

Summary
Bacteria use a wide range of immune pathways to counter phage infection. A subset of these genes shares homology with components of eukaryotic immune systems, suggesting that eukaryotes horizontally acquired certain innate immune genes from bacteria. Here we show that proteins containing a NACHT module, the central feature of the animal nucleotide-binding domain and leucine-rich repeat-containing gene family (NLRs), are found in bacteria and defend against phages. NACHT proteins are widespread in bacteria, provide immunity against both DNA and RNA phages, and display the characteristic C-terminal sensor, central NACHT, and N-terminal effector modules. Some bacterial NACHT proteins have domain architectures similar to the human NLRs that are critical components of inflammasomes. Human disease-associated NLR mutations that cause stimulus-independent activation of the inflammasome also activate bacterial NACHT proteins, supporting a shared signaling mechanism. This work establishes that NACHT module-containing proteins are ancient mediators of innate immunity across the tree of life.
Keywords: NLR, bacteriophage, NACHT, STAND, innate immunity, phage defense, inflammasome
Graphical Abstract
Introduction
Bacteria are in constant conflict with viruses called bacteriophages (phages) and have evolved elaborate antiphage signaling systems to halt infections. These phage defense systems are typically multi-gene operons encoding proteins that cooperate to sense infection and inhibit virion production through diverse mechanisms1. The best understood antiphage systems are restriction-modification and CRISPR/Cas; however, there are many additional antiphage genes/systems that we are only beginning to understand2–7. Most antiphage systems are a form of innate immunity, meaning they protect against a wide variety of phages and do not require previous exposure, unlike CRISPR/Cas systems which are a form of adaptive immunity. Bacteria typically encode multiple antiphage systems, often on mobile genetic elements, which are shared across the pangenome. This arsenal of antiphage systems creates a “pan-immune system”1, which depends on the ability of antiphage genes to function well in diverse host cells, protect against disparate phages, possess potential addiction modules, and encode most of their essential components in one gene/operon.
The endeavor to catalogue antiphage signaling systems from the bacterial pan-immune system has led to an unexpected finding: some bacterial antiphage proteins are homologous to core components of the human immune system. One clear example are bacterial cyclic-oligonucleotide-based antiphage signaling systems (CBASS6,8–11), which encode proteins homologous to the human cGAS and STING proteins. Other examples are bacterial Viperins12 and bacterial Gasdermins13, which are homologous to human Viperin and Gasdermin D, respectively. These genes are antiviral in both humans and bacteria, and bioinformatic evidence supports that these genes share a common ancestor6,11–13. It appears that the pervasive horizontal gene transfer of antiphage systems between bacteria may have also resulted in metazoans horizontally acquiring antiphage genes from bacteria, which were then adapted to fight viruses in eukaryotic cells. We therefore hypothesized that additional components of the metazoan innate immune system originated from antiphage signaling systems and searched for those genes in bacteria.
Results
A bacterial NACHT protein is antiphage
Antiphage systems frequently cluster together into “defense islands” throughout bacterial genomes14. This phenomenon has been used to identify novel antiphage systems by interrogating genes of unknown function that are co-located with known antiphage systems2,3,7. We investigated genes of unknown function located near CBASS systems and identified a gene encoding a NACHT protein module in Klebsiella pneumoniae MGH 35 (Figure 1A and B, WP_015632533.1). We named this gene bacterial NACHT module-containing protein 1 (bNACHT01), and it did not appear to be in an operon with any other genes. We hypothesized that bNACHT01 was antiphage and tested that hypothesis by expressing bNACHT01 from its endogenous promoter in E. coli, then challenging those bacteria with diverse phages. bNACHT01 conferred over a 100-fold increase in protection against phage T4 and over a 1000-fold increase in protection against phages T5 and T6 (Figure 1C and D, Figure S1C–E). We further confirmed that bNACHT01 expression is not impacted by phage infection and is constitutively expressed (Figure S1B).
Figure 1. A bacterial NACHT domain-containing protein is antiphage.
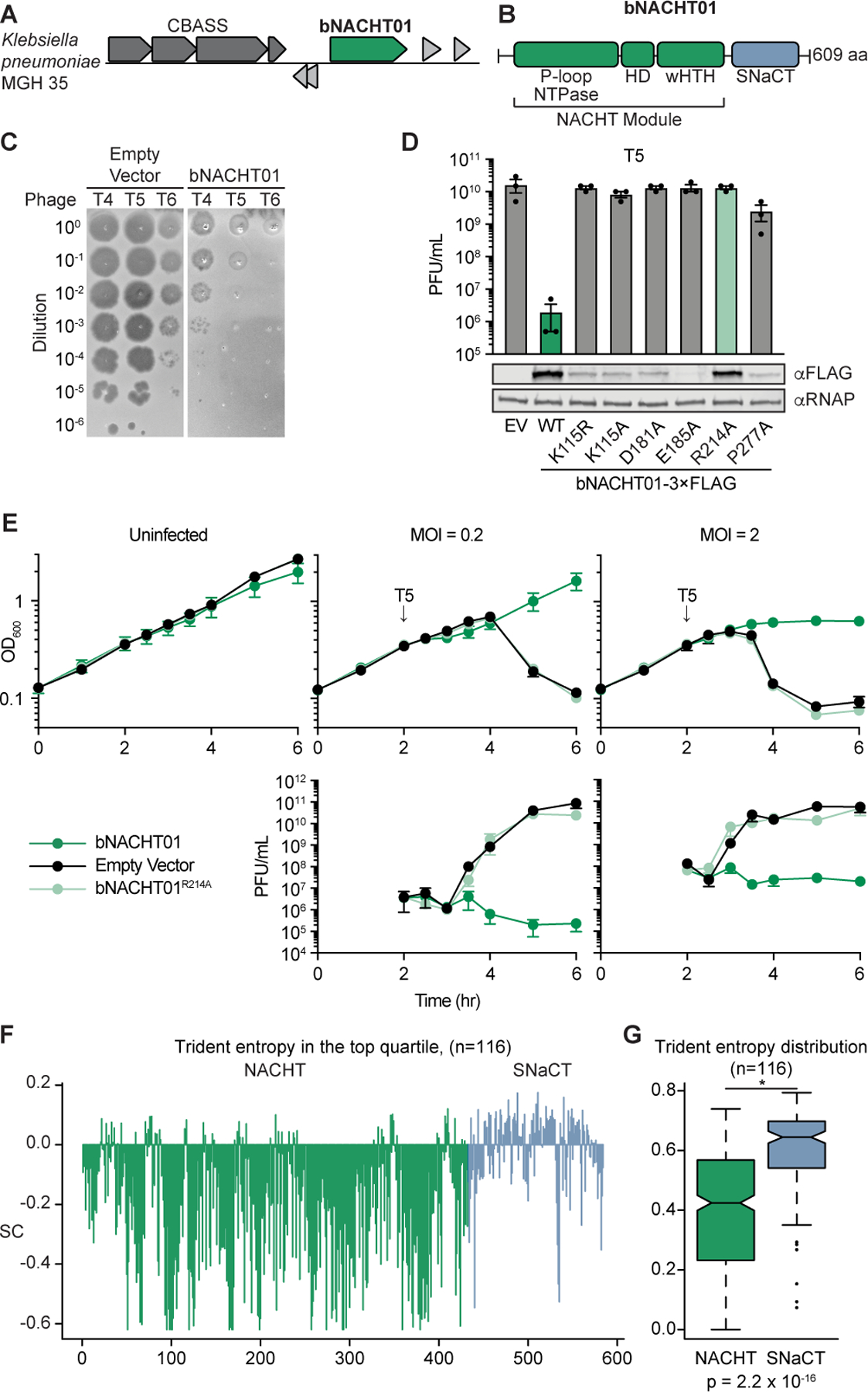
(A) Genome context of bNACHT01, which is located near a CBASS system in Klebsiella pneumoniae MGH 35.
(B) Schematic of bNACHT01 (WP_015632533.1) protein domains, annotated by alignment to the NACHT module of NLRC4. The P-loop NTPase domain is also known as a nucleotide-binding domain (NBD), the helical domain (HD), and the winged helix-turn-helix (wHTH, also called WHD for winged helical domain) are indicated. See Figure S1 for a protein alignment of bNACHT01 with eukaryotic NACHT modules.
(C) Efficiency of plating of indicated phages infecting E. coli expressing bNACHT01 or an empty vector (EV). Data are representative images of n = 3 biological replicates.
(D) Above: Efficiency of plating of phage T5 infecting E. coli expressing the indicated genotype. Data represent the mean ± standard error of the mean (s.e.m.) of n = 3 biological replicates, shown as individual points. See Figure S1 for efficiency of plating of phage T4 and T6. Below: Western blot analysis of E. coli expressing empty vector or FLAG-tagged bNACHT01 of the indicated genotype. Representative image of n = 2 biological replicates.
(E) Above: Growth curve of E. coli expressing the indicated plasmid. Arrows indicate the time each culture was infected with phage T5 at the indicated multiplicity of infection (MOI). Below: Efficiency of plating of the phage present in each sample at the indicated time points. Data represent the mean ± s.e.m. of n = 3 biological replicates.
(F) Scaled Trident entropy (SC) values (see STAR Methods) for individual residues of bNACHT01-like proteins. The trident entropy for each column of the alignment, including both the NACHT module and SNACT domain, is scaled with respect to the top quartile. Positions with values greater than 0 are those with diversity in the top quartile.
(G) Distribution of trident entropy (S) values across NACHT and SNaCT modules in bNACHT01-like proteins showing significantly different mean Trident entropy. Values were compared using a two-sample t-test.
Within three hours post-infection at a multiplicity of infection (MOI) of 0.2, phage replicated and lysed cultures of bacteria expressing an empty vector. However, bacteria expressing bNACHT01 restricted phage virion production and continued growing (Figure 1E). When cultures were infected at an MOI of 2, bacteria expressing bNACHT01 continued to restrict virion production, but the OD600 was stagnant and did not show continued growth or overt bacterial lysis (Figure 1E).
The NACHT module of bNACHT01 shares core features with NACHT modules in eukaryotes, including a Walker A motif that binds NTPs in those proteins15–17 (Figure 1B and Figure S1). Mutation of the conserved lysine residue within the bNACHT01 Walker A motif (K115) to arginine or alanine abrogated phage defense, but also decreased protein expression (Figure 1D). We therefore tested a range of previously published NACHT-inactivating mutations to find an inactivating mutation that did not impact bNACHT01 expression18. Mutation of R214 to alanine maintained expression of the protein but abrogated phage defense (Figure 1D and E and Figure S1C–E). Based on alignments to NOD1/2, this residue is expected to sense the ɣ-phosphate of ATP, indicating that NTP binding may be required for antiviral function (Figure S1A)18.
We next interrogated a multiple sequence alignment of bacterial NACHT proteins defined by bNACHT01 to better understand the mechanism of phage defense. This analysis revealed that the NACHT module is relatively stable in its sequence conservation; however, the region to the C-terminus of the NACHT module appears to be rapidly diversifying (Figure 1F and G). We named this region the Short NACHT-associated C-Terminal domain (SNaCT). Three SNaCT domain models illustrating the observed diversity are provided in Figure S1K. Deletion of the SNaCT domain or mutation to a conserved aspartate at the N-terminus of the domain abrogated bNACHT01 phage defense (Figure S1G–I). We used trident entropy scores to compare the degree of amino acid conservation for the NACHT and SNaCT domains and confirmed that the mean entropy score is higher for SNaCT domain (Figure 1F and G, see STAR Methods for a description of entropy calculation)19. The rapid diversification of the SNaCT domain is a hallmark of a host-pathogen “arms race”, where evolutionary pressure from interactions between an immune sensor and pathogen selects for amino acid substitutions that change protein-protein interactions. In eukaryotic NACHT proteins, the C-terminus is often the “sensor” or “receptor” region of the protein that responds to infection stimuli20. The C-terminal LRR region of a subset of these maintains the protein in an autoinhibited state that is alleviated by stimulant-induced conformational changes. The predicted structure of bNACHT01 (Figure S1J) shows the C-terminal SNaCT domain occludes the NTP-binding region, suggesting that the C-terminus serves a similar function for bNACHT01 as for some animal NACHT proteins. It is therefore paradoxical that bNACHT01 is both capable of detecting a variety of unrelated phages (T4, T6, and T5) and is rapidly diversifying in the sensor region. These data may suggest that the bNACHT01 SNaCT domain is evolutionarily diverging under pressure from constant antagonism by phage-encoded proteins that enable immune evasion (e.g., phage encoded bNACHT inhibitors).
Diversity and ubiquity of NACHT module-containing proteins in bacteria
The NACHT protein module was first discovered in eukaryotes where it is often found in proteins that mediate immunity and inflammation. The best understood metazoan NACHT proteins belong to the nucleotide-binding domain and leucine-rich repeat-containing gene family (NLRs)21, which have a core nucleotide-binding and oligomerization domain (NOD) whose function is fulfilled by a NACHT module. Mammalian NLRs are immune components that play an important role in the formation of inflammasomes (such as NAIP/NLRC4, NLRP1, and NLRP3), transcriptional regulation (CIITA), and other inflammatory responses17. NACHT modules are also found in Fungi where HetE/D proteins can mediate self/non-self discrimination after two hyphal cells have fused their cytosols, so called heterokaryon incompatibility22–24.
NACHT proteins have been rigorously investigated in eukaryotes, but little is known about their roles in bacteria. The NACHT protein module belongs to the large family of STAND NTPases, which describes many divergent proteins. Both active and inactive (with disrupted Walker A/B motifs) STAND NTPase domains were previously identified computationally in predicted antiviral conflict systems that are enriched in multicellular bacteria25,26. STAND NTPases were also observed in the Antiviral ATPase/NTPase of the STAND superfamily (AVAST) antiphage signaling system3,27, as well as in prophage-encoded antiphage systems4, but NACHT domains were not specifically recognized in these studies.
We undertook an exhaustive bioinformatic analysis of NACHT module-containing proteins across publicly available genomes of both eukaryotes and prokaryotes. We started with identifying NACHT modules based on amino acid sequence and predicted structural features. The NACHT protein module belongs to the STAND-Cdc6-Orc family of AAA+ NTPases, which in turn belong to the ASCE division of P-loop NTPases. STAND NTPase modules are unified structurally by a characteristic C-terminal extension to the core AAA+ domain in the form of winged helix-turn-helix (wHTH) domain, also referred to as a Winged Helix Domain (WHD), which they share with Cdc6-Orc AAA+ and transposase ATPases28–30. Within the STAND clade, the NACHT subclade is unambiguously separated from other STANDs based on characteristic motifs, including the D[GAS]hDE signature (a small amino acid directly C-terminal to the Mg2+-coordinating aspartate within the Walker B motif, followed by two acidic residues one position away), as well as signatures in the region N-terminal to the NTPase domain and the above-mentioned C-terminal wHTH extension (Figure S1)28,29.
Our analysis identified approximately 15,000 unique bacterial NACHT proteins (Table S1). They are encoded by about 9–10% of complete, published bacterial genomes (Figure S2 and Table S2). NACHT proteins are found throughout the bacterial superkingdom, including in the genomes of pathogenic bacteria, members of the human gut microbiome, and other important bacteria from environmental niches. Some bacterial phyla show a much higher tendency than average to encode NACHT proteins (Figure S2): we found that 58% of the cyanobacteria, 25% of the actinobacteria and 24% of the deltaproteobacteria encode NACHT proteins (Figure S2D). Cyanobacteria also tend to display a large number of paralogous versions per genome – for instance, a record number of 23 paralogous NACHT proteins are seen in Rivularia sp. PCC 7116 (Figure S2A). Moreover, organisms with 3 or more NACHT proteins have a significant tendency to be multicellular bacteria (Figure S2B, p-value=1.0089e-12). Notably, the multiple copies of the NACHT proteins in these organisms tend to possess distinct effector and sensor domains, suggesting that they are not merely duplications representing iterations of the same theme, but a diversified biochemical repertoire potentially optimized to deal with the unique immune challenges related to multicellularity. This situation mirrors the previously described class of immunity and apoptosis mechanisms shared by a range of multicellular bacteria25,26.
The NACHT modules from a representative subset of these proteins were aligned and related proteins were grouped into clades (Figure 2, Figure S3, and Table S2). By aligning proteins based on the NACHT module, this analysis was independent of fused protein domains on each polypeptide. Our analysis also included NACHT module-containing proteins from Archaea and eukaryotes, which allowed us to group related proteins from different domains of life into a total of 25 major clades and establish evolutionary relationships (Figure 2, Figure S3, and Tables S2 and S3). Phylogenetic analysis suggests that proteins with more closely related NACHT modules tend to have similar C-terminal sensor domains, possibly due to the need for the C-termini to coevolve with the NACHT domain to maintain autoinhibitory function. Conversely, the effector domains to which the NACHT module is fused can vary more dramatically between species of the same genus or bacterial lineage, suggesting that the effector domains may be in an arms race with viral inhibitors that target them.
Figure 2. NACHT module-containing proteins in bacteria are widespread and diverse.
