

Abstract
Fucose is a signaling carbohydrate that is attached at the end of glycan processing. It is involved in a range of processes, such as the selectin-dependent leukocyte adhesion or pathogen-receptor interactions. Mass-spectrometric techniques, which are commonly used to determine the structure of glycans, frequently show fucose-containing chimeric fragments that obfuscate the analysis. The rearrangement leading to these fragments – often referred to as fucose migration – has been known for more than 25 years, but the chemical identity of the rearrangement product remains unclear. In this work, we combine ion-mobility spectrometry, radical-directed dissociation mass spectrometry, cryogenic-ion IR spectroscopy, and density-functional theory calculations to deduce the product of the rearrangement in the model trisaccharides Lewis x and blood group H2. The structural search yields the fucose moiety attached to the galactose with an α(1 → 6) glycosidic bond as the most likely product.
Keywords: Carbohydrates, Mass-spectrometry, Gas-phase ions, IR spectroscopy, Density functional calculations
Graphical Abstract
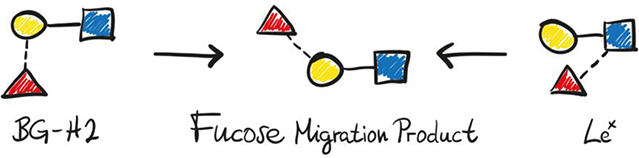
Mass-spectrometric techniques frequently show fucose-containing chimeric fragments that obfuscate the analysis of glycans. Here, ion-mobility spectrometry, radical-directed dissociation mass spectrometry, cryogenic-ion IR spectroscopy, and density-functional theory calculations are used to deduce the product of the internal glycan rearrangement of two model trisaccharides.
Introduction
Glycans are responsible for a range of key-biological functions, such as nutrient storage, molecular signaling, and cell-cell communications.[1] The widespread use of glycans is enabled by their large structural heterogeneity, owed to the non-trivial assembly rules of isomeric monosaccharides, and their large conformational flexibility. This structural complexity poses a challenge for analytical methods aimed at determining glycan molecular structure. Colluded with a lack of a simple amplification mechanism, this renders glycomics lagging behind genomics and proteomics.[2]
Today, the structural analysis of glycans is typically performed by sophisticated tandem mass spectrometry (MS) techniques. The tandem MS spectrum of a glycan usually contains glycosidic and cross-ring fragments, which can be used to reconstruct the topology of the precursor.[3] The assignment, however, is often hampered by the appearance of non-native chimeric fragments, which are caused by an internal rearrangement of particular monosaccharide building blocks. Fucose, a C6-deoxy l-monosaccharide involved in the extracellular communication, such as the regulation of selectin-dependent leukocyte adhesion,[4] and the viral docking to a host cell,[5] is particularly prone to migration along and across the glycan sequence.[6] This phenomenon has been known for more than 25 years, however, the mechanism as well as the structural triggers of the rearrangement remain unknown.[7] In addition, the migration is not limited to fucose, but has also been observed for other biologically relevant saccharides such as rhamnose,[8] mannose,[9] and recently, xylose.[7b] The implications of hexose migration for mass spectrometry-based glycomics can therefore be compared to that of peptide scrambling[10] for proteomics: both are erratic rearrangement reactions that can only be controlled and predicted sufficiently when the underlying molecular processes are understood in great detail.
In recent years, the capabilities of MS for glycan analysis have been significantly enhanced by the introduction of orthogonal gas-phase techniques. Ion mobility-mass spectrometry (IM-MS) adds a structural dimension to the mass-to-charge ratio in form of a rotationally-averaged collision-cross section[11] and action infrared spectroscopy probes the spatial structure of the ion in form of an IR spectrum.[12] In the past, we have applied both techniques to identify the glycosidic bond regio- and stereochemistry in glycans[12b] and glycoconjugates,[13] and to unravel the structure of reaction intermediates in glycosylation reactions.[14] Radical-mediated dissection of glycans in tandem MS has shown to be structure sensitive to discriminate isomeric glycans.[15] In radical-directed dissociation (RDD) MS, a radical, generated upon UV photodissociation, migrates along the glycan core to produce diverse dissociation products, such as cross-ring cleavages for cations, increasing its sensitivity compared to collision-induced dissociation.[16]
Recently, we demonstrated the migration of fucose in the Lewis x (Lex) and blood group antigen H2 (BG-H2) trisaccharides. They share a d-galactopyranose β(1→4)d-N-acetylglucosoaminepyranose core (Galβ(1 → 4)GlcNAc), but differ by the attachment of the fucosyl moiety (Figure 1A). In Lex, the fucose is attached via an α(1→3) glycosidic bond to the reducing GlcNAc, whereas in BG-H2 the fucose is linked by an α(1→2) bond to the galactose. Cryogenic IR spectroscopy revealed identical mid-IR fingerprints for the protonated [Lex+H]+ and [BG-H2+H]+ trisaccharide ions, which indicates that fucose migration can occur at ambient conditions and does not require collision-induced dissociation.[7c] In a subsequent work, we further showed that the rearrangement is promoted by a mobile proton, whereas lithium, sodium and adducts with an immobilized proton inhibit migration.[17] However, the final structure of the rearrangement product and the triggers that initiate the migration remain elusive to date. In this work, we combine structural data from IM-MS, cryogenic IR spectroscopy and radical-directed dissociation mass spectrometry (RDD) with high-throughput density-functional theory (DFT) modeling to elucidate the molecular structure of the rearrangement product, and discuss possible mechanisms leading to its formation. Our data shows that the glycan rearrangement of the intact analyte ions occurs at the front end of the respective instruments, which has important consequences for the MS-based analysis of glycans.
