

Abstract
The relationship between drug-target residence time and the post-antibiotic effect (PAE) provides insight into target vulnerability. To probe the vulnerability of bacterial acetyl-CoA carboxylase (ACC), a series of heterobivalent inhibitors were synthesized based on pyridopyrimidine 1 and moiramide B (3) which bind to the biotin carboxylase and carboxyltransferase ACC active sites, respectively. The heterobivalent compound 17, which has a linker of 50 Å, was a tight binding inhibitor of E. coli ACC (Kiapp 0.2 nM) and could be displaced from ACC by a combination of both 1 and 3, but not by just 1. In agreement with the prolonged occupancy of ACC resulting from forced proximity binding, the heterobivalent inhibitors produced a PAE in E. coli of 1-4 h in contrast to 1 and 3 in combination or alone, indicating that ACC is a vulnerable target and highlighting the utility of kinetic, time dependent effects in drug mechanism of action.
Keywords: ACC, residence time, post-antibiotic effect, avidity, heterobivalent, linkers
Graphical Abstract
INTRODUCTION
Drug activity is controlled by both the thermodynamics and kinetics of drug-target complex formation and breakdown, and there is extensive evidence that the life-time of the drug target complex can have a profound impact on drug pharmacology.1-6 The translation of extended target occupancy to prolonged drug activity depends on factors such as target vulnerability and the rate of target turnover,6 and previously, we demonstrated that the correlation between drug-target residence time and post-antibiotic effect (PAE) could provide direct insight into target vulnerability. Using this approach, we found that the ribosome and the LpxC enzyme from Pseudomonas aeruginosa were highly vulnerable, based on the observation of a strong positive correlation between residence time and PAE.7-11 To apply this approach to other systems requires the development of slowly dissociating inhibitors for the target of interest.
We have now extended our studies to acetyl-CoA carboxylase (ACC), which initiates fatty acid synthesis by generating malonyl-CoA from acetyl-CoA and HCO3−(Figure 1).
Figure 1. The acetyl-CoA carboxylase (ACC) reaction and ACC inhibitors.

A) Biotin carboxylase (BC) catalyzes the ATP-dependent carboxylation of biotin which is covalently tethered to biotin carboxyl carrier protein (BCCP). The carboxybiotin is then transferred to the carboxyltransferase domain (CT) by BCCP, where acetyl-CoA is carboxylated to form malonyl-CoA. B) Pyridopyrimidine 1 (IC50 < 5 nM)12 and aminooxazole 2 (IC50 125 nM)13 are biotin carboxylase (BC) inhibitors. The natural product moiramide B (3, IC50 6 nM) inhibits the carboxyltransferase (CT) domain.14
ACC is a multienzyme complex composed of biotin carboxylase (BC), biotin carboxyl carrier protein (BCCP), and carboxyltransferase (CT). Biotin, which is covalently attached to BCCP, is carboxylated by the BC domain in an ATP-dependent reaction,15 after which BCCP transfers carboxybiotin to the CT domain where acetyl-CoA is carboxylated.14,16,17 Unlike eukaryotic ACC, which is encoded on a single polypeptide, the prokaryotic ACC consists of four proteins that form a non-covalent multienzyme complex.18 These structural differences enable the selective inhibition of bacterial ACC, which is an attractive target for broad-spectrum antibiotic development.19,20 Compounds that inhibit either the BC or CT domains have been developed and have antibacterial activity against both Gram-positive and negative pathogens, including Staphylococcus aureus, Streptococcus pneumoniae, Escherichia coli, and Haemophilus influenza.12-14,21,22
We hypothesized that bivalent inhibitors of ACC, designed to simultaneously engage both the BC and CT active sites, would have increased affinity and potentially longer residence times on ACC due to avidity effects compared to the individual BC and CT inhibitors, providing an opportunity to evaluate the vulnerability of ACC by exploring the correlation between residence time and PAE. This strategy builds on the work of Waldrop and coworkers,23 who previously reported bivalent ACC inhibitors based on an aminooxazole BC inhibitor 2 and the natural product CT inhibitor moiramide B 3 (Figure 1), which displayed broad-spectrum antibacterial properties and impeded the development of bacterial resistance.
