

Summary
Cell classes in the human retina are highly heterogeneous with their abundance varying by several orders of magnitude. Here, we generated and integrated a multi-omics single-cell atlas of the adult human retina, including more than 250,000 nuclei for single-nuclei RNA-seq and 137,000 nuclei for single-nuclei ATAC-seq. Cross-species comparison of the retina atlas among human, monkey, mice, and chicken revealed relatively conserved and non-conserved types. Interestingly, the overall cell heterogeneity in primate retina decreases compared with that of rodent and chicken retina. Through integrative analysis, we identified 35,000 distal cis-element-gene pairs, constructed transcription factor (TF)-target regulons for more than 200 TFs, and partitioned the TFs into distinct co-active modules. We also revealed the heterogeneity of the cis-element-gene relationships in different cell types, even from the same class. Taken together, we present a comprehensive single-cell multi-omics atlas of the human retina as a resource that enables systematic molecular characterization at individual cell-type resolution.
Keywords: human retina, single-cell multi-omics, cross-species analysis, gene regulation
Graphical abstract
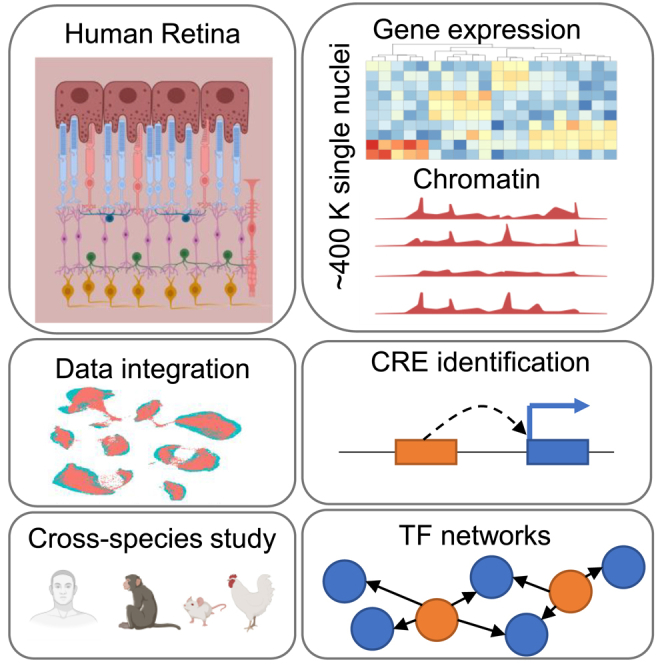
Highlights
-
•
Targeted enrichment of rare cell types in the human retina
-
•
Cross-species analysis reveals different conservation levels among cell types
-
•
Cis- and trans-regulatory elements identified through integrative analysis
-
•
Heterogeneity in regulatory elements for the same gene in different cell types
Liang et al. report a comprehensive multi-omics single-cell atlas with nearly 400,000 nuclei and 69 cell types in adult human retina. They identify regulatory elements that are specific to cell classes and cell types through integrative analysis. The dataset enables molecular characterization of the human retina at individual cell-type level.
Introduction
The vertebrate retina is a multi-layer neuronal structure that converts light to electrical signals that are transmitted to the brain.1 The retina is composed of six major neuronal classes, rod, cone, bipolar, horizontal, amacrine, and retinal ganglion cells (RGCs), along with several non-neuronal cell types, such as Müller glia.1,2 Although the overall structure and major cell classes of the retina are similar across all vertebrates, there are significant differences in cytoarchitecture, synaptic connections, number of cell types in each class, and the retinal vascular bed between species as well as a complete absence of the macula in most non-primate species.3,4 Recent single-cell transcriptomic studies of the retina have consistently indicated that the number and classification of amacrine and RGC types vary greatly across different species, providing a potential underlying mechanism of the retinal structure, circuitry, and functional divergence across species.5,6,7,8 Thus, conclusions drawn from studies on model organisms may not reflect the situation of the human retina. The human retina is composed of a highly heterogeneous population of cells, and it contains an estimated 60 neuronal types based on morphology, function, and most recently, single-cell transcriptional profiles.5,9,10,11 The abundance of each cell type is highly variable and ranges from over 60% to less than 0.05% of the cell population. As a result, a combination of both an increase in the number of cells profiled and targeted enrichment of rare cell types is required to build a comprehensive cell atlas of the human retina.
Beyond single-cell transcriptomics, obtaining the epigenomic landscape at single-cell resolution is critical for gaining insights into the dynamics of gene expression regulation and identifying candidate regulatory elements and variants that affect transcription. Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) has emerged as an ideal method for profiling open chromatin regions, which contain most of the active regulatory elements for gene expression, in cells.12 Recent advancements have made it possible to perform ATAC-seq to profile open chromatin regions for individual cells.13,14 By combining single-nuclei ATAC-seq data with single-nucleus transcriptomics data, it is possible to systematically map gene cis-regulatory elements (CREs) for each cell type and elucidate the gene-regulatory landscape of the retina. These putative regulatory elements will be useful for identifying and prioritizing non-coding mutations associated with retinal disorders.15,16 Furthermore, a large portion of genome-wide association study hits have mapped to potential gene-regulatory regions rather than the coding region of the genome.17,18 Thus, obtaining a high-quality chromatin landscape along with transcriptomic profiles at single-cell resolution is a critical step toward a better characterization of human retina biology and diseases at the molecular level. Several recent approaches reported single-cell level open chromatin profiles with human retina19,20,21,22; however, none of these studies included extensive cell-type classification, so in general, the open chromatin profiling for retina is still at the cell class level, instead of cell-type level.
In this report, we built a single-cell multi-omics map of the human retina by profiling transcriptomes of more than 250,000 nuclei and open chromatin from more than 137,000 nuclei. In total, more than 70 cell types, including 68 neuronal types, were identified from this dataset. By integrating the single-nuclei ATAC-seq (snATAC-seq) and single-nuclei RNA sequencing (snRNA-seq) data, open chromatin profiles were identified for each cell type. Cross-species comparison between humans, monkeys, mice, and chicken revealed various levels of similarity for the different cell types among these species. Counter-intuitively, we found that the sequence conservation of differentially accessible regions (DARs) could not explain the relative conservation of the cell types across species. Candidate CREs were identified and most of them showed strong cell-type specificity. We also showed that the regulatory relationship between genome regions and genes is only partially shared among cell types, which suggested that evaluation of gene regulation at cell-type level is required. Last, we constructed TF-target regulons and demonstrated that the TFs could potentially work in modules. We were able to annotate the TF modules and infer the hub regulators for the modules. In summary, our study has generated a comprehensive single-cell multi-omics atlas that enables the in-depth characterization of the human retina at specific cell-type resolution.
Results
The single-cell multi-omics atlas of human retina
To generate a comprehensive cell atlas and identify most cell types of the human retina, we performed snRNA-seq and snATAC-seq on six and 23 healthy donors, respectively (Tables 1 and 2). It is known that more than 60 cell types exist in the primate retina.5 Since the distribution of retinal types is not uniform, some of the cell types are exceedingly rare. For instance, at least 18 RGC types have been classified in the human retina based on morphology,23 but the total number of RGCs only accounts for approximately 1% of the cell population in the retina.24 In addition, the proportions of RGC types differ significantly, and some RGC types, such as intrinsically photosensitive RGCs (ipRGCs), make up less than 0.01% of retinal cells.25 Similarly, it has been estimated that more than 30 types of Amacrine cells (ACs) exist in the primate retina.5 Therefore, to profile these rare cell types, enriching RGCs and ACs before profiling could be very necessary.
Table 1.
Information of the donors from whom the retina tissue was obtained and profiled
| Donor-ID | Ethnicity | Sex | Age | Total cell number | Central | Peripheral | Peripheral (sorted) |
|---|---|---|---|---|---|---|---|
| D001-12 | European | F | 78 | 17,664 | 0 | 0 | 17,664 |
| 19D013 | European | F | 78 | 45,905 | 41,345 | 0 | 4,560 |
| 19D014 | European | M | 84 | 45,118 | 38,497 | 0 | 6,621 |
| 19D015 | Hispanic | M | 73 | 101,089 | 57,579 | 8,283 | 35,227 |
| 19D016 | European | M | 83 | 19,645 | 19,645 | 0 | 0 |
| 17D013 | European | M | 65 | 5,470 | 0 | 0 | 5,470 |
Table 2.