Figure 1.

A) BG-H2 and Lex trisaccharides represented using symbol nomenclature for glycans (SNFG). B) Arrival time distribution of activated (harsh conditions, 190 V cm−1) and non-activated (soft conditions, 30 V cm−1) intact precursor standards of [Lex+H]+ and [BG-H2+H]+ in the heatmap representation. Upon activation, DTCCSHe of the [BG-H2+H]+ ion decreases from 146 Å2 to 143 Å2, but DTCCSHe of [Lex+H]+ of 143 Å2 remains unchanged. C) IR spectra of the intact [Lex+H]+ and [BG-H2+H]+ standards recorded using warm (room temperature) or cold (cooled to approx. 90K) ion trap. The spectrum of [Lex+H]+ remains unchanged, but the spectrum of [BG-H2+H]+ changes (guided by dashed lines) when the temperature of the trap is changed. D) Radical fragmentation spectra afforded by photodissociation of the precursor m/z 895 and RDD of the generated radical glycan m/z 768 (indicated with an arrow). The spectra of the BG-H2-based tetrasaccharide show more pronounced differences between soft and harsh conditions compared to the spectra of the Lex-based tetrasaccharide.
Results and Discussion
The three utilized instruments have been described in great detail in previous publications,[16, 18] and therefore only relevant details are discussed here. In the IM-MS experiment, molecular ions are generated by nano-electrospray ionization and transferred into the instrument via a capillary inlet. Before being injected to a drift tube for the mobility separation, the ions are accumulated and stored in an hourglass ion funnel. This section can be used to collisionally activate the ions below their dissociation threshold. We used two values of the electric field, both sub-CID, to mimic the source conditions in commercial mass spectrometers. We defined harsh conditions by applying an electric field of up to 190 V cm−1, and soft conditions by applying an electric field strength of only 30 V cm−1. After the separation and subsequent m/z-selection, arrival time distributions (ATD) of the ions are recorded, the ion mobilities are extracted, and DTCCSHe are calculated using the Mason-Schamp equation.[19]
RDD experiments were performed using a modified Thermo Fisher LTQ linear ion trap. Prior to analysis, the molecules are derivatized with a para-iodoaniline linker to provide a photocleavable C–I bond using established protocols.[15a] This modification is technically achieved by adding an additional galactose monosaccharide at the reducing end of the original trisaccharide, such that the Lex and BG-H2 motives are preserved (Figure 1D). In a multistep MS workflow, the parent ions are generated by the nano-electrospray ionization, either collisionally activated below their dissociation threshold (harsh conditions) or treated gently during desolvation (soft conditions), m/z-selected and irradiated using fourth harmonic (266 nm) pulses from a Continuum Minilite II Nd:YAG laser. The generated radical ions of interest are isolated and activated via CID (with a relative energy of 20) to yield the respective mass spectrum.
In cryogenic-ion IR spectroscopy, the ions are again generated by nano-electrospray ionization. To avoid excessive activation, soft source conditions are used to transfer the intact ions to the gas phase.[7c] Next, the m/z-selected species are accumulated in a hexapole ion trap, which can be maintained under room temperature (warm trap) or cooled to 90 K (cold trap). These ions are then captured by superfluid helium nanodroplets (of approx. size of 105 atoms) traversing the trap. Inside the droplet, which acts as a transparent thermostat, the ion is rapidly cooled down to 0.4 K. Downstream the apparatus, these droplets are irradiated by the Fritz Haber Institute IR free-electron laser (FHI-FEL). Absorption of resonant FEL photons causes molecular vibrations leading to helium droplet evaporation. Subsequently, the ion is released from the droplet, which is recorded using the time-of-flight (ToF) spectrometer. The ion count recorded at the ToF as a function of the frequency yields the IR spectrum of the ion in cryogenic conditions.
