

ABSTRACT
Optimum formulation of Biological-E’s protein subunit CORBEVAX™ vaccine was selected in phase-1 and -2 studies and found to be safe and immunogenic in healthy adult population. This is a phase-3 prospective, single-blinded, randomized, active controlled study conducted at 18 sites across India in 18–80 year-old subjects. This study has two groups; (i) immunogenicity-group, participants randomized either to CORBEVAX™ (n = 319) or COVISHIELD™ arms (n = 320). (ii) Safety-group containing single CORBEVAX™ arm (n = 1500) and randomization is not applicable. Healthy adults without a history of COVID-19 vaccination or SARS-CoV-2 infection were enrolled into immunogenicity arm and subjects seronegative to SARS-CoV-2 infection were enrolled into the safety arm. The safety profile of CORBEVAX™ vaccine was comparable to the comparator vaccine COVISHIELD™. Majority of reported AEs were mild in nature in both arms. The CORBEVAX™ to COVISHIELD™ GMT-ratios at day-42 time-point were 1·15 and 1·56 and the lower limit of the 95% confidence interval for the GMT-ratios was determined as 1·02 and 1·27 against Ancestral and Delta strains of SARS-COV-2 respectively. Both COVISHIELD™ and CORBEVAX™ vaccines showed comparable seroconversion post-vaccination against anti-RBD-IgG response. The subjects in CORBEVAX™ cohort also exhibited higher interferon-gamma secreting PBMC’s post-stimulation with SARS-COV-2 RBD-peptides than subjects in COVISHIELD™ cohort.
KEYWORDS: Covid-19, vaccine, receptor binding domain, SARS-Cov-2, spike protein, protein subunit
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections has led to a global COVID-19 pandemic (WHO COVID-19 Situation report − 51) and there has been widespread impact on health, including substantial mortality among older people and those with preexisting health conditions.1 It also severely affected global economy.
Vaccines play an important role in increasing population immunity and preventing severe form of the disease. Global efforts to develop and test vaccines against SARS-CoV2 have resulted in approval of several vaccine candidates with varied efficacy.2 WHO has so far granted emergency use listing to 10 COVID-19 vaccines and 5 others are under assessment. Out of 10 WHO recognized vaccines, only Novavax COVID-19 Vaccine (NVX-CoV2373) is a subunit vaccine and all others are either inactivated virus, nucleic acid-based or viral vector-based vaccines. NVX-CoV2373 is based on of full-length, pre-fusion trimers of spike glycoprotein of prototype Ancestral sequence. Biological E developed a protein subunit vaccine (known as CORBEVAX™) that consists of Receptor Binding Domain (RBD) from the spike protein of SARS-COV-2 Wuhan-Hu1 strain as the antigen that is formulated with aluminum hydroxide (Al3+) and CpG1018 as adjuvants after preclinical evaluation.3–8 The recombinant protein is expressed in yeast and uses a technology similar to the one for producing the recombinant hepatitis B vaccine that has been widely accepted for decades by populations in low- and middle-income countries.9 Therefore, CORBEVAX™ is potentially well suited as a COVID-19 vaccine for global health and to address both vaccine equity and hesitancy.
The optimum dose of the candidate vaccine, CORBEVAX™, was determined in phase 1/2 studies conducted in adults. Final formulation used in the later phases of clinical trials consists of 25 mcg of RBD protein, 750 mcg of Al3+ (in Aluminum Hydroxide) and 750 mcg of CpG1018 per 0·5 mL dose. The optimum formulation showed good safety profile with minimal reactogenicity and high humoral immune response in terms of anti-RBD IgG titers and neutralizing antibody titers against Ancestral, Beta and Delta strains of SARS-COV-2 as well as desired Th1 skew of the cellular immune response.10 In the current phase-3 study using the optimum formulation of CORBEVAX™, we report the safety and immunogenic superiority of CORBEVAX™ vaccine over COVISHIELD™ vaccine.
Methods
Study design and study population
This is an ongoing phase-3 prospective, single-blinded, randomized, active controlled study conducted at 18 sites across India between September 2021 and December 2021. The study was conducted in accordance with the principles defined in the Declaration of Helsinki, International Conference on Harmonisation guidelines (Good Clinical Practices), and the local regulatory guidelines. The Investigational Review Board or Ethics Committee at each study site approved the protocol. All participants provided written informed consent before enrollment into the study. Participants were healthy adults, aged between 18 and 80 years. This study has two groups; one is immunogenicity group to assess the immunogenic superiority of CORBEVAX™ vaccine over COVISHIELD™ vaccine. Other group is safety only to assess the safety of CORBEVAX™ vaccine. Subjects enrolled into immunogenicity group were also assessed for safety. A total of 6485 subjects were screened, of which 2139 subjects were enrolled into the study (639 subjects in immunogenicity group and 1500 subjects in safety group). Subjects in immunogenicity group were further randomized in 1:1 ratio to receive either CORBEVAX™ vaccine (n = 319) or COVISHIELDTM vaccine (n = 320). Randomization is not applicable to single-arm safety group. Safety data until day-56 and immunogenicity data at day-42 compared to day-0 (baseline) are presented in this manuscript.
Participants were seronegative to anti-SARS-CoV-2 IgG antibody prior to randomization into immunogenicity arm, whereas in safety arm subjects, they were randomized irrespective of their serostatus for SARS-CoV-2. Other key eligibility criteria applicable to all participants were: virologically negative to SARS-CoV-2 infection confirmed by RT-PCR test on screening visit (day-1 to -3); seronegative to HIV 1 and 2; HBV and HCV infection on screening visit (day-1 to -3). Health status assessed during the screening period was based on the medical history and clinical laboratory findings, vital signs, and physical examination. All those who were with axillary temperature of more than 38·0°C, part of any other clinical trial, with a history of vaccination with any investigational vaccine against COVID-19 disease, known allergy to vaccine components, or were on immunosuppressants, and immunodeficient conditions were excluded from the study. Complete list of eligibility criteria were provided as supplementary information.
During the conduct of this study, there were no major protocol deviations reported at any of the study sites. Few subjects reported for their visits out of window period, but these deviations were not found to be significant and all deviations were notified to ethics committees of the respective study sites.
Randomisation and masking
Participants enrolled into immunogenicity arm were randomized equally either to receive CORBEVAX™ vaccine or COVISHILED™ vaccine. Randomization occurred after all screening-related activities were completed and prior to the first dose of study vaccine using the interactive web response system (IWRS) platform is a proprietary of JSS medical research. A subject was considered randomized when he/she has met all the eligibility criteria and have received the randomization number from IWRS. A randomization scheme was generated by using a validated system. This is also a single-blind study where study participants randomized into immunogenicity arm are kept blinded of the vaccination group to which they have been assigned, but the investigator and study staff are aware of the assigned group (CORBEVAX™ or COVISHIELD™).