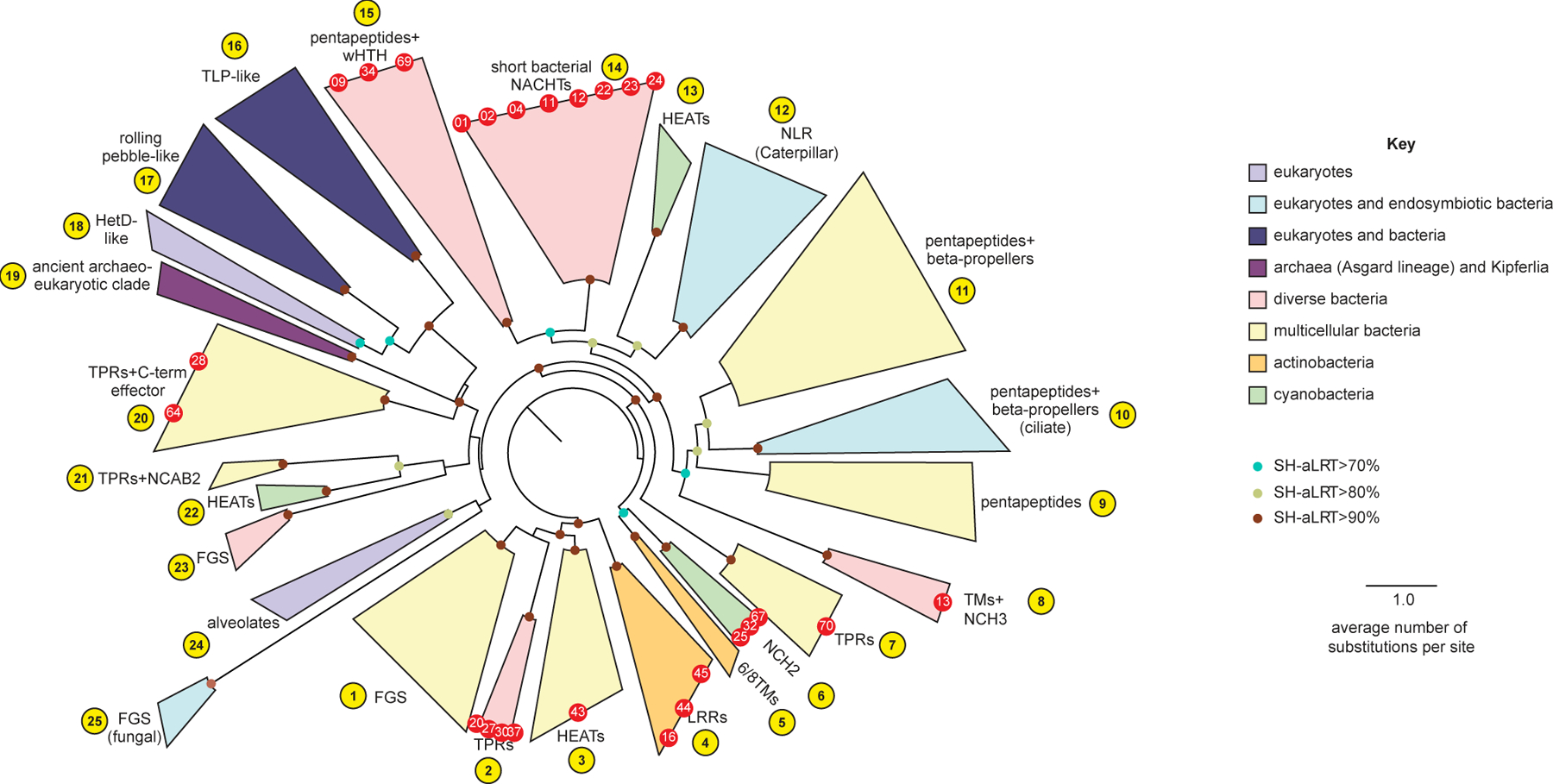
A sequence-based phylogenetic tree of NACHT modules was generated using NACHT module-containing proteins from eukaryotes and prokaryotes. The NACHT module, not accessory domains, were used for tree building. Clades are color-coded based on the indicated key and numbered arbitrarily in yellow circles. Red dots indicate the bacterial NACHT proteins from each clade that were selected for analysis in this study. Bootstrap values are provided where applicable. See Figure S2 for bNACHT gene distribution, Figure S3 for representative domain architectures from each clade, and Table S4 for the most common domain architectures found in each clade. Additional details on genes used to construct the phylogenetic tree can be found in Table S2, and Table S1 contains a full list of all NACHT module-containing proteins identified. See Table S3 for tree topology tests used to validate proposed evolutionary relationships.
The C-terminal regions of bacterial NACHT proteins can be placed into different, broad categories: the antigen receptor or infection signal recognition-type, those that have transmembrane (TM) domains, those with short C-terminal extensions, or those with a combination of these features (Tables S2 and S4). There are two types of antigen receptor-type domains: FGS (Formylglycine-generating enzyme sulfatase) domains and supersecondary structure-forming tandem repeats [e.g., LRR (Leucine Rich Repeat), TPR (Tetratricopeptide Repeat), HEAT (Huntington, Elongation Factor 3, PR65/A, TOR)]. Antigen receptor-type and TM domains are found across many different clades; however, bacterial NACHT proteins with short C-terminal extensions that lack supersecondary structure forming elements, such as bNACHT01, are predominantly found in the monophyletic clade 14 (Figure 2 and Figure S3). These and other characteristics that predominate each NACHT clade are annotated (Figure S3) and a quantification of frequency of NACHT protein architecture is found in Supplementary Table S4.
The N-terminal regions of bacterial NACHT proteins encode many enzymatic domains that have previously been associated with biological conflict, including nucleases (RNases and DNases), peptidases, nucleotide signal-generating or degrading domains, and NAD+-targeting enzymes (TIR and Sirtuin)31–34. Other domains also include predicted “adaptors” that lack a predicted enzymatic function but may mediate interactions with other factors such as Effector Associated Domains, Death-like domains, RNA-binding domains, transcription regulatory domains, and cyclic nucleotide sensors. (Tables S1, S2, and S4).
The tripartite domain architecture we observe in bacterial NACHT proteins is consistent with the domain architectures previously observed in eukaryotic NACHT proteins28,29. The central NACHT in mammalian NLRs is flanked by an N-terminal “effector” domain that coordinates signaling and a C-terminal “sensor” domain that often consists of supersecondary structure-forming tandem repeats, such as LRRs. The similarity of the tripartite domain architecture in bacterial and mammalian NACHT proteins suggest that the sensor and effector domains of canonical eukaryotic NLRs represents a broader organizational strategy across the tree of life28. Therefore, we suggest that these bacterial NACHT proteins are NLR-related, even though many of them lack the leucine-rich repeats required to be classified as true NLRs. These observations further imply that the role of the NACHT module is to act as a signaling hub that transduces detection of an invader signal into diverse biochemical outputs, enabling the host to respond to a threat.
While most of the effector domains on bacterial NACHTs are found at the N-terminus of the protein, before the core NACHT module, some occur at the extreme C-terminus of the protein, after tandem repeat or FGS domains. Published structures of metazoan NLRs suggest that, in these cases, the C-terminal effectors are likely in a similar position as the N-terminal effectors in the folded polypeptide. We predict that the toroidal or helical elements of the C-terminus may allow the effector domain to maintain its normal spatial location, despite being located at the opposite terminus of the protein.
Evolutionary history of NACHT proteins
Phylogenetic relationships between prokaryotic and eukaryotic NACHT proteins suggest these genes have horizontally transferred from prokaryotes to eukaryotes on multiple occasions. The clades in Figure 2 are categorized by the organisms that are most represented in that clade. This includes eukaryotes, a mixture of eukaryotes and bacteria, as well as various groups of prokaryotic organisms. Eukaryotic NACHT modules are found in multiple clades (Figure 2 and Figure S3), suggesting that NACHT modules were acquired on several distinct occasions and subsequently experienced lineage-specific expansions28. Fungi have acquired NACHT proteins from multiple horizontal gene transfer events and one of these resulted in the expansion of the heterokaryon incompatibility NACHT proteins (Clade 18, HetE/D-like). In the mammalian lineage, there are three distinct clades of NACHT modules. The first of these is TEP1, named for the telomerase associated protein 1 (previously named TP-1), found in clade 16, and was acquired early in eukaryotic evolution from bacteria. The second is the Rolling pebbles clade, a sister group of the fungal HetD/E (clade 17, e.g., human neural development protein TANC2). The third, typified by the mammalian NLR/Caterpillar NACHT proteins, (clade 12) represents a separate transfer from bacteria.
Despite these postulated horizontal gene transfer events being ancient, Clade 12 includes extant prokaryotic NACHT proteins encoded by Rickettsiales. The Rickettsiales are an order of obligate intracellular bacteria that have coevolved extensively with animals. Similarly, clade 10 includes NACHTs expanded in fungi that were horizontally acquired from their bacterial endosymbionts. Thus, our phylogenetic analysis suggests that metazoan and fungal hosts acquired their NACHT genes involved in immune mechanisms from obligate intracellular symbionts/pathogens. This likely origin of metazoan and fungal NLRs and NLR-related proteins, from intracellular bacteria, stands in contrast to the potential horizontal transfer of STING from an extracellular bacterial symbiont6,10.
Multiple bacterial NACHT proteins provide a broad spectrum of antiphage immunity
The bNACHT01 protein was potently antiphage (Figure 1 and Figure S1); however, our bioinformatic analysis demonstrated that there are many additional clades of NACHT proteins in bacteria (Figure 2 and Figure S3). To measure the breadth of antiphage activity of NACHT proteins, we expressed 27 representative NACHT module-encoding genes in our E. coli-based phage resistance assay (Figures 3, S5, and S6, and Table S4). Representatives were selected based on protein domain, similarity of domain architecture to eukaryotic NACHT proteins, and phylogenetic distance of the source genome to E. coli (to recapitulate native host cell conditions). Specifically, we focused on proteins primarily from the family Enterobacteriaceae to increase the likelihood that proteins would be functional in our E. coli heterologous system and/or recognize E. coli-specific phages. Bacterial NACHT proteins were expressed from promoters in their native genomic context and were only rarely within poorly conserved operons (Table S5). Bacteria were challenged with a diverse panel of double stranded DNA (T2–T6, λvir), single-stranded DNA (M13), and positive-sense single stranded RNA (MS2, Qβ) phages. We also included a previously characterized CBASS system from Vibrio cholerae and a restriction modification system from E. coli as positive controls in these experiments. Diverse bacterial NACHT proteins from different clades exhibited robust antiphage activity across a wide range of phages (Figures 3, S5, and S6). Intriguingly, some bacterial NACHT proteins defended against both DNA and RNA phages.
Figure 3. Bacterial NACHT proteins are antiphage.
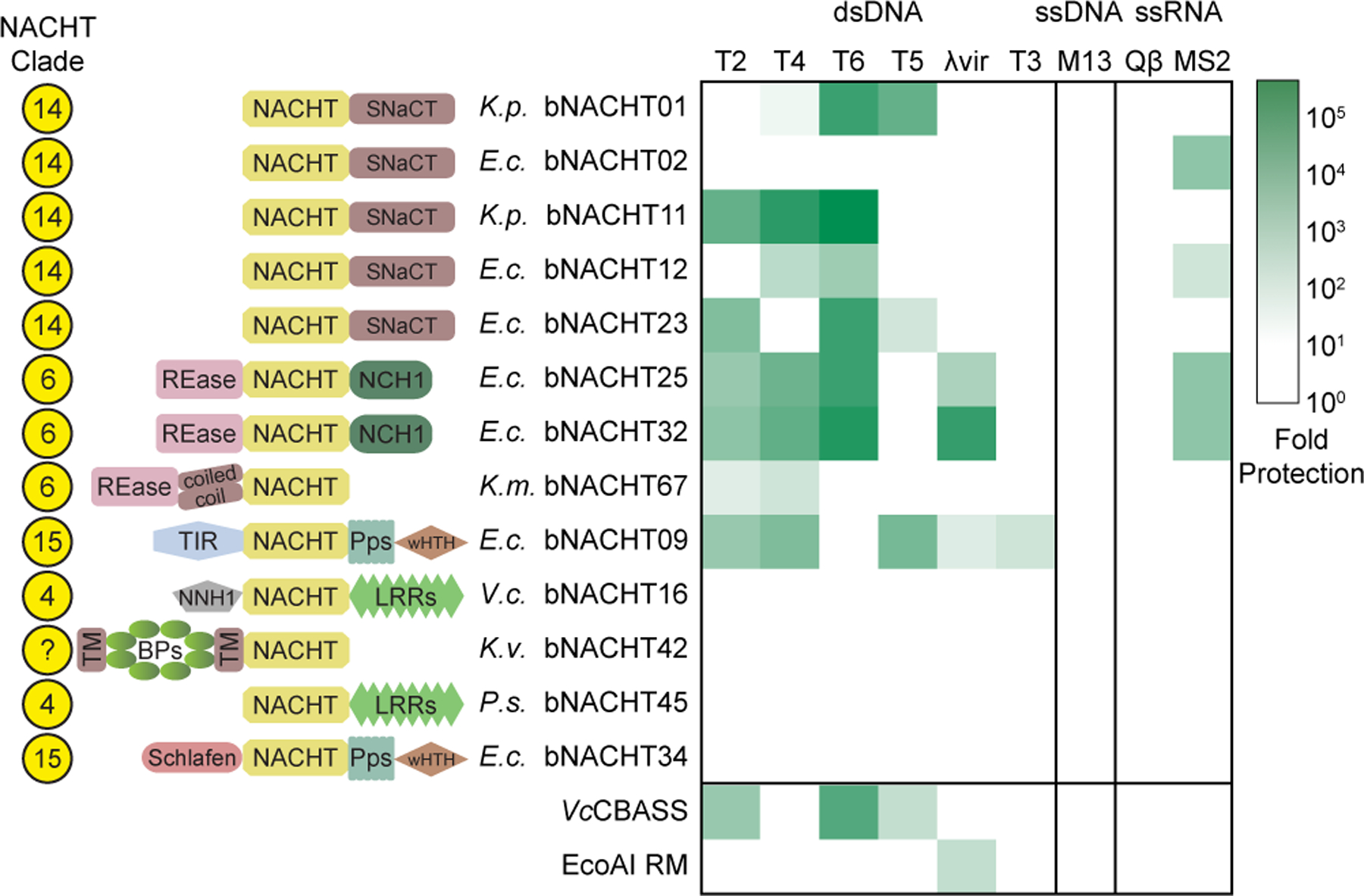
Heat map of fold defense provided by the indicated bNACHT gene for a panel of diverse phages. E. coli expressing the indicated defense system was challenged with phages and fold defense was calculated for each defense system-phage pair by dividing the efficiency of plating (in PFU/mL) on empty vector by efficiency of plating on defense system-expressing bacteria. The NACHT clade, domain architecture, and species of origin for each bNACHT are shown. bNACHT genes displayed in this figure are a subset of the 27 candidates interrogated, selected based on their robust antiphage activity or the diversity of domain architectures sampled. Vibrio cholerae CBASS (VcCBASS) and E. coli UPEC-36 restriction modification system (EcoAI RM) were included as positive controls. Data represent the mean of n = 3 biological replicates. Escherichia coli (E.c.), Klebsiella michiganensis (K.m.), Klebsiella pneumoniae (K.p.), Klebsiella variicola (K.v.), Pseudomonas sp. LAIL14HWK12:I6 (P.s.), Vibrio campbellii (V.c.). Domain abbreviations as described in Figure S3. See Table S5 and Figures S4 and S5 for details on all 27 bNACHT genes analyzed. See Figure S6 for raw efficiency of plating data.
Our interrogation of a wide range of bacterial NACHT proteins demonstrates that bacterial NACHT proteins are related to metazoan NLRs because they share several features: (1) a broad role in antipathogen immune activity, (2) conserved sequence and structural characteristics of their NACHT modules, and (3) a characteristic tripartite protein architecture (C-terminal supersecondary structure-forming tandem repeats, central NACHT, and N-terminal effector). These findings extend the scope of STAND NTPases involved in viral defense beyond the previously identified AVAST systems3,27 to include NACHT proteins.
NACHT proteins are activated in response to phage infection
We next sought to understand how bNACHT proteins restrict virion production and selected bNACHT09 and bNACHT25 for in-depth investigation because their N-terminal effector domains could be readily identified as TIR and REase domains, respectively. We first confirmed that both proteins functioned in single-copy when expressed under their native promoters from the chromosome of E. coli and conferred similar magnitudes of protection to phages T4–6 when compared to low-copy plasmid-based expression (Figure 4A and C, Figure S5B and C). Next, we investigated TIR domain activation upon phage infection for bNACHT09. TIR domains in plant resistosomes and other bacterial antiphage systems degrade NAD+32,33,35 upon activation. We therefore measured NAD(H) in bacteria expressing bNACHT09 and found a dramatic reduction in NAD(H) levels upon phage infection. The decrease in NAD(H) was dependent on phage and the catalytic glutamate of the TIR domain (Figure 4B). We then performed a similar analysis of effector activation for bNACHT25, which encodes a predicted N-terminal PD-(D/E)XK family endonuclease domain. Infection of bNACHT25-expressing bacteria with MS2 phage resulted in rapid destruction of plasmid DNA that was dependent on the predicted catalytic aspartate (Figure 4D). Taken together, these data demonstrate that phage infection results in activation of the N-terminal effector domains of bNACHT proteins.
Figure 4. Bacterial NACHT effector modules are activated by phage.
