

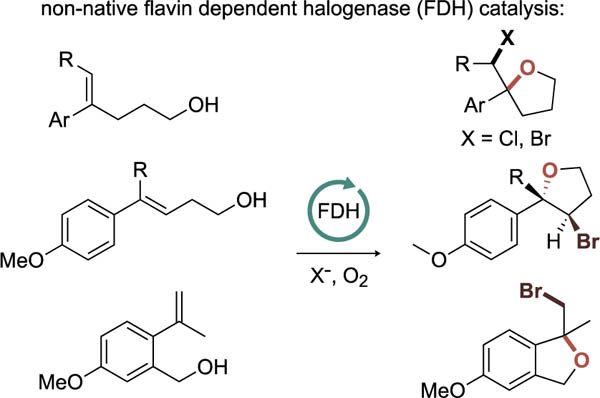
Abstract
Of the different classes of halogenases characterized to date, flavin dependent halogenases (FDHs) are most associated with site-selective halogenation of electron rich arenes and enol(ate) moieties in the biosynthesis of halogenated natural products. This capability has made them attractive biocatalysts, and extensive efforts have been devoted to both discovering and engineering these enzymes for different applications. We have established that engineered FDHs can catalyze different enantioselective halogenation processes, including halolactonization of simple alkenes with a tethered carboxylate nucleophile. In this study, we expand the scope of this reaction to include alcohol nucleophiles and a greater diversity of alkene substitution patterns to access a variety of chiral tetrahydrofurans. We also demonstrate that FDHs can be interfaced with ketoreductases to enable halocyclization using ketone substrates in one-pot cascade reactions and that the halocyclization products can undergo subsequent rearrangements to form hydroxylated and halogenated products. Together, these advances expand the utility of FDHs for enantio- and diastereoselective olefin functionalization.
Keywords: halogenase, flavin, halocyclization, chemoenzymatic synthesis, enzyme cascade
Graphical Abstract
Body
In nature, flavin-dependent halogenases (FDHs) catalyze halogenation of electron rich arenes and enol(ate) moieties using halide anions (typically X- = Cl- and Br-) as a halogen source and dioxygen as a terminal oxidant.1 These mild reaction conditions and the high site selectivity of FDHs toward their native substrates and even many non-native substrates have made them attractive biocatalysts.2–4 Our group and others have established that variants of these enzymes can be engineered to halogenate different non-native substrates or different sites on a given substrate with high (i.e. >90%) selectivity.5 We also established that FDHs can catalyze enantioselective desymmetrization of methylenedianilines6 and atroposelective7 halogenation of 3-aryl-4(3H)-quinazolinones; however, these transformations share a similar mechanism involving electrophilic attack by an active site halogenating agent, likely HOX bound within the FDH active site, and ipso deprotonation (Figure 1A).8–10
Figure 1.
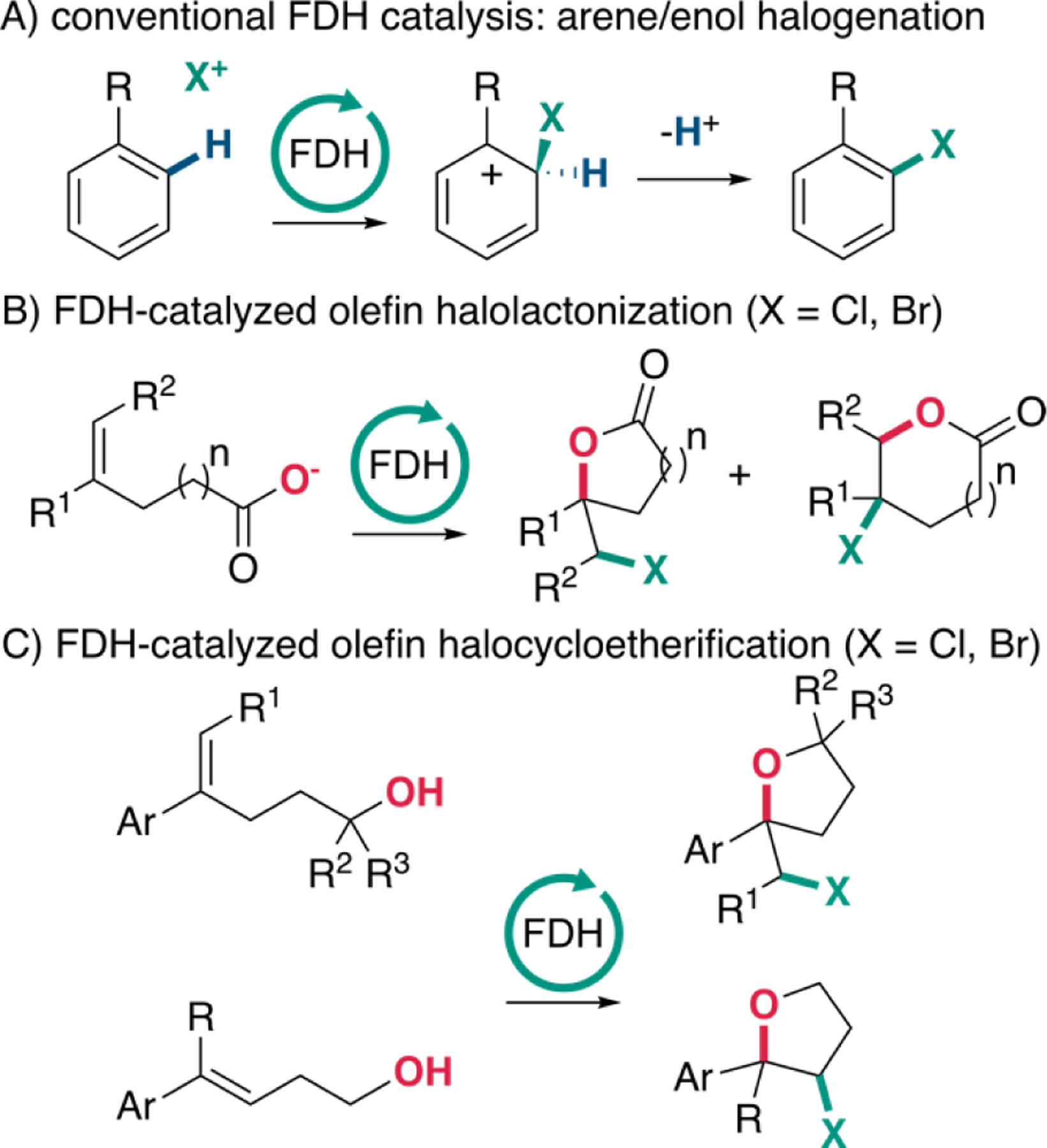
A) Conventional FDH catalysis: arene/enol halogenation. B) Previously reported FDH-catalyzed halolactonization with X = Cl, Br.12 C) FDH-catalyzed halocycloetherification with X = Cl, Br.
