

Summary
Based on evaluations of imputation performed on a genotype dataset consisting of about 11,000 sub-Saharan African (SSA) participants, we show Trans-Omics for Precision Medicine (TOPMed) and the African Genome Resource (AGR) to be currently the best panels for imputing SSA datasets. We report notable differences in the number of single-nucleotide polymorphisms (SNPs) that are imputed by different panels in datasets from East, West, and South Africa. Comparisons with a subset of 95 SSA high-coverage whole-genome sequences (WGSs) show that despite being about 20-fold smaller, the AGR imputed dataset has higher concordance with the WGSs. Moreover, the level of concordance between imputed and WGS datasets was strongly influenced by the extent of Khoe-San ancestry in a genome, highlighting the need for integration of not only geographically but also ancestrally diverse WGS data in reference panels for further improvement in imputation of SSA datasets. Approaches that integrate imputed data from different panels could also lead to better imputation.
Keywords: imputation, reference panel, GWAS, Africa, whole-genome sequence, non-reference discordance rate, TOPMed, AGR, imputation accuracy
Graphical abstract
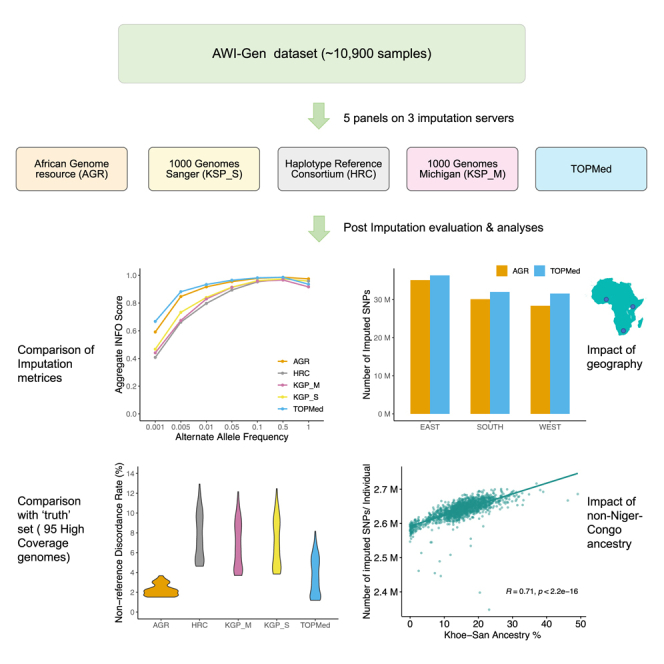
Highlights
-
•
TOPMed and AGR are currently the best panels for imputing sub-Saharan African datasets
-
•
Datasets from East, West, and South Africa show systematic differences in imputation
-
•
The AGR imputed dataset shows highest genotype concordance with high-coverage WGSs
-
•
Imputation accuracy is impacted by the extent of non-Niger-Congo ancestry in a genome
Sengupta et al. report a comprehensive evaluation of imputation performance of five widely used reference panels in a pan-African dataset. The study identifies several key factors such as sample size, geographic origin, and ancestral composition of the populations that need to be considered for selecting an optimal panel for genotype imputation of sub-Saharan African datasets.
Introduction
Imputation is a widely used technique to statistically predict unobserved genotypes in a single-nucleotide polymorphism (SNP) array dataset based on haplotypes inferred using a reference panel. This step not only adds to the genomic coverage for genome-wide association studies (GWASs) but also enables more efficient meta-analysis of independent GWASs by increasing the overlap between them.1 These reference panels generally consist of a set of curated whole-genome sequences (WGS), some of which are openly available while others are only accessible via specific imputation services.
As the size of a reference panel and its genetic proximity to the target populations have been shown to positively impact imputation performance, larger and more geographically representative panels have been introduced.1 These include globally focused panels such as the 1000 Genomes Project (KGP; n = 2,504),2 the Haplotype Reference Consortium (HRC; n = 32,470),3 Trans-Omics for Precision Medicine (TOPMed; n = 97,256),4 and panels that focus on representing a particular geographic region such as the African Genome Resource (AGR; n = 4,956),5 the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA; n = 883),6 and Genome Asia (n = 6,461).7 As is evident, the size of WGS datasets that are included in these panels range from about a thousand to around a hundred thousand individual genomes. Moreover, the degree of representation of different continents/geographic regions varies widely between these panels, and it has been shown that more representative and targeted panels can lead to better imputation compared with global panels.8,9 In the absence of a comprehensive evaluation of performance of current reference panels in imputing sub-Saharan African (SSA) populations, researchers have limited knowledge to inform their selection of panels for imputation of GWAS datasets.
To assess the performance of widely used reference panels in SSA populations, we imputed a dataset of ∼10,900 samples from four countries—Kenya (East), Ghana and Burkina Faso (West), and South Africa (South)10,11—using five imputation panels hosted on the Sanger, TOPMed, and Michigan imputation services. The sequence datasets included in global reference panels are predominantly based on West African populations and diaspora populations originating from this geographic region.2,4 Due to this geographic bias, the global panels lack the representation of diversity from Eastern, Central, and Southern Africa, which might impact the imputation performance in populations from these regions more adversely compared with populations with predominantly West African ancestry. In addition to enabling the evaluation of imputation performance based on a relatively large SSA GWAS dataset, the geographic spread of the participants, for the first time, enabled a comparison of imputation in datasets from different African regions.
Deep ancestral divisions among SSA populations often result in major genetic differences in populations inhabiting different geographic regions and sometimes also within a specific region.12,13 However, the impact of representation or the absence of these ancestries in the reference panels has not yet been assessed. Although our dataset has an overwhelming majority of one major African ethnolinguistic division (Niger-Congo speakers), three of the other major African divisions (Khoe-San, Nilo-Saharan, and Afro-Asiatic speakers) are represented in sizable proportions, as the dataset includes individuals from these divisions or individuals with significant gene flow from them.14 The list of ethnolinguistic groups in the Africa-Wits INDEPTH Partnership for Genomic Studies in African Populations (AWI-Gen) dataset is provided in Table S1, and more details can be found in Ramsay et al.10 This unique property of the dataset also enables an evaluation of the impact of ancestral diversity of SSA populations on imputation performance.
The number of SNPs imputed by a panel and the associated imputation statistics (e.g., INFO or R2 scores) are generally used to quantify the efficiency and quality of imputation. However, these statistics do not provide an assessment of the accuracy of the imputed genotypes, and a comparison of imputed datasets with high-quality WGSs is needed to estimate this. We sequenced 95 of the genotyped samples at high coverage (>30×) to generate data for a direct estimate of the imputation accuracy.
Meta-imputation with multiple panels is becoming a popular approach to improve imputation.15 These methods harness the content of individual panels to provide a more comprehensive imputation of the dataset. However, as these methods rely on factors such as the underlying linkage disequilibrium (LD) architecture and allele frequencies, they could be especially challenging in African populations that have generally smaller LD blocks and harbor a much higher number of rare variants.2,13,16 Based on a comparison with the WGS dataset, we assess the extent to which meta-imputation with multiple panels could boost the imputation of the SSA dataset.
