Tli to about the same level as the parental tli+ strain, but we were unable to detect antigen in tli-231, tli-663, and tli-763 cell lysates (Fig. 4A). We next asked whether truncated Tli provides any protection against Tle delivered from wild-type ECL inhibitor cells. Because the tli mutants also deploy Tle to intoxicate their siblings, we examined immunity function in a ΔtssM background to ensure that the mutants act only as target cells during coculture. The tli ΔtssM target cells are outcompeted between 50- and 100-fold by wild-type ECL, but they show no fitness disadvantage when cocultured with ECL ΔtssM mock inhibitor cells (Fig. 4B). This level of growth inhibition is comparable to that observed with Δtle Δtli target cells (Fig. 4B), which harbor a true null allele of tli. Thus, the transposon insertions completely abrogate immunity to Tle. Given that cytosolic immunity protein fails to protect, these results also indicate that Tli must be localized to the periplasm to neutralize toxic phospholipase activity.
FIG 4.

tli mutants produce Tli, but are not immune to Tle effector. (A) tli mutants carrying Tn5 insertions in the lipoprotein signal sequence still produce immunity protein. Protein samples from the indicated ECL strains were subjected to immunoblot analysis using polyclonal antisera to Tli. (B) Tn5 insertions ablate the immunity function of Tli. Wild-type ECL (tssM+) and mock (ΔtssM) inhibitor strains were mixed with the indicated tli ΔtssM target cells and spotted onto LB agar. After 4 h of coculture, inhibitor and target cells were enumerated as CFU on selective media. The competitive index equals the final ratio of inhibitor to target cells divided by the initial ratio. Presented data are the average ± standard error from three independent experiments.
ECL Δtli mutants fail to deploy active Tle.
Although the tli disruptions behave as null mutations in competition cocultures, we noted that the tli-231 insertion induces a modest permeability phenotype compared to the other alleles. This observation is curious because tli-231 is arguably the most disruptive to Tli structure and is therefore expected to cause a more profound phenotype. To explore this issue, we asked whether a Δtli null strain phenocopies the transposon mutants on X-Gluc indicator medium and surprisingly found that Δtli cells are not hyperpermeable (Fig. 5A). This result could reflect the acquisition of an inactivating mutation in tle during construction of the Δtli strain. However, the hyperpermeable phenotype is restored when truncated Tli proteins lacking the N-terminal 23 (Tli-Δ23) and 51 (Tli-Δ51) residues are expressed from a plasmid in Δtli cells (Fig. 5A). Further truncation of the immunity protein to Tli-Δ75, which approximates the fragment produced by the tli-231 allele, does not induce autopermeabilization (Fig. 5A). These data suggest that Δtli cells do not deploy active Tle. Accordingly, we found that tle+ Δtli cells behave like ΔtssM mock inhibitors when competed against Δtle Δtli target cells (Fig. 5B). The competitive fitness of the tle+ Δtli strain is restored to wild-type levels when the cells express wild-type Tli or Tli-Δ23, but not the Tli-Δ51 or Tli-Δ75 truncations (Fig. 5B). Although Tli-Δ51 fails to confer a competitive advantage (Fig. 5B), target cell growth in these cocultures is suppressed compared to that in experiments with tle+ Δtli inhibitors that harbor an empty vector plasmid (Fig. 5C). Quantification of viable Tli-Δ51-producing cells shows that their growth is also inhibited (Fig. 5D), indicating that intersibling intoxication obviates any competitive advantage gained by Tle delivery. This autoinhibition is not observed with Tli-Δ23-producing cells (Fig. 5D), although they exhibit a similar hyperpermeable phenotype on indicator medium (Fig. 5A).
FIG 5.
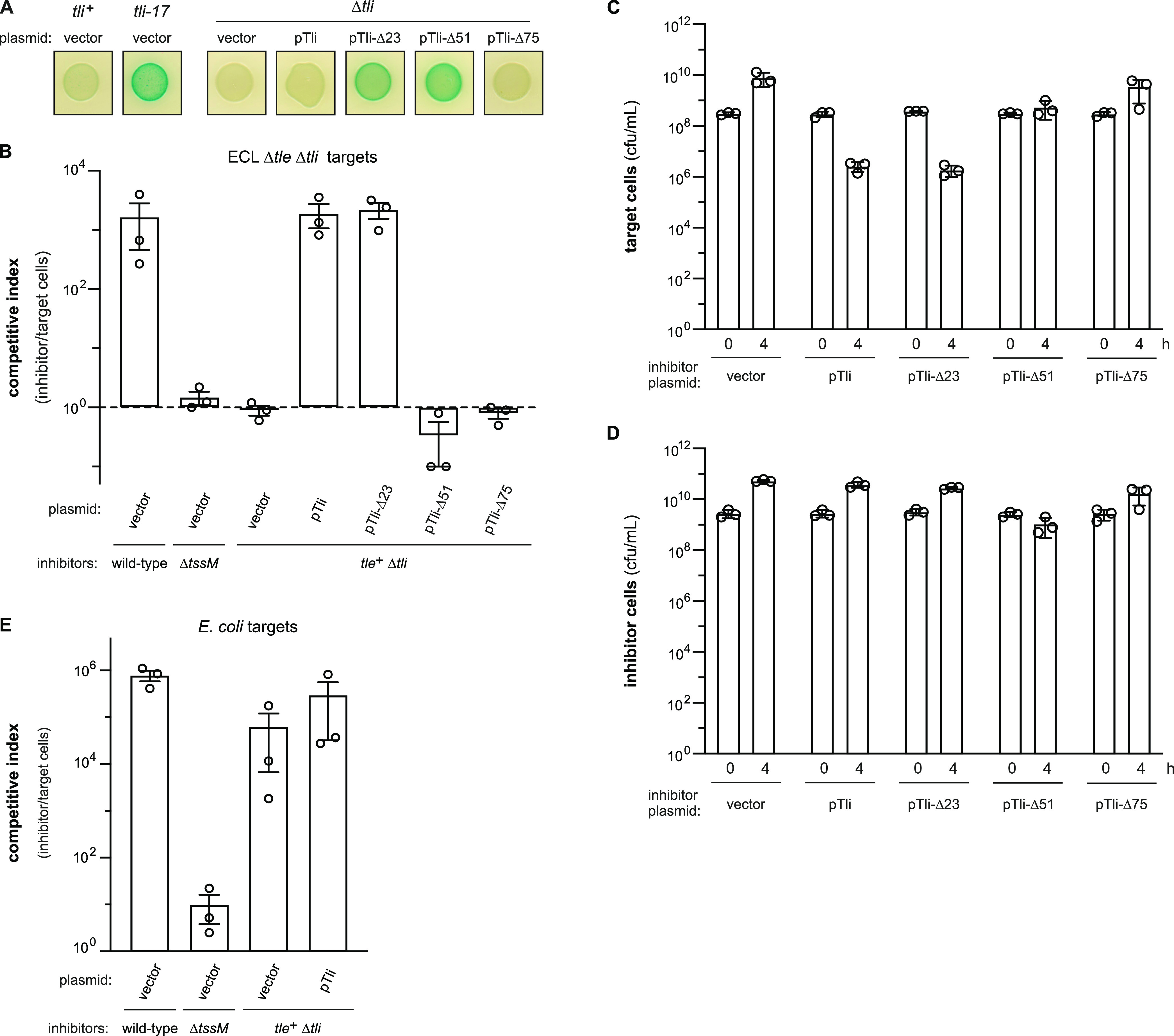
Δtli mutants fail to deploy active Tle. (A) ECL Δtli mutants do not exhibit hyper-permeability. The indicated strains were grown on LB-agar supplemented with X-gluc. Where indicated, strains carried an empty vector or were complemented with plasmids that produce Tli variants. (B) The indicated ECL inhibitor strains were mixed with ECL Δtle Δtli target cells and spotted onto LB-agar. After 4 h of co-culture, inhibitor and target cells were enumerated as colony forming units (cfu) on selective media. The competitive index equals the final ratio of inhibitor to target cells divided by the initial ratio. Presented data are the average ± standard error from three independent experiments. (C) Viable ECL Δtle Δtli target cell counts (cfu/mL) at 0 and 4 h for the cocultures shown in panel B. (D) Viable inhibitor cell counts (cfu/mL) at 0 and 4 h for the cocultures shown in panel B. (E) ECL inhibitor strains were mixed with E. coli X90 target cells and spotted onto LB-agar. Co-culture conditions and cfu enumerations were the same as described for panel B.