The arrival-time distributions (ATDs) of the [Lex+H]+ and [BG-H2+H]+ ions, which were recorded at soft and harsh conditions, are shown in the Figure 1B and S1. The [Lex+H]+ ions show highly similar ATD profiles under both conditions, and a collision cross-section in helium buffer gas, DTCCSHe, of 143 Å2. The ATD of the [BG-H2+H]+ ions, on the other hand, changes upon activation. Under soft conditions, the ions yield a DTCCSHe of 146 Å2, which decreases to 143 Å2 under harsh conditions. A structural compaction upon heating is not usually observed for glycan ions of that size, and the subtle drop in size instead is likely caused by structural isomerism yielding ions of similar size as [Lex+H]+.
To shed light on the potential rearrangement, RDD spectra of Lex- or BG-H2-based tetrasaccharides were recorded without (soft) and with sub-CID (harsh) activation (Figure 1D). The BG-H2-based structure generally shows more pronounced differences in fragment abundance between soft and harsh activation conditions than the Lex-based tetrasaccharide. In the respective RDD spectrum, the glycosidic fragment at m/z 512 increases in intensity at sub-CID conditions, whereas the glycosidic fragment at m/z 443 decreases, pointing towards a rearrangement within the core Gal-GlcNAc unit. In contrast, the ratios of the most intense ion fragments in the RDD spectrum of the Lex motif do not change significantly. Furthermore, in all RDD spectra, fucose migration to the linker or the additional galactose can be observed (m/z 404, denoted with an asterisk), which highlights the general ability of fucose to undergo migration reactions to different sites. While some of these sites could be easily identified by their masses, migration along the sequence might yield fragments with unchanged m/z value so that such rearrangement is camouflaged. To corroborate these results, we recorded the cryogenic-ion IR spectra (using the cold trap) of the two modified tetrasaccharides (Figure S2). The vibrational spectrum of the Lex-based tetrasaccharide appears as sub-spectrum of that of the BG-H2-based tetrasaccharide, which is readily visible in their amide region. The former shows only one band in the amide I region (main contribution from ν(C=O) of the GlcNAc), which, in addition to another blue-shifted band, is mirrored in the spectrum of the latter. With only one C=O oscillator in these molecules, the two bands in the IR spectrum of the BG-H2-based tetrasaccharide, strongly shifted by approx. 50 cm−1, can be associated to the presence of two isomeric structures.
Finally, we recorded the IR spectra of the two trisaccharides at different trap temperatures. We hypothesize that accumulation of the ions in the cold trap should slow the rearrangement process enough to record the IR spectra of the precursor ions. The IR spectra of the [Lex+H]+ ions obtained in a warm or cold trap show little difference, while the IR signatures of the [BG-H2+H]+ ions are clearly sensitive to the trap’s temperature (Figure 1C). In the warm trap, the reproducible spectrum of [BG-H2+H]+ (Figure S3) is similar to that of [Lex+H]+ (Figure 1C). Quenching of the isomerization kinetics by cooling of the trap leads to several additional features in the IR spectra. Both spectra of [Lex+H]+ exhibit a complex feature in the region of C-C and C-O stretching modes between 1025 – 1175 cm−1, and a narrow band in the amide I region at 1660 cm−1. By contrast, the spectrum of the [BG-H2+H]+ ions accumulated in the cold trap lacks the band at 1060 cm−1 and instead displays additional peaks at 1100 cm−1 and 1140 cm−1 as well as a broad feature in the amide I region between 1640 cm−1 and 1700 cm−1. Upon contact with the helium droplets, the ensemble of conformers present in the ion trap is shock-frozen, which leads to kinetic trapping of entropically-favored states. Because ions equilibrated in the ion trap maintained at room temperature are expected to present more conformations than in the ion trap maintained at 90K, it is conceivable that the IR spectrum recorded using a warm trap is more populated than the IR spectrum recorded using a cold trap. However, in the case of [BG-H2+H]+, the richer features in the amide I region of GlcNAc, which is particularly sensitive to the chemical environment, are observed for the cold-trap settings. This effect cannot be assigned to multiple conformations of the same ion. Instead, it can be explained by trapping of chemically different structures – precursor, product structures, and potential reaction intermediates – each of which displays a unique IR fingerprint. In other words, the low temperature of the trap may slow down the glycan rearrangement process and reveal the vibrational signature of the precursor ion.
In summary, the three MS-based experiments can be interpreted as the fucose rearrangement from [BG-H2+H]+ to the [Lex+H]+ ion. The measurements of [BG-H2+H]+ show strong dependence on the activation condition, while the [Lex+H]+ appears to be insensitive. However, at this point we cannot exclude the alternative scenario, in which both glycans rearrange to a third common – yet unknown – structure. In this case, the fucose of the [Lex+H]+ ion is more labile and promptly undergoes the rearrangement, while the [BG-H2+H]+ ion is stable enough to be preserved under soft conditions.
