

Abstract
The common aldehyde dehydrogenase 2 (ALDH2) alcohol flushing variant known as ALDH2*2 affects ~8% of the world’s population. Even in heterozygous carriers, this missense variant leads to a severe loss of ALDH2 enzymatic activity and has been linked to an increased risk of coronary artery disease (CAD). Endothelial cell (EC) dysfunction plays a determining role in all stages of CAD pathogenesis, including early-onset CAD. However, the contribution of ALDH2*2 to EC dysfunction and its relation to CAD are not fully understood. In a large genome-wide association study (GWAS) from Biobank Japan, ALDH2*2 was found to be one of the strongest single nucleotide polymorphisms associated with CAD. Clinical assessment of endothelial function showed that human participants carrying ALDH2*2 exhibited impaired vasodilation after light alcohol drinking. Using human induced pluripotent stem cell-derived ECs (iPSC-ECs) and CRISPR/Cas9-corrected ALDH2*2 iPSC-ECs, we modeled ALDH2*2-induced EC dysfunction in vitro, demonstrating an increase in oxidative stress and inflammatory markers and a decrease in nitric oxide (NO) production and tube formation capacity, which was further exacerbated by ethanol exposure. We subsequently found that sodium/glucose cotransporter-2 inhibitors (SGLT2i) such as empagliflozin mitigated ALDH2*2-associated EC dysfunction. Studies in ALDH2*2 knock-in mice further demonstrated that empagliflozin attenuated ALDH2*2-mediated vascular dysfunction in vivo. Mechanistically, empagliflozin inhibited Na+/H+-exchanger 1 (NHE-1) and activated AKT kinase and endothelial nitric oxide synthase (eNOS) pathways to ameliorate ALDH2*2-induced EC dysfunction. Taken together, our results suggest that ALDH2*2 induces EC dysfunction and that SGLT2i may potentially be used as a preventative measure against CAD for ALDH2*2 carriers.
One Sentence Summary:
ALDH2*2 iPSC-ECs recapitulate endothelial dysfunction in ALDH2*2 carriers, and SGLT2i could be a preventative therapy.
INTRODUCTION
Coronary artery disease (CAD) is the leading cause of global mortality, accounting for an estimated 9 million annual deaths, according to the World Health Organization (WHO) (1). The global prevalence of CAD is rising due to an aging population and the lack of novel strategies to effectively prevent and treat CAD (1). Precision medicine for CAD patients with genetic variants holds promise as a new approach toward more effective prevention and treatment by understanding their physiological mechanisms and causal relevance (2).
CAD is a complex disease caused by the interplay among multiple genetic and environmental factors (3). Hundreds of genomic loci have previously been associated with CAD by genome-wide association studies (GWAS) (4). A single nucleotide polymorphism (SNP) in the aldehyde dehydrogenase 2 (ALDH2) gene, ALDH2*2, also named rs671, causes a substitution of glutamate to lysine at position 504 (E504K) that alters the access of substrates to the catalytically active site of ALDH2 compared to the normal ALDH2*1 allele (5). ALDH2 is a critical enzyme in the second step of alcohol metabolism. It is vital in detoxifying toxic aldehydes such as oxidative stress-derived aldehydes and alcohol-derived acetaldehyde. ALDH2*2 carriers have diminished ALDH2 enzymatic activity, and this deficiency is associated with a variety of neurologic, cardiovascular, and dermatologic disorders, aberrant drug metabolism, and different types of cancers (6). A recent meta-analysis of 12 case-control studies with 3,305 cases and 5,016 controls shows that ALDH2*2 is associated with a 48% increased risk of CAD (7). To date, however, the underlying mechanisms for the association between ALDH2*2 and the onset of CAD remain unclear, which creates a barrier to the prevention and treatment of ALDH2*2-related CAD. In addition, alcohol consumption is a well-known risk factor for CAD (8). Although ethanol is not a primary contributor to CAD in studies of non-Asian subjects, it may have a substantial effect on ALDH2*2 carriers of Asian descent (9, 10). Thus, it is important to determine the combined effect of ALDH2*2 and alcohol consumption on CAD to develop preventative strategies to reduce CAD risk among ALDH2*2 carriers.
Endothelial cells (ECs), important constituents of blood vessel walls, play a critical role in maintaining cardiovascular homeostasis by regulating vascular tone and structure. EC dysfunction is implicated in a wide variety of human cardiovascular diseases (11). Coronary EC dysfunction occurs early in the development of CAD and can impact all phases of the disease from initiation to atherothrombotic complication (12). ALDH2*2 has been associated with EC dysfunction by increasing inflammation via nuclear factor kappa B (NF-κB), c-Jun, and mitogen-activated protein kinases (MAPK) pathways (13), decreasing nitric oxide (NO) production via the dimethylarginine dimethylaminohydrolase-1 (DDAH1) and asymmetric dimethylarginine (ADMA) pathway (14), and elevating oxidative damage via reactive oxygen species (ROS) and toxic aldehydes such as 4-hydroxynonenal (4-HNE) (15). However, the detailed mechanism underlying ALDH2*2-induced EC dysfunction remains unclear. Here, we focused on elucidating the molecular and cellular mechanisms underlying ALDH2*2-related EC dysfunction in human cells and identifying strategies to mitigate EC dysfunction in patients with this genetic variant.
RESULTS
ALDH2*2 is strongly associated with coronary artery disease in East Asians
ALDH2*2 (rs671) has been associated with an increased risk of CAD in case-control studies in East Asians (7). However, these studies have small sample sizes. To further understand the effect of ALDH2*2 on CAD, we performed a more extensive GWAS analysis in 29,319 CAD cases and 183,134 controls from Biobank Japan. ALDH2*2 and 8730 other SNPs (fig. S1A, table S1) passed the genome-wide significance threshold (P < 5×10−8). Among these SNPs, rs78069066 in the intron of MAPK-activated protein kinase 5 (MAPKAPK5) (P = 1.9×10−92), rs4646776 in the intron of ALDH2 (P = 3.7×10−92), and ALDH2*2 in the exon 12 of ALDH2 (P = 1.1×10−91) were the most significant SNPs associated with CAD (Fig. 1A). To identify other functionally relevant variants with high linkage disequilibrium (LD) with ALDH2*2, we generated a regional association plot of variants located around ± 50 kb region of ALDH2 gene loci (fig. S1B). Only one SNP, rs4646776, which was identified by our initial GWAS analysis, showed a high LD relationship with ALDH2*2.
Figure 1. ALDH2*2 is strongly associated with coronary artery disease (CAD) and induces endothelial dysfunction.

(A) Manhattan plot of GWAS on CAD using the Biobank Japan cohort. CAD cases, n = 29,319; healthy control, n = 183,134. The different colors of each block are used to distinguish data from each chromosome. The X-axis indicates each chromosome number (all autosomal), and the Y-axis shows the −log10 (p-value) of risk loci. The dotted line defines the significance threshold of P = 5 × 10−8. The strongest genetic loci associated with CAD are labeled in the plot. (B–C) Line plots show the individual changes of heart rate (B) and reactive hyperemia index (C) before and after drinking alcohol in human participants with WT (Black color) or ALDH2*1/*2 (red color) alleles. All data are represented as mean ± SEM, n = 9 (WT), n = 9 (ALDH2*1/*2); “after” data compared to the “before” data, *P < 0.05, ****P < 0.0001. Statistical analyses for B-C were performed using a paired Student’s t-test.
Due to the association of ALDH2*2 with many human diseases (6), we also conducted a phenome-wide association study (PheWAS) of ALDH2*2 with 47 human diseases from Biobank Japan. CAD was the most significant disease associated with ALDH2*2 (P = 1.1×10−91), followed by other conditions including esophageal cancer (P = 6.2×10−25) (fig. S1C). Epidemiological studies have also shown that ALDH2*2 increases the risk of esophageal cancer (16), which support our results indicating an association of ALDH2*2 with CAD as well.
ALDH2*2 carriers exhibit clinical EC dysfunction with alcohol consumption
Due to the pivotal role of ECs in CAD, we hypothesized that the ALDH2*2 variant contributed to EC dysfunction and the development of CAD. To determine whether ALDH2*2 carriers exhibited clinical EC dysfunction, an EndoPAT device was used to assess endothelial vasodilator function in a non-invasive manner. The device records endothelium-mediated changes in response to brachial artery occlusion and vasodilation, and calculates a Reactive Hyperemia Index (RHI), which measures NO-dependent changes in vascular tone and correlates with the measurement of endothelial vasodilator function in the coronary arteries (17, 18). RHI value positively correlates with endothelial function, and an RHI value of < 1.67 is associated with endothelial dysfunction and risk of cardiovascular disease, including CAD (19). EndoPAT analysis was performed on human participants carrying either ALDH2*1 wild-type (WT) (n = 9) or heterozygous ALDH2 variants (ALDH2*1/*2) (n = 9) before or immediately after a single alcoholic beverage (fig. S1, D and E). Although there was no change of heart rate before or after alcohol consumption in the WT group (before: 71 ± 4 bpm; after: 71 ± 3 bpm), ALDH2*1/*2 subjects showed a significantly increased heart rate after alcohol ingestion (before: 69 ± 4 bpm; after: 86 ± 5 bpm; P < 0.0001) (Fig. 1B, fig. S1, D and E, table S2), which was consistent with the results from previous studies (20–22). Whereas there was an increase in RHI after one drink in the WT group (before: 1.73 ± 0.15; after: 1.97 ± 0.13; P = 0.01), the ALDH2*1/*2 group showed the opposite trend in RHIs (before: 1.79 ± 0.16; after: 1.48 ± 0.19; P = 0.02) (Fig. 1C, fig. S1, D and E, table S2). The average RHI value was below 1.67 in the ALDH2*1/*2 group after one drink, indicating ALDH2*2 carriers exhibited acute EC dysfunction after one standard alcoholic beverage.
A recent study in Asians showed that ALDH2*2 increased the risk of atrial fibrillation in habitual drinkers (10), suggesting there may be synergetic effects of ALDH2*2 and chronic alcohol consumption on cardiac arrhythmias. To investigate whether ALDH2*2 was associated with higher risk of CAD in chronic drinkers, we examined genetic information and drinking status of human participants from the UK Biobank (23). The data was analyzed for the combinatorial effects of alcohol consumption and ALDH2*2 on CAD. Among the 424,507 participants in the UK Biobank who currently consume alcohol, 73,423 were defined as chronic drinkers by the self-reported criterion that they “drink more nowadays compared to 10 years ago”. Using logistic regression analysis, we found that chronic alcohol consumption in ALDH2*2 carriers led to an approximately 4-fold greater risk for CAD than their wild-type ALDH2*1 counterparts, suggesting that chronic alcohol consumption increases the rate of CAD among ALDH2*2 carriers (fig. S1F). Thus, these data suggest that ALDH2*2 variant carriers who consume alcohol are more susceptible to EC dysfunction and CAD.
iPSC-ECs with ALDH2*2 show impaired EC function
To uncover the precise genotype-phenotype relationship, we utilized iPSC-derived endothelial cells (iPSC-ECs) to model ALDH2*2-associated EC dysfunction in vitro. We recruited a cohort of 10 age- and sex-matched East Asian individuals who carried either the WT or ALDH2*1/*2 allele as previously described (n=5 per group, fig. S2A) (24). The ALDH2 genotype was confirmed with Sanger sequencing (fig. S2B). Following a chemically defined and small molecule-based generation of endothelial differentiation protocol (fig. S2C) (25), iPSCs were differentiated into iPSC-ECs as a monolayer. Immunostaining assays and fluorescence-activated cell sorting (FACS) showed that both WT and ALDH2*1/*2 iPSC-ECs expressed the endothelial marker, CD31, with high purity (Fig. 2, A and B). To examine whether ALDH2*2-associated EC dysfunction is associated with the differential expression of ALDH2, we performed a Western blot on the 10 WT and ALDH2*1/*2 iPSC-EC lines. We observed a decrease in ALDH2 protein in ALDH2*1/*2 iPSC-ECs compared to WT iPSC-ECs (Fig. 2, C and D), consistent with previous studies showing a reduction in ALDH2 protein expression in the liver of human subjects carrying ALDH2*2 and in Aldh2E487K knock-in mice (26, 27). As expected, ALDH2 enzymatic activity was also reduced in ALDH2*1/*2 cell lysates relative to WT and human umbilical vein endothelial cell (HUVEC) controls (Fig. 2E).