Inspired by the wide range of transformations known to proceed via electrophilic attack of different nucleophiles by electrophilic halogen species,11 we recently discovered that engineered FDHs can also catalyze enantioselective halolactonization of olefins (Figure 1B)12. This finding significantly broadened the catalytic repertoire of FDHs and revealed that their active sites can tolerate intermediate and transition state structures that are topologically distinct from those associated with electrophilic aromatic substitution. This activity also provided a starting point for assessing whether different olefin geometries, nucleophiles, and linkers between the olefins and pendant nucleophiles,13 could be tolerated by FDHs to further broaden the scope of enzymatic halocyclization.
Because our initial report only explored carboxylate nucleophiles, which are deprotonated and therefore highly nucleophilic under the conditions required for biocatalysis,14 we were particularly interested in evaluating the reactivity of less activated nucleophiles. Chemoenzymatic halocycloetherification has been achieved using vanadium-dependent haloperoxidases, but racemic products are obtained from this approach.15 With some notable exceptions,16,17 however, enantioselective catalysis of halocycloetherification remains challenging using small molecule catalysts,18–23 so we hoped that FDHs might be able to address this outstanding methodology challenge. Herein, we report that FDHs can indeed catalyze halocyclooetherification of electron rich styrenes bearing pendant alcohols to generate chiral tetrahydrofurans in high yield and with high enantioselectivity (Figure 1C). We also show that 2-(4-methoxyphenyl)-2-(bromomethyl)tetrahydrofurans can rearrange via nucleophilic attack on a phenonium intermediate leading to net chemoenzymatic oxidative rearrangements of the starting olefins. Finally, we demonstrate that alcohol nucleophiles can be generated from the parent ketones and cyclized in a ketoreductase/FDH cascade. These results further broaden the substrate scope of FDH-catalyzed halocyclization and establish that FDHs can be used to access chiral tetrahydrofurans and dihydrobenzofurans that are commonly found in a range of natural products and other biologically active compounds.24–26
We previously found that variants of the FDH RebH provided good conversion and enantioselectivity for halolactonization of 4-arylpent-4-enoic acids,12 so we analyzed the activity of a panel of 50 RebH variants and other WT FDHs27 toward the corresponding alcohol 1 (Table 1, Figure S1). Despite the lower nucleophilicity of its pendant alcohol, 1 undergoes 5-exo-trig cyclization to give 1a in good yields using several previously reported FDHs. While variant 4V+S28 was optimal for halolactonization,12 improved selectivity was observed for variant 4PL29 (Table 1, entries 1 and 3). Low selectivity for the opposite product enantiomer was observed for variant 6TL T52V,29 providing a starting point for directed evolution to improve this sense of asymmetric induction (Table 1, entry 2). Notably, improved yields were obtained using 4PL when the reaction time was decreased to 2 h. While glutathione and catalase, HOX and H2O2 scavengers, respectively, had only a minor effects on enantioselectivity, they were included in further reactions due to their beneficial effects on halolactonization reactions12 and to avoid any potential substrate-dependent background reactions involving these reactive oxygen species, which can form during FDH catalysis.27,30
Table 1.
Yields and enantioselectivities for bromoetherification of 1 using representative FDHsa
| ||||
|---|---|---|---|---|
| Entry | FDH | Time (h) | Yield (%)a | e.r.b |
| 1 | 4V+S | 18 | 66 | 79:21 |
| 2 | 6TL T52V | 18 | 48 | 36:64 |
| 3 | 4PL | 18 | 68 | 91:9 |
| 4 | 4PL | 2 | 88 | 91:9 |
Reactions conducted using 1 mM 1, 5 mM NaBr, 5 mol% FDH, 100 μM NAD and FAD, 20 mM glucose, 1 mM glutathione, 2.5 μM flavin reductase (RebF),31 9 U/mL glucose dehydrogenase (GDH), and 35 U/mL catalase (Cat.) in 200 mM tricine, pH 7.5.
Yields determined by LC/MS relative to internal standard; selectivities determined by chiral HPLC.