To determine which of these two migration mechanisms – the interconversion between the two precursors, or the rearrangement towards a third unknown structure – occurs, we applied methods of computational chemistry to identify a conformer of the isomer that matches the experimental DTCCSHe and IR spectrum. Previously, the DFT-based searches were successful in the structural assignment of glycoconjugates,[13] RNA rearrangement products,[20] and amino acids complexes.[21] The details of the search protocol, which has been implemented in the CarPpy python package,[22] are discussed in the SI. Briefly, the conformational spaces of the amide-protonated Lex and BG-H2 ions, with both the α- and β-anomers and cis (-c-) and trans orientation (-t-) of the amide bond, were sampled independently with CREST in combination with the xTB package,[23], and reoptimized using the PBE0 hybrid density functional.[24] The GFN2-xTB and PBE0 energy functions were selected, respectively, for the initial sampling and subsequent refinement, due to their performance against mono- and disaccharide benchmark sets.[25] Finally, we computed the harmonic free energy and projection approximation CCS (PACCS) of 306 lowest-energy structures.[26] The resulting free-energy hierarchy is shown in Figure 2A, and the anharmonic spectra of the most stable anomers of [BG-H2+H]+ and [Lex+H]+ are shown in Figure 2B and Figure S4.
Figure 2.

A) The PACCS vs relative free energy (ΔF) of conformers of the [Lex+H]+ and [BG-H2+H]+ at PBE0+D3/6–311+G(d,p) level of theory. The dashed line indicates the experimental CCS, and the free energies are plotted relative to the lowest-free energy conformer of [β-c-BG-H2+H]+. The most stable anomers of each ion are highlighted. B) The comparison of the experimental and vibrational spectrum of the most stable anomers of [BG-H2+H]+. The comparison of the experimental and vibrational spectrum of the most stable anomers of [Lex+H]+ is shown in Figure S4.
The theory predicts that the two most stable anomers are [β-c-BG-H2+H]+ (ΔF=0.0 kcal mol−1), and [α-t-BG-H2+H]+ (ΔF=1.5 kcal mol−1), whereas the lowest-free energy structures of [α-t-Lex-H]+ and [β-t-Lex+H]+ are 4.1 and 3.5 kcal mol−1 less stable, respectively. The PACCS of the [β-t-Lex+H]+ of 133 Å2 is substantially smaller than the experimental value of 143 Å2, and 9 Å2 lower than the PACCS of the [β-c-BG-H2+H]+ (142 Å2). Furthermore, the change is substantially larger than the contraction of 3 Å2 that occurs upon activation of the [BG-H2+H]+ ion. Furthermore, the simulated IR spectra of the [Lex+H]+ ion (Figure S3) show a poor agreement with the experimental spectrum, as the theory does not predict the correct shape of the band in the 1075–1125 cm−1 region. It also misplaces the position of the protonated amide band, predicting it at 1650 and 1700 cm−1 for the β- and α-anomers, respectively. The simulated spectrum of the [α-t-BG-H2+H]+ and [β-c-BG-H2+H]+ ions (Figure 2B), on the other hand, resembles to some degree the amide I features observed in the cold-trap spectrum. Specifically, the protonated amide bands of the β- and α-anomers at 1640 and 1700 cm−1, respectively, match the additional shoulders in the broad feature visible in the amide region. Thus, in addition to the rearrangement product, also the [BG-H2+H]+ ions are observed in the IR spectrum recorded using the cold trap. However, the [Lex+H]+ ion is not the product of this rearrangement and there is abundant evidence sorting this claim: 1) the α- and β-anomers of [Lex+H]+ are thermodynamically less stable than the [BG-H2+H]+ isomers; 2) the [Lex+H]+ ions are too compact and the predicted compaction of CCS is much larger than the observed 3 Å2 and 3) the predicted IR spectrum of [Lex+H]+ matches poorly to the experimental spectrum. Based on these grounds, we conclude that the rearrangement must proceed towards a third, unknown structure.
To reveal the identity of the rearrangement product, we have repeated the structural search for all conceivable constitutional isomers of the trisaccharide. We have investigated each type of linkage between the fucose and the Galβ(1 → 4)GlcNAc core in both configurations, both orientations of the amide group, and in both anomeric forms of the reducing GlcNAc, which in total equals to 56 additional types of structures (Table S1). The relative free energies of the most stable conformers, along with the difference between PACCS and DTCCSHe, are summarized in Table 1, and Figures S5–S36 in the SI.
Table 1.