Donor information and number of nuclei sequenced from each sample for single-nuclei ATAC-seq
| Donor-ID | Ethnicity | Sex | Age | Total cell number |
|---|---|---|---|---|
| 19D013 | European | F | 78 | 21,085 |
| 19D014 | European | M | 84 | 7,334 |
| 19D016 | European | M | 83 | 16,255 |
| 19_D007 | Caucasian | M | 66 | 3,577 |
| 19_D003 | Caucasian | M | 86 | 2,606 |
| 19_D005 | Hispanic | M | 53 | 6,527 |
| 19_D006 | Caucasian | M | 90 | 5,526 |
| 19_D008 | Hispanic | F | 64 | 4,489 |
| 19_D009 | Hispanic | M | 68 | 4,815 |
| 19_D010 | Caucasian | F | 88 | 5,334 |
| 19_D011 | Caucasian | F | 90 | 7,137 |
| 19_D019 | Caucasian | F | 82 | 5,570 |
| D005_13 | Caucasian | M | 80 | 4,266 |
| D009_13 | Asian | M | 85 | 3,854 |
| D013_13 | Caucasian | M | 69 | 4,480 |
| D017_13 | Caucasian | M | 65 | 5,295 |
| D018_13 | Caucasian | F | 91 | 1,693 |
| D019_13 | Caucasian | M | 82 | 6,281 |
| D021_13 | Caucasian | M | 86 | 2,408 |
| D026_13 | Caucasian | M | 81 | 6,632 |
| D027_13 | Caucasian | M | 77 | 4,606 |
| D028_13 | Caucasian | M | 67 | 8,018 |
NeuN is a nuclear envelope protein encoded by the RBFOX3 gene that has been reported to be highly expressed in a subset of neuronal cells.26 In the mouse retina, NeuN was shown to be highly expressed in RGCs and ACs but lowly expressed in photoreceptors.27 Antibody staining showed that NeuN showed a similar pattern in the human retina to the mice retina (Figure S1). Thus, we devised a strategy to profile cells from the retina, as illustrated in Figure 1A. Specifically, the retina was first separated into two regions, the macular/foveal and the peripheral. For the macular/foveal region, as cell proportion was relatively even, we directly profiled nuclei without enrichment. In contrast, for the retinal peripheral region, nuclei are fractioned based on the NeuN staining. As shown in Figure S1B, nuclei with high NeuN signal (top 5%, denoted as NeuNT) and moderately high NeuN signal (from 5% to 10%, denoted as NeuNM) are collected for snRNA profiling. The NeuNT group showed the highest enrichment of RGCs and ACs and the NeuNM fraction is enriched of ACs but not RGCs compared with the unenriched peripheral retina sample (Figure S1C). Given that snRNA-seq, instead of snATAC-seq, is the major approach for us to perform the cell-type classification, we decided to only collect snATAC-seq data from the macula region, which already has a relatively balanced cell-type distribution (Figure 1A).
Figure 1.

Overview of the single-cell multi-omics atlas of human adult retina
(A) The study design of this work. The retina samples were first split into the central and peripheral parts and rare cell enrichment was performed for peripheral retina. The snRNA-seq and snATAC-seq data were first processed separately and then integrated for analysis.
(B) Two-dimensional embeddings (UMAP) for snRNA-seq (left) and snATAC-seq (right) data. Each data point represents a cell, and the color represents the annotated cell class.
(C) The number of each cell class from the snRNA-seq (top) and snATAC-seq (bottom).
(D) The gene expression and gene accessibility of reported retinal cell class marker genes in the snRNA-seq and snATAC-seq data.
(E) Heatmap of the correlation between the open chromatin profiles of each retinal cell class and those of other human tissues.
We first performed the clustering and cell-type annotation on the snRNA-seq and snATAC-seq separately (Figures 1B and 1C) and all the major cell types showed well isolated tight clusters. In addition, cluster heterogeneity is visible for some cell classes with multiple types, such as ACs and bipolar cells (BCs). For both datasets, rod cells remain the most abundant major type (Figure 1C), while we were also able to profile more than 50,000 ACs and BCs, and 10,000 RGCs. Known markers were used to perform the cell-type annotation and as is shown in Figure 1D, we visualized the expression and gene body accessibility of the same set of markers (PDE6A for rod, NETO1 for BC, ARR3 for cone, ONECUT1 for HC, RLBP1 for MG, RBPMS for RGC, TFAP2B for AC, GFAP for astrocyte, and CD74 for microglia). In both datasets, all the markers showed quite distinct and expected distribution among cell types. To further evaluate the data quality of the snATAC-seq data, we split the data based on major cell types and compare them with published bulk ATAC-seq data (Figure 1E) of human tissues, including aorta, adrenal gland, pancreas, stomach, thyroid gland, hippocampus neuron, hippocampus glia, cerebellum, spinal cord, macula, and retina.28 The datasets primarily formed three groups, with one exclusively for the bulk retina, bulk macula, bulk spinal cord, and single-cell retina data, one for the hippocampus and the cerebellum, and the other for all the rest of the tissues. Interestingly, although all are from the central nervous system, the spinal cord data grouped close to the retina while the cerebellum and hippocampus data did form an individual cluster. This observation highlights the substantial epigenetic differences between the retina and the brain. Moreover, the bulk retina clustered closer to the photoreceptor cells while the bulk macula is closer to interneurons, which is expected and likely driven by the different cell-type proportions of the bulk and macula samples.
Transcriptomic and open chromatin profiles of retinal cell types
To obtain the profiles for both the transcriptome and open chromatin modality for retinal cell types, we adopted a strategy that we first annotate types using the RNA-seq data solely, because of the larger number of cells, and then use the annotated RNA-seq data as a reference to annotate the ATAC-seq data (Figure 2A). To determine the optimal parameters for clustering, a combination of two parameters, the “number of nearest neighbors” and the “Leiden resolution,” that gives the highest Silhouette score is identified (Figure S2). As a result, we identified 36 ACs, 14 BCs, 2 HCs, 13 RGCs, three photoreceptors (rod and two cone types), and two non-neuronal cells (MG and astrocyte) in our snRNA-seq data, totaling 70 cell types (Figure S3). We then compared this classification result with a previously published human retina transcriptome atlas.9 BCs and HCs showed high consistency between the two independent datasets with adjusted-rand-index (ARI) close to 1, while ACs showed some differences (we detected 10 more AC subtypes in the current dataset). Through the cross-dataset comparison, we are able to identify several well-studied AC types, such as the starburst AC (AC7), the CAI/CAII ACs (AC34), and VG3 AC (AC28), which all showed one-to-one matching. RGCs have the most differences (Figure S4). This is likely because both datasets are underpowered and have reached sufficient coverage for the rarest RGC types. Nevertheless, two OPN4 positive clusters in our dataset showed one-to-one matching to the two ipRGC clusters of the previous study, and their marker genes, EOMES and LMO2, were also consistent (Figure S5). It has been estimated that ipRGCs accounted for approximately 1% of all the RGCs, which sets the limit of resolution in our presented dataset. We also found substantial differences in the relative proportion of cell types between the central and peripheral retina (Figure S6). Given that the major peripheral retina nuclei underwent anti-NeuN enrichment in our snRNA-seq profiling experiments, we also investigated if this step accounts for some levels of the peripheral/central cell proportion differences (Figure S7). We showed that AC types with higher expression of RBFOX3 (NeuN) tend to have a higher relative proportion in the peripheral retina compared with the central retina, while RGC types did not show this effect. Thus, we cannot rule out the possibility that the anti-NeuN enrichment contributes to the different proportions of ACs between the two retina regions observed in our data.
Figure 2.

Classification and multi-omics integration of retinal cell types
(A) The analysis strategy to perform classification and multi-omics integration of retinal cell types. We perform sub-clustering solely using snRNA-seq data first and leverage published single-cell RNA-seq data of human or model organisms to assist the annotation of the cell types. We then annotate the snATAC-seq data using the annotated snRNA-seq data as the reference.
(B) Two-dimensional embeddings (UMAP) for bipolar cells of the ATAC (top) and RNA (bottom) modality.
