

Abstract
N-heterocycles are privileged pharmaceutical scaffolds in drug discovery and development. We disclose here divergent intermolecular coupling strategies that can access diverse N-heterocycles directly from olefins. The radical-to-polar mechanistic switching is key for the divergent cyclization processes. These distinctive annulations result in the coupling of alkenes with simple bifunctional reagents for divergent N-heterocycle syntheses.
Graphical Abstract

Saturated N-heterocycles such as pyrrolidines and piperidines are top ranked motifs in FDA approved medicines, bioactive pharmaceutical and natural products (Scheme 1a).1 Syntheses of these scaffolds based on widely abundant and structurally diverse olefins represent an attractive strategy.2 In this regard, intramolecular olefin cyclization,3 dipolar cycloaddition of olefins4 and the development of vinylsulfonium salts have been instrumental for inspiring new strategies to access these N-heterocycles.5,6 Alternatively, bifunctional reagents such as the SnAP/SLAP reagents,7 oxaziridines,8 N-allyl-N-halotosylamide,9 diaziridinones,10 and more recently, sulfilimines11 and redox-active amino esters12 have attracted significant attention from the synthetic community for N-heterocycles syntheses.13,14 Despite these advances, divergent olefin annulations that can access both pyrrolidine and piperidine cores from common bifunctional precursors remain an unsolved challenge.
Scheme 1.
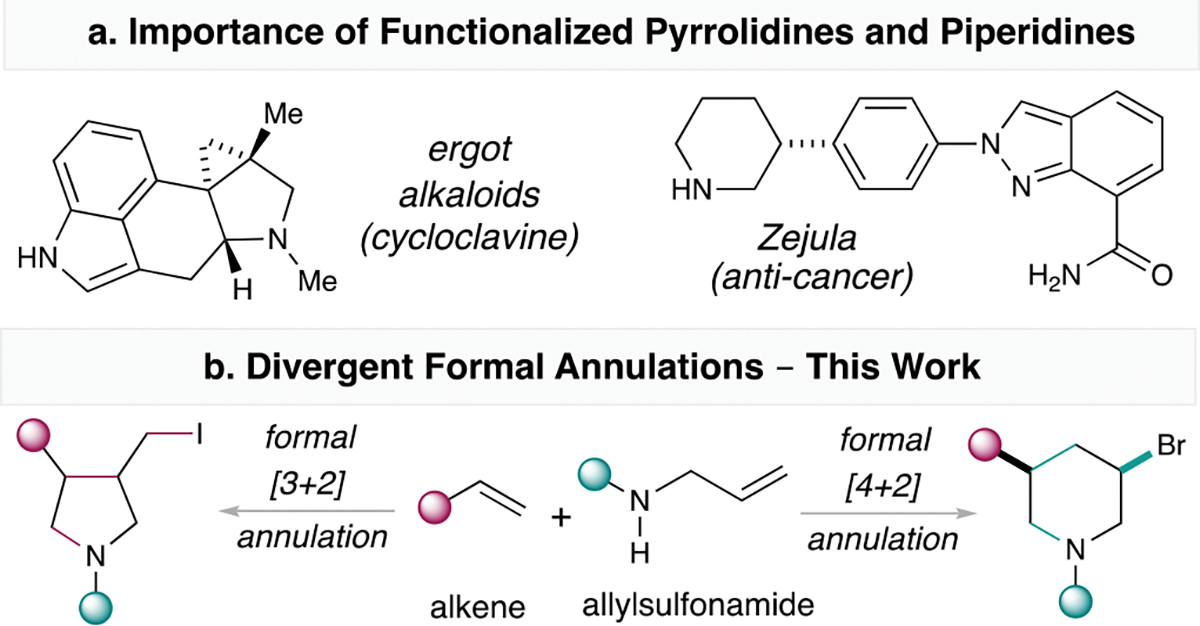
Background and proposed divergent annulations.
Inspired by these works, we have developed a series of bifunctional reagents that can couple with olefins.15 We have previously demonstrated that simple sulfonamides and NIS, under visible light promotion, can also couple with olefins to form aminohalide products.16 We were curious if this strategy can be adopted here for allylsulfonamides to generate similar olefin aminohalide products. If divergent cyclization strategies can be achieved with slight modification of conditions, then divergent olefin annulations can render access to both pyrrolidines and piperidines.17 Herein, we detail our development of such divergent coupling processes to access these highly useful N-heterocyclic scaffolds (Scheme 1b).