Figure 2. ALDH2*2 iPSC-ECs show increased oxidative stress and monocyte adhesion and decreased NO production at baseline.

(A) Representative brightfield and immunofluorescence images of WT and ALDH2*1/*2 iPSC-ECs for endothelial marker CD31 (green). Scale bar: 100 μm. (B) Representative fluorescence-activated cell sorting (FACS) analysis of WT and ALDH2*1/*2 iPSC-ECs at passage 1 with CD31 antibody. (C) Representative immunoblot for ALDH2 in WT and ALDH2*1/*2 iPSC-ECs. GAPDH was used as loading control. (D) Bar graph shows quantification of ALDH2 protein by immunoblot in WT and ALDH2*1/*2 iPSC-ECs. Data are represented as relative fold-change to loading control, GAPDH. (E) Enzymatic activity of ALDH2 in lysates from WT and ALDH2*1/*2 iPSC-ECs. HUVEC were used as a positive control. WT vs ALDH2*1/*2: ****P < 0.0001; HUVEC vs ALDH2*1/*2: $ $ $ $P < 0.0001. (F) 4-hydroxynonenal (4-HNE) adducts in WT and ALDH2*1/*2 iPSC-ECs were assessed by Western blot with GAPDH as loading control. (G) Quantification of 4-HNE Western blot analysis from two replicate experiments. (H) Total baseline reactive oxygen species (ROS) in WT and ALDH2*1/*2 iPSC-ECs as measured by the fluorogenic dye, CM-H2DCFDA. (I) Quantification of NO production in WT and ALDH2*1/*2 iPSC-ECs using the fluorogenic dye, DAF-FM. (J) Representative images of adherent THP-1 monocytes labeled with Calcein AM (green) in WT and ALDH2*1/*2 iPSC-ECs. (K) Quantification of monocyte adhesion in WT and ALDH2*1/*2 iPSC-ECs. Black bar: WT iPSC-ECs; Red bar: ALDH2*1/*2 iPSC-ECs. All data represent five individual iPSC lines from two technical replicates per group. Data are expressed as mean ± SEM. *P < 0.05, calculated by Student’s t-test (Fig. 2. E, H, I, J and L) or two-way ANOVA with Bonferroni correction (Fig. 2E).
We next measured the amount of toxic aldehyde 4-hydroxynonenal (4-HNE) in iPSC-ECs because enhanced production of toxic aldehydes such as 4-HNE has been shown to increase inflammation, oxidative stress, and cell death in patients with acute CAD (28, 29). We found increased 4-HNE (P < 0.05) in ALDH2*1/*2 cell lysates compared to WT controls (Fig. 2, F and G). We therefore evaluated the functional characteristics of iPSC-ECs from both WT and ALDH2*1/*2 carriers. Compared to WT, ALDH2*1/*2 iPSC-ECs had increased cellular ROS (Fig. 2H and fig. S2D) and a decreased capacity to generate NO (Fig. 2I and fig. S2E), which are implicated in vascular dysfunction (30, 31). In addition, ALDH2*1/*2 iPSC-ECs had increased monocyte adhesion compared to WT iPSC-ECs, suggesting increased endothelial inflammation in the ALDH2*2 variant (Fig. 2, J and K). ALDH2*1/*2 iPSC-ECs also showed a decreased capacity to form tubular networks versus WT iPSC-ECs (fig. S2, F and G). Thus, our data suggest a causal effect of ALDH2*2 on EC dysfunction.
Ethanol exacerbates ALDH2*2-induced EC dysfunction
As ALDH2 is the major enzyme for metabolizing acetaldehyde from ethanol, we hypothesized that ALDH2*2 carriers were more susceptible to the deleterious effect of ethanol on EC function. First, we determined there were no differences in the mRNA expression of alcohol-metabolizing enzymes alcohol dehydrogenase 5 (ADH5) and ALDH2 in WT and ALDH2*1/*2 iPSC-ECs (fig. S3A). We then treated WT and ALDH2*1/*2 iPSC-ECs with 5 mM ethanol, the concentration of ethanol in human blood after one standard alcoholic beverage (32). Consistent with previous findings showing that ALDH2*2 alone impairs endothelial function, ethanol further increased the generation of ROS (Fig. 3A), decreased NO production (Fig. 3B), increased monocyte adhesion (Fig. 3, C and D), and impaired tube formation compared to both WT (vehicle and ethanol-treated) and vehicle-treated ALDH2*2 iPSC-ECs (Fig. 3, E and F). These results suggest that ethanol exacerbates ALDH2*2-induced EC dysfunction.
Figure 3. Ethanol exacerbates ALDH2*2-induced endothelial dysfunction.

(A) Measurement of cellular ROS released from WT and ALDH2*1/*2 iPSC-ECs using the fluorogenic dye, CM-H2DCFDA, in the presence or absence of 5 mM ethanol. (B) Nitric oxide (NO) production in WT and ALDH2*1/*2 iPSC-ECs using the fluorogenic dye, DAF-FM, in the presence or absence of 5mM ethanol. (C) Representative images of adherent THP-1 monocytes labeled with Calcein AM (green) to WT and ALDH2*1/*2 iPSC-ECs in the presence or absence of 5 mM ethanol. Scale bar: 1mm. (D) Quantification of monocyte adhesion in WT and ALDH2*1/*2 iPSC-ECs in the presence or absence of 5 mM ethanol. (E) Representative brightfield images of capillary-like networks formed by WT and ALDH2*1/*2 iPSC-ECs in the presence or absence of 5 mM ethanol. Scale bar: 1mm. (F) Quantitative data from the tube-formation assay in WT and ALDH2*1/*2 iPSc-ECs in the presence or absence of 5 mM ethanol. Black bar: WT iPSC-ECs; Red bar: ALDH2*1/*2 iPSc-ECs. All data represent five individual iPSC lines from two technical replicates per group. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, calculated by a two-way ANOVA with Bonferroni correction.
CRISPR/Cas9-corrected iPSC-ECs reverse ALDH2*2-induced EC dysfunction
To confirm the role of ALDH2*2 in EC dysfunction, we next corrected ALDH2*2 in three ALDH2*1/*2 iPSC lines using CRISPR/Cas9 and compared EC function in three sets of WT cell lines, ALDH2*1/*2-corrected isogenic lines (Iso_GG) and their uncorrected counterparts. The missense ALDH2 variant (GA) in ALDH2*1/*2 lines was corrected and confirmed by Sanger sequencing (Fig. 4, A and B). Next, the Iso_GG iPSC lines were differentiated into ECs, and all showed high EC purity (fig. S3, B and C). As expected, ALDH2 enzyme activity was restored after gene correction (Fig. 4C). The EC dysfunction observed in ALDH2*1/*2 iPSC-ECs was also reversed in the genome-edited Iso_GG iPSC-ECs, as evidenced by reduced ROS concentrations (Fig. 4D) and monocyte adhesion (Fig. 4, E and F), and increased NO production (Fig. 4G) and tube formation (fig. S3, D to G). Ethanol treatment did not affect ALDH2 expression in corrected cells (fig. S3, H and I). Similarly, ethanol-induced exacerbation of EC dysfunction was also mitigated in Iso_GG iPSC-ECs (Fig. 4, G to I, fig. S3, D to G). These findings indicate a causal role of ALDH2*2 in EC dysfunction.
Figure 4. Genome-corrected isogenic ALDH2 iPSc-ECs rescue EC dysfunction phenotype.