Procedure
Biological E’s CORBEVAX™ vaccine is based on recombinant RBD protein, which is produced in Pichia Pastoris culture as secretory protein consisting of residues 331–549 of the spike protein of SARS-CoV-2 Wuhan-Hu1 (GenBank Accession Number: QHD43416.1).
It was evident from the published literature that RBD of the spike protein can generate excellent and sustainable immune response in terms of neutralizing antibody (nAb)-titers against SARS-COV-2 virus.4–8,10 Moreover, it was well established that SARS-CoV-2 uses RBD of the spike protein for viral entry into the host cell. So, inducing immune responses against RBD protein can potentially neutralize the binding site of the virus with host cell receptor, making it an essential target for vaccine development. In CORBEVAXTM vaccine, the RBD subunit (25 µg) is co-formulated along with aluminum Hydroxide (750 µg) and CpG1018 (750 µg).10 The active comparator used in immunogenicity arm of the study is COVISHIELD™ (ChAdOx1 nCoV- 19) is a COVID-19 vaccine. This vaccine is based on recombinant, replication-deficient chimpanzee adenovirus vector encoding the SARS-CoV-2 Spike (S) glycoprotein, produced in genetically modified human embryonic kidney (HEK) 293 cells (CDSCO.gov). In India, it is manufactured by the Serum Institute of India and approved to active immunization of individuals ≥ 18 years old for the prevention of COVID-19.
A 0·5-mL dose of the candidate COVID-19 vaccine (CORBEVAX™, composition: RBD Antigen [25 µg] + Aluminium Hydroxide [750 µg] + CpG 1018 [750 µg]) or COVISHIELD™ vaccine (5 × 1010 viral particles) was administered via an intramuscular (IM) injection into the deltoid muscle of the non-dominant arm in a two-dose schedule with 28-day interval between doses. No prophylactic medication was prescribed either before or after vaccination. Follow-ups were scheduled at day-42, day-56, day-118 (3 months post second dose) and day-208 (6 months post second dose). Study is ongoing and subjects are under the follow-up period.
Participants were evaluated for the absence of SARS-CoV-2 infection with RUPCR®SARS-CoV-2 RT qPCR diagnostic kit. This kit detects the presence of SARS-CoV-2 RNA (envelope, RNA-dependent RNA polymerase and nucleocapsid genes) from participant’s sample through Real-Time Polymerase chain reaction, performed at Dr.Dangs laboratory, India. Serology test for seronegative status was performed by Chemiluminescent Immunoassay using LIAISON® anti-SARS CoV-2 Human S1/S2 IgG ELISA kit11 supplied by Diasorin spA., at Dr.Dangs laboratory, India. These eligibility tests were performed during screening period (day-3 to day-1).
Outcomes
The primary outcome of the study was demonstration of immunogenic superiority of BE’s CORBEVAX™ vaccine against COVISHIELD™ vaccine in terms of GMTs of anti-SARS-CoV-2 virus neutralizing antibodies at day-42 (14 days after second dose). Secondary outcome was demonstration of immune response against the Delta variant in terms virus neutralizing antibodies (VNA) at day-42, measuring Anti-RBD antibody concentration in terms of GMCs and to descriptively assess the safety, tolerability and reactogenicity of CORBEVAX™ vaccine during the entire study period. The exploratory end-point included cellular immune response assessment in a subset of subjects via ELISPOT method.
Safety assessments
The safety assessments of the study include solicited and unsolicited, non-serious and serious adverse events (AEs), and medically attended AEs (MAAEs) reported in the study from the time of first dose of the vaccine. Participants were observed for 1-h post vaccination to assess reactogenicity. Solicited local and systemic reactions were recorded for 7 consecutive days (day 0–6), captured through subject diary after each vaccine dose. Solicited local AEs were pain, redness, swelling, itchy or warmth at injection site. Solicited systemic AEs were fever, headache, chills, Myalgia, arthralgia, fatigue, nausea, urticaria, rhinorrhea, irritability, hypotonic-hyporesponsive episodes, somnolence, seizure and acute allergic reaction.
Unsolicited local and systemic adverse events (AEs) were recorded during the post-vaccination follow-up period until 28 days after each dose. Serious adverse events (SAEs), medically attended adverse events (MAAEs) and adverse events of special interest (AESIs), if any, were collected during the entire study duration. Local and systemic reactions were scored by severity (mild, moderate, severe and life threatening) and the erythema and swelling or induration by the maximum diameter per day. Relatedness of study vaccine was also assessed for all reported AEs.
Sample size calculation
The study is formally powered to evaluate immunogenic superiority of CORBEVAX™ against COVISHIELD™ vaccine as primary endpoint and also to assess immunogenic noninferiority against COVISHIELD™ vaccine as a secondary endpoint for hypothesis testing between group-1 and group-2. As per the Summary of Product Characteristic (SmPC), COVISHIELD™ is known to offer a mean geometric concentration (GMC) of 22,222.73 (CI: 20360.50, 24255.3) 28 days after the second dose. This study was formally powered for 90% to primarily demonstrate immunogenic superiority with an alpha value set at 2.5% (one-sided). Superiority threshold (ratio) was set at >1.0 for statistical interpretation. Superiority to be inferred if the lower limit of the two-sided 95% CI for the ratio of two means will be above a ratio of 1.0 as primary endpoint evaluation. Accordingly, a sample size of 320 subjects equally randomized to group-1 and group-2 (including 10% dropout allocation, total n = 640) was to ensure a power of not less than 90% to demonstrate primarily immunogenic superiority of CORBEVAX™ against COVISHIELD™ vaccine and secondarily for immunogenic non-inferiority.
Immunogenicity analysis
Sera samples were collected from all the subjects in the immunogenicity cohort at day-0 (pre-vaccination) and at day-42 (14 days after second vaccine dose) time points. Following measurements were conducted to assess humoral and cellular immune response in both CORBEVAX™ and COVISHIELD™ vaccinated subjects.
Humoral immune responses were evaluated by measuring anti-RBD IgG levels and SARS-COV-2 (prototype) neutralizing antibody titers from pre (day-0) and post (day-42) vaccination sera samples.
Cellular immune response (IFN-gamma) was assessed by enzyme-linked immune sorbent spot (ELISPOT) assay using PBMCs isolated from post vaccination samples (day-42).