(C) Heatmap representing the similarity between the differentially expressed genes and the differentially accessible genes from each bipolar cell type.
(D) Demonstration of the consistency between the differentially accessible regions (DARs, left) and their nearest genes (right) among bipolar cell types.
(E) Phylogenetic tree representing the overall similarity of bipolar cell types among four species: human (gray), monkey (yellow), mouse (blue), and chicken (green).
(F) Boxplot showing the PhastCons score of DARs of each bipolar cell type. The DARs were partitioned to “gene body,” “intergenic,” or “promoter” before the visualization. The center line of the boxplot shows the median of the data; the box limits show the upper and lower quartiles; the whiskers show 1.5 times interquartile ranges.
With the annotated types from the snRNA-seq data, we performed data integration to co-embed the snRNA-seq and snATAC-seq data followed by the label transfer using the Seurat “transfer anchors” framework.29 As is shown in Figure 2B, BC types were well separated in the co-embedding space, and the predicted labels of the snATAC-seq cells match well with snRNA-seq clusters. To further evaluate the reliability of this approach, we first identified differentially expressed genes (DEGs) among cell types using the snRNA-seq data and snATAC-seq data (imputed gene expression) separately and then computed the pairwise Jaccard similarity between the two DEG lists (Figure 2C). The assumption is that if the label prediction is accurate, signatures detected from one modality of a type should be largely consistent with the ones from the other modality. For most types, the two lists generally showed one-to-one matching (Figure 2C, Figure S8). We also visualized the DAR peaks of BC types and showed that their nearest genes displayed a similar pattern in the BC snRNA-seq data. Both results supported the reliability of the snATAC-seq annotation of our data. Thus, we obtained the open chromatin profile of the retina at the individual cell-type level. With this dataset, we computed the DEGs and DARs for cell classes and types (Table S1). The DARs for rarer cell types, mostly ACs and RGCs, were not reported by previous studies.
Cross-species comparison between our dataset and the cell atlas of monkey,5 mouse,6,8,30,31 and chicken7 is performed. Given that no ATAC-seq-based study on human or any model organisms has reached type level, only snRNA-seq data is used. We first co-embedded the data of all four species into the same latent space, followed by clustering them based on their average distances in the space (Figure 2D). Examination of the result suggests that the cross-species matching of cell types is largely consistent with a previous study,7 while more accurate matching can be achieved for some cell types. For example, the BP-19 from the chicken BCs has been annotated as the chicken rod bipolar cell (RBC). In our phylogenetic tree, this type formed a cluster with the RBCs of the other three species, indicating that the RBC is a highly conserved type, while this BP-19 clustered together with human and monkey flat midget bipolar cells in the previous study.7 We observed that human and monkey BCs and HCs generally formed one-to-one matching, while ACs show more complex patterns (Figures 2E, S9). It is worth noting that even for BCs, some interesting matching patterns are observed. For example, human DB3b and DB5 clustered only with the monkey counterpart, indicating that they could be primate-specific types. We were also able to identify marker genes of DB3b (MEIS2) and DB5 (QPCT) for human and monkey BCs. These genes were either not detected or appeared non-specific in mouse or chicken BCs (Figure S10). Similarly, some mouse or chicken bipolar cell clusters are without closely related clusters from human or monkey data, indicating they are likely to be species specific as well. We were then interested that whether the relative conservation of cell types is related to the sequence conservation of their signature genes. We computed the PhastCons scores,32 an evaluation of the sequence conservation, for each of the DARs of bipolar cell types, and then visualized the scores of the DARs after partitioning them based on the genome distribution: gene body, promoter region, and intergenic regions (Figure 2F). The sequence conservation in promoter regions is constantly higher than those of the other regions. Interestingly, it was not observed that highly conserved types, such as RBCs, showed a higher sequence conservation score in DAR. This result suggested that activity rather than the sequence of the regulatory elements drive the difference among cell types.
Identification of CREs through integrative analysis of snRNA-seq and snATAC-seq data
The integration of the snRNA-seq and snATAC-seq allows the imputation of gene expression levels for each cell in the snATAC-seq data. Thus, we leveraged this information to compute the correlation between each gene and relatively proximal peaks (Figure 3A). For each gene, open chromatin regions detected by snATAC-seq within 250 kb from the gene transcription start site are tested (TSS). With a cutoff at 0.3 for the Pearson’s correlation coefficient, more than 35,000 peak-gene pairs were identified as putative enhancers (Table S2, Figure 3B), and they generally showed a closer distance to the gene TSS and a higher sequence conservation (Figure 3C). We also compared the putative enhancer list with the major cell-type DARs and found that the list enriches DARs compared with other proximal peaks (within 250 kb) (Figure 3D). Also, as is shown in Figure 3E, both these putative enhancers and their linked genes displayed high levels of cell-type specificity.
Figure 3.

Identification of putative enhancers by linking peak to genes
(A) Demonstration of the idea of correlating the gene expression with the proximal peak accessibility. When observing the relationship between a peak and a gene among cells, it is possible that they are positively, negatively, or neutrally correlated.
(B) Volcano plot showing the overall distribution of peak-gene links in two dimensions: their Pearson’s correlation coefficient and negative log-transformed q-values (p values with false discovery rate correction). Red data points highlight the links we considered as putative enhancers.
(C) Boxplot showing the comparison between the putative enhancers and other proximal peaks on their distance to linked gene and sequence conservation. Wilcoxon tests were performed to evaluate the difference between the groups. The center line of the boxplot shows the median of the data; the box limits show the upper and lower quartiles; the whiskers show 1.5 times interquartile ranges.
(D) Bar plot showing the comparison between the putative enhancers and other proximal peaks on their overlapping with major cell class DARs.
(E) Heatmap showing the relative expression of genes linked to peaks and their linked genes across different cell types. The gene expression level and peak accessibility level were both normalized between 0 and 1.
(F) The peak-gene correlation between chr12:6959945-6962406 and GNB3 in all bipolar cell types. For each panel, each data point represents a donor and the red dashed-line represents the linear fit of the correlation between the peak and the gene. The Pearson’s correlation coefficients were labeled on each panel.
Correlating peak accessibility to gene expression across cells has been incorporated in different analysis frameworks as a method to identify regulatory regions.33,34 However, it has not been well discussed what groups of cells should be used as the input. A simple question could be whether to use all the cells as the input or only use cells from the same cell type as the input. To obtain a core set of putative enhancers, we used all the cells as the input, and we reason that this approach will allow for detecting stronger signals since a much larger number of cells are used. However, given the complexity of the retinal types, we hypothesized that while some peaks have consistent activities, other peaks might display different or even opposite activities among cell types of the same class. Thus, we performed the peak-gene correlation analysis for each BC type separately. To account for the sparsity of the data, especially the ATAC-seq data, we computed the correlation at the pseudo-bulk level, by combining all the cells of the same type from one sample to a single pseudo-bulk data point, followed by the calculation of Pearson’s correlation coefficient. For each peak-gene pair, the mean and standard deviation of Pearson’s correlation coefficient across the cell types is calculated (Figure S11A). The coefficient did not show clear biases at the cell-type level due to the different cell numbers sampled for each cell (Figure S11B). Both constant and highly heterogeneous peak-gene pairs have been identified (Table S3). As an example of a heterogeneous peak-gene link (chr12:6959945-6962406; GNB3; Figure 3F), seven of the BC types showed a positive correlation while six showed a negative. As an example of constant link (chr3: 61548584-61550601; PTPRG; Figure S11C), 10 of 13 BC types showed a positive correlation with coefficients above 0.2 while the other three showed positive to neutral level correlation. With these results, we showed cases that the activity of CREs could differ among cell types of the same cell class, therefore underscoring the importance of obtaining high-resolution chromatin profiling.
Integrative analysis of transcription factor functions and cell-type specificity in the retina
We then inferred transcription factor (TF) activities for retina cells leveraging both the transcriptome and open chromatin information (Figure S12). We first created “meta cells” for each type based on their similarity in latent space (20 cells into one meta cell) and used the meta cells as the input. Our approach was based on the SCENIC pipeline,35 with a modification that we trimmed the initial TF-target regulon based on the appearance of the TF motif in proximal peaks of the potential target gene (Figure S12). We then scored the TF activity for each meta cell using the AUCell method (Figure 4A). It could be observed that most of the TFs (200) showed certain levels of cell-type specificity (Figure 4A). The number of target genes differed drastically across the TFs, with a median of 196 targets per TF. For each target gene, the median of the number of TFs was six (Figure 4B). The full TF-target list can be found in Table S4.
