

Abstract
Background
Cuproptosis, a novel identified cell death form induced by copper, is characterized by aggregation of lipoylated mitochondrial enzymes and the destabilization of Fe–S cluster proteins. However, the function and potential clinical value of cuproptosis and cuproptosis-related biomarkers in colorectal cancer (CRC) remain largely unknown.
Methods
A comprehensive multi-omics (transcriptomics, genomics, and single-cell transcriptome) analysis was performed for identifying the influence of 16 cuproptosis-related markers on clinical status, molecular functions and tumor microenvironment (TME) in CRC. A novel cuproptosis-related scoring system (CuproScore) based on cuproptosis-related markers was also constructed to predict the prognosis of CRC individuals, TME and the response to immunotherapy. In addition, our transcriptome cohort of 15 paired CRC tissue, tissue-array, and various assays in 4 kinds of CRC cell lines in vitro were applied for verification.
Results
Cuproptosis-related markers were closely associated with both clinical prognosis and molecular functions. And the cuproptosis-related molecular phenotypes and scoring system (CuproScore) could distinguish and predict the prognosis of CRC patients, TME, and the response to immunotherapy in both public and our transcriptome cohorts. Besides, the expression, function and clinical significance of these markers were also checked and analyzed in CRC cell lines and CRC tissues in our own cohorts.
Conclusions
In conclusion, we indicated that cuproptosis and CPRMs played a significant role in CRC progression and in modeling the TME. Inducing cuproptosis may be a useful tool for tumor therapy in the future.
Keywords: Colorectal Cancer, Cuproptosis, Molecular Phenotypes, CuproScore, Immunotherapy
Graphical Abstract
1. Background
Colorectal cancer (CRC) is one of the most common malignancies in the world, ranking second for incidence (more than 1,900,000 cases) and third for mortality (more than 930,000 cases) worldwide in 2020 [1]. Recently, with the development of comprehensive treatments in CRC, including curative surgery, neoadjuvant chemoradiotherapy, systemic chemotherapy, targeted therapy, the prognosis of CRC individuals has been significantly improved [2], [3]. Furthermore, tumor immunotherapy, especially immune checkpoint inhibitors (ICIs), has shown exciting clinical success in various solid tumor treatments [4]. Unfortunately, the prognosis for individuals diagnosed with CRC at an advanced stage remains poor, and only a few CRC individuals with microsatellite instability high, approximately 4–5% of metastatic colorectal cancer (mCRC) population, could be benefiting from ICIs [5]. Thus, more reliable and accurate biomarkers and potential therapeutic targets should be explored to improve the diagnosis, prognosis, and therapeutic effect of CRC individuals.
Cell death is an inevitable and complicated process in both normal and tumor cells. Different from the other well-known types of regulated cell death (RCD), such as apoptosis [6], autophagy [7], pyroptosis [8], and ferroptosis [9], cuproptosis is a copper-induced cell death reported in a recent study by Tsvetkov et al. [10], characterized by excess intracellular copper, lipoylated mitochondrial enzymes accumulation and Fe-S cluster proteins loss. Besides, cuproptosis has also been considered to have a close association with the tumor metabolism, such as the tricarboxylic acid (TCA) cycle. The metabolic reprogramming occurs throughout the process of malignant transformation, to meet the demands of the tumor microenvironment (TME) and unique tumor cell growth [11], [12]. In addition, the process of metabolic reprogramming also plays central role in CRC to enable tumor proliferation in the hypoxia condition and the tumor immune escape [13], [14], [15]. In previous studies, the functions of several cuproptosis-related molecules (CPRMs) in CRC have been found. DLAT, PDHA1, and PDHB may serve as components of the key enzyme pyruvate dehydrogenase complex in the TCA cycle, which converts pyruvate to acetyl-CoA to drive the TCA cycle [16], [17]. SLC31A1, ATP7A, and ATP7B acted as copper ion transporters in cancer cells [18], [19], [20], [21]. CDKN2A encoded a cell cycle inhibitory protein that typically prevented abnormal cell growth and proliferation [22], [23], [24]. Therefore, cuproptosis is likely to be involved in tumorigenesis and TME in CRC. However, there is few study deep exploring the role of cuproptosis in CRC, and whether these CPRMs could regulate the pathogenesis of CRC remains largely unknown [25], [26].
Therefore, to get a better understanding of cuproptosis in the tumorigenesis and clinicopathological characteristics in CRC individuals, we presented a multi-omic analysis of the CPRMs in CRC, including the clinical status, molecular functions, and tumor immune microenvironment. Our results reveal that cuproptosis plays a significant role in the tumorigenesis of CRC with potential influence on the tumor microenvironment. CuproScore is a potentially powerful scoring system for predicting prognosis and the response to immunotherapies in CRC individuals. These findings will help understand cuproptosis in regulating CRC profiles and provide new directions for therapeutic intervention in the future.
2. Methods
2.1. Collection of data
The pan-cancer RNA sequencing (RNA-seq), corresponding clinical characteristics, somatic mutation, and copy number variation data in the Cancer Genome Atlas (TCGA) datasets were downloaded from the Genomic Data Commons (GDC, https://portal.gdc.cancer.gov/). The RNA-seq data of the normal tissues in the Genotype-Tissue Expression (GTEx) were downloaded from the University of California Santa Cruz (UCSC, https://xenabrowser.net/datapages/). All the RNA-seq data were converted from count values to Transcripts Per Kilobase of exon model per Million mapped reads (TPM) values. Microsatellite Instability(MSI) data of TCGA colorectal cancer patients [27] was obtained by R package ‘TCGAbiolinks’ [28]. The Consensus Molecular Subtypes (CMS) [29] of CRC cohort in TCGA COAD and READ datasets were calculated by R package ‘CMScaller’ (https://github.com/Lothelab/CMScaller) [30]. Besides, information of metastatic or recurrent CRC patients who received 5-Fluorouracil (5-Fu), FOLFOX6, FOLFIRI, Cetuximab, PD-L1, or CTLA-4 treatments were respectively obtained in the Gene-Expression Omnibus (GEO) database (GSE62080, GSE19860, GSE108277, and GSE81005, https://www.ncbi.nlm.nih.gov/geo/) and all the RNA-seq and clinical characteristics were obtained by R package ‘GEOquery’ [31]. In addition, the single-cell RNA-seq data of the GSE132465 cohort was also downloaded from the GEO database. The RNA-seq data of the CRC cell lines were downloaded from the Cancer Cell Line Encyclopedia (CCLE, https://sites.broadinstitute.org/ccle/) [32]. The accessible immunohistochemical images of CRC individuals were obtained from the Human Protein Atlas (HPA, https://www.proteinatlas.org/) database [33]. Furthermore, the complete RNA-seq data and corresponding clinical characteristics of the IMvigor210 cohort [34] (the research cohort of anti-PD-L1 immunotherapy in bladder cancer) and Liu’s cohort [35] (the research cohort of anti-PD-1 immunotherapy in melanoma) were obtained from the link (IMvigor210 cohort, http://research-pub.gene.com/IMvigor210CoreBiologies/; Liu’s cohort, https://github.com/vanallenlab/schadendorf-pd1).
2.2. Generation of cuproptosis-related molecules
16 cuproptosis-related molecules (FDX1, LIPT1, LIAS, DLD, DBT, GCSH, DLST, DLAT, PDHA1, PDHB, SLC31A1, ATP7A, ATP7B, CDKN2A, GLS, and MTF1) were obtained from the previous study [10], including components of the lipoic acid pathway, components of the pyruvate dehydrogenase (PDH) complex and copper transporters. The biological information of these genes was shown in Supplementary Table S1.
2.3. Analysis of cuproptosis-related genes in pan-cancer
For differential expression analysis, the R package ‘DESeq2’ [36] was used to compare the cuproptosis-related gene expression levels between normal and tumor tissue among pan-cancer. The prognostic values of cuproptosis-related genes were determined by univariable Cox regression among CRC and other types of cancer. The R package ‘maftools’ was used to draw a waterfall chart of cuproptosis-related gene somatic mutation and estimate the Tumor Mutation Burden (TMB) of CRC individuals in TCGA datasets [37]. The correlation between cuproptosis-related gene expression and copy number in pan-cancer individuals was calculated by Spearman rank correlation analysis. The gain or loss of copy numbers of cuproptosis-related genes were determined by the total number of genes with copy numbers changing at the focal and arm levels. The correlation of cuproptosis-related gene expression levels was calculated by the R packages “igraph” and “reshape2”, and the protein-protein interaction (PPI) network was conducted to reveal the interaction of proteins among the proteins coding between the 16 cuproptosis-related genes by the STRING database (http://www.string-db.org/). Molecular pathways involved in these cuproptosis-related genes were estimated by the R package ‘ClusterProfiler’ [38] for the gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis.
2.4. Identification of the enrichment scores of gene features
The cuproptosis-related pathway was identified based on the 16 cuproptosis-related genes. The cuproptosis-related pathway enrichment scores of each sample were evaluated by R package ‘GSVA’ [39] through the single-sample gene set enrichment Analysis (ssGSEA) algorithm. Besides, the biological functions of each tumor sample were also quantified by GSVA, and the specific pathway signatures were derived and downloaded from the Hallmark gene sets in the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb/). Then, the ssGSEA pathway scores of cuproptosis together with several other gene features, including Apoptosis, TGF-β signal pathway, Glycolysis, Glutathione metabolism et al., were calculated by R package ‘GSVA’ through ssGSEA algorithm from MSigDB database [40].
2.5. Construction of cuproptosis molecular phenotypes
Based on the expression level of 16 cuproptosis-related molecules, the unsupervised clustering analysis was used to identify the cuproptosis molecular phenotypes in colorectal cancer. The consensus clustering algorithm was applied to determine the optimal clustering numbers and phenotypes of CRC individuals and tumor cells in the TCGA-COREAD and the GSE132465 cohorts by the R package ‘ConsensusClusterPlus’ [41]. Then, Principal Component Analysis (PCA) was used to show the cuproptosis-related molecule expression difference between different consensus phenotypes. In the single-cell levels, the differences in the expression distribution of the 16 cuproptosis-related molecules were analyzed and visualized by the R package “Seruat” [42]. For the bulk RNA-seq data, the overall survival statistical difference between different phenotypes were calculated and visualized by R packages ‘survival’, ‘survminer’, and the variations in the expression levels of 16 cuproptosis-related molecules and the clinical characteristics between different phenotypes were shown by R package ‘ComplexHeatmap’ [43].
