

Abstract
This narrative review paper provides an up-to-date overview of the potential of novel synthetic and semisynthetic compounds as antibacterials that target virulence traits in resistant strains. The review focused on research conducted in the last five years and investigated a range of compounds including azoles, indoles, thiophenes, glycopeptides, pleuromutilin derivatives, lactone derivatives, and chalcones. The emergence and spread of antibiotic-resistant bacterial strains is a growing public health concern, and new approaches are urgently needed to combat this threat. One promising approach is to target virulence factors, which are essential for bacterial survival and pathogenesis, but not for bacterial growth. By targeting virulence factors, it may be possible to reduce the severity of bacterial infections without promoting the development of resistance. We discuss the mechanisms of action of the various compounds investigated and their potential as antibacterials. The review highlights the potential of targeting virulence factors as a promising strategy to combat antibiotic resistance and suggests that further research is needed to identify new compounds and optimize their efficacy. The findings of this review suggest that novel synthetic and semisynthetic compounds that target virulence factors have great potential as antibacterials in the fight against antibiotic resistance.
Keywords: antibiotic resistance, virulence factors, novel synthetic compounds, biofilms, antibacterial activity
1. Introduction
Antibiotic resistance is a major global health challenge, threatening the efficacy of currently available antibiotics [1]. The emergence and spread of multidrug-resistant bacteria underscore the urgent need for new antibacterial agents that can overcome resistance mechanisms [2]. Estimates indicate that, annually, over 2 million infections caused by resistants strains occur worldwide, with as many as approximately 30,000 fatal outcomes in the USA alone and USD 5 billion in health care assets allocated to this issue. At the beginning of the 21st century, a list of pathogenic microorganisms that showed different levels of resistance to antimicrobial agents was released, and it included Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp. The list is also known as the famous register of ESKAPE pathogens. Traditional antibiotics target bacterial growth, which can lead to the development of resistance through the acquisition of mutations or the transfer of resistance genes [3]. In contrast, compounds that target bacterial virulence traits, such as biofilm formation, quorum sensing, and motility, may be less prone to the development of resistance [4]. Bacterial infections remain a significant challenge in public health, and the emergence of antimicrobial resistance (AMR) has further complicated the treatment of bacterial infections [5]. Despite the availability of many antibiotics, the prevalence of bacterial infections caused by multidrug-resistant bacteria has increased alarmingly. One strategy to overcome AMR is to target the virulence traits of bacterial pathogens, which are distinct from traditional antibiotic targets [6]. Virulence factors are the attributes that enable pathogens to cause disease in a host, such as adhesion, invasion, colonization, and the secretion of toxins and enzymes [7]. Therefore, the inhibition of virulence factors is a promising approach to combating bacterial infections [8].
The virulence traits of resistant bacteria have received increasing attention in recent years. Biofilm formation is one of the important virulence factors that contribute to bacterial resistance [9]. Biofilms are complex communities of microorganisms that are encased in a self-produced extracellular matrix, which confers resistance to antibiotics and immune defense mechanisms [10]. Bacteria can cause disease by producing agents known as virulence traits, which are specific compounds produced by bacteria that allow them to evade the host’s immune system response. Virulence traits such as quorum sensing, motility, and iron acquisition have also been reported to be involved in the pathogenicity and antibiotic resistance of bacterial pathogens. As well as adhesins, invasins, and antiphagocytic factors, toxins, hemolysins, and proteases are among the agents that cause harm to the host [11].
A variety of natural and synthetic compounds have been reported to possess anti-virulence activity against resistant bacteria. Among them, azoles, indoles, thiophenes, glycopeptides, pleuromutilin derivatives, lactone derivatives, and chalcones have been found to exhibit promising antivirulence activity [12,13,14,15]. These compounds target various virulence factors and interfere with the pathogenicity of bacterial pathogens (Figure 1), thus enhancing the efficacy of antibiotics and reducing the emergence of resistance.
Figure 1.

Group of compounds that target virulence traits in resistant bacteria.
Chalcones are a class of natural and synthetic compounds that have been shown to inhibit bacterial biofilm formation and quorum sensing [16]. Azoles, including pyrazoles, oxadiazoles, and triazoles, have been extensively studied as antifungal agents, but recent studies have shown that they also have antibacterial activity against resistant strains [17]. Coumarins have been found to inhibit bacterial quorum sensing and motility [18], while indoles and thiophenes have also shown potential as quorum sensing inhibitors [19]. Quinolines have been proposed as inhibitors of bacterial type II topoisomerases and have shown activity against multidrug-resistant bacteria [20]. Terpenoids, including triterpenoids and other scaffolds, have been mainly studied as semisynthetic analogues with promising antibacterial activity [21]. Glycopeptides, such as vancomycin and teicoplanin, have been widely used as antibiotics, but semisynthetic analogues have been developed to overcome resistance mechanisms [22]. Pleuromutilin derivatives, including retapamulin and lefamulin, have been approved for clinical use and have shown efficacy against resistant strains [23]. Finally, albocyclin and other lactone derivatives have shown activity against Gram-positive and Gram-negative bacteria, including resistant strains [24].
Targeting virulence traits is an attractive strategy to combat resistant bacteria. The use of compounds that target virulence factors can complement traditional antibiotic therapies, leading to enhanced efficacy and reduced resistance. Therefore, continuous research on the development of anti-virulence compounds and their mechanisms of action is crucial in the fight against AMR. In this review, we discuss recent studies on synthetic and semisynthetic compounds with antibacterial activity, focusing on their ability to target virulence traits in resistant strains. We also highlight the potential of these compounds to overcome antibiotic resistance mechanisms and suggest directions for future research. The presented review article mainly summarizes the progress in this field in the last 5 years, but its also covers some older important data. The fresh perspectives of compounds newly identified as potential therapeutics targeting virulence factors are presented, along with the established antimicrobial properties of certain novel compounds and the repurposing of existing antibacterial/antifungal therapeutics.
2. Indoles
Indoles, a widespread naturally occurring class of alkaloid compounds, are not only important bacterial intercellular signal molecules, but also a crucial component of the amino acid tryptophan. They are a particularly intriguing class of compounds covering a range of pharmacological activities, including antiinflammatory, antihistaminic, antitumor, antioxidative, and antidiabetic properties. Research covering this topic is quite important due to the versatile nature of indole compounds, which may lead to numerous chemical modifications, i.e., presenting possibilities for drug development. With respect to the subject of this review, their antibacterial potential, particularly their targeting of virulence factors, is thoroughly elaborated herein.
In the following section, we present the antibacterial potential of selected indole-derived compounds against clinically relevant strains, namely the causative agents of urinary and skin infections as well as gastroenteritis-causing bacteria. According to Balcerek et al. [25], commercial compounds, such as 5-halo-1H-indole-2-carboxylic acids, (Figure 2) were efficient against a panel of bacterial strains, particularly Listeria monocytogenes. The obtained results indicated that this activity may be used for the development of medicines in the treatment of listeriosis in cases when resistance/allergy is present. Along with these results, assays also showed that indol-2-one (Figure 2) with a morpholinosulfonyl component acted as a potent inhibitor of the DNA gyrase of both Gram-positive and Gram-negative bacteria, with activity against S. aureus even better than ciprofloxacin (IC50 values 18.75 µM and 26.43 µM, respectively) [26]. Furthermore, Alzahrani et al. [27] showed that novel derivatives of the compound thiazolo-indolin-2-one exerted rather promising antibacterial activity, with a noteworthy ability to affect virulence traits such as biofilm formation in S. aureus (ATCC 29213) and P. aeruginosa (biofilm inhibition concentration (BIC50) of 1.95 µg/mL and 3.9 µg/mL, respectively). As for the ability to affect traits of A. baumanii, literature data indicate that d-pyrimido[4,5-b] indole derivatives show inhibitory potential against this pathogen in the range of 0.25–1 gmL−1 [28]. Furthermore, 3-amino indoles (Figure 2), 4-hydroxy-2-pyridone derivatives containing indolyl, 2-hydrazino2-imidazoline, and bis-indolyl methane Schiff bases have also been identified as potential antimicrobial agents that may also inhibit the growth of MDR A. baumanii. Recent research conducted by Raorane et al. [29] showed that halogenated indole 5-iodoindole (Figure 2) promptly affected the development and motility of A. baumannii, disrupted its biofilm formation, and eventually eradicated this pathogenic microorganism as effectively as ciprofloxacin and gentamicin. This was achieved via the development of ROS, which had a profound influence on the integrity of the plasma membrane, eventually leading to a loss of bacterial viability. Furthermore, the tested compound turned out to be verry effective against Escherichia coli and S. aureus but did not influence the viability of P. aeruginosa. According to Kim et al. [30], indole and its derivatives also proved efficient in the inhibiting single-species and multi-species biofilms of the acne-forming bacterial skin strains Cutibacterium acnes and S. aureus, with 3,3′-diindolylmethane as the most potent inhibitor. The obtained results indicated that indole-derived compounds may be useful in developing efficient skin treatments related to the tested bacteria.
Figure 2.