We report comprehensive evaluations of imputation performance of widely used reference panels in a pan-African dataset and identify several key factors that need to be considered when deciding on an optimal panel for genotype imputation for SSA GWAS datasets.
Results
Comparison of imputed datasets
The AWI-Gen study consists of about 12,000 participants living in four countries from East, West, and Southern Africa.10,11 About 10,900 of these participants were genotyped using the H3Africa Custom SNP array (https://chipinfo.h3abionet.org/) and form our core dataset. The sample size and geographic location of the AWI-Gen participants are shown in Figure 1A, and the overall study design is presented in Figure 1B. We imputed the AWI-Gen dataset remotely using five reference panels—KGP (KGP_S), AGR, and HRC panels hosted at the Sanger Imputation Server (SIS), the TOPMed panel hosted at the TOPMed Imputation Server (TIS), and the KGP panel (KGP_M) hosted at the Michigan Imputation Server (MIS). The details of the imputation panels compared in our study are presented in Table 1.
Figure 1.

Summary of the AWI-Gen dataset and study schema
(A) The participants from the AWI-Gen cohort are sampled from different regions across Africa—Kenya (East), Ghana and Burkina Faso (West), and South Africa (South). Numbers below the circles on the map show approximate sample sizes.
(B) The schematic representation of the study design summarizing the main steps implemented to compare the datasets imputed using the five widely used reference panels: AGR, African Genome Resource hosted at the Sanger Imputation Server (SIS); KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the Michigan Imputation Server (MIS); TOPMed, hosted at the TOPMed Imputation Server (TIS).
Table 1.
Summary of imputation reference panels compared in our study
| Reference panel | Samples | Sites (millions) | Ancestry distribution | Panel content | Phasing and imputation |
|---|---|---|---|---|---|
| AGR | 4,956 | 93 | predominantly Africana | chr1-22 and X; biallelic SNPs only | EAGLE2+ PBWT |
| TOPMed | 97,256 | 308 | multi-ethnic | chr1-22 and X; SNPs and indels | EAGLE2+ Minimac4 |
| KGP_S | 2,504 | 85 | multi-ethnic | chr1-22 and X; SNPs and indels | EAGLE2+ PBWT |
| KGP_M | 2,504 | 49 | multi-ethnic | chr1-22 and X; SNPs and indels | EAGLE2+ Minimac4 |
| HRC | 32,470 | 40 | predominantly European | chr1-22 and X; SNPs only | EAGLE2+ PBWT |
Panel codes: AGR, African Genome Resource hosted at the Sanger Imputation Server (SIS); KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the Michigan Imputation Server; TOPMed, hosted at the TOPMed Imputation Server.
Populations from Uganda, Ethiopia, Egypt, and Nama from South Africa and the 1000 Genomes Project.
The comparison of the number and quality of SNPs that were imputed using these panels are summarized in Figure 2. As expected, TOPMed, the largest of these panels, imputed the highest number of SNPs (Figure 2A) and showed the highest average INFO (or R2) scores (Figure 2B). The differences between TOPMed and other panels were most prominent for extremely rare variants (alternate allele frequency [AAF] < 0.001), both in the number of imputed SNPs and average INFO scores (or R2) (Figures 2B and 2C). Despite being many folds smaller, the AGR emerged as a close competitor to TOPMed for AAFs over 0.001 and outperformed other panels in average INFO score (or R2) distribution (Figure 2B). While other panels impute a similar number of SNPs compared with TOPMed and AGR for AAF bins greater than 0.01 (Figure 2C), the difference in INFO score (or R2) remains conspicuous across a much wider part of the allele frequency spectrum (Figure 2B). Comparisons of imputed SNP density per Mb and average INFO score (or R2) per Mb further show the pattern of differences between panels to run more or less consistently along chromosomes (Figures 2D and S1).
Figure 2.

Evaluation of the AWI-Gen dataset imputed by the five reference panels
(A) Total number of SNPs imputed by different panels and their distribution across all R2 or INFO score bins.
(B) Average INFO score (or R2) across allele frequency bins.
(C) Number of imputed SNPs across allele frequency bins.
(D) SNP density per Mb for chromosome 1, related to Figure S1.
All evaluations are based on 10,903 individuals. AGR, African Genome Resource hosted at the SIS; KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the MIS; TOPMed, hosted at the TIS.
As GWAS datasets invariably undergo post-imputation quality control (QC) based on INFO score or R2 filtering, to obtain estimates that align better to actual GWAS datasets, we performed an additional set of comparisons including only those SNPs that were imputed with INFO scores (or R2) over 0.6. Overall, the same pattern of inter-panel differences was observed in the INFO score filtered datasets (Figure S2). Clear differences were observed in the imputed datasets generated using KGP panels at the SIS (KGP_S) and the MIS (KGP_M). For our dataset, the KGP_S panel imputed considerably more SNPs and also showed a higher average INFO score (or R2) than the KGP_M panel at lower allele frequency bins. The predominantly European data-based HRC panel, which has been one of the most successful and widely used panels for imputation in European ancestry populations, was outperformed by all other panels in our dataset.
Union and intersection of SNPs imputed by different panels
Next, we investigated the overlap between the set of SNPs that were imputed by all the panels. In total, ∼76 million SNPs (union of all SNPs) were imputed, of which ∼19 million SNPs were imputed by all five panels as a common subset irrespective of the reference panel used (Figure 3A). To assess whether the predicted genotypes for these SNPs were also the same, we performed pairwise comparison of the allele frequencies in the datasets imputed by all five panels. Although a large majority of SNPs had very similar allele frequencies, differences were not uncommon (Table S2). For instance, over 86,000 of these SNPs showed over 0.01 AAF differences between TOPMed and AGR imputed datasets. A further 6 million SNPs were imputed by at least four of the five panels.
Figure 3.

Overlap between SNPs imputed by the five reference panels
(A) UpSet plot showing panel-specific and shared SNPs between the imputed datasets.
(B) SNPs with allele frequency (AF) >0.005 that were imputed uniquely by each panel.
(C) SNPs reported in GWAS catalog that were imputed uniquely by each panel.
All the evaluations are based on 10,903 individuals. AGR, African Genome Resource hosted at the SIS; KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the MIS; TOPMed, hosted at the TIS.
We also explored the SNPs uniquely imputed by a panel or an intersection of panels. The TOPMed panel imputed the highest number of SNPs (18.3 million) that were unique to imputation by a single panel, followed by the AGR, which imputed about 9.6 million SNPs not imputed by other panels. In addition, these two panels both imputed 5.3 million SNPs that the others did not. Despite the inherent intersection between the sequence datasets used to build some of the panels (for instance, KGP is included in both the HRC and AGR panels), exclusive imputation of 4 million SNPs by the KGP_S and 0.4 million SNPs by KGP_M shows that differences in panel curation and imputation algorithms can lead to noticeable differences in the content imputed. The subset of SNPs with INFO scores (or R2) >0.6 showed a very similar pattern of intersection (Figure S3), with ∼15.6 million SNPs imputed as a common subset irrespective of the reference panel used.