We started our work with styrene 1 and allylsulfonamide 2 as the standard substrate. Using 2 equivalents of NIS, we were excited that the piperidine product 3 was formed in 37% yield as a single diastereomer (Scheme 2, entry 1).18 Although increasing solvent concentration raised the yield to 49%, we could not improve upon this level regardless of condition changes based on NIS loading (Scheme 2, entries 2 and 3). Our stoichiometry screening of NIS found that instead of piperidine 3, the pyrrolidine 4a was formed exclusively when the NIS stoichiometry was lowered to 1 equivalent (Scheme 2, entries 3–5).19 Additionally, lowering solvent concentration at low NIS loading improved the pyrrolidine formation. Furthermore, exclusion of oxygen (under N2) further eliminated a ketone byproduct (oxygen trapping byproduct) to afford the optimal conditions for pyrrolidine 4a in 79% yield as a 2:1 stereoisomeric mixture (Scheme 2, entries 6–9). The optimization of the piperidine product was conducted with NBS.20 After 1 h, addition of hexafluoroisopropanol (HFIP) and removal of the external light source afforded the optimal conditions for piperidine 3 (Scheme 2, entries 10–12).21 Our control reactions showed that although light was not essential for the piperidine formation, it was beneficial for the overall reaction efficiency.22
Scheme 2.
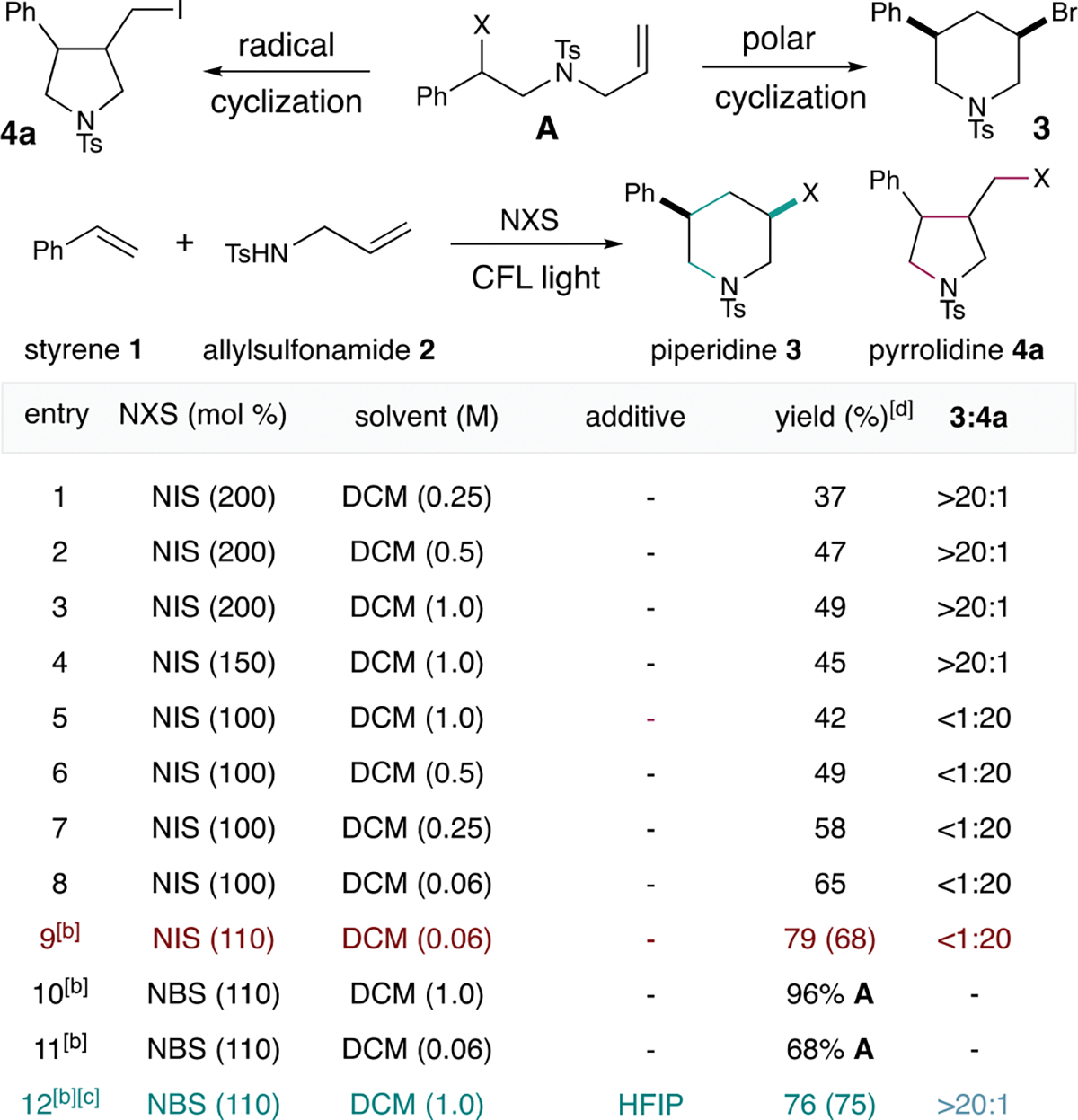
Reaction optimization studies.[a] [a]Standard reaction conditions: for pyrrolidine, 2 (0.25 mmol), 1 (200 mol%), NIS (110 mol%), DCM (4.0 mL), CFL light, N2, 16 h. For piperidine, 2 (0.25 mmol), 1 (200 mol%), NBS (110 mol%), DCM (0.25 mL), CFL light, N2, 1 h, then HFIP (0.5 mL), 36 h. [b]under an atmosphere of N2. [c]1 h under light before addition of HFIP. [d]Yields in parenthesis were isolated yields. The pyrrolidine 4a was isolated as an exocyclic olefin 4 after being treated to these conditions: NaH (1.25 mmol), THF (0.5 mL), 0 °C to rt, 18 h.