In brief, anti-RBD IgG concentration was measured by using validated ELISA method, conducted at Dang’s Lab, India. The antibody concentrations were reported in ELISA units/mL for each subject and Geometric Mean Concentrations were calculated for both time-points for both cohorts. Percent seroconversion was also calculated at day-42 time point for both cohorts. Neutralizing antibody titers (nAb titers) were measured against Wild-type SARS-COV-2 strain (Victoria isolate 01/2020) or Delta strain (isolate from India) using micro neutralisation assay (MNA) at the Translational Health Science and Technology Institute (THSTI), India.12 The nAb testing was conducted as per methods described previously.7 Geometric mean titers were calculated at scheduled time-points, and fold rise from the pre-vaccination values was calculated along with GMFR. For serum samples that did not demonstrate a minimum 50% neutralization of the virus at the initial dilution of 10-fold (the lower limit of quantitation (LLOQ) of the assay), titers were assigned as LLOQ/2. For key geometric mean titers (GMT)/Geometric mean concentration (GMC) values, 95% CI were also calculated. Seroconversion was assessed based on increase in anti-RBD IgG concentration. Subjects were considered seroconverted based on anti-RBD IgG concentration ratio of day-42 to day-0 sera samples. For subjects with nAb titers below the LLOQ at day-0, subjects were considered to be seroconverted if the ratio was ≥ 4 and for subjects with nAb titers > LLOQ at day-0, subjects were considered to be seroconverted if the ratio was ≥ 2.
Cellular immune response was assessed by ELISPOT method conducted at THSTI, India. Whole blood samples were collected post two-dose vaccination and PBMCs were isolated and stored frozen. The PBMCs were subsequently stimulated with various stimulants; SARS-COV-2 RBD peptides for specific response, DMSO for nonspecific response and PHA for assay validity criteria. Post-stimulation, the number of PBMCs that secrete cytokine Interferon-gamma were identified and quantified by ELISPOT technique and the Spot Forming Units (SFUs) per million PBMCs were calculated for each subject sample. Additional information on methodology was provided in supplementary section.
Statistical analyses
For the purposes of analysis, recruited subjects were further identified as total vaccinated cohort (TVC) and the according to protocol (ATP) cohort. All the demographic and primary safety analyses have been based on TVC population, defined as subjects who entered into the study and have received at least one single intramuscular dose of study vaccination.
ATP population is defined as population, who have blood samples available for immunogenicity analysis at all protocol specified time points from both CORBEVAX™ and COVISHIELD™ vaccinated cohorts. This has been the primary analysis population for immunogenicity assessment. The geometric mean titers (nAb) were calculated post-vaccination against both Ancestral and Delta strains and then the ratio of the GMT’s for CORBEVAX™ to COVISHIELD™ cohort. Variances for each cohort were calculated from Log10 converted nAb titer values for each subject. Then the lower bound (LB) of the 95% confidence interval (CI) for the ratio of GMT’s were calculated via standard statistical methods. Superiority was concluded, if the lower limit of the one-sided 95% confidence interval (CI) for the ratio of two GMTs) is > 1.0.
All data were summarized descriptively and data listings were based on all subjects enrolled in the study. By default, descriptive statistics for quantitative measurements included the number of subjects (n), mean, standard deviation (SD), minimum, median (IQR) and maximum. Safety data were summarized by System Organ Class and Preferred term. Serious adverse events, related adverse events, adverse events leading to death or withdrawal, solicited adverse events, medically attended adverse events and adverse events of special interest were summarized separately. In addition, adverse events were also summarized by severity. All analyses were conducted using SAS® Version 9·4 or higher. A significant vaccine response rate was defined as an initially seronegative subject at pre-vaccination who showed a rise in antibody concentration ≥ 4-fold post-vaccination
Results
Participants
A total of 6485 subjects were screened and 2140 subjects were randomized in the study (Figure 1). One subject randomized into immunogenicity arm did not receive the vaccination (subject’s choice). So, in total, 2139 subjects were enrolled into either immunogenicity arm (n = 639) or safety arm (n = 1500). Immunogenicity arm has two groups, receiving two doses of CORBEVAX™ vaccine (n = 319) or COVISHIELD™ vaccine (n = 320). Total vaccinated subjects with CORBEVAX™ vaccine including subjects from both immunogenicity and safety arm were n = 1819. All subjects in the study were of Indian origin. Demographic characteristics were comparable between subjects vaccinated with CORBEVAX™ or COVISHIELD™. Median age of CORBEVAX™ vaccinated cohort was 34 (IQR (Q1:Q3), 27·0:43·0) and COVISHIELD™ vaccinated cohort was 32 (IQR (Q1:Q3), 26·0:41·0) in years. Male: female ratio was 1283 (70·5%): 536 (29·5%) and 242 (75·6%): 78 (24·4%) in CORBEVAX™ and COVISHIELD™ vaccinated cohorts, respectively. Other demographic and baseline characteristics of vaccinated (CORBEVAX™ or COVISHIELD™) are present in Table 1.
Figure 1.
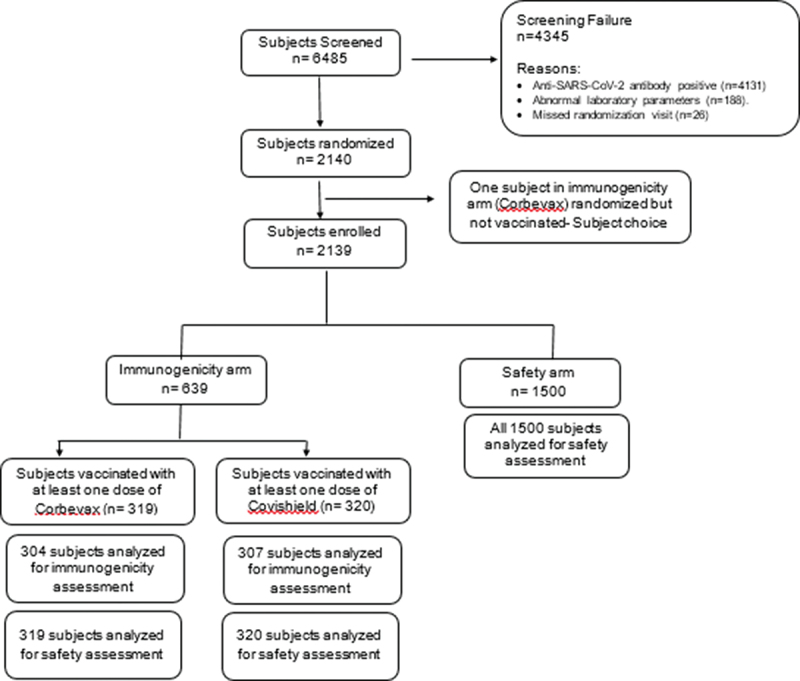
Subject disposition (consort diagram).