2.6. Evaluation of TME immunological characteristics
Immunomodulators, including Major Histocompatibility Complex (MHC), immunoinhibitors, immunostimulators, and chemokines were collected from the previous study [44]. The anti-cancer immune system response has been proven with seven steps: (1) Release of cancer cell antigens, (2) Cancer antigen presentation, (3) Priming and activation, (4) Trafficking of immune cells to tumors, (5) Infiltration of immune cells into tumors, (6) Recognition of cancer cells by T cells, and (7) Killing of cancer cells. In this study, the gene data and the enrichment scores of each step in TCGA-COREAD cohorts were downloaded from the tracking tumor immune phenotype website (http://biocc.hrbmu.edu.cn/TIP/index.jsp) [45], [46], [47]. Besides, the ssGSEA [44], and CIBERSORT [48] methods were used to estimate the immune cell infiltration of CRC individuals in the TCGA-COREAD cohorts and the Immune, Stromal, ESTIMATE scores, and tumor purity were calculated by R package ‘estimate’ [49]. The Hematoxylin-eosin (HE) staining immunophenotype pathology image data (Formalin-fixed paraffin-embedding) in TCGA datasets were obtained from the Cancer Digital Slide Archive (CDSA, https://cancer.digitalslidearchive.org/).
2.7. Construction of the CuproScore
PCA algorithm was applied to construct a scoring system, called Cuproscore, to evaluate the levels of 16 cuproptosis-related molecules in CRC individuals. The positive and negative functions of 16 cuproptosis-related genes were calculated by univariate Cox regression according to the Cox coefficient. Then PCA algorithm was performed to analyze the expression levels of 16 cuproptosis-related molecules, and the principal component 1(PC1) was determined to present as the scores. The CuproScore formula is:
| CuproScore = PC1i - PC1j |
(PC1i means the PC1 of positive Cox coefficient cuproptosis molecules, and PC1j means the PC1 of negative Cox coefficient cuproptosis molecules). Then, the predictive values of CuproScore for the prognosis, molecular functions, and immunotherapy response of CRC individuals were further evaluated.
2.8. Molecular and clinical significance of the CuproScore
Based on the median of CuproScore, the CRC patients in the TCGA-COREAD cohort were divided into the high and low CuproScore groups. Then the differences in the prognosis, mutation feature, molecular functions, and TME immunological characteristics between the two groups were also estimated by the same methods as previously introduced. The immunophenoscores, to predict the response to immunotherapy with CTLA-4 and PD-1 blockers in the TCGA datasets, were downloaded from the Cancer Immunome Database (TCIA) database(https://tcia.at/home) [44]. To further explore the predictive value of CuproScore in anti-PD-L1, anti-PD1, and anti-CTLA4 immunotherapy, the CuproScore algorithm was also applied in the TCGA, IMvigor210, and Liu’s cohorts. And the individuals with stable disease (SD) or progressive disease (PD) as non-responders, while those with complete response (CR) or partial response (PR) as responders.
2.9. High-throughput transcriptome analysis of CRC individuals
The study was approved by the Ethical Committee of Ruijin Hospital with a clinical trial registration number NCT04714814. Among the prospective patient cohort collected, 15 CRC individuals for the High-throughput transcriptome analysis were strictly screened in our center, without any medical treatment before recruitment. Detailed information about them is listed in Supplementary Table S2. Total RNA was isolated from the surgically discarded normal and tumor colorectal tissues of these 15 CRC individuals. Then, the cDNA library was constructed, and high-throughput sequencing was performed at Shanghai Jiao Tong University School of Medicine (Shanghai, China) using the HiSeq 4000 (Illumina, San Diego, CA). And all the sequencing data (including gene expression and clinical information) of our study has been deposited into Mendeley Data (a public and community-supported repository). Anyone with this share link can see our raw data and will be able to download our files (https://data.mendeley.com/datasets/8kgftsbf5k/draft?a=e8eebcba-a487–412c-a387-c315f31300f7).
2.10. Immunohistochemistry (IHC)
Cancer tissue-array including a total of 73-paired normal/tumor CRC individuals was also recruited from Ruijin Hospital (Shanghai, China). IHC training was performed according to standard protocols and the optimum sections of tissue specimens were obtained and deparaffinized. IHC was performed as the following antibodies: CDKN2A (A11651, ABclonal), DLAT (A14530, ABclonal), FDX1 (A9815, ABclonal), ATP7B (19786–1-AP, Proteintech) and GCSH (A13695, ABclonal). Each tissue-array was scored by three independent pathologists using a semi-quantitative method based on the German semi-quantitative scoring system [50].
2.11. Cell lines and cell culture
NCM460, HT29, SW620, HCT116, SW480, and RKO cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). All cells were stored at the Shanghai Institute of Digestive Surgery and cultured in the corresponding medium with 10% fetal bovine serum (FBS), 0.1 mg/ mL streptomycin, and 100 U/ mL penicillin under the atmosphere at 37 °C with 5% CO2.
2.12. Cell viability assay
The cuproptosis inducer Elesclomol+Cu2+ (1:1 ratio, APExBIO) was dissolved in dimethyl sulfoxide (DMSO) to a total concentration of 40 mM. The working concentrations were diluted to 0, 0.75, 1.5, 3, 6, 12, 25, 50, 100, and 200 nM, and three wells were applied for each concentration. After being treated with gradient concentrations of Elesclomol+Cu2+, cell viability was measured by Cell Counting Kit 8 (CCK8, APExBIO), following the standard protocols. Then, the half-maximal inhibitory concentration (IC50) values of each cell line were determined using nonlinear regression.
2.13. RNA isolation and real-time PCR
RNA Isolation and Real-Time PCR were performed as previous methods [51]. The sequences of primers used for qPCR analysis were listed in Supplementary Table S3. The relative mRNA expression levels of the 16 cuproptosis-related were calculated by the 2−ΔΔCt method and normalized against that of GAPDH.
2.14. Western bolt
Western blot was performed as previous methods [51]. Primary antibodies: β-Tubulin (AC030/A12289, ABclonal 1:2000), FDX1 (A9815, ABclonal, 1:1000), SLC31A1 (A0773, ABclonal, 1:1000), DLAT (12362 S, Cell Signaling Technology, 1:1000) and LIAS (11577–1-AP, Proteintech, 1:1000). Secondary antibody: anti-rabbit (30000–0-AP, Proteintech, 1:5000), anti-mouse (10283–1-AP, Proteintech, 1:5000).
2.15. Statistical analysis
All the statistical analysis was conducted by the R software (version: 3.4.0) in this study. All P values of statistical data were based on two-sided statistical tests. P < 0.05 was considered to be statistically significant. Spearman correlation coefficients were used to determine the correlation between variables. To compare two or more groups, Kruskal-Wallis and one-way ANOVA tests were used. The "surv_cutpoint" function in the R package ‘Survminer’ was used to evaluate the critical value of each data set. The Kaplan-Meier method was used to generate survival curves for the subgroups of each data set, and the log-rank (Cox) test was used to determine statistically significant differences.
3. Results
3.1. Pan-cancer analysis of cuproptosis-related molecules (CPRMs)
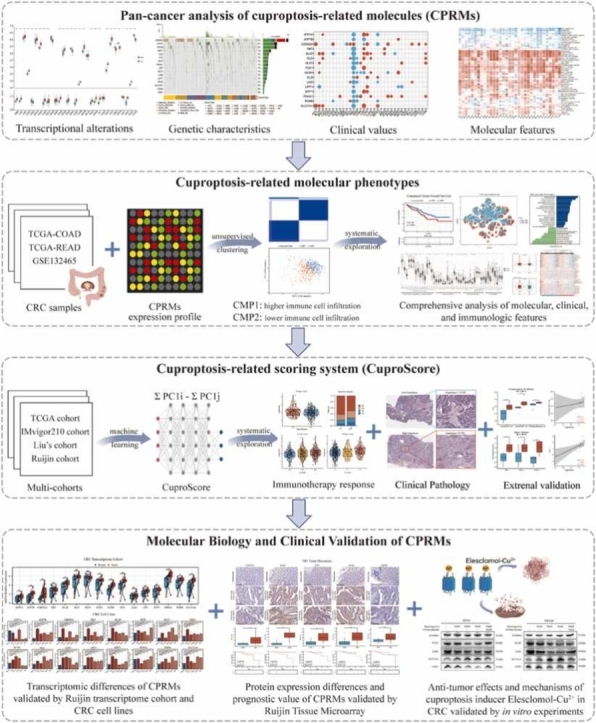
The schematic flowchart of the study was presented in Fig. 1. To explore the potential value of cuproptosis in tumors, the pan-cancer analysis of 16 cuproptosis-related molecules (CPRMs) was performed and the main results were as follows: Firstly, to evaluate the transcriptional changes between the normal and tumor tissues, the variations in the mRNA expression levels of the 16 CPRMs were shown in Fig. 2A and all CPRMs were highly statistical significances in different tumor types in the TCGA pan-cancer cohorts, especially CDKN2A. Secondly, to estimate the genetic variation status of the 16 CPRMs in pan-cancer samples, a waterfall diagram shows the somatic mutation frequency and types of cuproptosis-related genes in Fig. 2B. We found that CDKN2A had the highest mutation frequency of up to 4%, followed by ATP7A (2%) and ATP7B (2%) and the missense mutation was the most common type. The relationship between copy number variation (CNV) and mRNA expression levels of the 16 CPRMs in pan-cancer showed a positive correlation in most cancer types, especially in bladder urothelial carcinoma (TCGA-BLCA), Colon carcinoma (TCGA-COAD), Rectal carcinoma (TCGA-READ), and Lung squamous cell carcinoma (TCGA-LUSC) in Fig. 2 C. Then the prognostic values of cuproptosis-related genes in pan-cancer were also evaluated and the results showed that the CPRMs appeared to be the heterogeneous prognostic values in different cancers, such as most CPRMs played the protective roles in Kidney renal clear cell carcinoma (TCGA-KIRC) while mostly risky roles in brain lower-grade glioma (TCGA-LGG) in Fig. 2D. Besides, in Fig. 2E, we calculated the cuproptosis pathway score (ssGSEA) based on the 16 CPRMs and found there were also statistically significant differences in the score between the normal and tumor samples in pan-cancer, similarly to the situation of mRNA expression levels. Finally, to estimate the potential values of the cuproptosis pathway score, a systematic analysis was performed and a mostly negative correlation between the TME infiltrating cells and cuproptosis pathway score was found in pan-cancer except for the ovarian serous cystadenocarcinoma (TCGA-OV). In the molecular biology analysis, many pathways, such as adipogenesis, protein secretion, PI3K signaling, fatty acid metabolism, and oxidative phosphorylation were shown positively correlated with the cuproptosis pathway score in Fig. 2F.