The observed decrease in the yield of 1a at longer reaction times suggested that degradation of this compound could be occurring. Analysis of the reaction mixtures revealed that 1-(p-methoxyphenyl)-5-hydroxypentan-2-one (1b) was indeed forming (Figure 2A, Figure S2), which was surprising since it lacks a halogen substituent and requires structural rearrangement of the starting olefin. While this reaction was avoided by simply quenching the reactions following complete halogenation of the olefin starting material, it was facilitated by heating the completed haloetherification reaction mixtures to 50 °C. Related reactivity was reported for cyclic acetals of α-haloacetophenones, which undergo rearrangement to the corresponding 1-aryl-5-bromopentan-2-ones via a proposed phenonium intermediate (Figure 2B).32 In this reaction, the ZnBr2 is believed to serve as a Lewis acid to assist with abstraction of the Br substituent and could also help to mediate the nucleophilic attack on the phenonium intermediate. We found that 1a produced via FDH catalysis could be efficiently converted to brominated compound 1c under these conditions (Figure 2C). While we hoped that reaction of trisubstituted alkene 8 might lead to chiral α-substituted ketones, racemic 8c was obtained from this reaction despite the fact that moderate enantioselectivity is observed for the bromoetherification step (vide infra), suggesting that the rearrangement process is not stereospecific.
Figure 2.
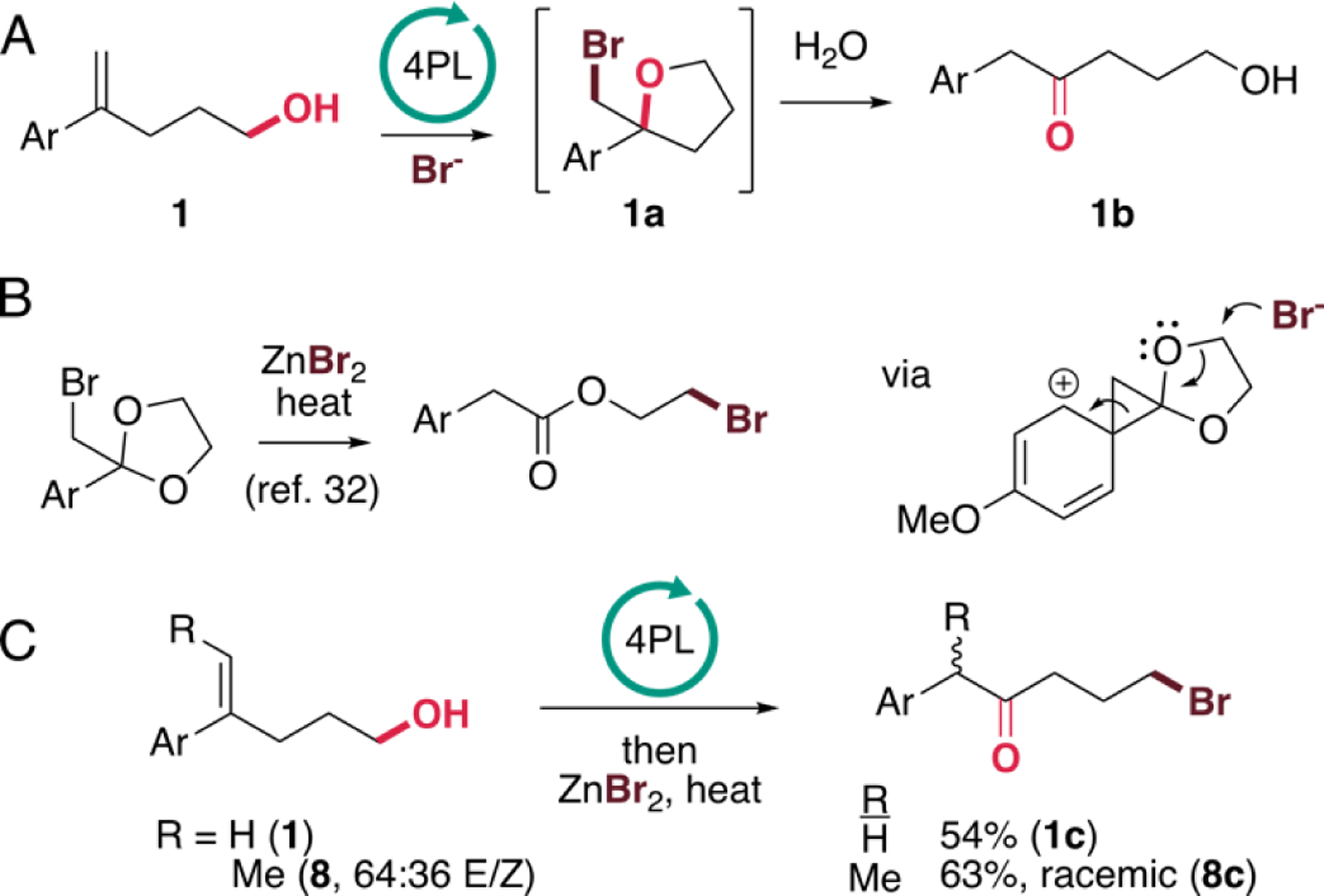
A) Chemo-enzymatic bromoetherification/rearrangement/hydroxylation of 1a using biocatalysis conditions shown in Table 1. B) Reported Zn(II)-catalyzed rearrangement of cyclic acetals.32 C) Chemoenzymatic bromoetherification/rearrangement/bromination of 1 and 8 (64:36 E/Z).