The relative free energy in kcal mol−1 at 300 K with respect to the lowest-free energy α anomer of Fucα(1 → 6)Galγ(1 →)4GlcNAcα and the difference between calculated PACCS and experimental DTCCSHe=143 Å2 of the lowest free-energy conformer of each possible fucose position around the Galβ(1 → 4)GlcNAc core. SNFG symbols show the bonds connectivity but omit the bond configuration. Cells with the data for the Lex and BG-H2 ions are highlighted in red and blue, respectively. The lowest free-energy conformer of [α-t-α16+H]+ is highlighted in gray.
| a-anomer | p-anomer | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bond configuration | α | β | α | β | |||||||
| Linkage | Amide | SNFG | △F | ACCS | △F | ACCS | △F | ACCS | △F | ACCS | |
|
| |||||||||||
| GlcNAc | 1–1 | trans |
|
1.0 | 1 | 2.3 | 2 | 6.0 | 0 | 4.6 | 0 |
| 1–1 | cis | 7.6 | −12 | 7.7 | −15 | 6.6 | −4 | 6.6 | −9 | ||
| 1–2 | trans |
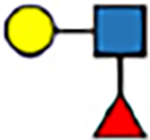
|
10.9 | −11 | 14.9 | −11 | 14.0 | −10 | 17.5 | −13 | |
| 1–2 | cis | 13.3 | 6 | 18.8 | −5 | 15.1 | 3 | 17.7 | 0 | ||
| 1–3 | trans |
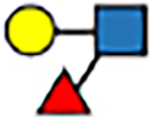
|
8.3 | −5 | 8.7 | −8 | 7.7 | −7 | 7.7 | −4 | |
| 1–3 | cis | 11.8 | −3 | 10.1 | −4 | 9.1 | −1 | 8.2 | −2 | ||
| 1–6 | trans |
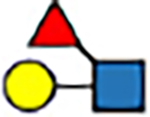
|
5.9 | −3 | 5.9 | −8 | 7.6 | −7 | 5.4 | −7 | |
| 1–6 | cis | 9.3 | 1 | 7.1 | −3 | 8.1 | 3 | 4.9 | −3 | ||
|
| |||||||||||
| Gal | 1–2 | trans |
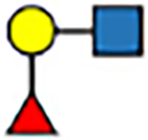
|
5.7 | −5 | 6.3 | −5 | 6.3 | 0 | 7.4 | 7 |
| 1–2 | cis | 7.0 | −3 | 7.4 | −5 | 4.2 | −1 | 5.0 | −3 | ||
| 1–3 | trans |
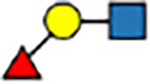
|
8.2 | 0 | 4.2 | −10 | 8.6 | 3 | 8.9 | 2 | |
| 1–3 | cis | 9.4 | 1 | 8 9 | 5 | 7.2 | 2 | 6.8 | 6 | ||
| 1–4 | trans |
|
5.8 | 2 | 4.4 | −3 | 7.7 | 3 | 7.3 | −1 | |
| 1–4 | cis | 10.1 | −12 | 6.5 | 0 | 9.1 | 0 | 4.5 | 0 | ||
| 1–6 | trans |
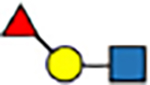
|
0.0 | 0 | 4.1 | 1 | 4.4 | 0 | 7.0 | −4 | |
| 1–6 | cis | 7.0 | 0 | 7.9 | −10 | 6.2 | −5 | 8.7 | 2 | ||
The expanded structural search confirms that neither [Lex+H]+ nor [BG-H2+H]+ is the thermodynamically most stable trisaccharide isomer. Instead, the most stable isomer features the fucose moiety attached by a α(1 → 6) glycosidic bond to the galactose. This trans-amide bond α-anomer ([α-t-α16+H]+, Figure S37) is more stable by 5.7 kcal mol−1 than the most stable conformer of the BG-H2/Lex pair, and its PACCS matches the experimental value well. Two other structures, which have fucose connected by either the α(1 ↔ 1)α or α(1 ↔ 1)β linkages to the reducing-end GlcNAc, are less stable by 1.0 and 2.3 kcal mol−1 respectively, and all other structures are at least 4 kcal mol−1 less stable than the global minimum. In case of the β-anomers, there are six isomers within ΔF=1.0 kcal mol−1 above the minimum, and the [β-t-α16+H]+ ion is among them. Most notably, the fucose attached to the N-acetyl group of the GlcNAc, which has been reported as the potential fucose rearrangement product,[27] is at least 10.9 kcal mol−1 less stable than the [α-t-α16+H]+ ion, and has a PACCS 11 Å2 smaller than the experimental value. Therefore, theory predicts three possible rearrangement products, which have fucose attached by the α(1 → 6), α(1 ↔ 1)α, or α(1 ↔ 1)β linkage to the Galβ(1 → 4)GlcNAc core. However, in the RDD and cryogenic-ion IR analysis of the Lex- and BG-H2-based tetrasaccharides, the (1 ↔ 1) position is blocked by the label but still rearrangement is observed when induced with sub-CID or warm trap conditions. Therefore, the (1 ↔ 1) position is unlikely to be the rearrangement product of Lex and BG-H2 trisaccharides, which leaves only one possible candidate for the rearrangement product – the fucose linked with the α(1 → 6) glycosidic bond to the terminal galactose.