To further unpack the panel-specific imputation, we compared the allele frequency distribution of the SNPs that were imputed by only one of the panels (Figure 3B). Although the majority of these SNPs were extremely rare, several hundred thousand SNPs were common enough to be included in modest-to-large GWASs. Moreover, these uniquely imputed SNPs included several known associations reported in the GWAS catalog (Figure 3C), suggesting that the choice of panels can also impact the SNPs that can be replicated by a study. Probably as a function of its widespread use in GWAS studies, the HRC exclusively imputes the largest number of SNPs that have been previously reported as GWAS associations. Therefore, this panel, although suboptimal according to other imputation evaluation parameters, can be more useful in replication studies.
Impact of geography and non-Niger-Congo ancestry gene flow on imputation
Previous studies like the KGP2 and Gurdasani et al.12 have shown that the number of SNPs observed in different African populations vary in a geographically stratified manner. The inclusion of samples from East, West, and South Africa in the AWI-Gen study provided an opportunity to investigate whether the number of SNPs imputed in genotype datasets of similar size also vary within these three SSA regions. The number of SNPs imputed by TOPMed and AGR (the two best-performing panels) for a similar number of individuals from these three regions is presented in Figure 4A. For both the panels, we observed the East African dataset to have the highest number of imputed SNPs, followed by South and West Africa. The higher number of imputed SNPs in East African Niger-Congo speakers compared with South African speakers is consistent with the previous observation of higher SNP content in WGSs of Bantu speakers from Uganda compared with South African Bantu speakers.12 Similarly, the KGP dataset shows that East African Luhya (LWK) contains more SNPs compared with the West African Yoruba (YRI).2 Our imputed datasets mirror this trend of a higher SNP count in East compared with West Africans (for both AGR and TOPMed). However, while AGR imputed a few million more SNPs in the South Africa compared with the West Africa, the difference is much less pronounced for the dataset imputed on the TOPMed panel (Figure 4A). Further work will be required to estimate the extent to which imputed dataset sizes are impacted by the presence of South African genomes in the AGR and their absence from TOPMed. The comparisons for SNPs with INFO scores (or R2) >0.6 show the same trend as observed for the overall estimates (Figure S4).
Figure 4.

Impact of geography and non-Niger-Congo ancestry gene flow on imputation
(A) Total number of imputed SNPs in samples from East, West, and South Africa by TOPMed and AGR.
(B) Correlation between the number of SNPs imputed per individual by the AGR and the level of Khoe-San ancestry in South African participants. The regression line, along with correlation coefficient (R) and p value (Pearson correlation), is shown.
(C) Inverse correlation between the number of SNPs imputed per individual by AGR and the level of East African non-Niger-Congo (EA non-NC) ancestry (Afro-Asiatic, Nilo-Saharan, or Eurasian) in the EA participants. The regression line, along with correlation coefficient (R) and p value (Pearson correlation), is shown. The ancestry proportions were inferred using ADMIXTURE (see Figure S5). Ancestry-based variation for the dataset imputed using the TOPMed panel is shown in Figure S6.
(D) Violin plot comparing the distribution of non-reference discordance rate (NDR) between genotypes imputed using AGR vs. TOPMed in the East, West, and South African populations. Each regional subset (i.e., East, West, and South African populations) consist of ∼2,000 participants. The NDR is almost constant across the dataset for West African participants, while the NDR shows substantial variation among the South African participants. Panel codes: AGR, African Genome Resource hosted at the SIS; TOPMed, hosted at the TIS.
Many extant African populations are the result of admixture of two or more ancestries from major African ethnolinguistic divisions (e.g., Khoe-San and Bantu speakers). Khoe-San is a collective term for populations across Southern Africa that predate the expansion of Bantu speakers; these populations descend from the earliest human population divergence and thus harbor greater genetic diversity and higher SNP content.17,18,19,20 While some of these ancestries are well represented in all reference panels, some are included in a limited set of panels, and still others are absent from all the current panels. Therefore, these ancestral differences arguably have the potential to influence how well a genome can be imputed. We assessed whether gene flow from ancestries that are relatively less represented in imputation panels (such as Khoe-San, Afro-Asiatic, and Nilo-Saharan ancestries) impact the number of SNPs imputed in a dataset. Based on a global ancestry inference approach (using ADMIXTURE21), we estimated the Khoe-San and Niger-Congo (the two major ancestries prevalent in this geographic region) contributions to the genomes of the AWI-Gen South African participants from Soweto (one of the study sites from South Africa). The admixture plot at K = 3 is shown in Figure S5A. Figure 4B shows a clear correlation (R = 0.71, p < 2.2e−16) between the level of Khoe-San ancestry and the imputed SNP count per individual by the AGR panel. This can be reasoned by the fact that the Khoe-San genomes are known to be more diverse and have, on average, higher SNP content compared with all other human populations.19 A similar analysis showed the level of non-Niger-Congo (mainly originating from Afro-Asiatic, Nilo-Saharans, and Eurasian) ancestry in the East African participants (Figure S5B) to be inversely correlated (R = −0.76, p < 2.2e−16) to the number of imputed SNPs in an individual (Figure 4C). The level for non-Niger-Congo ancestry shown here is based at K = 3, though the estimates might differ for other values of K (Figure S5B). The non-Niger-Congo ancestry in East Africans might also include a trace level of rain forest forager-related ancestry in some of the participants. Interestingly, the Ethiopian populations in the African Genome Variation Project (AGVP) dataset that correspond to these two ancestries contain fewer SNPs compared with Bantu speakers from Uganda.12 However, large pan-African high-coverage WGS datasets will be required to assess if there are systematic differences in SNP count between East African ancestral groups and also whether the differences observed in our data correspond to inherent genomic features or the varied ability of existing panels to impute genotype for such ancestral groups. Nevertheless, the much higher genomic diversity of African populations compared with the neighboring Eurasian populations leads us to speculate that the observed differences among the East African participants could be primarily driven by underlying differences in levels of the Eurasian component that is included under the non-Niger-Congo ancestry. The same trend was observed in the SNP set imputed by the TOPMed panel (Figure S6). Therefore, the impact of ancestry concurs broadly with the current understanding of overall diversity and SNP counts in SSA ancestral groups.
We also investigated the level of discordance of genotypes imputed by AGR and TOPMed for East, West, and South African populations. This was done by estimating non-reference discordance rates (NDRs) between AGR and TOPMed for individuals from each geographic region separately. Data for the South African participants showed the highest NDR between the two panels (NDR = 4.89% ± 1.26%), followed by East Africans (NDR = 3.61% ± 0.74%). The West African participants showed the lowest discordance (NDR = 2.71% ± 0.14%) (Figure 4D). To identify the possible source of systematic differences in NDRs between the two panels, we assessed the relationship between the level of non-Niger-Congo ancestry in the individuals from East and South Africa and their respective NDR scores. The observation of a very strong correlation in both South African (R = 0.99, p < 2.2e−16; Figure S7A) and East African (R = 0.93, p < 2.2e−16; Figure S7B) datasets suggests that genotype discordance between imputed datasets generally increase with increase in non-Niger-Congo ancestry in a genome. Therefore, the same panel might perform differently in populations from different geographic regions of the continent.