Chemical structures of 5-halo-1H-indole-2-carboxylic acid, indol-2-one, 3-amino indole, and 5-iodoindole.
The eradication of the nosocomial pathogen Enterococcus faecalis has been shown to be quite challenging in recent years, with biofilm development and resistance to antibiotics as the two main causes. Hence, new treatments are urgently needed, particularly those affecting these two traits. A study by Tatta et al. [31] showed that the indole terpenoid compound rhodethrin (Figure 3) in combination with chloramphenicol disrupted the overall formation of biofilm, which may lead to the easier and more effective treatment of vancomycin-resistant E. faecalis.
Figure 3.
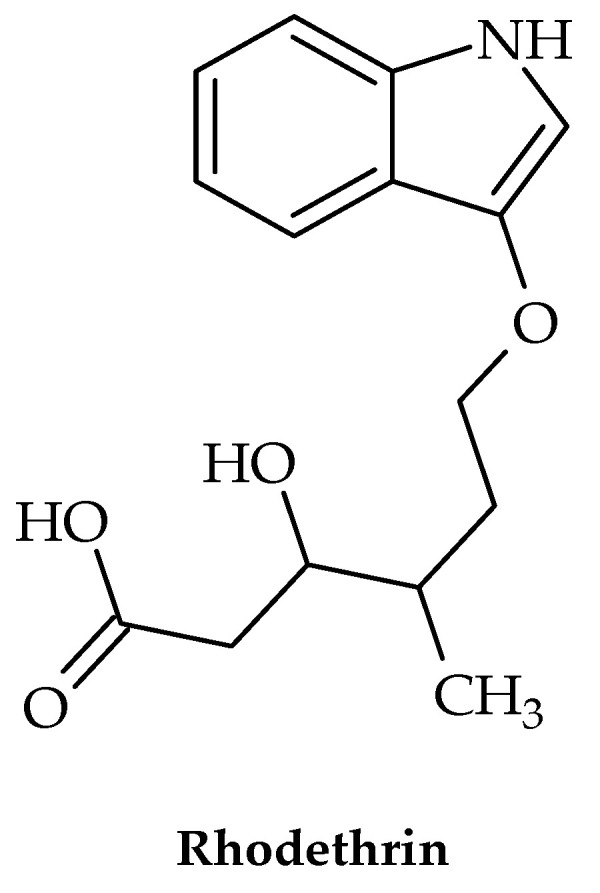
Chemical structure of rhodethrin.
Nosocomial urinary infections related to catheter application are most often caused by Proteus mirabilis. Due to biofilm development, they have been increasingly harder to treat, leading to a demand for novel and efficient treatments. Hence, Amer et al. [32] developed new Foley catheters impregnated with indole compounds (indole extract from the supernatant of the rhizobacterium Enterobacter sp. Zch127) in order to disrupt the biofilm formation of Proteus mirabilis. The results showed a reduction in the formation of biofilm of 60–70% in terms of biomass, which was confirmed by the expression of virulence genes responsible for biofilm formation, while genes that regulate the formation of capsular polysaccharides were not affected. The catheters were considered safe for use, since they had no cytotoxic effects on fibroblasts. Along with nosocomial P. mirabilis, uropathogenic E. coli is a common inhabitant of the human urinary tract, leading to recurrent infections. The recurrence rate depends on the pathogen’s ability to infiltrate the urinary epithelium and evade host defense mechanisms. Boya et al. [33] demonstrated that 4-chloroindole, 5-chloroindole, and 5-chloro 2-methyl indole may profoundly impact biofilm formation at an average dose of 20 g/mL by as much as 67%, along with their ability to reduce bacterial motility, necessary for colony dispersal. A more in-depth study showed that the tested compounds affected the expression of genes related to adhesion and toxin production, which may be of importance in managing clinical manifestations of these health conditions.
Due to their ability to regulate internal environments by removing toxic substances, efflux pumps are an important target when considering the development of new drugs. According to Cernicchi et al. [34], indole derivatives could also have wide-ranging applications in this area, which could increase their use in clinical practice.
Along with the fact that indoles are highly active against pathogenic microorganisms of clinical relevance, they have also been shown to be very efficient in targeting virulence traits of Agrobacterium tumefaciens. This may be rather important with respect to the economy, since this microorganism is known as a plant pathogen causing significant lossess in various crops. As Ahmed et al. [35] demonstrated in their study, among 83 indole derivatives that were tested against A. tumefaciens, 4-chloroindole, 6-iodoindole, and 5-chloro-2-methyl indole inhibited its growth at doses as high as 50 μg mL−1. Furthermore, they also affected virulence factors such as swimming motility, the production of exopolysaccharide and exoprotease, and cell surface hydrophobicity and biofilm formation.
Besides issues with various crops, the aquaculture sector also faces a serious problem resulting from bacterial infections. In terms of money, losses resulting from vibriosis—a disease caused by Vibrio campbelli—are quite substantial. This has inevitably led to the development of novel and sustainable strategies required for managing problems in the aquaculture industry. One of these is the evaluation of indole analogs’ activity against V. campbellii, probably the main bacterial pathogens in aquaculture. Out of 44 tested compounds, 17 halogenated indoles (including 6-bromoindole, 7-bromoindole, 4-fluoroindole, 5-iodoindole, and 7-iodoindole) have been shown to affect the virulence traits of V. campbellii. Furthermore, they have been found to increase the survival of brine shrimp, used as a valid in vivo system model, by over 80% at 10 mM, as well as to affect virulence traits such as swimming motility and biofilm formation (at concentrations of 10 mM and 100 mM), whereas only mild inhibition was achieved with the tested concentrations regarding protease activity. The absence of hemolytic activity was observed using the tested concentrations [36]. Similar antibacterial virulence-targeting activity was previously obtained for Vibrio tasmaniensis LGP32 and Vibrio crassostreae J2-9, used as two model infections of bivalves [36], which indicates that this strategy may be very useful in developing antivirulence therapy. The control of Vibrio parahaemolyticus, a potential cause of gastroenteritis brought on by the consummation of raw sea food, is also becoming increasingly important, since a certain amount of healthcare expenses have been directed towards treating this condition. In their study, Sathiyamoorthi et al. [37] demonstrated that halogenated indole derivatives (4-chloroindole, 7-chloroindole, 4-iodoindole, and 7-iodoindole) strongly influence some of the virulence factors of V. parahaemolyticus: for example, 4-chloroindole inhibited biofilm formation by 80% at a MIC of 50 g/mL, whereas 100 g/mL terminated its viability within the first 30 min of activity. As it turned out, the position of the halogenated substituent in indole core determines its extraordinary activity.
Though these results did not highlight the potential of indole compounds to target bacterial virulence factors, recent data published by Li et al. [38] showed that 5-methylindole instantly eradicated several bacterial strains, including S. aureus, E.faecalis, E. coli, P. aeruginosa, methicillin-resistant S. aureus, K. pneumoniae, and Mycobacterium tuberculosis.
3. Azoles
Azole derivatives are heterocyclic compounds comprised of a nitrogen atom and at least one other non-carbon atom (such as nitrogen, sulfur, or oxygen) as part of the ring. They encompass a wide number of derivatives, such as thiadiazole, oxadiazole, triazole, imidazole, isoxazole, and pyrazole. Mainly known as antifungal agents, azole derivatives demonstrate many other biological properties, including antidiabetic, immunosuppressant, antiinflammatory, and anticancer activities. Even though they were initially used for the treatment of fungal infections, various azole-containing compounds have been shown to inhibit the growth of bacteria as well, via a different mode of action. In fungi, azoles mainly inhibit the production of ergosterol—an essential component of the fungal plasma membrane—whereas in bacteria, their activity is based on the fact that the attachment of azole to bacterial flavohemoglobin (protein) eventually leads to the increasing production of ROS, which have fatal effects on bacterial viability [39].
Due to their versatility in chemical structure and biological activities, azoles have been widely investigated in pharmacochemistry, but they still present surprises. According to Srikanth et al. [28], azole compounds are highly efficient against A. Baumanii, which is of great importance considering that this multi-drug-resistant pathogenic microorganism belongs to the infamous ESKAPE group. In particular, naphthalimide-containing nitroimidazoles with decyl-piperazine exerted strong activity against A. baumannii (MIC 0.013 MmL−1) and, combined with norfloxacin, eradicated even the resistant strains. Additionaly, ammonium containing imidazoles also showed antimicrobial potential. The same study demonstrated that the type of the modification as well as the substituent determines the level of antimicrobial properties. Thus, the presence of 4-Br-phenol modification increased activity against A. baumanii, whereas a hydrophobic n-butyl chain on the phenyl ring decreased activity against the same pathogen. The absence of a halogen molecule is generally reflected through a decrease in bioactivity.
Along with this growing trend of repurposing already available therapeutics, Olaifa et al. [40] also investigated the ability of itraconazole and fluconazole (Figure 4) to target specific virulence factors. The ability to disrupt biofilm formation in A. baumanii was demonstrated in the abovementioned study, which clearly indicated that azole compounds may very well be underinvestigated in terms of their antibacterial and virulence-targeting potential. This was also previously demonstrated by Qiu et al. [41]—using Streptococcus mutans clinical isolates as model organisms, clotrimazole and econazole (Figure 5) inhibited its growth at 12.5 and 25 mgL−1, respectively. Furthermore, they were able to inhibit biofilm production, which undoubtedly demonstrated that these antifungal medicines may also target bacterial virulence factors.
Figure 4.

Chemical structures of itraconazole and fluconazole.
Figure 5.

Chemical structures of clotrimazole and econazole.
Numerous data dealing with the antibacterial potential of antifungal drugs have been presented in the last two years. Even though these drugs do not target virulence factors, the results are noteworthy, favoring the repurposing of antifungal drugs as novel antibacterials. For example, Nasr et al. [42] demonstrated that a pyrazole derivative (der. 30) proved to be more effective against Pneumocystis vulgaris and K. pneumoniae than sulfisoxazole and gentamycin. Among 4-(4-formyl-3-phenyl-1H-pyrazol-1-yl)benzoic acid derivatives, some of the identified compounds showed antibacterial activity against A. baumanii with an MIC of 4 µg/mL [28]. Furthermore, according to Gomes et al. [43], of twenty-one freshly synthesized 1,4-naphthoquinones linked to 1,2,3-1H-triazoles, four (9e, 9h, 9i and 9j) proved to possess antibacterial activity against S. mutans from oral cavities with IZs of 18.66–29.00 mm. The results also showed no toxic effects for these compounds, which possibly increases their potential for application in practice. 1,2,4-triazolidine-3-thiones (Figure 6) exerted antibacterial activity against the ESKAPE list of pathogenic bacteria. Furthermore, binaphthyl-1,2,3-triazole peptidomimetics were efficient against A. baumannii with an MIC of 4 g/mL. Along with this, cationic biaryl 1,2,3-triazolyl peptidomimetic derivatives moderately inhibited the growth of A. baumannii [28]. Antibacterial but not virulence-targeting activity was also demonstrated in a study by Sapijanskaite-Banevic et al. [44]. In order to create substituted 1-phenyl-5-oxopyrrolidine (Figure 6) derivatives with benzimidazole, oxadiazole, triazole, dihydrazone, and dithiosemicarbazide moieties in the structure, p-aminobenzoic acid (Figure 6) was employed. Using different assays, the antimicrobial activity of each drug was assessed in vitro against S. aureus, Bacillus cereus, L. monocytogenes, Salmonella enteritidis, E. coli, and P. aeruginosa. This work demonstrated the potent bactericidal effects of benzimidazoles and derivatives of amino acids, with some of the compounds exceeding the activity of ampicillin. In the field of medicinal chemistry, combining two or more pharmacological groups into a single molecule is a new approach to drug discovery [45]. As demostrated by Dawoud et al. [46], a novel group of heterocyclic compounds merged using a indazolylthiazole moiety was evaluated for their antimicrobial potential. The obtained results showed that four of the compounds exhibited antibacterial effects, with the strongest activity observed against Streptococcus mutans and P. aeruginosa. Furthermore, these novel compounds showed virulence-targeting activity, with high antibiofilm potential. Srikanth et al. [28] also suggested that aminothiazolyl berberine (Figure 6) affects the activity of the DNA gyrase of MDR A. baumannii strains, exerting remarkable activitiy at an MIC of 2 nmol/mL. Among oxazole/benzisoxazole-based compounds, N-(2-(1H-imidazol-4-yl)ethyl)-2-(2,3-dihydroxyphenyl)-N-hydroxy-5-methyloxazole-4-carboxamide showed antibacterial activity against A. Baumannii, with an MIC of 2 µg/mL (strains UNT190 and UNT197) [28].
Figure 6.