To test whether the ECL Δtli strain deploys other T6SS-1 effectors, we performed competition assays with E. coli target bacteria, which have previously been shown to be inhibited by ECL Δtle mutants (29). Wild-type ECL outcompetes E. coli almost 106-fold on agar medium, and most of this growth advantage is lost when T6SS-1 is inactivated with the ΔtssM mutation (Fig. 5E). ECL Δtli mutants retain most of this inhibition activity against E. coli target cells (Fig. 5E), demonstrating that tli is not required to deliver other T6SS-1 effectors. Together, these results indicate that Tle-mediated growth inhibition depends on a cytosolic pool of Tli.
Tli is required for the toxic activity of Tle.
Tli could be required to stabilize Tle, or perhaps the immunity protein acts as a chaperone to load effector into the T6SS-1 apparatus. We first asked whether Tli stabilizes the lipase effector, but were unable to detect Tle antigen in either wild-type (tle+ tli+) or Δtli cell lysates (Fig. 6A). Given the low endogenous level of Tle, we expressed tle and tle-tli from plasmid vectors (pTle and pTle-Tli) in ECL Δtle Δtli cells to examine effector stability. Immunoblotting showed that full-length Tle is produced from both constructs, though some minor degradation products are apparent in the absence of Tli (Fig. 6A). Despite producing detectable lipase, cells harboring the pTle plasmid have no inhibition activity against Δtle Δtli target bacteria (Fig. 6B). In contrast, the pTle-Tli construct confers a significant growth advantage under the same conditions (Fig. 6B). We also tested whether Tli is required for effector secretion, but were unable to detect Tle antigen in culture supernatants even when expressed from a plasmid vector. Therefore, we asked whether Tli is still required for growth inhibition activity when phospholipase export is ensured by fusion to the VgrG β-spike protein. The ECL T6SS-1 locus encodes two VgrG proteins (VgrG1 and VgrG2) (Fig. 3A), each of which is sufficient to support secretion activity (29). We reasoned that the lipase domain could be fused to VgrG2, because other Enterobacter strains encode “evolved” VrgG2 homologs that carry effector domains at their C termini (see Fig. S1 in the supplemental material). AlphaFold2 modeling indicates that Tle is composed of two domains connected by a flexible linker (Fig. 7A). The novel N-terminal domain of Tle is presumably required for T6SS-mediated export, and its C-terminal domain adopts an α/β-hydrolase fold that resembles neutral lipases from fungi. Using the AlphaFold2 model as a guide, we fused the linker region and phospholipase domain of Tle to VgrG2 (Fig. 7B), then asked whether the VgrG2-lipase chimera restores growth inhibition activity to a VgrG-deficient mutant. Because ECL Δtle-tli-vgrG1-vgrG2 cells do not produce functional VgrG, they cannot assemble the T6SS-1 apparatus (29) and do not kill E. coli target bacteria during coculture (Fig. 7C). Complementation of Δtle-tli-vgrG1-vgrG2 cells with the VgrG2-lipase construct restores growth inhibition activity against E. coli target cells (Fig. 7C). Moreover, this inhibition activity is quantitatively similar to that of an ECL ΔvgrG1 inhibitor strain (Fig. 7C), which produces VgrG2 and Tle as separate polypeptides. These results suggest that the VgrG2-lipase chimera supports the same T6SS-1 activity as wild-type VgrG2. However, the VgrG2-lipase construct has no growth inhibition activity against ECL Δtle Δtli target cells (Fig. 7D), indicating that the fused lipase domain is inactive. We next tested whether addition of the tli gene to the fusion construct promotes phospholipase activity (Fig. 7B). Although the resulting pVgrG2-lipase/Tli construct is somewhat less effective against E. coli target cells (Fig. 7C), it inhibits the growth of ECL Δtle Δtli target cells to the same extent as an ECL ΔvgrG1 inhibitor strain (Fig. 7D). We also showed that mutation of the predicted catalytic Ser341 residue in the lipase domain abrogates growth inhibition activity against ECL Δtle Δtli cells (Fig. 7D), but not E. coli target cells (Fig. 7C), indicating that the VgrG2-lipase fusion supports the delivery of other T6SS-1 effectors. Together, these results demonstrate that the Tle phospholipase domain is only active when coproduced with its cognate immunity protein.
FIG 6.

Tli is required for Tle-mediated growth inhibition. (A) Cell extracts from the indicated ECL strains were subjected to immunoblot analysis using polyclonal antisera against the Tle lipase domain. (B) The indicated ECL inhibitor strains were mixed with ECL Δtle Δtli target cells and spotted onto LB agar. After 4 h of coculture, inhibitor and target cells were enumerated as CFU on selective media. The competitive index equals the final ratio of inhibitor to target cells divided by the initial ratio. Presented data are the average ± standard error from three independent experiments.
FIG 7.
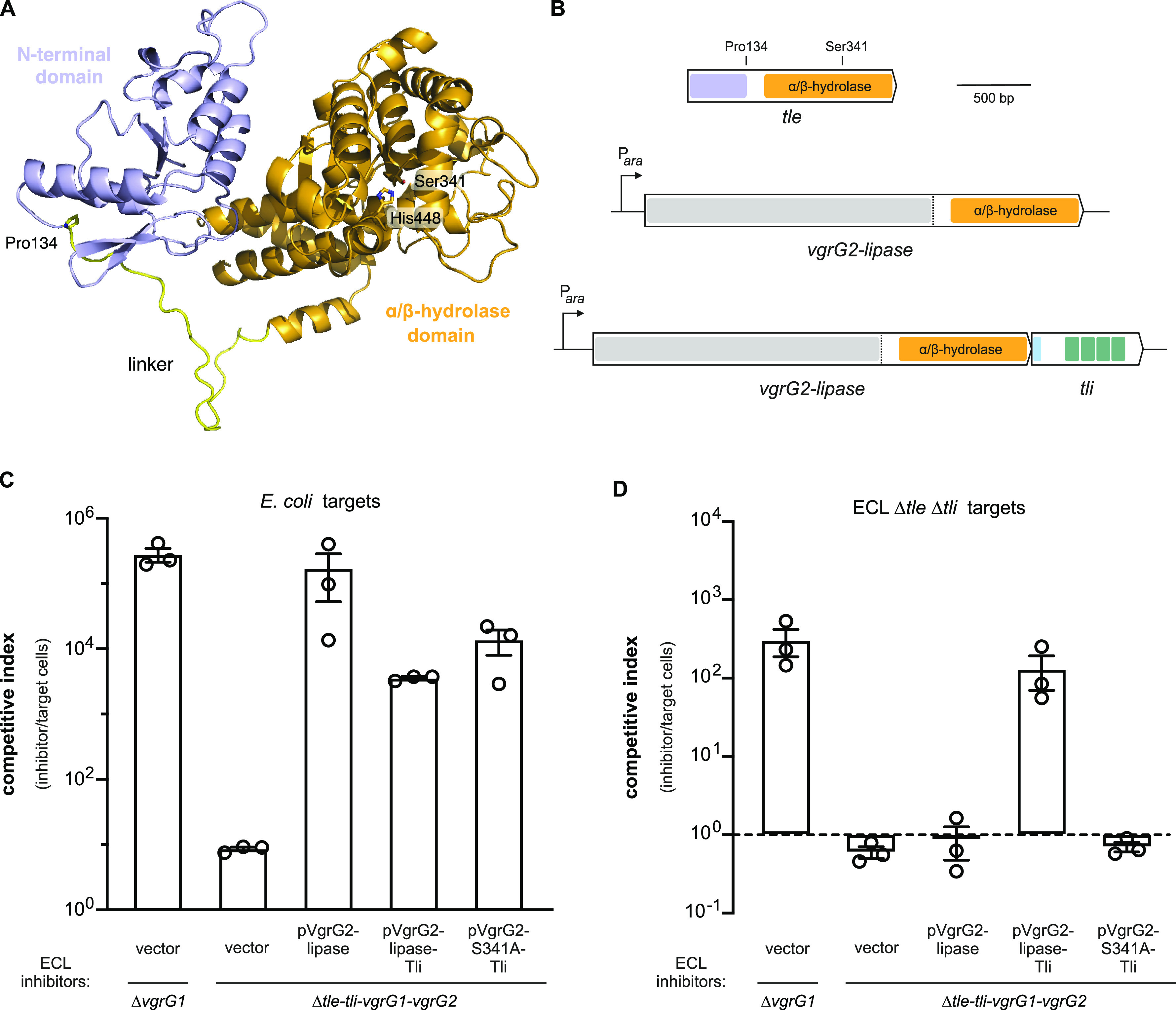
Tli is required to activate Tle prior to T6SS-mediate export. (A) AlphaFold2 model of Tle. The C-terminal α/β-hydrolase domain is rendered in light orange and the linker region in yellow. The locations of predicted catalytic residues Ser341 and His448 are indicated. (B) VgrG2-lipase domain fusion schematic. Coding sequence for the Tle linker region (beginning at Pro134) and lipase domain was fused to vgrG2 under the control of an arabinose-inducible promoter. A matched construct that also includes the wild-type tli gene was also generated. (C) The VgrG2-lipase fusion protein supports T6SS-1 activity. The indicated ECL inhibitor strains were mixed with E. coli X90 target cells and spotted onto LB agar supplemented with l-arabinose. After 4 h of coculture, inhibitor and target cells were enumerated as CFU on selective media. The competitive index equals the final ratio of inhibitor to target cells divided by the initial ratio. Presented data are the average ± standard error from three independent experiments. (D) ECL inhibitor strains were mixed with ECL Δtle Δtli target cells and spotted onto LB agar supplemented with l-arabinose. Coculture conditions and CFU enumerations were the same as described for panel C.