Figure 4.

Inference of TF activity and annotation of TF modules
(A) Heatmap representing the TF activities (row) for each meta cell (column). The TF activities were first calculated using the AUCell framework and then normalized between 0 and 1.
(B) Basic metrics of the TF-target regulons. The upper and bottom panels showed the distribution of number of targets per TF and numbers of TF regulators per target. Median values are labeled.
(C) Heatmap depicting the TF-TF correlations. Hierarchical clustering was first made, and modules were detected by cutting the tree to K groups (K = 10 here).
(D) The activity of TFs in each module in each cell class. The TF modules were then annotated based on the observed specificity of the module. Previously studied TFs are also listed. The center line of the boxplot shows the median of the data; the box limits show the upper and lower quartiles; the whiskers show 1.5 times interquartile ranges.
We then computed the correlation between TFs using their activity as the input (Figure 4C) and grouped them into 10 modules. Each TF module is annotated based on its activity in classes (Figure 4D). We found that rather than being specific at individual cell class level, most of the TF modules accounted for combinations of cell classes, such as the ones for photoreceptor-bipolar (module 3), AC-RGC (module 1), non-photoreceptor (module 9), etc. There were also two modules (5/6) being less specific. The full list of the partition of TF modules and the TF activity at the major cell class level can be found in Table S5. It is worth mentioning that substructures did appear in some of the relatively large modules, accounting for a finer cell class driven separation, such as module 1 (Figure S13A). Finally, we investigated the TF-TF regulatory relationships by building a directed network for the TFs. The out-degree is a measurement of the number of direct targets for a TF in this network, and we reason that the TFs with high out-degree could be potential hub regulators (Figure S13B). We selected the TFs with out-degree larger than 15 and calculated the overlapping between their targets and the TFs in each of the 10 modules (Table S6 and Figure S13C). Some well-studied TFs were identified. For example, PAX6 appeared as a potential hub regulator for module 1, which was the AC/RGC module. Also, CRX, OTX2, and NRL appeared as potential hub regulators for module 2, which was the photoreceptor module. These examples highlight the reliability of the analysis, while other less studied TFs still require further study.
Discussion
Comprehensive single-cell multi-omics atlas of the human retina
In this study, we have generated a comprehensive single-cell multi-omics atlas of the human retina that includes snRNA-seq for more than 250,000 nuclei and snATAC-seq for more than 137,000 nuclei. To capture the rare cell types in the retina, we have sorted nuclei based on NeuN gradient and collected fractions that are enriched for ACs and RGCs, which will be insufficiently sampled without such enrichment. This combined strategy resulted in an atlas that is improved over the previous published dataset in several aspects. First, the cell portion is better normalized for cell types with high heterogeneity, including AC, BC, and RGC, comprising 21.5%, 20.8%, and 4.1% of the cell population for this dataset, respectively. Consequently, this allows the detection of rare cell types, such as the two ipRGC types, which are estimated to comprise only 0.01% of all retinal cells. Second, the number of cell types identified in this study is 69, including 67 neuronal types, which is higher than previously published studies on the human retina. This is likely to represent a nearly complete catalog of all cell types in the retina. Third, this study is further strengthened with the first large-scale snATAC-seq dataset in the retina with 137,000 nuclei profiled.
Cross-species comparison among cell types
As the key light-sensing organ, the evolution of the eye is fascinating and the morphology and structure of the eye across metazoans are highly diverse. Vertebrate animals share the camera-like eye structure, which contains a multi-layer neuronal structure, the retina, whose function is to capture the light and convert it to an electric signal that is transmitted to the brain. The major cell types in the retina, which includes photoreceptor, bipolar, horizontal, amacrine, and RGCs are conserved across vertebrate animals. With the human retina cell atlas and previously published studies on model organisms, we could conduct cross-species comparisons among human, monkey, chicken, and mouse at cell-type level. This approach was first taken in a previous study,7 and we applied a more comprehensive dataset and a more dedicated computational framework (LIGER). Interestingly, we found that the degree of conservation differs among different cell types with increased divergence observed as we move from the outer nuclear to the inner nuclear layer of the retina. Specifically, conservation decreased from the outer to the inner retina, with cell types at the outer nuclear layer of the retina (photoreceptors) being the most conserved and cell types in the retinal ganglion layer being the most divergent. This finding is consistent with a previous study5 and suggests that although there is a common mechanism of initial retinal photon capture, by photoreceptors, downstream processing of the visual signal by the interneurons, diverges between species. Moreover, this gradient of conservation was also shown to extend to the chick retina by Yamagata et al.7 Interestingly, the number of cell types observed in ACs and RGCs is negatively correlated with the position of the organism in the evolution tree. Large numbers of AC and RGC types have been identified in mice and chicken retinas compared with human and monkey retinas. A potential model to explain this phenomenon is that primates might rely more on the visual cortex for processing visual signals.
Our approaches performing the comparison is also worth mentioning. There has not been a standard, widely applied workflow designed aiming at computing the distance between cross-species cell types from scRNA-seq data. Regardless of which metric was selected, direct measurement of distance among cross-species cell types tended to group the cell types from the same species together. This was due to significant “batch effects,” which were stronger than normal batch effects driven by different sequencing technologies or by different groups that generated the data. Thus, we chose to first minimize the batch effects across the datasets through data integration using LIGER,36 which performed the best out of several methods, many of which failed in this type of task, according to Luecken et al.37 The distance among cell types was then computed in the reduced dimensions after integration. This approach allows us to capture the conserved signal relatively accurately across species, such as the chicken BP-19 case as mentioned above.
With multi-omics data at the cell-type level, we were able to test the hypothesis regarding the relationship between gene regulation landscapes and the conservation of subtypes. The first natural hypothesis we generated was that the sequence of regulatory elements for signature genes of conserved subtypes might also be more conserved. We found that the conservation of promoters of these genes could not explain the conservation of cell types, indicating the involvement of more complex regulatory processes for cells to gain their identity.
Integrative analysis for snRNA-seq and snATAC-seq to decipher key elements in gene regulation
CREs are important genomic regions that regulate the transcription of neighboring genes usually through regulating the recruitment of transcription initiation machinery. With the two modalities of gene expression and peak accessibility, we associated them to infer the potential regulatory functions of genomic regions. More than 35,000 peak-gene pairs were identified in our study and most of the peaks were also identified as DARs for at least one cell class. Besides, we investigated the regulatory function of genomic regions in different types within a major type. We showed that some genomic regions could constantly act as regulators for proximal genes, while others displayed higher heterogeneity. The underlying mechanism of this observation could be the robustness and redundancy of transcription regulation.38 The expression of one gene could be driven by different transcription-activating proteins (e.g., transcription factors) using different genome regions as harbors. Thus, the gene transcription regulation could be highly specific to individual cell types or even cell states. Although straightforward, such scenarios have rarely been highlighted in previous studies. Our finding highlights the importance of investigating CREs in the context of cell types. Also, this could only be investigated with a comprehensive dataset as we presented here, for cell-type level resolution is required for both modalities.
In addition to the identification of CREs, mapping chromatin accessibility can also help to study gene-regulatory networks (GRNs). Many efforts have been made, using scRNA-seq alone, to decipher the GRNs for transcription factors or simply build gene modules, with some of them leading to the discovery of functionally important TFs for the retinal tissue.39 One of the popular frameworks, SCENIC,35 used the motif information at the proximal regions of genes (e.g., 10 kb regions around TSS) for trimming the initially built GRNs. With the snATAC-seq data, the search space for motif existence becomes broader and more accurate. In this study, we leveraged the information on the TF-motif presence in open chromatin regions around gene TSS to perform the pruning of co-expression-based TF-target networks. With this approach, we were able to quantitatively assess the activity of TFs and learn their specificity across cell types. Furthermore, we could group the TFs into modules based on their activity in cells and annotate these modules. Multiple well-studied cell-type-specific TFs such as OTX2, PAX6, and NRL were included in the expected modules and were likely the key regulators for these modules. Besides, we noticed that some TFs, less studied in the context of the retina, appeared to be potential master regulators, such as RORA, TCF4, FOXN3, BACH2, etc. Some retinal defects related to these TFs have been reported in mice according to the MGI database (https://www.informatics.jax.org/), for example, TCF4 heterozygous KO led to abnormal retina morphology and BACH2 KO led to abnormal optic disk morphology. Our study could serve as a prioritization step of the TFs as targets of in vivo functional genomics screening in the retina, with technologies including CRISPR screening or perturb-seq, using the mouse retina or preferably, human retina organoids. Given the technical challenge in in vivo screening approaches, a prioritized list of targets may facilitate the experiment when lower throughputs were more technically preferable.