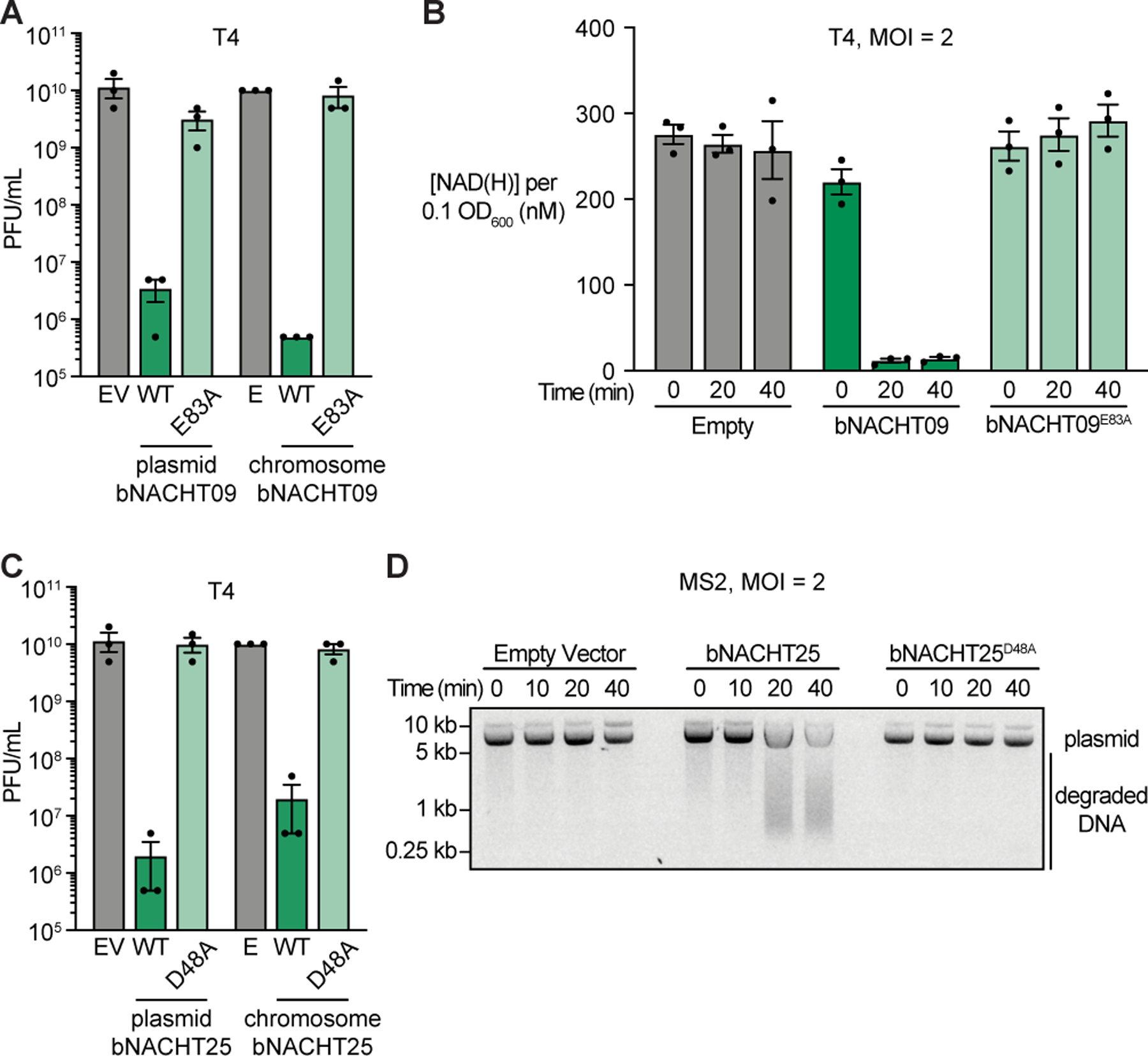
(A) Efficiency of plating of phage T4 infecting E. coli expressing the indicated genotype from a low copy plasmid or from the chromosome. See Figure S5 for efficiency of plating data for phages T5 and T6.
(B) Measurement of [NAD(H)] in each sample when normalized to an OD600 of 0.1 from E. coli expressing the indicated chromosomal genotype at the indicated time points after infection with phage T4.
(C) Efficiency of plating of phage T4 infecting E. coli expressing the indicated genotype from a low copy plasmid or from the chromosome. See Figure S5 for efficiency of plating data for phages T5 and T6. For A–C, Empty (E) indicates E. coli with chromosomal expression of a Kanamycin resistance cassette and gfpmut3. Data represent the mean ± s.e.m. of n = 3 biological replicates, shown as individual points.
(D) Visualization of plasmid integrity in E. coli expressing the indicated plasmid at the indicated time points after infection with MS2. Data are a representative image from n = 3 biological replicates.
Phage proteins alter bacterial NACHT phage defense
We sought to understand how phages alter bacterial NACHT protein signaling by generating phage mutants that evade defense (suppressor mutant phage). Phage T5 was selected for analysis. Wild-type T5 plaque formation is robustly inhibited by bNACHT01 (Figure 1D and E); however, when bacteria were infected with a high number of T5 PFU, suppressor mutants capable of escaping bNACHT01 and forming a plaque were isolated (Figure 5A). These mutants were extremely rare, appearing at an average rate of one suppressor for every 5 × 107 PFU of wild-type phage (Table S6). Accordingly, genome sequencing revealed that every suppressor phage encoded at least two mutations that affected the same ORFs: one mutation that altered orf008 (Genbank: AAX11945.1), an SH3-like fold β-barrel protein, and the other that altered orf015 (Genbank: AAX11952.1, Table S6), a 5-stranded β-meander protein. The majority of the suppressor mutations identified were missense mutations, although some included frameshifts, nonsense, and promoter mutations (Table S6). Genes orf008 and orf015 are encoded in the “first-step transfer” region of the T5 genome, a 10 kb section that is injected first into host bacteria and coordinates injection pausing for approximately five minutes before the remainder of the genome is injected36. During those first minutes of infection, other pre-early genes of the first-step transfer region remodel core processes and shut down signaling within the host cell37. We were unable to identify homologs of orf008 or orf015 outside of the Tequintavirus genus (T5-like phages).
Figure 5. Phage proteins modulate host immune responses.
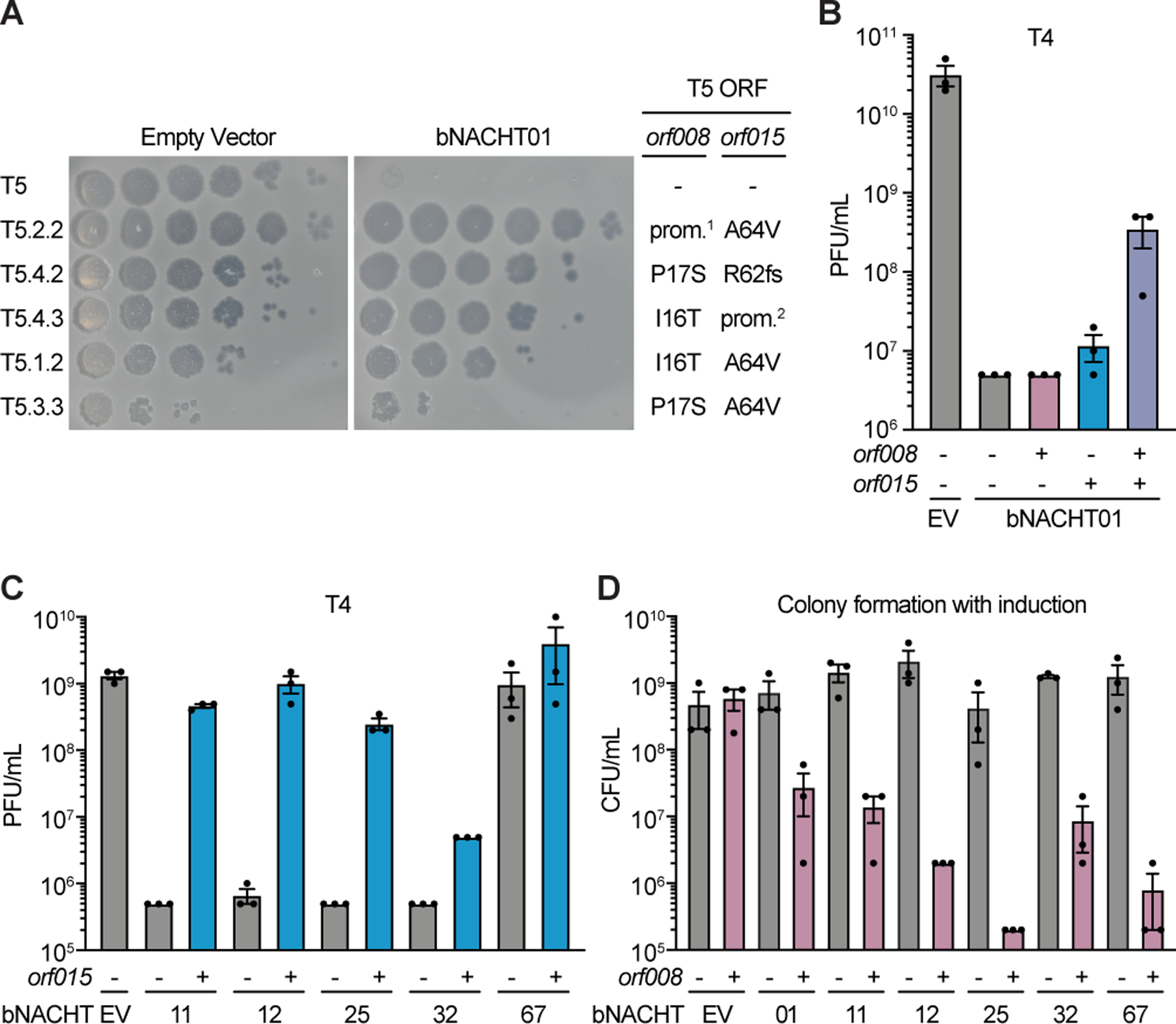
(A) Efficiency of plating of wild-type or suppressor T5 phage when infecting E. coli expressing the indicated plasmid. Impact of orf008 and orf015 suppressor mutations are indicated. Data are representative images of n = 3 biological replicates. Wild-type alleles (−); mutations in the promoter region of orf008 (prom.1); frame shift mutations at the indicated position (fs); and mutations deleting orf009–012 predicted to disrupt the promoter of orf015 (prom.2) are indicated. See Table S6 for rates of suppressor phage isolation, suppressor mutations identified in orf008 and orf015, and a complete list of mutations identified.
(B) Efficiency of plating of phage T4 when infecting E. coli expressing bNACHT01 or an empty vector (EV) on one plasmid and the indicated phage T5 gene(s) on a second plasmid. See Figure S5 for efficiency of plating of phage T6.
(C) Efficiency of plating of phage T4 when infecting E. coli expressing the indicated bNACHT gene on one plasmid and phage T5 orf015 on a second plasmid. See Figure S7 for efficiency of plating of phages T2 and T6.
(D) Quantification of colony formation of E. coli expressing the indicated bNACHT system on one plasmid and orf008 on a second plasmid. See Figure S7 for colony formation in the presence of orf015. For B–D, expression of orf008, orf015, and mCherry is IPTG-inducible. (−) symbols denote induction of an mCherry negative control. (+) symbols denote induction of the indicated phage gene. Data represent the mean ± s.e.m. of n = 3 biological replicates, shown as individual points.
We hypothesized that the low frequency of isolating suppressor phages reflect that T5 must encode mutations in both orf008 and orf015 to evade bNACHT01-mediated protection. To measure the impact of these genes on bNACHT01 antiphage activity, we constructed an assay where bNACHT01 was co-expressed with either orf008, orf015, or both phage genes, then challenged with phages T4 and T6 (Figure 5B and Figure S7A). bNACHT01 provided 1000-fold protection against phage T4 in this assay, and expression of either orf008 or orf015 individually had a modest impact on the efficiency of plating. However, expression of both genes together resulted in a 100-fold recovery of phage T4 virulence (Figure 5B). These data suggest that orf008 and orf015 act together to allow phage to evade bNACHT01.
Relatively few bacterial NACHT proteins protected E. coli against phage T5 (Figure 3 and Figure S5), and we hypothesized that orf008 and orf015 might be broadly responsible for T5 immune evasion. To test this, we selected bNACHT genes that defended against phage T4, but not phage T5, then repeated our assay for measuring the impact of these phage genes. Expression of orf015 significantly decreased the protection by bNACHT11, 12, 25, and 32 against phage T4 (Figure 5C). Similar results could be obtained for a subset of these genes when phage T2 and T6 were used (Figure S7B and C). We next analyzed the effect of orf008 on bNACHT activity. Interestingly, we found that bacterial growth was inhibited when orf008 was co-expressed with bNACHT genes but not when co-expressed with empty vector (Figure 5D). The growth inhibition was specific to orf008; orf015 did not alter bacterial growth when expressed with bNACHT genes (Figure S7D). Because orf008 resulted in growth inhibition, we did not measure its impact on phage defense beyond bNACHT01 (Figure 5B). These data demonstrate that two phage genes alter activity of a wide variety of bNACHT genes and provide evidence for a complicated relationship between phage genes and bNACHT-based host defense systems, where orf008 activates and orf015 inhibits bNACHT proteins.
Human disease mutations activate bacterial NLRs
Human NLR protein activation has potent signaling consequences and rare, monoallelic mutations in patients cause serious diseases that include bare lymphocyte syndrome, Crohn’s/inflammatory bowel disease, and autoimmune conditions16,38. A subset of these diseases are inflammasomopathies, which are point mutations in NLRs that result in stimulus-independent hyperactivation of inflammasome signaling. Patients encoding H443P mutations in NLRC4 display familial cold autoinflammatory syndrome (FCAS)39 and H443L mutations also result in NLRC4 activation in cells15. Histidine 443 is a highly conserved and defining residue located within the wHTH (WHD) domain of the NACHT module (Figure S7E). In NLRC4 H443 is thought to interact with ADP to stabilize an inactive conformation15.
Given the high degree of conservation between human and bacterial NACHT modules, we hypothesized that mutations which hyperactivate human NLRs might also hyperactivate bacterial NLR-related proteins. Structure-guided alignments between NLRC4 and bNACHT25 identified the analogous residue to H443 and mutations at this location were constructed in bNACHT25 (Figure S7). The effector domain of bNACHT25 is a predicted Mrr-like restriction endonuclease (REase). When activated, bNACHT25 cleaves DNA, resulting in toxicity to the host cell and/or destruction of the DNA phage chromosome (Figure 3, 4, S4, and Table S5). For this reason, we expressed wild-type bNACHT25 and mutant alleles using an inducible system. Expression of the histidine mutant, but not wild-type bNACHT25, resulted in potent bacterial growth inhibition (Figure 6A). An additional mutation predicted to disrupt the nuclease activity of the effector domain (D48A) rescued the growth inhibition of hyperactive bNACHT25 (Figure 6A). We next interrogated bNACHT16 because this gene is similar to human NLR proteins, encoding leucine-rich repeats (LRRs) at the C-terminus (Figure 3, Figure S4, Table S5). Introduction of mutations at the H443 equivalent residue of bNACHT16 also resulted in bacterial growth inhibition, consistent with NLR hyperactivation (Figure 6C). Histidine to leucine mutations, shown to synthetically activate NLRC415, or histidine to proline mutations, found in patients with inflammasomopathies39, both inhibited growth equivalently (Figure 6). Our findings for bNACHT16 suggest that even though we did not observe a phage protection phenotype, the protein is expressed and capable of effector activation in E. coli, despite originating in Vibrio campbellii. bNACHT16 may therefore be unable to respond to the phages tested due to a lack of the appropriate stimulus or host components required for phage sensing.
Figure 6. Human disease-associated mutations hyperactivate bacterial NACHT proteins.
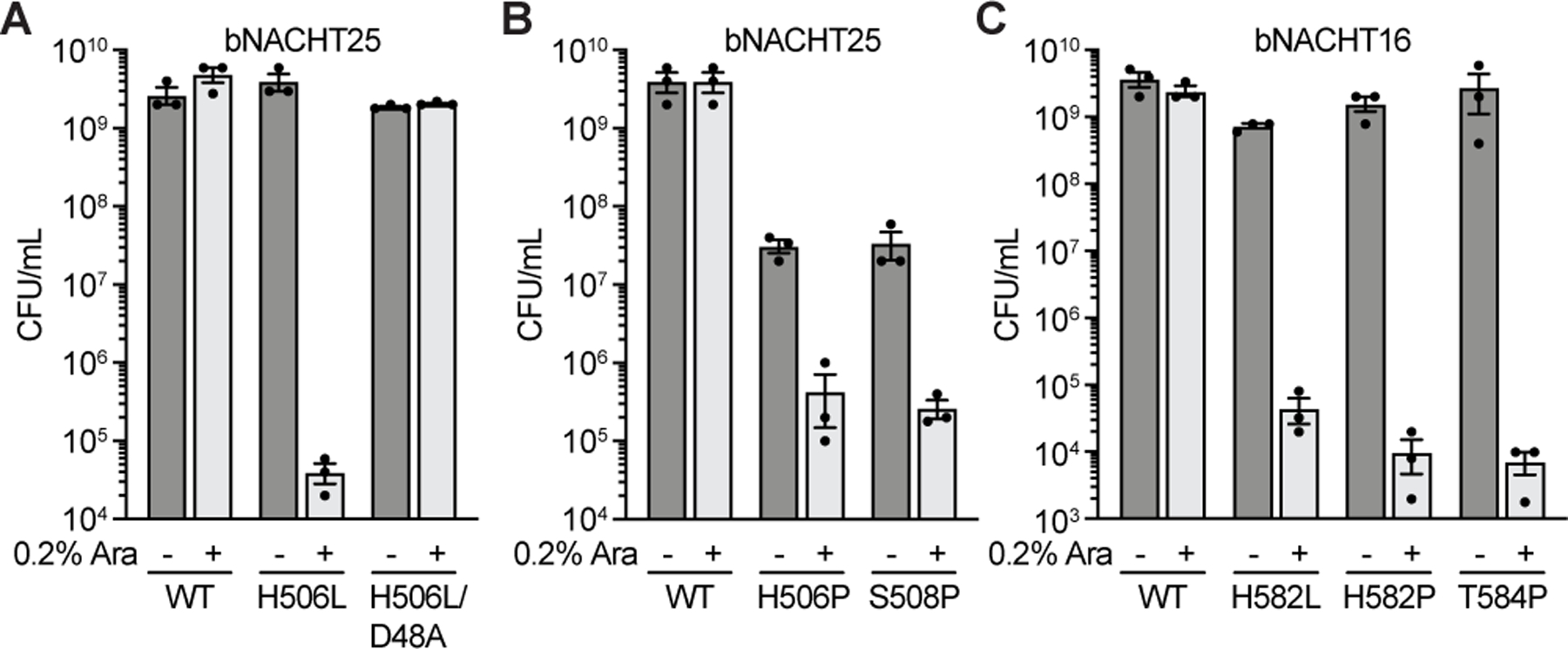
(A)–(B) Quantification of colony formation of E. coli expressing wild-type (WT) bNACHT25 or alleles with the indicated mutations.