To simplify spectroscopic analysis and characterization, we developed elimination conditions using NaH in THF to convert product 4a to the exocyclic olefin product 4 with no additional flash column chromatography needed. With the optimal conditions in hand, we examined a series of styrene derivatives bearing para- and ortho-substituents such as halogen and hydroxyl that were all excellent coupling partners to provide the desired products (Scheme 3, products 5-7). Heteroaromatic allylsulfonamides containing pyrazole or thiophene were also tolerated to form 8 and 9. 2-Vinylnaphthalene was converted to pyrrolidine 10 in excellent yield. Both trans- and cis-β-methylstyrene converged to the trans-pyrrolidine 11 in reasonable yields, consistent with a radical based olefin addition process. A mono-substituted aliphatic olefin was also effective, affording the pyrrolidine 12 in slightly diminished yield. Even a tri-substituted olefin was able to provide the desired pyrrolidine 13 in good yield. Moreover, a range of exocyclic olefins all proceeded smoothly to the corresponding spirocyclic pyrrolidines (Scheme 3, products 14-19). In cases, hetero- and macrocyclic 1,1-disubstituted olefins were viable substrates, demonstrating the robustness of this protocol in forming spirocyclic N-heterocycles. The propargylic sulfonamides were also able to provide the iodinated alkene structures 20-23 with reasonable yields. In these case, the vinyl radicals resulting from the 5-exo-dig cyclization could undergo iodination to form 20-23. Sterically-hindered sulfonamide proceeded to the elimination product 24.
Scheme 3.
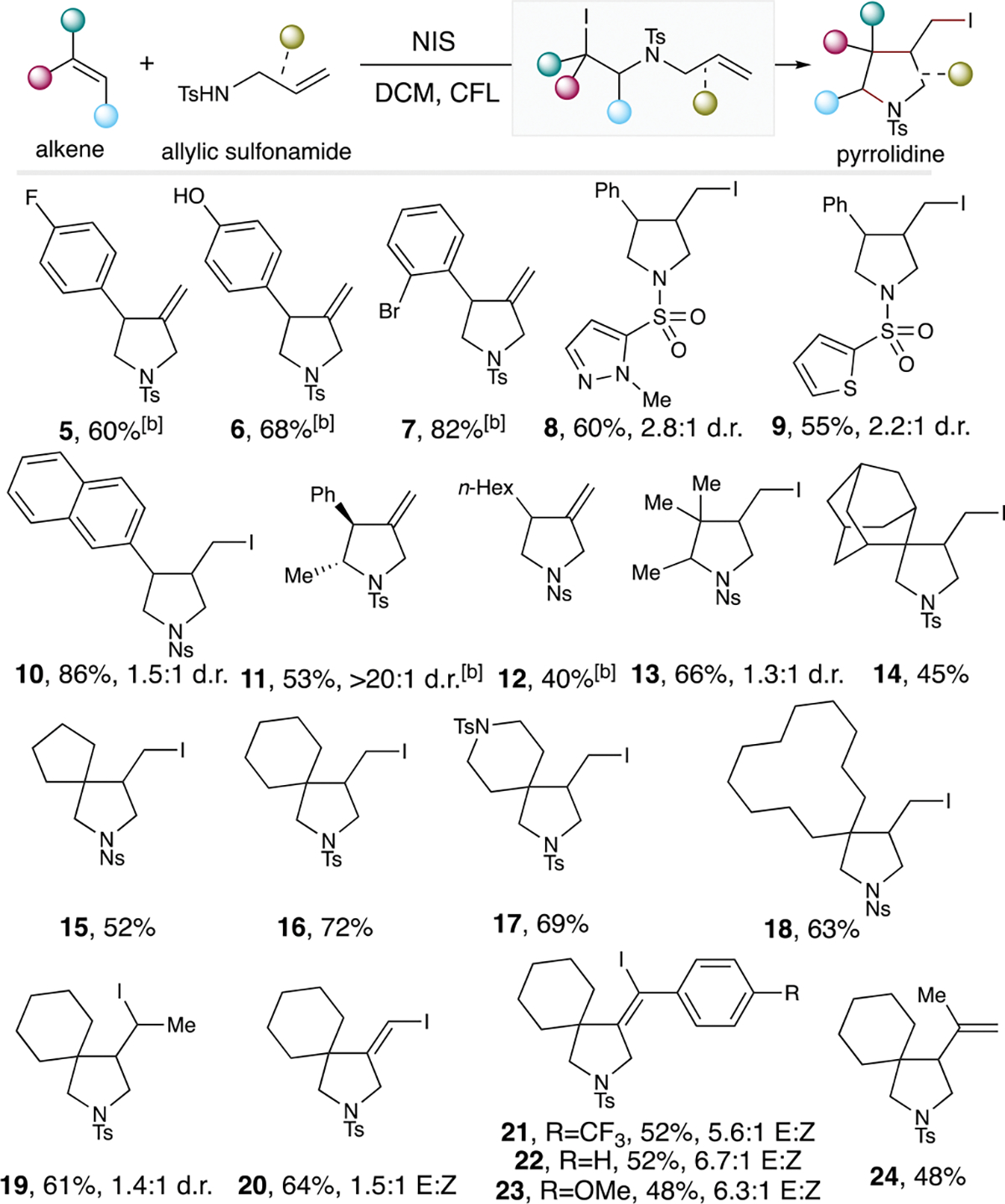