DISCUSSION
Here, we identify ECL transposon mutants that exhibit increased permeability to a chromogenic β-glucuronidase substrate. The transposon insertions disrupt tli, which encodes an immunity protein that neutralizes the previously described Tle phospholipase effector (29). Thus, unmitigated Tle activity damages cellular membranes, allowing chromogenic substrate to enter the cytosol and/or β-glucuronidase to escape into the extracellular medium. This hyperpermeability is dependent on T6SS-1 activity, indicating that the mutants are intoxicated by Tle delivered from neighboring siblings rather than their own internally produced phospholipase. This phenomenon is similar to the observations of Russell et al., who found that deletion of the tli5PA (PA3488) immunity gene from Pseudomonas aeruginosa PAO1 leads to increased cell permeability through unopposed PldDPA phospholipase activity (30). Moreover, P. aeruginosa autointoxication requires a functional H2-T6SS locus (30), indicating that the phenotype is due to intersibling exchange of phospholipase. However, in contrast to P. aeruginosa, ECL Δtli null mutants fail to deploy active phospholipase and consequently do not exhibit hyperpermeability. Instead, the strongest phenotypes are associated with mutations that prevent targeting of Tli to the periplasm, yet still allow the immunity protein to accumulate to wild-type levels. These findings indicate that Tli has different functions, depending on its subcellular localization. Tli in the periplasm acts to neutralize incoming phospholipase effectors, whereas cytoplasmic Tli produced from alternative translation initiation sites is required for the activation and/or export of Tle. Because we cannot ascertain whether Tle is secreted from Δtli mutants, it is possible that cytosolic Tli acts a chaperone to load the effector into the T6SS apparatus. However, the immunity protein probably has another function because growth inhibition activity remains tli-dependent when phospholipase delivery is achieved through fusion to VgrG2. We propose that Tli activates the effector domain prior to T6SS-mediated export. AlphaFold2 modeling suggests that the lipase catalytic core is not altered upon binding to Tli, although dramatic conformational changes are predicted for lipase helices α5, α10, and α11 (see Fig. S2A and B in the supplemental material). Helix α5 is pulled away from the lipase domain through predicted interactions with the first ankyrin repeat of Tli, allowing α10 and α11 to migrate into the vacated position (Fig. S2A and B). In the absence of Tli, helix α5 forms a “lid” that covers the active site and precludes access to solvent (Fig. S2A and B). Similar lid structures are found in many other lipases, and these helices are typically displaced to expose the active site upon binding to membranes in a process termed “interfacial activation” (31). Thus, the predicted conformational changes in the lid could underlie lipase activation, but because Tli blocks lipase activity, the model may correspond to the neutralized conformation. We also modeled lipase interactions with immunity proteins produced from the various tli alleles identified in this study. Lipase interactions with Tli-64, Tli-663, and Tli-673 are nearly identical to those with wild-type Tli (Fig. S2C, E, and F), but Tli-231, which lacks N-terminal ankyrin repeats, does not displace helix α5 of the lipase domain (Fig. S2D). The implications of the latter model for lipase activation are not clear given that we are unable to detect Tli antigen in tli-231 mutants. Though the mechanism remains unknown, it seems likely that Tle activation requires direct binding interactions with cytosolic Tli. Because presumably only the effector protein is exported, this model implies that there must be an additional mechanism to dissociate the Tle-Tli complex prior to loading into the T6SS apparatus.
Russell et al. first described and delineated five families of T6SS phospholipase effectors, which they designated Tle1 through Tle5 (30). Although not identified in that study, TleECL from ECL contains the GxSxG catalytic motif found in the Tle1, Tle2, Tle3, and Tle4 effector families. AlphaFold2 modeling suggests that TleECL adopts an α/β-hydrolase fold, with the GxSxG sequence forming a classic nucleophilic “elbow” in the turn between β4 and α7. A DALI server search indicates that TleECL is not closely related to known T6SS effectors and instead is most similar to a family of neutral lipases secreted by fungi (32–34). This conclusion is supported by direct comparisons of the TleECL model with the crystal structures of Tle1PAO1 and Tle4PAO1 from Pseudomonas aeruginosa PAO1. Superimposition of core α/β-hydrolase domains reveals that the flanking secondary elements are distinct between the effectors (Fig. S3). Moreover, Tle1PAO1 and Tle4PAO1 have modules that are absent from TleECL. Tle1PAO1 contains a putative membrane-anchoring domain (35), and the α/β-hydrolase core of Tle4PAO1 is interrupted by three inserted segments that form a cap domain to cover the active site (36). We note that the TleECL model also differs substantially from AlphaFold2 predictions for Tle2 (VasL) from Vibrio cholerae and Tle3 from Burkholderia thailandensis E264. Database searches reveal that TleECL is commonly encoded by Enterobacter and Cronobacter T6SS loci, and it shares significant sequence identity with the Slp effector from Serratia marcescens (see Table S1 in the supplemental material) (37). Homologous lipase domains are more broadly distributed across Erwinia, Dickeya, Burkholderia, and Vibrio species, where they are fused to predicted VgrG, PAAR, and DUF4150 domain proteins (Table S1). Related lipase domains also form the C termini of rearrangement hot spot (Rhs) proteins in various Pseudomonas and Vibrio isolates. Rhs proteins constitute another important class of T6SS effectors that carry variable C-terminal toxin domains (1, 38–40). Finally, a handful of Deltaproteobacteria encode predicted CdiA proteins with similar lipase domains (Table S1), indicating that this effector family can also be delivered through the T5SS pathway. Notably, these lipase sequences are linked to putative immunity genes that encode ankyrin repeat proteins (Table S1), raising the possibility that all of these effectors are activated by their cognate immunity proteins.
Although it remains to be determined whether other lipases require activation, a similar phenomenon has been reported for the PAAR domain-containing PefD effector of Proteus mirabilis HI4320. Mobley and coworkers found that disruption of the corresponding pefE immunity gene, which encodes a predicted β-propeller protein, abrogates the growth inhibition activity of PefD in cocultures (41). PefD function is also dependent on pefF and pefG, which encode an armadillo repeat protein and a thioredoxin-like oxidoreductase, respectively (41, 42). AlphaFold2 indicates that PefD contains an intricate array of six disulfide bonds (https://www.uniprot.org/uniprotkb/B4EVA8/entry#structure), suggesting that PefE and PefF could be chaperones that hold the effector in the proper conformation for PefG-catalyzed disulfide bond formation. However, PefD and the related Tse7 effector from P. aeruginosa are toxic DNases that must be transferred into the cytoplasm to inhibit growth (41, 43). Given that any disulfides would likely be reduced by glutathione and thioredoxin after delivery into the target cell, perhaps these bonds promote export rather than enzymatic activity. Proper disulfide formation is clearly important for other T6SS effectors that function in the periplasm. Peptidoglycan amidase and pore-forming effectors from Serratia marcescens both depend on DsbA (44), which is a highly conserved oxidoreductase that catalyzes disulfide formation in the periplasm of most Gram-negative bacteria (45). T5SS/CdiA effector domains are also known to exploit target cell proteins to promote toxicity. CdiA from uropathogenic E. coli delivers a novel tRNA anticodon nuclease domain that is only active when bound to the cysteine biosynthetic enzyme CysK (26, 46, 47). Other CdiA proteins carry BECR-fold RNases that interact with elongation factor Tu (EF-Tu) to cleave the 3′-acceptor stems of specific tRNA molecules (48–51).
Given that our screen ultimately selected for hyperpermeable cells, it is perhaps surprising that we did not recover other mutations that are known to disrupt the cell envelope. The first “leaky” mutants were isolated in screens for E. coli and Salmonella enterica serovar Typhimurium cells that release periplasmic RNase I and alkaline phosphatase into culture media (52–55). These early studies uncovered a critical role for TolA in Gram-negative cell envelope integrity (52). Later, E. coli lamB cells were used to identify mutations in ompC and ompF that allow high-molecular-weight maltodextrins to enter the periplasm (56, 57). The same strategy also led to the discovery of lptD (58), which encodes an essential β-barrel protein that inserts lipopolysaccharide into the outer membrane (59–62). More recently, Bernhardt and coworkers used a cell-impermeable β-galactosidase substrate to identify over 100 E. coli genes that contribute to envelope integrity (63). Although the approach taken by Paradis-Bleu et al. is essentially the same as in our study, they note that disruption of outer membrane integrity alone is probably sufficient for substrate entry in their screen (63). In contrast, ECL lacks outer and inner membrane transporters for β-glucuronides (64), and therefore both membranes must be permeabilized for substrate entry. These observations suggest that T6SS-delivered Tle damages the outer and inner membranes of target bacteria and that the loss of Tli immunity function produces the most striking phenotype on indicator media.