To validate this prediction, we have compared the calculated spectra of several different low-energy ions with the experimental IR spectrum (Figure 3A). The computed spectrum of the [α-t-α16+H]+ provides an excellent match in the amide I region, which explains the narrowing of the band that happens in combination with the warm trap. In addition, the 1000–1200 cm−1 region qualitatively agrees with the experimental data. The spectrum shows several bands at 1040, 1075, 1100 and 1130 cm−1, which are also present in the experimental IR spectrum, whereas none of the spectra of other low-energy structures provide a comparable match. The small discrepancies in the IR spectrum, such as the missing band at 1060 cm−1 and a small shoulder at 1700 cm−1, are likely caused by the presence of small amounts of the β-anomer which features more chemically different ions of similar stability. Therefore, we conclude that the unknown rearrangement product observed in the three MS-based experiments has the fucose connected with the α(1 → 6) glycosidic bond to the terminal galactose. This assignment, based on DFT calculations, is validated by three independent factors: the thermodynamic stability of the ion, its CCS, and its IR fingerprint.
Figure 3.

A) The comparison of the experimental and anharmonic theoretical spectra for four low-energy ions of different chemical structures. B) The proposed closed-ring reaction mechanism of the [α-t-BG-H2]+ leading to the formation of the [α-t-α16+H]+. C) The proposed open-ring reaction mechanism of the [α-t-BG-H2]+ leading to the formation of the [α-t-α16+H]+.
Finally, we shall discuss the hypothetical rearrangement mechanism that leads to the formation of the proposed structure. The proposed mechanisms are based on previously published work and have been adjusted to the Lex/BG-H2 system. The detailed reaction paths, energetics, and experimental validation of one of the mechanisms are to be considered in a follow-up study. There are two mechanisms in the literature, one that involves fucose transfer in the closed form, and one that incorporates the ring-opening step.[28] First, we note that the rearrangement product retains the α-configuration of the glycosidic bond, which can be achieved by two subsequent SN2-like reactions, proceeding through a less stable intermediate (Figure 3B). This mechanism does not suffer from the entropy penalty associated with the ring-opening mechanism. During the first step of the rearrangement of [BG-H2+H]+, the protonated amide acts as a proton donor, which protonates the Fucα(1 → 2)Gal glycosidic bond through a chain of hydroxy groups. The anomeric carbon of the fucose moiety is then attacked from the β-side by the carbonyl oxygen in the amide group via an SN2 mechanism, which leads to a stable intermediate similar to that reported by Boons et al.[27] Next, the rotation around the Galβ(1 → 4)GlcNAc bond is followed by a second SN2 reaction, a nucleophilic attack of the O6 of the galactose at the α-side of the anomeric carbon of the fucose moiety, which recovers the α-configuration of the product. In the proposed mechanism, the first fucose migration to the amide group constitutes the rate-determining step. In [Lex+H]+, the fucose moiety is already in the proximity of the amide group. As a result, this first step of the rearrangement is accelerated, making the ion more prone to the rearrangement – so prone in fact that none of the three experiments records the true spectrum of the Lex trisaccharide. Alternatively, the rearrangement might proceed via the ring-opening mechanism originally proposed by Harvey et. al,[28] which involves a protonation-induced fucose-ring opening step (Figure 3C). In the [BG-H2+H]+, the resulting carbocation is the target for the nucleophilic attack by the adjacent O3 group, which yields a stable intermediate three fused rings. The fucose transfer to O3 is then completed by the ring closing of O5. In the next step, the rotation around the Galβ(1 → 4)GlcNAc would move O6 in the proximity of the fucose and the protonated amide group. Following the same protonation-induced ring-opening mechanism, the fucose moiety can transfer to O6 on the galactose. In this mechanism, [Lex+H]+ is the intermediate, which explains why the spectrum of the Lex ion is not recorded.
Conclusion
In summary, using three MS-based methods – ion mobility-mass spectrometry, radical-directed dissociation MS and cryogenic-ion IR spectroscopy – we investigated the structure and stability of the intact, protonated ions of the human blood group epitopes Lex and BG-H2 and identified the final rearrangement product of fucose migration. First, our data confirm that collision-induced dissociation is not a prerequisite for fucose migration. Similar observations have been made for three unique instruments that were used to characterize the resulting ions, which shows that the substantial structural alterations (i.e. fucose migration) of the ions occur in the front end of the respective instruments, during or immediately after ionization. This implies that similar phenomena are common in most commercial instruments, which usually involve harsher conditions than the custom-built devices used here. Second, the large-scale DFT calculations reveal the structure of the rearrangement product. Surprisingly, this product is neither Lex nor BG-H2. Instead it is a third structure which is common to both of them and carries the fucose moiety attached via the α(1 → 6) glycosidic bond to the terminal galactose. Regardless of the activation conditions, the CCS and spectral fingerprint of this structure match well with that recorded for Lex. This indicates that the rearrangement barrier leading to this structure is particularly low, so low that none of the experiments probes the true Lex structure. The rearrangement of [BG-H2+H]+, on the other hand, requires more activation energy and can therefore be trapped intact in the mass spectrometer. This difference in migration barrier highlights that structural motifs which appear similar at first glance may behave very different in tandem MS experiments – which has broad implications for the MS–based sequencing of glycans and glycopeptides.