Pyrrolidine substrate scope studies.[a] [a]Standard reaction conditions were used and for the detailed procedure, please refer to the Supporting Information. [b]These exocyclic olefins were obtained after subjecting the isolated iodo-pyrrolidine products to the following conditions: NaH (1.25 mmol), THF (0.5 mL), 0 °C to rt, 18 h.
For the piperidine substrate scope, a series of styrene derivatives bearing p-F, -Br, -tBu and -ester were excellent substrates to afford the desired piperidines in good yields with exceptional stereoselectivity (Scheme 4, products 25-29). Similarly, heteroaromatic-containing piperidines 30 and 31 were generated in reasonable yields. 2-Vinylnaphthalene, β-methylstyrenes, indene and other substituted sulfonamides were all suitable substrates to form the piperidines 32-35 in good yields and diastereomeric ratios. A steroid-derived olefin also proceeded to the desired product 36 in good yields and excellent diastereoselectivity. For more challenging aliphatic alkenes, the standard procedure afforded low yields. In these cases, a two-step protocol was necessary by first isolating the corresponding bromo-intermediates 37a, 38a, and 39a, and then subjecting them to a suitable Lewis acid for the ensuing cyclization (please refer to the Supporting Information for details). In this regard, mono- and 1,1-di-substituted olefins could then proceed to the desired piperidine products 37 and 38. Even tri-substituted olefin could couple with 2-methylallylsulfonamide to 39a, prior to cyclization to form the dihydro-piperidine 39, albeit in marginal efficiency.
Scheme 4.
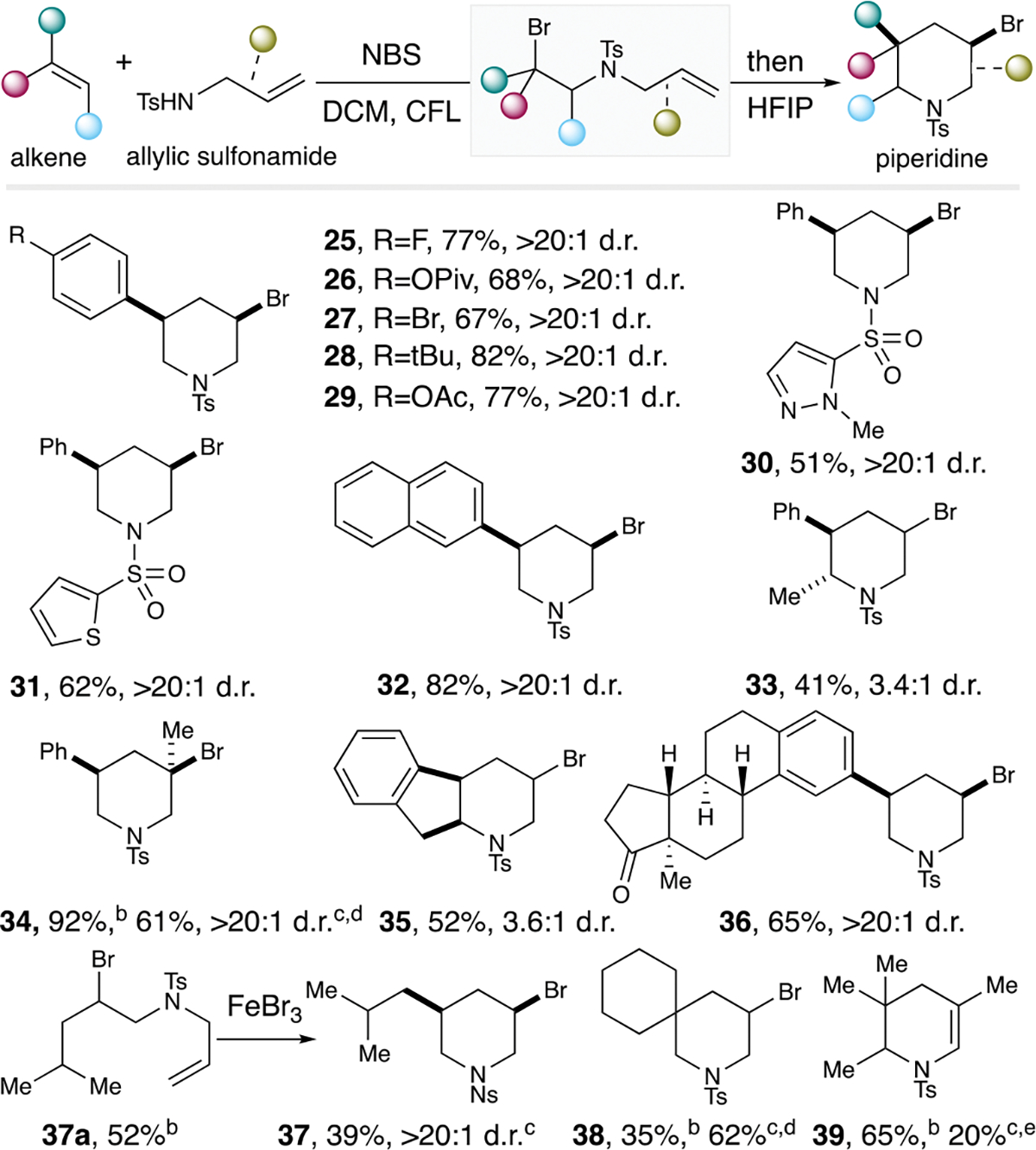
Piperidine substrate scope studies.[a] [a]For detailed information, please refer to the Supporting Information. [b]yield for the bromo-intermediate. [c]yield for the cyclization from the bromo-intermediate. [d]AlBr3. [e]Zn(OTf)2.