Chemical structures of 1,2,4-triazolidine-3-thione, 1-phenyl-5-oxopyrrolidine, p-aminobenzoic acid, and aminothiazolyl berberine.
4. Thiophenes
Thiophenes and related derivatives are rather versatile heterocyclic compounds with various applications in medicine and drug discovery. With a wide range of bioactive properties, they have been shown to possess remarkable anti-inflammatory, antianxiety, antimicrobial, antioxidant, and other activities. Furthermore, they have long been present on the market as commercial therapeutics, for example, tipeptidine, dorzolamide, and citizolam. However, only data relating to the scope of this review (i.e., antibacterial activity) are presented here. According to Rando et al. [47], 5,5′-dinitro-2-(2,3-diaza-4-(2′-tienyl)buta-1,3-dienyl)thiophene (Figure 7) possesses promising antituberculosis activity, as it inhibited the growth of pathogenic Mycobacterium avium and M. kansasei. This compound showed notable levels of mutagenicity as well, which limits its potential for application in clinical practice. Moreover, antimicrobial activity against S. aureus was observed by Scotti et al. [48], achieved by targeting RNA polymerase.
Figure 7.

Chemical structures of 5,5′-dinitro-2-(2,3-diaza-4-(2′-tienyl)buta-1,3-dienyl)thiophene, 2-amino-3-carbethoxy-6-N methyl piperidino thiophene, and thiophene-2-carboxamide.
According to Ramalingam et al. [49], 2-amino-3-carbethoxy-6-N methyl piperidino thiophene (Figure 7) was used as a starting point for the synthesis of novel compounds to be tested for their antibacterial potential using the Kirby–Baurer method. Among the tested compounds, twelve were found to be potent against B. subtilis and E. coli.
The most recent research of Metwally et al. [50] indicated that thiophene-2-carboxamide (Figure 7) derivatives showed antibacterial properties, with S. aureus, B. subtilis, E. coli, and P. aeruginosa being the most susceptible to the activity of the tested compounds. While the results showed no indication of targeting virulence factors, the very fact that the novel synthesized compounds possessed microbicidal activity with no targeted virulence factors whatsoever suggests that there is still hope for old-fashioned drugs as antimicrobials.
The types of activity against pathogenic bacteria presented by indoles, azoles, and thiophenes are presented in Table 1.
Table 1.
Selected compounds and their type of activity against pathogenic bacteria.
| Group of Compounds | Compound | Bacteria | Type of Activity | Reference |
|---|---|---|---|---|
| Indole | 5-halo-1H-indole-2-carboxylic acids | Listeria monocytogenes | Inhibits the growth of bacteria | [25] |
| Indole | indol-2-one with morpholinosulfonyl | Staphylococcus aureus | Inhibitor of DNA gyrase | [26] |
| Indole | thiazolo-indolin-2-one |
S. aureus (ATCC 29213) P. aeruginosa (ATCC 9027) |
Inhibits biofilm formation | [27] |
| Indole | d-pyrimido[4,5-b] indole | Acinetobacter baumanii | Inhibits the growth of bacteria | [28] |
| Indole | 3-amino indoles | Multi-drug resistant A. baumanii |
Inhibits the growth of bacteria | [28] |
| Indole | 4-hydroxy-2-pyridone derivatives containing indolyl | Multi-drug resistant A. baumanii |
Inhibits the growth of bacteria | [28] |
| Indole | 2-hydrazino2-imidazoline | Multi-drug resistant A. baumanii |
Inhibits the growth of bacteria | [28] |
| Indole | bis-indolyl methane | Multi-drug resistant A. baumanii |
Inhibits the growth of bacteria | [28] |
| Indole | 5-iodoindole | A. baumanii, Escherichia coli, S. aureus | Inhibits the growth of bacteria, decreases motility, disrupts biofilm formation | [29] |
| Indole | 3,3′-diindolylmethane |
Cutibacterium acnes
S. aureus |
Inhibits the growth of bacteria | [30] |
| Indole | indole terpenoid compound rhodethrin | Enterococcus faecalis | Inhibits biofilm formation | [31] |
| Indole | indole extract from the supernatant of the rhizobacterium Enterobacter sp. Zch127 | Proteus mirabilis | Inhibits biofilm formation | [32] |
| Indole | 4-chloroindole, 5-chloroindole, 5-chloro 2-methyl indole |
E. coli | Decreases bacterial motility, disrupts biofilm formation | [33] |
| Indole | 4-chloroindole, 6-iodoindole, 5-chloro-2-methyl indole |
Agrobacterium tumefaciens | Decreases swimming motility, the production of exopolysaccharide and exoprotease, and cell surface hydrophobicity and biofilm formation | [35] |
| Indole | indole | V. tasmaniensis LGP32 and V. crassostreae J2-9 | Decreases swimming motility, inhibits biofilm formation |
[36] |
| Indole | 4-chloroindole, 7-chloroindole, 4-iodoindole, and 7-iodoindole | V. parahaemolyticus | Inhibits biofilm formation | [37] |
| Azole | naphthalimide-containing nitroimidazoles | A. baumannii | Inhibits the growth of bacteria | [28] |
| Azole | itraconazole and fluconazole | A. baumannii | Inhibits biofilm formation | [40] |
| Azole | clotrimazole, econazole | Streptococcus mutans | Inhibits biofilm formation | [41] |
| Azole | pyrazole 30 | Pneumocystis vulgaris Klebsiella pneumoniae | Inhibits the growth of bacteria | [42] |
| Azole | 1,4-naphthoquinones linked to 1,2,3-1H-triazoles—compounds (9e, 9h, 9i, and 9j) | S. mutans | Inhibits the growth of bacteria | [43] |
| Azole | binaphthyl-1,2,3-triazole peptidomimetics | A. baumannii | Inhibits the growth of bacteria | [28] |
| Azole | heterocycle compounds with indazolylthiazole moiety (compounds 2, 3, 7, and 8) |
S. mutans, P.aeruginosa | Inhibits biofilm production | [46] |
| Azole | N-(2-(1H-imidazol-4-yl)ethyl)-2-(2,3-dihydroxyphenyl)-N-hydroxy-5-methyloxazole-4-carboxamide | A. baumannii | Inhibits the growth of bacteria | [28] |
| Thiophene | 5,5′-dinitro-2-(2,3-diaza-4-(2′-tienyl)buta-1,3-dienyl)thiophene |
Mycobacterium avium
M. kansasei |
Inhibits the growth of bacteria | [47] |
| Thiophene | 2 -amino-3-carbethoxy-6-N methyl piperidino thiophene |
B. subtilis
E. coli |
Inhibits the growth of bacteria | [49] |
| Thiophene | thiophene-2-carboxamide |
S. aureus, B. subtilis, E. coli, P. aeruginosa |
Inhibits the growth of bacteria | [50] |
5. Pleuromutilin Derivatives
Pleuromutilin (Figure 8), a diterpenoid secondary metabolite with a tricyclic structure, was initially discovered in Pleurotus passeckerianus and P. mutilis mushrooms in 1951 [51]. This compound and its derivatives demonstrated strong antibacterial efficacy against Gram-positive bacteria, mycoplasma, and chlamydia [52] by interacting with the peptidyl transferase core (PTC) of bacterial ribosomes and blocking protein synthesis [53,54].
Figure 8.

Chemical structures of pleuromutilin and lefamulin.
Pleuromutilins bind to the PTC and compete for binding with the 16-atom macrolide and peptidyltransferase inhibitor carbomycin (but not with the 14-atom macrolide erythromycin) [55], inhibiting the formation of peptide bonds [56]. The X-ray crystallography of ribosome-drug complexes was used to identify the precise nature of pleuromutilin binding to the ribosome [57].
Four semi-synthetic derivatives of pleuromutilin have so far received approval for use in the treatment of infectious disorders, including lefamulin (Figure 8) for the treatment of adult community-acquired bacterial pneumonia (CABP) [58], tiamulin and valnemulin for use in veterinary medicine, and retapamulin for use as an antibiotic in the treatment of human skin infections [59,60]. Lefamulin is the only pleuromutilin derivative that has been demonstrated to inhibit the S. aureus cfr (chloramphenicol–florfenicol resistance gene) strain [61].
Chemists have worked very hard to create pleuromutilin derivatives due to its unique mechanism of action and promising antibacterial properties [62].
The gene cluster for pleuromutilin has been described and functionally characterized with regard to its production [63]. The creation of new pleuromutilin-based antibiotics will be aided by the identification of new pleuromutilin derivatives [64].
Lefamulin interferes with the peptidyl transferase center of the 50S ribosome by specifically binding at the A- and P-sites, blocking the formation of peptide bonds. This interferes with the production of bacterial proteins [54].
Lefamulin uses a special induced fit mechanism to close the binding pocket within the ribosome, ensuring the tight binding of the drug to the target site, even though this mechanism of action is similar to that of the oxazolidinones and can actually compete with the phenicols for the same binding site [54]. This is a unique strategy for preventing bacterial peptide chain elongation, especially with the creation of the first peptide bond; however, lefamulin is ineffective once elongation has begun [65]. With the exception of M. pneumoniae, lefamulin presents bacteriostatic characteristics against the majority of species [66]. Lefamulin has exhibited action against all aerobic Gram-positive organisms, except E. faecalis [54].
Likewise, methicillin-resistant S. aureus (MRSA), heterogeneous VISA (hVISA), vancomycin-resistant S. aureus (VRSA), penicillin-resistant S. pneumoniae (PRSP), MDR S. pneumoniae, and vancomycin-resistant E. faecalis (VRE) are among the resistant Gram-positive organisms against which lefamulin is effective [54,67,68].
Tiamulin and valnemulin (Figure 9) attach to the bacterial 50S ribosomal subunit to prevent protein synthesis. It has been shown that these medications interact with 23S RNA’s domain V and are potent inhibitors of peptidyl transferase, leaving distinct chemical traces at the nucleotides A2058-9, U2506, and U2584-5. All of these nucleotides are at or near the PTC and have been linked to the binding of several antibiotics. The majority of them are well conserved both phylogenetically and functionally [69].
Figure 9.