Although there is no structure of the entire bacterial ACC multienzyme complex, structures are available of individual components as well as of related bacterial biotin-dependent carboxylase holoenzymes and the eukaryotic fatty acid synthase complex.24-28 Collectively, the structural data indicates that the BC and CT active sites can be separated by distances of anywhere from 40 to 80 Å,29 and thus we designed bivalent compounds with linkers of up to 80 Å compared to the bivalent compounds previously reported in which the two pharmacophores were separated by ~12 and 20 Å linkers.23 In addition, we replaced the BC inhibitor 2 with the pyridopyrimidine 1 (Figure 1) which inhibits the BC domain with an IC50 value of <5 nM, which is at least 25-fold more potent than 2. We subsequently synthesized a library of heterobivalent inhibitors based on 1 ranging from ~40 to ~110 Å in length, which demonstrated increased affinity for ACC compared to their parent warheads. Using a spin-column LC-MS/MS-based competition assay, the bivalent inhibitors could be displaced when treated with a combination of both 1 and 3, but not with a 100-fold excess of either 1 or 3 alone indicating very high affinity binding and/or long residence time on the enzyme. We propose that this is an example of ‘forced proximity’ where the binding of one ligand brings the second ligand into close proximity with the target, increasing its local concentration.30 Consistent with this observation, the heterobivalent inhibitors produced a PAE in E. coli, in contrast to either 1 or 3 suggesting that ACC is a vulnerable target for antibiotic discovery.
RESULTS
Heterobivalent Inhibitor Synthesis
The overall design of the novel heterobivalent ACC inhibitors involved linking the pyridopyrimidine BC inhibitor 1, to the CT inhibitor moiramide B 3, via a polyethylene glycol (PEG) linker of various lengths using copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC).31,32 We employed CuAAC to facilitate compound synthesis,33 and PEG units to increase the overall solubility of the heterobivalent compounds.34,35 The pyridopyrimidine 1 possesses better biochemical potency and antibacterial activity than the aminooxazole 2 (IC50 <5 vs 125 nM, MIC 16 vs >64 μg/mL, respectively), and it had already been demonstrated that 3 could be easily modified without alteration in antibacterial activity.21-23 In the X-ray crystal structure of 1 bound to BC, the dibromo ring of the inhibitor is solvent exposed (Figure 2), and thus we synthesized a small library of analogs with linker attachments at the meta and para positions of 1 to determine the most amenable position for synthetic modification (Figure 3, full SAR available in Table S1). While incorporation of a methoxy group at either position reduced the biochemical and antibacterial activity compared to 1, modification of the meta- position was found to have 4-fold smaller impact, and thus this position was chosen as the starting point for linker attachment.
Figure 2. Structure of 1 bound to the BC domain.

A) Residues surrounding 1 in the BC active site. B) Surface representation indicates that the dibromo ring of 1 is solvent exposed and suggests that the meta position (m) is more amenable to modification than the para position (p). The figure was made with PyMol,36 using PDB ID 2V58.12
Figure 3. Initial SAR of 1.

Meta position derivatives of 1 (S7, S11) retain more potency than para position derivatives (S8, S12). This trend is consistent through the initial SAR, the full scope which is in Table S1.
While it would have been an optimal point of divergence, late stage alkylation to introduce the linker failed. As a result, we had to redesign the synthesis of compound 1 analogs so that the linker was installed at an earlier stage of the synthesis. We found that linkers could be successfully installed onto intermediate 6, and that the resulting compounds (8) could be subjected to the Friedlaender synthesis to generate the desired derivatives of 1 with various linker lengths that incorporated a terminal alkyne for CuAAC (Scheme 1).
Scheme 1. Synthesis of the meta-substituted pyridopyrimidine alkynes.