To understand the mechanisms of these divergent annulation protocols better, our time studies revealed a rapid buildup of the amino-halogen intermediate A prior to its consumption towards product formations (Scheme 2). We also conducted N NMR studies by mixing NIS and sulfonamide 2 under dark conditions and observed shifting of the sulfonamide N peak correlating to NIS stoichiometry, suggesting the potential formation of a halogen bond-based charge transfer complex prior to N-centered radical formation (see Supporting Information). To rule out the potential hydrogen bonding scenario from the sulfonamide to NIS, we also used N-methylallyl sulfonamide for similar N NMR studies; however, the N NMR peak corresponding to the sulfonamide nitrogen completely disappeared presumably due to the low sensitivity of the N NMR. The rapid formation of A is consistent with prior literature rate studies demonstrating that the rate constant for the carbon radical halogenation process is measured to be ~10 faster than the 5-exo-trig radical cyclization.25 This explains why varying concentration as displayed in the optimization studies had a minimal impact on the selectivity of 3 vs. 4a (Scheme 2, entries 1–8). However, stoichiometry of NIS could have a significant impact on product selectivity, especially at high concentration, as additional NIS can elicit halogen bonding with intermediate A for polar cyclization to form 3. At low NIS loading (1.0 equiv.), photochemical scission of the C–I bond in A could proceed followed by radical cyclization to form pyrrolidine 4a. To validate this, we subjected 40 in DCM under CFL light and observed the formation of the pyrrolidine 4a in 76% yield (Scheme 5a).
Scheme 5.