Chemical structures of tiamulin, valnemulin, and retapamulin.
These two compounds can bind alongside the macrolide erythromycin but compete with the macrolide carbomycin, which is a peptidyl transferase inhibitor, according to competitive footprinting. In order to impede the proper placement of the CCA ends of tRNAs for peptide transfer, these two chemicals interact with the rRNA in the peptidyl transferase slot on the ribosomes. Although ribosomal protein uL3 is located adjacent to the tiamulin binding site without coming into contact with the medication, tiamulin only interacts with rRNA residues [70]. Accordingly, tiamulin binds to the 50s subunit’s A site, and the acetic acid tail extends to the P site, interfering with the formation of peptide bonds [70].
Retapamulin (Figure 9) is a pleuromutilin antibiotic that blocks the formation of the 50S ribosomal unit in bacteria, hence inhibiting the production of proteins [71]. It is effective against Gram-positive pathogens, and since 2007, a topical preparation has been licensed in the US for treating skin and soft tissue infections in adults and children older than 9 months [72].
Retapamulin has demonstrated remarkable in vitro and in vivo action against MRSA and MSSA strains of S. aureus in prior studies [73] and has also shown good outcomes against mupirocin-resistant MRSA [72].
6. Albocyclin and General Lactone Derivatives
A class of substances known as lactones is commonly present in nature [74]. Chemically, they can be categorized as variously sized intramolecular esters of hydroxycarboxylic acids. The most prevalent are the lactones with five- and six-membered rings due to the stability of the ring structure [75]. However, alternative ring sizes of lactones can also be extracted from natural sources or produced chemically [76].
Lactones are a very fascinating group that demonstrates various significant biological characteristics as a result of its diversity [77].
The main structure of the lactones group has recently been modified to create new analogs with stronger or different responses. These new analogs can exhibit a toxic effect on the cells of pathogenic bacteria and serve as an alternative to the widely used antibiotics [78].
It is known that bacteriostatic properties are exhibited by substances in which the lactone moiety is present in a small ring, e.g., xanthatin [79], a bicyclic lactone isolated from Xanthium pensylvanicum and X. strumarium, which is active against S. aureus, including MRSA-resistant methicillin strains [80].
Several strains of Streptomyces produce albocycline—a 14-membered macrolactone (Figure 10) [81]. This compound has shown in vitro antimicrobial activity against MRSA and VRSA equipotent to vancomycin [82,83]. Despite this, albocycline may represent a solution for the treatment of infections caused by S. aureus species.
Figure 10.

Chemical structure of albocycline.
A structural motif in the macrolide family of antibiotics, the 14-membered macrolactone of albocycline indicates that it targets the bacterial ribosome and thereby inhibits translation [84].
Albocycline, however, blocks the incorporation of radiolabeled N-acetylglucosamine ([3H]GlcNAc) into the peptidoglycan (PG), the protective polymer surrounding bacterial cells, according to research by Tomoda et al. The first component of bacterial PG production, N-acetylglucosamine (UDP-GlcNAc), accumulates as a result of albocycline’s inhibition [82,85].
Due to albocycline’s non-toxicity in mice and humans, in vivo investigations have suggested increased interest in the drug for potential therapeutic uses. Using human HepG2 hepatocellular liver cancer cells, the authors of [85] showed that albocycline was not harmful to human cells at a final concentration of less than 64 g/mL [83].
7. Glycopeptides
A class of non-ribosomal cyclic or polycyclic peptides known as glycopeptide antibiotics prevents the formation of Gram-positive bacterial cell walls. These substances function as substrate binders (of cell-wall precursors) as opposed to active-site enzyme inhibitors, unlike other antimicrobial classes [86,87,88].
By attacking lipid II (which represents a peptidoglycan-repeat unit that is related to the lipid transporter), glycopeptide antibiotics prevent Gram-positive bacteria from synthesizing PG. As a result, the lipid transporter shared by peptidoglycan and wall teichoic acid (WTA) biosynthesis, bactoprenol phosphate, cannot be recycled [89]. With each contributing almost 50% of the dry cell-wall weight, PG and WTA are two important parts of the cell wall. Through host attachment, colonization, infection, biofilm development, and the recruitment of penicillin-binding proteins (PBPs) to the septum during cell division, WTA plays a significant role in the pathogenicity of microbes [90]. Consequently, it serves as a desirable target for the creation of new antibiotics [89].
The oldest member of the class is vancomycin (Figure 11), while the more recent lipoglycopeptide derivatives oritavancin, teicoplanin, telavancin, and dalbavancin (Figure 12, Figure 13, Figure 14 and Figure 15) were developed specifically to boost antibacterial activity, sometimes via secondary modes of action.
Figure 11.

Chemical structure of vancomycin.
Figure 12.

Chemical structure of oritavancin.
Figure 13.

Chemical structure of teicoplanin.
Figure 14.

Chemical structure of telavancin.
Figure 15.

Chemical structure of dalbavancin.
The transglycosylation stage of PG production, which is necessary to replenish the lipid transporter, is prevented by glycopeptide antibiotic binding to lipid II. Therefore, for instance, when vancomycin is added to S. aureus during growth, Park’s nucleotide, a cytoplasmic PG-precursor, accumulates [91]. Vancomycin binding to lipid II is an efficient way to suppress both PG and wall teichoic acid biosynthesis in S. aureus [89], since C55 is present in a surprisingly low number of copies per bacterium [92] and is a shared transporter needed in these processes [93].
Vancomycin is used to treat acute infections caused by Gram-positive organisms. By attaching to the D-Ala-D-Ala terminus of lipid II, a PG precursor tethered to the cell membrane by the lipid transporter bactoprenol-phosphate, vancomycin suppresses the formation of PG (C55-P). To stop C55-P regeneration, vancomycin-bound lipid II is sequestered from the PG biosynthesis transglycosylation step. Vancomycin’s sequestration of lipid II causes the cytoplasmic buildup of Park’s nucleotide [91], a cytoplasmic PG precursor, because C55 is present in bacteria in low concentrations [94]. When the dipeptide is swapped out for a depsipeptide D-Ala-D-Lac, vancomycin is unable to attach to the D-Ala-D-Ala terminus of lipid II in VRE [95].
As a result of investigating the structure–activity relationship of chloroeremomycin to combat vancomycin resistance, oritavancin was discovered [96], a semi-synthetic lipoglycopeptide that has potent antimicrobial effects against vancomycin-resistant organisms such as VRE and S. aureus resistant to vancomycin (VRSA) [97].
Oritavancin is currently a top therapeutic option for treating serious infections brought on by Gram-positive organisms that are multi-drug-resistant, such MRSA [89]. Oritavancin’s chemical composition differs from vancomycin’s due to the inclusion of a N-alkylated chlorobiphenyl side chain in the drug sugar’s epivancosamine. In general, adding a hydrophobic side chain to the glycopeptide disaccharide greatly increases the medications’ overall effectiveness and revives their activity against vancomycin-resistant bacteria. By using solid-state NMR to structurally characterize the binding site of these disaccharide-modified glycopeptides in S. aureus [98] and E. faecium [99] intact whole cells, it was discovered that the drug’s hydrophobic side chain creates a secondary binding site. The lipoglycopeptides can target the cross-linked PG-bridge structure using this secondary binding site to aid in binding [100]. Oritavancin’s binding to the developing PG prevents transpeptidase from effectively recognizing the PG template, which is necessary for effective PG cross-linking during cell-wall synthesis [101].
Teicoplanin is used to treat multidrug-resistant Gram-positive bacteria, such as MRSA and Enterococci, that are responsible for life-threatening infectious illnesses [102]. This glycopeptide antibiotic was initially isolated from Actinoplanes teichomyceticus, which was identified in 1978 from an Indian soil sample [103].
Teicoplanin shares structural similarities with vancomycin but differs in that it does not contain a lipid. Both antibiotics work by forming hydrogen bonds with the D-Ala-D-Ala C-terminus of the pentapeptide substrate to prevent the formation of the peptidoglycan chains that make up bacterial cell walls [104]. The hydrophobic lipid chain of this pentapeptide substrate is also known to interact with teicoplanin, placing the antibiotic next to the peptidoglycan [102,105].
Derivatives of teicoplanin have also been shown to form nanoscale aggregates in aqueous solution [106], thereby achieving increased binding power [107].
The oral and topical routes of administration for teicoplanin may result in poor permeability across the epithelial lining due to this concentration-dependent aggregation, and the aggregated form may reduce effective concentrations on certain sites, necessitating a higher dose and ultimately causing bacteria to develop resistance [108].
Another lipoglycopeptide derivative of vancomycin is telavancin (TD-6424). This was developed as a cutting-edge treatment for MRSA and other resistant Gram-positive bacterial infections [109]. The United States Food and Drug Administration (USFDA) granted telavancin approval in 2009 for the treatment of difficult skin and skin structure infections (cSSSIs) caused by Gram-positive bacteria, including MRSA, S. aureus, Streptococcus agalactiae, S. pyogenes, the S. anginosus group, and E. faecalis [110,111].
Two modes of action for telavancin have been suggested. Telavancin achieves bactericidal activity by interacting with the C-terminal d-alanyl-d-alanine residue on bacterial cell-wall peptidoglycan precursors, just like vancomycin. This interaction significantly alters the phases of cell-wall formation that include the polymerization of peptidoglycan (transglycosylation) and subsequent cross-linking (transpeptidation) [112]. Telavancin is 10-times more effective than vancomycin at inhibiting peptidoglycan production in intact MRSA cells because it strongly inhibits peptidoglycan generation at the transglycosylase stage.
Furthermore, a second mechanism of action has been mentioned. The depolarization of the bacterial cell membrane is involved, which affects how the cell membrane functions. Given that so few other glycopeptides are thought to function in this way, this dual method of action is of special importance [109]. The interaction of the lipophilic decylaminoethyl moiety of telavancin with the lipid bilayer of the bacterial cell membrane is thought to be the process by which telavancin disrupts cell membranes, albeit this is not fully understood [113]. Telavancin’s affinity for lipid II, a molecule found in bacterial cell membranes, is facilitated by this lipophilic substance.
By disrupting the bacterial cell-wall transglycosylation pathway rather than the bacterial cell-wall transpeptidation mechanism, where vancomycin preferentially binds, telavancin is able to enter the bacterial cell with ease [114].
According to reports, lipid II binding is necessary for telavancin to cause membrane depolarization in S. aureus. This might not, however, accurately reflect the crucial phase of bacterial membrane disruption [115]. The loss of potassium ions and cytoplasmic adenosine triphosphate (ATP) may also be related to membrane depolarization. Telavancin’s faster bactericidal impact compared to vancomycin may be caused by this alternative method of action, which only affects bacterial cell membranes and not mammalian cells [112].
Dalbavancin is a semisynthetic derivative of teicoplanin. It is active against most pathogenic Gram-positive organisms, including Streptococcus spp., E. faecalis, E. faecium, MSSA, MRSA, and vancomycin-intermediate S. aureus. However, it has poor activity against vancomycin-resistant S. aureus and VRE [116].
Similarly to vancomycin and other glycopeptides, dalbavancin inhibits cell-wall formation by interacting with the D-alanyl-D-alanine terminus in the bacterial cell-wall peptidoglycan and blocking cross-linking.
In the USA and Europe, acute bacterial skin and skin structure infections (ABSSSIs) are the only conditions for which dalbavancin is currently licensed [117].
8. Chalcones
Due to the hues of the majority of naturally occurring chalcones, the name “chalcone” was derived from the Greek word “chalcos”, which means “bronze”. 1,3-diaryl-2-propen-1-one (Figure 16), also referred to as chalconoid, is a chemical building block shared by all chalcone molecules. The trans isomer is thermodynamically more stable than the cis isomer. Through the use of plants and herbs for the treatment of many diseases, such as cancer, inflammation, and diabetes, chalcones have been applied therapeutically for thousands of years. Several chalcone-based substances have received clinical use authorization.
Figure 16.