Reagents and conditions: (a) TBSCl, Imidazole, DMAP, CH2Cl2, 22°C, 84%; (b) NBS, benzoyl peroxide, CCl4, reflux, 76%; (c) NaCN, DMF, H2O, 0°C to 22°C, 79%; (d) K2CO3, DMF, 80°C, 59-90%; (e) NaH, 2-ethoxyethanol, reflux, 17-54%.
Linkers were synthesized in increments of ~10 Å and attached to 1 before cyclization (Scheme 1), yielding analogs of 1 appended with alkyne groups of increasing length. Linker synthesis involved PEGylation of a tosylated alkyne, in which the tosyl group both served as a convenient leaving group and a UV active group for reaction monitoring the reactions (Scheme 2).
Scheme 2. Synthesis of alkyne linkers.

Reagents and conditions: (a) NaH, DMF, 0°C to 22°C, 25-64%; (b) triethylamine, 4-toluenesulfonyl chloride, CH2Cl2, 0°C to 22°C, 62-86%.
The corresponding azide counterpart for the CuAAC reaction was generated in a straightforward fashion beginning with 11-bromoundecanoic acid. Displacement of the bromide with sodium azide delivered carboxylic acid 12. Boc deprotection of 13 using TFA yielded the amine that could then be coupled to carboxylic acid 12 using HATU DIPEA to deliver the desired moiramide azide analog (14) in 69% yield (Scheme 3).
Scheme 3. Synthesis of the moiramide B azide analogue.

Reagents and Conditions: (a) NaN3, DMSO, 22°C, 90%; (b) 1. TFA: CH2Cl2, 0°C; 2. HATU, DIPEA, DMF, 22°C, 69%.
Subsequently the monovalent analogues of 1 and 3 were coupled using CuAAC to generate a library of heterobivalent compounds (Scheme 4). In addition, a heterobivalent compound (17-p) with the linker attached to the para-position of 1 was developed as a control to assess the specificity of the heterobivalent molecules for the ACC complex.
Scheme 4. Synthesis of the heterobivalent inhibitors.

Reagents and conditions: (a) CuSO4, sodium ascorbate, 9:1 DMF:H2O, 22°C, 12-35%.
Biochemical and Antibacterial Activity
Inhibition of the ACC complex was determined for the parent compounds (1 and 3), the monovalent inhibitor analogues (9a-9f, 9c-p and 14), and the heterobivalent inhibitors (15-20) using a fluorescence assay in which the formation of ADP displaced a fluorescent ADP analog from an antibody leading to change in polarization. The fluorescence polarization assay was more sensitive than the standard coupled assays,12 and enabled routine reactions to be run with only 1 nM compared to 10 nM. Previous studies have shown that both efflux and cell penetration is an issue for the ACC inhibitors, and so antibacterial activity was evaluated towards both wild-type and efflux pump mutant strains of E. coli in the presence of polymyxin B nonapeptide (PMBN).37
The IC50 values for 1 and 3 were similar to reported values, while the IC50 values of the monovalent alkyne analogs of 1 increased with the attachment size of the linkers (Table 1). Specifically, whereas 1 had an IC50 value of 1 nM, respectively, the IC50 values of the m-substituted alkyne analogs (9) ranged from ≤ 1 nM (hexyne linker 9a) to 126 ± 36 nM (PEG 18 linker 9g). As expected, the regioisomer of 9c with the linker at the para- position (9c-p) had an IC50 value of >500 nM compared to 65 nM consistent with the initial SAR (Table S1). In addition, 14, the monovalent azido analogue of 3, had an IC50 value of 74 ± 12 nM compared to the value of 5 nM for 3 (Table 1).