Fig. 1.

Schematic flowchart of the study design.
Fig. 2.

Transcriptional alterations, genetic characteristics and clinical values of CPRMs in CRC. A. The thermogram shows the different mRNA expression levels of 16 CPRMs between the normal and tumor samples in pan-cancer from TCGA cohorts. T-test: * P < 0.05, * * P < 0.01, * ** P < 0.001. B. The waterfall diagram shows the somatic mutation frequency of 16 CPRMs in pan-cancer. C. The bubble chart shows the correlation between CNV and mRNA expression levels of 16 CPRMs in pan-cancer using Spearman correlation analysis. D. The bubble chart shows the relationship between prognosis and mRNA expression levels of 16 CPRMs in pan-cancer using the Log-rank test. E. The boxplot shows the different cuproptosis ssGSEA scores between the normal and tumor samples in pan-cancer. T-test: * P < 0.05, * * P < 0.01, * ** P < 0.001. F. The thermogram shows the correlation between mRNA expression levels of 16 CPRMs and 28 TME infiltrating cells or 50 common molecular biology pathways in pan-cancer. Spearman correlation analysis: * P < 0.05, * * P < 0.01, * ** P < 0.001.
3.2. Transcriptional alterations, genetic characteristics and clinical values of CPRMs in CRC
To further investigate the mRNA expression levels of 16 CPRMs in CRC bulk samples, a differential analysis was performed between the intestinal epithelium tissues and colorectal cancer tissues in GTEx and TCGA-COREAD cohorts. The results showed that each mRNA expression level of the 16 CPRMs was statistically different between normal and tumor tissues in Fig. 3 A, most of which were up-regulated in tumor samples, except DBT and DLST. In addition, we further explored the protein expression of the 16 CPRMs in HPA database, and found that mRNA expression levels of most CPRMs were consistent with their protein expression, except ATP7B, DBT, PDHB and DLST (Supplementary Fig. S1). Thus, it could be necessary to collect more samples and perform more studies to clarify the expression levels of CPRMs in CRC. Given that the cell lines may play an important role in vitro experiments, radar charts were applied to show the mRNA expression levels of 16 CPRMs in diverse CRC cell lines, demonstrating most CPRMs had sufficient expression abundance in CRC cell lines (Fig. 3B). Then the somatic mutation frequency of CPRMs in 538 CRC individuals in the TCGA-COREAD cohorts was estimated in Fig. 3 C and found that ATP7A (4%) and ATP7B (3%) ranked top mutation frequency compared to others. However, the gain frequency of CNV in ATP7A was the least among CPRMs, while ATP7B ranked first in Fig. 3D-E. And these findings are consistent with the previous report [52]. To further explore the clinical values of CPRMs in CRC, the univariate Cox regression analysis indicated that CDKN2A, DLAT, DLD, and PDHB might have prognostic values in CRC individuals. CDKN2A tended to be a risk factor, and others contributed to the protective factors in Fig. 3 F. And the Kaplan-Meier curves showed the disease-specific survival (DSS), overall survival (OS), and progression-free survival (PFS) statistical discrepancy between high and low CDKN2A or DLAT expression groups in the TCGA-COREAD cohorts (Fig. 3 G). We also found that LIAS, DLST, DLAT, and SLC31A1 had lower mRNA expression levels in the advanced TNM stage CRC compared to the early stage, while ATP7B, CDKN2A, and GLS had a higher level in stage IV CRC (Supplementary Fig. S2A). Furthermore, based on the several cohorts related to drug resistance and drug resistance prediction, we found the expression levels of many CPRMs were likely to be related to the chemotherapy, targeted therapy or immunotherapy resistance in CRC individuals (Fig. 3H). DBT, DLAT, DLST, MTF1, and PDHB may play a different role in resistance depending on therapy, which was meaningful to further explore. For the analysis of the molecular function, these CPRMs were mainly enriched in the oxidoreductase activity and metabolism-related pathways (Supplementary Fig. S2B). For the molecular correlates, DLAT could be the hub molecule and had a positive correlation with other CPRMs (Supplementary Fig. S2C-D). For the immune checkpoints analysis, there was a significant correlation between the CPRMs and immune checkpoint-related gene expression levels (Supplementary Fig. S2E). For the ssGSEA and CIBERSORT analysis, the immune cell infiltration degree was negatively related to the expression levels of most CPRMs, except DBT, DLST, MTF1 and SLC31A1 (Fig. 3I). For the GSVA analysis, ATP7B, CDKN2A and PDHA1 had a negative correlation with most specific well-defined biological states or processes based on Hallmark gene sets, while other CPRMs were the opposite (Fig. 3I).
Fig. 3.

Expression and clinical significance of CPRMs in CRC. A. The boxplot shows the mRNA expression levels of 16 CPRMs between the normal and tumor CRC samples in GTEX and TCGA-COREAD cohorts. T-test: * P < 0.05, * * P < 0.01, * ** P < 0.001. B. The radar charts show the mRNA expression levels of 16 CPRMs in different CRC cell lines in CCLE datasets. C. The waterfall diagram shows the mutation frequency of 16 CPRMs in 538 patients with colorectal cancer in the TCGA-COREAD cohorts. D. The vertical lollipop chart shows the frequencies of CNV gain, loss, and non-CNV among CPRMs in the TCGA-COREAD cohorts. E. The doughnut chart shows the locations of CNV alterations in CPRMs on 23 chromosomes. F. The forest map shows the results of Univariate regression analysis on the average survival rate of 16 CPRMs in the TCGA-COREAD cohorts. G. The Kaplan-Meier curves show disease-specific survival, overall survival, and progression-free interval of CDKN2A and DLAT in the TCGA-COREAD cohorts using the log-rank test. H. The boxplots show the relationship between mRNA expression levels of CPRMs and the chemotherapy, targeted therapy or immunotherapy resistance in CRC individuals. T-test: * P < 0.05, * * P < 0.01, * ** P < 0.001. I. The thermogram shows the correlation between mRNA expression levels of 16 CPRMs and TME infiltrating cells or common molecular biology pathways in CRC individuals. Spearman correlation analysis: * P < 0.05, * * P < 0.01, * ** P < 0.001.
3.3. Identification of two cuproptosis-related molecular phenotypes (CMP) by unsupervised learning in CRC
To explore the role of cuproptosis in CRC individuals, we identified two cuproptosis-related molecular phenotypes (307 cases in CMP1 and 311 cases in CMP2) by unsupervised learning analysis using the R package ‘ConsensusClusterPlus’ (Fig. 4A). The PCA analysis confirmed that the two phenotypes could be distinguished by the expression levels of the 16 CPRMs (Fig. 4B). And patients in CMP1 had a worse prognosis than those in CMP2 in Fig. 4C. The expression levels of cuproptosis molecules and the clinical characteristics were visualized using a thermogram in Fig. 4D and there were significant differences in the expression levels of 15 CPRMs except DLST between the two phenotypes. To further understand the biological discrepancy between the two phenotypes, the GSVA analysis was performed and the results showed that the biological discrepancy mainly focused on the immune-, mechanism- and tumorigenesis-related pathways, such as Interleukin, PD-1, and CTLA4 signaling, Energy, Fatty acid, Glutathione, and Metal ions metabolism, and Cell cycle, p53, Wnt and Kras signaling in Fig. 4E. In addition, there were also significant differences in the ssGSEA enrichment scores between the two phenotypes, including several Cell Death Pathways, Immune Pathways, Tumorigenesis Pathways, and Metabolism Pathways in Fig. 4F.
Fig. 4.

Identification of two cuproptosis-related molecular phenotypes by unsupervised learning in CRC. A. The consensus matrix thermogram defines two cuproptosis phenotypes (k = 2) and shows their correlation area by unsupervised learning. B. The principal component analysis of CPRMs shows a remarkable difference in transcriptomes between the two subtypes. C. The Kaplan-Meier curves for overall survival of two consensus phenotypes in TCGA using the log-rank test. D. The thermogram shows the differences in clinicopathologic features and mRNA expression levels of CPRMs between the two cuproptosis phenotypes. T-test: * P < 0.05, * * P < 0.01, * ** P < 0.001. E. The bar chart shows the results of GSVA analysis of the differentially expressed genes between the two consensus phenotypes. F. The boxplot shows the differences in pathway ssGSEA scores between the two cuproptosis phenotypes. T-test: nsP > 0.05, * P < 0.05, * * P < 0.01, * ** P < 0.001.
3.4. Variations in somatic mutation frequency and immune cell infiltration in two cuproptosis phenotypes
To explore the variations in somatic mutation frequency between two cuproptosis phenotypes, somatic mutation analysis was performed, and the results showed the different mutation frequencies of the common mutant genes in CRC between the CMP1 and CMP2, such as APC (72% vs 80%) and KRAS (45% vs 37%) in Fig. 5 A. Then the forest plot was drawn to show the different gene mutation distributions and samples in CMP1 were associated with higher mutation rates of several mutant genes, including DKK4, CCDC15, and NTAN1(Fig. 5B). Besides, TMB levels were calculated and analyzed in both phenotypes, indicating that TMB levels were significantly higher in CMP1 subtype in Fig. 5C. Based on the results of the above functional enrichment analysis, the immune process and pathways were significantly different between the two phenotypes. Thus, ESTIMATE, CIBERSORT, ssGSEA, and Cancer-immunity Cycle analysis were utilized to calculate the immune cell infiltration degree in each CRC sample based on the RNA-Sequencing data and explore the relationship between cuproptosis phenotype and TME. For the ESTIMATE analysis, samples in CMP1 had the higher immune and ESTIMATE scores which indicated that more immune components were contained (Fig. 5D). For the CIBERSORT and ssGSEA analysis, it could be seen that samples in CMP1 had a higher infiltration degree of several immune cells with dominant anti-tumor activity, such as T cells, CD8+ T cells, central memory CD4+ T cell, T follicular helper cell, Type 17 T helper cell, NK cells, CD56dim natural killer cell, Monocyte, and Neutrophil (Fig. 5E). Anti-tumor immune response has been proven as a series of step-by-step events referred to as the cancer-immune cycle. Our results indicated that these anti-tumor steps were mainly active in CMP1, including Step4 (Trafficking of immune cells to tumors): T cell recruiting, CD4 T cell recruiting, B cell recruiting, and Step5 (Infiltration of immune cells into tumors), while only Th22 cell recruiting in Step4 was enriched in CMP2 (Fig. 5E). These results further confirmed the potential role of cuproptosis in tumor immune cell infiltration. Moreover, we found that CMP1 group had a higher expression of immunoinhibitors, including PD-1, LAG3, and TGFB1 (Fig. 5F). MHC, immunostimulators, and chemokines were also more highly expressed in CMP1 (Supplementary Fig. S3). In conclusion, our results further confirmed that more infiltration of immune cells was contained in the tumor tissue of CMP1 group, while less in that of CMP2 group.