In summary, we report a comprehensive single-cell multi-omics atlas for the human adult retina as part of the HCA project. The utilization of well-characterized retinal tissue in this study increases the translational value of the resulting dataset. This atlas enables in-depth integrative analysis at individual cell-type resolution, making it a highly valuable and robust resource for the research community.
Limitations of the study
As is elaborated in the main text, the detection of RGCs was still underpowered in our dataset. This is likely because we only applied enrichment using the peripheral retinal samples, and ACs were the more dominant NeuN-positive cell class, not RGCs. The insufficient collection of RGCs also leads to a decrease in the robustness of the integration of the RNA-ATAC modalities (Figure S8). When some RGC types were rare in numbers and some (e.g., ON/OFF-MGCs) were much more abundant, the co-embedding will largely be driven by the abundant cell types and compromise the integration performance on rarer types. A full investigation of RGCs would need further efforts.
Given the rich size and comprehensiveness of these data, it is admitted that our dataset has certain limits in reflecting the diversity of human genetics. A major part of the profiled nuclei originated from Caucasian males, and from senior people. This study then failed to capture information from younger groups or people from other genetic backgrounds. We also did not consider sex as a factor that could alternate the transcriptome to certain levels for the analysis. Again, efforts have been made for postmortem phenotyping to ensure that the retinal tissues were in healthy states, and we have not observed biases such as a proportion decrease in RGCs or any cell types among age groups (Figure S14), yet it is not practical to rule out the aging driven effect in this way. These limits should be considered by potential users of our presented data.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-NeuN | Millipore | MAB377X; RRID:AB_2149209 |
| Anti-RBPMS | Novus Biological | NBP2-20112 |
| Biological samples | ||
| Frozen adult post-mortem human retinas | UTAH LIONS Eye Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Rnase inhibitor | Takara Bio | 2323B |
| MACS BSA Stock Solution | Miltenyi Biotec | 130-091-376 |
| Protease inhibitor | Roche | 11697498001 |
| Tween 20 | Bio-Rad | 1662404 |
| Trizma Hydrochloride Solution, pH 7.4 | Sigma-Aldrich | T2194 |
| Sodium Chloride Solution, 5M | Sigma-Aldrich | 59222C |
| Magnesium Chloride Solution, 1M | Sigma-Aldrich | M1028 |
| Nonidet P40 Substitute IGEPAL CA-630 | Sigma-Aldrich | I8896 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3′ Kit v3.1 | 10x Genomics | |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10x Genomics | |
| Deposited data | ||
| snRNA-seq and snATAC-seq data | This paper |
https://data.humancellatlas.org/explore/projects/9c20a245-f2c0-43ae-82c9-2232ec6b594f https://cellxgene.cziscience.com/collections/af893e86-8e9f-41f1-a474-ef05359b1fb7 |
| Software and algorithms | ||
| Cell Ranger | 10x Genomics | 3.0.2 |
| Cell Ranger ATAC | 10x Genomics | 1.2.0 |
| SoupX | Young and Behjati.40 | https://github.com/constantAmateur/SoupX |
| Seurat | Hao et al.41 | https://satijalab.org/seurat/ |
| DoubletFinder | McGinnis et al.42 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| scVI | Lopez et al.43 | https://scvi-tools.org/ |
| scPred | Alquicira-Hernandez et al.44 | https://github.com/powellgenomicslab/scPred |
| LIGER | Welch et al.36 | https://github.com/welch-lab/liger |
| Pysam | https://pysam.readthedocs.io/en/latest/api.html | |
| Bedtools | https://bedtools.readthedocs.io/en/latest/ | |
| macs2 | Zhang et al.45 | https://pypi.org/project/MACS2/ |
| Deeptools | Ramírez et al.46 | https://deeptools.readthedocs.io/en/develop/ |
| SCENIC | Aibar et al.35 | https://github.com/aertslab/SCENIC |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Rui Chen (ruichen@bcm.edu).
Materials availability
Since the primary samples were limited and were used for data collection, human tissues used in this study will not be available to be shared upon requests. This study does not report other unique materials. For further information, please contact the lead contact.
Experimental model and subject details
The samples used for this study were collected from 25 individuals from the Utah Lions Eye Bank. Detailed information for these donors was included in Table 1 and 2. All donor eye samples were collected within 6 h postmortem. Donors’ eyes were subsequently phenotyped to ensure that they were absent of any disease pathology. Details for the postmortem phenotyping and dissection were according to a previously described standardized protocol.47 In brief, only samples from donors with no record of diabetes, retinal degeneration, macular degeneration, or any other retinal diseases were used in this study. Postmortem phenotyping with the spectral domain–optical coherence tomography (SD-OCT) and fundus imaging, to confirm that there were no drusen, atrophy, or other disease pathology, was done utilizing our standardized approach.48 Although both donor’s eyes were phenotyped for each subject, to ensure that disease pathology was absent for a given subject, only one donor eye was used. For this study, the fovea and macula samples were collected separately, using a 4 mm and 6 mm disposable biopsy punch, respectively, and were flash-frozen in liquid nitrogen. All samples were then stored at -80 °C before nuclei isolation.
All tissues were de-identified under HIPAA Privacy Rules. Institutional approval for the consent of patients for their tissue donation was obtained from the University of Utah and conformed to the tenets of the Declaration of Helsinki.
Method details
Nuclei isolation and sorting
Nuclei were isolated by pre-chilled fresh-made RNase-free lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.02% NP40). The frozen tissue was resuspended and triturated to break the tissue structure in lysis buffer and homogenized with a Wheaton™ Dounce Tissue Grinder. Isolated nuclei were stained with mouse anti-NeuN monoclonal antibody (1:5000, Alexa Flour 488 Conjugate MAB377X, Millipore, Billerica, Massachusetts, United States) in pre-chilled fresh-made wash buffer (1% BSA in PBS, 0.2U/μl RNAse inhibitor) for 30min at 4°C. After being centrifuged at 500g, the pellet was resuspended in wash buffer and filtered with a 40μm Flowmi Cell Strainer. DAPI (4′,6-diamidino-2-phenylindole, 10 μg/ml) was added before loading the nuclei for fluorescent cytometry sorting.
Stained nuclei were sorted with a FACS Aria II flow sorter (Becton Dickinson, San Jose, CA), (70μm nozzle). Sorting gates were based on flow analysis of events and strengths of the DAPI signal, as well as the FITC signal. Samples were sorted at a rate of 50 events per second, based on side scatter (threshold value >200). Fluorescence detection used a 450-nm/40-nm-band pass barrier filter for the DAPI, and a 530-nm/30-nm-band pass filter for FITC. For the NeuNT group, the nuclei with the strongest 5% FITC signal were collected into Eppendorf tubes with 3μl pre-chilled wash buffer.
Single-nuclei sequencing
All single-nuclei RNA or ATAC sequencing in this study was performed at the Single Cell Genomics Core at Baylor College of Medicine. Single-nuclei cDNA library preparation and sequencing were performed following the manufacturer's protocols (https://www.10xgenomics.com). Single-nuclei suspension was loaded on a Chromium controller to obtain single-cell GEMS (Gel Beads-In-Emulsions) for the reaction. The snRNA-seq library was prepared with Chromium Next GEM Single Cell 3' Kit v3.1 (10x Genomics). The snATAC-seq library was prepared with Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 (10x Genomics). The library was then sequenced on Illumina Novaseq 6000 (https://www.illumina.com).
Immunofluorescence (IF) staining
Healthy human donor retinal tissue was dissected and fixed in 4% PFA for 48 h, cryo-protected with 30% sucrose overnight at 4°C, and then embedded in OCT to be flash-frozen. Cryosections from a peripheral region of the human retina with 10 μm thickness were used for the IF.