Estimating the accuracy of the imputed genotypes
To assess the accuracy of the genotypes imputed by different panels, we sequenced 95 samples from the AWI-Gen South African dataset at high depth (>30×) and compared this with the imputed datasets for these individuals. The overlap (sharing of sites) between the WGS and the imputed dataset generated using the five panels is shown in Figure 5A and Table 2. All panels other than HRC were able to impute over 70% of the SNPs detected in these genomes, with AGR and TOPMed showing the highest overlap. The partitioning of SNPs that were shared by the imputed dataset and the WGS, by allele frequency bins (Figure S8), further shows the AGR to better represent rare-to-moderate-frequency SNPs (<0.05) compared with TOPMed, while TOPMed marginally better represents more common SNPs. AGR had the lowest alternate allele mismatch between WGSs and imputed data, markedly lower than all the other panels, including TOPMed (Table 2).
Figure 5.

Comparison of imputed genotypes and genotypes inferred using WGS data
(A) Number of sites that were shared by the imputed and WGS datasets for the 95 individuals. The red line on top shows the number of SNPs in the WGS data.
(B) Venn diagram showing the overlap of SNPs between the WGSs and datasets imputed using AGR and TOPMed panels.
(C) Violin plot summarizing the distribution of NDR for the five panels in the 95 individuals.
(D) Correlation between the overall genotype discordance (estimated by NDR) and the level of Khoe-San ancestry in the five imputed datasets. The regression line for each panel is shown in a different color. The inclusion of the representative Khoe-San population probably leads to a much lower discordance and a gentler slope in the AGR compared with other panels.
AGR, African Genome Resource hosted at the SIS; KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the MIS; TOPMed, hosted at the TIS.
Table 2.
Comparison of the subset of 95 whole-genome sequence data from South African participants with the five imputed datasets
| Parameters compared | AGR | TOPMed | KGP_S | KGP_M | HRC |
|---|---|---|---|---|---|
| Total number of SNPs imputed | 19,507,662 | 19,898,122 | 19,123,670 | 18,193,059 | 15,654,537 |
| Number of sites common between imputed and sequence data | 18,340,523 | 18,190,569 | 16,245,629 | 16,065,601 | 14,089,125 |
| Number of sites unique to sequence data (fraction of sites in the file ∼22.8 M SNPs) (%) | 4,482,848 (19.6) | 4,632,802 (20.3) | 6,577,742 (28.8) | 6,757,770 (29.6) | 8,734,246 (38.3) |
| Number of sites unique to imputed data (fraction of sites in the file) (%) | 1,167,139 (6.0) | 1,707,553 (8.6) | 2,878,041 (15.0) | 2,127,458 (11.7) | 1,565,412 (10.0) |
| Number of reference allele (REF) mismatches | 0 | 0 | 1 | 4,912 | 0 |
| Number of alternate allele (ALT) mismatches | 637 | 8,536 | 14,083 | 10,814 | 12,794 |
Panel codes: AGR, African Genome Resource hosted at the Sanger Imputation Server (SIS); KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the Michigan Imputation Server; TOPMed, hosted at the TOPMed Imputation Server.
Figure 5B depicts the intersection between TOPMed, AGR, and the WGS dataset. Although TOPMed originally imputes a substantially larger number of SNPs than the AGR, the latter had a slightly better overlap with the SNPs present in the WGS data. The pattern of overlap between the panels also provided an indirect estimate for the maximum improvement that could be possible with a meta-imputation approach. An efficient combination of AGR and TOPMed can increase the coverage of the genome by up to 7%. However, this could also add up to 2.5 million (1.52 million from TOPMed + 0.97 million from the AGR) SNPs to the imputed dataset that are not present in the WGS data (Figure 5B). In the absence of additional very-high-coverage WGS data, it is difficult to distinguish whether the SNPs imputed by the panels but not present in the WGSs represent improvement, noise, or a mix of these two. Nevertheless, it can be assumed that while meta-imputation approaches can lead to considerable gain in the number of imputed SNPs, it may also introduce noise in the final dataset.
For the SNPs that were common to the imputed datasets and WGS, we next assessed the accuracy of the imputed genotypes by estimating the NDRs (against WGS) for these 95 participants for all the five panels. Overall, the AGR panel showed the highest concordance rates (i.e., lowest NDR = 2.23% ± 0.58%) followed by the TOPMed (NDR = 3.57% ± 1.88%) panel (Figure 5C). Furthermore, it needs to be noted that as the size of the imputed dataset and also the WGS overlapping dataset were different, the same NDR reflects different numbers of actual SNPs for each panel.
To investigate whether differences in ancestral composition lead to the large-scale differences in observed NDRs among the 95 individuals, we compared the level of the NDR with the level of Khoe-San ancestry in each individual. For all the imputed datasets, we observed an increase in the NDR with an increase in the level of overall Khoe-San ancestry in an individual (Figure 5D). The dataset imputed using the AGR panel not only showed the overall lowest NDR but also the lowest rise in NDR with the increase in Khoe-San ancestry. Based on these results, we postulate that the inclusion of 84 Nama genomes in this panel to be the source of the considerably better performance of AGR, especially in individuals with substantial Khoe-San ancestry.
The choice of an optimal INFO score (or R2) cutoff value is also an issue that researchers grapple with. To study the extent to which INFO score cutoffs concur with genomic coverage and accuracy, we performed two sets of analyses. In the first set, we compared the proportion of SNPs in each of the imputed datasets (AGR and TOPMed) that showed overlap with the WGS dataset at progressively higher INFO score (or R2) cutoffs (Figure S9). The results show the overlap of each panel with a WGS to decrease marginally as the cutoff increases from 0.30 to 0.60. Moreover, the core set of SNPs that were present in the WGS and imputed by both the panels remained largely robust to the INFO score (or R2) variations. In the second set, we studied the dynamics of NDRs with increase in INFO score (or R2) cutoff (Table 3). We observed an almost negligible change in NDR values even when using a rather stringent INFO score (or R2) cutoff of 0.8. As expected, the NDR shows a significant improvement when an INFO score (or R2) cutoff >0.9 is used, but using such a high imputation quality measure cutoff leads to the loss of about ∼10% (TOPMed) to ∼25% (HRC) of SNPs from the imputed datasets (Table S3). Therefore, while the reduction of discordance is possible with very stringent INFO scores (or R2) cutoffs, the gain in accuracy would be at the expense of having a substantial number of variants filtered out.
Table 3.