Fig. 5.

Variations in somatic mutation frequency and immune cell infiltration in two cuproptosis phenotypes. A. The waterfall diagrams show genetic alterations of common mutant genes between the two cuproptosis phenotypes. B. Forest plot shows variations in the somatic mutation frequencies between the two cuproptosis phenotypes. Chi-square test: * * P < 0.01, * ** P < 0.001. C. The violin plot shows variations in TMB scores between the two cuproptosis phenotypes. T-test: * P < 0.05. D. The violin plots show variations in TME scores between the two cuproptosis phenotypes using ESTIMATE analysis. T-test: * P < 0.05, * * P < 0.01. E. The thermogram shows variations in immune scores and the frequency of TME infiltrating cells between the two cuproptosis phenotypes. Wilcoxon test: * P < 0.05, * * P < 0.01, * ** P < 0.001. F. The boxplot shows variations in mRNA expression of different kinds of immune checkpoints between the two cuproptosis phenotypes. Wilcoxon test: nsP > 0.05, * P < 0.05, * * P < 0.01, * ** P < 0.001.
3.5. Single-cell transcriptome analysis of CRC cells based on cuproptosis-related molecular phenotypes
Considering the influence of different cell populations in bulk tissue, the differential analysis of the single-cell RNA cohort was performed to investigate the single-cell mRNA expression levels of 16 CPRMs in CRC cells. Consistent with the bulk-seq results, significant expression differences were demonstrated between 1070 normal and 17,469 tumor epithelial cells, as shown in Fig. 6 A. Besides, based on the results of the differential analysis, the t-SNE plots were presented to show the molecular expression of these CPRMs at the single-cell level in Fig. 6 C. Then, to explore whether the cuproptosis-related molecular classification could be also exactly reflected in the CRC single cells, 17,469 CRC epithelial cells were screened from the single-cell cohort, and then also identified two cuproptosis-related molecular phenotypes (7408 single cells in CMP1-like and 10,061 single cells in CMP2-like) by unsupervised learning analysis using the R package ‘ConsensusClusterPlus’ in Fig. 6B. To verify the variations in the proportion and number of molecular and clinical characteristics between the two cuproptosis-related single-cell molecular phenotypes, the stacked histograms were shown in Fig. 6D. For clinical significance, tumor cells in CMP1-like were predominantly derived from TNM stage III&IV, right-hemi colon cancer, poorly or mucinous differentiation CRC individuals. For the CMS subtype, tumor cells in CMP1-like were mainly derived from CMS1, CMS3, or CMS4 subtype CRC individuals through both single-cell and bulk levels. In contrast, tumor cells in CMP2-like were enriched in the CMS2 subtype which indicated a better prognosis. For MSI subtypes, tumor cells in CMP1-like were predominantly clustered into the MSI-H subtype, which intimated a higher probability of sensitivity to immunotherapy. For gene mutation type, tumor cells in CMP1-like were mainly clustered into the KRAS mutation group while tumor cells in CMP2-like were predominantly clustered into APC and TP53 mutation groups. Consistent with the functional enrichment analysis in bulk levels, we found the biological discrepancy was also focused on the immune-, mechanism- and tumorigenesis-related pathways between the two cuproptosis-related single-cell molecular phenotypes, such as ATP metabolic process, immune response, oxidative stress, IL-17 signaling, Glutathione metabolism, Glycolysis and Cell cycle pathways by GO and KEGG analysis in Fig. 6E-F.
Fig. 6.

Single-cell transcriptome analysis of CRC cells based on cuproptosis-related molecular classification. A. The volcano map shows the different single-cell mRNA expression levels of 16 CPRMs between 1070 normal and 17,469 tumor epithelial cells. B. The t-SNE visualization shows the different single-cell distributions between the two cuproptosis phenotypes. C. The t-SNE visualizations show the molecular expression of 16 CPRMs except MTF1. D. The stacked histogram shows variations in the proportion and number of molecular and clinical characteristics between the two cuproptosis single-cell phenotypes. E-F. The bar plots show the results of GO (E) and KEGG (F) analysis of the differentially expressed genes between the two cuproptosis single-cell phenotypes.
3.6. Construction and evaluation of a novel cuproptosis-related scoring system (CuproScore) in CRC
As the above results, cuproptosis may play an important role in the regulation of tumorigenesis, TME, and survival in colorectal cancer. Thus, based on the 16 CPRMs, a novel cuproptosis-related scoring system called CuproScore was constructed to quantify cuproptosis regulation in each individual using the PCA algorithm. In the TCGA-COREAD cohorts, samples in CMP1 had a higer CuproScore than those in CMP2 in Fig. 7A. Meanwhile, the high CuproScore group, divided by median or optimal cut-off score, all had the worse prognosis and the Kaplan-Meier curves were shown in Fig. 7B-C. ROC analysis of the CuproScore demonstrated that the AUCs from 1 to 6 year were all greater than 0.6, indicating that CuproScore may have a certain predictive effect on the survival of CRC patients (Supplementary Fig. S9). For the somatic mutations, different mutation frequencies of the common mutant genes were presented between high and low CuproScore groups, such as APC (72% vs 81%) and KRAS (44% vs 38%), as shown in Fig. 7D. Besides, we also found that the CuproScore of the high TMB group was higher than that of the low TMB group in Fig. 7E. For the molecular pathways, the GSVA analysis was further performed and the results showed that the biological discrepancy also mainly focused on the immune-, mechanism- and tumorigenesis-related pathways, such as Interleukin, TGF-β, and CTLA4 signaling; Energy, Fatty acid, copper homeostasis, and lipid metabolism; Cell cycle, p53, mTOR, Wnt, and Kras signaling in Fig. 7 F. For the TME, we evaluated the correlation between CuproScore and immune cell infiltration degree and found that CD56dim natural killer cell, monocyte, and central memory CD4 T cell had a statistically positive correlation with cuproptosis-related scores, while Th22 cell recruiting tended to be negative in Fig. 7 G. In addition, for the ESTIMATE analysis, the CuproScore also had a statistically positive relationship with TMEscore (Immunescore and ESTIMATEscore) in Fig. 7H. The HE staining immunophenotype also confirmed that the high CuproScore group displayed a higher lymphocyte infiltration and further suggested that there was a positive correlation between cuproptosis and immune cell infiltration in Fig. 7I.
Fig. 7.

Construction and evaluation of a novel cuproptosis-related scoring system (CuproScore) in CRC. A. The violin plot shows variations in CuproScore between the two cuproptosis phenotypes using the Wilcoxon test. B-C. The Kaplan-Meier curves show the significant difference in the survival rate between the high and low CuproScore groups in the TCGA database using median (B) or best (C) cut-off values. D. The waterfall diagrams show genetic alterations of common mutant genes between the high and low CuproScore groups. E. The violin plot shows variations in CuproScore between the high and low TMB groups using the Wilcoxon test. F. The bar chart shows the results of GSVA analysis of the differentially expressed genes between the high and low CuproScore groups. G. The lollipop chart shows the correlation between the CuproScore and the level of immune cell infiltration. H. The scatter plots show the correlation between the CuproScore and TME scores (ESTIMATE and Immune scores) using Spearman correlation analysis. I. The representative images show the variations in pathological HE staining between the high and low CuproScore groups.
3.7. Predictive value of CuproScore for immunotherapy response
Immunotherapies represented by anti-PD-L1, anti-PD1, and anti-CTLA4 have been widely applied in clinical treatments. Considering the molecular and clinical significance of CuproScore in CRC as described above, especially in immune cell infiltration, we next selected several immunotherapy-related cohorts to investigate whether the CuproScore could predict an effective response to these immunotherapies in different types of cancer. For the TCGA-COREAD cohorts, the results showed significant variations in CuproScore between different immunotherapy groups with high or low predicted immunotherapy sensitivity scores in Fig. 8 A. Then, for the IMvigor210 cohort, patients with high CuproScore had a significantly better prognosis compared to those with low CuproScore (Fig. 8B). In addition, patients with high CuproScore were more likely to get an effective clinical response to anti-PD-L1 immunotherapy (Fig. 8C-D). Furthermore, the CuproScore of the immune-inflamed phenotype was significantly higher than that of the immune-desert and immune-excluded phenotypes (Fig. 8E). And the result indicated the CuproScore could be negatively correlated with the rejection of treatment and the desert immunophenotype. It has been proven that the expression level of PD-L1 was strongly correlated with the efficacy of anti-PD-L1 immunotherapy. Thus, the relationship between the CuproScore and the tumor-infiltrating immune cells (IC) and tumor cells (TC) immune types was further analyzed in Fig. 8F-G and found the CuproScore was positively correlated with PD-L1 expression in tumor cells. For Liu’s cohort, consistent with the previous conclusions, the CuproScore could also predict the prognosis of melanoma individuals and their clinical response to the anti-PD-1 immunotherapy (Fig. 8H-I). Taken together, the CuproScore we constructed could be a potentially powerful scoring system for predicting prognosis and the response to immunotherapies in different cancers.
Fig. 8.