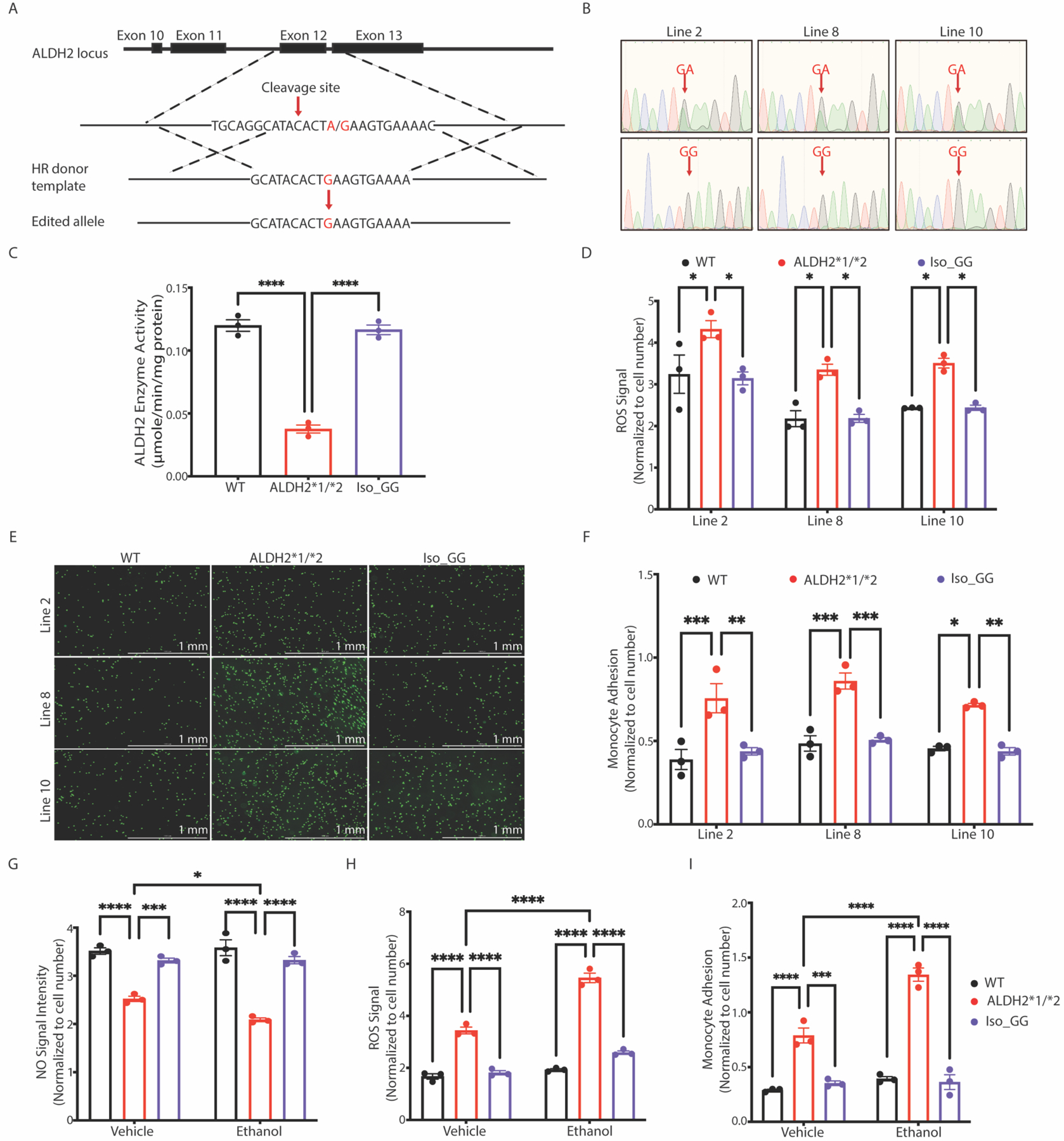
(A) CRISPR/Cas9-mediated genome modification to correct heterozygous ALDH2*1/*2 variant (GA) into isogenic WT (Iso_GG) iPSCs using homologous repair (HR). CRISPR/Cas9 was designed to specifically cleave near the nucleotide position 111803962 at exon 12 of the ALDH2 locus. The ALDH2*1/*2 allele (GA) is shown in red. (B) Sequencing chromatogram showing three Iso_GG iPSC lines generated from their respective ALDH2*1/*2 (GA) iPSC lines. The risk allele A is substituted by G after genome-editing as shown in the lower panel (indicated by red arrows). (C) Enzymatic activity of ALDH2 in lysates from WT, ALDH2*1/*2, and Iso_GG iPSC-ECs. (D) Cellular ROS in WT, ALDH2*1/*2, and Iso_GG iPSC-ECs. Cellular ROS were quantified using CM-H2DCFDA dye. (E) Representative images of adherent THP-1 monocytes (green) to WT, ALDH2*1/*2, and Iso_GG iPSC-ECs. Scale bar: 1mm. (F) Quantification of monocyte adhesion in WT, ALDH2*1/*2 and Iso_GG iPSC-ECs. (G) Nitric oxide (NO) production was determined in WT, ALDH2*1/*2, and Iso_GG iPSC-ECs using DAF-FM in the presence or absence of 5 mM ethanol. (H) Measurement of ROS in WT, ALDH2*1/*2, and Iso_GG iPSC-ECs using CM-H2DCFDA in the presence or absence of 5 mM ethanol. (I) Quantification of monocyte adhesion in WT, ALDH2*1/*2, and Iso_GG iPSC-ECs in the presence or absence of 5 mM ethanol. Black bar: WT iPSC-ECs; Blue bar: ALDH2*1/*2 iPSC-ECs; Violet bar: Iso_GG iPSC-ECs. Data in panel C to I represent two technical replicates from three individual iPSC lines. All data are expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, calculated by a one-way (Fig. C, D and F) or two-way ANOVA (Fig. G-I) with Bonferroni correction.
Transcriptional profiling reveals downregulation of the AKT/eNOS pathway in ALDH2*2 ECs
To elucidate the molecular mechanisms of EC dysfunction, we performed bulk RNA sequencing (RNA-seq) on two ALDH2*2-corrected isogenic iPSC-EC lines (Iso_GG) and their uncorrected counterparts (ALDH2*1/*2). A direct comparison of total RNA expression between ALDH2*1/*2 iPSC-ECs and Iso_GG controls revealed 1,460 differentially expressed genes (DEGs), of which 803 were upregulated and 657 were downregulated (Fig. 5A and table S3). Among these DEGs, 14 were related to angiogenesis genes such as secreted frizzled-related protein 1 (SFRP1) and protein tyrosine kinase 2 (PTK2), 21 were related to antioxidants or oxidants such as glutathione S-transferase pi 1 (GSTP1) and thioredoxin reductase 2 (TXNRD2), and 147 were related to inflammatory genes such as interleukin-18 (IL18) and intercellular adhesion molecule-1 (ICAM1) (Fig. 5B and table S4). ICAM1 is essential in mediating monocyte adhesion to endothelial cells (33). IL18 belongs to interleukin-1 family and is a prospective and independent marker of CAD risk (34). qPCR analysis confirmed expression of ICAM1 and IL18 was upregulated in ALDH2*1/*2 compared to Iso_GG iPSC-ECs. In addition, ethanol further increased the expression of ICAM-1 and IL18 in ALDH2*1/*2 iPSC-ECs, but not Iso_GG iPSC-ECs, suggesting ICAM1 and IL18 may contribute to the inflammatory phenotype in ALDH2*1/*2 iPSC-ECs (Fig. 5C). GSTP1 is a critical enzyme in reducing oxidative stress through increasing glutathione availability, and mutations in this gene have been associated with greater susceptibility to CAD (35, 36). The expression of GSTP1 was downregulated in ALDH2*1/*2 iPSC-ECs and further decreased in ethanol-treated ALDH2*1/*2 iPSC-ECs, indicating GSTP1 may play a role in the ALDH2*2-mediated increase in oxidative stress (Fig. 5C). SFRP1, an endogenous modulator of canonical wingless and int-1 (Wnt) signaling pathway with implications in cardiac neovascularization after injury (37), was also downregulated in ALDH2*1/*2 iPSC-ECs, and further decreased in ethanol-treated ALDH2*1/*2 iPSC-ECs (Fig. 5C), indicating that SFRP1 may be involved in ALDH2*2-dependent angiogenesis defects.
Figure 5. Transcriptional profiling of ALDH2 iPSC-ECs implicates AKT/eNOS pathway in EC dysfunction.

(A) Volcano plot of differentially expressed genes (DEGs) between ALDH2*1/*2 and Iso_GG iPSC-ECs. Upregulated genes are highlighted in red and downregulated genes are shown in blue. FDR < 0.05 was set as a threshold of significance. (B) Heatmap of DEGs in oxidative stress, angiogenesis, and inflammatory pathways. Expression of representative genes is indicated on the bottom the heatmap. SFRP1: secreted frizzled-related protein 1; GSTP1: glutathione S-transferase pi 1; IL18: interleukin 18; ICAM1: intercellular adhesion molecule-1. (C) Quantitative PCR data from ALDH2*1/*2 and Iso_GG iPSC-ECs in the presence or absence of 5 mM ethanol. (D) Top 10 upregulated (red) and downregulated pathways (blue) in ALDH2*1/*2 iPSC-ECs compared to Iso_GG iPSC-ECs as determined by −log10 false discovery rate (FDR) using the KEGG pathway database. (E) Immunoblots for total AKT, p-AKT (Ser 473), total eNOS, and p-eNOS (Ser 1177) proteins in ALDH2*1/*2 and Iso_GG iPSC-ECs in the presence or absence of 5 mM ethanol. GAPDH was used as loading control. (F) Bar graph shows quantification of p-AKT/AKT ratio, p-eNOS/eNOS ratio in WT and ALDH2*1/*2 iPSC-ECs in the presence or absence of 5 mM ethanol. (G) Measurement of ROS and NO in ethanol-treated WT and ALDH2*1/*2 iPSC-ECs in the presence or absence of 4 μg/ml AKT activator, SC79. Vehicle: equal volume of EC medium. Black bar: WT iPSC-ECs; Red bar: ALDH2*1/*2 iPSC-ECs; Violet bar: Iso_GG iPSC-ECs. Data in panel C, F and G represent two independent technical replicates from three individual iPSC lines. All data are expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, calculated by a two-way ANOVA with Bonferroni correction.
Gene ontology analysis of DEGs showed multiple dysregulated pathways in ALDH2*1/*2 iPSC-ECs compared to Iso_GG iPSC-ECs (Fig. 5D). For example, genes associated with extracellular matrix (ECM)-receptor interaction, phosphoinositide 3-kinase (PI3K)-AKT signaling, relaxin signaling, MAPK signaling, and AMP-activated protein kinase (AMPK) signaling pathways were downregulated, whereas genes associated with nucleotide-binding oligomerization domain (NOD)-like receptor signaling, cellular senescence, calcium signaling, FoxO signaling, p53, and NF-kB signaling pathways were upregulated in ALDH2*1/*2 iPSC-ECs (Fig. 5D and table S5). AKT plays a vital role in vascular homeostasis and pathogenesis by mediating EC death, migration, and production or degradation of extracellular matrix (38). Our results further confirmed that the abundance of the active form of AKT, p-AKT (phosphorylation at serine 473), rather than total AKT, was decreased in ALDH2*1/*2 iPSC-ECs (Fig. 5, E and F). The abundance of p-AKT was further reduced by ethanol treatment in ALDH2*1/*2 iPSC-ECs, indicating that both the ALDH2*2 variant and ethanol could limit AKT activity. Because endothelial nitric oxide synthase (eNOS) is activated by AKT-dependent phosphorylation in endothelial cells (39), we used Western blot analysis to demonstrate that the active form of eNOS, p-eNOS (phosphorylation at serine 1177) was decreased in ALDH2*1/*2 iPSC-ECs, and further decreased by ethanol treatment (Fig. 5, E and F). We confirmed the relevance of this pathway by treating cells with SC79, a pan-AKT activator that specifically enhances AKT phosphorylation and activation (40). We found that increased AKT activity led to increased NO production and decreased ROS generation in both Iso_GG and ALDH2*1/*2 iPSC-ECs (Fig. 5G). Thus, these findings suggest that inhibition of the AKT/eNOS pathway mediates ALDH2*2-induced EC dysfunction.
SGTL2i counteracts ALDH2*2-associated EC dysfunction
Human iPSC-derived cells provide a unique platform to screen for compounds that have human-specific drug responsiveness (41). With ROS, NO, and inflammation as primary targets, we searched cardiovascular drugs approved by the Food and Drug Administration (FDA) and identified seven drugs that regulate these pathways (Fig. 6A). Three of these were sodium-glucose cotransporter-2 (SGTL2) inhibitors (empagliflozin, canagliflozin, and dapagliflozin), which have previously been shown to induce NO production and reduce ROS generation in ECs (42, 43). We used Alda-1, an activator of both WT ALDH2 and of ALDH2*2 (44), as a positive control. Our analysis revealed that all SGLT2i showed a dose-dependent decrease in ROS generation and increase in NO production in ALDH2*1/*2 iPSC-ECs (Fig. 6, B and C). Aspirin and metformin promoted NO generation, but they also increased ROS production. Meanwhile, fenofibrate and Alda-1 required a large dose to effectively reduce ROS. Alda-1 did not have a beneficial effect in Iso_GG iPSC-ECs, which was likely due to the high dose of Alda-1, as shown by a decrease of NO in Iso_GG iPSC-ECs (Fig. 6, B and C). Empagliflozin was selected for use in all subsequent experiments as it was efficacious for increasing NO production and reducing ROS generation at the lowest dose. A dose of 5 μM of empagliflozin was selected based on the cellular experiments for rescuing capacity. We then treated iPSC-ECs with 5 μM empagliflozin in the presence of ethanol (5 mM) and found empagliflozin not only decreased ROS (fig. S4A) and increased NO production (fig. S4B), but also reduced monocyte adhesion (fig. S4, C and D) and increased EC tube formation in ALDH2*1/*2 samples (fig. S4, E and F). Empagliflozin also reduced monocyte adhesion in Iso_GG iPSC-ECs (fig. S4, C and D), indicating that empagliflozin may reduce the pro-inflammatory phenotype even in the absence of the ALDH2*2 variant. Taken together, these results suggested that empagliflozin improves EC dysfunction in ALDH2*1/*2 iPSC-ECs.