With conditions to avoid this non-selective rearrangement process in hand, we next investigated the substrate scope of FDH-catalyzed halocycloetherification (Chart 1). Reactions involving 1,1-disubstituted alkenes generally proceed with good yield and selectivity as long as the styrene possesses an electron donating group para to the olefin. For example, while p-methoxyphenyl substituted substrate 1 gives a 91% yield of 1a and 91:9 e.r., phenyl substituted substrate 2 provides no detectable products under the reaction conditions; only unreacted starting material remains. This trend was previously observed for the analogous halolactonization reactions, but low yields of cyclized products were still obtained for phenyl- and p-fluorophenyl-substituted substrates.12 We attributed the lower yields for these relatively electron deficient substrates to the difficulty of accessing the transition state required for the concerted nucleophile-assisted alkene activation pathway that such substrates have been shown to follow.14 The lack of reaction for the corresponding alcohols suggests that while the concerted pathway could still occur to some extent with a more potent carboxylate nucleophile, it was no longer possible with alcohols, and only substrates with electron donating groups that would allow for a stepwise pathway were suitable for FDH catalysis. Alternatively, if these reactions proceed via bromiranium intermediates,13 substrates like 2 that lack electron donating groups would also be expected to react less readily. Substrate 3, which has a methoxy group in the ortho position, was also problematic and gave 3a in low yield and e.r. Substrates 4-7 all cyclize in greater than 90% yield and >92:8 e.r., and p-acetamide substituted substrate 7 provided particularly high selectivity for 7a. This reaction was scale up to 28 mL scale with 2 mM substrate loading to provide 86% yield of 7a (14 mg) with 99:1 e.r. Trisubstituted substrate 8 (64:36 E/Z) also cyclizes in good yield to give 8a as a 72:28 mixture of diastereomers (largely reflecting the E/Z ratio of the starting material) and moderate enantioselectivity. Substrate 9 gives 2,2’,5,5’-tetrasubstituted tetrahydrofuran 9a in moderate yield and selectivity. Finally, chloroetherification of substrate 1 was also achieved to give 1d in 70% yield and 99:1 e.r. using NaCl in place of NaBr under otherwise standard reaction conditions.
Chart 1.
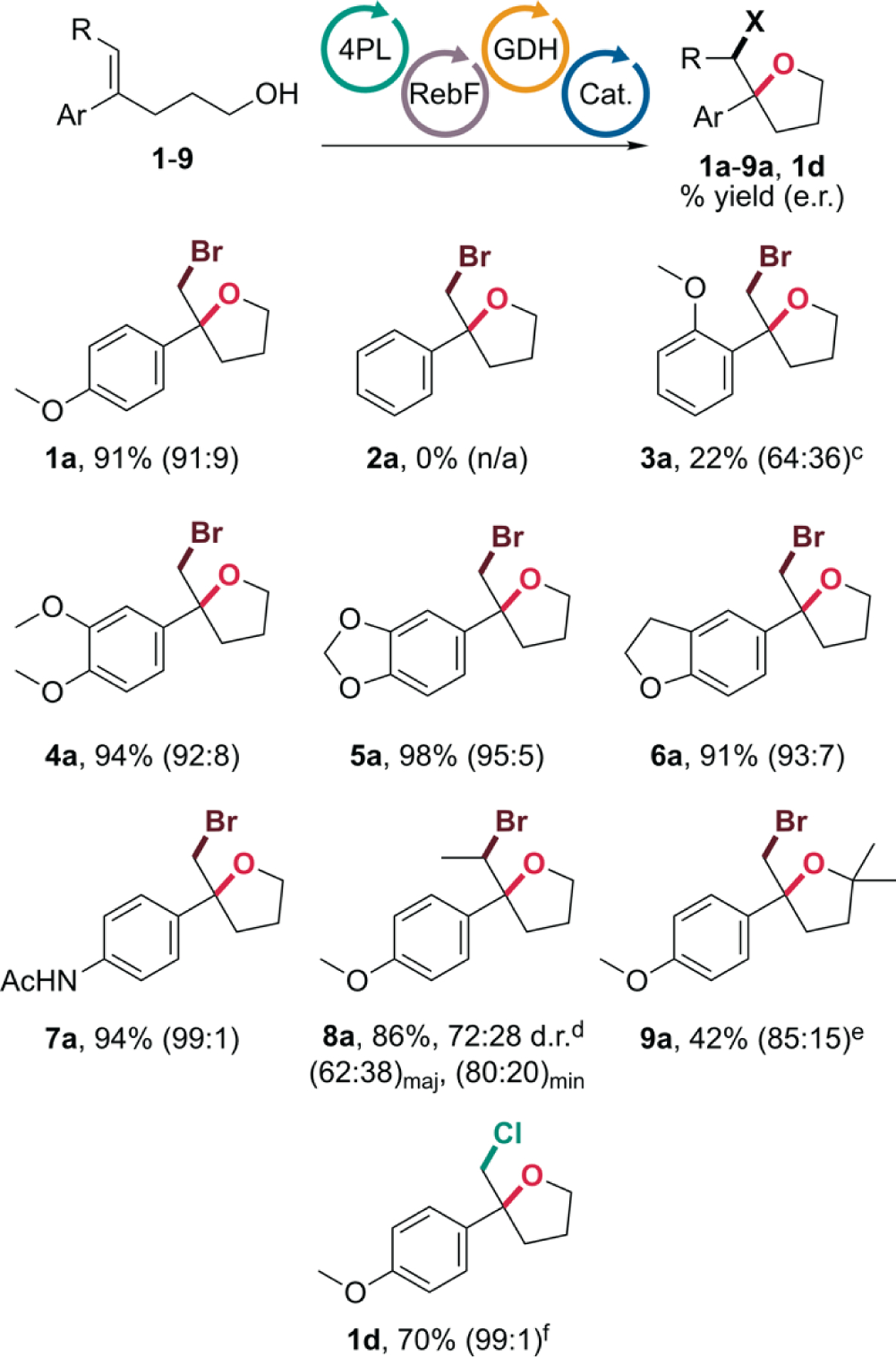
Representative scope of FDH-catalyzed haloetherification of 1,1-disubstituted alkenesa,b
aReactions conducted at room temperature for times specified in the supporting information using 1 mM substrate, 5 mM NaBr, 5 mol% FDH, 100 μM NAD and FAD, 20 mM glucose, 1 mM glutathione, 2.5 μM RebF, 9 U/mL GDH, and 35 U/mL catalase in 200 mM tricine, pH 7.5. bYields determined by LC/MS relative to internal standard; selectivities determined by chiral HPLC. cVariant 4PL-F111S was used in place of 4PL. dA 64:36 mixture of E/Z-8 was used.e Variant 2RFQ-F111S was used in place of 4PL. f5 mM NaCl used in place of NaBr.