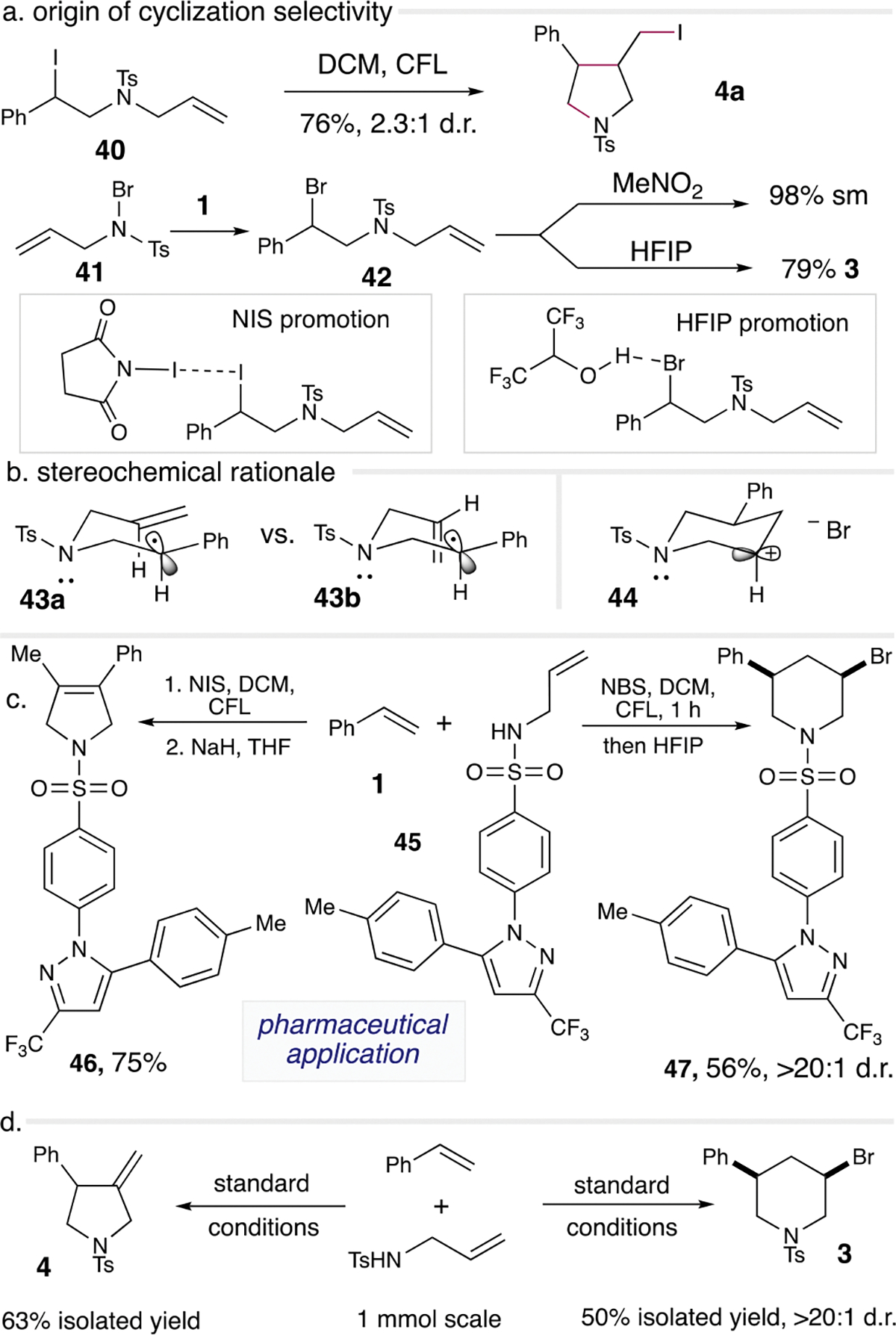
Mechanistic studies and synthetic utilities.
For the piperidine protocol, we observed the formation of 41 in 54% yield when mixing only 2 and NBS in DCM for 1 h under dark conditions, suggesting that 41 is a viable intermediate for the generation of the nitrogen-centered radical to couple with styrene to form 42 (Scheme 5a). Ionic addition of 41 to styrene will likely lead to the opposite regioisomer of 42.26 We subjected 42 to HFIP that resulted in the formation of piperidine 3 in 79% yield. On the other hand, nitromethane as a solvent additive resulted in only starting material recovery, suggesting that HFIP also functions as an activating reagent similar to the role NIS (2.0 equiv.) played to induce the polar cyclization pathway. The low stereoselectivity observed for the pyrrolidine formation was likely due to the small energy difference of the two transition states 43a vs. 43b during this stereochemistry-determining step (Scheme 5b). For the piperidine as shown in 44, the stereochemistry-determining step is the bromide addition step where pseudo-equatorial addition is preferred.
To extend the synthetic utility, we used these annulation protocols to modify known FDA-approved medicines. In this case, Celebrex-derived allylsulfonamide 45 was subjected to our standard reaction conditions (Scheme 5c). The coupling of 45 and 1 under the pyrrolidine protocol, followed by the standard NaH-mediated elimination conditions afforded a mixture of exo- and endocyclic olefin products. Conducting the elimination reaction at slightly higher temperature (rt) afforded only 46 in 75% yield with complete isomerization to the more stable endocyclic product. The piperidine protocol generated 47 in 56% yield as a single diastereomer. This data suggests that these tunable annulation protocols are viable for late-stage pharmaceutical diversifications. Larger scale reactions of these annulation processes resulted in the formation of the respective pyrrolidine 4 and piperidine 3 with reasonable efficiency and reproducibility (Scheme 5d).