A total of 6485 subjects were screened, and 2140 subjects were randomized to either immunogenicity arm or safety arm. Anti-SARS-CoV-2 antibody positive status and abnormal laboratory parameters were the reasons for screen failure of the subjects. One subject randomized in the immunogenicity arm refused to receive the study vaccine. So, in total, 2139 subjects received at least one dose of the study vaccine. In the immunogenicity arm (n = 639), subjects received either CORBEVAX™ vaccine (n = 319) or COVISHIELD™ vaccine (n = 320). All subjects were analysed for safety assessment in this arm. Immunogenicity assessment was performed in n = 304 and n = 307 subjects in CORBEVAX™ or COVISHIELD™ vaccinated groups, respectively. All 1500 subjects in safety arm were included for safety analysis.
n, number; RBD; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Table 1.
Demographic characteristics of study participants (ITT population).
| Parameter | Statistic/Category n (%) |
CORBEVAX™ (N = 1819) |
COVISHIELD™ (N = 320) |
|---|---|---|---|
| Age (years) | |||
| N | 1819 | 320 | |
| Mean | 36·2 | 34·8 | |
| Median | 34·0 | 32·0 | |
| Range (Min: Max) | (18·0:79·0) | (18·0:77·0) | |
| IQR (Q1:Q3) | (27·0:43·0) | (26·0:41·0) | |
| Age Group | |||
| 18–44 | 1427 (78·5%) | 263 (82·2%) | |
| 45–59 | 291 (16·0%) | 38 (11·9%) | |
| 60–80 | 101 (5·6%) | 19 (5·9%) | |
| Nationality | |||
| Indian | 1819 | 320 | |
| Gender | |||
| Male | 1283 (70·5%) | 242 (75·6%) | |
| Female | 536 (29·5%) | 78 (24·4%) | |
| Height (cm) | |||
| N | 1819 | 320 | |
| Mean | 164·0 | 163·3 | |
| Median | 165·0 | 164·0 | |
| Range (Min:Max) | (135·0:190·0) | (136·0:185·0) | |
| IQR (Q1:Q3) | (158·0:170·0) | (158·0:169·0) | |
| Weight (kg) | |||
| N | 1819 | 320 | |
| Mean | 64·6 | 63·6 | |
| Median | 65·3 | 64·5 | |
| Range (Min:Max) | (32·3:103·0) | (36·0:89·9) | |
| IQR (Q1:Q3) | (58·4:70·5) | (55·6:72·0) | |
| BMI | |||
| N | 1819 | 320 | |
| Mean | 24·0 | 23·9 | |
| Median | 23·7 | 23·8 | |
| Range (Min:Max) | (12·5:42·3) | (15·0:40·6) | |
| IQR (Q1:Q3) | (21·8:25·6) | (21·1:25·8) |
Safety findings
Safety data was presented for subjects enrolled into immunogenicity arm (n = 639) including CORBEVAX™ vaccinated cohort (n = 319) and COVISHIELD™ vaccinated cohort (n = 320) and a safety cohort (n = 1500) exclusively enrolled for safety assessment of CORBEVAX™ vaccine.
Safety assessment of immunogenicity group
Out of total 639 enrolled subjects, 68/319 (21·3%) subjects reported 100 events and 136/320 (42·5%) subjects reported 192 events in CORBEVAX™ and COVISHIELD™ arms, respectively. CORBEVAX™ appeared to cause fewer local and systemic adverse reactions/events. The safety profile of CORBEVAX™ was comparable to the comparator vaccine COVISHIELD™ in terms of overall AE rates, related AEs and medically attended AEs. All the reported adverse events were mild to moderate in their intensity, and most of the reported adverse events were related to the study vaccine. Summary of AEs occurred in immunogenicity cohorts by system organ class (SOC) and preferred term (PT), severity grade and causality is listed in Table 2. Summary of local and systemic AEs by SOC and PT in immunogenicity arm and safety arm were reported as Supplementary Table S1 and S2, respectively.
Table 2.
Summary of AEs by SOC and PT – CORBEVAX™ and COVISHIELDTM vaccinated cohort.
| SOC/PT | CORBEVAX™ (N = 1819) |
COVISHIELD™ (N = 320) |
||||
|---|---|---|---|---|---|---|
| N (1%) n | Severity | Causality | N (1%) n | Severity | Causality | |
| OVERALL | 621 (34.1) 1313 | 136 (42·5%) (192) | ||||
| Gastrointestinal disorders | 82 (4.51%) 102 | 3 (0·94%) (4) | ||||
| Nausea | 43 (2·36%) 45 | Mild | Related (30) | 3 (0·94%) (4) | Mild | Related |
| Unrelated (15) | ||||||
| Abdominal pain upper | 21 (1·15%) 22 | Mild | Related (2) | |||
| Unrelated (20) | ||||||
| Vomiting | 18 (0.99%) 18 | Mild | Unrelated | |||
| Diarrhoea | 10 (0.55%) 10 | Mild | Unrelated | |||
| Mouth ulceration | 6 (0.33%) 6 | Mild | Unrelated | |||
| Toothache | 1 (0.05%) 1 | Mild | Unrelated | |||
| General disorders and administrative site conditions | 516 (28.37%) 828 | 107 (33·44%) (137) | ||||
| Asthenia | 5 (0.27%) 5 | Mild | Related (1) | 1 (0·31%) (1) | Mild | Related |
| Unrelated (4) | ||||||
| Chills | 15 (0.82%) 15 | Mild | Related (14) | 5 (1·56%) (5) | Mild | Related |
| Unrelated (1) | ||||||
| Fatigue | 110 (6.05%) 113 | Mild (109) | Related (108) | 8 (2·5%) (8) | Mild | Related |
| Moderate (4) | Unrelated (5) | |||||
| Injection site pain | 300 (16.49%) 319 | Mild | Related | 48 (15%) (50) | Mild | Related |
| Injection site pruritus | 52 (2.86%) 55 | Mild | Related | 6 (1·88%) (6) | Mild | Related |
| Injection site swelling | 24 (1.32%) 24 | Mild | Related | 5 (1·56%) (5) | Mild | Related |
| Injection site warmth | 3 (0.16%) 3 | Mild | Related | |||
| Irritability | 1 (0.05%) 1 | Mild | Related | 1 (0·31%) (1) | Mild | Related |
| Injection site irritation | 1 (0.05%) 1 | Mild | Related | |||
| Pyrexia | 200 (11.00%) 210 | Mild (205) | Related (187) | 50 (15·63%) (52) | Mild (51) | Related (45) |
| Moderate (5) | Unrelated (23) | Moderate (1) | Unrelated (7) | |||
| Injection site erythema | 63 (3.46%) 65 | Mild | Related | 8 (2·5%) (8) | Mild | Related |