Predictive value of CuproScore for immunotherapy response. A. The violin plots show variations in CuproScore between groups with high or low immunotherapy sensitivity scores in the TCGA-COREAD cohorts. B. The Kaplan-Meier curve shows the significant difference in the survival rate between the high and low CuproScore groups in the IMvigor210 cohort using the best cut-off value. C. The violin plot shows variation in CuproScore between groups with different anti-PD-L1 responsiveness. D. The stacked histogram shows variations in the proportion of different anti-PD-L1 responsiveness between the high and low CuproScore groups. E. The violin plot shows variations in CuproScore among three immune phenotypes. F-G. The violin plots show variations in CuproScore among PD-L1 expression of different IC (F) and TC (G). H-I. The Kaplan-Meier curve (H) and stacked histogram (I) show the potential predictive value of CuproScore for anti-PD-L1 immunotherapy in Liu’s dataset.
3.8. Validation of the clinical and molecular value of CuproScore in our transcriptome cohort
Considering over-reliance on the public datasets could destabilize the results, we constructed our transcriptome cohort (15 CRC individuals from Ruijin hospital) to further verify the value of CuproScore. The CuproScore of each sample was calculated based on the above scoring system, and the median CuproScore was utilized to classify samples into high or low CuproScore groups. The clinical characteristics of all these samples were presented in Supplementary Table S2. Consistent with the above results in the TCGA-COREAD cohorts, CRC individuals in the high CuproScore group tended to have higher immune cell infiltrations, including active CD4 T cells, active CD8 T cells, and CD56bright natural killer cells (Fig. 9A). Meanwhile, the immune checkpoint expression levels, such as PD-1, PD-L1, and CTLA4, were also higher in the high CuproScore group (Fig. 9B), and CuproScore had a statistically positive correlation with the immune cell infiltrations (Fig. 9 C) and immune checkpoint expression (Fig. 9D). Then, we further compared the immune checkpoint expression and immune cell infiltration degree in high and low groups of only II stage-CRC patients. And the same tendency of CuproScore in distinguishing immune checkpoint and immune cell infiltration between the same stage of CRC was found despite the small sample size (Supplementary Fig. S6). For the GO and KEGG analysis, the differentially expressed genes between the high and low CuproScore groups were similarly enriched in the immune-, mechanism- and tumorigenesis-related pathways, such as regulation of immune response, T/B cell receptor signaling pathway; Glutamic acid, Metal ion homeostasis, and Methionine metabolism; Cell cycle, and p53 signaling pathways (Fig. 9E-F). For the GSEA analysis, several inflammation-related functions, including Chemokine and IL17 signaling pathways, were up-regulated (Fig. 9G). The HE staining immunophenotype also confirmed that the high CuproScore group displayed a higher lymphocyte infiltration (Fig. 9H).
Fig. 9.

Validation of the clinical and molecular value of CuproScore in our transcriptome cohort. A-B. The boxplot shows the differences in immune cell infiltration (A) and immune checkpoint expression levels (B) between low and high CuproScore groups in CRC individuals from our transcriptome cohort (Ruijin hospital). Wilcoxon test: * P < 0.05, * * P < 0.01, * ** P < 0.001. C-D. The scatter charts with regression lines show the correlation between CuproScore and immune cell infiltration (C), and immune checkpoint expression levels (D) using Spearman correlation analysis. E-F. The bar charts show the results of GO (E) and KEGG (F) enrichment analysis between the high and low CuproScore groups by GSVA. G. The string diagrams show the enrichment pathways between low and high CuproScore groups using GSEA analysis based on the differential expression genes. H. The representative images show the variations in pathological HE staining between the high and low CuproScore groups from our transcriptome cohort.
3.9. Validation of the expression and clinical significance of 16 CPRMs in our cohorts
To address the scientificity and reliability of the above results, the transcriptome cohort, cell lines, and tissue-array of our center (Ruijin hospital) were applied to verify the expression levels and clinical significance of 16 CPRMs in CRC. For the transcriptome cohort, the differential analysis was performed between the paired normal and tumor tissues from 15 CRC individuals in our center. The results showed that the mRNA expression levels of GCSH, DLAT, PDHA1, SLC31A1, ATP7B, CDKN2A, and GLS were statistically different between normal and tumor tissues in Fig. 10 A and all of them were up-regulated in tumor samples consistent with the above results in the TCGA-COREAD cohorts. Then, for the cell line level, the normal colon cell line (NCM460) and five CRC cell lines (HT29, SW620, HCT116, SW480, and RKO) were utilized to verify the 16 CPRMs expression levels in Fig. 10B. Different from the results in TCGA and Ruijin cohorts, the expression levels of DBT and DLST were significantly up-regulated, while ATP7B was downregulated in CRC cell lines, compared to normal colon cell lines. Furthermore, the protein expression levels and clinical significance of hub CPRMs, including FDX1, DLAT, CDKN2A, GCSH and ATP7B were further verified in the tissue-arrays of our center by IHC (Fig. 10C). Consistently, the results showed that the expression of FDX1, DLAT, CDKN2A, GCSH and ATP7B all increased in tumor tissues. Meanwhile, DLAT and CDKN2A were also associated with the prognosis of CRC individuals in our center.
Fig. 10.

Validation of the expression and clinical significance of 16 CPRMs in our cohorts. A. The boxplot shows the different mRNA expression levels of 16 CPRMs between the normal and tumor CRC samples from our transcriptome cohort. T-test: * P < 0.05, * * P < 0.01, * ** P < 0.001. B. The boxplots show the different mRNA expression levels of 16 CPRMs between the normal colon cell line (NCM460) and five CRC cell lines (HT29, SW620, HCT116, SW480, and RKO) using real-time PCR. One-way ANOVA: nsP > 0.05, * P < 0.05, * * P < 0.01, * ** P < 0.001. C. Representative immunohistochemistry images show the different protein expression levels of CDKN2A, DLAT, FDX1, GCSH and ATP7B in CRC tissues and corresponding normal tissues from the CRC tissue-array of Ruijin hospital. The boxplots display the differences in IHC scores for CDKN2A, DLAT, FDX1, GCSH and ATP7B between CRC tissues and corresponding normal tissues from the CRC tissue-array. Wilcoxon test: * ** P < 0.001. The Kaplan-Meier curves show overall survival of CDKN2A, DLAT, FDX1, GCSH and ATP7B in the CRC tissue-array using the log-rank test.
3.10. The effects of cuproptosis inducer Elesclomol-Cu2+ on 16 CPRMs in CRC cell lines
To explore the role of 16 CPRMs in cuproptosis, CRC cell lines HT29 (TP53-R273H mutated), HCT116 (TP53-wt), SW480 (primary-derived) and SW620 (metastatic-derived) were treated with different working concentrations of the cuproptosis inducer Elesclomol-Cu2+. The copper-chelating agent D-penicillamine was acted as the cuproptosis inhibitor and used to perform the restoration experiments of cuproptosis. The results of the cell viability assay indicated that Elesclomol-Cu2+ could effectively inhibit the cell proliferation of HT29 and SW620 underlying a dose-dependent manner. The IC50 of HT29 and SW620 was around 30 nM, while the IC50 of HCT116 and SW480 was around 20 nM (Supplementary Fig. S4). Based on their IC50, different concentrations of Elesclomol-Cu2+ (15, 30, and 45 nM for HT29/SW620; 10, 20, and 30 nM for HCT116/SW480) were applied to treat corresponding cell lines for 24 h. In addition, after different concentration of Elesclomol-Cu2+ treatment for 24 h, 30 µM concentration of D-penicillamine was then used to treat these cell lines for 12 h. Then, the mRNA expression levels of 16 CPRMs in these cell lines were investigated by Real-Time PCR. The results indicated that the mRNA expression levels of 11 CPRMs (FDX1, LIPT1, LIAS, DLD, DBT, GCSH, DLAT, PDHA1, PDHB, ATP7A, and ATP7B) were decreased and 5 CPRMs (DLST, SLC31A1, CDKN2A, GLS, and MTF1) were increased after being treated with Elesclomol-Cu2+ in Fig. 11A-D. However, the mRNA expression level of DLD was not statistically different in the SW480 and SW620 cell line. After the D-penicillamine treatment, the variations in the mRNA expression levels of most CPRMs in these cell lines were reversed and seemed to be returned to normal levels, while a few of them had no significant statistical difference. In addition, consistent with the mRNA expression results, the different protein expression levels of the hub CPRMs (DLAT, LIAS, FDX1 and SLC31A1) were further confirmed by Western Blot and shown in Fig. 11E-F. In summary, the potential effects of cuproptosis inducer Elesclomol-Cu2+ on 16 CPRMs in CRC could be verified by in-vitro experiments.
Fig. 11.

The effects of cuproptosis inducer Elesclomol-Cu2+ on 16 CPRMs in CRC cell lines. A-D. The boxplots show the different mRNA expression levels of 16 CPRMs in HT29 (A), HCT116(B), SW620 (C) and SW480 (D) cell lines after being treated with Elesclomol-Cu2+ and D-penicillamine using real-time PCR. E-F. The western blots and boxplots show the four protein expression levels of hub CPRMs (DLAT, LIAS, FDX1 and SLC31A1) in HT29 (E) and SW620 (F) cell lines after being treated with Elesclomol-Cu2+ and D-penicillamine using Western Blot. One-way ANOVA: nsP > 0.05, * P < 0.05, * * P < 0.01, * ** P < 0.001.
4. Discussion
Recently, besides apoptosis, new forms of regulated cell death have been identified, including necroptosis [53], pyroptosis [54], Autophagy [55], etc. Among them, heavy metal-related RCD has drawn more and more attention in this field, such as ferroptosis and cuproptosis [10], [56]. Heavy metals are essential for normal function, however, like a double-edged sword, either too little or too much can lead to cell death. Tresv demonstrated that cuproptosis, copper-induced RCD, was featured by aggregation of lipoylated mitochondrial enzymes and the destabilization of Fe–S cluster proteins [10]. Several studies have found that cuproptosis-related genes could be closely associated with the development of cancers, such as glioma [57] and hepatocellular carcinoma [58]. However, how copper specifically induces cell death and the potential value of cuproptosis and cuproptosis-related molecules in CRC clinical treatments remain unknown.