(C) Quantification of colony formation of E. coli expressing bNACHT16 with the indicated mutations. See Figure S7 for an alignment of NLRC4, bNACHT16, and bNACHT25. For A–C, gene expression was induced with arabinose. Symbols denote induction (+) or lack of induction (−). Data represent the mean ± s.e.m. of n = 3 biological replicates, shown as individual points.
The disease-associated mutation at S445 also results in hyperactivation of NLRC440,41. To test whether we could recapitulate the effects of mutation to this residue in bacteria, we mutated the corresponding residue in bNACHT25 (S508P) and bNACHT16 (T584P). Overexpression of both the bNACHT25 and the bNACHT16 mutants resulted in inhibition of bacterial growth (Figure 6B and C). These data demonstrate that NACHT modules in humans and bacteria can be hyperactivated by similar mutations, suggesting these proteins have a similar mechanism of effector domain activation.
Our analysis of orf008 and orf015 from phage T5 demonstrated that these proteins alter bNACHT-dependent phage resistance and growth (Figure 5). To further characterize the effect of these two phage genes on bNACHT activity, we co-expressed orf008 and orf015 with a hyperactive allele of bNACHT25, then measured bacterial growth. As predicted from our previous experiments demonstrating orf008-mediated activation of bNACHT proteins (Figure 5), orf008 did not appreciably alter colony formation as hyperactive bNACHT25 already leads to growth inhibition (Figure 6A, Figure 7). However, the orf015 gene was sufficient to rescue the growth inhibition of the bNACHT25 hyperactive allele (Figure 7A). These data are consistent with the impact of orf015 on bNACHT25-mediated resistance during infection (Figure 5C). Similar results were also obtained for bNACHT01 when this protein was hyperactivated by overexpression, a method used to activate other antiphage systems (Figure 7B)42.
Figure 7. Phage proteins alter the activity of hyperactive bacterial NACHT proteins.
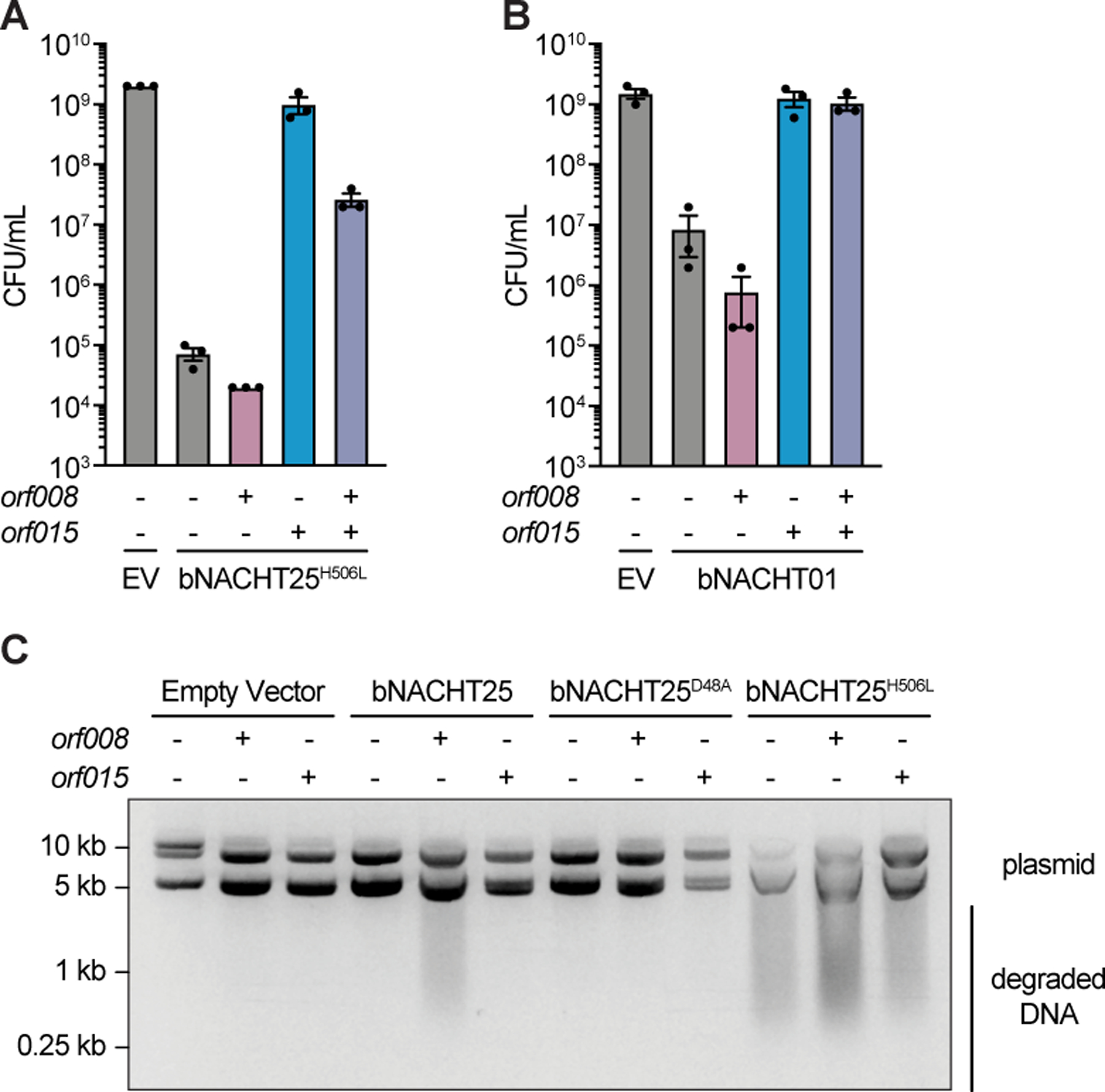
(A)–(B) Quantification of colony formation of E. coli expressing a bNACHT gene with the indicated genotype on one plasmid and the indicated phage T5 gene(s) on a second plasmid. Data represent the mean ± s.e.m. of n = 3 biological replicates, shown as individual points.
(C) Visualization of plasmid integrity in E. coli expressing a bNACHT gene with the indicated genotype on one plasmid and the indicated phage T5 gene on a second plasmid. For A–C, expression of orf008, orf015, and sfGFP was induced with IPTG. Symbols denote induction of an sfGFP negative control gene (−) or induction of the indicated phage gene (+). Expression of the indicated bNACHT gene or empty vector (EV) was arabinose-inducible. Data are an image representative of n = 3 biological replicates.
To analyze the impact of orf008 and orf015 on bNACHT25 effector activation, we returned to our assay for plasmid integrity and measured REase activity of bNACHT25 during co-expression. These experiments showed that induced expression of hyperactive alleles of bNACHT25 result in plasmid destruction, which is inhibited by orf015. Conversely, expression of orf008 with wild-type bNACHT25 results in REase activation (Figure 7C). These data demonstrate that in the absence of phage, orf015 interrupts, and orf008 activates, bNACHT signaling.
Discussion
Here we identify that NACHT module-containing proteins are abundant and widespread in the genomes of bacteria where they are potent phage defense systems. Bacterial and animal NACHT proteins are highly similar in their overall domain architecture, the predicted structure of their NACHT module, and their role in immune signaling. These data establish bacterial NACHT proteins are related to eukaryotic NLRs. In support of a shared molecular mechanism of NACHT module activation, point mutations that hyperactivate NACHT modules in human cells also hyperactive NACHT modules in bacteria15,39,40. Hyperactivated alleles of bacterial NACHT proteins inhibited growth of bacteria. Further, phage infection also appeared to inhibit growth of NACHT protein-expressing bacteria, suggesting that these systems may inhibit phage replication via abortive infection. Abortive infection is a form of programmed cell death that interrupts the viral lifecycle by prematurely destroying a host component essential to virion production43. In this way, the antimicrobial signaling outcome of bacterial NLR-related proteins may also be similar to mammalian inflammasomes, which initiate a caspase-dependent programmed cell death called pyroptosis when activated44. We anticipate that further understanding of the molecular mechanisms of bacterial NACHT protein signaling will provide valuable insights into human NLRs.
Our expansive bioinformatic analysis found that bacteria encode the largest diversity of NACHT module sequences compared to other superkingdoms, which suggests that this protein module first evolved in bacteria before being acquired into the genomes of eukaryotes. However, not all eukaryotic NACHT module sequences are monophyletic and each often clusters with distinct groups of bacterial NACHT proteins, implying that horizontal gene transfer of NACHT modules from prokaryotes to eukaryotes has occurred on multiple occasions. Evidence for one transfer event is found in NACHT module Clade 12, which groups mammalian NLRs (aka Caterpillar genes) with bacterial NACHT proteins from Rickettsiales, an order of intracellular bacteria. This observation suggests that metazoans acquired their NLRs from Rickettsiales. A similar horizontal gene transfer event has been suggested for the innate immune gene STING; however, the most probable bacterial source for that event is the Bacteroidetes6. Both Bacteroidetes (living extracellularly as a symbiont) and Rickettsia (living intracellularly) have intimate interactions with eukaryotes yet distinctly different lifestyles. The shared evolutionary history of NACHT genes may enable future investigators to take advantage of studying bacterium-phage interactions to learn about cryptic aspects of human NLR signaling.
Fungi also encode NACHT proteins that are uniquely suited to their lifestyle. The HET-D and HET-E proteins from the filamentous fungus Podospora anserina are NACHT proteins that mediate kin recognition after two cells have fused their cytoplasms. When kin cells expressing these proteins fuse, the subsequent heterokaryon survives; however, when non-kin cells expressing HET-D or HET-E fuse, the NACHT protein initiates programmed cell death45,46. HET-E/D recognize allelic differences in the HET-C protein to distinguish kin, i.e., self from non-self47. This phenomenon is known as heterokaryon incompatibility. In related systems, heterokaryon incompatibility has been shown to restrict the spread of endogenous viruses between non-kin fungi48,49. Thus, in fungi, as in animals and bacteria, NLR-related proteins are part of the innate immune system.
NLRs within the mammalian inflammasome require additional factors to induce cell death. NLRC4 requires the pore-forming protein Gasdermin D to execute cell death (pyroptosis)44,50. Gasdermin D homologs can also be found in fungi, where they mediate heterokaryon incompatibility, and in bacteria, where they mediate antiphage signaling13,51,52. However, bacterial NLR-related proteins interrogated here do not require Gasdermin D homologs for signaling although in a few bacteria they are encoded in operons also coding for NACHT proteins (Table S2). Heterokaryon incompatibility loci are highly polymorphic across fungi and there are many more than het-d/e (NLRs) and rcd-1 (Gasdermin D)22,53. These observations suggest that heterokaryon incompatibility loci, like bacterial antiphage systems, may be an important repository for identifying mammalian innate immune genes.
Bacterial NACHT proteins are the first example of an innate immune antiphage system in bacteria capable of defending against RNA phages. While adaptive immune systems like CRISPR can be programmed to defeat RNA phages54, this may not represent their natural function. Bacterial NLRs capable of recognizing RNA phages also recognize DNA phages, suggesting that the stimulus recognized is highly conserved between disparate viruses. We do not yet know what the stimulus might be, or if the stimulus is the same for all bacterial NACHT proteins. However, we are able to synthetically activate these proteins using mutations that hyper-activate mammalian NLRs. Many NACHT-associated effector domains are highly conserved and found across multiple known and predicted antiphage systems but remain as yet biochemically uncharacterized. Given that they cannot be readily activated in the absence of a phage (which might be unknown), synthetic activation might prove highly useful to study the large array of effector domains fused to the N-terminus of bacterial NACHTs. Some noteworthy examples include: (i) the Schlafen RNase domain found at the N-terminus of bNACHT34 that is related to human Schlafen proteins involved in HIV1 restriction55; (ii) The PNPase domain that is predicted to degrade nucleotides or NAD+ by removal of the base11; (iii) bacterial domains related to the Death-superfamily domains found in metazoan apoptosis25,26.
Our data support a unifying role for proteins encoding NACHT modules and related STAND NTPases as mediators of innate immunity across the tree of life. NACHT module-encoding NLRs in mammals initiate inflammation and are potently antimicrobial. Fungal NACHT proteins mediate heterokaryon incompatibility, which can stop viral transmission. Here, we demonstrate that bacterial NACHT proteins are antiphage. Land plants also show an expansion of the antibacterial and antiviral R (NB-ARC) proteins that contain another clade of STAND NTPase modules, i.e., the AP-ATPase, which is a sister-group of the NACHT clade. Further, a contemporary analysis of bacterial STAND NTPases outside the NACHT clade by Gao et al. shows that those proteins use a similar tripartite domain architecture to recognize structural motifs of specific phage proteins27. Thus, it appears that the NACHT and related STAND modules define an architectural theme that is especially suited for immune signaling and apoptosis. One potential explanation is that these modules can serve as switches that combine sensing of infection signals (either pathogen or endogenous molecules), activation-threshold setting, signal transduction, and effector deployment, all in a single protein56,57. Understanding the unique qualities of the NACHT module is an exciting area for future investigation.
Limitations of the Study
There are important limitations to our study. First, NLRs are defined as Nucleotide-binding leucine-rich repeat containing proteins21, not by the presence of a NACHT domain. As our analysis focused solely on NACHT domain-containing proteins, we are unable to draw conclusions about NLRs that encode other STAND NTPases such as the AP-ATPase domains found in plants. Second, we have measured phage resistance using a heterologous system in E. coli, which limits our ability to test bNACHT proteins from outside the Enterobacteriaceae family as these are often poorly expressed or unable to recognize E. coli phages. This limits our ability to interrogate the Rickettsiales NACHT proteins found in clade 12. Despite this limitation, we were able to test many of the most common domain architectures found in Enterobacteriaceae (Table S4). We found these genes are antiphage when expressed from the chromosome, but have been unsuccessful in investigating E. coli strains natively expressing NACHT proteins because these strains are refractory to genetic manipulation and encode many redundant antiphage systems that obscure interpretation. Third, we do not understand the effector mechanism of bNACHT01 and other clade 14 proteins that lack N-terminal effector domains. These proteins inhibit growth upon activation; however, without a predicted effector signaling outcome we cannot conclusively show that growth inhibition is cell death, and thus abortive infection. Nevertheless, other bNACHT proteins with catalytic effector domains are likely to limit infection through abortive infection by destroying NAD(H) (TIR domain, bNACHT09) and degrading the genome (REase domain, bNACHT25).
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Correspondence and requests for materials should be addressed to and will be fulfilled by the lead contact, Aaron Whiteley (aaron.whiteley@colorado.edu).
Materials availability
Strains, plasmids, and phages used in this study are available upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions
E. coli strains used in this study are listed in Table S7. E. coli were cultured in LB medium (1% tryptone, 0.5% yeast extract, and 0.5% NaCl) shaking at 37 °C and 220 rpm in 1–3 mL of media in 14 mL culture tubes, unless otherwise indicated. Where applicable, carbenicillin (100 μg/mL), chloramphenicol (20 μg/mL), and tetracycline (15 μg/mL) were added. We defined “overnight” bacterial cultures as 16–20 hours post-inoculation from a glycerol stock or single colony. All strains were frozen for storage in LB plus 30% glycerol at −70 °C. E. coli OmniPir was used for construction and propagation of all plasmids. E. coli MG1655 (CGSC6300) was used to collect all experimental data.
