

Abstract
Hereditary sensory and autonomic neuropathy type IV (HSAN) is a rare and debilitating disorder highlighted by congenital absence of pain and anhidrosis. Orthopedic sequelae include physeal fractures, Charcot joint development, excessive joint laxity, soft tissue infections and recurrent painless dislocations, all of which often present in a delayed fashion. While there is no accepted guideline on management of these patients, several case studies have highlighted the importance of early diagnosis and cautioned against surgical intervention in these patients due to their inability to perceive pain and comply with post-operative restriction. The purpose of this case report is to present the clinical course of a patient with HSAN IV and the unique orthopedic challenges it presented. While some of her orthopedic injuries healed appropriately following treatment, others have gone on to have devastating complications and progressive joint destruction.
Level of Evidence: IV
Keywords: pediatric, neuropathy, charcot, HSAN
Introduction
Hereditary sensory and autonomic neuropathy type IV (HSAN), also known as congenital indifference to pain with anhidrosis (CIPA), is a rare and incompletely understood diagnosis.1-4 At the molecular level, HSAN is caused by mutations in the nerve growth factor receptor tyrosine kinase 1 (NTKR1) which leads to abnormal peripheral nerve formation and function.5 At the clinical level, HSAN IV is associated with complete absence of pain response to noxious stimuli and inability to thermo-regulate, which is the etiology of its wide spectrum of clinical manifestations.6,7 Precise incidence of this condition is not well known, with existing literature limited to small case series and case reports only.
Diagnosis of HSAN IV is difficult in the early years of life, as these patients can be otherwise developmentally normal without other congenital anomalies.1-4 Early signs of HSAN IV may include oral/buccal trauma from repetitive biting or onychophagia with resultant self-mutilation.1,8 Secondary to anhidrosis and impaired thermoregulation, patients may also present with recurrent hyperthermia or febrile seizures at a young age.9-11
As HSAN IV patients age and increase their activity level with ambulation, orthopedic manifestations become apparent.12-18 Painless limb swelling may reveal diaphyseal fractures and painless joint swelling may reveal significant physeal injuries or early Charcot changes. Early recognition of orthopedic injuries in HSAN IV patients is paramount to prevent further damage to the extremity.
We report a case here of a 10-year-old child with HSAN IV whose orthopedic manifestations included recurrent hip dislocations, proximal patellar sleeve fracture, proximal humerus physeal fracture, distal radius physeal injury, and bilateral Charcot ankle. Her treatment remains exceedingly difficult and has presented unique complications.
Case Report
The patient, a Caucasian female, was initially diagnosed at 18 months with HSAN IV after presenting with febrile seizures and painless recurrent left hip dislocations. Genetic and neurologic evaluation at that time confirmed the diagnosis, with heterozygosity for L213P mutation and partial deletion of the NTRK1 gene. For her hip dislocations, she underwent a period of intermittent hip abduction bracing over the course of two years and then was not seen again by our pediatric orthopedic team until she was 6 years old.
At age 6 the patient returned to our orthopedic clinic for evaluation of a left proximal patellar sleeve fracture, having already undergone attempted ORIF at an outside hospital which was complicated by hardware failure and wound healing issues with underlying deep infection. She developed recurrent knee effusions and an incompetent extensor mechanism. Ultimately, she underwent proximal pole of the patella excision and primary repair of her quadricep tendon with prolonged non-weightbearing and immobilization. Once allowed to mobilize, it was noted that her patella tracked in a laterally dislocated position throughout range of motion (Figure 1), albeit without any pain or functional limitation to the patient. Family preference was to avoid further surgery to this knee and defer patellar stabilization surgery given she had returned to functional ambulation. She eventually underwent lateral retinacular release and medial imbrication approximately 3 years later in efforts to improve her patellar tracking.
Figure 1.

Axial CT scan demonstrating a laterally subluxated patella following quad tendon repair.
At age 7, painless effusions were noted of the patient’s bilateral ankles. Radiographs and MRI demonstrated evidence of early degenerative changes and chronic ligamentous injuries, suggestive of excessive joint laxity and early Charcot changes (Figure 2). Joint aspirates in clinic were performed to ensure no underlying infection which all returned negative. Patient was fitted with solid ankle bilateral AFOs with attempts to maintain joint alignment to prevent further collapse and degeneration of her foot and ankle.
Figure 2.

Ankle XRs and T2 weighted sagittal MRI highlighting talar collapse and early Charcot changes.