Table 1:
Biochemical and antibacterial activity of the monovalent inhibitors
| Compound | IC50 (nM)a | MIC (μM)b | |||||
|---|---|---|---|---|---|---|---|
| WTc | WTc + PMBNf |
ΔacrABd | ΔacrABd + PMBNf |
ΔtolCe | ΔtolCe + PMBNf |
||
| 1 | ≤ 1 | ≥ 25 | 3.13 | 3.13 | 0.78 | 1.56 | 0.78 |
| 3 | 4.5 ± 1.8 | > 25 | 3.13 | 25 | 0.39 | 1.56 | 0.1 |
| 9a | ≤ 1 | > 25 | 12.5 | > 25 | 0.78 | 1.56 | 0.78 |
| 9b | 19 ± 4 | > 25 | > 25 | > 25 | 3.13 | 3.125 | 1.56 |
| 9c | 32 ± 6 | > 25 | > 25 | > 25 | 6.25 | 6.25 | 6.25 |
| 9c-p | 849 ± 166 | NDg | NDg | >25 | >25 | >25 | >25 |
| 9d | 98 ± 21 | > 25 | > 25 | > 25 | 25 | 25 | 12.5 |
| 9e | 82 ± 17 | > 25 | > 25 | > 25 | > 25 | 25 | 12.5 |
| 9f | 64 ± 11 | > 25 | > 25 | > 25 | > 25 | 25 | 12.5 |
| 9g | 126 ± 36 | > 25 | > 25 | > 25 | > 25 | > 25 | 12.5 |
| 14 | 74 ± 12 | > 25 | 12.5 | > 25 | 1.56 | 1.56 | 0.2 |
ACC activity was determined using a fluorescence polarization assay. Reactions were performed in triplicate and the errors are standard deviation from the mean.
MIC values were determined by the microbroth dilution method. Experiments were performed in triplicate.
Wild-type, E. coli strain (K-12, MG1655).
E. coli ΔacrAB, knockout of the acrAB inner efflux pump.
E. coli ΔtolC, knockout of the tolC outer efflux pump.
PMBN was present at final concentration of 6 μM (8 μg/mL).
ND, not determined.
As observed previously for 1 and 3, no antibacterial activity was observed against the wild-type strain of E. coli unless PMBN was included in the media and/or when the acrAB or tolC efflux pumps were disabled.12,14,23,37 Antibacterial activity of the monovalent analogs of 1 followed a similar trend to the IC50 values for enzyme inhibition, with MIC values increasing with linker length, while 14, the azido analog of 3 had antibacterial activity that was similar to 3. A checkerboard MIC assay was also performed for 1 and 3 against E. coli ΔtolC in the presence and absence of PMBN to explore potential synergy of the ACC inhibitors. The fractional inhibitory concentration (FIC) index values for 1 and 3 showed additive effects for E. coli ΔtolC where the lowest FIC index value was 0.5. However, in the presence of PMBN an FIC index value of 0.38 (0.025 μM 3, 0.1 μM 1) was observed indicating synergistic activity.
In contrast to the monovalent warheads, the heterobivalent inhibitors (15-20) increased in potency as the linker length increased (Table 2). Compound 15 with the shortest linker had an IC50 of 1.8 nM for inhibition of ACC and an MIC value of 0.78 μM against E. coli ΔtolC in the presence of PMBN. Compounds 16 to 20, with linkers that increased from n = 3 to 15, respectively, exhibited higher biochemical potency than the parent compounds and were tight binding inhibitors. Ki values calculated using the Morrison equation gave values of 0.2-0.3 nM for 16 to 20 which was at the limit of the enzyme assay.38 As observed for the monovalent compounds, the antibacterial activity of the bivalent ACC inhibitors generally followed biochemical potency and were more active towards the E. coli ΔtolC strain (+/−PMBN) compared to the wild-type or ΔacrAB strains. The lowest MIC observed for E. coli ΔtolC in the presence of PMBN was 0.39 μM for 17, which is the same as that seen for 1 although higher than the MIC of 3 (0.1 μM). As observed for the monovalent analogs of 1, 17-p, the para-analog of 17, was 10-fold less potent in the biochemical assay, and 2-fold less active in the antibacterial assay than 17.
Table 2.