| Pain (Generalized body pain) | 10 (0.55%) 10 | - | Related (2) | 1 (0·31%) (1) | Mild | Unrelated |
| Unrelated (8) | ||||||
| Injection site rash | 7 (0.38%) 7 | Mild | Related | |||
| Immune system disorders | 7 (0.38%) 7 | 1 (0·31%) (1) | ||||
| Urticaria | 7 (0.38%) 7 | Mild | Related | |||
| Dermatitis contact | 0 (0·0%) 0 | - | - | 1 (0·31%) (1) | Mild | Unrelated |
| Infections and infestation | 5 (0.27%) 5 | 1 (0·31%) (1) | ||||
| Dengue fever | 1 (0.05%) 1 | Severe | Unrelated | |||
| Nasopharyngitis | 4 (0.22%) 4 | Mild | Unrelated | |||
| Upper respiratory tract infection | 0 (0·0%) 0 | - | - | 1 (0·31%) (1) | Mild | Unrelated |
| Injury, poisoning and procedural complications |
1 (0·3%) (4) | 0 (0·0%) (0) | ||||
| Femur fracture | 1 (0.05%) 1 | Severe | Unrelated | 0 (0·0%) (0) | ||
| Traumatic hematoma | 1 (0.05%) 1 | Moderate | Unrelated | 0 (0·0%) (0) | ||
| Skin abrasion (Road traffic accident) |
1 (0·05%) 2 | Mild | Unrelated | 0 (0·0%) (0) | - | - |
| Musculoskeletal and connective tissue disorders | 166 (9.13%) 170 | 18 (5·63%) (24) | ||||
| Arthralgia | 4 (0.22%) 4 | Mild | Related (2) | 4 (1·25%)(6) | Mild | Related |
| Unrelated (2) | ||||||
| Back pain | 2 (0.11%) 2 | Mild (1) | Related (1) | 1 (0·31%) (1) | Mild | Unrelated |
| Moderate (1) | Unrelated (1) | |||||
| Myalgia | 162 (8.91%) 164 | Mild (159) | Related (160) | 16 (5·0%) (17) | Mild | Related |
| Moderate (5) | Unrelated (4) | |||||
| Nervous system disorders | 135 (7.42%) 143 | 21 (6·56%) (23) | ||||
| Headache | 129 (7.09%) 134 | Mild (133) | Related (104) | 21 (6·56%) (21) | Mild (20) | Related (13) |
| Moderate (1) | Unrelated (30) | Moderate (1) | Unrelated (8) | |||
| Loss of consciousness | 1 (0.05%) 2 | Moderate | Unrelated | 0 (0·0%) (0) | - | - |
| Somnolence | 4 (0.22%) 4 | Mild | Related | 1 (0·31%) (2) | Mild | Related |
| Syncope | 1 (0.05%) 2 | Moderate | Unrelated | 0 (0·0%) (0) | - | - |
| Seizure | 1 (0.05%) 1 | Moderate | Unrelated | |||
| Skin and subcutaneous tissue disorder | 8 (0.44%) 14 | 2 (0·63%) (2) | ||||
| Pruritus | 6 (0.33%) 6 | Mild (4) | Related | 0 (0·0%) (0) | - | |
| Moderate (2) | ||||||
| Rash | 7 (0.38%) 7 | Mild (3) | Related | 2 (0·63%) (2) | Mild (1) | |
| Moderate (4) | Moderate (1) | Related | ||||
| Acne | 1 (0.05%) 1 | Mild | Related | |||
| Respiratory, thoracic and mediastinal disorders | 36 (1.98%) 40 | |||||
| Cough | 24 (1.32%) 24 | Mild | Related (1) | 0 (0·0%) (0) | ||
| Unrelated (23) | - | - | ||||
| Nasal obstruction | 3 (0.16%) 3 | Mild | Related (1) | 0 (0·0%) (0) | - | - |
| Oropharyngeal pain | 6 (0.33%) 6 | Unrelated (2) Unrelated |
||||
| Mild | ||||||
| Rhinorrhoea | 3 (0.16%) 3 | Mild | Related | 0 (0·0%) (0) | - | - |
| Sneezing | 2 (0.11%) 2 | Mild | Related (1) Unrelated (1) |
0 (0·0%) (0) | - | - |
| Throat irritation | 2 (0.11%) 2 | Mild | Unrelated | 0 (0·0%) (0) | - | - |
Percentages were calculated using column header count as denominator.
95% CI was calculated by Clopper-Pearson Method.
N1: Subject Count, N: Sample Size, n: Event Count, NE: Not Estimable.
General Note.
• All AE’s were represented as: Subject count (Percentage of subjects) [95% CI] Event Count.
• Solicited Local and Systemic AEs were recorded during 7 days (day-0–6) after each dose.
• Unsolicited adverse event reported at any time, until 28 days after the each dose.
Safety assessment of safety group
Out of total 1500 enrolled subjects, 553/1500 (36·9%) subjects reported 1213 events. The most commonly reported adverse events were Injection site pain [285 AEs in 267 (17·8%) subjects], Pyrexia [192 AEs in 184 (12·3%) subjects], Myalgia [158 AEs in 156 (10·4%) subjects], Headache [119 AEs in 115 (7·7%) subjects] and Fatigue [112 AEs in 109 (7·3%) subjects]. All the reported adverse events were mild to moderate in their intensity and most of the reported adverse events were related to the study vaccine (Table 2). Two serious AEs were reported in the safety group, which was of grade-3 severity and were diagnosed to be Dengue fever and Femur fracture. Causality of the events Dengue fever and femur fracture with the study vaccine (CORBEVAX™) is considered as not related by Principal Investigator and sponsor. There were no adverse events reported in the first 60 min post vaccination and no deaths were reported in the study.
No marked changes overtime were noted in the vital signs recorded. AEs were observed and physical examination results did not indicate any safety issues of concern. Majority of adverse events are mild to moderate in intensity and no AESI were reported in the study. Summary of AEs occurred in safety cohort by system organ class (SOC) and preferred term (PT), severity grade and causality is listed in Table 2.
Summary of local and systemic AEs by SOC and PT occurred in immunogenicity cohorts and in safety cohort are listed as Supplementary Table S1 and S2, respectively. Most of the systemic events are resolved within 1−2 days. Most cases of fever resolved with antipyretic medications in 1−2 days. For fever occurring beyond 7th day of each dose of vaccination, an RTPCR test for COVID-19 infection was done. None of them were positive for COVID-19 infection.