We also found that 1,2-disubstituted alkenes 10 and 11 undergo haloetherification to give 10a and 11a as single diastereomers (Figure 3A). The analogous halolactonization reactions provided only trace conversion, so this finding demonstrates that FDHs can accommodate 5-endo-trig cyclization and provides access to 2,3-disubstituted and 2,2’,3-trisubstituted tetrahydrofurans. While 4PL provided these products in 72% and 84% yield, respectively, modest enantioselectivity was observed in both cases. Variants 2RFQ F111S29 and 3LR33 provided increased selectivity at the expense of yield (Figure 3A). Finally, α-methyl styrene 12,17 which possesses a pendant benzyl alcohol nucleophile, undergoes cyclization catalyzed by 4PL to give chiral 1,3-dihydroisobenzofuran 12a in 74% yield and 78:22 e.r. (Figure 3B).
Figure 3.
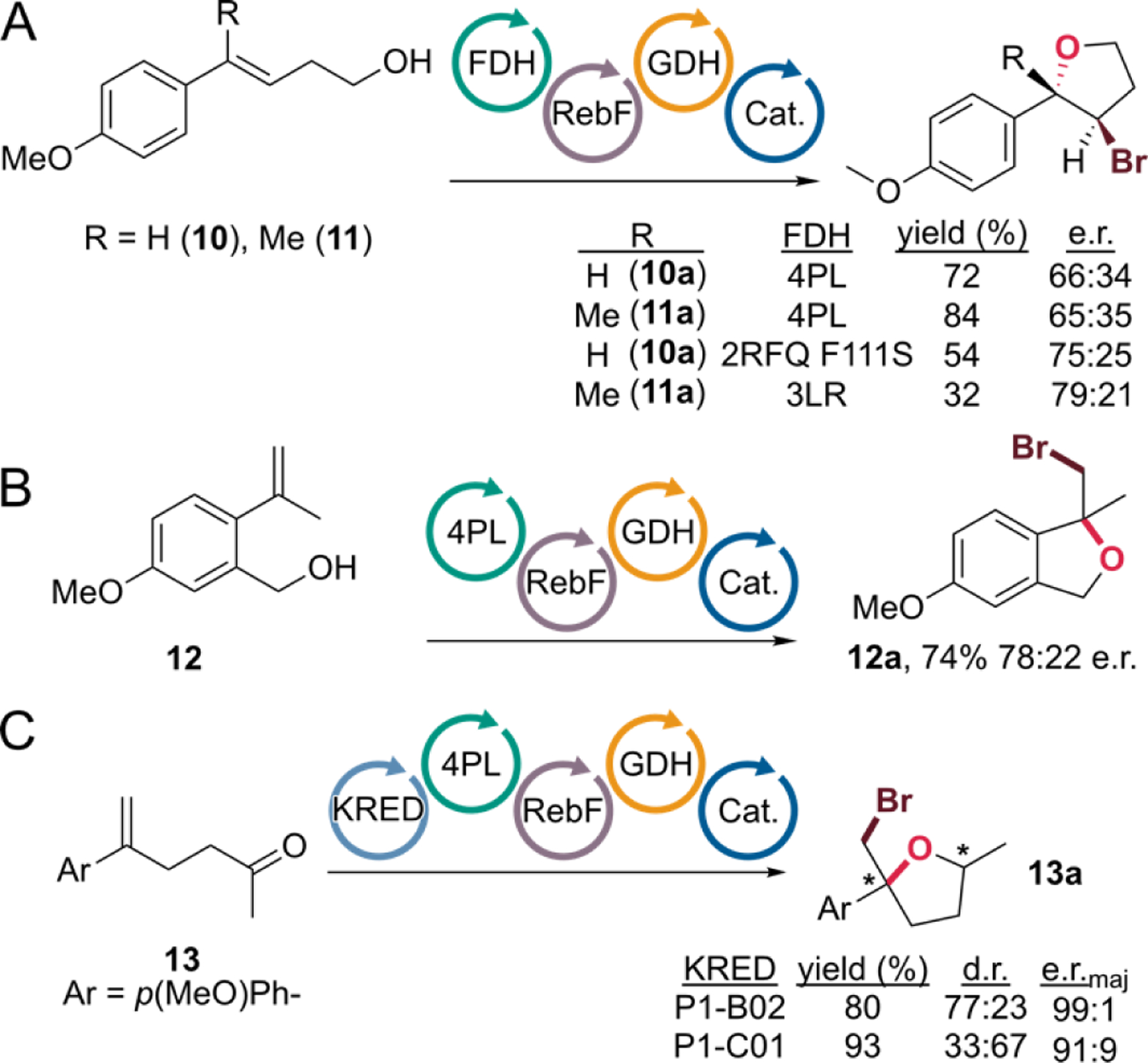
FDH-catalyzed bromoetherification A) via 5-endo-trig cyclization, B) involving a pendant benzyl alcohol nucleophile, and C) starting from ketones using a ketoreductase/FDH cascade. e.r.maj indicates the e.r. of the major diastereomer of 13a. See supporting information for reaction details.