In summary, we have developed effective and tunable annulation protocols for the synthesis of privileged pyrrolidine and piperdine scaffolds. The strategy disclosed here centers on controlling the halogenation reagents, concentration, and solvent media for selective radical or polar cyclization pathways. These protocols are also applicable for pharmaceutical diversifications. Most importantly, we demonstrated that divergent olefin annulations can be achieved from simple bifunctional reagents.
Supplementary Material
Acknowledgments
We thank the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R15GM139156 for supporting this work. We thank the University of Toledo for an internal seed grant from the Summer Research Awards and Fellowship Programs for supporting our initial work. We thank Dr. Yong W. Kim (University of Toledo) for NMR assistance.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Vitaku E, Smith DT, Njardarson JT, J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews on olefin difunctionalizations: Derosa J, Tran VT, van der Puyl VA, Engle KM, Aldrichimica. Acta. 2018, 51, 21–32.Wickham LM, Giri R, Acc. Chem. Res. 2021, 54, 3415–3437.Siu JC, Fu N, Lin S, Acc. Chem. Res. 2020, 53, 547–560.
- 3.For a selected review and early examples: Chemler SR, Fuller PH, Chem. Soc. Rev. 2007, 36, 1153–1160.Schlummer B, Hartwig JF, Org. Lett. 2002, 4, 1471–1474.Ney JE, Wolfe JP, Angew. Chem. Int. Ed. 2004, 43, 3605–3608.Michael FE, Cochran BM J. Am. Chem. Soc. 2006, 128, 4246–4248.Fuller PH, Kim J-W, Chemler SR, J. Am. Chem. Soc. 2008, 130, 17638–17639.Nguyen TM, Nicewicz DA, J. Am. Chem. Soc. 2013, 135, 9588–9591.
- 4.Pandey G, Banerjee P, Gadre SR, Chem. Rev. 2006, 106, 4484–4517 [DOI] [PubMed] [Google Scholar]
- 5.(a) Yar M, McGarrigle EM, Aggarwal VK, Angew. Chem., Int. Ed. 2008, 47, 3784–3786. [DOI] [PubMed] [Google Scholar]; (b) Matlock JV, Svejstrup TD, Songara P, Overington S, McGarrigle EM, Aggarwal VK, Org. Lett. 2015, 17, 5044–5047. [DOI] [PubMed] [Google Scholar]; (c) Juliá F, Yan J, Paulus F, Ritter T, J. Am. Chem. Soc. 2021, 143, 12992–12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For a selected review and example on C-H functionalization to access N-heterocycles: Campos KR, Chem. Soc. Rev. 2007, 36, 1069–1084.Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS, Nature 2016, 531, 220–224.
- 7.(a) Vo C-VT, Mikutis G, Bode JW, Angew. Chem. Int. Ed. 2013, 52, 1705–1708. [DOI] [PubMed] [Google Scholar]; (b) Siau W-Y, Bode JW, J. Am. Chem. Soc. 2014, 136, 17726–17729. [DOI] [PubMed] [Google Scholar]
- 8.(a) Michaelis DJ, Shaffer CJ, Yoon TP, J. Am. Chem. Soc. 2007, 129, 1866–1867. [DOI] [PubMed] [Google Scholar]; (b) Williamson KS, Yoon TP, J. Am. Chem. Soc. 2010, 132, 4570–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Tsuritani T, Shinokubo H, Oshima K, Org. Lett. 2001, 3, 2709–2711. [DOI] [PubMed] [Google Scholar]; (b) Engl S, Reiser O, Org. Lett. 2021, 23, 5581–5586. [DOI] [PubMed] [Google Scholar]; (c) Crespin LNS, Greb A, Blakemore DC, Ley SV, J. Org. Chem. 2017, 82, 13093–13108. [DOI] [PubMed] [Google Scholar]
- 10.Yuan W, Du H, Zhao B, Shi Y, Org. Lett. 2007, 9, 2589–2591. [DOI] [PubMed] [Google Scholar]
- 11.(a) Cheng Q, Bai Z, Tewari S, Ritter T, Nature Chem. 2022, 14, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baskaran P, Li W, Nature Chem. 2022, 14, 850–852. [DOI] [PubMed] [Google Scholar]
- 12.Murray PRD, Liebler IN-M, Hell SM, Villalona E, Doyle AG, Knowles RR, ACS Catal. 2022, 12, 13732–13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For other selected recent examples of bifunctional reagents in olefin couplings: Lu D-F, Zhu C-L, Jia Z-X, Xu H, J. Am. Chem. Soc. 2014, 136, 13186–13189.Kaneko K, Yoshino T, Matsunaga S, Kanai M, Org. Lett. 2013, 15, 2502–2505.McAtee RC, Noten EA, Stephenson CRJ, Nat. Commun. 2020, 11, 2528.Ni H-Q, Kevlishvili I, Bedekar PG, Barber JS, Yang S, Tran-Dubé M, Romine AM, Lu H-X, McAlpine I, Liu P, Engle KM, Nat. Commun. 2020, 11, 6432.Holst DE, Wang DJ, Kim M, Guzei IA, Wickens ZK Nature 2021, 596, 74–79.Lei H, Jr JH. Conway, C. C. Cook, T. Rovis, J. Am. Chem. Soc. 2019, 141, 11864–11869.