Dynamics of non-reference discordance rate (NDR) with increase in INFO or R2 score cutoffs
| INFO score | AGR | TOPMed | KGP_S | KGP_M | HRC |
|---|---|---|---|---|---|
| >0 | 2.23 ± 0.58 | 3.57 ± 1.88 | 7.01 ± 2.37 | 6.74 ± 2.32 | 7.64 ± 2.28 |
| >0.3 | 2.23 ± 0.58 | 3.59 ± 1.90 | 7.01 ± 2.37 | 6.71 ± 2.34 | 7.64 ± 2.28 |
| >0.4 | 2.23 ± 0.58 | 3.59 ± 1.90 | 6.99 ± 2.37 | 6.67 ± 2.35 | 7.63 ± 2.29 |
| >0.5 | 2.21 ± 0.58 | 3.58 ± 1.90 | 6.93 ± 2.36 | 6.61 ± 2.35 | 7.55 ± 2.28 |
| >0.6 | 2.18 ± 0.58 | 3.56 ± 1.90 | 6.80 ± 2.35 | 6.52 ± 2.34 | 7.37 ± 2.27 |
| >0.7 | 2.11 ± 0.57 | 3.50 ± 1.88 | 6.54 ± 2.31 | 6.23 ± 2.33 | 7.00 ± 2.24 |
| >0.8 | 1.95 ± 0.53 | 3.39 ± 1.82 | 6.01 ± 2.20 | 5.91 ± 2.24 | 6.30 ± 2.12 |
| >0.9 | 1.55 ± 0.43 | 2.97 ± 1.63 | 4.70 ± 1.86 | 4.66 ± 1.94 | 4.73 ± 1.76 |
Panel codes: AGR, African Genome Resource hosted at the Sanger Imputation Server (SIS); KGP_S, 1000 Genomes Project hosted at the SIS; HRC, Haplo-type Reference Consortium hosted at the SIS; KGP_M, 1000 Genomes Project hosted at the Michigan Imputation Server; TOPMed, hosted at the TOPMed Imputation Server.
We also compared the allele frequencies observed in the WGS dataset with the allele frequencies in the imputed dataset (for the 95 individuals) for each panel. AGR once again showed the best correlation value and the narrowest diagonal band along with the least number of deviant SNPs, closely followed by the TOPMed panel (Figure S10). Taken together, the WGS-based comparisons consistently show the AGR to be the most accurate, while the TOPMed was the second best. Both were much better than the other panels.
Discussion
The under-representation of African ancestry populations in current GWASs has been flagged as a major concern, and the need to improve this representation has been highlighted repeatedly in recent literature.16,22 Currently, only 2% of individuals included in GWASs are individuals of “African ancestry,” and the vast majority of these African ancestry populations in genetic studies are related to Western African, e.g., African Americans or Afro-Caribbeans (72%–93% in the GWAS catalog and ≥90% in gnomAD).23,24 Even among the limited SSA GWAS datasets that are around, many have been imputed using panels that are not optimal for these populations. Therefore, evaluating the accuracy of imputation in SSA populations is a vital step toward utilizing the full potential of these datasets and improving their inclusion in global GWASs.
Imputation performance of five widely used reference panels was assessed in the pan-African AWI-Gen dataset of ∼10,900 samples. As expected, the largest panel, TOPMed, and the panel that has the highest proportional representation of SSA populations, the AGR, emerged as the best-performing panels. In our dataset, both these panels clearly outperformed global panels such as the KGP and HRC for imputation of SSA datasets. The relatively weaker performance of HRC compared with the KGP was recently observed in a study on African American populations.25 Reiterating the importance of genetic proximity, despite a much larger size, the HRC panel was outperformed by all other panels including both KGP panels. We also observed notable differences in imputed SNP count and INFO score (or R2) for the KGP panel accessed via the SIS and the MIS. These differences could be driven by panel size variation (the KGP_M panel is almost half the size of the KGP_S panel) as well as the use of different imputation algorithms (SIS uses PBWT, while MIS employs MiniMac4). Due to the unavailability of the actual panels for evaluation, we were unable to assess the relative contribution of these factors. However, as observed in previous studies,26,27 both these factors probably contribute to the differential imputation performance of the KGP panel hosted at the SIS and the MIS. Irrespective of panel size, each panel uniquely imputed a considerable number of SNPs. Although a large proportion of these are extremely rare and therefore have less relevance for modest GWASs, the presence of tens of thousands of modest-frequency SNPs in these uniquely imputed datasets shows that GWAS association, replication of signals, and polygenic risk score-based assessment could be impacted by the choice of reference panels.
Several studies have highlighted substantial genetic differentiation within populations from East, West, and South Africa.12,13 However, the systematic evaluation of imputation differences among geographic regions in Africa is not straightforward, as it requires a homogeneous dataset containing samples from three geographic regions that are comparable in all technical aspects such as genotyping platform, sample size, and QC standards. As our full dataset was already genotyped and QCed together, we only selected sample sets of almost equal size from one site each in East, West, and South Africa to represent these regions. Despite the increase in overall representation of African ancestry in some of the more recent reference panels, there is a bias toward West African origin populations and a lack of representation of other African regions and ancestries in them. Our study, for the first time, shows evidence for noticeable variation in the imputation of genotype datasets from different parts of the continent. We were also able to show that the level of specific ancestries such as East African and Khoe-San ancestry in an individual can lead to considerably higher or lower numbers of imputed SNPs. Moreover, the representation of these ancestries in the panel could be critical in determining the content that can be imputed for some populations. For instance, the presence of Khoe-San genomes in the AGR seems to have a positive impact on the imputation of Southern African samples, as demonstrated by much greater differences in imputation of West and South African populations compared with TOPMed. Moreover, the complex pattern flow of African ancestry to the Caribbean Islands and different parts of North and South America28 suggests that this lack of representation could also impact the imputation performance and accuracy for some of the diaspora populations. Therefore, a careful assessment of the sampling geography and ancestry is essential for choosing the optimal imputation panel. It is also critical to focus on efforts to ensure representation of each of these unique African ancestries in the imputation panels in addition to increasing sample sizes and African representation.
For an end user, an estimate of how accurately a reference panel imputes data in their population is as important as the total number of SNPs that are imputed and the imputation scores. Our evaluation based on comparison with a truth dataset consisting of 95 of our South African samples shows that although TOPMed imputes substantially more rare SNPs, the AGR has higher accuracy. Therefore, while TOPMed is generally considered the best imputation panel, the AGR panel may be a better alternative for specific African genetic association studies, especially when they have limited power for testing rare variations due to smaller sample sizes or include individuals from a particular geographic region/ancestry better represented in that panel. Due to the large-scale differences in size of the panels compared, which could intrinsically bias the NDR estimates against larger panels, we did not include sites that were reference/reference across the WGS dataset in our comparisons. Therefore, NDR estimates derived using alternative approaches, such as those based on comparison of individual-level gVCF files, might differ from the results presented here.
As an indirect estimate of the level of difference that might be observed if reference/reference sites in the WGS were included in NDR estimation, we have noted the number of sites (Figure 5B) that were reference/reference in the WGS dataset but had at least one non-reference allele in each of the imputed datasets. The observation of a considerably higher number of such sites for the TOPMed panel (1.45 million) compared with the AGR panel (0.93 million) hints that the consideration of reference/reference sites in the comparisons could further augment the difference in discordance between these panels and the WGS, as the discordance estimates for TOPMed would increase by much larger values compared with the AGR.
The comparison of imputed and truth datasets also revealed that meta-imputation of datasets generated using the two best-performing panels has the potential to further improve the coverage of the genome in the imputed data. However, as around a million variants that were absent in the WGS were imputed by each of these panels, further exploration and evaluation are required to ascertain the accuracy when implementing this approach in SSA datasets.
Based on the evaluation of five popular imputation panels, we recommend a careful consideration of several factors such as sample size, geographic origin, and ancestry composition of the reference panel and target population to inform the choice of the most suitable panel for SSA datasets. While TOPMed, currently the largest and most diverse panel, outperforms other panels in most of the metrics, higher concordance with WGS data makes the AGR a promising alternative for SSA populations. We anticipate that our in-depth comparisons will assist researchers when prioritizing reference panels based on the characteristics of their specific dataset and that it will also inform strategies for future improvement of SSA-focused reference panels.