For the IF, sections were fixed with 4% PFA for 15 min at room temperature, and after being washed with PBS 3 times, 5min per time, they were blocked for 3 h with blocking buffer (10% normal goat serum in PBS + 0.1% Triton X-100) at room temperature. Sections were then incubated with primary antibodies (anti-NeuN: 1:50, MAB377X, Millipore, Billerica, Massachusetts, United States; anti-RBPMS:1:1000, Novus Biologicals NBP2-20112) diluted in blocking buffer. The sections were washed and stained with species-specific fluorophore-conjugated secondary antibody in blocking buffer (1:500) for 2hrs at room temperature, then washed and stained with DAPI for 10 min. After washing with PBS 3 times, the sections were mounted with AquaMount Slide Mounting Media (Thermo Scientific 13800). IF stained slides were visualized with Zeiss Axio Imager M2m.
Quantification and statistical analysis
Sequencing read alignment
Reads from snRNA-seq were demultiplexed and then aligned to the ‘Ref_cellranger.hg19.premrna’ human reference (from 10x Genomics) using Cell Ranger (version 3.0.2, 10x Genomics). Reads from snATAC-seq were demultiplexed and then aligned to the refdata-cellranger-atac-hg19-1.2.0 human reference (from 10x Genomics) using Cell Ranger (version 1.2.0, 10x Genomics).
snRNA-seq quality control, clustering, and differential expression
The gene expression matrices generated by Cell Ranger were filtered for quality control purposes. We first used the soupX package40 to correct the feature counts to alleviate the effect of ambient RNA. For each matrix, genes detected in less than 5 cells were removed; cells with total UMI counts less than 800 or more than 8000 were removed, and the top 5% of cells with the highest mitochondrial gene expression proportion were removed. This processing was performed using the Seurat R package.41 Unlike single-cell RNA-seq, snRNA-seq data had a much lower mitochondrial gene expression proportion, and thus, we removed high-mitochondrial-content cells based on the rank of cells but not a fixed threshold of mitochondrial gene expression proportion. We then performed doublet removal on the remaining cells using DoubletFinder.42 The key parameters of DoubletFinder were set such that pN equals 0.25, the estimated doublet ratio was 0.1, and pK was estimated automatically with ‘find.pK’ function. Details of cell filtering of each QC-filtering step are reported in Table S7.
The expression matrices after the initial QC were combined into one single scanpy49 anndata object. To account for batch effects originating from sample collection processes, the scVI framework43 was applied with standard parameters. The low dimensional embeddings generated were then used to perform leiden clustering,50 and major cell classes were annotated based on known marker genes. Major cell classes were then split for sub-clustering, which was also performed with scVI with the same process as described above. To avoid under- or over-clustering in type discovery, we adapted a parameter-searching approach to finding the best combination of neighbors and resolution for clustering, as described in Figure S2. After sub-clustering, we checked the expression of marker genes for all major cell classes and used that to remove doublets for a second round. For example, if an AC cluster had high rod marker gene expression, it was removed.
For the differential expression analysis, we used the default Wilcoxon method implemented in the Seurat package. The ‘logfc.threshold’ was set to 0.25 and ‘min.pct’ was set to 0.2. Only genes with an adjusted p value less than 0.05 were retained as differentially expressed genes (DEGs). The DEGs for major cell classes were computed using other classes as background, while the DEGs for types were computed using other types from the same class as background.
Obtaining additional RGC data from other local datasets
To increase the number of RGCs for building a more comprehensive atlas, besides the cells from the six donors primarily used, we also used RGCs from other locally sequenced datasets. These RGCs were identified using a supervised single-cell annotation framework, scPred.44 Annotated snRNA-seq data, including rod, cone, MG, AC, BC, HC, and RGC, were downsampled to 10,000 cells per cell type, as input for training the scPred model. 70% of cells were used for training and the rest were used for evaluating the performance of the model. The model was then applied to query datasets for automatically annotating cells to major classes, with the prediction threshold set to 0.8.
Matching cell types between the current study and previous ones using random forest classifiers
Random forest based classifiers had been shown to perform well in previous primate retina studies.5 To match cell types between the current study and a previous human retina study, the top 2000 highly variable genes were selected from the human expression matrix and were used to build a classifier on the monkey expression matrix. 500 cells for each cell type were used to train the classifier and the rest were used to evaluate the classifier. Cell types that had small numbers of cells were split by 70% and 30% as training and test set. The random forest classifier was built using the ‘randomForest’ package in R, using ‘ntree’ equal to 1001. For each cell, if the highest prediction voting rate was less than a threshold (20%), it was labeled as ‘unassigned’. Sankey plot was used to visualize the matching of the data, performed in python with the plotly.graph_objects package.
Cross-species integration of snRNA-seq data
The expression matrices and annotations for single-cell RNA-seq data for mouse, chicken, and monkey retinas were obtained from previous publications.5,7,8,30 All the gene names were converted to human gene nomenclatures, and the matrices from the three species were then merged into a single matrix, for each cell type. At this step, we only retained the genes with 1:1 matching orthologs across species. The data were then integrated using the LIGER framework,36 implemented as a Seurat wrapper. A standard workflow was applied, using the top 5000 highly variable genes, with the k value set to 20 and lambda set to 5. The data was projected to an integration space with 20 dimensions. The coordinates of cells from the same type were averaged and then used to calculate the distance matrix (across each species) using the ‘Dist’ function of the ‘amap’ R package.51 Euclidean distance was used. The distance matrix was then used to build the phylogeny tree using the ‘nj’ function of the ‘ape’ R package.52
snATAC-seq quality control, clustering, and differential accessible analysis
The snATAC-seq data was mainly analyzed using the Signac33 framework. The data output from Cell Ranger ATAC was used as the input. Data from each donor were first combined based on a consensus set of genomic regions and then filtered with QC metrics after merging to a single object. Cells with less than 3000 fragments or more than 20000 fragments were removed for downstream analysis. We also used another two QC criteria implemented in Signac, nucleosome signal and TSS enrichment for filtering nuclei (nucleosome signal less than 4 and TSS enrichment larger than 2 for the nuclei to be retained). Details of nuclei QC-filtering could be found in Table S7. Thus, we resulted in an initial peak-by-nuclei matrix.
Dimensionality reduction was performed using the ‘RunTFIDF’ and ‘RunSVD’ functions with default parameters and then clustered using ‘FindClusters’ function, also with default parameters. The clusters were annotated to major cell types by checking the genome accessibility of known marker genes. Also, with the annotation information, peaks were identified with macs245 called by each cell class. With the macs2 output, we updated the peak-by-nuclei matrix, and all the downstream analyses were based on this updated version.
Differential accessible analysis for finding differential peaks was performed with FindAllMarkers of the Seurat package, setting the minimal detection percentage (min.pct) equal to 0.1. For calculating the conservation of each peak, the peak coordinates were first converted to a GenomicRanges object, and we used the R package GenomicScores53 to score the conservation of each peak using the ‘phastCons 100 species’ system.
Comparing snATAC-seq data with bulk ATAC-seq data
Public ATAC-seq data in bigwig format were downloaded from ENCODE portal54 (ENCFF158OVK for pancreas, ENCFF507OEP for aorta, ENCFF111XAE for adrenal gland, ENCFF961DZL for thyroid gland, ENCFF492WGC for stomach, ENCFF160VHY for cerebellum, and ENCFF577GPG for motor neurons of spinal cord), and GEO (GSE137311, for retina and macula; GSE96949 for hippocampus neuron and glia). Conversion from hg38 aligned coordinates to hg19 versions was performed using CrossMap when needed. snATAC-seq data (bam files) were split based on cell classes using Pysam (https://github.com/pysam-developers/pysam) based on cell barcodes. The bam files for each cell type were deduplicated using the ‘MarkDuplicates’ function in picard tools (http://broadinstitute.github.io/picard/). They were then converted to bigwig format, normalized by total reads, and scaled to 100 million reads using the ‘genomecov’ function in bedtools.55 The genome coverage for all the public bulk ATAC-seq data or grouped snATAC-seq data were transformed to a data frame using the ’multiBigwigSummary’ function of the deeptools software,46 using a window size of 10 kb. Spearman correlation among samples was computed using ‘plotCorrelation’ function of deeptools.