While substrates 1-12 involve achiral alcohol nucleophiles, secondary alcohols introduce the possibility of using FDHs to control the diastereoselectivity of bromoetherification involving chiral nucleophiles.19 We envisioned that such alcohols could be accessed via reduction of the corresponding ketones using a ketoreductase (KRED).34 These enzymes are NAD-dependent and require reduced cofactor supplied by a cofactor regeneration system like the glucose/glucose dehydrogenase (GDH) system that we use to drive FAD reduction or by oxidation of a sacrificial alcohol like isopropanol (IPA). We therefore anticipated that KREDs could be used in a one-pot cascade reaction with FDHs to enable sequential ketone reduction/bromoetherification with the respective enzymes controlling the enantioselectivity of the former and the diastereoselectivity of the latter. Indeed, we found that simply adding different KREDs to our standard reaction conditions with a 10-fold excess of NAD relative to our usual loading and with IPA in place of DMSO as a co-solvent led to high yields of bromoetherification catalyzed by 4PL starting from ketone 13 (Figure 3C). Depending on the KRED used, modest selectivity for either diastereomer could be obtained, and high enantioselectivity for the major diastereomer was observed.
The FDH variants with haloetherification activity described in this study were identified from an initial screen of 50 WT and engineered FDHs on substrate 1 (Figure S2) followed by secondary screens of a set of these enzymes on individual substrates (Figures S4-S11). Four engineered FDHs (4PL-F111S, 2RFQ-F111S, 4PL, and 3LR) were sufficient to provide the haloetherification yields and selectivities outlined above (Figure S12). The first three of these enzymes contain the F111S/L and N470S mutations that imparted halolactonization activity to RebH variants and were predicted by docking simulations to favor the formation of the pro-R form of the halolactonization product in previously reported docking simulations.12 Observation of the (R)-7a by X-ray crystallography (Figure S13) is consistent with these simulations and suggests that alcohol substrates that undergo 5-exo-trig cyclization could bind analogously to the corresponding carboxylic acid substrates. Variant 3-LR doesn’t possess any of these mutations, but it was only used in the 5-endo-trig cyclization of 11, which provided a low yield of 11a relative to formation of 10a by 2RFQ F111S (Figure 3A). This finding, and the fact that FDHs exhibited very different selectivity toward substrates 10 and 11 (Figures S10/S11), suggests that the methyl substituent of 11 hinders 5-endo-trig cyclization but that mutations in 3-LR somehow facilitate this reaction with modest enantioselectivity. Further analysis of FDH activity and selectivity toward different halocyclization reactions will be required to understand the subtleties of this non-native reactivity.
Conclusion
This study establishes that FDH-catalyzed halocyclization can be extended to substrates bearing alcohol nucleophiles in addition to the previously reported carboxylate nucleophiles. It also shows that these enzymes can tolerate different styrene substitution patterns to enable the synthesis of di-, tri-, and tetra-substituted tetrahydrofurans. Good yields and enantioselectivity are obtained as long as styrenes with electron donating aromatic groups are used, and promising diastereoselectivity was obtained for a trisubstituted olefin. Related efforts to catalyze bromoetherification of 1,1-disubstituted alkenes via 5-exo-trig cyclization using chiral Lewis bases revealed that geminal dimethyl substituents on the pendant alcohol were required to achieve even modest enantioselectivity.19 Olefin-to-olefin bromiranium ion exchange was proposed to degrade the selectivity of these reactions, but such exchange would not be possible within the FDH active site, perhaps explaining the high selectivities for simple styrenes in the current study. This feature should aid efforts to engineer FDHs with improved haloetherification selectivity on tri-substituted olefins, substrates that undergo 5-endo-trig cyclization, and additional chloroetherification reactions, all which give modest selectivity using existing FDHs. Extending the scope of FDH-catalyzed halocyclization to substrates lacking electron donating groups will likely require more significant sculpting of the active site to accommodate the nucleophile-assisted alkene activation pathway proposed for such substrates.14 We also characterized novel rearrangements of the initial bromoetherification products that give rise to hydroxylated and brominated ketones, providing initial hints at how enzymatic bromination could be used to further elaborate compounds in a chemoenzymatic sense. Likewise, we showed that KREDs could be used to simplify access to the alcohol nucleophiles required for bromoetherification involving secondary alcohols, showing how these enzymes can be readily integrated with FDHs in cascade processes. Together, these results highlight the utility of FDHs and (chemo)enzymatic sequences and cascades for olefin functionalization.