E. coli OmniPir was constructed from OmniMAX 2 T1R E. coli (Thermo Fisher Scientific) and pGRG36pir-116 as previously described58. Briefly, the pir116 gene was integrated at the Tn7 attachment site by conjugating pGRG36pir-116 into OmniMAX E. coli, cultivating bacteria at the permissive temperature with arabinose induction, then curing the plasmid at 42 °C. Integration of pir116 was confirmed by PCR and retention of the F′ plasmid was confirmed by tetracycline resistance. E. coli MG1655 F+ strain was constructed by isolating the F′ plasmid from OmniPir following a previously described protocol59. Briefly, 3 mL of an overnight culture was pelleted and resuspended in 200 μL resuspension buffer (50 mM glucose, 10 mM EDTA, 10 mM Tris-Cl, pH 8.0). 400 μL Buffer P2 (0.2 M NaOH, 1% sodium dodecyl sulfate) was then added and the sample was incubated for 5 minutes at 25 °C. 300 μL of ammonium acetate (7.5 M) and 300 μL chloroform were then added. The sample was incubated at 4 °C for 10 minutes, and pelleted spinning at 21,000 × g for 10 minutes at 25 °C. The supernatant was transferred to a new tube containing 400 μL precipitation solution (30% polyethylene glycol 8000, 1.5 M NaCl) and incubated for 15 minutes at 4 °C. After incubation, DNA was pelleted by centrifugation for 5 minutes at 15,000 × g at 25 °C. The supernatant was discarded, the DNA pellet was resuspended in 100 μL UltraPure water and allowed to dissolve at 4 °C for 2 hours. Purified plasmid was then electroporated into electrocompetent MG1655, followed by selection with tetracycline.
MMCG medium (47.8 mM Na2HPO4, 22 mM KH2PO4, 18.7 mM NH4Cl, 8.6 mM NaCl, 22.2 mM Glucose, 2 mM MgSO4, 100 μM CaCl2, 3 μM Thiamine, Trace Metals at 0.1× (Trace Metals Mixture T1001, Teknova, final concentration: 5mM Ferric chloride, 2mM Calcium chloride, 1mM Manganese chloride, 1mM Zinc Sulfate, 0.2mM Cobalt chloride, 0.2mM Cupric chloride, 0.2mM Nickel chloride, 0.2mM Sodium molybdate, 0.2mM Sodium selenite, 0.2mM Boric acid)) with appropriate antibiotics was used to collect all experimental data. When experiments required bacteria expressing two plasmids, strains were grown using reduced antibiotic concentrations to enhance growth rate (MMCG with 20 μg/mL carbenicillin and 4 μg/mL chloramphenicol).
When growing strains that required induction, 100 μM IPTG or 0.2% arabinose was used to induce, as appropriate.
Phage amplification and storage
The phages used in this study are listed in Table S7. Phages were amplified via either liquid or plate amplification using a modified double agar overlay60. For liquid amplification, 5 mL mid-log cultures of E. coli MG1655 in LB plus 10 mM MgCl2, 10 mM CaCl2, and 100 μM MnCl2 were infected with phage at an MOI of 0.1 and grown, shaking, for 2–16 hours. The supernatant was harvested and filtered through a 0.2 μm spin filter to remove bacterial contamination. For plate amplification, 400 μL of mid-log MG1655 were mixed with 3.5 mL LB soft agar mix (LB with 0.35% agar and 10 mM MgCl2, 10 mM CaCl2 and 100 μM MnCl2) and 100–1,000 PFU. Plates were then incubated for 16 hours at 37 °C. 5 mL of SM buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl pH 7.5, 0.01% gelatin) was added to the plate and allowed to soak out the phages for 1 hour before SM buffer was collected and passed through a 0.2 μm filter or treated with 1–3 drops of chloroform to remove viable bacteria. All phages were stored at 4 °C in SM buffer or LB.
Validation of phages used in this study
All phages were first tested for F plasmid-dependent infection, which confirmed that only M13, MS2, and Qβ required the F plasmid for successful infection of MG1655, as previously reported61,62.
Genomes of dsDNA phage were purified as previously described63. Briefly, 450 μL of phage lysate (>107 PFU/mL) was treated with DNAse I (final concentration 3 × 10−3 U/μL) and RNAse A (final concentration 3 × 10−2 μg/μL) and incubated for 1.5 hours at 37 °C to remove extracellular nucleic acids. EDTA was added (final concentration 20 mM) to stop the reaction. Phage genomes were subsequently isolated and purified using the Qiagen DNeasy cleanup kit, starting at the proteinase K digestion step63. Purified phage genomes were sequenced using 200Mbp Illumina sequencing (SeqCenter). Reads were mapped to the following NCBI Genome accessions using Geneious software’s Map to Reference feature: AP018813.1 (T2), NC_047864.1 (T3), NC_000866.4 (T4), AY587007 (T5), NC_054907.1 (T6), and NC_001416.1 (λvir).
To purify the RNA genomes of MS2 and Qβ, 172.8 μL of phage lysate (>106 PFU/mL) was treated with DNAse I (final concentration 3 × 10−3 U/μL) in DNase I buffer (10mM Tris-HCl pH 7.6, 2.5 mM MgCl2, 0.5 mM CaCl2) and incubated for 1 hour at 37 °C. EDTA was added to a final concentration 20 mM to stop the reaction. RNA was extracted following the PureLink™ RNA Minikit (Invitrogen) protocol for RNA clean-up and purification from liquid samples with omission of the on-column DNase treatment. RNA was eluted in 30 μL nuclease-free water.
Qβ RNA was sequenced directly using RNA sequencing, 12M reads with rRNA depletion and omitted DNase treatment (SeqCenter). Reads were mapped to NCBI Genome accession AB971354.1 using Geneious software’s Map to Reference feature, default settings.
MS2 cDNA was synthesized using the Invitrogen SuperScript III First-Strand Synthesis System. Briefly, 4 μL of phage RNA was combined with dNTP mix (final concentration 1 mM), random hexamers (final concentration 5 ng/μL), a primer that anneals to the 3′ end of the genome (final concentration 0.2 μM, oAC0025: gccaaaacagccaagctttgggtggtaactagccaagcag), and 3 μL of nuclease-free water. The RNA/primer mix was incubated at 65 °C for 5 minutes, and from this step the rest of the protocol was followed as described in the manufacturer instructions. The MS2 genome was amplified in 3 overlapping fragments from the First-Strand cDNA using OneTaq PCR using previously reported primers64. Amplified MS2 genome was prepared for Illumina sequencing using a modification of the Nextera kit protocol as previously described65. Illumina sequencing was performed using a MiSeq V2 Micro 300-cycle kit (CU Anschutz Genomics and Microarray Core). Reads were mapped to NCBI Genome accession NC_001417 using Geneious software’s Map to Reference feature, default settings.
METHOD DETAILS
Plasmid construction
The plasmids used in this study are listed in Table S7. DNA manipulations and cloning were performed as previously described8. Briefly, genes of interest were amplified from phage or bacterial genomic DNA using Q5 Hot Start High Fidelity Master Mix (NEB, M0494L), or synthesized as GeneFragments (Genewiz) flanked by ≥ 18 base pairs of homology to the vector backbone. Ligation of genes into restriction-digested, linearized vectors was accomplished using modified Gibson Assembly66. Gibson reactions were transformed via heat shock or electroporation into competent OmniPir and plated onto appropriate antibiotic selection. Where possible, bNACHT coding sequences and endogenous regulatory regions were amplified from the genomic DNA of E. coli strains from the ECOR collection67. All other bNACHT gene inserts were ordered as GeneFragments (Genewiz). bNACHT point mutations were generated by amplifying out the gene of interest in two parts from a plasmid template, with the desired mutation occurring in the overlapping region between the two amplicons. Inserts for expression of all orf008 and orf015 alleles were amplified from appropriate phage genomic DNA. Unless otherwise indicated, all enzymes were purchased from New England Biolabs.
For all vectors using the pLOCO2 backbone, pAW1382 was amplified and purified from OmniPir. Purified plasmid was then linearized using SbfI-HF and NotI-HF or FseI-HF. Gibson ligation was used to circularize the plasmid with a new insert.
For all vectors using the pTACxc backbone, pAW1608 was amplified and purified from OmniPir. Purified plasmid was then linearized using BamHI-HF and NotI-HF. Gibson ligation was used to circularize the plasmid with the new insert.
For all vectors using the pBAD30x backbone, pAW1367 was amplified and purified from OmniPir. Purified plasmid was then linearized using EcoRI-HF and HindIII-HF. Gibson ligation was used to circularize the plasmid with the new insert.
Sanger sequencing (Genewiz) was used to validate the correct sequence within the multiple cloning site. Additionally, all plasmids expressing bNACHT genes were sequence verified by Illumina sequencing (CU Boulder Sequencing Facility). A NextSeq V2 Mid Output 150-cycle kit was used to sequence the plasmids. Reads were mapped to the predicted plasmid sequence using the Map to Reference feature of Geneious Prime (default settings).
Construction of E. coli expressing bNACHT genes on the chromosome
MG1655 strains expressing bNACHT alleles at the chromosomal lacZ locus were constructed by Lambda red methodology, as previously described68. Sequences were inserted by replacing the lacZ coding sequence, eg. replacing ATG…TAA. Synthesis by overlap extension (SOE) PCR was used to generate dsDNA products that contained in order: homology to 50 bp immediately upstream of the MG1655 lacZ gene, a kanamycin resistance cassette amplified from pKD468, GFPmut3 or the indicated bNACHT allele with its endogenous regulatory regions, and homology to 50bp immediately downstream of the MG1655 lacZ gene. Purified PCR products were transformed into electrocompetent MG1655 expressing pKD46 and Lambda red was induced with 0.2% arabinose for 2 hours at 30 °C. Cultures were then plated on LB plus kanamycin (25 μg/mL) and grown overnight at 37 °C. Resulting colonies were patched onto LB plus kanamycin (50 μg/mL), and LB plus IPTG (500 μM) and X-Gal (40 μg/mL) to screen for integration of the kanamycin resistance cassette and deletion of the lacZ gene, respectively. PCR was used to confirm insertion of the bNACHT genes at the lacZ locus.
Efficiency of plating/phage resistance analysis
A modified double agar overlay was used to measure the efficiency of plating (EOP) of phages60,69. Briefly, overnight cultures of E. coli MG1655 expressing the indicated plasmids cultured in MMCG plus appropriate antibiotics were diluted 1:10 into the same media and cultivated for an additional two hours to reach mid-log phase (OD600 0.1–0.8). 400 μL of the mid-log culture was mixed with 3.5 mL MMCG (0.35% agar), plus an additional 5 mM MgCl2 and 100 μM MnCl2. The mixture was poured onto an MMCG (1.6% agar) plate and cooled for ~15 minutes. 2 μL of a phage dilution series in SM buffer was spotted onto the overlay and allowed to adsorb for 10 minutes before incubating the plate overnight at 37 °C.
Plaque formation was enumerated the following day. For instances with a hazy zone of clearance rather than individual plaque formation, the lowest phage concentration at which clearance was observed was counted as ten plaques. In instances where no clearance or plaque formation was visible, 0.9 plaques at the least dilute spot were used as the limit of detection.
Fold protection was calculated using the inverse of EOP. The PFU of a given phage lysate was measured on sensitive host bacteria, expressing an empty vector, then divided by the PFU for the same phage lysate measure on test bacterial strains. In this way, a 10-fold decrease in EOP is a 10-fold increase phage protection.
bNACHT22 is included in Figure S4 but not selected for inclusion in Figure 3. Although we did observe a decrease in T3 PFU for this system, we did not observe an expected decrease in T3 plaque size, which undermined our confidence in this result.
Time course of phage infection
Overnight cultures of the appropriate strains were inoculated in 30 mL MMCG plus 5 mM MgCl2, and 100 μM MnCl2 to an OD600 of 0.1. Cultures were then cultivated shaking at 37 °C for two hours and infected with phage at the indicated MOI. Culture OD600 was measured at indicated times.
To enumerate PFU, 250 μL of culture was harvested at each time point and centrifuged at 20,000×g for 5 minutes at 4 °C. The supernatant was transferred to a new Eppendorf tube and 20–50 μL of chloroform was added to kill any remaining bacteria. Phage lysates were tittered using the Efficiency of Plating assay described above.
Validation of bNACHT01 expression
For bNACHT01 mutant expression analysis, 5 mL of the indicated strains were grown to mid-logarithmic phase in MMCG and 5×108 CFU were pelleted. For analysis of bNACHT01 expression in response to phage infection, overnight cultures of the appropriate strains were inoculated in 10 mL MMCG plus 5 mM MgCl2 and 100 μM MnCl2 to an OD600 of 0.1. Cultures were then cultivated shaking at 37 °C for two hours and infected with phage at an MOI of 2. One milliliter of sample was collected at indicated time points and pelleted. Bacterial pellets were washed with water and resuspended in 50 μL of 1× LDS buffer (106 mM Tris-HCl pH7.4, 141 mM Tris Base, 2% w/v Lithium dodecyl sulfate, 10% v/v Glycerol, 0.51 mM EDTA, 0.05% Orange G). Samples were then incubated at 95 °C for 10 minutes followed by a 5-minute centrifugation at 20,000×g to remove debris. Samples in LDS were loaded at equal volumes and resolved using SDS-PAGE, then transferred to PVDF membranes charged in methanol. Membranes were blocked in Licor Intercept Buffer for one hour at 24 °C, followed by incubation with primary antibodies diluted in Intercept buffer overnight at 4 °C with rocking. αFLAG antibody (Sigma) was used at 1:10,000 to detect bNACHT01–3×FLAG and αE. coli RNA polymerase B antibody (Biolegend) was used at 1:5,000 as a loading control. Blots were then incubated with Licor infrared (800CW/680RD) αRabbit/Mouse secondary antibodies at 1:30,000 dilution in TBS-T (0.1% Triton-X) for one hour at 24 °C and visualized using the Licor Odyssey CLx. Representative images were assembled using Adobe Illustrator CC 2021.
Identification of bacterial NACHTs
We started with an initial sequence library of known NACHT modules from prior studies26,28,70. Upon identification of additional homologs these were then integrated into the initial library for further large-scale sequence analysis as described below. We iterated this procedure for several rounds, and eventually generated an exhaustive collection of NACHT module homologs. To detect distant relationships, iterative sequence profile searches were conducted using the PSI-BLAST (RRID:SCR_001010)71 and JACKHMMER (RRID:SCR_005305)72 programs with profile-inclusion threshold of expect (e)-value at 0.005 against the non-redundant database of National Center for Biotechnology Information (NCBI) clustered down to 50%. Clustering of proteins based on bit score density and length of aligned sequence was performed using the BLASTCLUST program (ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html). Remote homology searches were performed using profile-profile comparisons with HHpred program (RRID:SCR_010276)73 against profile libraries comprised of the PFAM (RRID: SCR_004726)74 and PDB (RRID:SCR_012820)75 databases as well as an in-house library of profiles of conserved domains. Multiple sequence alignments were built using the Kalign (RRID:SCR_011810)76 and Muscle (RRID:SCR_011812)77 programs followed by manual adjustments based on profile–profile alignment, secondary structure prediction, and structural alignment. Secondary structures were predicted using the JPred (RRID:SCR_016504)78 and RoseTTa Fold79 programs.
Searches for establishing taxonomic counts of NACHT domains from lineages across the tree of life and viruses was performed using a custom database of 14785 completely sequenced genomes (6847 bacteria) using known NACHT domains as queries for PSI-BLAST searches run for 3 iterations with an inclusion threshold of 0.0001. The detected candidates were then run through a confirmation step with the RPS-BLAST program to obtain the final count of NACHT proteins.
Phylogenetic analysis
The input multiple alignment for this analysis contained 437 proteins and 1112 aligned columns, spanning NACHT domains from across the Tree of Life. Phylogenetic analysis was conducted using the maximum likelihood method implemented in the IQtree program (RRID: SCR_017254)80 under multiple parameter regimes using: 1) the Q.pfam substitution matrix derived from alignments in the Pfam database and 1 invariant site category with 8 gamma distributed sites; 2) the LG substitution matrix with 1 invariant site category with 8 gamma distributed sites; 3) with a 20-profile mixture model. Bootstrap values were calculated using the Shimodaira-Hasegawa-like approximate likelihood ratio (SH-aLRT) and the bootstrap proportion-RELL approximation tests81,82 The trees were rendered using the FigTree program (RRID:SCR_008515) (http://tree.bio.ed.ac.uk/software/figtree/). Clades with tree topologies evidencing HGT events (Figure 2, Figure S3) were further tested with a range of tree topology tests. Briefly, this involved construction of the complete set of possible tree topologies within a clade. A subset of these were randomly selected for testing by a range of tree topology tests including approximately unbiased (AU), Kishino-Hasegawa, Shimodaira-Hasegawa, and expected likelihood weights with the IQtree program80,83–85 (see Table S3 for a complete list of tests). Trees passing all tests were visually inspected for adherence to the proposed HGT events, and all screened trees retained the proposed HGT topology (Table S3).
Tests for association with multicellularity
Tests for significance of the bNACHT proteins with bacterial multicellularity used the hypergeometric distribution implemented in the phyper command of the R language as previously described, using the available curated database of multicellularity25. Bacterial multicellularity is defined as reported in the literature (for review, see Lyons and Kolter, 2015)86. These include presence of obligate colonial aggregates; namely, the rosettes of planctomycetes, cooperating bacteroid aggregates with branching structures, aggregating cells forming fruiting bodies like the Myxobacteria in the deltaproteobacteria, filaments with differentiated cells (cyanobacteria) and hyphal filamentous aggregates (actinobacteria).
Domain detection
To establish the domain architectures of the NACHT proteins, they were first searched for previously known domains using the RPS-BLAST program with the Pfam database and a custom database including all of domains detected by the Aravind group and augmentations of the Pfam profiles to improve detection. Unknown regions were then investigated. Profile-profile searches were performed with the HHpred program against libraries of profiles based on non-redundant PDB structures, the Pfam database, and a custom collection of profiles of domains not detected by Pfam. Kalign with default parameters and Mafft with maxiterate= 3000, globalpair, op= 1.9 and ep= 0.5 were used to generate input multiple sequence alignments (MSA), followed by refinements using HHpred profile-profile matches or HMM-align. For specific cases structural modeling was performed using the RoseTTAFold program, which uses a “three-track” neural network, utilizing patterns of sequence conservation, distance inferred from coevolutionary changes in MSAs, and coordinate information79. MSAs of related sequences (>30% similarity) were used to initiate HHpred searches for the initial step of correlated position and contact identification to be used by the neural networks.