Biochemical and antibacterial activity of the heterobivalent inhibitors
| Compound | IC50 (nM)a | MIC (μM)b | |||||
|---|---|---|---|---|---|---|---|
| WTc | WTc + PMBNf |
ΔacrABd | ΔacrABd + PMBNf |
ΔtolCe | ΔtolCe+ PMBNf |
||
| 15 | 1.81 ± 0.4 (IC50) | >25 | > 25 | > 25 | 12.5 | > 25 | 0.78 |
| 16 | 0.26 ± 0.09 (Ki) | >25 | > 25 | > 25 | 6.25 | 3.13 | 0.78 |
| 17 | 0.25 ± 0.06 (Ki) | >25 | > 25 | > 25 | 3.13 | 3.13 | 0.39 |
| 17-p | 3.53 ± 0.4 (IC50) | NDg | NDg | >25 | >25 | 6.25 | 0.78 |
| 18 | 0.31 ± 0.1 (Ki) | >25 | > 25 | > 25 | 3.13 | 3.13 | 0.78 |
| 19 | 0.18 ± 0.05 (Ki) | >25 | > 25 | > 25 | 6.25 | 6.25 | 1.56 |
| 20 | 0.33 ± 0.4 (Ki) | >25 | > 25 | > 25 | 12.5 | 12.5 | 1.56 |
ACC activity was determined using the fluorescence polarization assay. Data were analyzed using the Morrison equation if the compound displayed tight binding behavior (IC50 < 0.5 nM). The experiment was performed in triplicate and the errors are standard deviation from mean.
MIC values were determined by the microbroth dilution method. Experiments were performed in triplicate.
Wild-type, E. coli strain (K-12, MG1655).
ΔacrAB, knockout of inner efflux pump acrAB of E. coli
ΔtolC, knockout of outer efflux pump tolC of E. coli
PMBN was present at final concentration of 6 μM (8 μg/mL).
ND=Not Determined.
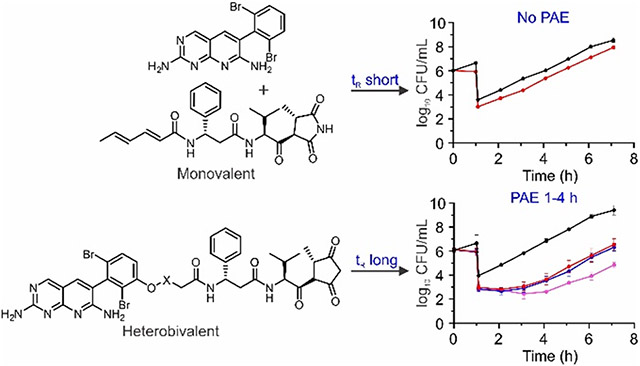
The time-dependent antibacterial activity of the ACC inhibitors was evaluated by determining the post-antibiotic effect (PAE) against E. coli ΔtolC in the presence of PMBN. While the parent compounds 1 and 3 displayed no PAE either when used alone (data not shown), or when used in the synergistic and additive FIC combinations (Figure S1), all the heterobivalent ACC inhibitors displayed a PAE (Figure 4, Table 3). In addition, consistent with the lower potency of the p-substituted analogs of 1, the PAE of 17-p (Figure S2), was shorter than that of 17, further supporting the proposal that the activity of the compounds stems from specific inhibition of ACC. Finally, time kill assays demonstrated that all compounds were bacteriostatic, similar to the individual warheads (Figure S3).39
Figure 4. Post antibiotic effect of the ACC inhibitors against E. coli ΔtolC.

A) PAE of 15; B) PAE of 16; C) PAE of 17; D) PAE of 18; E) PAE of 19, F) PAE of 20. Cultures of E. coli ΔtolC (106 CFU/mL) were treated with 6 μM (8 μg/mL) PMBN and 0x, 8x, 16x, or 32x MIC of each inhibitor for 1 h followed by 1:1000-fold dilution into fresh cation-adjusted Mueller-Hinton (CaMH) media at 37 °C. Samples (100 μL) of the diluted cultures were then plated on Muller-Hinton agar plates every hour, and CFUs enumerated following incubation of the plates at 37 °C for 16 h. The experimental data points are the mean values from duplicate, independent measurements, and the error bars represent the standard deviation from the mean.