Figure 6. Drug screening for treatment of ALDH2*2-induced oxidative stress and NO impairment using iPSC-ECs.
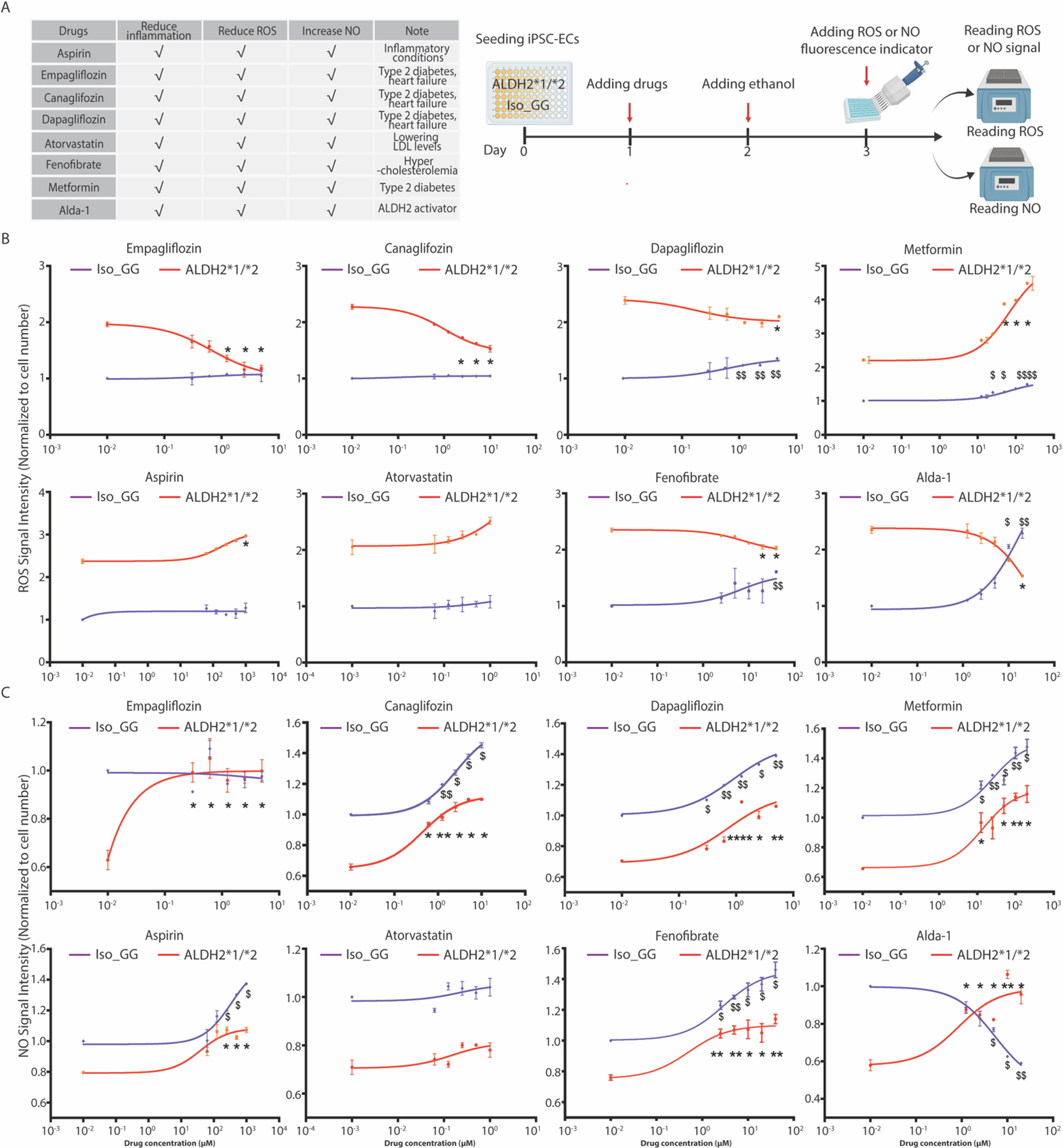
(A) Table listing the eight compounds assessed and their current uses, including known effects on inflammation, ROS and NO (left panel). These include seven medications approved by the Food and Drug Administration (FDA) and the ALDH2 activator Alda-1. Schematic workflow of the experimental design (right panel). These drugs were assessed for their impact on ROS and NO production in Iso_GG and ALDH2*1/*2 iPSC-ECs. iPSC-ECs were treated at day one. 5mM ethanol was added the following day, and then NO and ROS signals were measured at day 3. (B–C) Dose-response curves based on ROS generation, indicated by CM-H2DCFDA (B), and NO production indicated by DAF-FM (C) in WT and ALDH2*1/*2 iPSC-ECs with drug treatment. Six different dosages were used for each drug, including a vehicle control (DMSO). Due to a wide range of drug dosage used in WT and ALDH2*2 iPSC-ECs, the power of 10 format was used on the X-axis to plot drug response. 0.01 μM (10−2 μM) instead of 0 μM was used to indicate response to vehicle control (DMSO) and the concentrations of NO or ROS in Iso_GG or ALDH2*1/*2 iPSC-ECs were normalized to vehicle (DMSO) treatment. Data in panel B and C represent three individual iPSC lines from two technical replicates for each group. Data were expressed as mean ± SEM. Non-linear regression model was applied to plot drug dose-response. Iso_GG iPSC-ECs with drug treatment compared to Iso_GG with vehicle (DMSO) treatment: $P < 0.05, $ $P < 0.01, $ $ $P < 0.001, $ $ $ $P < 0.0001. ALDH2*1/*2 iPSC-ECs with drug treatment compared to ALDH2*1/*2 with vehicle (DMSO) treatment: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, calculated by Student’s t-test.
Empagliflozin rescues endothelial dysfunction in ALDH2*2 mice
Next, we examined whether empagliflozin rescued endothelial dysfunction in mice in which a single human variant, E504K, was knocked into one copy of WT mouse Aldh2 allele (heterozygous ALDH2*1/*2) (26). The ALDH2*1/*2 mice were divided into three experimental groups: ALDH2*1/*2 mice with the daily saline intraperitoneal injection (vehicle group), ALDH2*1/*2 mice with the daily saline intraperitoneal injection (vehicle group), ALDH2*1/*2 mice challenged with the daily ethanol intraperitoneal injection (20%, 1 g/kg per day) combined with osmotic pump providing polyethylene glycol (PEG) in 50% dimethyl sulfoxide (DMSO) (EtOH group), and ALDH2*1/*2 mice with the daily ethanol intraperitoneal injection combined with osmotic pump delivering empagliflozin (10 mg/kg per day) (Empa group) (Fig. 7A). After daily saline or ethanol intraperitoneal injection for 21 days, aortas were isolated from the three groups and from saline-injected WT mice (WT group) to examine their vascular structure and function. First, we examined the structure and function of isolated aortas from untreated WT and ALDH2*1/*2 mice using Masson’s trichrome staining. The results showed no differences in aorta area and wall thickness between WT and ALDH2*1/*2 mice (Fig. 7, B to D). We next investigated whether ALDH2*1/*2 mice had differences in aortic contraction and relaxation compared with WT mice using a wire myograph (fig. S5A). The aortic rings isolated from the ALDH2*1/*2 mice demonstrated a lower basal endothelial-dependent relaxation response to acetylcholine and a lower basal contractile response to endothelin-1 (ET-1) compared with the aortic rings isolated from the WT mice (fig. S5, B and C). The maximal endothelial-dependent relaxation induced by acetylcholine was 91.8 ± 1.4% in the WT group and 81.1 ± 4.0% in the ALDH2*1/*2 group. The maximal contraction induced by ET-1 was 0.44 ± 0.03 mN/mg in the WT group and 0.35 ± 0.01 mN/mg in the ALDH2*1/*2 group (fig. S5, B and C). These data showed that the ALDH2*1/*2 mice have a reduced response to endothelial-dependent relaxation and contraction at baseline.
Figure 7. Empagliflozin alleviates vascular dysfunction in ALDH2*1/*2 heterozygous mice.

(A) Schematic of the experimental design to examine the effects of empagliflozin on vascular function in ethanol-treated ALDH2*1/*2 mice. Mice were divided into 4 groups: saline-injected WT mice, saline-injected ALDH2*1/*2 (Vehicle group), ethanol- and solvent (PEG/DMSO) control-treated ALDH2*1/*2 (EtOH group), and ethanol- and Empa-treated ALDH2*1/*2 mice (Empa group). (B) Representative brightfield images of Masson’s trichome staining of mouse aortas from the 4 experimental groups above. (C–D) Quantification of aortic area and wall thickness. *P < 0.05, **P < 0.01, ****P < 0.0001, calculated by a one-way ANOVA with Bonferroni correction. (E–F) Concentration-response curve for acetylcholine-induced aortic relaxation (E) and ET-1-induced aortic contraction (F) in WT, Vehicle, EtOH, and Empa groups. Data represent six mice for each group. Data are expressed as mean ± SEM. In Fig. 7E and F, Vehicle vs WT group: #P < 0.05, ##P < 0.01; EtOH vs Vehicle group: $P < 0.05, $ $P < 0.01; EtOH vs Empa group: *P < 0.05, **P < 0.01, calculated by Student’s t-test.
We next examined whether ethanol could induce further endothelial-dependent vascular dysfunction in the ALDH2*1/*2 mice (45). We found a 24% increase in aortic area and 83% increase in wall thickness in the EtOH group relative to aortas from the vehicle group. In contrast, aortas from mice with sustained empagliflozin administration, in addition to ethanol treatment, limited vascular remodeling and reduced aorta area and wall thickness (Fig. 7, B to D). Aortic rings from the EtOH group exhibited a 42% decrease in acetylcholine-induced endothelial-dependent maximal relaxation and an 18% decrease in ET-1–induced endothelial-modified maximal contraction relative to the vehicle group (Fig. 7, E and F, and Table 1). Maximal relaxation induced by acetylcholine was 81.1 ± 4.0% in the vehicle group and 47.0 ± 13.2% in the EtOH group, whereas maximal contraction induced by ET-1 was 0.35 ± 0.01 mN/mg in the vehicle group and 0.29 ± 0.02 mN/mg in the EtOH group (Fig. 7, E and F, and Table 1). Sustained treatment with empagliflozin restored vascular function of the aortic rings in ALDH2*1/*2 mice by enhancing acetylcholine-induced relaxation response (maximal relaxation: 91.2 ± 1.7%) and ET-1-induced contraction response (maximal contraction: 0.45 ± 0.04%), similar to WT mice (Fig. 7, E and F, and Table 1).
Table 1.