Patient was seen again at age 8, when she presented to our emergency department with 2-week history of left shoulder redness, swelling and difficulty with range of motion. Radiographs demonstrated a displaced Salter Harris 1 proximal humerus fracture. There was no reported preceding trauma and patient was in no pain. Given her young age and concern about potential complications with pin fixation with her HSAN IV diagnosis, decision was made to pursue non-operative management with sling immobilization. She was followed closely over the next 18 months. An attempt at callus formation and healing was noted at her 4 week and 8 week follow up radiographs, however by 6 months there was noted to be continued motion across her physis with progressive metaphyseal resorption noted at 20 months post initial presentation (Figure 3).
Figure 3.

Left shoulder XRs at injury (A), 4 weeks (B), 8 weeks (C), 3 months (D), 6 months (E), 1 year (F) and 18 months (G), demonstrating attempted healing but ultimate nonunion with significant bone loss after proximal humerus physeal injury.
Painless right wrist swelling was noted in clinic at age 9, which correlated with a chronic appearing distal radius physeal injury and associated longitudinal growth disturbance as noted by positive ulnar variance (Figure 4). She underwent distal ulna epiphysiodesis to prevent further angular deformity of the wrist with pinning of the distal radius.
Figure 4.

Wrist XRs (A) showing distal radius physeal injury with significant ulnar positivity that underwent distal ulna epiphysiodesis (B).
At most recent follow up examination patient was 10 years old and had severely limited use of her left arm, with active forward flexion of 30 degrees and abduction to 30 degrees. An MRI was obtained at that point to better evaluate bone stock for possible surgical fixation but there was noted to be approximately 6 cm bone defect of the proximal humerus (Figure 5). For this reason, fixation efforts were deferred and continued conservative cares were chosen. Regarding her ankles, she was ambulatory with the use of AFOs. Her left knee had good range of motion, but her patella was noted to dislocate laterally when flexed beyond 45 degrees. Her right distal radius was healing but with continued positive ulnar variance not causing functional difficulty.
Figure 5.
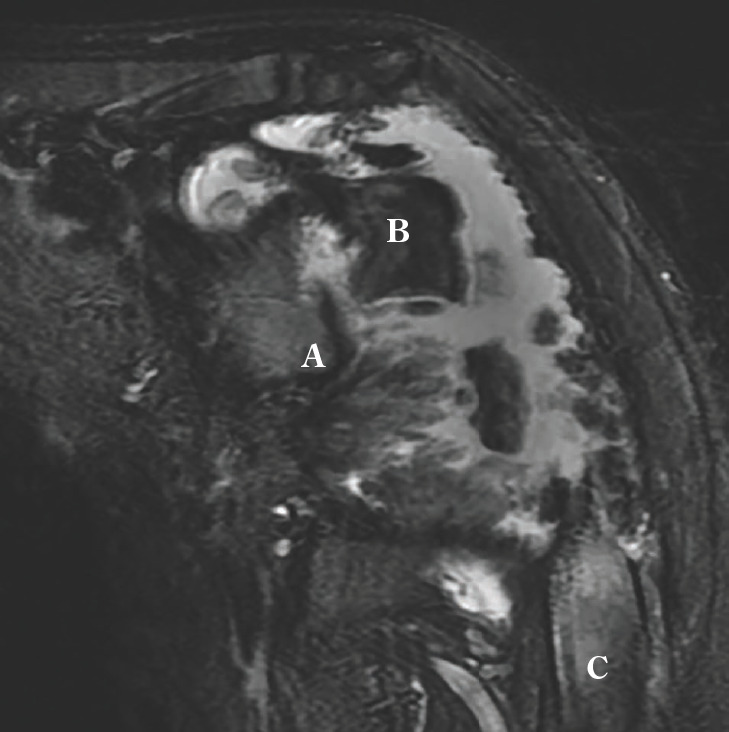
Coronal T2 weighted left shoulder MRI showing large area of metaphyseal bone loss between residual epiphysis (B) and diaphysis (C). Glenoid (A).
Discussion
Hereditary sensory and autonomic neuropathy has subtypes, with HSAN IV representing the rarest form of HSANs. HSAN IV is an autosomal recessive disease characterized by recurrent episodic fever, anhidrosis, absence of reaction to noxious stimuli, self-mutilating behavior, and often cognitive delay.1-4 At the genetic level, the etiology of HSAN IV is a loss of function mutation in the NTRK1 gene located on chromosome 1 (1q21-q22).5 Defects in this signaling pathway lead to apoptosis of various NGF dependent neurons during development. On the molecular level, electron microscopic studies of the radial and sural nerves of HSAN IV patients have shown a reduction in number or complete absence of small myelinated and unmyelinated fibers with very few Schwann cells present.19 Examination of the skin of these patients reveals a lack of sympathetic innervation of the eccrine sweat glands, which manifests as anhidrosis. Immunohistochemistry studies of the skin reveals absent C and A-delta fibers in the skin, which parallels the findings of electron microscopy.