Limitations of the study
As some of these panels are only available for use on a specific imputation platform that implements a particular algorithm, there is a limit to the combinations of panels and algorithms that could be tested. Also, the TOPMed imputation service provides outputs in genomic GRCh38 only, while the AGR on the SIS only generates results in GRCh37. A LiftOver step is needed to make these datasets comparable, which might lead to some loss of information. Furthermore, the imputation quality assessment metrics provided by these two services, although similar, are not exactly the same, and this could have influenced our evaluations. While the other panels are fixed, TOPMed periodically undergoes updates; therefore, the outcomes might change for more recent versions of the panel. The AWI-Gen dataset was genotyped on the H3Africa SNP array, so the trends presented here may vary for datasets genotyped on other genotyping arrays. As the AGR imputes only biallelic SNPs, we have not included insertions or deletions (indels) and structural variants in our evaluations. Moreover, sex chromosomes were also not included in the current analysis. These factors could also have impacted the genome-wide estimates. Although we do not expect the reported trends to substantially differ between arrays, type of variants (i.e., SNPs and indels), or autosomes and sex chromosomes, further investigations of these categories will be required for a more comprehensive picture. The relatively small sample size of the high-coverage sequence dataset (just about 1%) and its restriction to one geographic region limits the assessment of imputation accuracy.
STAR★Methods
Key resources table
Resource availability
Lead contact
The lead contact for this paper is Ananyo Choudhury (Ananyo.Choudhury@wits.ac.za)
Materials availability
This study did not generate new unique reagents.
Method details
AWI-gen genotype dataset
AWI-Gen is a cohort of ∼12,000 SSA participants sampled from different sites across Africa- Kenya (East), Ghana and Burkina Faso (West) and South Africa (South). More details on this cohort are available in Ramsay et al. and Ali et al.10,11 Of these, about 10,900 samples were genotyped on the 2.3 M SNP H3Africa array (https://chipinfo.h3abionet.org/) at Illumina FastTrack™ Microarray services (Illumina, San Diego, USA). The Illumina pipeline was used to perform the genotype calling. The following quality control (QC) steps were performed for the genotype dataset using PLINKv1.930 - removal of SNPs showing missingness >0.05, minor allele frequency (MAF) < 0.01, and Hardy-Weinberg equilibrium (HWE) p-value <0.0001. Additionally, duplicates, indels, X chromosome, Y chromosome and mitochondrial SNPs, and SNPs that did not match the GRCh37 references alleles were also removed. Samples which had a missing SNP genotyping rate >0.05, showed sex inconsistencies between the recorded and genetic sex and were potential duplicates (PIHAT > 0.9) were excluded. The clean final dataset included 1,729,661 SNPs and 10,903 individuals.
Pre-imputation phasing and imputation
We used five different widely used reference panels to evaluate which performed the best to impute the African genomes. Three of these panels – the African Genome Recourse (AGR), 1000 Genomes Project (KGP_S), Haplotype Reference Consortium (HRC) are hosted at Sanger Imputation Server (SIS) (https://imputation.sanger.ac.uk/), TOPMed panel hosted at TOPMed Imputation Server (TIS) (https://imputation.biodatacatalyst.nhlbi.nih.gov/) and lastly the 1000 Genomes Project (KGP_M) hosted at Michigan Imputation Server (MIS) (https://imputationserver.sph.umich.edu/index.html). For the datasets imputed at SIS, the EAGLE231 + positional Burrows–Wheeler transform (PBWT)32 pipeline was used for pre-phasing and imputation. For the datasets imputed at TIS and MIS on the other hand, EAGLE2 + Minimac429 pipeline was used. The VCF files were downloaded from online servers post imputation and processed using VCFtools33 to remove monomorphic SNPs, SNPs showing missingness >0.05 and HWE p value <0.00001.
Imputation performance evaluation and accuracy
To evaluate the imputation accuracy, we noted the number of SNPs imputed as well as the imputation quality for each panel. The SIS (that implements PWBT algorithm) provides an INFO score (an estimate of the ratio of statistical information about the population allele frequency in the imputed genotypes and in the true genotypes) for imputation quality assessment, while TIS and MIS (that implements Minimac4) provide R2 values (squared correlation between the true and estimated dose of an allele across all imputed samples) for the same. Though by definition, the two evaluation metrics are not the same, and cannot be compared directly, studies have shown that the matrices provided by these two imputation programs often show a high correlation.1,8,38 Hence, for all the imputed datasets, we used an evaluation metrics cut-off of 0.6 irrespective of the imputation algorithm used.
We calculated the total number of imputed SNPs and number of well imputed SNPs (i.e. SNPs with INFO score or R2 >0.6) across all chromosomes for each imputed dataset. As the frequency of an allele can impact the quality of imputation, we calculated the allele frequency using VCFtools and classified the SNPs into seven frequency bins accordingly (0.00–0.001, 0.001–0.005, 0.005–0.01, 0.01–0.05, 0.05–0.1, 0.1–0.5 and 0.5–1.0) and calculated the total number of SNPs, number of well imputed SNPs and aggregate INFO score or R2 per frequency bin. To compare the density and quality of imputed SNPs across chromosomes in the five imputed datasets, we calculated the number of imputed (and well imputed) SNPs and aggregate INFO score or R2 value per 1MB for all the chromosomes in each dataset. The AGR and HRC panels cannot impute INDELs, so we restricted the said analyses to ‘SNPs only’ for KGP_S, KGP_M and TOPMed.
Overlap between imputed datasets
To enable a comparison of imputed datasets generated by the five panels, the TOPMed imputed dataset that was in build GRCh38 was converted to GRCh37 using UCSC liftOver tool.35 The R package UpSetR39 was used to estimate the overlap between these datasets. Allele frequencies of SNPs that were uniquely imputed by each panel were estimated using VCFtools. To compare the occurrence of known GWAS associations among the uniquely imputed SNP sets, we downloaded the GWAS catalog24 (accessed in May 2022), converted it to GRCh37 using UCSC liftOver35 and studied the overlap of these SNPs to the uniquely imputed SNP sets.
To identify cases where the panels even if imputing the same SNPs do not impute the same genotypes across the dataset, we calculated the allele frequency of the SNPs imputed by all five reference panels and compared it among all possible pairs of the imputed datasets, and identified the SNPs that had allele frequency differences greater than 0.01.
Assessing the impact of geography
Since our dataset had representation from three geographic regions across sub-Saharan Africa-East (Kenya), West (Ghana and Burkina Faso) and South (South Africa), we investigated whether the number of SNPs imputed by the reference panels also vary within these three regions. To avoid bias in sample size, we selected representative datasets of similar size from the three SSA regions. Overall, the Eastern region had the lowest sample size in our dataset (n = 1766). Therefore, a similar number of individuals were selected from the other two regions: West (n = 1856) and South (n = 1777). These subsets were extracted separately using BCFtools v.1.534 from the full datasets that were imputed by the AGR and TOPMed panels. SNPs with minor allele count less than one were removed and the residual SNPs were used to estimate the number of imputed SNPs for East, West and South African datasets.