Table 3:
Post-antibiotic effect (PAE) against E. coli ΔtolC + PMBN
| Compound | MIC (μM) | PAE (h)a | ||
|---|---|---|---|---|
| 8x | 16x | 32x | ||
| 15 | 0.78 | 1.9 ± 0.2 | 2.2 ± 0.3 | 3.8 ± 0.2 |
| 16 | 0.78 | 1.0 ± 0.2 | 1.2 ± 0.3 | 2.6 ± 0.09 |
| 17 | 0.39 | 1.7 ± 0.3 | 1.7 ± 0.2 | 3.4 ± 0.2 |
| 18 | 0.78 | 1.2 ± 0.4 | 1.8 ± 0.1 | 3.4 ± 0.02 |
| 19 | 1.56 | 2.1 ± 0.1 | 2.6 ± 0.07 | 3.6 ± 0.07 |
| 20 | 1.56 | 1.9 ± 0.3 | 3.8 ± 0.3 | 4.6 ± 0.2 |
The PAE was calculated using a standard procedure in which the time required for the bacteria to recover 1 log after washing out the inhibitor was compared to the culture treated with the vehicle (DMSO). Experiments were performed in triplicate, and the reported values are the average of the three independent experiments with the errors representing the standard deviation from the mean.
We attempted to measure the residence times of the inhibitors on ACC using a traditional jump dilution activity assay. However, this assay was unable to measure activities at the very low enzyme concentrations thought to be needed to ensure complete dissociation of the inhibitors (<0.1 nM). We therefore used a competition-based assay, in which the heterobivalent enzyme-inhibitor complexes were incubated with high concentrations of the parent compounds prior to separating free from bound inhibitor using size exclusion spin columns.11 Following centrifugation, the bound inhibitor was extracted using acetonitrile to denature the enzyme and then quantified by LC-MS/MS.
The ACC complex (1 μM) was incubated with an equimolar concentration of the heterobivalent inhibitor 17 for 1 h at 22 °C, and then subjected to rapid gel filtration using a gel filtration centrifuge SpinTrap column. Quantitative LC/MSMS was used to determine the concentration of the inhibitor in the flow through following addition of buffer, (50% MeCN, 50% 10 mM ammonium formate pH 8.0) to denature the protein. In the control experiments, the concentration of 17 was in the flow through was shown to be similar to the protein concentration determined using a Bradford assay. The experiment was repeated following 1 h incubation of the ACC-heterobivalent complex by addition of 100 μM of either 1 or 3, or 50 μM of 1 and 3 together. Whereas even a 100-fold excess of 1 or 3 is unable to displace 17 from ACC, the combination of both 1 and 3 causes rapid dissociation (Figure 5). Experiments with 15, 16, 18-20, gave similar results in which little or no dissociation of the bivalent inhibitor occurred following incubation of the ACC-inhibitor complex with 1 alone whereas rapid dissociation occurred in the presence of both 1 and 3 (Figure S4).
Figure 5. Competition assay.

ACC 1 μM was incubated with 1 μM of 17 for 1 h at 22 °C after which either 100 μM of 1 or 3 alone, or 50 μM of 1 and 3 were added. Samples were subject to gel filtration using SpinTrap columns at different times after addition or the monovalent compounds, and the concentration of 17 in the flow through was quantified by LC/MSMS after addition of buffer. No dissociation of 17 is observed when 1 or 3 alone is incubated with the ACC:17 complex (blue and purple). However, rapid dissociation of 17 was observed when 1 and 3 were incubated together with the ACC:17 complex (red). Fitting of the data to a single exponential function in GraphPad Prism gave a rate of inhibitor dissociation of 0.3 min−1 when both 1 and 3 were used, giving a residence time of 17 on ACC of 3.3 min.