MATERIALS AND METHODS
Bacterial strain and plasmid constructions.
The bacterial strains and plasmids used in this study are listed in Table 1 and Table 2, respectively, and oligonucleotides are listed in Table S2 in the supplemental material. All bacteria were grown at 37°C in lysogeny broth (LB): 1% tryptone, 1% yeast extract, and 0.5% NaCl. Media were supplemented with antibiotics at the following concentrations: ampicillin (Amp), 150 μg/mL; gentamicin (Gent), 12 μg/mL; kanamycin (Kan), 50 μg/mL; rifampin (Rif), 200 μg/mL; spectinomycin (Spec), 15 μg/mL; tetracycline (Tet), 100 μg/mL; and trimethoprim (Tmp), 100 μg/mL (where indicated). The ECL cdi-gusA reporter strain ZR71 was constructed by Tn7-mediated transposition. E. coli gusA was amplified with oligonucleotide primers CH3025/CH3026 and ligated to pUC18T-mini-Tn7T-Gm (65) using EcoRI/SacI restriction sites to generate plasmid pZR42. The ECL cdiB promoter region was amplified with CH3146/CH3147 and ligated to pZR42 using KpnI/EcoRI restriction sites to generate pCH3468. pCH3468 and pTNS2 (66) were introduced into wild-type ECL by conjugation, and gentamicin-resistant (Gentr) clones were screened for cdi-gusA transposition into the glmS locus. The same mating procedure was used to generate strain CH3469 (ECL Δtli glmS::cdi-gusA) from strain CH14588. The ΔtssM1::kan allele (from pCH11050) was introduced into ECL strains carrying plasmid pKOBEG using Red-mediated recombination as described previously (27, 29). ECL Δtli::kan (CH14587) and Δtle-tli-vgrG1-vgrG2::kan (CH1360) mutants were constructed using pCH371, which is a pRE118 (67) derivative that carries the pheS(A294G) counterselectable marker instead of sacB. The pheS(A294G) allele was first amplified with DL2217/DL2194 and fused to the tet promoter in pBR322 using EcoRV/SphI restriction sites to generate plasmid pDAL912. The Ptet-pheS(A294G) fragment was then amplified with DL2195/DL2196 and combined with pRE118-derived fragments generated with DL2197/DL2198 and D2199/DL1667 using overlap extension PCR (OE-PCR). The final PCR product was introduced into pRE118 using Red-mediated recombination to generate plasmid pDAL6480. A chloramphenicol resistance (Cmr) cassette was amplified with CH5269/CH5270, digested with XbaI/NsiI, and ligated to XbaI/SbfI-digested pDAL6480 to generate plasmid pCH377. The FLP recombination target (FRT)-flanked Kanr cassette from pKAN/pCH70 (68) was subcloned into pCH377 via SacI/KpnI restriction sites to generate pCH371. The pCH1359 Δtle-tli-vgrG1-vgrG2 knockout construct was generated by sequential cloning of CH3373/CH3374 (SacI/BamHI) and CH3197/CH3198 (XhoI/KpnI) fragments into pCH371. The pCH3884 Δtli::kan knockout construct was generated by sequential cloning of ZR39/ZR40 (SacI/BamHI) and CH3377/CH3378 (XhoI/KpnI) fragments into pCH371. Plasmids pCH1359 and pCH3884 were introduced into ECL by conjugation, followed by selection for Kanr Cmr exconjugants. Clones were cultured in M9 minimal medium supplemented with 0.4% d-glucose, 2 mM d/l-chlorophenylalanine, and 50 μg/mL kanamycin and then screened for Kanr Cms clones that had lost the pheS(A294G) marker through homologous recombination. The Kanr cassette was removed from CH14587 using the FLP recombinase encoded on pCP20 (69) to generate strain CH14588.
TABLE 1.
Bacterial strains used in this study
| Strain | Descriptiona | Reference or source |
|---|---|---|
| CH1360 | ECL Δtle-tli-vgrG1-vgrG2::kan Kanr | This study |
| CH2016 | E. coli X90(DE3) Δrna ΔslyD::kan Kanr | 73 |
| CH3469 | ECL Δtli glmS::cdi-gusA Gentr | This study |
| CH11196 | ECL ΔtssM1::kan Kanr | 27 |
| CH11199 | ECL Δhcp1::spc Specr | 29 |
| CH11248 | ECL araC-PBAD::cdiB ΔaraBAD::kan Specr Kanr | 27 |
| CH11396 | ECL ΔtssM1 | 29 |
| CH11876 | ECL Δtle::kan Kanr | 29 |
| CH11895 | ECL Δtle Δtli::spc Specr | 29 |
| CH12384 | ECL ΔvgrG1::kan Kanr | 28 |
| CH14384 | ECL Δtle Δtli::spc rif Specr Rifr | This study |
| CH14587 | ECL Δtli::kan Kanr | This study |
| CH14588 | ECL Δtli | This study |
| E2072 | E. coli F– λ– mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 deoR recA1 endA1 galU galK rpsL nupG ΔdapA::pir-lacIq-Gentr-FRT8 mob[recA::RP4-2 Tc::Mu-Kanr] leu+ Strr Gentr Kanr | 75 |
| ECL | Enterobacter cloacae subsp. cloacae ATCC 13047 | ATCC |
| MFD pir | E. coli MG1655 RP4-2-Tc::[ΔMu1::aac(3)IV-ΔaphA-Δnic35-ΔMu2::zeo] dapA::(erm-pir) ΔrecA Aprr Zeor Ermr | 76 |
| X90 | E. coli F′ lacIq lac′ pro′/ara Δ(lac-pro) nal1 argE(Amb) rif thi-1 Rifr | 77 |
| ZR71 | ECL glmS::cdi-gusA Gentr | This study |
| ZR135 | ZR71 tli-231; Tn5 insertion after tli nucleotide 231; Gentr Specr | This study |
| ZR137 | ZR71 tli-21; Tn5 insertion after tli nucleotide 21; Gentr Specr | This study |
| ZR138 | ZR71 tli-57; Tn5 insertion after tli nucleotide 57; Gentr Specr | This study |
| ZR139 | ZR71 tli-663; Tn5 insertion after tli nucleotide 663; Gentr Specr | This study |
| ZR140 | ZR71 tli-17; Tn5 insertion after tli nucleotide 17; Gentr Specr | This study |
| ZR148 | ZR71 tli-231 ΔtssM::kan Gentr Specr Kanr | This study |
| ZR149 | ZR71 tli-21 ΔtssM::kan Gentr Specr Kanr | This study |
| ZR150 | ZR71 tli-57 ΔtssM::kan Gentr Specr Kanr | This study |
| ZR151 | ZR71 tli-663 ΔtssM::kan Gentr Specr Kanr | This study |
| ZR152 | ZR71 tli-17 ΔtssM::kan Gentr Specr Kanr | This study |
| ZR161 | ZR71 tli-31; Tn5 insertion after tli nucleotide 31; Specr Gentr | This study |
| ZR163 | ZR71 tle-1106; Tn5 insertion after tle nucleotide 1106; Specr Gentr | This study |
| ZR165 | ZR71 tli-673; Tn5 insertion after tli nucleotide 673; Specr Gentr | This study |
| ZR170 | ZR71 tli-64; Tn5 insertion after tli nucleotide 64; Specr Gentr | This study |
| ZR200 | ZR71 ΔtssM::kan Gentr Kanr | This study |
Aprr, apramycin resistant; Ermr, erythromycin resistant; Kanr, kanamycin resistant; Gentr, gentamicin resistant; Rifr, rifampin resistant; Specr, spectinomycin resistant; Strr, streptomycin resistant; Zeor, zeocin resistant.
TABLE 2.