Analysis of differential diversity of the NACHT module and the SNaCT domain
The analysis of the Shannon entropy (H) for a given multiple sequence alignment was performed using the equation:
P is the fraction of residues of amino acid type i and M is the number of amino acid types. The Shannon entropy for the ith position in the alignment is ranges from 0 (only one residue at that position) to 4.32 (all 20 residues equally represented at that position).
Trident entropy was used as the metric used to analyze the differential divergence of the NACHT module and the SNaCT domain in clade 14. This measure simultaneously unites three distinct elements (hence trident) of positional variability19 namely: 1) residue diversity; 2) Biochemical diversity among residues; 3) Gapiness of an alignment column. The first t(x) is measured using normalized Shannon entropy (see above); the second r(x) is measured using dissimilarity between two amino acids based on Karlin’s formula using a substitution matrix computed from the alignment; the third g(x) measures the number of gaps in the column. The three united as a product (S=t(x)a.r(x)b.g(x)c, with each factor scaled with an exponent. The respective exponents used here are: a=1, b=½ and c=3. The analysis of the entropy values which were thus derived were performed in the R language.
bNACHT gene selection
bNACHT proteins were selected for screening by considering relatedness of the source genome to E. coli and protein domain diversity. For each gene tested, we included the coding sequence of the bNACHT gene, as well as any other genes in the operon. We also included the endogenous regulatory elements of each system, using bPROM87 to predict bacterial promoters and ARNOLD to predict terminators88. We included at least 100 nucleotides to the 3′ and 5′ region of the gene of interest, to ensure that even unidentified regulatory elements would be included.
DNA degradation measurements
Overnight cultures of the appropriate strains were diluted in 20 mL MMCG plus carbenicillin (100 μg/mL), 5 mM MgCl2, and 100 μM MnCl2 to an OD600 of 0.1. Cultures were then grown shaking at 37 °C to an OD600 of 0.6–0.8 and infected with MS2 at an MOI of 2. 7. 5×109 CFU of each sample were harvested at the indicated time points, pelleted by centrifugation at 4,000×g for 10 minutes at 4 °C, and plasmid DNA extracted using a standard plasmid miniprep protocol (Qiagen). 10 μL DNA sample was combined with 2 μL of 6x DNA loading dye (final concentration 3.3 m M Tris-HCl pH 8, 2.5% Ficoll-400, 10 mM EDTA, 0.05% Orange G) and run for 30 min at 130 V on a 1% agarose gel (1% agarose, 40 mM Tris, 20 mM acetic acid, 1 mM EDTA, SYBR Safe DNA stain). Gels were imaged using an Azure Biosystems Azure 200 Bioanalytical Imaging System.
To measure impact of orf008 and orf015 on DNA degradation, overnight cultures of the appropriate strains were inoculated in 20 mL MMCG plus carbenicillin (50 μg/mL) and chloramphenicol (10 μg/mL) to an OD600 of 0.1. Cultures were then cultivated shaking at 37 °C to an OD600 of 0.6–0.8 before the addition of 0.2% arabinose and 500 μM IPTG to induce expression of bNACHT25 and phage orfs, respectively. Cultures were then harvested after an additional two hours of growth and analyzed as described above.
NAD(H) degradation measurements
Concentrations of NAD(H) were measured using the Promega NAD/NADH-Glo Assay following the manufacturer instructions. Briefly, overnight cultures of the appropriate strain were diluted in 25 mL MMCG to an OD600 of 0.1 and grown for 2 hours at 37 °C. Cultures were then split into 1.5 mL aliquots and infected with phage T4 at an MOI of 2 in 14 mL culture tubes. At the indicated time points, 500 μL culture was harvested by centrifugation at 21,000×g for 5 minutes at 4 °C. Bacterial pellets were resuspended in 50 μL PBS (Corning) and incubated with 50 μL 0.2 M NaOH with 0.1% dodecyltrimethylammonium bromide at 24 °C for 8 minutes to lyse. 100 μL HCl/Tris solution was added to neutralize the sample and incubated at 24 °C for 5 minutes. 50 μL sample was transferred to a white opaque 96-well plate (Pierce) and mixed with 50 μL NAD/NADH-Glo™ Detection Reagent. Luminescence was measured after 30 min incubation at 25 °C using a Tecan Spark multimode microplate reader.
The amount of NAD(H) present in each sample was calculated based on the NAD(H) standard curve for each experiment and normalized to the amount of NAD(H) present in an equivalent volume of sample with an OD600 of 0.1 to allow for accurate comparisons between samples. Each biological sample was analyzed in technical triplicate.
Growth inhibition measurements
The impact of bNACHT expression with and without orf008 and orf015 alleles on bacterial growth was quantified using a colony formation assay. E. coli was cultivated overnight in MMCG with appropriate antibiotics. Cultures were diluted in a 10-fold series into MMCG and 5 μL of each dilution was spotted onto a MMCG agar plate containing the appropriate antibiotics, as well as IPTG and/or arabinose as appropriate. Spotted bacteria were allowed to dry for ~10 minutes before the plates were incubated overnight at 37 °C.
Growth inhibition was measured the following day by enumerating the colony forming units of each strain, reported as CFU/mL for the starting culture. For instances where bacteria were growing but no individual colonies could be counted, the lowest bacterial concentration at which growth was observed was counted as ten CFU. In instances where no growth was visible, 0.9 CFU at the least dilute spot was used as the limit of detection.
Phage suppressor generation and amplification
T5 phages able to evade bNACHT01-mediated protection were generated by mixing 400 μL of mid-log bacteria expressing bNACHT01 in MMCG plus 100 μg/mL carbenicillin with wild-type T5 at an MOI ~10 and pouring the mixture onto a MMCG agar plate. Individual plaques were isolated and spot-plated onto E. coli MG1655 expressing bNACHT01 to confirm that phages were able to replicate in the presence of bNACHT01 and to plaque-purify each clone. Phage bNACHT01 suppressors were generated using three separate wild-type T5 stocks amplified from individual plaque purifications. Phage T5 suppressors were subsequently plate amplified on E. coli MG1655 expressing bNACHT01 in MMCG.
Genome sequencing and analysis of phage suppressors
Suppressor phage genomes were extracted as described above. Extracted phage genomes were prepared for Illumina sequencing using a modification of the Nextera kit protocol as previously described65. Illumina sequencing was performed using a MiSeq V2 Micro 300-cycle kit (CU Boulder Sequencing Facility). Reads were mapped to Genome accession AY587007 (empirically determined to be most similar to the T5 phage used in this study) using Geneious software’s Map to Reference feature. Reads were trimmed to remove the Nextera adapter sequences before mapping (sequence trimmed: AGATGTGTATAAGAGACAG) using the “Trim primers” option, with otherwise default settings. Sequences were mapped using default settings, selecting “map multiple best matches to all locations” to accommodate repetitive T5 sequences.
Geneious was also used for variant detection from the reference T5 genome. Variants that were present in ≥75 percent of reads from the suppressor phage genome but not the parent phage genome were identified as potential suppressor mutations.
Effect of phage genes on bNACHT protection against phage
Bacterial strains were cultured overnight in MMCG plus 20 μg/mL carbenicillin and 4 μg/mL chloramphenicol. Cultures were then diluted 1:10 into the same media with or without 100 μM IPTG and grown for 4 more hours to reach mid-log phase. Phage resistance was measured as described above, with the addition of IPTG to the MMCG top agar (0.35%) to continue inducing conditions.
QUANTIFICATION AND STATISTICAL ANALYSES
All experiments were performed in biological triplicate using cultures grown on three separate days. Data was plotted using Graphpad Prism 9 at an n of 3 with error bars indicating standard error of the mean. Illumina sequencing results were analyzed using Geneious Prime Software. Geneious Prime was also used to generate alignments, using MAFFT alignment89 and default settings. Figures were created using Adobe Illustrator CC. For statistical analysis of trident entropy, see STAR methods section titled “Analysis of differential diversity of the NACHT module and the SNaCT domain”.
Supplementary Material
Figure S1. Protein alignment of the NACHT modules from bNACHT01 and other NACHT proteins, related to Figure 1. (A) Protein alignment of the NACHT modules of NLRC4 (Mus musculus; NP_001028539), bNACHT01 (Klebsiella pneumoniae MGH 35; WP_015632533.1), NAIP (Homo sapiens; NP_001333799.1), CIITA (Homo sapiens; NP_000237.2), NOD1 (Homo sapiens; NP_006083.1), HET-E (Podospora anserina; Q00808), and TEP1 (Homo sapiens; NP_009041.2). The secondary structure of NLRC4 as determined using structure 4KXF in the PDB15, is indicated above with alpha helices depicted as cylinders and beta sheets depicted as arrows. Secondary structure elements are color-coded to represent the NBD, HD, and WHD of NLRC4. Amino acid residues are color-coded based on conservation such that darker colors indicate a higher degree of conservation in this alignment. Black boxes indicate the Walker A and Walker B motifs. Residues mutated and analyzed for expression in Figure 1D are highlighted in red. The D[GAS]hDE motif within the Walker B region that distinguishes NACHT modules from other STAND NTPases is indicated.
(B) Western blot analysis of E. coli expressing empty vector or FLAG-tagged bNACHT01 at 0, 10, and 20 minutes post-infection with phage T5. Representative image of n = 2 biological replicates.
(C)–(E) Efficiency of plating of the indicated phage infecting E. coli expressing the indicated genotype. Data represent the mean ± standard s.e.m. of n = 3 biological replicates, shown as individual points.
(F) Schematic of bNACHT01 (WP_015632533.1) protein domains, annotated by alignment to the NACHT module of NLRC4. The P-loop NTPase domain is also known as a nucleotide-binding domain (NBD), the helical domain (HD), and the winged helix-turn-helix (wHTH, also called WHD for winged helical domain) are indicated.
(G)–(I) Efficiency of plating of the indicated phage infecting E. coli expressing the indicated genotype. Data represent the mean ± standard s.e.m. of n = 3 biological replicates, shown as individual points.
(J) Predicted structure of bNACHT01. Aspartate represents the conserved residue D449.
(K) Predicted structures of 3 different SNaCT domain families. N represents the N-terminus of the domain.
Figure S2. Distribution of NACHT proteins in bacterial taxa, related to Figure 2. (A) Quantification of the number of individual NACHTs found within a single genome across different bacterial taxa. The maximum for the x-axis is 23. Plantomycetota, Verrumicrobiota, Chlamydiota (PVC).
(B) Relative distribution of taxa that have organisms with 3 or more NACHTs in a single genome.
(C) Propensity of organisms within the indicated taxa to encode a NACHT module- containing protein.
(D) Percent of organisms in the indicated taxa to encode at least one NACHT module- containing protein.
(E) Distribution of the number of NACHT proteins per organism, which can be fitted to the equation y = x−2.05. For A–E, a custom database of complete bacterial genomes was used to analyze distribution of NACHT proteins in diverse bacterial taxa.
Figure S3. NACHT module-containing proteins in bacteria are widespread and diverse, related to Figure 2. A sequence-based phylogenetic tree of NACHT modules was generated using NACHT module-containing proteins from eukaryotes and prokaryotes. The NACHT module, not accessory domains, were used for tree building. Clades are color-coded based on the indicated key and numbered arbitrarily in yellow circles. Red dots indicate the bacterial NACHT proteins from each clade that were selected for analysis in this study. Bootstrap values are provided where applicable. The scale bar indicates average number of substitutions per site. See Table S4 for the most common domain architectures found in each clade. The genes used to construct this phylogenetic tree can be found in Table S2 and Table S1 contains a full list of all NACHT domain-containing proteins identified. See Table S3 for tree topology tests used to validated proposed evolutionary relationships. Representative domain architectures for each clade are provided, including NCBI Protein accession number, species of origin, and gene name in green where appropriate. 3′,5′ cyclic nucleotide-generating cyclase (cNMP), Ankyrin repeats (ANKs), Bacterial Death-like domain-3 (bDLD3), Beta-propeller repeats (BPs), Bacterial transglutaminase-like cysteine protease (BTCLP), Carbon-nitrogen hydrolase (CN-hydrolase), CoA-dependent acyltransferase (CoA-acyltrans), Deoxyribohydrolase (DrHyd), Effector associated constant component (EACC), Effector-associated domain (EAD), Formylglycine-generating enzyme sulfatase (FGS), FGS C-terminal domain (FGS-C), Forkhead-associated domain (FHA), Fish-specific NACHT-associated domain (FISNA), Glycosyltransferase 4 (GT4), Huntington, Elongation Factor 3, PR65/A, TOR (HEAT), Higher eukaryotes and prokaryotes nucleotide-binding domain (HEPN), Homing endonuclease (HNH), Helix-turn-helix (HTH), Leucine-rich repeat (LRR), Membrane occupation and recognition nexus (MORN), , NACHT N-terminal helical domain (NNH), NACHT C-terminal helical domain (NCH), NACHT C-terminal α / β domain (NCAB), NACHT C-terminal cysteine and histidine-containing domain (NCC-H), Protein kinase domain (Pkinase), Polyribonucleotide nucleotidyl transferase (PNPase), Pentapeptide repeat (Pp), Restriction endonuclease (REase), Receiver domain (Rec), Rhodanese domain (RHOD), Sirtuin (SIR2), Second messenger oligonucleotide or dinucleotide synthetase domain (SMODS), Telomerase associated protein 1 (TEP1), Toll/interleukin receptor (TIR), Transmembrane (TM) number indicates how many copies of this motif, Tetratricopeptide repeat (TPR), N-terminal domain of tumor necrosis factor receptor type 1 associated death domain protein (TRADD-N), UvRC and Intron-encoded endonuclease domain (URI), Zinc ribbon (ZnR). Red slash indicates catalytically inactive domain.
Figure S4. Domain architectures and details of experimentally tested NACHT proteins, related to Figure 3. Diagram of the N-terminal domains, central NACHT modules, and C-terminal domains of each experimentally tested bNACHT protein. The clade, species of origin, and length of each protein in amino acids (aa) are also indicated. Leucine-rich repeat (LRR), Huntington, Elongation Factor 3, PR65/A, TOR (HEAT), Restriction endonuclease (REase), Toll/interleukin receptor (TIR), Transmembrane (TM), NACHT N-terminal helical domain 1 (NNH1), NACHT C-terminal helical domain 1 (NCH1).
Figure S5. Expanded screen for bacterial NACHT protein phage defense and comparison of defense conferred by plasmid- or chromosomally-expressed bacterial NACHTs, related to Figure 3. (A) Heat map of fold defense provided by the indicated bNACHT gene for a panel of diverse phages. Data are as described in Figure 3 and show all experimentally interrogated bNACHT genes. Data were not collected for bacterial NACHT proteins that exhibited insufficient growth under these experimental conditions, indicated as “X”. Data represent the mean of n = 3 biological replicates. See Table S5 and Figure S4 for details on all 27 bNACHT genes analyzed. See Figure S6 for raw efficiency of plating data.
(B)–(C) Efficiency of plating of the indicated phage infecting E. coli expressing the indicated genotype. Empty (E) indicates E. coli with chromosomal expression of a Kanamycin resistance cassette and gfpmut3. Data represent the mean ± standard s.e.m. of n = 3 biological replicates, indicated as individual points.
Figure S6. Efficiency of plating of phages infecting bacteria expressing NACHT proteins, related to Figure 3. Efficiency of plating of a phage panel infecting E. coli expressing the indicated bNACHT gene. The negative control is empty vector, which expresses an inactive gfp gene. Positive controls are V. cholerae CBASS (VcCBASS) and E. coli UPEC-36 (EcoAI RM) restriction modification system (Z and AA). Data represent the mean ± s.e.m. of n = 3 biological replicates, indicated as individual points.
Figure S7. Effect of orf008 and orf015 expression on bNACHT01 activity and comparison of bNACHT modules to the NACHT domain of NLRC4, related to Figures 5 and 6. (A) Efficiency of plating of phage T6 when infecting E. coli expressing bNACHT01 or empty vector (EV) on one plasmid and the indicated phage T5 gene(s) on a second plasmid.
(B) Efficiency of plating of phage T2 when infecting E. coli expressing the indicated bNACHT gene on one plasmid and phage T5 orf015 on a second plasmid.
(C) Efficiency of plating of phage T6 when infecting E. coli expressing the indicated bNACHT gene on one plasmid and phage T5 orf015 on a second plasmid.
(D) Quantification of colony formation of E. coli expressing the indicated bNACHT system on one plasmid and phage T5 orf015 on a second plasmid. For A–D, expression of orf008, orf015, or sfGFP is IPTG-inducible. (−) symbols denote induction of an sfGFP negative control. (+) symbols denote induction of orf015. Data represent the mean ± s.e.m. of n = 3 biological replicates, shown as individual points.
(E) Protein alignment of the NACHT modules of NLRC4 (Mus musculus; NP_001028539), bNACHT16 (Vibrio campbellii CAIM 519; WP_005534681.1), and bNACHT25 (E. coli ECOR11; WP_001702659.1). The secondary structure of NLRC4 as determined by structure 4KXF in the PDB15, is indicated above with alpha helices depicted as cylinders and beta sheets depicted as arrows. Secondary structure elements are color coded and labeled as in Figure S1. Amino acid residues are color-coded based on conservation in the sequence alignment. Black boxes indicate Walker A and B motifs. The conserved histidine and serine/threonine in the WHD is highlighted in red.