Assessing the impact of non-Niger-Congo ancestry
To investigate the impact of non-Niger-Congo ancestry on the number of imputed SNPs, we used the previously described subset of individuals from East (n = 1766) and South Africa (n = 1777). These individuals were extracted from the pre-imputation genotype dataset and converted to plink format. Both South African and East African dataset was merged with a genotype dataset from Schlebusch et al.17 and Gurdasani et al.12 separately, using PLINKv1.9. For both the merged datasets, SNPs with high missingness and very low allele frequency were removed, and pruned for SNPs in high linkage disequilibrium using PLINK v1.9 (window size of 50 SNPs, with a window slide of 5 SNPs and r2 > 0.5). To estimate the proportions of the non-Niger-Congo ancestry, we used an unsupervised clustering algorithm implemented in ADMIXTURE (v1.3)21 on the merged dataset at K = 3. For the subset of individuals from East Africa, we estimated the proportion of non-Niger-Congo (Afro-Asiatic/Nilo-Saharan/Eurasian) ancestry for each individual. Similarly, for the subset of individuals from South Africa, the level of Khoe-San ancestry was inferred.
To estimate the number of imputed SNPs in each individual, we extracted one individual at a time from the imputed dataset (for both AGR and TOPMed panels) using BCFtools v.1.5 and removed all SNPs with minor allele count equal to zero and an INFO score or R2 value less than 0.6. Next, we performed a regression analysis between the non-Niger-Congo ancestry proportion and imputed SNP count by individual for both East and South Africa.
We further investigated whether the non-Niger-Congo ancestry observed in East and South African individuals have any impact on the genotypes imputed by AGR and TOPMed panels. The BCFtools v.1.5 ‘stats’ module was implemented to estimate the genotype concordance by samples between the AGR-South Africa versus TOPMed-South Africa (n = 1777) and AGR-East Africa versus TOPMed-East Africa (n = 1766). The TOPMed GRCh37 version (generated using liftOver) was used so that the genotypes could be compared directly. The non-reference Discordance Rate (NDR) is calculated using the formula NDR = (Err+Era+Eaa)/(Mra+Maa+Err+Era+Eaa), where Err, Era and Eaa are the homozygous reference, heterozygous and homozygous alternative genotypes mismatch counts, while Mra and Maa are the heterozygous and homozygous alternative genotypes match counts.40 Next, we plotted the NDR of each individual against the proportion of Khoe-San ancestry for South African individuals and Afro-Asiatic/Nilo-Saharan/Eurasian ancestry for East African individuals.
Comparison with 95 high-coverage whole genome sequences
A subset of 95 high-coverage WGS were used as the gold standard to compare the quality of imputation by the five reference panels. The 95 samples of HiSeq/NextGen 151 bp read pairs were aligned with BWA (v0.7.17-r1188).36 After alignment additional improvements were applied to the BAM file. Duplicates were marked with MarkDuplicates included in the GATK package (v4.1.3).37 Base quality recalibration (BaseRecaibrator, ApplyBQSR in GATK v4.1.3) was used to assure high quality base scores. Per sample calling with GATKs HaplotypeCaller (v4.1.3) was conducted followed by joint genotyping on the complete set using CombineGVCFs (v4.1.3) and GenotypeGVCFs (v4.1.3). Quality control was applied using GATK’s VarianRecalibrator which uses existing truth sets to filter out low quality calls. All code for this process can be found here: https://github.com/grbot/varcall. The processed VCF file was subjected to another round of QC by removing SNPs showing missingness greater than 0.05 and minor allele count less than 1. The INDELs and sex chromosomes were also removed. The final QCed file had 95 individuals and 22,823,371 variants. These 95 individuals were then extracted from all the 5 imputed datasets. For the TOPMed panel, the GRCh37 version of the imputed dataset was used. INDELs and SNPs with minor allele count less than one were removed.
The genotype concordance by samples between the sequence data and imputed data of 95 individuals was estimated using the BCFtools stats module. To investigate the change in NDR levels with the levels of INFO score or R2 value threshold, we repeated the above analysis at different INFO score or R2 value cut-offs – 0.3, 0.4, 0.5, 0.6, 0.7, 0.8 and 0.9, and calculated the mean (±SD) NDR for five imputed datasets. We also investigated whether there is any correlation between the NDR levels and the proportion of Khoe-San ancestry in these individuals.
To assess how well the frequency of SNPs in the imputed datasets correlate with the sequence data, for each imputed dataset, the SNPs common to both the imputed and sequence data were considered and frequencies in the two datasets were calculated using VCFtools and compared. We also estimated the SNP overlap (compared to WGS data) by allele frequency bin for AGR and TOPMed.
Acknowledgments
The authors acknowledge the AWI-Gen field workers, phlebotomists, laboratory scientists, administrators, data personnel, and all other staff who contributed to the data and sample collections, processing, storage, and shipping and the participants, without whom this work would not have been possible. The AWI-Gen Collaborative Center is funded by the National Human Genome Research Institute (NHGRI), Office of the Director (OD); the Eunice Kennedy Shriver National Institute Of Child Health & Human Development (NICHD); the National Institute of Environmental Health Sciences (NIEHS); the Office of AIDS Research (OAR); and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), of the National Institutes of Health, under award number U54HG006938 and its supplements, as part of the H3Africa Consortium. Additional funding was granted by the Department of Science and Technology (now the Department of Science and Innovation), South Africa, award number DST/CON 0056/2014. G.B., A.M., and N.M. are funded by the H3ABioNet grant (U24HG006941) as part of H3Africa. The authors thank the anonymous reviewers for their constructive comments and valuable suggestions that helped in improving the quality of the manuscript.
Author contributions
D.S., A.C., and M.R. designed the study. D.S., G.B., and S.H. performed the initial processing and curation of the data. D.S. performed the data analysis with analytic support and advice from A.C., A.M., G.B., M.M., N.M., S.H., and M.R. D.S. and A.C. drafted the manuscript with input and additional editing from all co-authors. All authors critically evaluated and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100332.