Plasmids used in this study
| Plasmid | Descriptiona | Reference |
|---|---|---|
| pBR322 | Cloning vector; Ampr Tetr | 78 |
| pCH70 | pBluescript with FRT-flanked Kanr cassette ligated into SmaI restriction site; Ampr Kanr | 79 |
| pCH371 | pCATKAN-pheS(A294G); Cmr Kanr | This study |
| pCH377 | pCAT-pheS(A294G); Cmr | This study |
| pCH417 | pCH450::vgrG2(ECL)-CT/vgrI(Eh49162); Tetr | This study |
| pCH450 | pACYC184 derivative with E. coli araC and araBAD promoter for arabinose-inducible expression; Tetr | 68 |
| pCH494 | pCH450K::tli; Tetr | 29 |
| pCH495 | pCH450K::tli(Δ23); Tetr | This study |
| pCH1359 | pCATKAN-pheS(A294G)-Δtle-tli-vgrG1-vgrG2; Cmr Kanr | This study |
| pCH3128 | pCH450::tle-tli; Tetr | This study |
| pCH3291 | pET21::tli(Δ23); Ampr | This study |
| pCH3468 | pUC18T-mini-Tn7T-Gm with the ECL cdi promoter region fused to E. coli gusA within mini-Tn7; Ampr Gentr | This study |
| pCH3763 | pCH450::vgrG2-lipase(S341A)-tli; Tetr | This study |
| pCH3884 | pCATKAN-pheS(A294G)-Δtli; Cmr Kanr | This study |
| pCH5036 | pCH450K::tli(Δ51); Tetr | This study |
| pCH5037 | pCH450K::tli(Δ75); Tetr | This study |
| pCH6118 | pET21b::cdiB(STEC3); Ampr | This study |
| pCH9062 | pCH450::vgrG2-lipase-tli; Tetr | This study |
| pCH9063 | pCH450::vgrG2-lipase; Tetr | This study |
| pCH9384 | pBluescript with FRT-flanked Specr cassette ligated into SmaI restriction site; Ampr Specr | 38 |
| pCH11050 | pCH70 with regions upstream and downstream of ECL tssM1; Ampr Kanr | 27 |
| pCH11198 | pCH450::tssM; Tetr | This study |
| pCH14212 | pCH450::tle; Tetr | This study |
| pCH15269 | pSH21P::lipase; Ampr | This study |
| pCP20 | Temp-sensitive replication origin, expresses FLP recombinase; Ampr Cmr | 80 |
| pDAL912 | pBR322::pheS(A294G); Ampr | This study |
| pDAL6480 | pRE118-pheS(A294G); Kanr | This study |
| pKOBEG | Temp-sensitive replication origin, expresses the Bet-Gam-Exo proteins from phage λ; Cmr | 69 |
| pLG99 | Tn5 transposase expression vector harboring a mini-Tn5 derivative with PrhaB-out; Ampr Tmpr | 70 |
| pLG99-spec | pLG99 with Specr cassette cloned into Tn5; Ampr Specr | This study |
| pRE118 | Vector plasmid for sacB allelic exchange; Kanr | 67 |
| pSCBAD | pBBR1 derivative that carries araC and PBAD promoter; Tmpr | 72 |
| pTNS2 | Expresses Tn7 transposase, Ampr | 66 |
| pUC18T-mini-Tn7T-Gm | Tn7 transposase expression vector harboring mini-Tn7 with Gentr cassette; Ampr Gentr | 65 |
| pZR42 | pUC18T-mini-Tn7T-Gm with E. coli gusA; Ampr Gentr | This study |
Ampr, ampicillin resistant; Cmr, chloramphenicol resistant; Kanr, kanamycin resistant; Tmpr, trimethoprim resistant; Tetr, tetracycline resistant; Gentr, gentamicin resistant; Specr, spectinomycin resistant.
The Tn5 delivery vector pLG99 (70) was modified to introduce a spectinomycin resistance (Specr) cassette. An EcoRV/SpeI fragment from pSPM/pCH9384 (28) was ligated to a pLG99 vector prepared by NsiI (followed by end-filling with T4 DNA polymerase) and AvrII digestion. The ECL tli coding sequence was amplified with primer pairs, CH3418/CH3419 (wild-type tli), CH3719/CH3419 (tli-Δ23), CH5137/CH3419 (tli-Δ51), and CH5318/CH3419 (tli-Δ75), and the products were ligated to pCH450K (68) using KpnI/XhoI restriction sites to generate plasmids pCH494, pCH495, pCH5036, and pCH5037, respectively. ECL tle was amplified with primers CH4469/ZR248 and ligated to pCH450 using EcoRI/XhoI restriction sites to generate plasmid pCH14212. tli was then amplified with CH4762/CH3419 and ligated via BstXI/XhoI to pCH14212 to generate plasmid pCH3128. Primers CH3719/CH4154 were used amplify tli-Δ23 for ligation into pET21K via KpnI/XhoI to generate pCH3291, which overproduces cytosolic Tli with a C-terminal His6 epitope. Primers CH5087/ZR248 were used to amplify the lipase domain coding region of tle for ligation into pSH21P via SpeI/XhoI to generate plasmid pCH15269, which overproduces the domain with an N-terminal His6 epitope. ECL tssM was amplified with CH3023/CH3024 and ligated to pCH450 using NcoI/XhoI restriction sites to generate plasmid pCH11198. The cdiB gene from E. coli STEC_O31 was amplified with ZR443/ZR444 and ligated to pET21b using EcoRI/XhoI restriction sites to generate plasmid pCH6118.
To construct the VgrG2-lipase fusion, vgrG2 from ECL was amplified with CH4452/CH5205 and ligated to pCH450 using EcoRI/KpnI restriction sites. vgrG-CT/vgrI sequences were amplified from Enterobacter hormaechei ATCC 49162 using CH5203/CH5204 and appended to the vgrG2 construct using KpnI/PstI restriction sites to generate pCH417. Plasmid pCH417 was amplified with CH4452/CH5770 to add a SacI linker, and lipase domain/tli modules from CH5772/ZR248 and CH5772/CH3419 amplifications were ligated via SacI/XhoI to generate plasmids pCH9063 and pCH9062, respectively. The Ser341Ala substitution was introduced into the lipase domain by OE-PCR using primers CH4762/CH4763 in conjunction with CH5772/CH3419 to generate plasmid pCH3763.
Transposon mutagenesis and mutant screening.
Plasmid pLG99::spec was introduced into ECL strain ZR71 cells via conjugation, and Specr clones were selected on tryptone broth agar (1% tryptone, 0.5% NaCl, 1.5% agar) supplemented with 100 μg/mL Spec and 100 μg/mL of 5-bromo-4-chloro-1H-indol-3-yl β-d-glucopyranosiduronic acid (X-Gluc). Transposon insertion sites were identified by arbitrarily primed PCR. The Tn5 right arm was amplified with Tn5-Right1/CH2020 or Tn5-Right1/CH2022. Products were reamplified using nested primers Tn5-Right2/CH2021. The Tn5 left arm was amplified using primers Tn5-Left1/CH2020 and Tn5-Left1/CH2022. The resulting reactions were amplified with nested primers Tn5-Left2/CH2021. Second-round reactions were sequenced using oligonucleotides Tn5-Right3 and Tn5-Left3 as primers.
X-Gluc permeability and β-glucuronidase assays.
β-Glucuronidase activity was quantified as described by Miller (71). Cells were harvested from tryptone broth (TB) agar and suspended in 100 mM sodium phosphate (pH 7.0) at an optical density at 600 nm (OD600) of 1.0. A drop of toluene and a drop of 0.1% SDS were added to 100 μL of the cell suspension, followed by vortexing for 15 s. The suspension was then incubated at 37°C for 90 min without caps to allow evaporation of toluene. Cell suspensions and substrate solution (1 mg/mL p-nitrophenyl β-d-glucuronide in 100 mM sodium phosphate [pH 7.0] and 5 mM β-mercaptoethanol) were equilibrated to 28°C. Cell lysate (100 μL) was added to 600 μL of substrate solution and incubated at 28°C for 30 min. Reaction mixtures were quenched with 700 μL of 1 M sodium carbonate. Quenched reaction mixtures were centrifuged at 14,000 × g for 10 min, supernatant was removed, and absorbance was measured at 420 nm. β-Glucuronidase activity was quantified in Miller units, where enzyme units = 1,000 (A420)/(OD600 of 1.0 at 0.1 mL for 30 min). Overnight cultures were adjusted to an OD600 of 3.0, and 10 μL was spotted onto TB agar supplemented with 100 μg/mL X-Gluc and incubated overnight at 37°C. Culture plates were imaged using a light scanner.
Competition cocultures.