Immunogenicity findings
Humoral and cellular immune responses were evaluated from immunogenicity arm (n = 639) of the study aimed to test immunogenic superiority of CORBEVAX™ vaccine (n = 319) compared to COVISHIELD™ vaccine (n = 320). Paired anti-RBD IgG concentration data at day-0 and day-42 were available in 304 subjects of CORBEVAX™ cohort and in 307 subjects of COVISHIELD™ cohort. Anti-RBD IgG concentrations (GMCs) increased significantly in both CORBEVAX™ and COVISHIELD™ vaccinated groups after the administration of two doses of vaccine compared to baseline (CORBEVAX™: 1439 EU/ml at day 0 Vs 24,478 EU/ml at day 42; COVISHIELD™: 1503 EU/ml at day 0 Vs 16,203 EU/ml at day 42). However, the total antibody response against the RBD antigen is significantly higher in CORBEVAX™ cohort as compared to COVISHIELD™ cohort (24478 EU/ml vs. 16,203 EU/ml at day 42) (Table 3). Percent Seroconversion (SCR) was also calculated from the ratio of anti-RBD IgG concentration at day-42 time point to day-0 time-point i.e. post vs. pre-vaccination. SCR was 91% in CORBEVAX™ vaccinated cohort and 88% in COVISHIELD™ vaccinated cohort.
Table 3.
Summary of Anti-RBD IgG concentration and nAb titers at day-0 and day-42 time points.
| Summary of Anti-RBD IgG concentration | |||||
|---|---|---|---|---|---|
| Day-0 Testing |
Day-42 testing |
||||
| Vaccine arm | GMC; EU/mL Number of subjects (N) |
95% CI | GMC, EU/mL Number of subjects (N) |
95% CI | Ratio of CORBEVAX™ to COVISHIELD™ |
| CORBEVAX™ | 1439 N = 304 |
1268 –1633 | 24478 N = 304 |
21075 –28431 | 1.51 |
| COVISHIELD™ | 1503 N = 307 |
1316–1716 | 16203 N = 307 |
14428–18196 | |
| Summary of nAb titers against Ancestral and Delta strains | |||||
| CORBEVAX™ -Ancestral | 85 N = 303 |
75–96 | 2123 N = 301 |
1801 –2514 | 1.15 |
| COVISHIELD™ -Ancestral | 75 N = 307 |
65–86 | 1833 N = 304 |
1632 – 2089 | |
| CORBEVAX™ -Delta | ND | ND | 874 N = 301 |
724–1055 | 1.56 |
| COVISHIELD™ -Delta | ND | ND | 562 N = 304 |
482–657 | |
Neutralizing antibody (nAb) titers (GMTs) were assessed against the Ancestral and the Delta strain at baseline and day 42 in both CORBEVAX™ (n = 303) and COVISHIELD™ (n = 307) vaccinated cohorts. GMTs based on MNT50 when assessed against Ancestral strain were 85 (95% CI 75–96) and 75 (95% CI 65–86) at baseline and increased significantly at day 42 to 2123 (95% CI 1801 –2514) and 1833 (95% CI 1632–2089) in CORBEVAX™ and COVISHIELD™ cohorts, respectively. GMTs based on MNT50 when assessed against delta strain were also significantly higher in CORBEVAX™ cohort (874; 95% CI 724–1055) as compared to COVISHIELD™ cohort (562; 95% CI 482–657). The CORBEVAX™ to COVISHIELD™ GMT ratios for day-42 time-point were 1·15 and 1·56, respectively, against Ancestral and Delta strains of SARS-COV-2 respectively. Using standard statistical techniques, the lower limit of the 95% confidence interval was determined as 1·02 and 1·27 for the GMT ratios against Ancestral and Delta strains, respectively. Taken together, at day-42 (14 days after second vaccine dose) neutralizing antibody titers post-CORBEVAX™ vaccination is superior to COVISHIELD™ against both the Ancestral strain and Delta strains (Table 3).
Comparison of cellular responses in terms of ELISPOT data is depicted in Figure 2 in a randomly selected subset of subjects in CORBEVAX™ and COVISHIELD™ cohorts. The CORBEVAX™ cohort had higher Interferon-gamma secreting PBMC’s post stimulation with SARS-COV-2 RBD peptides than COVISHIELD™ cohort in terms of average SFU’s and median SFU’s as summarized in Table 4.
Figure 2.
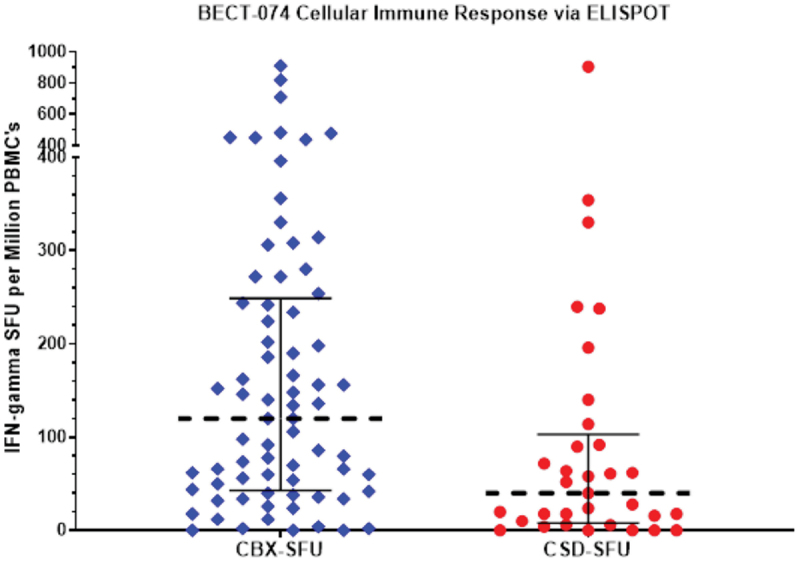
Cellular immune responses measured via ELISPOT.
Cellular immune responses (IFN-gamma) measured by ELISPOT method were assessed in a randomly selected subset of subjects in CORBEVAX™ and COVISHIELD™ vaccinated cohorts. The CORBEVAX™ cohort had higher interferon-gamma secreting PBMCs post stimulation with SARS-COV-2 RBD peptides than COVISHIELD™ cohort in terms of average SFUs and median SFUs.
IFN: Interferon gamma; SFU: Spot forming units; PBMC: Peripheral blood mononuclear cells; CBX- CORBEVAX™ vaccinated cohort; CSD- COVISHIELD™ vaccinated cohort; ELISPOT: Enzyme linked immunosorbent assay.
Table 4.