Table S1. All bioinformatically identified NACHT proteins, related to Figure 2.
Table S2. (a) Diverse NACHT proteins used in phylogenetic analysis and (b) comparison of the prevalence of different defense systems, related to Figure 2.
Table S3. Tests for alternative tree topologies, related to Figure 2.
Table S4. Common domain architectures found in each NACHT clade, related to Figure 2.
Table S5. Experimentally investigated bacterial NACHT proteins, related to Figure 3.
Table S6. (a) Rate of suppressor isolation for T5 infecting bNACHT01, (b) suppressor mutations in orf008 and orf015, and (c) all T5 suppressor mutations, related to Figure 5.
Table S7. (a) E. coli strains and plasmids, and (b) bacteriophages used in this study, related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-FLAG | Sigma-Aldrich | Cat#F7425; RRID: AB_439687 |
| Mouse monoclonal anti-E. coli RNA polymerase B | Biolegend | Cat#663006; RRID: AB_2565555 |
| IRDye® Goat 680RD anti-Mouse | Li-Cor | Cat#926–68070; RRID: AB_10956588 |
| IRDye® Goat 800CW anti-Rabbit | Li-Cor | Cat#926–32211; RRID: AB_621843 |
| Bacterial and virus strains | ||
| See Table S7 for a complete list of bacterial strains | ||
| See Table S7 for a complete list of virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| NAD (β-Nicotinamide Adenine Dinucleotide) | Gold Biotechnology | Cat#N-030–1 |
| Carbenicillin | Gold Biotechnology | Cat#C-103–50 |
| Chloramphenicol | Gold Biotechnology | Cat#C-105–25 |
| Tetracycline Hydrochloride | Gold Biotechnology | Cat#T-101–25 |
| Kanamycin Monosulfate | Gold Biotechnology | Cat#K-120–10 |
| IPTG (Isopropyl-beta-D-thiogalactoside) | Gold Biotechnology | Cat#I2481C |
| X-Gal (5-Bromo-4-chloro-3-indolyl β-D-galactopyranoside) | Gold Biotechnology | Cat#X4281C |
| PBS (Phosphate Buffered Saline) | Corning | Cat#21–040-CMX12 |
| DNase I (RNase-free) | New England BioLabs | Cat#M0303S |
| RNase A (Bovine ribonuclease A from pancreas) | VWR Chemicals | Cat#E866–5ML |
| EDTA (Ethylenediaminetetraacetic acid) Disodium, dihydrate | Gold Biotechnology | Cat#E-210 |
| Critical commercial assays | ||
| NAD/NADH-Glo Assay | Promega | Cat#G9071 |
| DNeasy Cleanup Kit | Qiagen | Cat#69506 |
| PureLink RNA Minikit | Invitrogen | Cat#12183018A |
| SuperScript III First-Strand Synthesis System | Invitrogen | Cat#18080051 |
| Tagment DNA Enzyme and Buffer Small Kit | Illumina | Cat#20034197 |
| NEBNext® Multiplex Oligos for Illumina® | New England BioLabs | Cat#E7335S |
| NEBNext® dsDNA Fragmentase | New England BioLabs | Cat#M0348S |
| Deposited data | ||
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| oAC0025: gccaaaacagccaagctttgggtggtaactagccaagcag | This Study | |
| Recombinant DNA | ||
| See Table S7 for a complete list of plasmids | ||
| Software and algorithms | ||
| Geneious Prime® 2022.2.2 | Biomatters Ltd. | RRID: SCR_010519 |
| PSI-BLAST | Altschul et al.71 | RRID: SCR_001010 |
| JACKHMMER | Potter et al.72 | RRID: SCR_005305 |
| BLASTClust | NCBI | RRID: SCR_016641 |
| HHpred | Zimmermann et al.73 | RRID: SCR_010276 |
| PFAM | Mistry et al.74 | RRID: SCR_004726 |
| PDB | Berman et al.75 | RRID: SCR_012820 |
| Kalign | Lassmann et al.76 | RRID: SCR_011810 |
| Muscle | Edgar et al.77 | RRID: SCR_011812 |
| JPred | Drozdetskiy, et al.78 | RRID: SCR_016504 |
| RoseTTa Fold | Baek et al.79 | |
| IQ-TREE | Minh et al.80 | RRID: SCR_017254 |
| FigTree | tree.bio.ed.ac.uk | RRID: SCR_008515 |
| MAFFT | Katoh et al.89 | RRID: SCR_011811 |
| Adobe Illustrator 2021 | Adobe | RRID: SCR_010279 |
| GraphPad Prism 9.2.0 | GraphPad Software | RRID: SCR_002798 |
| Other | ||
Highlights.
Bacteria encode proteins with NACHT modules that are related to metazoan NLRs
Bacterial NLR-related proteins are widespread and defend against DNA and RNA phages
Phages encode proteins that activate and inhibit NLR-related proteins
Metazoan NLRs likely originated from horizontal gene transfer from bacteria
In Brief.
A superfamily of proteins containing a NACHT domain participates in phage defense processes in bacteria, and functional similarity to NOD-like receptors in eukaryotes suggests conservation of these molecular defense strategies across the domains of life
Acknowledgements
The authors would like to thank Russell Vance for advice; Aidan Litt for assistance with cloning; the CU Boulder Department of Biochemistry Shared Instruments Pool core facility (RRID:SCR_018986), Annette Erbse, and its staff; Amber Scott and Kevyn E. Jackson at the University of Colorado Boulder BioFrontiers Institute’s Next Generation Sequencing Core (RRID:SCR_019308) for technical assistance; Brian Kvitko for generously sharing strains and plasmids; and members of the Whiteley lab for advice and helpful discussion. This work was funded by the National Institutes of Health through the NIH Director’s New Innovator Award DP2AT012346 (A.T.W.), a Mallinckrodt Foundation Grant (A.T.W.), and the Boettcher Foundation’s Webb-Waring Biomedical Research Program (A.T.W.). E.M.K was supported in part by the NIH T32 Signaling and Cellular Regulation training grant (T32 GM008759 and T32 GM142607). J.A.V. was supported in part by the Wuttke and Beckman Foundation Biochemical Sciences Research Award, the Biological Sciences Initiative funded by the University of Colorado Boulder, and an Undergraduate Research Opportunities Program Student Assistantship Grant funded by the University of Colorado Boulder. L.A.W. is supported in part by the Biological Sciences Initiative funded by the University of Colorado Boulder. A.M.B. and L.A. are supported by the Intramural Research Program of the National Library of Medicine at the NIH.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Footnotes
Declaration of interests
The authors declare no competing interests.
References
- 1.Bernheim A, and Sorek R (2020). The pan-immune system of bacteria: antiviral defence as a community resource. Nat. Rev. Microbiol 18, 113–119. 10.1038/s41579-019-0278-2. [DOI] [PubMed] [Google Scholar]
- 2.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, and Sorek R (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120. 10.1126/science.aar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao L, Altae-Tran H, Böhning F, Makarova KS, Segel M, Schmid-Burgk JL, Koob J, Wolf YI, Koonin EV, and Zhang F (2020). Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084. 10.1126/science.aba0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rousset F, Depardieu F, Miele S, Dowding J, Laval A-L, Lieberman E, Garry D, Rocha EPC, Bernheim A, and Bikard D (2022). Phages and their satellites encode hotspots of antiviral systems. Cell Host Microbe 30, 740–753.e5. 10.1016/j.chom.2022.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vassallo CN, Doering CR, Littlehale ML, Teodoro GIC, and Laub MT (2022). A functional selection reveals previously undetected anti-phage defence systems in the E. coli pangenome. Nat. Microbiol 7, 1568–1579. 10.1038/s41564-022-01219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burroughs AM, and Aravind L (2020). Identification of Uncharacterized Components of Prokaryotic Immune Systems and Their Diverse Eukaryotic Reformulations. J. Bacteriol 202, e00365–20, /jb/202/24/JB.00365–20.atom. 10.1128/JB.00365-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Millman A, Melamed S, Leavitt A, Doron S, Bernheim A, Hör J, Garb J, Bechon N, Brandis A, Lopatina A, et al. (2022). An expanded arsenal of immune systems that protect bacteria from phages. Cell Host Microbe 30, 1556–1569.e5. 10.1016/j.chom.2022.09.017. [DOI] [PubMed] [Google Scholar]
- 8.Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, Danilchanka O, King DS, Lee ASY, Mekalanos JJ, et al. (2019). Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199. 10.1038/s41586-019-0953-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen Daniel, Melamed Sarah, Millman Adi, Shulman Gabriela, Oppenheimer-Shaanan Yaara, Kacen Assaf, Doron Shany, Amitai Gil, and Sorek Rotem (2019). Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574, 691–695. 10.1038/s41586-019-1605-5. [DOI] [PubMed] [Google Scholar]
- 10.Morehouse BR, Govande AA, Millman A, Keszei AFA, Lowey B, Ofir G, Shao S, Sorek R, and Kranzusch PJ (2020). STING cyclic dinucleotide sensing originated in bacteria. Nature 586, 429–433. 10.1038/s41586-020-2719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burroughs AM, Zhang D, Schäffer DE, Iyer LM, and Aravind L (2015). Comparative genomic analyses reveal a vast, novel network of nucleotide-centric systems in biological conflicts, immunity and signaling. Nucleic Acids Res. 43, 10633–10654. 10.1093/nar/gkv1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernheim A, Millman A, Ofir G, Meitav G, Avraham C, Shomar H, Rosenberg MM, Tal N, Melamed S, Amitai G, et al. (2021). Prokaryotic viperins produce diverse antiviral molecules. Nature 589, 120–124. 10.1038/s41586-020-2762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson AG, Wein T, Mayer ML, Duncan-Lowey B, Yirmiya E, Oppenheimer-Shaanan Y, Amitai G, Sorek R, and Kranzusch PJ (2022). Bacterial gasdermins reveal an ancient mechanism of cell death. Science 375, 221–225. 10.1126/science.abj8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makarova KS, Wolf YI, Snir S, and Koonin EV (2011). Defense Islands in Bacterial and Archaeal Genomes and Prediction of Novel Defense Systems. J. Bacteriol 193, 6039–6056. 10.1128/JB.05535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, Wang R, Zhang Y, Martinon F, Miao D, et al. (2013). Crystal Structure of NLRC4 Reveals Its Autoinhibition Mechanism. Science 341, 172–175. 10.1126/science.1236381. [DOI] [PubMed] [Google Scholar]
- 16.Kim YK, Shin J-S, and Nahm MH (2016). NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J 57, 5. 10.3349/ymj.2016.57.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandall CF, Ziehr BK, and MacDonald JA (2020). ATP-Binding and Hydrolysis in Inflammasome Activation. Molecules 25, 4572. 10.3390/molecules25194572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zurek B, Proell M, Wagner RN, Schwarzenbacher R, and Kufer TA (2012). Mutational analysis of human NOD1 and NOD2 NACHT domains reveals different modes of activation. Innate Immun. 18, 100–111. 10.1177/1753425910394002. [DOI] [PubMed] [Google Scholar]
- 19.Valdar WSJ (2002). Scoring residue conservation. Proteins Struct. Funct. Bioinforma 48, 227–241. 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 20.Tenthorey JL, Haloupek N, López-Blanco JR, Grob P, Adamson E, Hartenian E, Lind NA, Bourgeois NM, Chacón P, Nogales E, et al. (2017). The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 358, 888–893. 10.1126/science.aao1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ting JP-Y, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. (2008). The NLR Gene Family: A Standard Nomenclature. Immunity 28, 285–287. 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dyrka W, Lamacchia M, Durrens P, Kobe B, Daskalov A, Paoletti M, Sherman DJ, and Saupe SJ (2014). Diversity and Variability of NOD-Like Receptors in Fungi. Genome Biol. Evol 6, 3137–3158. 10.1093/gbe/evu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saur IML, Panstruga R, and Schulze-Lefert P (2021). NOD-like receptor-mediated plant immunity: from structure to cell death. Nat. Rev. Immunol 21, 305–318. 10.1038/s41577-020-00473-z. [DOI] [PubMed] [Google Scholar]
- 24.Uehling J, Deveau A, and Paoletti M (2017). Do fungi have an innate immune response? An NLR-based comparison to plant and animal immune systems. PLOS Pathog. 13, e1006578. 10.1371/journal.ppat.1006578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaur G, Burroughs AM, Iyer LM, and Aravind L (2020). Highly regulated, diversifying NTP-dependent biological conflict systems with implications for the emergence of multicellularity. eLife 9, e52696. 10.7554/eLife.52696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur G, Iyer LM, Burroughs AM, and Aravind L (2021). Bacterial death and TRADD-N domains help define novel apoptosis and immunity mechanisms shared by prokaryotes and metazoans. eLife 10, e70394. 10.7554/eLife.70394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao LA, Wilkinson ME, Strecker J, Makarova KS, Macrae RK, Koonin EV, and Zhang F (2022). Prokaryotic innate immunity through pattern recognition of conserved viral proteins. Science 377, eabm4096. 10.1126/science.abm4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leipe DD, Koonin EV, and Aravind L (2004). STAND, a Class of P-Loop NTPases Including Animal and Plant Regulators of Programmed Cell Death: Multiple, Complex Domain Architectures, Unusual Phyletic Patterns, and Evolution by Horizontal Gene Transfer. J. Mol. Biol 343, 1–28. 10.1016/j.jmb.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 29.Koonin EV, and Aravind L (2000). The NACHT family – a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem. Sci 25, 223–224. 10.1016/S0968-0004(00)01577-2. [DOI] [PubMed] [Google Scholar]
- 30.Zhang D, Burroughs AM, Vidal ND, Iyer LM, and Aravind L (2016). Transposons to toxins: the provenance, architecture and diversification of a widespread class of eukaryotic effectors. Nucleic Acids Res. 44, 3513–3533. 10.1093/nar/gkw221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aravind L, Iyer LM, and Burroughs AM (2022). Discovering Biological Conflict Systems Through Genome Analysis: Evolutionary Principles and Biochemical Novelty. Annu. Rev. Biomed. Data Sci 5, annurev-biodatasci-122220–101119. 10.1146/annurev-biodatasci-122220-101119. [DOI] [PubMed] [Google Scholar]
- 32.Ofir G, Herbst E, Baroz M, Cohen D, Millman A, Doron S, Tal N, Malheiro DBA, Malitsky S, Amitai G, et al. (2021). Antiviral activity of bacterial TIR domains via immune signaling molecules. Nature 600, 116–120. 10.1038/s41586-021-04098-7. [DOI] [PubMed] [Google Scholar]
- 33.Essuman K, Summers DW, Sasaki Y, Mao X, Yim AKY, DiAntonio A, and Milbrandt J (2018). TIR Domain Proteins Are an Ancient Family of NAD+-Consuming Enzymes. Curr. Biol 28, 421–430.e4. 10.1016/j.cub.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imai S, Armstrong CM, Kaeberlein M, and Guarente L (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 35.Horsefield S, Burdett H, Zhang X, Manik MK, Shi Y, Chen J, Qi T, Gilley J, Lai J-S, Rank MX, et al. (2019). NAD+ cleavage activity by animal and plant TIR domains in cell death pathways. Science 365, 793–799. 10.1126/science.aax1911. [DOI] [PubMed] [Google Scholar]
- 36.Davison J (2019). Phage T5 two-step injection. biorxiv. 10.1101/866236. [DOI] [Google Scholar]
- 37.Davison J (2015). Pre-early functions of bacteriophage T5 and its relatives. Bacteriophage 5, e1086500. 10.1080/21597081.2015.1086500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong Y, Kinio A, and Saleh M (2013). Functions of NOD-Like Receptors in Human Diseases. Front. Immunol 4. 10.3389/fimmu.2013.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura A, Sasaki Y, Abe T, Kano H, and Yasutomo K (2014). An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med 211, 2385–2396. 10.1084/jem.20141091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romberg N, Vogel TP, and Canna SW (2017). NLRC4 inflammasomopathies. Curr. Opin. Allergy Clin. Immunol 17, 398–404. 10.1097/ACI.0000000000000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volker-Touw C. m. l., de Koning H. d., Giltay JC, de Kovel C. g. f., van Kempen T. s., Oberndorff K. m. e. j, Boes M. l., van Steensel M. a. m., van Well G. t. j., Blokx W. a. m., et al. (2017). Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC4-related autoinflammatory disease, expansion of the phenotype. Br. J. Dermatol 176, 244–248. 10.1111/bjd.14757. [DOI] [PubMed] [Google Scholar]
- 42.Severin GB, Ramliden MS, Hawver LA, Wang K, Pell ME, Kieninger A-K, Khataokar A, O’Hara BJ, Behrmann LV, Neiditch MB, et al. (2018). Direct activation of a phospholipase by cyclic GMP-AMP in El Tor Vibrio cholerae. Proc. Natl. Acad. Sci 115, E6048–E6055. 10.1073/pnas.1801233115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopatina A, Tal N, and Sorek R (2020). Abortive Infection: Bacterial Suicide as an Antiviral Immune Strategy. Annu. Rev. Virol 7, 371–384. 10.1146/annurev-virology-011620-040628. [DOI] [PubMed] [Google Scholar]
- 44.Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 45.Aanen DK, Debets AJM, Glass NL, and Saupe SJ (2010). Biology and Genetics of Vegetative Incompatibility in Fungi. In Cellular and Molecular Biology of Filamentous Fungi (John Wiley & Sons, Ltd; ), pp. 274–288. 10.1128/9781555816636.ch20. [DOI] [Google Scholar]
- 46.Glass NL, and Dementhon K (2006). Non-self recognition and programmed cell death in filamentous fungi. Curr. Opin. Microbiol 9, 553–558. 10.1016/j.mib.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Espagne E, Balhadère P, Penin M-L, Barreau C, and Turcq B (2002). HET-E and HET-D Belong to a New Subfamily of WD40 Proteins Involved in Vegetative Incompatibility Specificity in the Fungus Podospora anserina. Genetics 161, 71–81. 10.1093/genetics/161.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi GH, Dawe AL, Churbanov A, Smith ML, Milgroom MG, and Nuss DL (2012). Molecular Characterization of Vegetative Incompatibility Genes That Restrict Hypovirus Transmission in the Chestnut Blight Fungus Cryphonectria parasitica. Genetics 190, 113–127. 10.1534/genetics.111.133983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paoletti M, and Saupe SJ (2009). Fungal incompatibility: Evolutionary origin in pathogen defense? BioEssays 31, 1201–1210. 10.1002/bies.200900085. [DOI] [PubMed] [Google Scholar]