Effects of empagliflozin on vascular function in ALDH2*2 mice using wire myograph.
| WT | Vehicle | EtOH | Empa | |
|---|---|---|---|---|
| Maximal Contractile Force (mN/mg) | 0.44 ± 0.03 | 0.35 ± 0.01# | 0.29 ± 0.02$ | 0.45 ± 0.04%* |
| Maximal Relaxation (%) | 91.8 ± 1.4% | 81.1 ± 4.0%# | 47.0 ± 13.2%$ | 91.2 ± 1.7%* |
Data correspond to results presented in Fig. 7 (E and F). The maximal contractile force was recorded in response to 1 μM endothelin-1 (ET-1), and the relaxation maximal relaxation (%) was recorded in response to 1 mM acetylcholine in mouse aortas isolated from WT, vehicle, EtOH, and Empa groups. WT group, wild-type Aldh2 mice; vehicle group, solvent (PEG/DMSO) control– treated ALDH2*1/*2 mice; EtOH group, alcohol-treated ALDH2*1/*2 mice; Empa group, ethanol- and Empa-treated ALDH2*1/*2 mice. Data represent six mice for each group. Data are expressed as means ± SEM.
Vehicle versus WT group: #P < 0.05
EtOH versus vehicle group: $P < 0.05
EtOH versus Empa group: *P < 0.05, calculated by a one-way ANOVA with Bonferroni correction.
ALDH2*2 has been associated with a higher incidence of endothelial dysfunction-related diseases and exacerbates coronary endothelial damage and function in type 1 diabetic mice (13, 46, 47). We therefore investigated the effect of empagliflozin on vascular function in ALDH2*1/*2 mice with diabetes mellitus induced by streptozotocin (STZ) treatment (fig. S5D). Four weeks after induction of diabetes mellitus, aortas were isolated, and endothelial function was examined. Whereas the aortas from diabetic ALDH2*1/*2 mice (STZ group) demonstrated a 24.1% increase in wall thickness and 16.5% increase in aortic area, relative to the vehicle group (fig. S5, E and F), empagliflozin administration diminished the progress of vascular remodeling by reducing wall thickness and area by ~8.7% and ~14.5%, respectively in comparison to the STZ group (fig. S5, E and F). Functional studies demonstrated that aortas isolated from STZ group had a 29.8% decrease in acetylcholine-induced endothelial-dependent relaxation relative to the vehicle group (fig. S5G). Here empagliflozin administration (STZ + Empa group) restored acetylcholine-induced endothelial-dependent relaxation and further enhanced ET-1-induced endothelial-modified contraction relative to the STZ group (fig. S5, G and H).
These data showed that at baseline, the aortic contractile response to ET-1 and the endothelial-dependent relaxation response to acetylcholine were reduced in heterozygous ALDH2*1/*2 mice compared to the WT mice despite having normal vascular structure. Ethanol exposure or diabetes mellitus induced further endothelial dysfunction by decreasing the vascular tone and increasing vascular wall thickness and aorta area in ALDH2*1/*2 mice. Sustained co-treatment with empagliflozin restored vascular function and attenuated vascular remodeling in ethanol-treated or diabetic ALDH2*1/*2 mice.
Empagliflozin does not directly activate ALDH2 enzymatic activity
To determine whether empagliflozin improved ALDH2*2-related endothelial dysfunction through direct activation of ALDH2 enzyme, we first performed induced-fit docking studies of tested drugs on ALDH2 in silico. We used the co-crystal structure of ALDH2 with a known activator, Alda-1 (PDB ID: 3INJ) and docked empagliflozin at the same site (fig. S6, A to C) (48). Alda-1 was previously reported to activate the WT ALDH2 enzyme over 2-fold and increase the activity of ALDH2*1/*2 to WT values (44). In silico analysis suggested that empagliflozin occupied the Alda-1-binding site in ALDH2 in a similar manner to Alda-1 (fig. S6, A to C), suggesting that empagliflozin may activate ALDH2 through a mechanism similar to Alda-1.
To verify these results, we assessed ALDH2 enzyme activity in vitro in the presence of empagliflozin or Alda-1 using recombinant ALDH2 protein isolated from bacteria containing one copy of ALDH2*1/*2 variant and one copy of WT variant (ALDH2*1/*2) or two copies of the WT variant (WT). ALDH2*1/*2 enzyme activity exhibited approximately 45% of WT activity in the absence of activators, with ~1.04 μmol/min/mg of enzyme activity for ALDH2*1/*2 versus ~2.30 μmol/min/mg for WT. Empagliflozin did not enhance the activity of either WT (P = 0.94) or ALDH2*1/*2 (P = 0.99) (fig. S6D). These results suggest that empagliflozin is not a direct ALDH2 activator in vitro.
We have previously shown that Alda-1 can inhibit 4-HNE-induced ALDH2 inactivation (44). We therefore examined whether empagliflozin can also limit 4-HNE inactivation of ALDH2. In the presence of 50 μM 4-HNE, both WT and ALDH2*1/*2 were inactivated. As shown previously (44), 1 μM Alda-1 increased enzyme activity, but 5 μM empagliflozin had no effect (fig. S6E). Thus, our results indicate that empagliflozin may not directly regulate ALDH2 activity but may instead improve ALDH2*2-induced endothelial dysfunction indirectly.
Empagliflozin improves ALDH2*2-related EC dysfunction via NHE-1/AKT/eNOS pathway
Many studies have shown that the target of empagliflozin, SGLT2, is not expressed in cardiac cells, and that empagliflozin exhibits its beneficial effects on cardiovascular disease via off-target mechanisms (49). We also confirmed that mRNA for SGLT genes SGLT1 and SGLT2 were not detected in either ethanol-free or ethanol-treated Iso_GG and ALDH2*1/*2 iPSC-ECs (fig. S7A), suggesting that the benefits of empagliflozin treatment for ALDH2*2-induced endothelial dysfunction may be via off-target effects. The Na+/H+ exchanger isoform-1 (NHE-1), sodium voltage-gated channel alpha subunit 5 (SCN5A), and L-type Ca2+ channels, are three targets through which empagliflozin may exert its beneficial effects on cardiovascular cells (50). We first examined the expression of these channels in iPSC-ECs. The Ct values of SCN5A, and L-type Ca2+ channel genes such as calcium voltage-gated channel subunit alpha1 C (CACAN1C), calcium voltage-gated channel subunit alpha1 D (CACAN1D) are greater than 33, indicating expression of the SCN5A, and L-type Ca2+ channel genes are low in iPSC-ECs. In contrast, the Ct value for NHE1 is approximately 25, indicating higher expression (fig. S7B). Thus, we focused on the NHE-1 channel for further studies.
NHE-1 is a membrane protein that exchanges intracellular H+ and extracellular Na+ and plays critical roles in regulating intracellular pH and volume homeostasis. Previous studies showed that activated NHE-1 drove ROS formation in human ECs (51), suggesting that NHE-1 could be involved in ALDH2*2-related endothelial dysfunction. To test this hypothesis, we examined NHE activity (Δ[H+]/s) and cytoplasmic Na+ in Iso_GG and ALDH2*1/*2 iPSC-ECs. NHE-1 activity in ALDH2*1/*2 iPSC-ECs was higher compared with Iso_GG iPSC-ECs (Iso_GG: −0.55 ± 0.02; ALDH2*1/*2: −0.66 ± 0.02) (Fig. 8A). In addition, the concentration of cytoplasmic Na+ in ALDH2*1/*2 iPSC-ECs higher than in the Iso_GG group (Iso_GG: 0.79 ± 0.01 versus ALDH2*1/*2: 0.87 ± 0.002), which also indicated higher NHE-1 activity (Fig. 8B). Empagliflozin decreased Δ[H+]/s (Empa: −0.56 ± 0.02) and cytoplasmic Na+ (Empa: 0.80 ± 0.03) in ALDH2*1/*2 iPSC-ECs, suggesting that empagliflozin may regulate endothelial NHE-1 activity (Fig. 8, A and B). Furthermore, cariporide, an NHE-1 inhibitor, was applied to iPSC-ECs in the presence or absence of empagliflozin. As expected, cariporide reduced NHE-1 activity (Car: −0.55 ± 0.02), and cytoplasmic Na+ (Car: 0.78 ± 0.02) in ALDH2*1/*2 iPSC-ECs (Fig. 8, A and B). However, there was no further reduction of NHE-1 activity in ALDH2*1/*2 iPSC-ECs (Car + Empa: –0.54 ± 0.01) and Na+ (Car + Empa: 0.76 ± 0.03) when empagliflozin and cariporide were combined, suggesting that empagliflozin directly targets NHE-1 (Fig. 8, A and B, fig. S7, C to E).
Figure 8. Empagliflozin improves ALDH2*2-related endothelial dysfunction through NHE-1/AKT/eNOS pathway.

(A–B) Bar graphs show quantification of NHE-1 activity (A) and intracellular Na+ (B) in Iso_GG and ALDH2*1/*2 iPSC-ECs in the presence or absence of Empa (5 μM), Car (10 μM), or Car (10 μM) plus Empa (5 μM) (Car + Empa). DMSO served as vehicle. (C) In silico induced-fit docking showing Empa (magenta) bound to NHE-1 with a similar binding site to Car (orange). (D–E) Cellular NO (D) and ROS (E) in Iso_GG and ALDH2*1/*2 iPSC-ECs in the presence or absence of 5 μM Empa, 10 μM Car, or Car + Empa. (F) Representative immunoblots show total AKT, p-AKT, total eNOS, and p-eNOS proteins in Iso_GG and ALDH2*1/*2 iPSC-ECs in the presence or absence of 5 μM Empa, 10 μM Car, or Car + Empa. DMSO was used as vehicle. GAPDH was used as loading control. (G–H) Bar graphs show quantification of p-AKT to total AKT ratios (G) and p-eNOS to total eNOS ratios (H) in Iso_GG and ALDH2*1/*2 iPSC-ECs in the presence or absence of Empa (5μM), Car (5mM), or Car + Empa. Data represent three individual iPSC lines from two technical replicates. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, calculated by one-way ANOVA with Bonferroni correction.
Next, in silico induced-fit docking studies were performed and the ability of empagliflozin to bind NHE-1 was assessed based on a comparison of the docking score and ligand stability to cariporide (52). The results showed that empagliflozin bound well to NHE-1 in its most stable binding pose (docking score was –5.20 for Empa and –5.58 for Car) (Fig. 8C). However, empagliflozin had a more stable interaction compared to cariporide. The total interaction energy was –200 kJ/mol for empagliflozin and –160 kJ/mol for Car based on a 100 ns simulation with a 10 ps sampling-rate (fig. S7, F and G, Movies S1–2). Thus, the data suggest that empagliflozin improves ALDH2*2-related EC dysfunction via targeting NHE-1.