In the current study, we aimed to comprehensively explore the role of cuproptosis in CRC. Pan-cancer analysis was applied to explore the molecular and clinical significance of 16 CPRMs in 33 cancer types, including transcriptional changes, genetic variation status, prognostic differences, and molecular functions. Then, TCGA (TCGA-COREAD), GEO (GSE62080, GSE19860, GSE108277, and GSE81005), and single-cell RNA sequencing (GSE132465) and immunotherapy-related (IMvigor210 and Liu’s) cohorts were integrated to systematically evaluate the association between cuproptosis and tumorigenesis, TME, and immunotherapy response in CRC. Two CMPs were identified in both bulk and single-cell RNA sequencing samples in CRC, which were closely related to clinicopathological and TME cell-infiltrating characteristics. Furthermore, a scoring system called CuproScore was established to quantify the expression rate of CPRMs in the individual tumor sample. We further confirmed these results using transcriptome cohort of CRC tissues, cell lines, and tissue-array of our center.
Copper is an essential microelement to maintain normal physiological [59]. At the same time, the copper imbalance is closely associated with a variety of human diseases, including Wilson's disease [60], cardiovascular disorders [61], and cancers [62]. Due to the unique energy and metabolic demands in tumors, copper has been proven to play dual roles in tumor initiation and tumor progression, especially in TME. Recently, Tsvetkov et al. have found a novel form of cell death caused by copper ionophores, called cuproptosis. 16 CPRMs have been identified to regulate the process of cuproptosis. Indeed, heavy metal-related RCD is not uncommon. Ferroptosis, an iron-dependent cell death, was firstly reported in 2012 [9] and a growing number of studies have evidenced the potential value of ferroptosis and ferroptosis-related molecules in tumors, especially in tumorigenesis, tumor therapy, and tumor immunity [63], [64], [65]. Distinct from ferroptosis, cuproptosis is another type of ion-metabolism-related RCD, which was dependent on mitochondrial respiration and the TCA cycle, rather than ROS or mitochondrial ROS [66]. In the current study, the pan-cancer analysis showed that the CPRMs expression was deregulated between normal and tumor tissues in 33 cancer species, which also closely associated with both clinical prognosis and molecular functions, consistent with the previous study [67].
Copper is mainly (50–70%) absorbed by the colonic epithelium [68]. Compared to the normal colonic epithelium, the copper metabolism has been reprogrammed and the expression levels of copper transport and metabolism molecules are also distinctly changed in CRC, leading to the severe imbalance of intratumoral copper and facilitating tumor progression, invasion, and metastasis [25]. Copper overload has been proven to be a profound inducer of cuproptosis [10]. Therefore, it could be more meaningful to explore the relationship between cuproptosis and CRC. In this study, most CPRMs were found up-regulated in CRC, and four of them (CDKN2A, DLAT, DLD, and PDHB) were demonstrated with prognostic value. Meanwhile, the expression levels of LIAS, DLST, DLAT, SLC31A1, and ATP7B were related to the TNM stages in CRC individuals. Furthermore, CPRMs expression levels were associated with the pathological characteristics, tumorigenesis-related pathways, tumor immune microenvironment, and tumor resistance in CRC. Surprisingly, we found that DLAT could be a potential marker in CRC, especially in drug-resistance. CRC individuals with higher DLAT expression are more likely resistant to chemotherapy and immunotherapy. Interestingly, prognostic analysis revealed that the expression of DLAT seems to be positively correlated with the prognosis of CRC individuals, serving as a protective factor. In the process of exploring the mechanisms of tumor occurrence and development, the discrepancies and even paradoxes in the expression and function of cancer genes were a common and critical phenomenon. Some explanations suggest that dual-role or pleiotropic effects of genes may be related to their efficacy in different tumor microenvironments and/or through different signaling pathways [69], [70]. Moreover, the present research fails to analyze the regulatory changes in gene alternative splicing and protein modification during tumor occurrence and development, thus overlooking the potential roles played by distinct alternative splicing isoforms and protein modifications in gene expression efficacy variances [71], [72], [73]. DLAT, a subunit of the mitochondrial pyruvate dehydrogenase complex [74], plays an important role in tumorigenesis and carbohydrate metabolism, acting as a core metabolic node that promotes the TCA cycle and mediates pyruvate oxidation in cancers [75]. The aggregation of DLAT may also lead to the cuproptosis in lung cancer cells [10]. In our study, the potential value of DLAT has been demonstrated in CRC individuals and weather DLAT may exhibit distinct alternative splicing isoforms and protein modifications in tumors, which warrants further exploration in future studies.
As a result of the integration of molecular changes, molecular classification has been emerging as a useful tool in clinical treatments for tumor patients. The status of MSI-H/dMMR or CMS of CRC patients is highly significant for therapy option and prognosis prediction [76]. Based on the robust advancement of Next Generation Sequencing technology and machine learning, by which we constructed the cuproptosis-related molecular subtyping in this study, CMP1, and CMP2. We claimed that there were obviously differences in molecular functions, TNM stages, and prognosis between these two phenotypes of CRC patients. The differentially expressed genes between two phenotypes were obviously enriched in the tumor immune-related and tumor metabolism-related pathways, indicating diverse TME existed in different subtypes. The results of single-cell transcriptome analysis also confirmed a similar tendency. Despite the recent progress in tumor immunotherapy, there is a great heterogeneity in the efficacy of immunotherapy in CRC individuals, which highlights that CRC may have a special TME different from other tumors. TME is a complex tissue structure composed of the extracellular matrix, blood vessels, immune cells, and fibroblasts [77]. Immune cells, the major cellular components of TME, include various types of anti-tumor immune cells (T cells, B cells, NK cells, and Dendritic cells) and pro-tumor immune cells (Tregs, MDSC, and Macrophages) [78], [79], which have been proven to play a significant role in tumor development, progression and therapeutic resistance. Therefore, it is meaningful to further explore the potential influence of cuproptosis on TME. TMB is an important indicator to evaluate the effect of immunotherapy and is positively correlated with immunotherapy response to tumor individuals [80]. In our results, we found that CRC individuals in CMP1 were presented with higher TMB than those in CMP2.
Increasing evidence has shown that the accumulative infiltration of T cells, NK cells, and Dendritic cells suggests a positive role in the immune defense of CRC [81], [82]. Besides, a recent study revealed that B cells also play an important role in therapeutic responses to ICIs by altering T cell activation and function [83]. A recent study has revealed a cuproptosis-related molecular pattern associated with the TME phenotype [52]. And we also observed more infiltration of immune cells (CD4+ T cell, CD8+ T cell, NK cell, B cell, Dendritic cell, and Neutrophil et.al.) in CMP1, indicating cuproptosis was involved in the tumor immune microenvironment and has a stronger immunoactivated effect of CRC individuals in CMP1. ICIs are a revolutionary breakthrough in tumor therapy, usually, higher expression of PD-1 or PD-L1 indicates better sensitivity to ICIs. We also found individuals in CMP1 have higher expression levels of ICIs, indicating better immunotherapy response. Unfortunately, the relationship between tumor immune microenvironment and the prognosis of tumor patients is still controversial, especially in CRC [84], [85]. In our study, we found that CRC individuals in CMP1 had a poor prognosis while higher immune cell infiltrations. To our mind, the possible reasons for this result are as follows: (1) CRC has a unique tumor and immune microenvironment different from other solid tumors, leading to the small proportion of CRC individuals benefiting from immunotherapy [86], [87]; (2) The immune microenvironment among tumor patients is highly variable, which is even heterogenous in different regions of the same tumor tissue [88]. Evidence showed that nearly one-third of tumors show different immune infiltration [89]; (3) Anti-tumor activity is dependent on tumor antigen presentation and tumor cell recognition [90]. It is necessary to clarify whether the infiltrated immune cells can recognize antigens and play a role in killing tumors. (4) With the detailed IHC analysis of solid tumors, three classifications of tumor immunophenotypes have emerged: ‘hot’ or ‘immune-inflamed’ tumors (with pronounced immune cell infiltrations in the tumor core), ‘immune-excluded’ tumors (showing immune infiltrates at the tumor boundaries), and ‘cold’ or immune-desert’ tumors (with no immune cell infiltration in tumor) [91], [92]. While recent studies have confirmed that the average immune cell infiltration around the tumor is higher than in the tumor core [93], [94], the concept of tumor immunophenotypes has been ignored in these studies. Meanwhile, the common transcriptome/bulk sequencing technique cannot effectively distinguish ‘immune-inflamed’ and ‘immune-excluded’ tumors. Several spatially resolved ways or techniques, such as spatial transcriptomics, will be performed to close the knowledge gap in the future.
To quantify the expression level of 16 CPRMs in each patient as much as possible, a scoring system, CuproScore, was also constructed based on the PCA algorithm, which was further comprehensively utilized in several tumors, including CRC, bladder and kidney cancer, to assess its clinical value. Our results demonstrated patients in CMP1 often presented higher CuproScore, consistent with the molecular phenotype of cuproptosis (CMP1 and CMP2), higher CuproScore was closely related to worse prognosis, higher TMB and more immune cell infiltration. Similar to the results in the TCIA dataset, higher CuproScore indicated better sensitivity to anti-PD-1, anti-PD-L1, or anti-CTLA-4 ICIs in the TCGA dataset. Unfortunately, there was no research cohort of immunotherapy available with open access in CRC. Thus, we checked the predictive capability of CuproScore in other tumor immunotherapy. The same trends of CuproScore were also observed in IMvigor210 and Liu’s cohorts. Therefore, we speculate that cuproptosis may be related to immunotherapy in different types of tumor patients and the combination of cuproptosis inducers and ICIs could be a potential therapeutic strategy. In summary, CuproScore is a reliable method to predictive immune infiltration and could distinguish which patients may benefit from immunotherapy in different types of tumors.