Here we present a patient with HSAN IV, with both confirmatory genetic and molecular studies. Her orthopedic manifestations included painless dislocations, physeal injuries and Charcot joint development, all of which are known orthopedic sequala of HSAN.12-18 Her orthopedic care was exceedingly difficult and presented unique challenges secondary to her insensitivity to pain.
As with our patient, some HSAN IV patients have limited neurocognitive involvement with an otherwise normal intelligence and healthy appearing child.3,4 Special scrutiny needs to be made to rule out non-accidental injury in these cases, as patients may have relatively unexplained injuries due to their absence of pain and recollection of the injury timing.
Repetitive microtrauma to insensate articular surfaces is known to cause progressive articular collapse and deformity as first described by Charcot.4,20 This same principle applies to our patient and others with HSAN. The inability to respond to pain is compounded by the child’s young age and inability to modify activities once an injury is identified on imaging. This makes Charcot joint prevention exceedingly difficult in HSAN patients and relies heavily upon vigilant parents and close monitoring. There must also be high suspicion for concomitant pyogenic arthritis, with sterile aspirates needed to rule out infection. Treatment of Charcot joints in HSAN IV patients is much like adult Charcot treatment: stabilization, and prevention of additional joint deterioration.21,22 Conservative treatment using air cast boots, wheelers, and custom-walking boots is recommended to achieve this. Casts must be adequately padded, and skin integrity must be diligently monitored to prevent skin ulcers.
HSAN IV patients are also known to have abnormal gait kinematics which is thought to lead to abnormal joint laxity and contact stresses,4 which certainly contributed to our patient’s chronic patella dislocation and Charcot joint progression. Behavioral monitoring with gait and postural training can be helpful in preventing undo stress on the developing limbs and spine.
Surgical treatment for orthopedic injuries in HSAN patients must be approached with caution, as postoperative immobilization and activity restrictions can be exceedingly difficult in these children. Further, an increased incidence of infection, non-union, hardware complications, wound healing complications, and avascular necrosis following surgery has been reported.12-18 Previous studies suggest K-wire fixation may not be sufficient in this cohort,14 as patients lack the pain reflex that aids in immobilization. Rigid external or intermedullary fixation is recommended if fracture non-union persists. When surgery is indicated, close post-operative wound monitoring and radiographic follow up is essential. Furthermore, general anesthesia can lead to increased risk of autonomic dysfunction in this cohort so special monitoring may be required.23
HSAN IV presents difficult and unique challenges for treating orthopedic surgeons. This case highlights the variety of orthopedic presentations seen in HSAN IV, including physeal injuries, wound healing complications, and Charcot joint. Parent education and fracture prevention techniques are paramount when treating these patients. Operative treatment remains difficult given the congenital absence of pain and ability to comply with post-operative immobilization.
References
- 1.Ashwin D. P. “Hereditary Sensory and Autosomal Peripheral Neuropathy-Type IV: Case Series and Review of Literature.”. Oral and Maxillofacial Surgery, 2015;19(no. 2):117–123. doi: 10.1007/s10006-015-0486-5. [DOI] [PubMed] [Google Scholar]
- 2.Capsoni S. “From Genes to Pain: Nerve Growth Factor and Hereditary Sensory and Autonomic Neuropathy Type V.”. European Journal of Neuroscience. 2014;39(no. 3):392–400. doi: 10.1111/ejn.12461. [DOI] [PubMed] [Google Scholar]
- 3.Schwartzlow C, Mohamed K. “Hereditary Sensory and Autonomic Neuropathies: Adding More to the Classification.”. Current Neurology and Neuroscience Reports. 2019;19(no. 8) doi: 10.1007/s11910-019-0974-3. [DOI] [PubMed] [Google Scholar]