Supplementary Material
Acknowledgements
K.G. thanks the Fonds National de la Recherche, Luxembourg, for funding the project GlycoCat (13549747). R.R.J. gratefully acknowledges the funding from the US National Science Foundation (CHE1904577). M.M. gratefully acknowledges the funding from the National Institute of General Medical Sciences (SC2GM135145). K.P. acknowledges generous funding by the European Research Council, ERC-2019-CoG-863934-GlycoSpec. Open Access funding enabled and organized by Projekt DEAL.
References
- [1].Varki A, Glycobiology 2017, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Agre P, Bertozzi C, Bissell M, Campbell KP, Cummings RD, Desai UR, Estes M, Flotte T, Fogleman G, Gage F, Ginsburg D, Gordon JI, Hart G, Hascall V, Kiessling L, Kornfeld S, Lowe J, Magnani J, Mahal LK, Medzhitov R, Roberts RJ, Sackstein R, Sarkar R, Schnaar R, Schwartz N, Varki A, Walt D, Weissman I, J. Clin. Investig. 2016, 126, 405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Ruhaak LR, Xu G, Li Q, Goonatilleke E, Lebrilla CB, Chem. Rev. 2018, 118, 7886–7930; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gray C, Flitsch SL, in Coupling and Decoupling of Diverse Molecular Units in Glycosciences (Eds.: Witczak ZJ, Bielski R), Springer International Publishing: Cham, 2018, pp. 225–267. [Google Scholar]
- [4].Li J, Hsu H-C, Mountz JD, Allen JG, Cell Chem. Biol. 2018, 25, 499–512. [DOI] [PubMed] [Google Scholar]
- [5].Watanabe Y, Bowden TA, Wilson IA, Crispin M, Biochim. Biophys. Acta - Gen. Subj. 2019, 1863, 1480–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Ernst B, Müller DR, Richter WJ, Int. J. Mass Spectrom. Ion Processes 1997, 160, 283–290; [Google Scholar]; b) Wuhrer M, Koeleman CA, Hokke CH, Deelder AM, Rapid Commun. Mass Spectrom. 2006, 20, 1747–L; [DOI] [PubMed] [Google Scholar]; c) Chen X, Flynn GC, Anal. Biochem. 2007, 370, 147–161; [DOI] [PubMed] [Google Scholar]; d) Broberg A, Carbohydr. Res. 2007, 342, 1462–1469; [DOI] [PubMed] [Google Scholar]
- e).Desai N, Thomas DA, Lee J, Gao J, Beauchamp JL, Chem. Sci. 2016, 7, 5390–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Wuhrer M, Deelder AM, van der Burgt YE, Mass Spectrom. Rev. 2011, 30, 664–680; [DOI] [PubMed] [Google Scholar]; b) Hecht ES, Loziuk PL, Muddiman DC, J. Am. Soc. Mass Spectrom. 2017, 28, 729–732; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mucha E, Lettow M, Marianski M, Thomas DA, Struwe WB, Harvey DJ, Meijer G, Seeberger PH, von Helden G, Pagel K, Angew. Chem. Int. Ed. 2018, 57, 7440–7443. [DOI] [PubMed] [Google Scholar]
- [8].Kováčik V, Hirsch J, Kováč P, Heerma W, Thomas-Oates J, Haverkamp J, Mass Spectrom J. 1995, 30, 949–958. [Google Scholar]
- [9].Wuhrer M, Koeleman CA, Deelder AM, Anal. Chem. 2009, 81, 4422–4432. [DOI] [PubMed] [Google Scholar]
- [10].Vachet RW, Bishop BM, Erickson BW, Glish GL, J. Am. Chem. Soc. 1997, 119, 5481–5488. [Google Scholar]
- [11].a) Both P, Green AP, Gray CJ, Šardzík R, Voglmeir J, Fontana C, Austeri M, Rejzek M, Richardson D, Field RA, Widmalm G, Flitsch SL, Eyers CE, Nat. Chem. 2014, 6, 65–74; [DOI] [PubMed] [Google Scholar]; b) Hofmann J, Hahm HS, Seeberger PH, Pagel K, Nature 2015, 526, 241–244. [DOI] [PubMed] [Google Scholar]