General structure of 1,3-diaryl-2-propen-1-ones.
Chalcones are a class of natural and synthetic compounds that have shown promising antivirulence properties against a variety of pathogenic bacteria. With the rise of antibiotic-resistant strains, there is an urgent need to develop alternative therapies that target virulence factors of bacteria, rather than traditional bactericidal approaches. In recent years, several studies have investigated the antivirulence potential of chalcones in resistant bacterial strains.
Several studies have investigated the activity of chalcones against multidrug-resistant Pseudomonas aeruginosa and found that they were able to inhibit the expression of virulence genes involved in quorum sensing, motility, and biofilm formation [118,119].
Furthermore, a study on Acinetobacter baumannii, a notorious multi-drug resistant pathogen, showed that chalcones exhibited significant antivirulence activity by modulating gene expression, biofilm formation, and virulence traits [120].
These studies suggested that chalcones have potential as antivirulence agents against resistant bacterial strains by targeting various virulence traits. However, further studies are needed to evaluate the efficacy of chalcones in vivo and their potential as a therapeutic option for antibiotic-resistant infections.
Overall, it can be concluded that, since virulence factors are essential for the infection of the host, techniques employed to prevent this process from initiating and search for novel bioactive compounds with these properties are rather appealing.
Along with the synthetic and hemi-synthetic compounds elaborated in this review article, numerous naturally derived compounds have also demonstrated great potential as efficient virulence-targeting compounds. Dehydroabietic acid showed considerable potential against several pathogenic microorganisms, especially Pseudomonas syringae pv. actinidiae, Xanthomonas oryzae pv. oryzae, and Xanthomonas axonopodis pv. citri [121]. Furthermore, for some natural compounds, a mode of action has even been proposed. For example, the exposure of Serratia marcescens to hordenine (25, 50, and 100 g/mL) reduced the synthesis of acyl-homoserine lactones and prevented the development of biofilms. It also increased the susceptibility of preformed biofilms to commercial antibiotic ciprofloxacin by lowering extracellular polysaccharide production and altering membrane permeability. Additionally, the presence of hordenine downregulated expression and affected genes associated with biofilm and QS [122], which may be explored in other matrices. Additionally, even though some compounds do not exert anti-QS activities per se, they can sometimes be easily modified into compounds that do exert various bioactivities, as was argued by Du et al. [123]. This comprehensive review article offered new research solutions; proposed novel strategies; and compared existing results, leading to new conclusions.
9. Future Perspectives
The use of novel synthetic and semisynthetic compounds that target virulence factors as antibacterials presents a promising avenue for combating antibiotic resistance. However, there are several challenges that need to be addressed in order to fully realize the potential of this approach. One of the challenges is the identification of new compounds with antibacterial potency that can target virulence factors. Despite recent progress in this area, many of the compounds that have been investigated are not yet ready for clinical use. The process of discovering, developing, and testing new compounds can be time-consuming and expensive, and there is a need for new screening methods and assays to identify potential candidates more efficiently. Another challenge is the optimization of the efficacy and safety of existing compounds. Many of the compounds that have been identified have shown promising results in vitro, but their efficacy in vivo and safety in humans need to be further evaluated. In addition, the development of resistance to these compounds is a potential concern, and efforts must be made to prevent or delay the emergence of resistance. Furthermore, there is a need for an improved understanding of the mechanisms of action of these compounds. Many of the compounds that target virulence factors have complex modes of action that are not yet fully understood. A deeper understanding of these mechanisms could lead to the development of more effective compounds, as well as the identification of new targets for antibacterial therapy.
There are ongoing clinical trials on novel antivirulence drugs that are trying to take the next steps forward within this area, evaluating the safety and efficacy of novel synthetic and semisynthetic compounds. These trials are being conducted by pharmaceutical companies, academic institutions, and government agencies around the world.
Despite the many challenges, the potential benefits of targeting virulence factors as a strategy to combat antibiotic resistance are significant. By reducing the severity of bacterial infections without promoting the development of resistance, this approach could help to extend the lifespan of existing antibiotics and reduce the need for new ones. In addition, the use of antibacterials that target virulence factors could help to reduce the burden of antibiotic-resistant infections, which are a major public health concern.
10. Conclusions
The development of novel antibacterials that target virulence factors is an area of active research aimed at addressing the global challenge of antibiotic resistance. The potential benefits of these compounds lie in their ability to attenuate bacterial pathogenesis without necessarily killing the bacteria, thus reducing selective pressure for resistance development. While the field is still in its early stages, the progress made so far is promising. The use of synthetic and semisynthetic compounds has emerged as an important strategy to combat antibiotic resistance. The compounds reviewed in this paper—chalcones, azoles, indoles, thiophenes, terpenoids, glycopeptides, pleuromutilin derivatives, and lactone derivatives—have shown potential as antibacterials mostly targeting virulence traits in resistant strains. Clinical trials evaluating the safety and efficacy of these compounds are ongoing, and their results will provide critical insights into the role of virulence-targeted antibacterials in the management of bacterial infections. However, given the complexity of bacterial pathogenesis and the evolution of resistance mechanisms, the development of novel antibacterials remains a challenging task. Further research is required to identify novel targets and to optimize the efficacy and safety of these compounds. Additionally, efforts are needed to overcome the regulatory and economic hurdles that often hinder the development and commercialization of novel antibacterial agents.
The development of novel antibacterials that target virulence factors offers a promising avenue for combating antibiotic resistance. While there is still much work to be carried out, the progress made so far suggests that these compounds have the potential to play an important role in the management of bacterial infections in the future.
Author Contributions
Conceptualization, D.S., K.L; investigation, D.S., J.P., T.C., M.S. and K.L.; resources, D.S and K.L.; writing—original draft preparation, D.S., J.P. and T.C.; writing—review and editing, M.S. and K.L. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (451-03-68/2022-14/200007).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Piddock L.J. Reflecting on the final report of the O’Neill review on antimicrobial resistance. Lancet Infect. Dis. 2016;16:767–768. doi: 10.1016/S1473-3099(16)30127-X. [DOI] [PubMed] [Google Scholar]
- 2.Rasko D.A., Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 3.Zhang F., Cheng W. The Mechanism of Bacterial Resistance and Potential Bacteriostatic Strategies. Antibiotics. 2022;11:1215. doi: 10.3390/antibiotics11091215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clatworthy A.E., Pierson E., Hung D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 5.Fischbach M.A., Walsh C.T. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annunziato G. Strategies to Overcome Antimicrobial Resistance (AMR) Making Use of Non-Essential Target Inhibitors: A Review. Int. J. Mol. Sci. 2019;20:5844. doi: 10.3390/ijms20235844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dörr T., Lewis K., Vulic M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 2009;5:e1000760. doi: 10.1371/journal.pgen.1000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flemming H.C., Wingender J. The biofilm matrix. Nat. Rev. Microbiol. 2010;8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 9.Kalia V.C. Quorum sensing inhibitors: An overview. Biotechnol. Adv. 2013;31:224–245. doi: 10.1016/j.biotechadv.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Bhat A., Ahmad I. Anti-virulence strategies: Stalling the micromachines of bacterial pathogens. Front. Microbiol. 2018;9:1456. doi: 10.3389/fmicb.2018.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeong J.W., Lee K.Y., Kim J.S. Natural products as sources of new fungicides. J. Microbiol. Biotechnol. 2009;19:1263–1270. [Google Scholar]
- 12.Gutiérrez-Barranquero J.A., Reen F.J., McCarthy R.R., O’Gara F. Deciphering the role of coumarin as a novel quorum sensing inhibitor suppressing virulence phenotypes in bacterial pathogens. Appl. Microbiol. Biotechnol. 2015;99:3303–3316. doi: 10.1007/s00253-015-6436-1. [DOI] [PubMed] [Google Scholar]
- 13.Frederich M., Tits M., Angenot L. Potential antimalarial activity of indole alkaloids. Trans R. Soc. Trop. Med. Hyg. 2008;102:11–19. doi: 10.1016/j.trstmh.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Singh R., Dubey A.K. Lactone derivatives as potential antibacterial agents. Med. Chem. Res. 2018;27:1589–1611. [Google Scholar]