Next, the effects of empagliflozin on ROS and NO production were examined in Iso_GG and ALDH2*1/*2 iPSC-ECs in the presence of cariporide. Whereas cariporide increased NO production and decreased ROS generation in ALDH2*1/*2 iPSC-ECs, there were no further changes with the addition of empagliflozin, suggesting empagliflozin ameliorates ALDH2*2-related NO reduction and oxidative stress by inhibiting NHE-1 activity (fig. 8, D and E). In addition, we determined whether empagliflozin can regulate AKT in ALDH2*1/*2 iPSC-ECs via NHE-1. Western blot analysis showed that the total AKT protein and the active form of AKT, p-AKT (phosphorylation at serine 473), were increased in both empagliflozin and cariporide-treated ALDH2*1/*2 iPSC-ECs (Fig. 8, F and G). Cariporide increased AKT activity more than empagliflozin, however, there was no further improvement of AKT activity in co-treatment of cariporide and empagliflozin, suggesting that empagliflozin regulates AKT activity by inhibiting NHE-1 (Fig. 8, F and G). We also found eNOS activity (p-eNOS/eNOS ratio) in ALDH2*1/*2 iPSC-ECs was significantly increased with empagliflozin or cariporide treatment (P < 0.0001 for Empa and P = 0.002 for Car) (Fig. 8H). The effect of eNOS activity and NO production was stronger with empagliflozin, which may be due to additional off-target effects of empagliflozin on eNOS (53). These findings suggested that empagliflozin alleviates ALDH2*2-induced oxidative stress and NO reduction via the NHE-1/AKT/eNOS pathway.
DISCUSSION
As the most common enzymopathy caused by a single SNP in humans, the ALDH2*2 variant has been linked to a broad spectrum of human diseases, including CAD, cancer, diabetes mellitus, and atherosclerosis (54). Most of these diseases are associated with vascular dysfunction (55). The endothelium is a delicate monolayer of cells lining all blood vessels and plays a pivotal role in the physiologic regulation of vascular tone and function (55). However, the role of endothelium in these diseases, especially in ALDH2*2-related CAD, remains unclear (56). In this study, we discovered that one standard drink of alcohol impaired endothelial vasodilator function in human subjects with the ALDH2*2 variant, but not in wild-type carriers. We did not observe baseline differences in EC function between WT and ALDH2*1/*2 human subjects, but this may be due to the complex mechanisms of homeostasis in the human body (57). Data from iPSC-ECs derived from human subjects revealed EC dysfunction in ALDH2*2 carriers by increased ROS and proinflammation and reduced NO production and tube formation, which have been linked to the onset and development of CAD (12). Our data also demonstrated the adverse effects of ALDH2*2 and alcohol consumption on EC dysfunction and CAD. Given the prevalence of CAD in ALDH2*2 carriers (58, 59), identification of ALDH2*2 in patients with CAD may be used to educate individuals and provide guidance regarding alcohol consumption.
We used transcriptome profiling to identify downregulation of the AKT pathway in ALDH2*2 iPSC-ECs and found it may play a crucial role in ALDH2*2-related EC dysfunction. We also found that impaired ALDH2 enzyme activity in the ALDH2*2 genotype led to elevated ROS and decreased NO through AKT and eNOS signaling, suggesting these pathways could also be targeted for intervention (28). One study has demonstrated that ALDH2 rescues myocardial ischemia/reperfusion injury by activating the AKT pathway (60). Our data also suggested that AKT activation by SC-79 may be an intervention to limit ALDH2*2-related CAD. However, overexpression of AKT has also been shown to have adverse effects on endothelial cells through the development of vascular malformations (61). Therefore, downstream factors of AKT will need to be identified to more precisely target its pathological role in ALDH2*2-related CAD.
We previously found that ALDH2*2 mice exhibited higher concentrations of pro-inflammatory cytokines that are further exacerbated with chronic moderate ethanol exposure of 1 g/kg/day (62) relative to WT mice (63). Here transcriptome profiling revealed that upregulation of AMPK, MAPK, and NF-κB signaling pathways were associated with ALDH2*2-related EC dysfunction. These pathways have been shown to mediate ALDH2-related inflammation and atherosclerotic plaque formation in mouse and cell models (13, 64), suggesting they may also be targeted to reduce or inhibit ALDH2*2-induced EC dysfunction.
Another strategy we explored was to directly increase the catalytic activity and stability of the ALDH2*2 with small molecules. We used the small molecule Alda-1 and showed a dose-dependent increase in NO and a decrease in ROS in ALDH2*1/*2 iPSC-ECs. However, at high dose, it has a toxic effect on ECs in culture. After searching for FDA-approved drugs with cardiovascular benefit, we found that SGLT2i improved endothelial dysfunction in ALDH2*1/*2 iPSC-ECs. Recently, SGLT2i, including empagliflozin were found to be beneficial effects for cardiovascular disease, including heart failure, myocardial infarction, and hypertension (42). In addition, a recent study showed the combined positive effect of Alda-1 and empagliflozin on diabetes-associated HFpEF in mice, indicating that empagliflozin could be repurposed for preventing and treating ALDH2*2-related CAD (65). However, a more comprehensive test in human subjects is needed to confirm that empagliflozin prevents EC dysfunction in ALDH2*2 carriers.
SGLT2 is mainly expressed in the kidney and has not been detected in cardiac cells (66). We confirmed that SGLT mRNAs were not expressed in iPSC-ECs, suggesting empagliflozin may improve EC function independently of its action on the sodium-glucose co-transporter. Although our in silico results and those of others (67) showed that empagliflozin could potentially activate ALDH2 enzyme activity, our subsequent in vitro data demonstrated that empagliflozin did not directly activate ALDH2 or limit 4-HNE-induced adducts. The benefits of empagliflozin on ALDH2*2-related endothelial dysfunction may therefore be due to other off-target effects (68).
Empagliflozin has been shown to activate AMPK (69), promote AKT/eNOS signaling (70), reduce ROS generation, and restore NO bioavailability in ECs (43). In addition, empagliflozin was previously found to alleviate TNFα-mediated inflammation in ECs (43). However, the direct targets of these beneficial effects of empagliflozin were unknown. Here, we discovered that empagliflozin rescued endothelial dysfunction by inhibiting NHE-1 activity AKT/eNOS signaling. Although several functional studies have indicated that empagliflozin can directly target NHE-1, additional experiments are needed to demonstrate physical binding of empagliflozin to NHE-1 (71–74). We also showed that the NHE-1 inhibitor cariporide could improve ALDH2*2-induced EC dysfunction. Earlier pilot studies have shown the beneficial effects of cariporide on cardiovascular disease including CAD (75). However, cariporide failed in phase III clinical trials for CAD patients due to increased mortality (75). More recently, one study showed that cariporide could improve vascular function ex vivo in saphenous veins from ALDH2*2 carriers (76), suggesting that NHE-1 may be a potential therapeutic target for follow up studies in either specific patient populations or with other small molecules.
Our study demonstrated clinical endothelial dysfunction in healthy control participants with ALDH2*2 variant and moderate alcohol consumption. We further examined the mechanisms of ALDH2*2-mediated endothelial dysfunction and found improved ALDH2*2-mediated endothelial dysfunction with empagliflozin treatment in both in vitro and in vivo models. In the future, clinical trials should investigate whether the ALDH2*2 variant and alcohol consumption exacerbate endothelial dysfunction in diabetics or those with existing CAD. More importantly, these studies should also test whether empagliflozin ameliorates ALDH2*2-mediated atherosclerosis in the setting of diabetes or CAD. Lastly, another limitation of this study is that only male participants were recruited. Future studies are needed to investigate the effects of the ALDH2*2 variant in females.
In summary, we found that even minimal alcohol consumption impaired endothelial function in human participants with the ALDH2*2 allele. Empagliflozin ameliorated this dysfunction in vitro and in mice by inhibiting NHE-1 activity and restoring the AKT and eNOS signaling. Given the crucial role of the endothelium in vascular function, these results suggest that targeting ALDH2*2 activity and its downstream effects may be an effective strategy to limit vascular disease in ALDH2*2 carriers (77).
MATERIALS AND METHODS
Study Design.
To investigate the function of the endothelium in ALDH2*2 carriers, we recruited18 healthy volunteers carrying WT or heterozygous variant (ALDH2*1/*2) at Stanford University under an Institutional Review Board-approved protocol (ID: 61417). A single hair was extracted to genotype the ALDH2 allele. The human participants were served one beer (Heineken, 12 fl oz, 5% ethanol/volume) to consume over a 30-min time period. The EndoPAT test was performed before and immediately after ethanol consumption. The sample size was determined to provide >80% power for detecting at least 20% difference of RHI between before and after ethanol consumption in ALDH2*2 carriers at the significance of 0.05. Volunteers’ endothelial function was assessed by digital plethysmography using the EndoPAT2000 system (Itamar Medical Ltd.), and a detailed clinical history was recorded (table S2). The genotype of these human participants was not known during the test, and the results were analyzed in a blinded fashion.
To study the molecular mechanisms underlying endothelial dysfunction in the ALDH2*2 carriers, a cohort of 10 age- and sex-matched East Asian individuals who carried either the WT or ALDH2*1/*2 allele were recruited as previously described (24) (table S6). These iPSC lines were obtained from Stanford University Cardiovascular Institute Biobank and assigned to WT (n = 5) or ALDH2*1/*2 (n = 5) groups based on the presence of the ALDH2 variant. Measurements of endothelial function from both wild-type and subjects with ALDH2*2 variant were not blinded. A minimum of two technical replicates were used for each experiment.
To examine whether empagliflozin rescued endothelial dysfunction in ALDH2 mutant mice, we tested our hypothesis by using: 1) ALDH2*1/*2 knock-in mice matched to C57/BL6 wild type ALDH2 mice; 2) ethanol-induced vascular dysfunction by injecting ethanol into the rodent peritoneum; 3) streptozotocin (STZ)-induced diabetic model by injecting STZ into the rodent peritoneum; and 4) empagliflozin, an SGLT2i, delivered by osmotic pump. The knock-in ALDH2*2 mice were generated by homologous recombination. A single nucleotide substitution (G to A) within the ALDH2 genomic fragment was introduced by site-directed mutagenesis corresponding to the position of human E504K mutation. The construction and phenotype of ALDH2*1/*2 knock-in mice have been described previously (26). An experimental group size of six or greater animals was used to detect at least a 20% difference in with a power of 95% and P < 0.05. All procedures in this study were performed in accordance with the animal protocols approved by the Animal Care and Use Committee of Stanford University (ID: 34047). Wire myograph tests were performed by observers blinded to the treatment groups. All animals were randomized and the genotypes test and drug treatments were blinded.
Statistical analysis.
Data were analyzed using Prism (GraphPad) or Excel and reported as mean ± SEM. Comparisons were measured by one-way analysis of variance (ANOVA) or two-way ANOVA test with Bonferroni correction for experiments with more than two groups, or by two-tailed Student’s t-test for experiments with two groups. A p-value of < 0.05 was considered statistically significant. For GWAS in Fig. 1A and PheWAS in fig. S1, the SNP-phenotype association was analyzed using linear or logistic regression models, and age, gender, and principal components were adjusted as covariates. Further details can be found in the supplementary materials.
Supplementary Material
Acknowledgements
We thank N. Leeper and H. Zhu for assistance with the EndoPAT experimentation and analysis. We also thank all the recruited human participants for participating in this study. We thank B. Wu and M. Nishiga for helpful editing on the manuscript. We also thank G. Pan at Henry Ford Health System for his advice on establishing STZ-induced diabetic mice.
Funding:
This work was supported by National Institutes of Health (NIH) grant K99 HL150319 (HCG), K99 HL150216 (DTP), R01 AA11147 (DM-R), R35 GM119522 (ERG), R01 HL130020 (JCW), R01 HL126527 (JCW), R01 HL113006 (JCW), R01 HL146690 (JCW), P01 HL141084 (JCW), American Heart Association grant 20CDA35260261 (DTP), and Tobacco-Related Disease Research Program T31FT1392 (XY).