- [12].a) Schindler B, Barnes L, Renois G, Gray C, Chambert S, Fort S, Flitsch S, Loison C, Allouche A-R, Compagnon I, Nat. Commun. 2017, 8, 973; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mucha E, González Flórez AI, Marianski M, Thomas DA, Hoffmann W, Struwe WB, Hahm HS, Gewinner S, Schöllkopf W, Seeberger PH, von Helden G, Pagel K, Angew. Chem. Int. Ed. 2017, 56, 11248–11251; [DOI] [PubMed] [Google Scholar]; c) Masellis C, Khanal N, Kamrath MZ, Clemmer DE, Rizzo TR, J. Am. Soc. Mass Spectrom. 2017, 28, 2217–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kirschbaum C, Greis K, Mucha E, Kain L, Deng S, Zappe A, Gewinner S, Schöllkopf W, von Helden G, Meijer G, Savage PB, Marianski M, Teyton L, Pagel K, Nat. Commun. 2021, 12, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Mucha E, Marianski M, Xu F-F, Thomas DA, Meijer G, von Helden G, Seeberger PH, Pagel K, Nat. Commun. 2018, 9, 4174; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Marianski M, Mucha E, Greis K, Moon S, Pardo A, Kirschbaum C, Thomas DA, Meijer G, von Helden G, Gilmore K, Seeberger PH, Pagel K, Angew. Chem. Int. Ed. 2020, 59, 6166–6171; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Greis K, Kirschbaum C, Fittolani G, Mucha E, Chang R, von Helden G, Meijer G, Delbianco M, Seeberger PH, Pagel K, Eur. J. Org. Chem. 2022, e202200255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Zhang X, Julian RR, Int. J. Mass Spectrom. 2014, 372, 22–28; [Google Scholar]; b) Gao J, Thomas DA, Sohn CH, Beauchamp JL, J. Am. Chem. Soc. 2013, 135, 10684–10692. [DOI] [PubMed] [Google Scholar]
- [16].Riggs DL, Hofmann J, Hahm HS, Seeberger PH, Pagel K, Julian RR, Anal. Chem. 2018, 90, 11581–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lettow M, Mucha E, Manz C, Thomas DA, Marianski M, Meijer G, von Helden G, Pagel K, Anal. Bioanal. Chem. 2019, 411, 4637–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Warnke S, von Helden G, Pagel K, Proteomics 2015, 15, 2804–2812; [DOI] [PubMed] [Google Scholar]; b) Thomas DA, Chang R, Mucha E, Lettow M, Greis K, Gewinner S, Schöllkopf W, Meijer G, von Helden G, Phys. Chem. Chem. Phys. 2020, 22, 18400–18413. [DOI] [PubMed] [Google Scholar]
- [19].Gabelica V, Shvartsburg AA, Afonso C, Barran P, Benesch JLP, Bleiholder C, Bowers MT, Bilbao A, Bush MF, Campbell JL, Campuzano IDG, Causon T, Clowers BH, Creaser CS, De Pauw E, Far J, Fernandez-Lima F, Fjeldsted JC, Giles K, Groessl M, Hogan CJ Jr, Hann S, Kim HI, Kurulugama RT, May JC, McLean JA, Pagel K, Richardson K, Ridgeway ME, Rosu F, Sobott F, Thalassinos K, Valentine SJ, Wyttenbach T, Mass Spectrom. Rev. 2019, 38, 291–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Greis K, Kirschbaum C, Taccone MI, Götze M, Gewinner S, Schöllkopf W, Meijer G, von Helden G, Pagel K, Angew. Chem. Int. Ed. 2022, 61, e202115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Scutelnic V, Perez MAS, Marianski M, Warnke S, Gregor A, Rothlisberger U, Bowers MT, Baldauf C, von Helden G, Rizzo TR, Seo J, J. Am. Chem. Soc. 2018, 140, 7554–7560. [DOI] [PubMed] [Google Scholar]
- [22].Rahim S, Yaman M, Marianski M, Manuscript in preparation. [Google Scholar]
- [23].Pracht P, Bohle F, Grimme S, Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [DOI] [PubMed] [Google Scholar]
- [24].Adamo C, Barone V, J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar]
- [25].a) Marianski M, Supady A, Ingram T, Schneider M, Baldauf C, J. Chem. Theory Comput. 2016, 12, 6157–6168; [DOI] [PubMed] [Google Scholar]; b) Bannwarth C, Ehlert S, Grimme S, J. Chem. Theory Comput. 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- [26].von Helden G, Hsu MT, Gotts N, Bowers MT, J. Phys. Chem. 1993, 97, 8182–8192. [Google Scholar]
- [27].Sastre Toraño J, Gagarinov IA, Vos GM, Broszeit F, Srivastava AD, Palmer M, Langridge JI, Aizpurua-Olaizola O, Somovilla VJ, Boons G-J, Angew. Chem. Int. Ed. 2019, 58, 17616–17620. [DOI] [PubMed] [Google Scholar]
- [28].Harvey DJ, Mattu TS, Wormald MR, Royle L, Dwek RA, Rudd PM, Anal. Chem. 2002, 74, 734–740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.