Despite the recent developments in bioinformatics that have provided great convenience in biological science research, there are still limitations by this method, such as over-reliance on the public databases, lack of enough biological and clinical validation, and ignoring differences in ethnicities [52]. Therefore, to ensure our study is more scientific and convincing, we used the tissue transcriptome, the tissue-array, and the in vitro experiments of our cohort to verify the above results. According to our transcriptome cohort and HE staining immunophenotype results, CRC individuals in the high CuproScore group may have more immune cell infiltrations (CD4+ T cell, CD8+ T cell, and NK cell), higher expressions levels of immune checkpoint (PD-1, PD-L1, and CTLA-4) and more lymphocyte infiltrations of pathological specimens. Based on the results of enrichment analysis, the differentially expressed genes were similarly enriched in the immune-, mechanism- and tumorigenesis-related pathways. We also used the tissue-array and the in vitro experiments to verify the expression and clinical significance of 16 CPRMs in our center. It is noteworthy that DLAT and CDKN2A were associated with the prognosis of CRC individuals in both public and our cohorts. Considering the little understanding of the role of cuproptosis in cancers, whether cuproptosis inducers could be developed as effective anti-tumor therapies, such as Elesclomol-Cu2+, etc., remains largely unknown. Therefore, we applied Elesclomol-Cu2+ to trigger cuproptosis in two CRC cell lines, HT-29 and SW-620. And we detected significant lethal toxicity in both CRC cell lines by Elesclomol-Cu2+ at low concentration, revealing IC50 value was both around 30 nM. To evaluate whether these 16 CPRMs play key roles in cuproptosis of CRC, the expression levels of CPRMs were further tested after treatment with different concentrations of Elesclomol-Cu2+. Not surprisingly, most of these CPRMs were dysregulated at mRNA or protein levels, while the underlying mechanism remained unknown. Therefore, cuproptosis inducer could be a novel therapeutic strategy in CRC and these CPRMs could be potential targets for inducing tumor cell cuproptosis personalized anti-tumor therapy. As mentioned above, mitochondrial respiration was required for cuproptosis, while glucose-induced glycolysis showed less sensitivity to cuproptosis. Therefore, there is a hint to future tumor therapy that inhibition of glucose-related metabolism may sensitize tumor cells to cuproptosis while still impairing the tumor's malignant behavior. In addition, the Elesclomol, or other copper ionophores, may be of benefit to the tumors with high expression of lipoylated mitochondrial proteins, or with tolerance to conventional apoptosis-induced therapy. And we expect that our study could promote the development of new therapeutic or combined therapeutic strategies in the future.
Several limitations have been recognized in our study. The novel CMP and scoring system (CuproScore) were constructed based on the retrospective data from several databases, and further prospective studies are necessary to testify their clinical value. To minimize the influence of tumor heterogeneity on outcomes, we have used different databases, including public datasets, our transcriptome cohort, and tissue-array, there could be still some impact. Therefore, we are also collecting an expanded cohort of patients from multiple centers to validate these results. Although the tumor killing effects of Elesclomol-Cu2+ has been proven in our study, the effectiveness of the combination of cuproptosis inducer with other drugs, such as chemotherapy drugs, targeted drugs or ICIs, for CRC patients is still unclear and meaningful to be further clarified.
In summary, our study suggested that cuproptosis played an important role in CRC development. CuproScore was a reliable scoring system for remodeling tumor microenvironment and response to immunotherapy in CRC. Cuproptosis could be a promising therapeutic target and a useful tool for therapeutic interventions and possible combination treatments in CRC patients.
Ethics approval and consent to participate
The study was approved by the Ethical Committee of Ruijin Hospital with a clinical trial registration number NCT04714814. All participants gave their written informed consent.
Funding
This study was supported by grants from the National Nature Science Foundation of China (NSFC) (Grant No. 82273344, 82103207 and 82072614), Shanghai Science and Technology Commission (20JC1410100), Multicenter trial of Shanghai Jiao Tong University School of Medicine (Grant No. DLY201504), Research Physician Program of Shanghai Jiao Tong University School of Medicine (Grant No. 826304).
CRediT authorship contribution statement
S.Z., M.Z., and J.S. served as corresponding authors and designed the study. Y.S., X.F. and X.Y. contributed equally to this work and should be considered co-first authors. X.Y., S.L., X.Z., and L.H. charged the data curation and coordinated the research activities. Y.S. and X.F. collected the data, performed the major analysis and interpreted the data. S.Z. and J.S. supervised the study. S.Z., M.Z., and J.S. verified the replication and reproducibility of results. Z.S., Y.S., and X.F. drafted the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
We appreciate the patients who have participated in TCGA, GEO, and Ruijin cohorts.
Consent for publication
The authors give the consent to publish the work.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.06.011.
Contributor Information
Sen Zhang, Email: zs01d43@rjh.com.cn.
Minhua Zheng, Email: zmhtiger@yeah.net.
Jing Sun, Email: sj11788@rjh.com.cn.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Veenstra C.M., Krauss J.C. Emerging systemic therapies for colorectal cancer. Clin Colon Rectal Surg. 2018;31:179–191. doi: 10.1055/s-0037-1602238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gbolahan O., O'Neil B. Update on systemic therapy for colorectal cancer: biologics take sides. Transl Gastroenterol Hepatol. 2019;4:9. doi: 10.21037/tgh.2019.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomasek J., Kiss I. [Immunotherapy of Colorectal and Anal Carcinoma]. Klin Onkol 30: 62–65. [DOI] [PubMed]
- 6.Green D.R. Cancer and apoptosis: who is built to last? Cancer Cell. 2017;31:2–4. doi: 10.1016/j.ccell.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y.G., Codogno P., Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. 2021;22:733–750. doi: 10.1038/s41580-021-00392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rana N., Privitera G., Kondolf H.C., Bulek K., Lechuga S., De Salvo C., et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell. 2022;185:283–298. doi: 10.1016/j.cell.2021.12.024. e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsvetkov P., Coy S., Petrova B., Dreishpoon M., Verma A., Abdusamad M., et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–1261. doi: 10.1126/science.abf0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Reyes I., Chandel N.S. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21:669–680. doi: 10.1038/s41568-021-00378-6. [DOI] [PubMed] [Google Scholar]
- 12.Dey P., Kimmelman A.C., DePinho R.A. Metabolic codependencies in the tumor microenvironment. Cancer Discov. 2021;11:1067–1081. doi: 10.1158/2159-8290.CD-20-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nenkov M., Ma Y., Gassler N., Chen Y. Metabolic reprogramming of colorectal cancer cells and the microenvironment: implication for therapy. Int J Mol Sci. 2021;22 doi: 10.3390/ijms22126262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neitzel C., Demuth P., Wittmann S., Fahrer J. Targeting altered energy metabolism in colorectal cancer: oncogenic reprogramming, the central role of the TCA cycle and therapeutic opportunities. Cancers. 2020;12 doi: 10.3390/cancers12071731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin F., Yang R., Wei Y., Wang D., Zhu Y., Wang X., et al. HIF-1alpha-induced miR-23a approximately 27a approximately 24 cluster promotes colorectal cancer progression via reprogramming metabolism. Cancer Lett. 2019;440–441:211–222. doi: 10.1016/j.canlet.2018.10.025. [DOI] [PubMed] [Google Scholar]
- 16.Even S., Garrigues C., Loubiere P., Lindley N.D., Cocaign-Bousquet M. Pyruvate metabolism in Lactococcus lactis is dependent upon glyceraldehyde-3-phosphate dehydrogenase activity. Metab Eng. 1999;1:198–205. doi: 10.1006/mben.1999.0120. [DOI] [PubMed] [Google Scholar]
- 17.Yang C., Lee D., Zhang M.S., Tse A.P., Wei L., Bao M.H., et al. Genome-wide CRISPR/Cas9 library screening revealed dietary restriction of glutamine in combination with inhibition of pyruvate metabolism as effective liver cancer treatment. Adv Sci. 2022;9 doi: 10.1002/advs.202202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., Hodgkinson V., Zhu S., Weisman G.A., Petris M.J. Advances in the understanding of mammalian copper transporters. Adv Nutr. 2011;2:129–137. doi: 10.3945/an.110.000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masaldan S., Clatworthy S.A.S., Gamell C., Smith Z.M., Francis P.S., Denoyer D., et al. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol. 2018;16:322–331. doi: 10.1016/j.redox.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cutler D. Crucial copper transporter. Trends Pharm Sci. 2001;22:400. doi: 10.1016/s0165-6147(00)01779-x. [DOI] [PubMed] [Google Scholar]
- 21.Tadini-Buoninsegni F., Bartolommei G., Moncelli M.R., Inesi G., Galliani A., Sinisi M., et al. Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B. Angew Chem Int Ed Engl. 2014;53:1297–1301. doi: 10.1002/anie.201307718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatzistergos K.E., Williams A.R., Dykxhoorn D., Bellio M.A., Yu W., Hare J.M. Tumor suppressors RB1 and CDKN2a cooperatively regulate cell-cycle progression and differentiation during cardiomyocyte development and repair. Circ Res. 2019;124:1184–1197. doi: 10.1161/CIRCRESAHA.118.314063. [DOI] [PubMed] [Google Scholar]
- 23.Liggett W.H., Jr., Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998;16:1197–1206. doi: 10.1200/JCO.1998.16.3.1197. [DOI] [PubMed] [Google Scholar]
- 24.Clurman B.E., Groudine M. The CDKN2A tumor-suppressor locus--a tale of two proteins. N Engl J Med. 1998;338:910–912. doi: 10.1056/NEJM199803263381312. [DOI] [PubMed] [Google Scholar]
- 25.Aubert L., Nandagopal N., Steinhart Z., Lavoie G., Nourreddine S., Berman J., et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat Commun. 2020;11 doi: 10.1038/s41467-020-17549-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Z., Sha G., Zhang W., Zhang Z., Liu T., Wang D., et al. The huge potential of targeting copper status in the treatment of colorectal cancer. Clin Transl Oncol. 2023 doi: 10.1007/s12094-023-03107-7. [DOI] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D., et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guinney J., Dienstmann R., Wang X., de Reynies A., Schlicker A., Soneson C., et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eide P.W., Bruun J., Lothe R.A., Sveen A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci Rep. 2017;7 doi: 10.1038/s41598-017-16747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis S., Meltzer P.S. GEOquery: a bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 32.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347 doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 34.Balar A.V., Galsky M.D., Rosenberg J.E., Powles T., Petrylak D.P., Bellmunt J., et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389:67–76. doi: 10.1016/S0140-6736(16)32455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu D., Schilling B., Liu D., Sucker A., Livingstone E., Jerby-Arnon L., et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med. 2019;25:1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15 doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayakonda A., Lin D.C., Assenov Y., Plass C., Koeffler H.P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–1756. doi: 10.1101/gr.239244.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14 doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akbani R., Ng P.K., Werner H.M., Shahmoradgoli M., Zhang F., Ju Z., et al. A pan-cancer proteomic perspective on the cancer genome atlas. Nat Commun. 2014;5 doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587. doi: 10.1016/j.cell.2021.04.048. e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 44.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 45.Chen D.S., Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 46.Chen X., Chen H., Yao H., Zhao K., Zhang Y., He D., et al. Turning up the heat on non-immunoreactive tumors: pyroptosis influences the tumor immune microenvironment in bladder cancer. Oncogene. 2021;40:6381–6393. doi: 10.1038/s41388-021-02024-9. [DOI] [PubMed] [Google Scholar]