Footnotes
Competing interests: D.M.-R and C.H.C. hold patents related to Alda-1 activation of ALDH2. One of the patents is licensed to Foresee Pharmaceuticals, a company at which D. M.-R. is a consultant. J.C.W. is a co-founder of Greenstone Biosciences. M.C. is a consultant for Greenstone Biosciences.
Data and materials availability:
All data associated with this study are presented in the paper or the Supplementary Materials. RNA-seq data generated for this study are deposited at the Gene Expression Omnibus in the NCBI database under the accession number GSE184419. All Code is available at https://zenodo.org/record/7478648.
REFERENCES
- 1.Khan MA, Hashim MJ, Mustafa H, Baniyas MY, Al Suwaidi S, AlKatheeri R, Alblooshi FMK, Almatrooshi M, Alzaabi MEH, Al Darmaki RS, Lootah S, Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 12, e9349 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leopold JA, Loscalzo J, Emerging Role of Precision Medicine in Cardiovascular Disease. Circ Res 122, 1302–1315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talmud PJ, Gene-environment interaction and its impact on coronary heart disease risk. Nutr Metab Cardiovasc Dis 17, 148–152 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Kessler T, Schunkert H, Coronary Artery Disease Genetics Enlightened by Genome-Wide Association Studies. JACC Basic Transl Sci 6, 610–623 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson HN, Weiner H, Hurley TD, Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J Biol Chem 280, 30550–30556 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross ER, Zambelli VO, Small BA, Ferreira JC, Chen CH, Mochly-Rosen D, A personalized medicine approach for Asian Americans with the aldehyde dehydrogenase 2*2 variant. Annu Rev Pharmacol Toxicol 55, 107–127 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li YY, Wang H, Wu JJ, Kim HJ, Yang XX, Geng HY, Gong G, ALDH2 gene G487A polymorphism and coronary artery disease: a meta-analysis including 5644 participants. J Cell Mol Med 22, 1666–1674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosoff DB, Davey Smith G, Mehta N, Clarke TK, Lohoff FW, Evaluating the relationship between alcohol consumption, tobacco use, and cardiovascular disease: A multivariable Mendelian randomization study. PLoS Med 17, e1003410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung CL, Chang SC, Chang SH, Chi PC, Lai YJ, Wang SW, Wu YJ, Yeh HI, Lin SJ, Chen CH, Mochly-Rosen D, Wang LY, Investigator MS, Genetic Polymorphisms of Alcohol Metabolizing Enzymes and Alcohol Consumption are Associated With Asymptomatic Cardiac Remodeling and Subclinical Systolic Dysfunction in Large Community-Dwelling Asians. Alcohol Alcohol 52, 638–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hung CL, Sung KT, Chang SC, Liu YY, Kuo JY, Huang WH, Su CH, Liu CC, Tsai SY, Liu CY, Lee AS, Pan SH, Wang SW, Hou CJ, Hung TC, Yeh HI, Variant Aldehyde Dehydrogenase 2 (ALDH2*2) as a Risk Factor for Mechanical LA Substrate Formation and Atrial Fibrillation with Modest Alcohol Consumption in Ethnic Asians. Biomolecules 11, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, Luo S, Li Z, Liu P, Han J, Harding IC, Ebong EE, Cameron SJ, Stewart AG, Weng J, Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol Rev 73, 924–967 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Matsuzawa Y, Lerman A, Endothelial dysfunction and coronary artery disease: assessment, prognosis, and treatment. Coron Artery Dis 25, 713–724 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan C, Xing JH, Zhang C, Zhang YM, Zhang LT, Wei SJ, Zhang MX, Wang XP, Yuan QH, Xue L, Wang JL, Cui ZQ, Zhang Y, Xu F, Chen YG, Aldehyde dehydrogenase 2 inhibits inflammatory response and regulates atherosclerotic plaque. Oncotarget 7, 35562–35576 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo YJ, Chen L, Bai YP, Li L, Sun J, Zhang GG, Yang TL, Xia J, Li YJ, Chen XP, The ALDH2 Glu504Lys polymorphism is associated with coronary artery disease in Han Chinese: Relation with endothelial ADMA levels. Atherosclerosis 211, 545–550 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Nannelli G, Terzuoli E, Giorgio V, Donnini S, Lupetti P, Giachetti A, Bernardi P, Ziche M, ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid Med Cell Longev 2018, 9765027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang SJ, Yokoyama A, Yokoyama T, Huang YC, Wu SY, Shao Y, Niu J, Wang J, Liu Y, Zhou XQ, Yang CX, Relationship between genetic polymorphisms of ALDH2 and ADH1B and esophageal cancer risk: a meta-analysis. World J Gastroenterol 16, 4210–4220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubinshtein R, Kuvin JT, Soffler M, Lennon RJ, Lavi S, Nelson RE, Pumper GM, Lerman LO, Lerman A, Assessment of endothelial function by non-invasive peripheral arterial tonometry predicts late cardiovascular adverse events. Eur Heart J 31, 1142–1148 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Michelsen MM, Mygind ND, Pena A, Aziz A, Frestad D, Host N, Prescott E, Steering PS Committee of the i, Peripheral Reactive Hyperemia Index and Coronary Microvascular Function in Women With no Obstructive CAD: The iPOWER Study. JACC Cardiovasc Imaging 9, 411–417 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Bonetti PO, Pumper GM, Higano ST, Holmes DR, Kuvin JT, Lerman A, Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. Journal of the American College of Cardiology 44, 2137–2141 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Minami J, Todoroki M, Ishimitsu T, Yamamoto H, Abe S, Fukunaga T, Matsuoka H , Effects of alcohol intake on ambulatory blood pressure, heart rate, and heart rate variability in Japanese men with different ALDH2 genotypes. J Hum Hypertens 16, 345–351 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Shin MJ, Cho Y, Davey Smith G, Alcohol Consumption, Aldehyde Dehydrogenase 2 Gene Polymorphisms, and Cardiovascular Health in Korea. Yonsei Med J 58, 689–696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YC, Peng GS, Tsao TP, Wang MF, Lu RB, Yin SJ, Pharmacokinetic and pharmacodynamic basis for overcoming acetaldehyde-induced adverse reaction in Asian alcoholics, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenet Genomics 19, 588–599 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Xue A, Jiang L, Zhu Z, Wray NR, Visscher PM, Zeng J, Yang J, Genome-wide analyses of behavioural traits are subject to bias by misreports and longitudinal changes. Nat Commun 12, 20211 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert AD, Kodo K, Liang P, Wu H, Huber BC, Riegler J, Churko J, Lee J, de Almeida P, Lan F, Diecke S, Burridge PW, Gold JD, Mochly-Rosen D, Wu JC, Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci Transl Med 6, 255ra130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu M, Efficient Differentiation of Human Pluripotent Stem Cells to Endothelial Cells. Curr Protoc Hum Genet, e64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambelli VO, Gross ER, Chen CH, Gutierrez VP, Cury Y, Mochly-Rosen D, Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci Transl Med 6, 251ra118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin S, Chen J, Chen L, Histen G, Lin Z, Gross S, Hixon J, Chen Y, Kung C, Chen Y, Fu Y, Lu Y, Lin H, Cai X, Yang H, Cairns RA, Dorsch M, Su SM, Biller S, Mak TW, Cang Y, ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc Natl Acad Sci U S A 112, 9088–9093 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CH, Ferreira JC, Gross ER, Mochly-Rosen D, Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev 94, 1–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur K, Bedi G, Kaur M, Vij A, Kaur I, Lipid peroxidation and the levels of antioxidant enzymes in coronary artery disease. Indian J Clin Biochem 23, 33–37 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C, The role of nitric oxide on endothelial function. Curr Vasc Pharmacol 10, 4–18 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Victor VM, Rocha M, Sola E, Banuls C, Garcia-Malpartida K, Hernandez-Mijares A, Oxidative stress, endothelial dysfunction and atherosclerosis. Curr Pharm Des 15, 2988–3002 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Cahill PA, Redmond EM, Alcohol and cardiovascular disease--modulation of vascular cell function. Nutrients 4, 297–318 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kevil CG, Patel RP, Bullard DC, Essential role of ICAM-1 in mediating monocyte adhesion to aortic endothelial cells. Am J Physiol Cell Physiol 281, C1442–1447 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Jefferis BJ, Papacosta O, Owen CG, Wannamethee SG, Humphries SE, Woodward M, Lennon LT, Thomson A, Welsh P, Rumley A, Lowe GD, Whincup PH, Interleukin 18 and coronary heart disease: prospective study and systematic review. Atherosclerosis 217, 227–233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conklin DJ, Haberzettl P, Prough RA, Bhatnagar A, Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am J Physiol Heart Circ Physiol 296, H1586–1597 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phulukdaree A, Khan S, Moodley D, Chuturgoon AA, GST polymorphisms and early-onset coronary artery disease in young South African Indians. S Afr Med J 102, 627–630 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Dufourcq P, Leroux L, Ezan J, Descamps B, Lamaziere JM, Costet P, Basoni C, Moreau C, Deutsch U, Couffinhal T, Duplaa C, Regulation of endothelial cell cytoskeletal reorganization by a secreted frizzled-related protein-1 and frizzled 4- and frizzled 7-dependent pathway: role in neovessel formation. Am J Pathol 172, 37–49 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiojima I, Walsh K, Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res 90, 1243–1250 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM, Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Nitulescu GM, Van De Venter M, Nitulescu G, Ungurianu A, Juzenas P, Peng Q, Olaru OT, Gradinaru D, Tsatsakis A, Tsoukalas D, Spandidos DA, Margina D, The Akt pathway in oncology therapy and beyond (Review). Int J Oncol 53, 2319–2331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paik DT, Chandy M, Wu JC, Patient and Disease-Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharmacol Rev 72, 320–342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopaschuk GD, Verma S, Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl Sci 5, 632–644 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uthman L, Homayr A, Juni RP, Spin EL, Kerindongo R, Boomsma M, Hollmann MW, Preckel B, Koolwijk P, van Hinsbergh VWM, Zuurbier CJ, Albrecht M, Weber NC, Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor alpha-Stimulated Human Coronary Arterial Endothelial Cells. Cell Physiol Biochem 53, 865–886 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D, Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321, 1493–1495 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kudo R, Yuui K, Kasuda S, Hatake K, [Effect of alcohol on vascular function]. Nihon Arukoru Yakubutsu Igakkai Zasshi 50, 123–134 (2015). [PubMed] [Google Scholar]