Summary of interferon-gamma secreting PBMCs as SFUs per million PBMCs in CORBEVAX™ and COVISHIELD™ cohorts.
| Median (IQR) | Average | |
|---|---|---|
| CORBEVAX™ (N = 73) | 120 (43–249) | 176 |
| COVISHIELD™ (N = 33) | 40 (8–103) | 99 |
Discussion
In this trial, immunogenicity of a novel subunit vaccine for COVID-19 vaccine CORBEVAX™ was studied for safety and immunogenicity against RBD domain of SARS-CoV-2. Results indicated that CORBEVAX™ is safe and well tolerated with no vaccine-related serious adverse events, MAAEs or AESI when administered to adult individuals confirming the favorable safety profile of CORBEVAX™ that was observed in phase-1 and -2 trials. High immune responses in terms of anti-RBD IgG specific binding and protective antibodies were observed after second dose of vaccination. Here, we also report immunogenic superiority of CORBEVAX™ over COVISHIELD™ vaccine , an adenoviral vector-based vaccine which is licensed in multiple countries, in terms of higher GMTs of neutralizing antibodies against both the SARS-COV-2 Ancestral strain and the Delta strain.
To establish relative immunogenicity of novel CORBEVAX™, we compared anti-RBD IgG antibody concentrations and neutralizing antibody tiers in individuals receiving CORBEVAX™ or COVISHIELD™. Both COVISHIELD™ and CORBEVAX™ induced marked anti-RBD IgG Abs and neutralizing antibodies against Ancestral and Delta strains. However, neutralizing antibody titers as well as IFN-gamma cellular immune responses induced by CORBEVAX™ vaccination is superior to COVISHIELD™. .
CORBEVAXTM is a RBD-based protein subunit vaccine containing adjuvants (Alum and CpG1018), whereas COVISHIELDTM is a recombinant adenovector expressing spike protein (Supplementary Table S3). Antigenic component of both the vaccines is derived from spike protein of SARS-CoV-2. However, higher immunogenicity of CORBEVAXTM may be due to selection of optimized composition of the vaccine and use of RBD as an antigen.10
In a study by Ambrosino et al., immunogenicity of protein subunit.COVID-19 vaccine that uses same adjuvants as CORBEVAXTM i.e. aluminum hydroxide and CpG1018, SCB-2019 was compared to four other approved vaccines and showed that the comparable or superior immune responses induced by SCB-2019 accurately predicted success of their phase-3 efficacy trial. Authors suggested that immunogenicity comparisons to original strain and variants of concern should be considered as a basis for authorization of new vaccines.13 In these lines, there is at least one regulatory authority approval of COVID-19 vaccine (inactivated SARS-CoV-2 virus adjuvanted with aluminum hydroxide and CpG1018, developer Valneva) solely based on immunogenicity comparisons with an active comparator.14 Immunogenic superiority of CORBEVAX™ over COVISHIELD™ and favorable safety profile further strengthens the real-life scenario of the capability of the vaccine to be used globally.
Neutralizing antibody (nAb) titers were measured against the Ancestral strain which mimics the infectivity of the wild-type SARS-CoV-2. However, as the pandemic has progressed, the Ancestral strain has undergone significant mutations, especially in Spike protein (variants of concern). Throughout the clinical development program of CORBEVAXTM (phase-1 to -3), appropriate variants of SARS-CoV-2 were used in nAb titer assay to determine the cross-neutralization potential post-vaccination. The CORBEVAXTM formulation demonstrated excellent and consistent cross-neutralization potential against beta and delta strains of SARS-CoV-2.10 Omicron and further sub-lineages of Omicron VOC are currently the dominant circulating strains in all geographies, and hence the most recently collected samples in this clinical trial were tested against the Omicron BA.1 VOC at 3-month time point after second dose of the CORBEVAXTM vaccine (unpublished data). Cross-neutralization data from different trials of CORBEVAXTM suggest that CORBEVAXTM vaccination will provide reasonable cross-neutralization of multiple SARS-CoV-2 VOCs. This has been confirmed by lack of severe disease and COVID-19-related hospitalization in any of the >3500 subjects that received CORBEVAXTM primary immunization regimen over >6 months of monitoring period post completion of vaccination.
Study limitations
This study has several limitations like efficacy of the vaccine against COVID-19 infection was not studied and long-term safety is yet to be established as interim results presented in this manuscript are available only until day-56. However, it is worth to mention that in the subjects from phase 1/2 study of CORBEVAX™ (n = 360) safety was established until 12 months and significantly higher neutralizing antibody titers (nAbs) persisted at least 6 months after second dose of the vaccination when compared to human convalescent serum (HCS).10 Last patient recruited in the study was in Dec 2021, and in India, omicron wave started in the mid of Dec 2021. So, study subjects might have not affected by the Omicron wave.
Overall, we conclude that CORBEVAX™ is safe, well tolerated and elicited good antibody and cellular immune responses that can offer significant protection against symptomatic infection from SARS-CoV-2 virus. The overall finding suggests that CORBEVAX™ may offer meaningful protection against symptomatic SARCoV2 infection, but this will need to be confirmed in studies with clinical endpoints.
Supplementary Material
Acknowledgments
We are thankful to all the study participants, the principal investigators, and the study staff at all the clinical sites. All authors wish to express their appreciation and gratitude for all front-line healthcare workers. In addition, we are thankful to the team at Dang’s Lab, New Delhi, led by Dr. Leena Chatterjee, Dr Arjun Dang, Mr Dinesh Kumar and Mr. Shakeeb Mohammad, performed the functions of study sample coordination (receipt, accessioning, aliquoting, labelling, storage, and dispatch) as well as conducted testing of all the ELISA assays (anti-RBD and cytokines) for all the samples. The study was funded by grants from BIRAC − a division of the Department of Biotechnology, Govt of India, and by the Coalition for Epidemic Preparedness Innovations. Dr. Maria Bottazzi and Dr. Peter Hotez, and their scientific team at the Centre for Vaccine Development at Baylor College of Medicine/Texas Children’s Hospital, created and produced the recombinant Pichia Pastoris strain expressing the RBD protein. Dynavax, Inc. supplied the adjuvant CpG1018 used in the CORBEVAX™ formulations. The clinical assay development team led by Dr. Arun Kumar at CEPI helped with neutralizing antibody titre assays in the supply of reagents and establishing assay consistency across multiple laboratories. The authors would like to thank Mr. Srinivas Kosaraju and Mr. Varma Bhupathiraju for the regulatory support and guidance. Authors would also like to thank Mr. Kamal Thammireddy, Mr. Kalyan Kumar P, Mr. Raju Esanakarra and Mr. Naga Ganesh B for their valuable support in the study conduct. Development of this vaccine candidate would not have been possible without the efforts of manufacturing, quality control, quality assurance and regulatory teams from Biological E. The authors would like to thank Scientific Advisory Board (SAB) and the Management of Biological E Limited for their support and valuable guidance. We would also like to thank the members of the DSMB for safety monitoring of the study data.