- 50.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 51.Clavé C, Dyrka W, Turcotte EA, Granger-Farbos A, Ibarlosa L, Pinson B, Vance RE, Saupe SJ, and Daskalov A (2022). Fungal gasdermin-like proteins are controlled by proteolytic cleavage. Proc. Natl. Acad. Sci 119, e2109418119. 10.1073/pnas.2109418119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daskalov A, Mitchell PS, Sandstrom A, Vance RE, and Glass NL (2020). Molecular characterization of a fungal gasdermin-like protein. Proc. Natl. Acad. Sci 117, 18600–18607. 10.1073/pnas.2004876117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van der Nest MA, Olson Å, Lind M, Vélëz H, Dalman K, Durling MB, Karlsson M, and Stenlid J (2014). Distribution and evolution of het gene homologs in the basidiomycota. Fungal Genet. Biol 64, 45–57. 10.1016/j.fgb.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 54.Strutt SC, Torrez RM, Kaya E, Negrete OA, and Doudna JA (2018). RNA-dependent RNA targeting by CRISPR-Cas9. eLife 7, e32724. 10.7554/eLife.32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jakobsen MR, Mogensen TH, and Paludan SR (2013). Caught in translation: innate restriction of HIV mRNA translation by a schlafen family protein. Cell Res. 23, 320–322. 10.1038/cr.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lisa M-N, Cvirkaite-Krupovic V, Richet E, André-Leroux G, Alzari PM, Haouz A, and Danot O (2019). Double autoinhibition mechanism of signal transduction ATPases with numerous domains (STAND) with a tetratricopeptide repeat sensor. Nucleic Acids Res. 47, 3795–3810. 10.1093/nar/gkz112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marquenet E, and Richet E (2007). How Integration of Positive and Negative Regulatory Signals by a STAND Signaling Protein Depends on ATP Hydrolysis. Mol. Cell 28, 187–199. 10.1016/j.molcel.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 58.Kvitko BH, Bruckbauer S, Prucha J, McMillan I, Breland EJ, Lehman S, Mladinich K, Choi K-H, Karkhoff-Schweizer R, and Schweizer HP (2012). A simple method for construction of pir + Enterobacterial hosts for maintenance of R6K replicon plasmids. BMC Res. Notes 5, 157. 10.1186/1756-0500-5-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herrick J, Heringa S, and Monroe J (2018). A Simple, Rapid Method for Extracting Large Plasmid DNA from Bacteria (figshare) 10.6084/M9.FIGSHARE.5872515.V2. [DOI] [Google Scholar]
- 60.Kropinski AM, Mazzocco A, Waddell TE, and Johnson RP (2009). Enumeration of Bacteriophages by Double Agar Overlay Plaque Assay. Bacteriophages 501, 69–76. 10.1007/978-1-60327-164-6_7. [DOI] [PubMed] [Google Scholar]
- 61.Loeb T, and Zinder ND (1961). A BACTERIOPHAGE CONTAINING RNA. Proc. Natl. Acad. Sci. U. S. A 47, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Novotny C, Knight WS, and Brinton CC (1968). Inhibition of Bacterial Conjugation by Ribonucleic Acid and Deoxyribonucleic Acid Male-Specific Bacteriophages. J. Bacteriol 95, 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppenheimer-Shaanan Y, and Sorek R (2020). Bacterial Retrons Function In Anti-Phage Defense. Cell 183, 1551–1561.e12. 10.1016/j.cell.2020.09.065. [DOI] [PubMed] [Google Scholar]
- 64.Harel N, Meir M, Gophna U, and Stern A (2019). Direct sequencing of RNA with MinION Nanopore: detecting mutations based on associations. Nucleic Acids Res. 47, e148. 10.1093/nar/gkz907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, and Kishony R (2015). Inexpensive Multiplexed Library Preparation for Megabase-Sized Genomes. PLOS ONE 10, e0128036. 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 67.Ochman H, and Selander RK (1984). Standard reference strains of Escherichia coli from natural populations. J. Bacteriol 157, 690–693. 10.1128/jb.157.2.690-693.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ledvina HE, Ye Q, Gu Y, Sullivan AE, Quan Y, Lau RK, Zhou H, Corbett KD, and Whiteley AT (2023). An E1–E2 fusion protein primes antiviral immune signalling in bacteria. Nature. 10.1038/s41586-022-05647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aravind L, Iyer LM, Leipe DD, and Koonin EV (2004). A novel family of P-loop NTPases with an unusual phyletic distribution and transmembrane segments inserted within the NTPase domain. Genome Biol. 5. 10.1186/gb-2004-5-5-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, and Finn RD (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, and Alva V (2018). A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol 430, 2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 74.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, et al. (2020). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lassmann T (2019). Kalign 3: multiple sequence alignment of large data sets. Bioinforma. Oxf. Engl, btz795. 10.1093/bioinformatics/btz795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edgar RC (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Drozdetskiy A, Cole C, Procter J, and Barton GJ (2015). JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baek M, Dimaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876. DOI: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, and Lanfear R (2020). IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol 37, 1530–1534. 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, and Gascuel O (2010). New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol 59, 307–321. 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 82.Kishino H, Miyata T, and Hasegawa M (1990). Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol 31, 151–160. 10.1007/BF02109483. [DOI] [Google Scholar]
- 83.Kishino H, and Hasegawa M (1989). Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol 29, 170–179. 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 84.Strimmer K, and Rambaut A (2002). Inferring confidence sets of possibly misspecified gene trees. Proc. R. Soc. B Biol. Sci 269, 137–142. 10.1098/rspb.2001.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shimodaira H (2002). An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol 51, 492–508. 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- 86.Lyons NA, and Kolter R (2015). On The Evolution of Bacterial Multicellularity. Curr. Opin. Microbiol 24, 21–28. 10.1016/j.mib.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Solovyev V, and Salamov A (2011). AUTOMATIC ANNOTATION OF MICROBIAL GENOMES AND METAGENOMIC SEQUENCES. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies, Li RW, ed. (Nova Science Publishers; ), pp. 61–78. [Google Scholar]
- 88.Naville M, Marchais A, and Gaudeffroy AG ARNold: finding terminators.
- 89.Katoh K, Misawa K, Kuma K, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Protein alignment of the NACHT modules from bNACHT01 and other NACHT proteins, related to Figure 1. (A) Protein alignment of the NACHT modules of NLRC4 (Mus musculus; NP_001028539), bNACHT01 (Klebsiella pneumoniae MGH 35; WP_015632533.1), NAIP (Homo sapiens; NP_001333799.1), CIITA (Homo sapiens; NP_000237.2), NOD1 (Homo sapiens; NP_006083.1), HET-E (Podospora anserina; Q00808), and TEP1 (Homo sapiens; NP_009041.2). The secondary structure of NLRC4 as determined using structure 4KXF in the PDB15, is indicated above with alpha helices depicted as cylinders and beta sheets depicted as arrows. Secondary structure elements are color-coded to represent the NBD, HD, and WHD of NLRC4. Amino acid residues are color-coded based on conservation such that darker colors indicate a higher degree of conservation in this alignment. Black boxes indicate the Walker A and Walker B motifs. Residues mutated and analyzed for expression in Figure 1D are highlighted in red. The D[GAS]hDE motif within the Walker B region that distinguishes NACHT modules from other STAND NTPases is indicated.
(B) Western blot analysis of E. coli expressing empty vector or FLAG-tagged bNACHT01 at 0, 10, and 20 minutes post-infection with phage T5. Representative image of n = 2 biological replicates.
(C)–(E) Efficiency of plating of the indicated phage infecting E. coli expressing the indicated genotype. Data represent the mean ± standard s.e.m. of n = 3 biological replicates, shown as individual points.
(F) Schematic of bNACHT01 (WP_015632533.1) protein domains, annotated by alignment to the NACHT module of NLRC4. The P-loop NTPase domain is also known as a nucleotide-binding domain (NBD), the helical domain (HD), and the winged helix-turn-helix (wHTH, also called WHD for winged helical domain) are indicated.
(G)–(I) Efficiency of plating of the indicated phage infecting E. coli expressing the indicated genotype. Data represent the mean ± standard s.e.m. of n = 3 biological replicates, shown as individual points.
(J) Predicted structure of bNACHT01. Aspartate represents the conserved residue D449.
(K) Predicted structures of 3 different SNaCT domain families. N represents the N-terminus of the domain.
Figure S2. Distribution of NACHT proteins in bacterial taxa, related to Figure 2. (A) Quantification of the number of individual NACHTs found within a single genome across different bacterial taxa. The maximum for the x-axis is 23. Plantomycetota, Verrumicrobiota, Chlamydiota (PVC).
(B) Relative distribution of taxa that have organisms with 3 or more NACHTs in a single genome.
(C) Propensity of organisms within the indicated taxa to encode a NACHT module- containing protein.
(D) Percent of organisms in the indicated taxa to encode at least one NACHT module- containing protein.
(E) Distribution of the number of NACHT proteins per organism, which can be fitted to the equation y = x−2.05. For A–E, a custom database of complete bacterial genomes was used to analyze distribution of NACHT proteins in diverse bacterial taxa.
Figure S3. NACHT module-containing proteins in bacteria are widespread and diverse, related to Figure 2. A sequence-based phylogenetic tree of NACHT modules was generated using NACHT module-containing proteins from eukaryotes and prokaryotes. The NACHT module, not accessory domains, were used for tree building. Clades are color-coded based on the indicated key and numbered arbitrarily in yellow circles. Red dots indicate the bacterial NACHT proteins from each clade that were selected for analysis in this study. Bootstrap values are provided where applicable. The scale bar indicates average number of substitutions per site. See Table S4 for the most common domain architectures found in each clade. The genes used to construct this phylogenetic tree can be found in Table S2 and Table S1 contains a full list of all NACHT domain-containing proteins identified. See Table S3 for tree topology tests used to validated proposed evolutionary relationships. Representative domain architectures for each clade are provided, including NCBI Protein accession number, species of origin, and gene name in green where appropriate. 3′,5′ cyclic nucleotide-generating cyclase (cNMP), Ankyrin repeats (ANKs), Bacterial Death-like domain-3 (bDLD3), Beta-propeller repeats (BPs), Bacterial transglutaminase-like cysteine protease (BTCLP), Carbon-nitrogen hydrolase (CN-hydrolase), CoA-dependent acyltransferase (CoA-acyltrans), Deoxyribohydrolase (DrHyd), Effector associated constant component (EACC), Effector-associated domain (EAD), Formylglycine-generating enzyme sulfatase (FGS), FGS C-terminal domain (FGS-C), Forkhead-associated domain (FHA), Fish-specific NACHT-associated domain (FISNA), Glycosyltransferase 4 (GT4), Huntington, Elongation Factor 3, PR65/A, TOR (HEAT), Higher eukaryotes and prokaryotes nucleotide-binding domain (HEPN), Homing endonuclease (HNH), Helix-turn-helix (HTH), Leucine-rich repeat (LRR), Membrane occupation and recognition nexus (MORN), , NACHT N-terminal helical domain (NNH), NACHT C-terminal helical domain (NCH), NACHT C-terminal α / β domain (NCAB), NACHT C-terminal cysteine and histidine-containing domain (NCC-H), Protein kinase domain (Pkinase), Polyribonucleotide nucleotidyl transferase (PNPase), Pentapeptide repeat (Pp), Restriction endonuclease (REase), Receiver domain (Rec), Rhodanese domain (RHOD), Sirtuin (SIR2), Second messenger oligonucleotide or dinucleotide synthetase domain (SMODS), Telomerase associated protein 1 (TEP1), Toll/interleukin receptor (TIR), Transmembrane (TM) number indicates how many copies of this motif, Tetratricopeptide repeat (TPR), N-terminal domain of tumor necrosis factor receptor type 1 associated death domain protein (TRADD-N), UvRC and Intron-encoded endonuclease domain (URI), Zinc ribbon (ZnR). Red slash indicates catalytically inactive domain.
Figure S4. Domain architectures and details of experimentally tested NACHT proteins, related to Figure 3. Diagram of the N-terminal domains, central NACHT modules, and C-terminal domains of each experimentally tested bNACHT protein. The clade, species of origin, and length of each protein in amino acids (aa) are also indicated. Leucine-rich repeat (LRR), Huntington, Elongation Factor 3, PR65/A, TOR (HEAT), Restriction endonuclease (REase), Toll/interleukin receptor (TIR), Transmembrane (TM), NACHT N-terminal helical domain 1 (NNH1), NACHT C-terminal helical domain 1 (NCH1).
Figure S5. Expanded screen for bacterial NACHT protein phage defense and comparison of defense conferred by plasmid- or chromosomally-expressed bacterial NACHTs, related to Figure 3. (A) Heat map of fold defense provided by the indicated bNACHT gene for a panel of diverse phages. Data are as described in Figure 3 and show all experimentally interrogated bNACHT genes. Data were not collected for bacterial NACHT proteins that exhibited insufficient growth under these experimental conditions, indicated as “X”. Data represent the mean of n = 3 biological replicates. See Table S5 and Figure S4 for details on all 27 bNACHT genes analyzed. See Figure S6 for raw efficiency of plating data.
(B)–(C) Efficiency of plating of the indicated phage infecting E. coli expressing the indicated genotype. Empty (E) indicates E. coli with chromosomal expression of a Kanamycin resistance cassette and gfpmut3. Data represent the mean ± standard s.e.m. of n = 3 biological replicates, indicated as individual points.
Figure S6. Efficiency of plating of phages infecting bacteria expressing NACHT proteins, related to Figure 3. Efficiency of plating of a phage panel infecting E. coli expressing the indicated bNACHT gene. The negative control is empty vector, which expresses an inactive gfp gene. Positive controls are V. cholerae CBASS (VcCBASS) and E. coli UPEC-36 (EcoAI RM) restriction modification system (Z and AA). Data represent the mean ± s.e.m. of n = 3 biological replicates, indicated as individual points.
Figure S7. Effect of orf008 and orf015 expression on bNACHT01 activity and comparison of bNACHT modules to the NACHT domain of NLRC4, related to Figures 5 and 6. (A) Efficiency of plating of phage T6 when infecting E. coli expressing bNACHT01 or empty vector (EV) on one plasmid and the indicated phage T5 gene(s) on a second plasmid.
(B) Efficiency of plating of phage T2 when infecting E. coli expressing the indicated bNACHT gene on one plasmid and phage T5 orf015 on a second plasmid.
(C) Efficiency of plating of phage T6 when infecting E. coli expressing the indicated bNACHT gene on one plasmid and phage T5 orf015 on a second plasmid.
(D) Quantification of colony formation of E. coli expressing the indicated bNACHT system on one plasmid and phage T5 orf015 on a second plasmid. For A–D, expression of orf008, orf015, or sfGFP is IPTG-inducible. (−) symbols denote induction of an sfGFP negative control. (+) symbols denote induction of orf015. Data represent the mean ± s.e.m. of n = 3 biological replicates, shown as individual points.
(E) Protein alignment of the NACHT modules of NLRC4 (Mus musculus; NP_001028539), bNACHT16 (Vibrio campbellii CAIM 519; WP_005534681.1), and bNACHT25 (E. coli ECOR11; WP_001702659.1). The secondary structure of NLRC4 as determined by structure 4KXF in the PDB15, is indicated above with alpha helices depicted as cylinders and beta sheets depicted as arrows. Secondary structure elements are color coded and labeled as in Figure S1. Amino acid residues are color-coded based on conservation in the sequence alignment. Black boxes indicate Walker A and B motifs. The conserved histidine and serine/threonine in the WHD is highlighted in red.
Table S1. All bioinformatically identified NACHT proteins, related to Figure 2.
Table S2. (a) Diverse NACHT proteins used in phylogenetic analysis and (b) comparison of the prevalence of different defense systems, related to Figure 2.
Table S3. Tests for alternative tree topologies, related to Figure 2.
Table S4. Common domain architectures found in each NACHT clade, related to Figure 2.
Table S5. Experimentally investigated bacterial NACHT proteins, related to Figure 3.
Table S6. (a) Rate of suppressor isolation for T5 infecting bNACHT01, (b) suppressor mutations in orf008 and orf015, and (c) all T5 suppressor mutations, related to Figure 5.
Table S7. (a) E. coli strains and plasmids, and (b) bacteriophages used in this study, related to STAR Methods.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.