Inhibitor and target cells were grown overnight in LB and then diluted 1:50 into fresh LB and grown at 37°C to mid-log phase. Cells were harvested by centrifugation at 3,400 × g for 2 min, resuspended to an optical density at 600 nm (OD600) of 3.0, and mixed at a 10:1 ratio of inhibitor to target cells. Cell mixtures (10 μL) were spotted onto LB agar (supplemented with 0.4% l-arabinose when using strains with arabinose-inducible constructs) and incubated at 37°C. A portion of each cell mixture was serially diluted into 1 × M9 salts and plated onto antibiotic-supplemented LB agar to enumerate inhibitor and target cells as CFU at t = 0 h. For the competition in Fig. 4B, inhibitor strains were scored using Tmpr (conferred by plasmid pSCBAD) (72), and targets were scored by Specr. For all other competitions, inhibitors were scored by Tetr (conferred by pCH450 derivatives), and E. coli and ECL target cells were enumerated by Rifr. After 4 h of coculture, cells were harvested from the agar surface with polyester-tipped swabs and resuspended in 800 μL of 1 × M9 salts. The cell suspensions were serially diluted in 1 × M9 salts and plated as described above to quantify endpoint CFU Competitive indices were calculated as the final ratio of inhibitor to target cells divided by the initial ratio of inhibitor to target cells.
Hcp secretion.
Overnight cultures of ECL were diluted 1:50 into 3 mL LB and grown to an OD600 of ~1.0 at 37°C. The cultures were then supplemented with 0.4% l-arabinose and incubated for 45 min. The cultures were centrifuged at 3,400 × g for 5 min, and the cell pellets were resuspended in 100 μL of urea lysis buffer (8 M urea, 150 mM NaCl, 20 mM Tris-HCl [pH 8.0]) for lysis. Supernatants were collected and centrifuged for an additional 5 min to remove any remaining cells. Supernatants were then passed through a 0.22-μm-pore low-binding polyvinylidene fluoride (PVDF) filter. The filtered culture supernatants were adjusted to 10% trichloroacetic acid and incubated on ice for 1 h. Samples were centrifuged at 21,000 × g for 10 min. Precipitates were resuspended twice in 1 mL ice-cold acetone and recollected by centrifugation at 21,000 × g for 5 min. Precipitates were air-dried at ambient temperature and then dissolved in 50 μL urea lysis buffer. Protein concentrations were estimated by Bradford assay, and ~5 μg was loaded onto 10% polyacrylamide SDS gels for electrophoresis and immunoblot analysis.
Antiserum generation and immunoblotting.
His6-tagged variants of Tli-Δ23 and the Tle lipase domain from ECL, and CdiB from E. coli STEC_O31 were overproduced in E. coli strain CH2016 from plasmids pCH3291, pCH15269, and pCH6118, respectively. Overnight cultures were diluted 1:100 in fresh LB supplemented with Amp and grown to mid-log phase at 37°C in a baffled flask. Expression was then induced with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1.5 mM for 90 min. Cultures were harvested by centrifugation and cell pellets frozen at −80°C. Cells were broken by freeze-thaw in urea lysis buffer supplemented with 0.05% Triton X-100 and 20 mM imidazole as described previously (73). Proteins were purified by Ni2+-affinity chromatography under denaturing conditions in urea lysis buffer, followed by elution in urea lysis buffer supplemented with 25 mM EDTA. The purified proteins were dialyzed against water and lyophilized to be used as antigens to generate rabbit polyclonal antisera (Cocalico Biologicals, Inc., Reamstown, PA).
Samples were analyzed by SDS-PAGE using Tris-tricine-buffered gels run at 110 V. For Hcp, CdiB, and Tle immunoblots, samples were run on 10% polyacrylamide gels for 1 h. Tli samples were run on 10% polyacrylamide gels for 2 h 15 min, and CdiA samples were run on 6% polyacrylamide gels for 3.5 h. Gels were soaked for 10 min in transfer buffer (25 mM Tris, 192 mM glycine [pH 8.6], 20% methanol [10% methanol for CdiA gels]) before electroblotting to PVDF membranes using a semidry transfer apparatus run at 17 V for 30 min. For CdiA, gels were electroblotted for 1 h. Membranes were blocked with 4% nonfat milk in 1 × phosphate-buffered saline (PBS) for 45 min at ambient temperature and then incubated with primary antibodies in 1 × PBS with 4% nonfat milk overnight. Rabbit polyclonal antisera for Hcp (29), Tle, Tli, CdiA (74) and CdiB were used at dilutions of 1:10,000, 1:5,000, 1:5,000, 1:10,000, and 1:12,500, respectively. After three 10-min washes in 1 × PBS, the membranes were incubated with IRDye 800CW-conjugated goat anti-rabbit IgG (1:125,000 dilution) (Li-Cor) in 1 × PBS for 45 min. Immunoblots were visualized using a Li-Cor Odyssey infrared imager.
ACKNOWLEDGMENTS
We thank Stephanie Aoki for the construction of plasmid pDAL6480 and Jiang Xiong for technical assistance.
This work was supported by grants GM117930 (C.S.H.) from the National Institutes of Health and MCB 1545720 (D.A.L. and C.S.H.) from the National Science Foundation. Z.C.R. was supported by the Tri-Counties Blood Bank Postdoctoral Fellowship.
Conceptualization, S.J.J., Z.C.R., D.A.L., and C.S.H.; Funding Acquisition, D.A.L. and C.S.H.; Investigation, S.J.J., Z.C.R., A.F.W., D.Q.N., and F. G.-S.; Methodology, Z.C.R. and C.S.H.; Supervision, D.A.L. and C.S.H.; Validation, S.J.J., D.Q.N., and F.G.-S.; Visualization, Z.C.R. and C.S.H.; Writing – Original Draft, Z.C.R. and C.S.H.; Writing – Review & Editing, S.J.J., D.A.L., and C.S.H.
Footnotes
Supplemental material is available online only.
Contributor Information
Christopher S. Hayes, Email: chayes@lifesci.ucsb.edu.
Laurie E. Comstock, University of Chicago
REFERENCES
- 1.Ruhe ZC, Low DA, Hayes CS. 2020. Polymorphic toxins and their immunity proteins: diversity, evolution, and mechanisms of delivery. Annu Rev Microbiol 74:497–520. doi: 10.1146/annurev-micro-020518-115638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. 2005. Contact-dependent inhibition of growth in Escherichia coli. Science 309:1245–1248. doi: 10.1126/science.1115109. [DOI] [PubMed] [Google Scholar]
- 3.Ruhe ZC, Subramanian P, Song K, Nguyen JY, Stevens TA, Low DA, Jensen GJ, Hayes CS. 2018. Programmed secretion arrest and receptor-triggered toxin export during antibacterial contact-dependent growth inhibition. Cell 175:921–933.e14. doi: 10.1016/j.cell.2018.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, Li M, Schwarz S, Wang WY, Merz AJ, Goodlett DR, Mougous JD. 2010. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7:25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. 2010. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci USA 107:19520–19524. doi: 10.1073/pnas.1012931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ. 2009. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci USA 106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunet YR, Zoued A, Boyer F, Douzi B, Cascales E. 2015. The type VI secretion TssEFGK-VgrG phage-like baseplate is recruited to the TssJLM membrane complex via multiple contacts and serves as assembly platform for tail tube/sheath polymerization. PLoS Genet 11:e1005545. doi: 10.1371/journal.pgen.1005545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. 2007. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci USA 104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shneider MM, Buth SA, Ho BT, Basler M, Mekalanos JJ, Leiman PG. 2013. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500:350–353. doi: 10.1038/nature12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silverman JM, Agnello DM, Zheng H, Andrews BT, Li M, Catalano CE, Gonen T, Mougous JD. 2013. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell 51:584–593. doi: 10.1016/j.molcel.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hernandez RE, Gallegos-Monterrosa R, Coulthurst SJ. 2020. Type VI secretion system effector proteins: effective weapons for bacterial competitiveness. Cell Microbiol 22:e13241. doi: 10.1111/cmi.13241. [DOI] [PubMed] [Google Scholar]