DISCUSSION
Target vulnerability quantifies the fractional drug target engagement required to induce the desired physiological response.6,10,11 High vulnerability targets require low levels of engagement while low vulnerability targets require high levels of engagement to produce the desired response. Target vulnerability is thus a critical factor in determining the drug exposure required for the pharmacodynamic response, and the lower drug levels required for a high vulnerability target are expected to translate into lower, less frequent drug doses and a widening of the therapeutic window. Since antibiotics are often given at high levels for sustained periods of time, approaches that reduce the required drug exposure are likely to improve the success rate of new drug approvals.7 Target vulnerability can be assessed using time-dependent measurements of drug activity, and in antibacterial space time-dependent cellular activity is assessed using the post-antibiotic effect (PAE), which is the delay in bacterial regrowth following compound washout. The PAE has important implications for dosing regimens and can have several origins, including the slow dissociation of the drug from the target. We have shown that the correlation between residence time and PAE can inform on target vulnerability and using this approach, we have shown that the LpxC enzyme from P. aeruginosa and the bacterial ribosome are highly vulnerable targets.7-10 In contrast, no PAE is observed in E. coli either for inhibitors of the E. coli LpxC or for β-lactam antibiotics, which in both cases is due to rapid target resynthesis and low target vulnerability.11 To extend this approach to other targets requires the availability of long residence time or irreversible inhibitors, and in the present work we have sought to develop long residence time inhibitors of acetyl-CoA carboxylase (ACC), which catalyzes the first committed step in fatty acid biosynthesis. ACC has two active sites which collectively act together to catalyze the carboxylation of acetyl-CoA to generate malonyl-CoA. Building on the work of Waldrop and coworkers,23 the goal of the present work was to develop heterobivalent inhibitors of ACC based on the premise that such compounds would have high affinity and potentially long residence time on the ACC multienzyme complex.
The importance of multivalent interactions in biological systems is well documented and includes the binding of viral particles and bacteria to mammalian cells, and the interaction of antibodies with antigens.40 In most cases polyvalent interactions have much higher affinity than the binding of equivalent monovalent ligands, although the magnitude of the effect depends on the cooperativity of the polyvalent interaction. For small molecules, bivalent ligands have been used extensively in G protein-coupled receptors, for example by linking pharmacophores that bind to orthosteric and allosteric binding sites.41 Bivalent ligands can exhibit both an increase in affinity and potentially slower off rates, which is due to the increase in local concentration which arises when a pharmacophore that dissociates from a binding site remains in ‘forced proximity’ as long as the companion pharmacophore is still bound.30,41 Such effects have been observed for multivalent inhibitors of voltage-gated sodium (NaV) channels, M2 muscarinic acetylcholine receptors, and muscarinic receptor antagonists and beta2-adrenoceptor agonists.42-45 In the case of NaV, the heterobivalent ligands display apparently irreversible binding when compounds are used that compete at only one active site.42,43
In the present work we have synthesized a series of tight binding heterobivalent ACC inhibitors based on the BC and CT inhibitors 1 and 3, respectively. The dibromo ring of 1 is solvent exposed and, in agreement with the structural data, modification of the meta-position of the dibromo ring is preferred compared to the para-position. In contrast to the individual pharmacophores, in which an increase in the length of the attached linker leads to a reduction in affinity, heterobivalent inhibitors based on 1 and 3 show an increase in affinity as the linker is lengthened from ~25 Å in 15 (IC50 1.8 nM). However, elucidating the precise structure activity relationship for inhibition of ACC is hindered since the apparent Ki values for compounds 16-20 (0.2-0.3 nM) are at the limit of the assay. In terms of antibacterial activity, the lowest MIC is observed for 17 (0.39 μM) which has a linker of ~ 50 Å. Importantly, the observation that the Ki value for the para-analogue of 17 (17-p) is at least 10-fold higher than that of 17 (3.53 v 0.25 nM, respectively), and also shows a 2-fold increase in MIC, supports the belief that the compounds interact specifically with the ACC binding sites.