- 15.Okano A., Isley N.A., Boger D.L. Peripheral modifications of [9]dodecanylglycylgramicidin A: Discovery of novel antimicrobial agents with high potency and selectivity against methicillin-resistant Staphylococcus aureus (MRSA) J. Am. Chem. Soc. 2017;139:944–955. [Google Scholar]
- 16.El-Messery S.M., Habib E.E., Al-Rashood S.T.A., Hassan G.S. Synthesis, antimicrobial, anti-biofilm evaluation, and molecular modelling study of new chalcone linked amines derivatives. J. Enzym. Inhib. Med. Chem. 2018;33:818–832. doi: 10.1080/14756366.2018.1461855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng X.M., Cai G.X., Zhou C.H. Recent developments in azole compounds as antibacterial and antifungal agents. Curr. Top. Med. Chem. 2013;13:1963–2010. doi: 10.2174/15680266113139990125. [DOI] [PubMed] [Google Scholar]
- 18.Deryabin D., Inchagova K., Rusakova E., Duskaev G. Coumarin’s Anti-Quorum Sensing Activity Can Be Enhanced When Combined with Other Plant-Derived Small Molecules. Molecules. 2021;26:208. doi: 10.3390/molecules26010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sethupathy S., Sathiyamoorthi E., Kim Y.G., Lee J.H., Lee J. Antibiofilm and Antivirulence Properties of Indoles Against Serratia marcescens. Front. Microbiol. 2020;11:584812. doi: 10.3389/fmicb.2020.584812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hooper D.C., Jacoby G.A. Topoisomerase Inhibitors: Fluoroquinolone Mechanisms of Action and Resistance. Cold Spring Harb. Perspect.Med. 2016;6(9):a025320. doi: 10.1101/cshperspect.a025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cappiello F., Loffredo M.R., Del Plato C., Cammarone S., Casciaro B., Quaglio D., Mangoni M.L., Botta B., Ghirga F. The Revaluation of Plant-Derived Terpenes to Fight Antibiotic-Resistant Infections. Antibiotics. 2020;9:325. doi: 10.3390/antibiotics9060325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Groesen E., Innocenti P., Martin N.I. Recent Advances in the Development of Semisynthetic Glycopeptide Antibiotics: 2014–2022. ACS Infect. Dis. 2022;8:1381–1407. doi: 10.1021/acsinfecdis.2c00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shang R., Wang S., Xu X., Yi Y., Guo W., Liu Y., Liang J. Chemical synthesis and biological activities of novel pleuromutilin derivatives with substituted amino moiety. PLoS ONE. 2013;8:e82595. doi: 10.1371/journal.pone.0082595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breijyeh Z., Karaman R. Design and Synthesis of Novel Antimicrobial Agents. Antibiotics. 2023;12:628. doi: 10.3390/antibiotics12030628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balcerek M., Szmigiel-Bakalarz K., Lewańska M., Günther D., Oeckler O., Malik M., Morzyk-Ociepa B. Experimental and computational study on dimers of 5-halo-1H-indole-2-carboxylic acids and their microbiological activity. J. Mol. Struct. 2023;1274:134492. doi: 10.1016/j.molstruc.2022.134492. [DOI] [Google Scholar]
- 26.Salem M.A., Ragab A., El-Khalafawy A., Makhlouf A.H., Askar A.A., Ammar Y.A. Design, synthesis, in vitro antimicrobial evaluation and molecular docking studies of indol-2-one tagged with morpholinosulfonyl moiety as DNA gyrase inhibitors. Bioorg. Chem. 2020;96:103619. doi: 10.1016/j.bioorg.2020.103619. [DOI] [PubMed] [Google Scholar]
- 27.Alzahrani Y.A., Ammar M., Abu-Elghait M.A., Salem M., Assiri T.E., Ali R. Development of novel indolin-2-one derivative incorporating thiazole moiety as DHFR and quorum sensing inhibitors: Synthesis, antimicrobial, and antibiofilm activities with molecular modelling study. Bioorg. Chem. 2022;119:105571. doi: 10.1016/j.bioorg.2021.105571. [DOI] [PubMed] [Google Scholar]
- 28.Srikanth D., Joshi S.V., Shaik M.G., Pawar G., Bujji S., Kanchupalli V., Chopra S., Nanduri S. A comprehensive review on potential therapeutic inhibitors of nosocomial Acinetobacter baumannii superbugs. Bioorg. Chem. 2022;124:105849. doi: 10.1016/j.bioorg.2022.105849. [DOI] [PubMed] [Google Scholar]
- 29.Raorane C.J., Lee J.H., Lee J. Rapid Killing and Biofilm Inhibition of Multidrug-Resistant Acinetobacter baumannii Strains and Other Microbes by Iodoindoles. Biomolecules. 2020;10:1186. doi: 10.3390/biom10081186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim Y.G., Lee J.H., Park S., Lee J. The Anticancer Agent 3,3′-Diindolylmethane Inhibits Multispecies Biofilm Formation by Acne-Causing Bacteria and Candida albicans. Microbiol. Spectr. 2022;10:e0205621. doi: 10.1128/spectrum.02056-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tatta E.R., Kumavath R. Attenuation of Enterococcus faecalis biofilm formation by Rhodethrin: A combinatorial study with an antibiotic. Microb. Pathog. 2022;163:105401. doi: 10.1016/j.micpath.2022.105401. [DOI] [PubMed] [Google Scholar]
- 32.Amer M.A., Ramadan M.A., Attia A.S., Wasfi R. Silicone Foley catheter simpregnated with microbial indole derivatives inhibit crystalline biofilm formation by Proteus mirabilis. Front. Cell. Infect. Microbiol. 2022;12:1010625. doi: 10.3389/fcimb.2022.1010625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boya B.R., Lee J.H., Lee J. Antibiofilm and Antimicrobial Activities of Chloroindoles Against Uropathogenic Escherichia coli. Front. Microbiol. 2022;13:872943. doi: 10.3389/fmicb.2022.872943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cernicchi G., Felicetti T., Sabatini S. Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives. Molecules. 2021;26:6996. doi: 10.3390/molecules26226996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed B., Jailani A., Lee J.H., Lee J. Effect of halogenated indoles on biofilm formation, virulence, and root surface colonization by Agrobacterium tumefaciens. Chemosphere. 2022;293:133603. doi: 10.1016/j.chemosphere.2022.133603. [DOI] [PubMed] [Google Scholar]
- 36.Zhang S., Yang Q., Fu S., Janssen C., Eggermont M., Defoirdt T. Indole decreases the virulence of the bivalve model pathogens Vibrio tasmaniensis LGP32 and Vibrio crassostreae J2-9. Sci. Rep. 2022;12:5749. doi: 10.1038/s41598-022-09799-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sathiyamoorthi E., Faleye O.S., Lee J.H., Raj V., Lee J. Antibacterial and Antibiofilm Activities of Chloroindoles Against Vibrio parahaemolyticus. Front. Microbiol. 2021;12:714371. doi: 10.3389/fmicb.2021.714371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z., Sun F., Fu X., Chen Y. 5-Methylindole kills various bacterial pathogens and potentiates aminoglycoside against methicillin-resistant Staphylococcus aureus. PeerJ. 2022;10:e14010. doi: 10.7717/peerj.14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nobre L.S., Todorovic S., Tavares A.F., Oldfield E., Hildebrandt P., Teixeira M., Saraiva L.M. Binding of azole antibiotics to Staphylococcus aureus flavohemoglobin increases intracellular oxidative stress. J. Bacteriol. 2010;192:1527–1533. doi: 10.1128/JB.01378-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olaifa K., Ajunwa O., Marsili E. Electroanalytic evaluation of antagonistic effect of azole fungicides on Acinetobacter baumannii biofilms. Electrochim. Acta. 2022;405:139837. doi: 10.1016/j.electacta.2022.139837. [DOI] [Google Scholar]
- 41.Qiu W., Ren B., Dai H., Zhang L., Zhang Q., Zhou X., Li Y. Clotrimazole and econazole inhibit Streptococcus mutans biofilm and virulence in vitro. Arch. Oral. Biol. 2017;73:113–120. doi: 10.1016/j.archoralbio.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 42.Nasr M., Bondock S., Eid S. Design, synthesis, antimicrobial evaluation and molecular docking studies of some new thiophene, pyrazole and pyridone derivatives bearing sulfisoxazole moiety. Eur. J. Med. Chem. 2014;84:491–504. doi: 10.1016/j.ejmech.2014.07.052. [DOI] [PubMed] [Google Scholar]
- 43.Gomes M.P., Correia E.M., Gomes M.W.L., Santos C.C.C., Barros C.S., Abreu F.V., de Antunes L.S., Ferreira V.F., Gonçalves M.C., Pinto C.E.C., et al. Antibacterial profile in vitro and in vivo of new 1,4-naphthoquinones tethered to 1,2,3-1h-triazoles against the planktonic growth of Streptococcus mutans. J. Braz. Chem. Soc. 2022;33:1028–1040. doi: 10.21577/0103-5053.20220014. [DOI] [Google Scholar]
- 44.Sapijanskaite-Banevic B., Palskys V., Vaickelioniene R., Šiugždaite J., Kavaliauskas P., Grybaite B., Mickevicius V. Synthesis and Antibacterial Activity of New Azole, Diazole and Triazole Derivatives Based on p-Aminobenzoic Acid. Molecules. 2021;26:2597. doi: 10.3390/molecules26092597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nechaeva O.V., Tikhomirova E.I., Zayarsky D.A., Bespalova N.V., Glinskaya E.V., Shurshalova N.F., Al Bayati B.M., Babailova A.I. AntiBiofilm Activity of Polyazolidinammonium Modified with Iodine Hydrate Ions against Microbial Biofilms of Uropathogenic Coliform Bacteria. Bull. Exp. Biol. Med. 2017;162:781–783. doi: 10.1007/s10517-017-3712-3. [DOI] [PubMed] [Google Scholar]
- 46.Dawoud N.T.A., El-Fakharany E.M., Abdallah A.E., El-Gendi H., Lotfy D.R. Synthesis, and docking studies of novel heterocycles incorporating the indazolyl thiazole moiety as antimicrobial and anticancer agents. Sci. Rep. 2022;12:3424. doi: 10.1038/s41598-022-07456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rando D.G., Doriguetto A.C., Tomich de Paula da Silva C.H., Ellena J., Sato D.N., Leite C.Q., Varanda E.A., Ferreira E.I. A duplicated nitrotienyl derivative with antimycobacterial activity: Synthesis, X-ray crystallography, biological and mutagenic activity tests. Eur. J. Med. Chem. 2006;41:1196–1200. doi: 10.1016/j.ejmech.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 48.Scotti L., Oliveira Lima E., da Silva M.S., Ishiki H., Oliveira Lima I., Oliveira Pereira F., Mendonça Junior F.J., Scotti M.T. Docking and PLS studies on a set of thiophenes RNA polymerase inhibitors against Staphylococcus aureus. Curr. Top. Med. Chem. 2014;14:64–80. doi: 10.2174/1568026613666131113151347. [DOI] [PubMed] [Google Scholar]
- 49.Ramalingam A., Sarvanan J. Synthesis, Docking and Antimicrobial Activity Studies of Some Novel Fused Thiophenes of Biological Interest. J. Young Pharm. 2020;12:118–124. doi: 10.5530/jyp.2020.12.24. [DOI] [Google Scholar]