- 12.Brunet YR, Khodr A, Logger L, Aussel L, Mignot T, Rimsky S, Cascales E. 2015. H-NS silencing of the Salmonella pathogenicity island 6-encoded type VI secretion system limits Salmonella enterica serovar Typhimurium interbacterial killing. Infect Immun 83:2738–2750. doi: 10.1128/IAI.00198-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caro F, Caro JA, Place NM, Mekalanos JJ. 2020. Transcriptional silencing by TsrA in the evolution of pathogenic Vibrio cholerae biotypes. mBio 11:e02901-20. doi: 10.1128/mBio.02901-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Storey D, McNally A, Åstrand M, Sa-Pessoa Graca Santos J, Rodriguez-Escudero I, Elmore B, Palacios L, Marshall H, Hobley L, Molina M, Cid VJ, Salminen TA, Bengoechea JA. 2020. Klebsiella pneumoniae type VI secretion system-mediated microbial competition is PhoPQ controlled and reactive oxygen species dependent. PLoS Pathog 16:e1007969. doi: 10.1371/journal.ppat.1007969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma J, Sun M, Pan Z, Song W, Lu C, Yao H. 2018. Three Hcp homologs with divergent extended loop regions exhibit different functions in avian pathogenic Escherichia coli. Emerg Microbes Infect 7:49. doi: 10.1038/s41426-018-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernard CS, Brunet YR, Gueguen E, Cascales E. 2010. Nooks and crannies in type VI secretion regulation. J Bacteriol 192:3850–3860. doi: 10.1128/JB.00370-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA 103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernard CS, Brunet YR, Gavioli M, Lloubès R, Cascales E. 2011. Regulation of type VI secretion gene clusters by sigma54 and cognate enhancer binding proteins. J Bacteriol 193:2158–2167. doi: 10.1128/JB.00029-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sana TG, Hachani A, Bucior I, Soscia C, Garvis S, Termine E, Engel J, Filloux A, Bleves S. 2012. The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and Fur and modulates internalization in epithelial cells. J Biol Chem 287:27095–27105. doi: 10.1074/jbc.M112.376368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majerczyk C, Schneider E, Greenberg EP. 2016. Quorum sensing control of type VI secretion factors restricts the proliferation of quorum-sensing mutants. eLife 5:e14712. doi: 10.7554/eLife.14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mougous JD, Gifford CA, Ramsdell TL, Mekalanos JJ. 2007. Threonine phosphorylation post-translationally regulates protein secretion in Pseudomonas aeruginosa. Nat Cell Biol 9:797–803. doi: 10.1038/ncb1605. [DOI] [PubMed] [Google Scholar]
- 22.Wäneskog M, Halvorsen T, Filek K, Xu F, Hammarlöf DL, Hayes CS, Braaten BA, Low DA, Poole SJ, Koskiniemi S. 2021. Escherichia coli EC93 deploys two plasmid-encoded class I contact-dependent growth inhibition systems for antagonistic bacterial interactions. Microb Genom 7:mgen000534. doi: 10.1099/mgen.0.000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majerczyk C, Brittnacher M, Jacobs M, Armour CD, Radey M, Schneider E, Phattarasokul S, Bunt R, Greenberg EP. 2014. Global analysis of the Burkholderia thailandensis quorum sensing-controlled regulon. J Bacteriol 196:1412–1424. doi: 10.1128/JB.01405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rojas CM, Ham JH, Deng WL, Doyle JJ, Collmer A. 2002. HecA, a member of a class of adhesins produced by diverse pathogenic bacteria, contributes to the attachment, aggregation, epidermal cell killing, and virulence phenotypes of Erwinia chrysanthemi EC16 on Nicotiana clevelandii seedlings. Proc Natl Acad Sci USA 99:13142–13147. doi: 10.1073/pnas.202358699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rojas CM, Ham JH, Schechter LM, Kim JF, Beer SV, Collmer A. 2004. The Erwinia chrysanthemi EC16 hrp/hrc gene cluster encodes an active Hrp type III secretion system that is flanked by virulence genes functionally unrelated to the Hrp system. Mol Plant Microbe Interact 17:644–653. doi: 10.1094/MPMI.2004.17.6.644. [DOI] [PubMed] [Google Scholar]
- 26.Aoki SK, Diner EJ, de Roodenbeke CT, Burgess BR, Poole SJ, Braaten BA, Jones AM, Webb JS, Hayes CS, Cotter PA, Low DA. 2010. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature 468:439–442. doi: 10.1038/nature09490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, Goulding CW, Hayes CS. 2014. CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure 22:707–718. doi: 10.1016/j.str.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitney JC, Beck CM, Goo YA, Russell AB, Harding BN, De Leon JA, Cunningham DA, Tran BQ, Low DA, Goodlett DR, Hayes CS, Mougous JD. 2014. Genetically distinct pathways guide effector export through the type VI secretion system. Mol Microbiol 92:529–542. doi: 10.1111/mmi.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donato SL, Beck CM, Ruhe ZC, Cunningham DA, Singleton I, Low DA, Hayes CS. 2020. The β-encapsulation cage of rearrangement hotspot (Rhs) effectors is required for type VI secretion. Proc Natl Acad Sci USA 117:33540–33548. doi: 10.1073/pnas.1919350117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, Wiggins PA, Wai SN, Mougous JD. 2013. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496:508–512. doi: 10.1038/nature12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan FI, Lan D, Durrani R, Huan W, Zhao Z, Wang Y. 2017. The lid domain in lipases: structural and functional determinant of enzymatic properties. Front Bioeng Biotechnol 5:16. doi: 10.3389/fbioe.2017.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brady L, Brzozowski AM, Derewenda ZS, Dodson E, Dodson G, Tolley S, Turkenburg JP, Christiansen L, Huge-Jensen B, Norskov L. 1990. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature 343:767–770. doi: 10.1038/343767a0. [DOI] [PubMed] [Google Scholar]
- 33.Lan D, Xu H, Xu J, Dubin G, Liu J, Iqbal Khan F, Wang Y. 2017. Malassezia globosa MgMDL2 lipase: crystal structure and rational modification of substrate specificity. Biochem Biophys Res Commun 488:259–265. doi: 10.1016/j.bbrc.2017.04.103. [DOI] [PubMed] [Google Scholar]
- 34.Lou Z, Li M, Sun Y, Liu Y, Liu Z, Wu W, Rao Z. 2010. Crystal structure of a secreted lipase from Gibberella zeae reveals a novel “double-lock” mechanism. Protein Cell 1:760–770. doi: 10.1007/s13238-010-0094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu H, Zhang H, Gao Z, Wang D, Liu G, Xu J, Lan K, Dong Y. 2014. Structure of the type VI secretion phospholipase effector Tle1 provides insight into its hydrolysis and membrane targeting. Acta Crystallogr D Biol Crystallogr 70:2175–2185. doi: 10.1107/S1399004714012899. [DOI] [PubMed] [Google Scholar]
- 36.Lu D, Zheng Y, Liao N, Wei L, Xu B, Liu X, Liu J. 2014. The structural basis of the Tle4-Tli4 complex reveals the self-protection mechanism of H2-T6SS in Pseudomonas aeruginosa. Acta Crystallogr D Biol Crystallogr 70:3233–3243. doi: 10.1107/S1399004714023967. [DOI] [PubMed] [Google Scholar]
- 37.Cianfanelli FR, Alcoforado Diniz J, Guo M, De Cesare V, Trost M, Coulthurst SJ. 2016. VgrG and PAAR proteins define distinct versions of a functional type VI secretion system. PLoS Pathog 12:e1005735. doi: 10.1371/journal.ppat.1005735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koskiniemi S, Lamoureux JG, Nikolakakis KC, t'Kint de Roodenbeke C, Kaplan MD, Low DA, Hayes CS. 2013. Rhs proteins from diverse bacteria mediate intercellular competition. Proc Natl Acad Sci USA 110:7032–7037. doi: 10.1073/pnas.1300627110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alcoforado Diniz J, Coulthurst SJ. 2015. Intraspecies competition in Serratia marcescens is mediated by type VI-secreted Rhs effectors and a conserved effector-associated accessory protein. J Bacteriol 197:2350–2360. doi: 10.1128/JB.00199-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. 2012. Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alteri CJ, Himpsl SD, Zhu K, Hershey HL, Musili N, Miller JE, Mobley HLT. 2017. Subtle variation within conserved effector operon gene products contributes to T6SS-mediated killing and immunity. PLoS Pathog 13:e1006729. doi: 10.1371/journal.ppat.1006729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alteri CJ, Himpsl SD, Pickens SR, Lindner JR, Zora JS, Miller JE, Arno PD, Straight SW, Mobley HL. 2013. Multicellular bacteria deploy the type VI secretion system to preemptively strike neighboring cells. PLoS Pathog 9:e1003608. doi: 10.1371/journal.ppat.1003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pissaridou P, Allsopp LP, Wettstadt S, Howard SA, Mavridou DAI, Filloux A. 2018. The Pseudomonas aeruginosa T6SS-VgrG1b spike is topped by a PAAR protein eliciting DNA damage to bacterial competitors. Proc Natl Acad Sci USA 115:12519–12524. doi: 10.1073/pnas.1814181115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mariano G, Monlezun L, Coulthurst SJ. 2018. Dual role for DsbA in attacking and targeted bacterial cells during type VI secretion system-mediated competition. Cell Rep 22:774–785. doi: 10.1016/j.celrep.2017.12.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collet JF, Cho SH, Iorga BI, Goemans CV. 2020. How the assembly and protection of the bacterial cell envelope depend on cysteine residues. J Biol Chem 295:11984–11994. doi: 10.1074/jbc.REV120.011201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diner EJ, Beck CM, Webb JS, Low DA, Hayes CS. 2012. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev 26:515–525. doi: 10.1101/gad.182345.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson PM, Beck CM, Morse RP, Garza-Sánchez F, Low DA, Hayes CS, Goulding CW. 2016. Unraveling the essential role of CysK in CDI toxin activation. Proc Natl Acad Sci USA 113:9792–9797. doi: 10.1073/pnas.1607112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones AM, Garza-Sánchez F, So J, Hayes CS, Low DA. 2017. Activation of contact-dependent antibacterial tRNase toxins by translation elongation factors. Proc Natl Acad Sci USA 114:E1951–e1957. doi: 10.1073/pnas.1619273114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michalska K, Gucinski GC, Garza-Sánchez F, Johnson PM, Stols LM, Eschenfeldt WH, Babnigg G, Low DA, Goulding CW, Joachimiak A, Hayes CS. 2017. Structure of a novel antibacterial toxin that exploits elongation factor Tu to cleave specific transfer RNAs. Nucleic Acids Res 45:10306–10320. doi: 10.1093/nar/gkx700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gucinski GC, Michalska K, Garza-Sánchez F, Eschenfeldt WH, Stols L, Nguyen JY, Goulding CW, Joachimiak A, Hayes CS. 2019. Convergent evolution of the barnase/EndoU/colicin/RelE (BECR) fold in antibacterial tRNase toxins. Structure 27:1660–1674.e5. doi: 10.1016/j.str.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J, Yashiro Y, Sakaguchi Y, Suzuki T, Tomita K. 2022. Mechanistic insights into tRNA cleavage by a contact-dependent growth inhibitor protein and translation factors. Nucleic Acids Res 50:4713–4731. doi: 10.1093/nar/gkac228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amouroux C, Lazzaroni JC, Portalier R. 1991. Isolation and characterization of extragenic suppressor mutants of the tolA-876 periplasmic-leaky allele in Escherichia coli K-12. FEMS Microbiol Lett 62:305–313. doi: 10.1111/j.1574-6968.1991.tb04461.x. [DOI] [PubMed] [Google Scholar]
- 53.Lazzaroni JC, Portalier RC. 1981. Genetic and biochemical characterization of periplasmic-leaky mutants of Escherichia coli K-12. J Bacteriol 145:1351–1358. doi: 10.1128/jb.145.3.1351-1358.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopes J, Gottfried S, Rothfield L. 1972. Leakage of periplasmic enzymes by mutants of Escherichia coli and Salmonella typhimurium: isolation of “periplasmic leaky” mutants. J Bacteriol 109:520–525. doi: 10.1128/jb.109.2.520-525.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weigand RA, Rothfield LI. 1976. Genetic and physiological classification of periplasmic-leaky mutants of Salmonella typhimurium. J Bacteriol 125:340–345. doi: 10.1128/jb.125.1.340-345.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benson SA, Decloux A. 1985. Isolation and characterization of outer membrane permeability mutants in Escherichia coli K-12. J Bacteriol 161:361–367. doi: 10.1128/jb.161.1.361-367.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benson SA, Occi JL, Sampson BA. 1988. Mutations that alter the pore function of the OmpF porin of Escherichia coli K12. J Mol Biol 203:961–970. doi: 10.1016/0022-2836(88)90121-0. [DOI] [PubMed] [Google Scholar]
- 58.Sampson BA, Misra R, Benson SA. 1989. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122:491–501. doi: 10.1093/genetics/122.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, He C, Zhang Y, Wang W, Dong C. 2014. Structural basis for outer membrane lipopolysaccharide insertion. Nature 511:52–56. doi: 10.1038/nature13464. [DOI] [PubMed] [Google Scholar]
- 60.Gu Y, Stansfeld PJ, Zeng Y, Dong H, Wang W, Dong C. 2015. Lipopolysaccharide is inserted into the outer membrane through an intramembrane hole, a lumen gate, and the lateral opening of LptD. Structure 23:496–504. doi: 10.1016/j.str.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Gu Y, Dong H, Wang W, Dong C. 2015. Trapped lipopolysaccharide and LptD intermediates reveal lipopolysaccharide translocation steps across the Escherichia coli outer membrane. Sci Rep 5:11883. doi: 10.1038/srep11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qiao S, Luo Q, Zhao Y, Zhang XC, Huang Y. 2014. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 511:108–111. doi: 10.1038/nature13484. [DOI] [PubMed] [Google Scholar]
- 63.Paradis-Bleau C, Kritikos G, Orlova K, Typas A, Bernhardt TG. 2014. A genome-wide screen for bacterial envelope biogenesis mutants identifies a novel factor involved in cell wall precursor metabolism. PLoS Genet 10:e1004056. doi: 10.1371/journal.pgen.1004056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang WJ, Wilson KJ, Xie H, Knol J, Suzuki S, Rutherford NG, Henderson PJ, Jefferson RA. 2005. The gusBC genes of Escherichia coli encode a glucuronide transport system. J Bacteriol 187:2377–2385. doi: 10.1128/JB.187.7.2377-2385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi KH, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, Schweizer HP. 2008. Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomallei. Appl Environ Microbiol 74:1064–1075. doi: 10.1128/AEM.02430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 67.Edwards RA, Keller LH, Schifferli DM. 1998. Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207:149–157. doi: 10.1016/s0378-1119(97)00619-7. [DOI] [PubMed] [Google Scholar]
- 68.Hayes CS, Sauer RT. 2003. Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol Cell 12:903–911. doi: 10.1016/s1097-2765(03)00385-x. [DOI] [PubMed] [Google Scholar]
- 69.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 70.Gallagher LA, Ramage E, Patrapuvich R, Weiss E, Brittnacher M, Manoil C. 2013. Sequence-defined transposon mutant library of Burkholderia thailandensis. mBio 4:e00604-13. doi: 10.1128/mBio.00604-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 72.Koskiniemi S, Garza-Sánchez F, Edman N, Chaudhuri S, Poole SJ, Manoil C, Hayes CS, Low DA. 2015. Genetic analysis of the CDI pathway from Burkholderia pseudomallei 1026b. PLoS One 10:e0120265. doi: 10.1371/journal.pone.0120265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garza-Sánchez F, Janssen BD, Hayes CS. 2006. Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem 281:34258–34268. doi: 10.1074/jbc.M608052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruhe ZC, Townsley L, Wallace AB, King A, Van der Woude MW, Low DA, Yildiz FH, Hayes CS. 2015. CdiA promotes receptor-independent intercellular adhesion. Mol Microbiol 98:175–192. doi: 10.1111/mmi.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zarzycki-Siek J, Norris MH, Kang Y, Sun Z, Bluhm AP, McMillan IA, Hoang TT. 2013. Elucidating the Pseudomonas aeruginosa fatty acid degradation pathway: identification of additional fatty acyl-CoA synthetase homologues. PLoS One 8:e64554. doi: 10.1371/journal.pone.0064554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrières L, Hémery G, Nham T, Guérout AM, Mazel D, Beloin C, Ghigo JM. 2010. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol 192:6418–6427. doi: 10.1128/JB.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beckwith JR, Signer ER. 1966. Transposition of the lac region of Escherichia coli. I. Inversion of the lac operon and transduction of lac by phi80. J Mol Biol 19:254–265. doi: 10.1016/s0022-2836(66)80003-7. [DOI] [PubMed] [Google Scholar]
- 78.Bolivar F, Rodriguez RL, Greene PJ, Betlach MC, Heyneker HL, Boyer HW, Crosa JH, Falkow S. 1977. Construction and characterization of new cloning vehicles. II. A multipurpose cloning system. Gene 2:95–113. doi: 10.1016/0378-1119(77)90000-2. [DOI] [PubMed] [Google Scholar]
- 79.Hayes CS, Bose B, Sauer RT. 2002. Proline residues at the C terminus of nascent chains induce SsrA tagging during translation termination. J Biol Chem 277:33825–33832. doi: 10.1074/jbc.M205405200. [DOI] [PubMed] [Google Scholar]
- 80.Pérez A, Canle D, Latasa C, Poza M, Beceiro A, Tomás MdM, Fernández A, Mallo S, Pérez S, Molina F, Villanueva R, Lasa I, Bou G. 2007. Cloning, nucleotide sequencing, and analysis of the AcrAB-TolC efflux pump of Enterobacter cloacae and determination of its involvement in antibiotic resistance in a clinical isolate. Antimicrob Agents Chemother 51:3247–3253. doi: 10.1128/AAC.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download jb.00113-23-s0001.pdf, PDF file, 0.03 MB (29.4KB, pdf)
Table S2. Download jb.00113-23-s0002.pdf, PDF file, 0.02 MB (24.7KB, pdf)
Fig. S1. Download jb.00113-23-s0003.pdf, PDF file, 1.3 MB (1.3MB, pdf)
Fig. S2. Download jb.00113-23-s0004.pdf, PDF file, 2.1 MB (2.1MB, pdf)
Fig. S3. Download jb.00113-23-s0005.pdf, PDF file, 2.1 MB (2.1MB, pdf)