- 14.For a review on bifunctional reagents: Huang H-M, Bellotti P, Ma J, Dalton T, Glorious F, Nat. Rev. Chem. 2021, 5, 301–321.
- 15.(a) Wu F, Kaur N, Alom N-E, Li W, J. Am. Chem. Soc. Au 2021, 1, 734–741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu F, Alom N-E, Ariyarathna JP, Naß J, Li W, Angew. Chem. Int. Ed. 2019. 58, 11676–11680. [DOI] [PubMed] [Google Scholar]; (c) Ariyarathna JP, Alom N-E, Roberts LP, Kaur N, Wu F, Li W, J. Org. Chem 2022, 87, 2947–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kaur N, Wu F, Alom N-E, Ariyarathna JP, Saluga SJ, Li W, Org. Biomol. Chem. 2019, 17, 1643–1654. [DOI] [PubMed] [Google Scholar]
- 16.Wu F, Ariyarathna JP, Kaur N, Alom N-E, Kennell ML, Bassiouni OM, Li W, Org. Lett. 2020, 22, 2135–2140. [DOI] [PubMed] [Google Scholar]
- 17.For a selected review: Jiang H, Studer A, Chem. Soc. Rev 2020, 49, 1790–1811.
- 18.For selected recent intermolecular examples for piperidine synthesis: Sandmeier T, Krautwald S, Carreira EM, Angew. Chem. Int. Ed. 2017, 56, 11515–11519.Larsen MA, Hennessy ET, Deem MC, Lam Y-H, Sauri J, Sather AC, J. Am. Chem. Soc. 2020, 142, 726–732.
- 19.For selected recent intermolecular examples for pyrrolidine synthesis: Jiang H, He J, Liu T, Yu J-Q, J. Am. Chem. Soc. 2016, 138, 2055–2059.Lee S, Lei H, Rovis T, J. Am. Chem. Soc. 2019, 141, 12536–12540.
- 20.Constantin T, Julia F, Leonori D, Chem. Rev. 2022, 122, 2292–2352. [DOI] [PubMed] [Google Scholar]
- 21.Motiwala HF, Armaly AM, Cacioppo JG, Coombs TC, Koehn KRK, Norwood IV VM, Aubé J, Chem. Rev 2022, 122, 12544–12747. [DOI] [PubMed] [Google Scholar]
- 22.For the pyrrolidine 4, dark conditions provided no product; and for piperidine 3, exact same conditions under dark conditions generated 55% product.
- 23.For a selected review on halogen bond: Bulfield D, Huber SM, Chem. Eur. J. 2016, 22, 14434–14450.For an interesting example: Sladojevich F, McNeil E, Börgel J, Zheng S-L, Ritter T, Angew. Chem. Int. Ed 2015, 54, 3712–3716.
- 24.For selected reviews on NCRs: Zard SA, Chem. Soc. Rev. 2008, 37, 1603–1618.Chen J-R, Hu X-Q, Lu L-Q, Xiao W-J, Chem. Soc. Rev. 2016, 45, 2044–2056.For selected recent examples: Wappes EA, Nakafuku KM, Nagib DA, J. Am. Chem. Soc. 2017, 139, 10204–10207.Hemric BN, Shen K, Wang Q, J. Am. Chem. Soc. 2016, 138, 5813–5816.Zhu Q, Graff DE, Knowles RR, J. Am. Chem. Soc. 2018, 140, 741–747.Maity S, Zhu M, Shinabery RS, Zheng N, Angew. Chem. Int. Ed. 2012, 51, 222–226.Maity A, Roychowdhury P, Herrera RG, Powers DC, Org. Lett 2022, 24, 2762–2766.
- 25.(a) Griller D, Ingold KU, Acc. Chem. Res 1980, 13, 317–323. [Google Scholar]; (b) Foldiak G, Schuler RH, J. Phys. Chem 1978, 82, 2756–2757. [Google Scholar]
- 26.Tan CK, Yeung Y-Y, Chem. Commun. 2013, 49, 7985–7996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.