- 4.Feldman David S. “Peripheral Arthropathy in Hereditary Sensory and Autonomic Neuropathy Types III and IV.”. Journal of Pediatric Orthopaedics, 2009;29(no. 1):91–97. doi: 10.1097/BPO.0b013e31818f9cc4. [DOI] [PubMed] [Google Scholar]
- 5.Indo Yasuhiro. “Molecular Basis of Congenital Insensitivity to Pain with Anhidrosis (CIPA): Mutations and Polymorphisms Intrka (NTRK1) Gene Encoding the Receptor Tyrosine Kinase for Nerve Growth Factor.”. Human Mutation. 2001;18(no. 6):462–471. doi: 10.1002/humu.1224. [DOI] [PubMed] [Google Scholar]
- 6.Axelrod FB, Pearson J. Congenital sensory neuropathies. Diagnostic distinction from familial dysautonomia. Am J Dis Child. 1984;138(10):947e54. [PubMed] [Google Scholar]
- 7.Pinsky L, Di George AM. Congenital familial sensory neuropathy with anhidrosis. J Pediatr. 1966;68:1–13. doi: 10.1016/s0022-3476(66)80417-1. [DOI] [PubMed] [Google Scholar]
- 8.Bodner L, Woldenberg Y, Pinsk V, et al. Orofacial manifestations of congenital insensitivity to pain with anhidrosis: a report of 24 cases. J Dent Child. 2002;69:293–296. [PubMed] [Google Scholar]
- 9.Iwanaga R, Matsuishi T, Ohnishi K, et al. Serial magnetic resonance images on a patient with congenital sensory neuropathy and complications resembling heat stroke. J Neurol Sci. 1996;142:79–84. doi: 10.1016/0022-510x(96)00152-9. [DOI] [PubMed] [Google Scholar]
- 10.Gupta B. Congenital insensitivity of pain with anhidrosis; clinical brief. Indian J Pediatr. 2003;70(1):109e11. doi: 10.1007/BF02722757. [DOI] [PubMed] [Google Scholar]
- 11.Nagasako EM, Oaklander AL, Dworkin RH. Congenital insensitivity to pain: an update. Pain. 2003;101:213–9. doi: 10.1016/S0304-3959(02)00482-7. [DOI] [PubMed] [Google Scholar]
- 12.Kayani Babar. “Orthopaedic Manifestations of Congenital Indifference to Pain with Anhidrosis (Hereditary Sensory and Autonomic Neuropathy Type IV).”. European Journal of Paediatric Neurology. 2017;21(no. 2):318–326. doi: 10.1016/j.ejpn.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Pérez-López L. M. “Update Review and Clinical Presentation in Congenital Insensitivity to Pain and Anhidrosis.”. Case Reports in Pediatrics. 2015;2015:1–7. doi: 10.1155/2015/589852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marik Ivo. “Hereditary Sensory and Autonomic Neuropathy Type IV Orthopaedic Complications.”. Journal of Pediatric Orthopaedics B. 2009;18(no. 3):138–140. doi: 10.1097/BPB.0b013e3283298815. [DOI] [PubMed] [Google Scholar]
- 15.Siddiqi Asad R. “Acute Hip Dislocation in a Patient with Congenital Insensitivity to Pain and Anhidrosis: A Case Report.”. PM&R. 2013;5 [Google Scholar]
- 16.Mindle J, Svensson O, Holmberg M, et al. Orthopedic aspects of familial insensitivity to pain due to a novel nerve growth factor beta mutation. Acta Orthopaedica. 2006;77:198–202. doi: 10.1080/17453670610045911. [DOI] [PubMed] [Google Scholar]
- 17.Kuo RS, Macnicol MF. Congenital insensitivity to pain: orthopaedic implications. J Pediatr Orthop B. 1996;5:292–5. doi: 10.1097/01202412-199605040-00013. [DOI] [PubMed] [Google Scholar]
- 18.Bronfen C, Bensahel H, Teule JG. Orthopedic aspects of congenital insensitivity to pain. Chir Pediatr. 1985;26:193–6. [PubMed] [Google Scholar]
- 19.Minde J, Toolanen G, Andersson T, et al. Familial insensitivity to pain (HSAN V) and a mutation in the NGFB gene. A neurophysiological and pathological study. Muscle Nerve. 2004 Dec;30(6):752–60. doi: 10.1002/mus.20172. [DOI] [PubMed] [Google Scholar]
- 20.Gupta R. A short history of neuropathic arthropathy. Clinical Orthopaedics and Related Research. 1993;296:43–49. [PubMed] [Google Scholar]
- 21.Fath M, Hassannein R, James JI. Congenital absence of pain. J Bone Joint Surg. 1983;65-B:186–188. doi: 10.1302/0301-620X.65B2.6186667. [DOI] [PubMed] [Google Scholar]
- 22.Bar-On E, Weigl D, Parvari R, Katz K, et al. Congenital insensitivity to pain. Orthopaedic manifestations. J Bone Joint Surg Br. 2002 Mar;84(2):252–7. doi: 10.1302/0301-620x.84b2.11939. [DOI] [PubMed] [Google Scholar]
- 23.Weingarten TN, Sprung J, Ackerman JD, et al. Anesthesia and patients with congenital hyposensitivity to pain. Anesthesiology. 2006 Aug;105(2):338–45. doi: 10.1097/00000542-200608000-00017. [DOI] [PubMed] [Google Scholar]