Funding Statement
BIRAC-division of Department-of-Biotechnology, Government-of-India, and Coalition-for-Epidemic-Preparedness-Innovations funded the study.
Authors contribution
ST and VP conceptualized the study and edited the manuscript for intellectual content. ST, SG, VY, RM and KT curated, accessed and verified the data and helped in interim report generation. VP, MK, SKM, SA, ASJ, GM and NG led the immunogenicity experiments. GM and NG contributed in performing and analyzing neutralizing antibody assays. AB, AZ and AA contributed in performing the ELISpot testing for cellular immune response assessment. CS and VRA were the key contributors of study conduct. ST was responsible for overall supervision of the project. All authors contributed to data interpretation, reviewing, and editing this manuscript.
Disclosure statement
ST, VP, KT, SG, VY, RM, PVS, MK, SKM, SA and ASJ are employees of Biological E Limited and they don’t have any incentives or stock options. All other participating authors declare no competing interests.
Data sharing agreement
Study data presented in the manuscript can be made available upon request and addressed to the corresponding author Dr. Subhash Thuluva at his e-mail: subhash.thuluva@biologicale.com.
Role of the funding source
BIRAC − a division of the Department of Biotechnology, Govt of India provided partial funding for the execution of trials. CEPI provided support for nAb titer testing in terms of reagents. Funding sources were not involved in the study conduct, data analysis/interpretation or writing the manuscript.
Supplemental data
Supplemental data for this article can be accessed on the publisher’s website at https://doi.org/10.1080/21645515.2023.2203632.
Trial registration
CTRI/2021/08/036074.
References
- 1.Sanyaolu A, Okorie C, Marinkovic A, Patidar R, Younis K, Desai P, Hosein Z, Padda I, Mangat J, Altaf M.. Comorbidity and its impact on patients with COVID-19. SN Compr Clin Med. 2020;2(8):1–11. doi: 10.1007/s42399-020-00363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586(7830):516–27. doi: 10.1038/s41586-020-2798-3. [DOI] [PubMed] [Google Scholar]
- 3.Chen W-H, Wei J, Kundu RT, Adhikari R, Liu Z, Lee J, Versteeg L, Poveda C, Keegan B, Villar MJ, et al. Genetic modification to design a stable yeast-expressed recombinant SARS-CoV-2 receptor binding domain as a COVID-19 vaccine candidate. Biochim Biophys Acta Gen Subj. 2021;1865(6):129893. doi: 10.1016/j.bbagen.2021.129893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollet J, Chen W-H, Versteeg L, Keegan B, Zhan B, Wei J, Liu Z, Lee J, Kundu R, Adhikari R, et al. SARS‑CoV-2 RBD219-N1C1: a yeast-expressed SARS-CoV-2 recombinant receptor-binding domain candidate vaccine stimulates virus neutralizing antibodies and T-cell immunity in mice. Human Vacc Immunother. 2021;17(8):2356–66. doi: 10.1080/21645515.2021.1901545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J, Liu Z, Chen W-H, Wei J, Kundu R, Adhikari R, Rivera JA, Gillespie PM, Strych U, Zhan B, et al. Process development and scale-up optimization of the SARS-CoV-2 receptor binding domain–based vaccine candidate, RBD219-N1C1. Appl Microbiol Biotechnol. 2021;105(10):4153–65. doi: 10.1007/s00253-021-11281-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen WH, Tao X, Agrawal AS, Algaissi A, Peng B-H, Pollet J, Strych U, Bottazzi ME, Hotez PJ, Lustigman S, et al. Yeast-expressed SARS-CoV recombinant receptor-binding domain (RBD219-N1) formulated with aluminum hydroxide induces protective immunity and reduces immune enhancement. Vaccine. 2020;38(47):7533–41. doi: 10.1016/j.vaccine.2020.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen WH, Pollet J, Strych U, Lee J, Liu Z, Kundu RT, Versteeg L, Villar MJ, Adhikari R, Wei J, et al. Yeast-expressed recombinant SARS-CoV-2 receptor binding domain RBD203-N1 as a COVID-19 protein vaccine candidate. Protein Expr Purif. 2022;190:106003. doi: 10.1016/j.pep.2021.106003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J, Wang W, Chen Z, Lu S, Yang F, Bi Z, Bao L, Mo F, Li X, Huang Y, et al. A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity. Nature. 2020;586(7830):572–7. doi: 10.1038/s41586-020-2599-8. [DOI] [PubMed] [Google Scholar]
- 9.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307(5947):178–80. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 10.Thuluva S, Paradkar V, Turaga K, Yerroju V, Mogulla R, Turaga K, Kyasani M, Manoharan SK, Medigeshi G, Singh J, et al. Selection of optimum formulation of RBD-based protein subunit COVID-19 vaccine (CORBEVAX™) based on safety and immunogenicity in an open-label, randomised Phase-1 and 2 clinical studies. EBioMedicine. 2022;83:104217. doi: 10.1016/j.ebiom.2022.104217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LIAISON® SARS-CoV-2 S1/S2 IgG . The fully automated serology test for the detection of SARS-CoV-2 IgG Antibodies. [accessed 2022. Mar 17]. https://www.diasorin.com/sites/default/files/allegati/liaisonr_sars-cov-2_s1s2_igg_brochure.pdf.pdf.
- 12.Bewley KR, Coombes NS, Gagnon L, McInroy L, Baker N, Shaik I, St-Jean JR, St-Amant N, Buttigieg KR, Humphries HE, et al. Quantification of SARS-CoV-2 neutralizing antibody by wild-type plaque reduction neutralization, microneutralization and pseudotyped virus neutralization assays. Nat Protoc. 2021;16(6):3114–40. doi: 10.1038/s41596-021-00536-y. [DOI] [PubMed] [Google Scholar]
- 13.Ambrosino D, Han HH, Hu B, Liang J, Clemens R, Johnson M, Siber G, Goldblatt D. Immunogenicity of SCB-2019 coronavirus disease 2019 vaccine compared with 4 approved vaccines. J Infect Dis. 2022;225(2):327–31. doi: 10.1093/infdis/jiab574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valneva reports positive phase 3 results for inactivated, adjuvanted COVID-19 vaccine candidate VLA2001. https://valneva.com/press-release/valneva-reports-positive-phase-3-results-for-inactivated-adjuvanted-covid-19-vaccine-candidate-vla2001/#:~:text=%7C%20DE-,Valneva%20Reports%20Positive%20Phase%203%20Results%20for%20Inactivated,COVID%2D19%20Vaccine%20Candidate%20VLA2001&text=VLA2001%20induced%20broad%20T%2Dcell,S%2C%20M%20and%20N%20proteins.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.