- 46.Pan G, Roy B, Palaniyandi SS, Diabetic Aldehyde Dehydrogenase 2 Mutant (ALDH2*2) Mice Are More Susceptible to Cardiac Ischemic-Reperfusion Injury Due to 4-Hydroxy-2-Nonenal Induced Coronary Endothelial Cell Damage. J Am Heart Assoc 10, e021140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan G, Deshpande M, Pang H, Palaniyandi SS, Precision medicine approach: Empagliflozin for diabetic cardiomyopathy in mice with aldehyde dehydrogenase (ALDH) 2*2 mutation, a specific genetic mutation in millions of East Asians. Eur J Pharmacol 839, 76–81 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD, Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol 17, 159–U154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uthman L, Baartscheer A, Schumacher CA, Fiolet JWT, Kuschma MC, Hollmann MW, Coronel R, Weber NC, Zuurbier CJ, Direct Cardiac Actions of Sodium Glucose Cotransporter 2 Inhibitors Target Pathogenic Mechanisms Underlying Heart Failure in Diabetic Patients. Front Physiol 9, 1575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen S, Coronel R, Hollmann MW, Weber NC, Zuurbier CJ, Direct cardiac effects of SGLT2 inhibitors. Cardiovasc Diabetol 21, 45 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uthman L, Li X, Baartscheer A, Schumacher CA, Baumgart P, Hermanides J, Preckel B, Hollmann MW, Coronel R, Zuurbier CJ, Weber NC, Empagliflozin reduces oxidative stress through inhibition of the novel inflammation/NHE/[Na(+)]c/ROS-pathway in human endothelial cells. Biomed Pharmacother 146, 112515 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Dong Y, Gao Y, Ilie A, Kim D, Boucher A, Li B, Zhang XC, Orlowski J, Zhao Y, Structure and mechanism of the human NHE1-CHP1 complex. Nat Commun 12, 3474 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li X, Preckel B, Hermanides J, Hollmann MW, Zuurbier CJ, Weber NC, Amelioration of endothelial dysfunction by sodium glucose co-transporter 2 inhibitors: pieces of the puzzle explaining their cardiovascular protection. Br J Pharmacol 179, 4047–4062 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen CH, Kraemer BR, Mochly-Rosen D, ALDH2 variance in disease and populations. Dis Model Mech 15, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I, The vascular endothelium and human diseases. Int J Biol Sci 9, 1057–1069 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mizuno Y, Hokimoto S, Harada E, Kinoshita K, Nakagawa K, Yoshimura M, Ogawa H, Yasue H, Variant Aldehyde Dehydrogenase 2 (ALDH2*2) Is a Risk Factor for Coronary Spasm and ST-Segment Elevation Myocardial Infarction. J Am Heart Assoc 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chia PY, Teo A, Yeo TW, Overview of the Assessment of Endothelial Function in Humans. Front Med (Lausanne) 7, 542567 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yokoyama A, Kato H, Yokoyama T, Tsujinaka T, Muto M, Omori T, Haneda T, Kumagai Y, Igaki H, Yokoyama M, Watanabe H, Fukuda H, Yoshimizu H, Genetic polymorphisms of alcohol and aldehyde dehydrogenases and glutathione S-transferase M1 and drinking, smoking, and diet in Japanese men with esophageal squamous cell carcinoma. Carcinogenesis 23, 1851–1859 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Higuchi S, Matsushita S, Imazeki H, Kinoshita T, Takagi S, Kono H, Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet 343, 741–742 (1994). [DOI] [PubMed] [Google Scholar]
- 60.Ma H, Guo R, Yu L, Zhang Y, Ren J, Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J 32, 1025–1038 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perry B, Banyard J, McLaughlin ER, Watnick R, Sohn A, Brindley DN, Obata T, Cantley LC, Cohen C, Arbiser JL, AKT1 overexpression in endothelial cells leads to the development of cutaneous vascular malformations in vivo. Arch Dermatol 143, 504–506 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Ghosh Dastidar S, Warner JB, Warner DR, McClain CJ, Kirpich IA, Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration. Biomolecules 8, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joshi AU, Van Wassenhove LD, Logas KR, Minhas PS, Andreasson KI, Weinberg KI, Chen CH, Mochly-Rosen D, Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol Commun 7, 190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhong S, Li L, Zhang YL, Zhang L, Lu J, Guo S, Liang N, Ge J, Zhu M, Tao Y, Wu YC, Yin H, Acetaldehyde dehydrogenase 2 interactions with LDLR and AMPK regulate foam cell formation. J Clin Invest 129, 252–267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pan G, Roy B, Giri S, Lanfear DE, Thandavarayan RA, Guha A, Ortiz PA, Palaniyandi SS, Aldehyde Dehydrogenase 2 Activator Augments the Beneficial Effects of Empagliflozin in Mice with Diabetes-Associated HFpEF. Int J Mol Sci 23, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Palmiero G, Cesaro A, Vetrano E, Pafundi PC, Galiero R, Caturano A, Moscarella E, Gragnano F, Salvatore T, Rinaldi L, Calabro P, Sasso FC, Impact of SGLT2 Inhibitors on Heart Failure: From Pathophysiology to Clinical Effects. Int J Mol Sci 22, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steven S, Oelze M, Hanf A, Kroller-Schon S, Kashani F, Roohani S, Welschof P, Kopp M, Godtel-Armbrust U, Xia N, Li H, Schulz E, Lackner KJ, Wojnowski L, Bottari SP, Wenzel P, Mayoux E, Munzel T, Daiber A, The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol 13, 370–385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iborra-Egea O, Santiago-Vacas E, Yurista SR, Lupon J, Packer M, Heymans S, Zannad F, Butler J, Pascual-Figal D, Lax A, Nunez J, de Boer RA, Bayes-Genis A, Unraveling the Molecular Mechanism of Action of Empagliflozin in Heart Failure With Reduced Ejection Fraction With or Without Diabetes. JACC Basic Transl Sci 4, 831–840 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J, Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol 15, 335–346 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakao M, Shimizu I, Katsuumi G, Yoshida Y, Suda M, Hayashi Y, Ikegami R, Hsiao YT, Okuda S, Soga T, Minamino T, Empagliflozin maintains capillarization and improves cardiac function in a murine model of left ventricular pressure overload. Sci Rep 11, 18384 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uthman L, Nederlof R, Eerbeek O, Baartscheer A, Schumacher C, Buchholtz N, Hollmann MW, Coronel R, Weber NC, Zuurbier CJ, Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc Res 115, 1533–1545 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ, Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 60, 568–573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trum M, Riechel J, Lebek S, Pabel S, Sossalla ST, Hirt S, Arzt M, Maier LS, Wagner S, Empagliflozin inhibits Na(+) /H(+) exchanger activity in human atrial cardiomyocytes. ESC Heart Fail, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zuurbier CJ, Baartscheer A, Schumacher CA, Fiolet JWT, Coronel R, Sodium-glucose co-transporter 2 inhibitor empagliflozin inhibits the cardiac Na+/H+ exchanger 1: persistent inhibition under various experimental conditions. Cardiovasc Res 117, 2699–2701 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Theroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, Tognoni G, White HD, Willerson JT, Jessel A, Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation 102, 3032–3038 (2000). [DOI] [PubMed] [Google Scholar]
- 76.Lin Y, Luo C, Shi Y, Ma R, Xia Q, Ding W, The Role of Cariporide in Protecting Saphenous Vein among Different Aldehyde Dehydrogenase Genotypes. Cardiology 145, 456–466 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Nannelli G, Ziche M, Donnini S, Morbidelli L, Endothelial Aldehyde Dehydrogenase 2 as a Target to Maintain Vascular Wellness and Function in Ageing. Biomedicines 8, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, Sakaue S, Matoba N, Low SK, Okada Y, Terao C, Amariuta T, Gazal S, Kochi Y, Horikoshi M, Suzuki K, Ito K, Koyama S, Ozaki K, Niida S, Sakata Y, Sakata Y, Kohno T, Shiraishi K, Momozawa Y, Hirata M, Matsuda K, Ikeda M, Iwata N, Ikegawa S, Kou I, Tanaka T, Nakagawa H, Suzuki A, Hirota T, Tamari M, Chayama K, Miki D, Mori M, Nagayama S, Daigo Y, Miki Y, Katagiri T, Ogawa O, Obara W, Ito H, Yoshida T, Imoto I, Takahashi T, Tanikawa C, Suzuki T, Sinozaki N, Minami S, Yamaguchi H, Asai S, Takahashi Y, Yamaji K, Takahashi K, Fujioka T, Takata R, Yanai H, Masumoto A, Koretsune Y, Kutsumi H, Higashiyama M, Murayama S, Minegishi N, Suzuki K, Tanno K, Shimizu A, Yamaji T, Iwasaki M, Sawada N, Uemura H, Tanaka K, Naito M, Sasaki M, Wakai K, Tsugane S, Yamamoto M, Yamamoto K, Murakami Y, Nakamura Y, Raychaudhuri S, Inazawa J, Yamauchi T, Kadowaki T, Kubo M, Kamatani Y, Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different dise1ases. Nat Genet 52, 669–679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ, LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sayed N, Liu C, Ameen M, Himmati F, Zhang JZ, Khanamiri S, Moonen JR, Wnorowski A, Cheng L, Rhee JW, Gaddam S, Wang KC, Sallam K, Boyd JH, Woo YJ, Rabinovitch M, Wu JC, Clinical trial in a dish using iPSCs shows lovastatin improves endothelial dysfunction and cellular cross-talk in LMNA cardiomyopathy. Sci Transl Med 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bonetti PO, Pumper GM, Higano ST, Holmes DR, Kuvin JT, Lerman A, Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. Journal of the American College of Cardiology 44, 2137–2141 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Wang X, Zeng C, Gong H, He H, Wang M, Hu Q, Yang F, The influence of nitroglycerin on the proliferation of endothelial progenitor cells from peripheral blood of patients with coronary artery disease. Acta Biochim Biophys Sin (Shanghai) 46, 851–858 (2014). [DOI] [PubMed] [Google Scholar]
- 83.Carpentier G, Berndt S, Ferratge S, Rasband W, Cuendet M, Uzan G, Albanese P, Angiogenesis Analyzer for ImageJ - A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci Rep 10, 11568 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jamal M, Ameno K, Tanaka N, Ito A, Takakura A, Kumihashi M, Kinoshita H, Ethanol and Acetaldehyde After Intraperitoneal Administration to Aldh2-Knockout Mice-Reflection in Blood and Brain Levels. Neurochem Res 41, 1029–1034 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Furman BL, Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr Protoc Pharmacol 70, 5 47 41–45 47 20 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Braun E, Gilmer J, Mayes HB, Mobley DL, Monroe JI, Prasad S, Zuckerman DM, Best Practices for Foundations in Molecular Simulations [Article v1.0]. Living J Comput Mol Sci 1, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee KH, Kim HS, Jeong HS, Lee YS, Chaperonin GroESL mediates the protein folding of human liver mitochondrial aldehyde dehydrogenase in Escherichia coli. Biochem Biophys Res Commun 298, 216–224 (2002). [DOI] [PubMed] [Google Scholar]
- 88.Chen CH, Ferreira JCB, Joshi AU, Stevens MC, Li SJ, Hsu JH, Maclean R, Ferreira ND, Cervantes PR, Martinez DD, Barrientos FL, Quintanares GHR, Mochly-Rosen D, Novel and prevalent non-East Asian ALDH2 variants; Implications for global susceptibility to aldehydes’ toxicity. EBioMedicine 55, 102753 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang JZ, Zhao SR, Tu C, Pang P, Zhang M, Wu JC, Protocol to measure contraction, calcium, and action potential in human-induced pluripotent stem cell-derived cardiomyocytes. STAR Protoc 2, 100859 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are presented in the paper or the Supplementary Materials. RNA-seq data generated for this study are deposited at the Gene Expression Omnibus in the NCBI database under the accession number GSE184419. All Code is available at https://zenodo.org/record/7478648.