Supplementary Material
Acknowledgements
This study was supported by the NIH (R01 GM115665). NMR data were acquired on a spectrometer funded by the NSF (MRI CHE-1920026) using a Prodigy probe that was partially funded by the Indiana Clinical and Translational Sciences Institute. MS data were acquired on a spectrometer funded by NSF Grant CHE1726633. We thank Codexis, Inc. for providing the KREDs used in this study and Harrison Snodgrass for helpful discussions. We thank Dr. Maren Pink in the Indiana University Molecular Structure Center for solving the crystal structure of compound 7a. We think the group of Professor Kevin Brown at IU for the use of different chiral columns. Support for the acquisition of the Bruker Venture D8 diffractometer through the Major Scientific Research Equipment Fund from the President of Indiana University and the Office of the Vice President for Research is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
supporting_information (PDF containing supplementary figures, complete experimental procedures, and relevant characterization)
References
- (1).Pée K.-H. van; Patallo E Flavin-Dependent Halogenases Involved in Secondary Metabolism in Bacteria. Applied Microbiology And Biotechnology 2006, 70 (6), 631–641. 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- (2).Gkotsi DS; Dhaliwal J; McLachlan MM; Mulholand KR; Goss RJ ScienceDirect Halogenases: Powerful Tools for Biocatalysis (Mechanisms Applications and Scope). Current Opinion In Chemical Biology 2018, 43, 119–126. 10.1016/j.cbpa.2018.01.002. [DOI] [PubMed] [Google Scholar]
- (3).Latham J; Brandenburger E; Shepherd SA; Menon BRK; Micklefield J Development of Halogenase Enzymes for Use in Synthesis. Chemical Reviews 2017, 118 (1), 232–269. 10.1021/acs.chemrev.7b00032. [DOI] [PubMed] [Google Scholar]
- (4).Weichold V; Milbredt D; Pée K.-H. van. Specific Enzymatic Halogenation-From the Discovery of Halogenated Enzymes to Their Applications In Vitro and In Vivo. Angewandte Chemie International Edition in English 2016, 55 (22), 6374–6389. 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- (5).Andorfer MC; Lewis JC Understanding and Improving the Activity of Flavin-Dependent Halogenases via Random and Targeted Mutagenesis. Annual Review of Biochemistry 2018, 87 (1), 159–185. 10.1146/annurev-biochem-062917-012042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Payne JT; Butkovich PH; Gu Y; Kunze KN; Park H-J; Wang D-S; Lewis JC Enantioselective Desymmetrization of Methylenedianilines via Enzyme-Catalyzed Remote Halogenation. Journal of the American Chemical Society 2018, 140 (2), 546–549. 10.1021/jacs.7b09573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Snodgrass HM; Mondal D; Lewis JC Directed Evolution of Flavin-Dependent Halogenases for Site- and Atroposelective Halogenation of 3-Aryl-4(3H)-Quinazolinones via Kinetic or Dynamic Kinetic Resolution. J Am Chem Soc 2022, 144 (36), 16676–16682. 10.1021/jacs.2c07422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Flecks S; Patallo EP; Zhu X; Ernyei AJ; Seifert G; Schneider A; Dong C; Naismith JH; Pée K.-H. van. New Insights into the Mechanism of Enzymatic Chlorination of Tryptophan. Angewandte Chemie International Edition in English 2008, 47 (49), 9533–9536. 10.1002/anie.200802466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ainsley J; Mulholland AJ; Black GW; Sparagano O; Christov CZ; Karabencheva-Christova TG Structural Insights from Molecular Dynamics Simulations of Tryptophan 7-Halogenase and Tryptophan 5-Halogenase. ACS Omega 2018, 3 (5), 4847–4859. 10.1021/acsomega.8b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Andorfer MC; Evans D; Yang S; He CQ; Girlich AM; Vergara-Coll J; Sukumar N; Houk KN; Lewis JC Analysis of Laboratory-Evolved Flavin-Dependent Halogenases Affords a Computational Model for Predicting Halogenase Site Selectivity. ChemRxiv [Preprint], Feb 16, 2022. Version 1, DOI: 10.26434/chemrxiv-2022-w059p. 10.26434/chemrxiv-2022-w059p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Podgorš A; Zupan M; Iskra J Oxidative Halogenation with “Green” Oxidants: Oxygen and Hydrogen Peroxide. Angewandte Chemie International Edition in English 2009, 48 (45), 8424–8450. 10.1002/anie.200901223. [DOI] [PubMed] [Google Scholar]
- (12).Mondal D; Fisher BF; Jiang Y; Lewis JC Flavin-Dependent Halogenases Catalyze Enantioselective Olefin Halocyclization. Nat Commun 2021, 12 (1), 3268. 10.1038/s41467-021-23503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Denmark SE; Kuester WE; Burk MT Catalytic, Asymmetric Halofunctionalization of Alkenes-A Critical Perspective. Angewandte Chemie International Edition in English 2012, 51 (44), 10938–10953. 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ashtekar KD; Vetticatt M; Yousefi R; Jackson JE; Borhan B Nucleophile-Assisted Alkene Activation: Olefins Alone Are Often Incompetent. Journal of the American Chemical Society 2016, 138 (26), 8114–8119. 10.1021/jacs.6b02877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Younes SHH; Tieves F; Lan D; Wang Y; Süss P; Brundiek H; Wever R; Hollmann F Chemoenzymatic Halocyclization of γ,Δ‐Unsaturated Carboxylic Acids and Alcohols. ChemSusChem 2019, 13 (1), 97–101. 10.1002/cssc.201902240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou P; Cai Y; Zhong X; Luo W; Kang T; Li J; Liu X; Lin L; Feng X Catalytic Asymmetric Intra- and Intermolecular Haloetherification of Enones: An Efficient Approach to (−)-Centrolobine. Acs Catal 2016, 6 (11), 7778–7783. 10.1021/acscatal.6b02048. [DOI] [Google Scholar]