- 50.Metwally H.M., Khalaf N.A., Abdel-Latif E. Synthesis, DFT investigations, antioxidant, antibacterial activity and SAR-study of novel thiophene-2-carboxamide derivatives. BMC Chem. 2023;17:6. doi: 10.1186/s13065-023-00917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kavanagh F., Hervey A., Robbins W.J. Antibiotic substances from basidiomycetes. Proc. Natl. Acad. Sci. USA. 1951;37:570–574. doi: 10.1073/pnas.37.9.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li B., Zhang Z., Zhang J.F., Liu J., Zuo X.Y., Chen F., Zhang G.Y., Fang H.Q., Jin Z., Tang Y.Z. Design, synthesis and biological evaluation of pleuromutilin-Schiff base hybrids as potent anti-MRSA agents in vitro and in vivo. Eur. J. Med. Chem. 2021;223:113624. doi: 10.1016/j.ejmech.2021.113624. [DOI] [PubMed] [Google Scholar]
- 53.Hogenauer G. The mode of action of pleuromutilin derivatives: Location and properties of pleuromutilin binding site on Escherichia coli ribosomes. Eur. J. Biochem. 1975;52:93–98. doi: 10.1111/j.1432-1033.1975.tb03976.x. [DOI] [PubMed] [Google Scholar]
- 54.Paukner S., Riedl R. Pleuromutilins: Potent drugs for resistant bugs-mode of action and resistance. Cold Spring Harb. Perspect. Med. 2017;7:a027110. doi: 10.1101/cshperspect.a027110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poulsen S.M., Karlsson M., Johansson L.B., Vester B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2001;41:1091–1099. doi: 10.1046/j.1365-2958.2001.02595.x. [DOI] [PubMed] [Google Scholar]
- 56.Hodgin L.A., Högenauer G. The mode of action of pleuromutilin derivatives. Effect on cell-free polypeptide synthesis. Eur. J. Biochem. 1974;47:527–533. doi: 10.1111/j.1432-1033.1974.tb03721.x. [DOI] [PubMed] [Google Scholar]
- 57.Gürel G., Blaha G., Moore P.B., Steitz T.A. U2504 determines the species specificity of the A-site cleft antibiotics: The structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009;389:146–156. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adhikary S., Duggal M.K., Nagendran S., Chintamaneni M., Tuli H.S., Kaur G. Lefamulin: A new hope in the field of community-acquired bacterial pneumonia. Cur. Pharm. Rep. 2022;8:418–426. doi: 10.1007/s40495-022-00297-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Veve M.P., Wagner J.L. Lefamulin: Review of a promising novel pleuromutilin antibiotic. Pharmacotherapy. 2018;38:935–946. doi: 10.1002/phar.2166. [DOI] [PubMed] [Google Scholar]
- 60.Yi Y., Zhang J., Zuo J., Zhang M., Yang S., Huang Z., Li G., Shang R., Lin S. Novel pyridinium cationic pleuromutilin analogues overcoming bacterial multidrug resistance. Eur. J. Med. Chem. 2023;251:115269. doi: 10.1016/j.ejmech.2023.115269. [DOI] [PubMed] [Google Scholar]
- 61.Deng Y., Tang D., Wang Q.R., Huang S., Fu L.Z., Li C.H. Semi-synthesis, antibacterial activity, and molecular docking study of novel pleuromutilin derivatives bearing cinnamic acids moieties. Arch. Pharm. 2019;352:e1800266. doi: 10.1002/ardp.201800266. [DOI] [PubMed] [Google Scholar]
- 62.Farney E.P., Feng S.S., Schafers F., Reisman S.E. Total synthesis of (+)-pleuromutilin. J. Am. Chem. Soc. 2018;140:1267–1270. doi: 10.1021/jacs.7b13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alberti F., Khairudin K., Venegas E.R., Davies J.A., Hayes P.M., Willis C.L., Bailey A.M., Foster G.D. Heterologous expression reveals the biosynthesis of the antibiotic pleuromutilin and generates bioactive semi-synthetic derivatives. Nat. Commun. 2017;8:1831. doi: 10.1038/s41467-017-01659-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yi Y., Yang S., Liu Y., Yin B., Zhao Z., Li G., Huang Z., Chen L., Liu F., Shang R., et al. Antibiotic resistance and drug modification: Synthesis, characterization and bioactivity of newly modified potent pleuromutilin derivatives with a substituted piperazine moiety. Bioorg. Chem. 2023;132:106353. doi: 10.1016/j.bioorg.2023.106353. [DOI] [PubMed] [Google Scholar]
- 65.Novak R. Are pleuromutilin antibiotics finally fit for human use? Ann. N. Y. Acad. Sci. 2011;1241:71–81. doi: 10.1111/j.1749-6632.2011.06219.x. [DOI] [PubMed] [Google Scholar]
- 66.Waites K.B., Crabb D.M., Duffy L.B., Jensen J.S., Liu Y., Paukner S. In vitro activities of lefamulin and other antimicrobial agents against macrolidesusceptible and macrolide-resistant Mycoplasma pneumoniae from the United States, Europe, and China. Antimicrob. Agents Chemother. 2017;61:e02008-16. doi: 10.1128/AAC.02008-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sader H.S., Biedenbach D.J., Paukner S., Ivezic-Schoenfeld Z., Jones R.N. Antimicrobial activity of the investigational pleuromutilin compound BC3781 tested against Gram-positive organisms commonly associated with acute bacterial skin and skin structure infections. Antimicrob. Agents Chemother. 2012;56:1619–1623. doi: 10.1128/AAC.05789-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Afshari A., Taheri S., Hashemi M., Norouzy A., Nematy M., Mohamadi S. Methicillin- and Vancomycin-Resistant Staphylococcus aureus and Vancomycin-Resistant Enterococci Isolated from Hospital Foods: Prevalence and Antimicrobial Resistance Patterns. Curr. Microbiol. 2022;79:326. doi: 10.1007/s00284-022-03022-0. [DOI] [PubMed] [Google Scholar]
- 69.Tang Y.Z., Liu Y.H., Chen J.X. Pleuromutilin and its derivatives-the lead compounds for novel antibiotics. Mini Rev. Med. Chem. 2012;12:53–61. doi: 10.2174/138955712798868968. [DOI] [PubMed] [Google Scholar]
- 70.Killeavy E.E., Jogl G., Gregory S.T. Tiamulin-Resistant Mutants of the Thermophilic Bacterium Thermus thermophilus. Antibiotics. 2020;9:313. doi: 10.3390/antibiotics9060313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klitgaard R.N., Ntokou E., Norgaard K., Biltoft D., Hansen L.H., Traedholm N.M., Kongsted J., Vester B. Mutations in the bacterial ribosomal protein l3 and their association with antibiotic resistance. Antimicrob. Agents Chemother. 2015;59:3518–3528. doi: 10.1128/AAC.00179-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patel A.B., Lighter J., Fulmer Y., Copin R., Ratner A.J., Shopsin B. Retapamulin Activity Against Pediatric Strains of Mupirocin-resistant Methicillin-resistant Staphylococcus aureus. Pediatr. Infect. Dis. J. 2021;40:637–638. doi: 10.1097/INF.0000000000003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh R., Gombosev A., Dutciuc T., Evans K., Portillo L.M., Hayden M.K., Gillen D., Peterson E., Tjoa T., Cao C., et al. Randomized Double-Blinded Placebo-Controlled Trial to Assess the Effect of Retapamulin for Nasal Decolonization of Mupirocin-Resistant Methicillin-Resistant Staphylococcus aureus Nasal Carriers. Open Forum. Infect. Dis. 2016;3((Suppl. S1)):301. doi: 10.1093/ofid/ofw172.167. [DOI] [Google Scholar]
- 74.Surowiak A.K., Balcerzak L., Lochyński S., Strub D.J. Biological Activity of Selected Natural and Synthetic Terpenoid Lactones. Int. J. Mol. Sci. 2021;22:5036. doi: 10.3390/ijms22095036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sartori S.K., Diaz M.A.N., Diaz-Muñoz G. Lactones: Classification, Synthesis, Biological Activities, and Industrial Applications. Tetrahedron. 2021;84:132001. doi: 10.1016/j.tet.2021.132001. [DOI] [Google Scholar]
- 76.Fan B.Z., Hiasa H., Lv W., Brody S., Yang Z.Y., Aldrich C., Cushman M., Liang J.H. Design, Synthesis and Structure-Activity Relationships of Novel 15-Membered Macrolides: Quinolone/Quinoline-Containing Sidechains Tethered to the C-6 Position of Azithromycin Acylides. Eur. J. Med. Chem. 2020;193:112222. doi: 10.1016/j.ejmech.2020.112222. [DOI] [PubMed] [Google Scholar]
- 77.Mazur M., Masłowiec D. Antimicrobial Activity of Lactones. Antibiotics. 2022;11:1327. doi: 10.3390/antibiotics11101327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kowalczyk P., Gawdzik B., Trzepizur D., Szymczak M., Skiba G., Raj S., Kramkowski K., Lizut R., Ostaszewski R. δ-Lactones-A New Class of Compounds That Are Toxic to E. coli K12 and R2-R4 Strains. Materials. 2021;14:2956. doi: 10.3390/ma14112956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yokoe H., Yoshida M., Shishido K. Total synthesis of (-)-Xanthatin. Tetrahedron Lett. 2008;49:3504–3506. doi: 10.1016/j.tetlet.2008.03.081. [DOI] [Google Scholar]
- 80.Matsuo K., Ohtsuki K., Yoshikawa T., Shisho K., Yokotani-Tomita K., Shinto M. Total synthesis of xanthanolides. Tetrahedron. 2010;66:8407–8419. doi: 10.1016/j.tet.2010.08.061. [DOI] [Google Scholar]
- 81.Nagahama N., Suzuki M., Awataguc S., Okuda T. Studies on a new antibiotic, albocycline. I. isolation, purification and properties. J. Antibiot. 1967;20:261–266. [PubMed] [Google Scholar]
- 82.Koyama N., Yotsumoto M., Onaka H., Tomoda H. New structural scaffold 14-membered macrocyclic lactone ring for selective inhibitors of cell wall peptidoglycan biosynthesis in Staphylococcus aureus. J. Antibiot. 2013;66:303–304. doi: 10.1038/ja.2012.122. [DOI] [PubMed] [Google Scholar]
- 83.Daher S.S., Franklin K.P., Scherzi T., Dunman P.M., Andrade R.B. Synthesis and biological evaluation of semi-synthetic albocycline analogs. Bioorg. Med. Chem. Lett. 2020;30:127509. doi: 10.1016/j.bmcl.2020.127509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wilson D.N. The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 2009;44:393. doi: 10.3109/10409230903307311. [DOI] [PubMed] [Google Scholar]
- 85.Liang H., Zhou G., Ge Y., D’Ambrosio E.A., Eidem T.M., Blanchard C., Shehatou C., Chatare V.K., Dunman P.M., Valentine A.M., et al. Elucidating the inhibition of peptidoglycan biosynthesis in Staphylococcus aureus by albocycline, a macrolactone isolated from Streptomyces maizeus. Bioorg. Med. Chem. 2018;26:3453–3460. doi: 10.1016/j.bmc.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barna J.C.J., Williams D.H. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol. 1984;38:339–357. doi: 10.1146/annurev.mi.38.100184.002011. [DOI] [PubMed] [Google Scholar]
- 87.Nagarajan R. Drugs and the Pharmaceutical Sciences. Volume 63 Marcel Dekker; New York, NY, USA: 1994. Glycopeptide antibiotics. [Google Scholar]