Supplemental information
Data and code availability
The data used in this study are available at the European Genome-phenome Archive (EGA) - Genotype Data: EGAD00010001996 and WGS data: EGAD00001006418. These datasets are available subject to controlled access through the Data and Biospecimen Access Committee of the H3Africa Consortium or via collaboration. The details for tools and codes used for conducting this research are provided in the key resource table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Das S., Abecasis G.R., Browning B.L. Genotype imputation from large reference panels. Annu. Rev. Genomics Hum. Genet. 2018;19:73–96. doi: 10.1146/annurev-genom-083117-021602. [DOI] [PubMed] [Google Scholar]
- 2.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K., et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gurdasani D., Carstensen T., Fatumo S., Chen G., Franklin C.S., Prado-Martinez J., Bouman H., Abascal F., Haber M., Tachmazidou I., et al. Uganda genome resource enables insights into population history and genomic discovery in Africa. Cell. 2019;179:984–1002.e36. doi: 10.1016/j.cell.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathias R.A., Taub M.A., Gignoux C.R., Fu W., Musharoff S., O’Connor T.D., Vergara C., Torgerson D.G., Pino-Yanes M., Shringarpure S.S., et al. A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nat. Commun. 2016;7 doi: 10.1038/ncomms12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GenomeAsia100K Consortium The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature. 2019;576:106–111. doi: 10.1038/s41586-019-1793-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schurz H., Müller S.J., van Helden P.D., Tromp G., Hoal E.G., Kinnear C.J., Möller M. Evaluating the accuracy of imputation methods in a five-way admixed population. Front. Genet. 2019;10:34. doi: 10.3389/fgene.2019.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Q., Liu W., Rosen J.D., Huang L., Pace R.G., Dang H., Gallins P.J., Blue E.E., Ling H., Corvol H., et al. Leveraging TOPMed imputation server and constructing a cohort-specific imputation reference panel to enhance genotype imputation among cystic fibrosis patients. HGG Adv. 2022;3 doi: 10.1016/j.xhgg.2022.100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsay M., Crowther N., Tambo E., Agongo G., Baloyi V., Dikotope S., Gómez-Olivé X., Jaff N., Sorgho H., Wagner R., et al. H3Africa AWI-Gen Collaborative Centre: a resource to study the interplay between genomic and environmental risk factors for cardiometabolic diseases in four sub-Saharan African countries. Glob. Health Epidemiol. Genom. 2016;1:e20. doi: 10.1017/gheg.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali S.A., Soo C., Agongo G., Alberts M., Amenga-Etego L., Boua R.P., Choudhury A., Crowther N.J., Depuur C., Gómez-Olivé F.X., et al. Genomic and environmental risk factors for cardiometabolic diseases in Africa: methods used for Phase 1 of the AWI-Gen population cross-sectional study. Glob. Health Action. 2018;11 doi: 10.1080/16549716.2018.1507133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurdasani D., Carstensen T., Tekola-Ayele F., Pagani L., Tachmazidou I., Hatzikotoulas K., Karthikeyan S., Iles L., Pollard M.O., Choudhury A., et al. The African genome variation Project shapes medical genetics in Africa. Nature. 2015;517:327–332. doi: 10.1038/nature13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choudhury A., Aron S., Botigué L.R., Sengupta D., Botha G., Bensellak T., Wells G., Kumuthini J., Shriner D., Fakim Y.J., et al. High-depth genome sequencing in diverse African populations informs migration history and human health. Nature. 2020;586:741–748. doi: 10.1038/s41586-020-2859-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sengupta D., Choudhury A., Fortes-Lima C., Aron S., Whitelaw G., Bostoen K., Gunnink H., Chousou-Polydouri N., Delius P., Tollman S., et al. Genetic substructure and complex demographic history of South African Bantu speakers. Nat. Commun. 2021;12:2080. doi: 10.1038/s41467-021-22207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu K., Das S., LeFaive J., Kwong A., Pleiness J., Forer L., Schönherr S., Fuchsberger C., Smith A.V., Abecasis G.R. Meta-imputation: an efficient method to combine genotype data after imputation with multiple reference panels. Am. J. Hum. Genet. 2022;109:1007–1015. doi: 10.1016/j.ajhg.2022.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bentley A.R., Callier S.L., Rotimi C.N. Evaluating the promise of inclusion of African ancestry populations in genomics. NPJ Genom. Med. 2020;5:5. doi: 10.1038/s41525-019-0111-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlebusch C.M., Skoglund P., Sjödin P., Gattepaille L.M., Hernandez D., Jay F., Li S., De Jongh M., Singleton A., Blum M.G.B., et al. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science. 2012;338:374–379. doi: 10.1126/science.1227721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlebusch C.M., Malmström H., Günther T., Sjödin P., Coutinho A., Edlund H., Munters A.R., Vicente M., Steyn M., Soodyall H., et al. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science. 2017;358:652–655. doi: 10.1126/science.aao6266. [DOI] [PubMed] [Google Scholar]
- 19.Schlebusch C.M., Sjödin P., Breton G., Günther T., Naidoo T., Hollfelder N., Sjöstrand A.E., Xu J., Gattepaille L.M., Vicente M., et al. Khoe-san genomes reveal unique variation and confirm the deepest population divergence in Homo sapiens. Mol. Biol. Evol. 2020;1:2944–2954. doi: 10.1093/molbev/msaa140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choudhury A., Sengupta D., Ramsay M., Schlebusch C. Bantu-speaker migration and admixture in southern Africa. Hum. Mol. Genet. 2021;30:R56–R63. doi: 10.1093/hmg/ddaa274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fatumo S., Chikowore T., Choudhury A., Ayub M., Martin A.R., Kuchenbaecker K. A roadmap to increase diversity in genomic studies. Nat. Med. 2022;28:243–250. doi: 10.1038/s41591-021-01672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin A.R., Teferra S., Möller M., Hoal E.G., Daly M.J. The critical needs and challenges for genetic architecture studies in Africa. Curr. Opin. Genet. Dev. 2018;53:113–120. doi: 10.1016/j.gde.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connell J., Yun T., Moreno M., Li H., Litterman N., Kolesnikov A., Noblin E., Chang P.-C., Shastri A., Dorfman E.H., et al. A population-specific reference panel for improved genotype imputation in African Americans. Commun. Biol. 2021;4:1269. doi: 10.1038/s42003-021-02777-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng T., Zhang P., Garrick D., Gao H., Wang L., Zhao F. Comparison of genotype imputation for SNP array and low-coverage whole-genome sequencing data. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.704118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stahl K., Gola D., König I.R. Assessment of imputation quality: comparison of phasing and imputation algorithms in real data. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.724037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patin E., Lopez M., Grollemund R., Verdu P., Harmant C., Quach H., Laval G., Perry G.H., Barreiro L.B., Froment A., et al. Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science. 2017;356:543–546. doi: 10.1126/science.aal1988. [DOI] [PubMed] [Google Scholar]
- 29.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loh P.-R., Danecek P., Palamara P.F., Fuchsberger C., A Reshef Y., K Finucane H., Schoenherr S., Forer L., McCarthy S., Abecasis G.R., et al. Reference-based phasing using the haplotype reference Consortium panel. Nat. Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durbin R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT) Bioinformatics. 2014;30:1266–1272. doi: 10.1093/bioinformatics/btu014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F., et al. The UCSC genome browser database: update 2006. Nucleic Acids Res. 2006;34:D590–D598. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Van der Auwera G.A., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D., et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2018 doi: 10.1101/201178. Preprint at. [DOI] [Google Scholar]
- 38.Browning B.L., Browning S.R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016;98:116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lex A., Gehlenborg N., Strobelt H., Vuillemot R., Pfister H. UpSet: visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 2014;20:1983–1992. doi: 10.1109/TVCG.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubinacci S., Ribeiro D.M., Hofmeister R.J., Delaneau O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat. Genet. 2021;53:120–126. doi: 10.1038/s41588-020-00756-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this study are available at the European Genome-phenome Archive (EGA) - Genotype Data: EGAD00010001996 and WGS data: EGAD00001006418. These datasets are available subject to controlled access through the Data and Biospecimen Access Committee of the H3Africa Consortium or via collaboration. The details for tools and codes used for conducting this research are provided in the key resource table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.