- (17).Han C; Feng X; Du H Asymmetric Halocyclizations of 2-Vinylbenzyl Alcohols with Chiral FLPs. Org Lett 2021, 23 (19), 7325–7329. 10.1021/acs.orglett.1c02361. [DOI] [PubMed] [Google Scholar]
- (18).Denmark SE; Burk MT Enantioselective Bromocycloetherification by Lewis Base/Chiral Brønsted Acid Cooperative Catalysis. Org Lett 2012, 14 (1), 256–259. 10.1021/ol203033k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Böse D; Denmark S Investigating the Enantiodetermining Step of a Chiral Lewis Base Catalyzed Bromocycloetherification of Privileged Alkenes. Synlett 2018, 29 (04), 433–439. 10.1055/s-0036-1590951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ke Z; Tan CK; Liu Y; Lee KGZ; Yeung Y-Y Catalytic and Enantioselective Bromoetherification of Olefinic 1,3-Diols: Mechanistic Insight. Tetrahedron 2016, 72 (21), 2683–2689. 10.1016/j.tet.2015.09.016. [DOI] [Google Scholar]
- (21).Müller CH; Rösner C; Hennecke U Enantioselective Haloetherifications Catalyzed by 1,1′‐Bi‐2‐naphthol (BINOL) Phosphates: From Symmetrical Alkenediols to Simple Alkenols. Chem Asian J 2014, 9 (8), 2162–2169. 10.1002/asia.201402229. [DOI] [PubMed] [Google Scholar]
- (22).Tay DW; Leung GYC; Yeung Y Desymmetrization of Diolefinic Diols by Enantioselective Amino‐thiocarbamate‐Catalyzed Bromoetherification: Synthesis of Chiral Spirocycles. Angewandte Chemie Int Ed 2014, 53 (20), 5161–5164. 10.1002/anie.201310136. [DOI] [PubMed] [Google Scholar]
- (23).Ke Z; Tan CK; Chen F; Yeung Y-Y Catalytic Asymmetric Bromoetherification and Desymmetrization of Olefinic 1,3-Diols with C 2-Symmetric Sulfides. J Am Chem Soc 2014, 136 (15), 5627–5630. 10.1021/ja5029155. [DOI] [PubMed] [Google Scholar]
- (24).Khanam H; Shamsuzzaman. Bioactive Benzofuran Derivatives: A Review. Eur J Med Chem 2015, 97, 483–504. 10.1016/j.ejmech.2014.11.039. [DOI] [PubMed] [Google Scholar]
- (25).Delost MD; Smith DT; Anderson BJ; Njardarson JT From Oxiranes to Oligomers: Architectures of U.S. FDA Approved Pharmaceuticals Containing Oxygen Heterocycles. J Med Chem 2018, 61 (24), 10996–11020. 10.1021/acs.jmedchem.8b00876. [DOI] [PubMed] [Google Scholar]
- (26).Lorente A; Lamariano-Merketegi J; Albericio F; Álvarez M Tetrahydrofuran-Containing Macrolides: A Fascinating Gift from the Deep Sea. Chem Rev 2013, 113 (7), 4567–4610. 10.1021/cr3004778. [DOI] [PubMed] [Google Scholar]
- (27).Fisher BF; Snodgrass HM; Jones KA; Andorfer MC; Lewis JC Site-Selective C–H Halogenation Using Flavin-Dependent Halogenases Identified via Family-Wide Activity Profiling. ACS Cent Sci 2019, 5 (11), 1844–1856. 10.1021/acscentsci.9b00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Payne JT; Poor CB; Lewis JC Directed Evolution of RebH for Site-Selective Halogenation of Large Biologically Active Molecules. Angewandte Chemie International Edition in English 2015, 54 (14), 4226–4230. 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Andorfer MC; Park H-J; Vergara-Coll J; Lewis JC Directed Evolution of RebH for Catalyst-Controlled Halogenation of Indole C–H Bonds. Chemical Science 2016, 7 (6), 3720–3729. 10.1039/c5sc04680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang Y; Chen L; Chen H; Huang T; Shi Q; Wang X; Wang Y; Tang M-C; Zhou N-Y; Lin S Aryl C-H Iodination: Are There Actual Flavin-Dependent Iodinases in Nature? Sci China Chem 2021, 64 (10), 1730–1735. 10.1007/s11426-021-1018-0. [DOI] [Google Scholar]
- (31).Payne JT; Andorfer MC; Lewis JC Regioselective Arene Halogenation Using the FAD-Dependent Halogenase RebH. Angewandte Chemie International Edition in English 2013, 52 (20), 5271–5274. 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Giordano C; Castaldi G; Uggeri F Synthesis of Anti-Inflammatory α-Arylalkanoic Acids by 1,2-Aryl Shift. Angewandte Chemie International Edition 1984, 23, 413–419. 10.1002/anie.198404131. [DOI] [Google Scholar]
- (33).Poor CB; Andorfer MC; Lewis JC Improving the Stability and Catalyst Lifetime of the Halogenase RebH By Directed Evolution. ChemBioChem 2014, 15 (9), 1286–1289. 10.1002/cbic.201300780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Voss M; Küng R; Hayashi T; Jonczyk M; Niklaus M; Iding H; Wetzl D; Buller R Multi‐faceted Set‐up of a Diverse Ketoreductase Library Enables the Synthesis of Pharmaceutically‐relevant Secondary Alcohols. Chemcatchem 2021, 13 (6), 1538–1545. 10.1002/cctc.202001871. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.