- 88.Zeng D., Debabov D., Hartsell T.L., Cano R.J., Adams S., Schuyler J.A., McMillan R., Pace J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect Med. 2016;6:a026989. doi: 10.1101/cshperspect.a026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh M., Chang J., Coffman L., Kim S.J. Hidden mode of action of glycopeptide antibiotics: Inhibition of wall teichoic acid biosynthesis. J. Phys. Chem. B. 2017;121:3925–3932. doi: 10.1021/acs.jpcb.7b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown S., Santa Maria J.P., Walker S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013;67:313–336. doi: 10.1146/annurev-micro-092412-155620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cegelski L., Kim S.J., Hing A.W., Studelska D.R., O’Connor R.D., Mehta A.K., Schaefer J. Rotational-echo double resonance characterization of the effects of vancomycin on cell wall synthesis in Staphylococcus aureus. Biochemistry. 2002;41:13053–13058. doi: 10.1021/bi0202326. [DOI] [PubMed] [Google Scholar]
- 92.Muller A., Klockner A., Schneider T. Targeting a cell wall biosynthesis hot spot. Nat. Prod. Rep. 2017;34:909–932. doi: 10.1039/C7NP00012J. [DOI] [PubMed] [Google Scholar]
- 93.Olademehin O.P., Shuford K.L., Kim S.J. Molecular dynamics simulations of the secondary-binding site in disaccharide-modified glycopeptide antibiotics. Sci. Rep. 2022;12:7087. doi: 10.1038/s41598-022-10735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brotz H., Bierbaum G., Reynolds P.E., Sahl H.G. The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur. J. Biochem. 1997;246:193–199. doi: 10.1111/j.1432-1033.1997.t01-1-00193.x. [DOI] [PubMed] [Google Scholar]
- 95.Arthur M., Molinas C., Bugg T.D., Wright G.D., Walsh C.T., Courvalin P. Evidence for in vivo incorporation of D-lactate into peptidoglycan precursors of vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992;36:867–869. doi: 10.1128/AAC.36.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicas T.I., Mullen D.L., Flokowitsch J.E., Preston D.A., Snyder N.J., Zweifel M.J., Wilkie S.C., Rodriguez M.J., Thompson R.C., Cooper R.D. Semisynthetic glycopeptide antibiotics derived from LY264826 active against vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1996;40:2194–2199. doi: 10.1128/AAC.40.9.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sweeney D., Stoneburner A., Shinabarger D.L., Arhin F.F., Belley A., Moeck G., Pillar C.M. Comparative in vitro activity of oritavancin and other agents against vancomycin-susceptible and -resistant enterococci. J. Antimicrob. Chemother. 2016;72:622–624. doi: 10.1093/jac/dkw451. [DOI] [PubMed] [Google Scholar]
- 98.Kim S.J., Tanaka K.S., Dietrich E., Far A.R., Schaefer J. Locations of the hydrophobic side chains of lipoglycopeptides bound to the peptidoglycan of Staphylococcus aureus. Biochemistry. 2013;52:3405–3414. doi: 10.1021/bi400054p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patti G.J., Kim S.J., Yu T.Y., Dietrich E., Tanaka K.S., Parr T.R., Jr., Far A.R., Schaefer J. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 2009;392:1178–1191. doi: 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim S.J., Matsuoka S., Patti G.J., Schaefer J. Vancomycin derivative with damaged D-Ala-D-Ala binding cleft binds to cross-linked peptidoglycan in the cell wall of Staphylococcus aureus. Biochemistry. 2008;47:3822–3831. doi: 10.1021/bi702232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim S.J., Cegelski L., Stueber D., Singh M., Dietrich E., Tanaka K.S., Parr T.R., Far A.R., Schaefer J. Oritavancin exhibits dual mode of action to inhibit cell-wall biosynthesis in Staphylococcus aureus. J. Mol. Biol. 2008;377:281–293. doi: 10.1016/j.jmb.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chun T., Pattem J., Gillis R.B., Dinu V.T., Yakubov G.E., Corfield A.P., Harding S.E. Self-association of the glycopeptide antibiotic teicoplanin A2 in aqueous solution studied by molecular hydrodynamics. Sci. Rep. 2023;13:1969. doi: 10.1038/s41598-023-28740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parenti F., Beretta G., Berti M., Arioli V. Teichomycins, new antibiotics from actinoplanes teichomyceticus nov. sp. J. Antibiot. 2006;31:276–283. doi: 10.7164/antibiotics.31.276. [DOI] [PubMed] [Google Scholar]
- 104.Reynolds P.E. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1989;8:943–950. doi: 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- 105.Vimberg V., Gazak R., Szűcs Z., Borbás A., Herczegh P., Cavanagh J.P., Zieglerova L., Závora J., Adámková V., Novotna G.B. Fluorescence assay to predict activity of the glycopeptide antibiotics. J. Antibiot. 2019;72:114–117. doi: 10.1038/s41429-018-0120-5. [DOI] [PubMed] [Google Scholar]
- 106.Pintér G., Batta G., Kéki S., Mándi A., Komáromi I., Takács-Novák K., Sztaricskai F., Röth E., Ostorházi E., Rozgonyi F., et al. Diazo transfer–click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: An aggregation and receptor binding study. J. Med. Chem. 2009;52:6053–6061. doi: 10.1021/jm900950d. [DOI] [PubMed] [Google Scholar]
- 107.Tollas S., Bereczki I., Sipos A., Rőth E., Batta G., Daróczi L., Kéki S., Ostorházi E., Rozgonyi F., Herczegh P. Nano-sized clusters of a teicoplanin ψ-aglycon-fullerene conjugate. Synthesis, antibacterial activity and aggregation studies. Eur. J. Med. Chem. 2012;54:943–948. doi: 10.1016/j.ejmech.2012.06.054. [DOI] [PubMed] [Google Scholar]
- 108.Corno G., Coci M., Giardina M., Plechuk S., Campanile F., Stefani S. Antibiotics promote aggregation within aquatic bacterial communities. Front. Microbiol. 2014;5:297. doi: 10.3389/fmicb.2014.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Higgins D.L., Chang R., Debabov D.V., Leung J., Wu T., Krause K.M., Sandvik E., Hubbard J.M., Kaniga K., Schmidt D.E., Jr., et al. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005;49:1127–1134. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Draghi D.C., Benton B.M., Krause K.M., Thornsberry C., Pillar C., Sahm D.F. Comparative surveillance study of telavancin activity against recently collected gram-positive clinical isolates from across the United States. Antimicrob. Agents Chemother. 2008;52:2383–2388. doi: 10.1128/AAC.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Food and Drug Administration FDA Labelling Information. [(accessed on 15 November 2010)];2009 Available online: https://www.fda.gov/files/food/published/Food-Labeling-Guide-%28PDF%29.pdf.
- 112.Das B., Sarkar C., Das D., Gupta A., Kalra A., Sahni S. Telavancin: A novel semisynthetic lipoglycopeptide agent to counter the challenge of resistant Gram-positive pathogens. Ther. Adv. Infect. Dis. 2017;2:49–73. doi: 10.1177/2049936117690501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Breukink E.J., Humphrey P.P.A., Benton B.M., Visscher I. Evidence for a multivalent interaction between telavancin and membrane-bound lipid II; Proceedings of the 46th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; San Francisco, CA, USA. 27–30 September 2006. [Google Scholar]
- 114.Benton B., Breukink E., Visscher I., Debabov D., Lunde C., Janc J., Humphrey P. Telavancin inhibits peptidoglycan biosynthesis through preferential targeting of transglycosylation: Evidence for a multivalent interaction between telavancin and lipid II. Int. J. Antimicrob. Agents. 2007;29:51–52. doi: 10.1016/S0924-8579(07)70166-8. [DOI] [Google Scholar]
- 115.Lunde C.S., Hartouni S.R., Janc J.W., Mammen M., Humphrey P.P., Benton B.M. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob. Agents Chemother. 2009;53:3375–3383. doi: 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dunne M.W., Puttagunta S., Sprenger C.R., Rubino C., Van Wart S., Baldassarre J. Extended –duration dosing and distribution of dalbavancin into bone and artic ular tissue. Antimicrob. Agents Chemother. 2015;59:1849–1855. doi: 10.1128/AAC.04550-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fazili T., Bansal E., Garner D., Gomez M., Stornelli N. Dalbavancin as sequential therapy for infective endocarditis due to Gram-positive organisms: A review. Int. J. Antimicrob. Agents. 2023;61:106749. doi: 10.1016/j.ijantimicag.2023.106749. [DOI] [PubMed] [Google Scholar]
- 118.Zhang Y., Sass A., Van Acker H., Wille J., Verhasselt B., Van Nieuwerburgh F., Kaever V., Crabbé A., Coenye T. Coumarin Reduces Virulence and Biofilm Formation in Pseudomonas aeruginosa by Affecting Quorum Sensing, Type III Secretion and C-di-GMP Levels. Front. Microbiol. 2018;9:1952. doi: 10.3389/fmicb.2018.01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ušjak D., Ivković B., Božić D.D., Bošković L., Milenković M. Antimicrobial activity of novel chalcones and modulation of virulence factors in hospital strains of Acinetobacter baumannii and Pseudomonas aeruginosa. Microb. Pathog. 2019;131:186–196. doi: 10.1016/j.micpath.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 120.Ušjak D., Dinić M., Novović K., Ivković B., Filipović N., Stevanović M., Milenković M.T. Methoxy-Substituted Hydroxychalcone Reduces Biofilm Production, Adhesion and Surface Motility of Acinetobacter baumannii by Inhibiting ompA Gene Expression. Chem. Biodivers. 2021;18:e2000786. doi: 10.1002/cbdv.202000786. [DOI] [PubMed] [Google Scholar]
- 121.Qi P., Wang N., Zhang T., Feng Y., Zhou X., Zeng D., Meng J., Liu L., Jin L., Yang S. Anti-Virulence Strategy of Novel Dehydroabietic Acid Derivatives: Design, Synthesis, and Antibacterial Evaluation. Int. J. Mol. Sci. 2023;24:2897. doi: 10.3390/ijms24032897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou J.W., Ruan L.Y., Chen H.J., Luo H.Z., Jiang H., Wang J.S., Jia A.Q. Inhibition of Quorum Sensing and Virulence in Serratia marcescens by Hordenine. J. Agric. Food Chem. 2019;67:784–795. doi: 10.1021/acs.jafc.8b05922. [DOI] [PubMed] [Google Scholar]
- 123.Du Y., Sun J., Gong Q., Wang Y., Fu P., Zhu W. New α-Pyridones with Quorum-Sensing Inhibitory Activity from Diversity-Enhanced Extracts of a Streptomyces sp. Derived from Marine Algae. J. Agric. Food Chem. 2018;66:1807–1812. doi: 10.1021/acs.jafc.7b05330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.