Integration of snATAC-seq and snRNA-seq data
For snATAC-seq data, the gene expression was modeled using the ‘GeneActivity’ implemented in Signac framework. For cell classes with known cell type heterogeneity (AC, BC, HC, Cone, and RGC), the integration was performed within that class, to annotate snATAC-seq data to types. Highly variable genes in both snRNA-seq data and GeneActivity matrices of snATAC-seq data were selected and the common ones were used as inputs for data integration. The GeneActivity matrices of snATAC-seq were first used as query data for further imputing the gene expression using the ‘transfer anchors’ method in the Seurat R package, with snRNA-seq as reference. The imputed gene expression matrices are used for the calculation of the peak-gene correlation at single-cell levels. We also directly predict labels for the ATAC-seq data for the purpose of cell type annotation.
Identification of CREs
The gene expression for each nucleus in the snATAC-seq data was inferred using the Seurat ‘transfer anchors’ method with the snRNA-seq data as the reference. Thus, for each nucleus, we have both the gene expression and peak accessibility information. These two pieces of information were used as inputs to infer the association between genes and peaks. To avoid the effect of the sparsity of the single-cell data, we created meta cells by averaging cells being neighbors in the low-dimensional embedding. Each meta cell is an average of 10 different cells and each cell is only merged into a single meta cell (https://github.com/qingnanl/SRAVG). We used the ‘LinkPeaks’ function in the Signac package to calculate the correlation between each gene and any peak within 250 kb of the gene TSS. We performed the analysis with all the nuclei as input to calculate an overall peak-gene association (Figures 3B–3E). For cell-type specific CRE inference, we performed the analysis in a ‘pseudo-bulk’ manner. We grouped the nuclei with the same sample (donor) and calculated the Pearson’s correlation coefficients between any gene and peaks within 250 kb of the TSS (Figure 3F).
Transcription factor regulon determination and scoring
The initial construction of TF regulons required gene co-expression analysis using the snRNA-seq data. Given the large size of our data, in consideration of computational efficiency, we created meta-cells by grouping cells within each cell type and used the average gene expression for this meta-cell. The size of each group was 20, so that the size of this meta-cell matrix could be reduced to about 5% of the original one. This approach was previously reported by Suo et al.56
The meta cell matrix was then used as the input of GINIE357 algorithm to construct an initial TF regulon network. The output of GINIE3 was a weight matrix regarding how likely each TF is a regulator of each target gene. We set the weight cutoff at 0.005 for deciding the TF-target relationship and thus we resulted in an initial TF-target network. We then used the peaks proximal to target TSS, which could be obtained from snATAC-seq data, to prune the network. We searched motifs in peaks that were 50kb upstream to the TSS and required at least one appearance for the TF-target pair to be retained. With the pruned regulons, we used the AUCell R package by the default parameters to calculate the TF activity score for each meta cell.
Acknowledgments
This work is supported by the Chan-Zuckerburg Foundation Grant CZF2019-002425. Single-nucleus RNA sequencing was performed at the Single Cell Genomics Core at BCM partially supported by NIH shared instrument grants (S10OD018033, S10OD023469, S10OD025240), P30CA125123, P30EY002520, and CPRIT Comprehensive Cancer Epigenomics Core Facility RP200504. The cell-sorting experiments are also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the CPRIT Core Facility Support Award (CPRIT-RP180672), the NIH (CA125123 and RR024574), and the assistance of Joel M. Sederstrom. Ascertainment, collection, processing, and phenotyping of donor eye tissue was partially supported by the Macular Degeneration Foundation, Inc., the Carl Marshall and Mildred Almen Reeves Foundation, and Ira G. Ross and Elizabeth Olmsted Ross Endowed Chair.
We want to thank Kaitlyn Xiong for offering critical advice. We want to thank Dr. Aboozar Monavarfeshani for important discussion regarding the NeuN enrichment technique.
Author contributions
R.C. and M.D. conceived and supervised the project. Q.L. and J.W. performed the data analysis. X.C. and Y.L. performed the single-nuclei dissociation, sequencing, and validation experiments. L.O., A.S., J.L., C.Z., M.F., and I.K. performed the sample collection and postmortem phenotyping. Q.L. and R.C. wrote the initial draft with input of all other authors. All authors approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 11, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100298.
Contributor Information
Margaret DeAngelis, Email: mmdeange@buffalo.edu.
Rui Chen, Email: ruichen@bcm.edu.
Supplemental information
Data and code availability
All sequencing data generated in this study is available at: https://data.humancellatlas.org/explore/projects/9c20a245-f2c0-43ae-82c9-2232ec6b594f; or on gene expression omnibus (GEO) with accession number GSE226108.
HCA portal for this data could be used for interactive visualization: https://cellxgene.cziscience.com/collections/af893e86-8e9f-41f1-a474-ef05359b1fb7.
Original code is available in: https://github.com/qingnanl/human-retina-multiomics.
References
- 1.Masland R.H. The neuronal organization of the retina. Neuron. 2012;76:266–280. doi: 10.1016/j.neuron.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolb H. Webvision; 2012. Gross anatomy of the eye.http://webvision.med.utah.edu/book/part-i-foundations/gross-anatomy-of-the-ey/ [PubMed] [Google Scholar]
- 3.Baden T., Euler T., Berens P. Understanding the retinal basis of vision across species. Nat. Rev. Neurosci. 2019;21:5–20. doi: 10.1038/s41583-019-0242-1. [DOI] [PubMed] [Google Scholar]
- 4.Hoon M., Okawa H., Della Santina L., Wong R.O.L. Functional architecture of the retina: development and disease. Prog. Retin. Eye Res. 2014;42:44–84. doi: 10.1016/J.PRETEYERES.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng Y.R., Shekhar K., Yan W., Herrmann D., Sappington A., Bryman G.S., van Zyl T., Do M.T.H., Regev A., Sanes J.R. Molecular classification and comparative taxonomics of foveal and peripheral cells in primate retina. Cell. 2019;176:1222–1237.e22. doi: 10.1016/j.cell.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran N.M., Shekhar K., Whitney I.E., Jacobi A., Benhar I., Hong G., Yan W., Adiconis X., Arnold M.E., Lee J.M., et al. Single-cell profiles of retinal ganglion cells differing in resilience to injury reveal neuroprotective genes. Neuron. 2019;104:1039–1055.e12. doi: 10.1016/J.NEURON.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamagata M., Yan W., Sanes J.R. A cell atlas of the chick retina based on single-cell transcriptomics. Elife. 2021;10:e63907. doi: 10.7554/ELIFE.63907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan W., Laboulaye M.A., Tran N.M., Whitney I.E., Benhar I., Sanes J.R. Mouse retinal cell atlas: molecular identification of over sixty amacrine cell types. J. Neurosci. 2020;40:5177–5195. doi: 10.1523/JNEUROSCI.0471-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan W., Peng Y.-R., van Zyl T., Regev A., Shekhar K., Juric D., Sanes J.R. Cell atlas of the human fovea and peripheral retina. Sci. Rep. 2020;10:9802. doi: 10.1038/s41598-020-66092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cowan C.S., Renner M., De Gennaro M., Gross-Scherf B., Goldblum D., Hou Y., Munz M., Rodrigues T.M., Krol J., Szikra T., et al. Cell types of the human retina and its organoids at single-cell resolution. Cell. 2020;182:1623–1640.e34. doi: 10.1016/J.CELL.2020.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orozco L.D., Chen H.-H., Cox C., Katschke K.J., Arceo R., Espiritu C., Caplazi P., Nghiem S.S., Chen Y.-J., Modrusan Z., et al. Integration of eQTL and a single-cell atlas in the human eye identifies causal genes for age-related macular degeneration. Cell Rep. 2020;30:1246–1259.e6. doi: 10.1016/J.CELREP.2019.12.082. [DOI] [PubMed] [Google Scholar]