- 47.Xiao Y., Ma D., Zhao S., Suo C., Shi J., Xue M.Z., et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin Cancer Res. 2019;25:5002–5014. doi: 10.1158/1078-0432.CCR-18-3524. [DOI] [PubMed] [Google Scholar]
- 48.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshihara K., Shahmoradgoli M., Martinez E., Vegesna R., Kim H., Torres-Garcia W., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4 doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koo C.L., Kok L.F., Lee M.Y., Wu T.S., Cheng Y.W., Hsu J.D., et al. Scoring mechanisms of p16INK4a immunohistochemistry based on either independent nucleic stain or mixed cytoplasmic with nucleic expression can significantly signal to distinguish between endocervical and endometrial adenocarcinomas in a tissue microarray study. J Transl Med. 2009;7 doi: 10.1186/1479-5876-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shao Y., Jia H., Li S., Huang L., Aikemu B., Yang G., et al. Comprehensive analysis of ferroptosis-related markers for the clinical and biological value in gastric cancer. Oxid Med Cell Longev. 2021;2021 doi: 10.1155/2021/7007933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Z., Zhao Q., Song W., Weng J., Li S., Guo T., et al. A novel cuproptosis-related molecular pattern and its tumor microenvironment characterization in colorectal cancer. Front Immunol (Orig Res) 2022;13 doi: 10.3389/fimmu.2022.940774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong Y.N., Guy C., Olauson H., Becker J.U., Yang M., Fitzgerald P., et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169:286–300. doi: 10.1016/j.cell.2017.03.020. e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z., Zhang Y., Xia S., Kong Q., Li S., Liu X., et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–420. doi: 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miyamoto S. Autophagy and cardiac aging. Cell Death Differ. 2019;26:653–664. doi: 10.1038/s41418-019-0286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shao Y., Jia H., Huang L., Li S., Wang C., Aikemu B., et al. An original ferroptosis-related gene signature effectively predicts the prognosis and clinical status for colorectal cancer patients. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.711776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ouyang Z., Zhang H., Lin W., Su J., Wang X. Bioinformatic profiling identifies the glutaminase to be a potential novel cuproptosis-related biomarker for glioma. Front Cell Dev Biol. 2022;10 doi: 10.3389/fcell.2022.982439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu J., Wang S., Li Z., Qin W., Tong Q., Liu C., et al. Comprehensive multiomics analysis of cuproptosis-related gene characteristics in hepatocellular carcinoma. Front Genet. 2022;13 doi: 10.3389/fgene.2022.942387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trammell R., Rajabimoghadam K., Garcia-Bosch I. Copper-promoted functionalization of organic molecules: from biologically relevant Cu/O2 model systems to organometallic transformations. Chem Rev. 2019;119:2954–3031. doi: 10.1021/acs.chemrev.8b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee S., Barin G., Ackerman C.M., Muchenditsi A., Xu J., Reimer J.A., et al. Copper capture in a thioether-functionalized porous polymer applied to the detection of Wilson's disease. J Am Chem Soc. 2016;138:7603–7609. doi: 10.1021/jacs.6b02515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fukai T., Ushio-Fukai M., Kaplan J.H. Copper transporters and copper chaperones: roles in cardiovascular physiology and disease. Am J Physiol Cell Physiol. 2018;315:C186–C201. doi: 10.1152/ajpcell.00132.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Z., Zhou R., Zhao Y., Pan Y., Liang H., Zhang J.S., et al. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif. 2019;52 doi: 10.1111/cpr.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu H., Ye D., Ren M., Zhang H., Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med. 2021;27:856–867. doi: 10.1016/j.molmed.2021.06.014. [DOI] [PubMed] [Google Scholar]
- 64.Xu G., Wang H., Li X., Huang R., Luo L. Recent progress on targeting ferroptosis for cancer therapy. Biochem Pharm. 2021;190 doi: 10.1016/j.bcp.2021.114584. [DOI] [PubMed] [Google Scholar]
- 65.Yang J., Xu J., Zhang B., Tan Z., Meng Q., Hua J., et al. Ferroptosis: at the crossroad of gemcitabine resistance and tumorigenesis in pancreatic cancer. Int J Mol Sci. 2021;22 doi: 10.3390/ijms222010944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tang D., Chen X., Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32:417–418. doi: 10.1038/s41422-022-00653-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bian Z., Fan R., Xie L. A novel cuproptosis-related prognostic gene signature and validation of differential expression in clear cell renal cell carcinoma. Genes. 2022;13 doi: 10.3390/genes13050851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arendsen L.P., Thakar R., Sultan A.H. The use of copper as an antimicrobial agent in health care, including obstetrics and gynecology. Clin Microbiol Rev. 2019;32 doi: 10.1128/CMR.00125-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adashek J.J., Kato S., Lippman S.M., Kurzrock R. The paradox of cancer genes in non-malignant conditions: implications for precision medicine. Genome Med. 2020;12 doi: 10.1186/s13073-020-0714-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liyanage C., Fernando A., Batra J. Differential roles of protease isoforms in the tumor microenvironment. Cancer Metastas-- Rev. 2019;38:389–415. doi: 10.1007/s10555-019-09816-2. [DOI] [PubMed] [Google Scholar]
- 71.Lin S., Xu H., Qin L., Pang M., Wang Z., Gu M., et al. UHRF1/DNMT1-MZF1 axis-modulated intragenic site-specific CpGI methylation confers divergent expression and opposing functions of PRSS3 isoforms in lung cancer. Acta Pharm Sin B. 2023;13:2086–2106. doi: 10.1016/j.apsb.2023.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin S., Wang X., Pan Y., Tian R., Lin B., Jiang G., et al. Transcription factor myeloid zinc-finger 1 suppresses human gastric carcinogenesis by interacting with metallothionein 2A. Clin Cancer Res. 2019;25:1050–1062. doi: 10.1158/1078-0432.CCR-18-1281. [DOI] [PubMed] [Google Scholar]
- 73.Carter B., Zhao K. The epigenetic basis of cellular heterogeneity. Nat Rev Genet. 2021;22:235–250. doi: 10.1038/s41576-020-00300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lacroix M., Rodier G., Kirsh O., Houles T., Delpech H., Seyran B., et al. E4F1 controls a transcriptional program essential for pyruvate dehydrogenase activity. Proc Natl Acad Sci USA. 2016;113:10998–11003. doi: 10.1073/pnas.1602754113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goh W.Q., Ow G.S., Kuznetsov V.A., Chong S., Lim Y.P. DLAT subunit of the pyruvate dehydrogenase complex is upregulated in gastric cancer-implications in cancer therapy. Am J Transl Res. 2015;7:1140–1151. [PMC free article] [PubMed] [Google Scholar]
- 76.Le D.T., Kim T.W., Van Cutsem E., Geva R., Jager D., Hara H., et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38:11–19. doi: 10.1200/JCO.19.02107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spill F., Reynolds D.S., Kamm R.D., Zaman M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol. 2016;40:41–48. doi: 10.1016/j.copbio.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Davidov V., Jensen G., Mai S., Chen S.H., Pan P.Y. Analyzing one cell at a TIME: analysis of myeloid cell contributions in the tumor immune microenvironment. Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.01842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chiu D.K., Tse A.P., Xu I.M., Di Cui J., Lai R.K., Li L.L., et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8 doi: 10.1038/s41467-017-00530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Dijk N., Funt S.A., Blank C.U., Powles T., Rosenberg J.E., van der Heijden M.S. The cancer immunogram as a framework for personalized immunotherapy in urothelial cancer. Eur Urol. 2019;75:435–444. doi: 10.1016/j.eururo.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mokarram P., Albokashy M., Zarghooni M., Moosavi M.A., Sepehri Z., Chen Q.M., et al. New frontiers in the treatment of colorectal cancer: Autophagy and the unfolded protein response as promising targets. Autophagy. 2017;13:781–819. doi: 10.1080/15548627.2017.1290751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 83.Helmink B.A., Reddy S.M., Gao J., Zhang S., Basar R., Thakur R., et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dennison J.B., Shahmoradgoli M., Liu W., Ju Z., Meric-Bernstam F., Perou C.M., et al. High intratumoral stromal content defines reactive breast cancer as a low-risk breast cancer subtype. Clin Cancer Res. 2016;22:5068–5078. doi: 10.1158/1078-0432.CCR-16-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Perez-Villamil B., Romera-Lopez A., Hernandez-Prieto S., Lopez-Campos G., Calles A., Lopez-Asenjo J.A., et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behavior. BMC Cancer. 2012;12 doi: 10.1186/1471-2407-12-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shi G., Yang Q., Zhang Y., Jiang Q., Lin Y., Yang S., et al. Modulating the tumor microenvironment via oncolytic viruses and CSF-1R inhibition synergistically enhances anti-PD-1 immunotherapy. Mol Ther. 2019;27:244–260. doi: 10.1016/j.ymthe.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Momen-Heravi F., Babic A., Tworoger S.S., Zhang L., Wu K., Smith-Warner S.A., et al. Periodontal disease, tooth loss and colorectal cancer risk: results from the Nurses' Health Study. Int J Cancer. 2017;140:646–652. doi: 10.1002/ijc.30486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Niemeijer A.N., Leung D., Huisman M.C., Bahce I., Hoekstra O.S., van Dongen G., et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat Commun. 2018;9 doi: 10.1038/s41467-018-07131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rosenthal R., Cadieux E.L., Salgado R., Bakir M.A., Moore D.A., Hiley C.T., et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479–485. doi: 10.1038/s41586-019-1032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saha D., Martuza R.L., Rabkin S.D. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32:253–267. doi: 10.1016/j.ccell.2017.07.006. e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen D.S., Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 92.Kather J.N., Weis C.A., Bianconi F., Melchers S.M., Schad L.R., Gaiser T., et al. Multi-class texture analysis in colorectal cancer histology. Sci Rep. 2016;6 doi: 10.1038/srep27988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mlecnik B., Bindea G., Angell H.K., Maby P., Angelova M., Tougeron D., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 94.Bindea G., Mlecnik B., Tosolini M., Kirilovsky A., Waldner M., Obenauf A.C., et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material