- 12.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for multimodal regulatory analysis and personal epigenomics. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/NMETH.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P., Chang H.Y., Greenleaf W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/NATURE14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/S41587-019-0206-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatia S., Bengani H., Fish M., Brown A., Divizia M.T., de Marco R., Damante G., Grainger R., van Heyningen V., Kleinjan D.A. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet. 2013;93:1126–1134. doi: 10.1016/J.AJHG.2013.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghiasvand N.M., Rudolph D.D., Mashayekhi M., Brzezinski J.A., Goldman D., Glaser T. Deletion of a remote enhancer near ATOH7 disrupts retinal neurogenesis, causing NCRNA disease. Nat. Neurosci. 2011;14:578–586. doi: 10.1038/NN.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rai V., Quang D.X., Erdos M.R., Cusanovich D.A., Daza R.M., Narisu N., Zou L.S., Didion J.P., Guan Y., Shendure J., et al. Single-cell ATAC-Seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol. Metab. 2020;32:109–121. doi: 10.1016/J.MOLMET.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S.S., Jagadeesh K., Dey K.K., Shen A.Z., Raychaudhuri S., Kellis M., Price A.L. Leveraging single-cell ATAC-seq to identify disease-critical fetal and adult brain cell types. bioRxiv. 2021 doi: 10.1101/2021.05.20.445067. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyu P., Hoang T., Santiago C.P., Thomas E.D., Timms A.E., Appel H., Gimmen M., Le N., Jiang L., Kim D.W., et al. Gene regulatory networks controlling temporal patterning, neurogenesis, and cell-fate specification in mammalian retina. Cell Rep. 2021;37:109994. doi: 10.1016/J.CELREP.2021.109994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finkbeiner C., Ortuño-Lizarán I., Sridhar A., Hooper M., Petter S., Reh T.A. Single-cell ATAC-seq of fetal human retina and stem-cell-derived retinal organoids shows changing chromatin landscapes during cell fate acquisition. Cell Rep. 2022;38:110294. doi: 10.1016/J.CELREP.2021.110294. [DOI] [PubMed] [Google Scholar]
- 21.Thomas E.D., Timms A.E., Giles S., Harkins-Perry S., Lyu P., Hoang T., Qian J., Jackson V.E., Bahlo M., Blackshaw S., et al. Cell-specific cis-regulatory elements and mechanisms of non-coding genetic disease in human retina and retinal organoids. Dev. Cell. 2022;57:820–836.e6. doi: 10.1016/j.devcel.2022.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S.K., Nair S., Li R., Kraft K., Pampari A., Patel A., Kang J.B., Luong C., Kundaje A., Chang H.Y. Single-cell multiome of the human retina and deep learning nominate causal variants in complex eye diseases. Cell Genom. 2022;2:100164. doi: 10.1016/J.XGEN.2022.100164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dacey D. In: The Cognitive Neurosciences Iii. Michael S. G., editor. MIT Press; 2004. 20 origins of perception: retinal ganglion cell diversity and the creation of parallel visual pathways; p. 281. [Google Scholar]
- 24.Kolb H. Webvision. 2015. Morphology and circuitry of ganglion cells; pp. 1–16.https://webvision.med.utah.edu/book/part-ii-anatomy-and-physiology-of-the-retina/morphology-and-circuitry-of-ganglion-cells/ [Google Scholar]
- 25.DO M.T.H., YAU K.-W. Intrinsically photosensitive retinal ganglion cells. Physiol. Rev. 2010;90:1547–1581. doi: 10.1152/PHYSREV.00013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf H.K., Buslei R., Schmidt-Kastner R., Schmidt-Kastner P.K., Pietsch T., Wiestler O.D., Blümcke I. NeuN: a useful neuronal marker for diagnostic histopathology. J. Histochem. Cytochem. 1996;44:1167–1171. doi: 10.1177/44.10.8813082. [DOI] [PubMed] [Google Scholar]
- 27.Lin Y.-S., Kuo K.-T., Chen S.-K., Huang H.-S. RBFOX3/NeuN is dispensable for visual function. PLoS One. 2018;13:e0192355. doi: 10.1371/JOURNAL.PONE.0192355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cherry T.J., Yang M.G., Harmin D.A., Tao P., Timms A.E., Bauwens M., Allikmets R., Jones E.M., Chen R., De Baere E., Greenberg M.E. Mapping the cis-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc. Natl. Acad. Sci. USA. 2020;117:9001–9012. doi: 10.1073/PNAS.1922501117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shekhar K., Lapan S.W., Whitney I.E., Tran N.M., Macosko E.Z., Kowalczyk M., Adiconis X., Levin J.Z., Nemesh J., Goldman M., et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166:1308–1323.e30. doi: 10.1016/j.cell.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/GR.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welch J.D., Kozareva V., Ferreira A., Vanderburg C., Martin C., Macosko E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177:1873–1887.e17. doi: 10.1016/J.CELL.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luecken M., Büttner M., Chaichoompu K., Danese A., Interlandi M., Mueller M., Strobl D., Zappia L., Dugas M., Colomé-Tatché M., et al. Benchmarking atlas-level data integration in single-cell genomics. bioRxiv. 2020 doi: 10.1101/2020.05.22.111161. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaugg J.B., Sahlén P., Andersson R., Alberich-Jorda M., de Laat W., Deplancke B., Ferrer J., Mandrup S., Natoli G., Plewczynski D., et al. Current challenges in understanding the role of enhancers in disease. Nat. Struct. Mol. Biol. 2022;29:1148–1158. doi: 10.1038/s41594-022-00896-3. [DOI] [PubMed] [Google Scholar]
- 39.Gautam P., Hamashima K., Chen Y., Zeng Y., Makovoz B., Parikh B.H., Lee H.Y., Lau K.A., Su X., Wong R.C.B., et al. Multi-species single-cell transcriptomic analysis of ocular compartment regulons. Nat. Commun. 2021;12:5675. doi: 10.1038/S41467-021-25968-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young M.D., Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9:giaa151. doi: 10.1093/GIGASCIENCE/GIAA151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/J.CELL.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/J.CELS.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alquicira-Hernandez J., Sathe A., Ji H.P., Nguyen Q., Powell J.E. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019;20 doi: 10.1186/S13059-019-1862-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/GB-2008-9-9-R137/FIGURES/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/NAR/GKW257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Owen L.A., Shakoor A., Morgan D.J., Hejazi A.A., McEntire M.W., Brown J.J., Farrer L.A., Kim I., Vitale A., DeAngelis M.M. The Utah protocol for postmortem eye phenotyping and molecular biochemical analysis. Invest. Ophthalmol. Vis. Sci. 2019;60:1204–1212. doi: 10.1167/iovs.18-24254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang Q., Dharmat R., Owen L., Shakoor A., Li Y., Kim S., Vitale A., Kim I., Morgan D., Liang S., et al. Single-nuclei RNA-seq on human retinal tissue provides improved transcriptome profiling. Nat. Commun. 2019;10:5743. doi: 10.1038/s41467-019-12917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/S13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lucas A. amap. https://cran.r-project.org/web/packages/amap/index.html
- 52.Paradis E., Schliep K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/BIOINFORMATICS/BTY633. [DOI] [PubMed] [Google Scholar]
- 53.Puigdevall P., Castelo R. GenomicScores: seamless access to genomewide position-specific scores from R and Bioconductor. Bioinformatics. 2018;34:3208–3210. doi: 10.1093/BIOINFORMATICS/BTY311. [DOI] [PubMed] [Google Scholar]
- 54.Sloan C.A., Chan E.T., Davidson J.M., Malladi V.S., Strattan J.S., Hitz B.C., Gabdank I., Narayanan A.K., Ho M., Lee B.T., et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016;44:D726–D732. doi: 10.1093/NAR/GKV1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/BIOINFORMATICS/BTQ033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suo S., Zhu Q., Saadatpour A., Fei L., Guo G., Yuan G.C., Yuan G.-C. Revealing the critical regulators of cell identity in the mouse cell atlas. Cell Rep. 2018;25:1436–1445.e3. doi: 10.1016/j.celrep.2018.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5:e12776. doi: 10.1371/JOURNAL.PONE.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this study is available at: https://data.humancellatlas.org/explore/projects/9c20a245-f2c0-43ae-82c9-2232ec6b594f; or on gene expression omnibus (GEO) with accession number GSE226108.
HCA portal for this data could be used for interactive visualization: https://cellxgene.cziscience.com/collections/af893e86-8e9f-41f1-a474-ef05359b1fb7.
Original code is available in: https://github.com/qingnanl/human-retina-multiomics.