The very high affinity of the compounds for ACC prevented the use of traditional jump dilution methods to quantify the residence time of the inhibitors on ACC since we were unable to measure enzyme activity at low enough concentrations where the inhibitors would dissociate from ACC. We therefore turned to a competition displacement assay to measure drug-target residence time. While a mixture of 1 and 3 is able to displace the heterobivalent inhibitors from ACC, consistent with the specific binding of these compounds to the enzyme, 1 or 3 alone are unable to displace the compounds from ACC. The inability of the individual warheads to displace the heterobivalent inhibitors from ACC supports the forced proximity effect for bivalent inhibitors which increases their residence time on the target.30 In keeping with the displacement experiments, which indicate that 17 has a long residence time on ACC, the bivalent inhibitors display a PAE of 1-2 h at 8xMIC towards E. coli which rises to 3-4 h at 32xMIC. In contrast no PAE is observed for either 1 or 3 either alone or in combination. These data indicate that ACC is a vulnerable target for antibacterial drug discovery.
While the bivalent compounds are useful tools for probing the relationship between residence time and PAE, they clearly do not adhere to the rule of five,46 and are not suitable for in vivo studies. In addition, antibacterial activity is only for observed for the ΔacrAB pump mutant in the presence of PMBN (6 μM, 8 μg/mL) while the MIC for the ΔtolC E. coli strains is decreased 4-30-fold by the addition of PMBN. For instance, the MIC for 17 against E. coli ΔtolC drops from 3.13 μM to 0.39 μM (4 μg/mL to 0.5 μg/mL) when PMBN is included in the assay. Previous studies on ACC inhibitors have reported similar observations,23,37 indicating that both penetration and efflux is an issue for these compounds, and the antibacterial activity of many antibiotics against E. coli is potentiated by PMBN such as azithromycin (10-30-fold) and rifampin (30-300-fold).47 The current generation of compounds utilized a single linker chemistry and further optimization will focus on the generation of more drug-like bivalent inhibitors with shorter linkers. In addition, the inclusion of CuAAC in the synthesis of the current series raises the potential for in cell self-assembly of two smaller precursors as observed for hetero-bifunctional proteolysis targeting chimeras.48,49 Ultimately, a structure of the prokaryotic ACC is needed to fully exploit the potential of this drug target and to optimize linker composition, orientation, and rigidity. In this regard the heterobivalent inhibitors may prove useful for stabilizing the multienzyme complex for structural studies which are currently in progress.
CONCLUSION
Although there is increasing recognition that drug-target kinetics play a major role in drug action, the ability to probe the relationship between prolonged target occupancy and time-dependent drug activity is hindered by the lack of strategies for rationally modulating drug-target residence time. Here we describe the synthesis of heterobivalent compounds that inhibit bacterial ACC, a target for antibacterial discovery, with sub-nM potency. ACC is a multienzyme complex that contains two active sites, and the bivalent inhibitors bind simultaneously to both active sites resulting in forced proximity binding which results in long residence time of the compounds on the enzyme. In contrast to the monovalent inhibitors, the bivalent compounds generate a post-antibiotic effect in E. coli, substantiating the relationship between residence time and prolonged drug activity following compound washout, and thereby demonstrating that ACC is a vulnerable target for intervention.
EXPERIMENTAL SECTION
General Chemistry.
All chemicals and reagents were purchased from Sigma-Aldrich, Fisher, Aces Pharma, AmBeed, or AA Blocks and used without further purification. Flash chromatography was performed using a Teledyne CombiFlash system using prepackaged silica cartridges from Teledyne. All reactions were run under inert atmosphere unless otherwise noted. RP-HPLC was performed with a Shimadzu LC-20AB pump, Shimadzu SIL-20A HT auto sampler, and Shimadzu SPD-M20A diode array detector using Shimadzu Prominence LC. RP-HPLC was performed using a Luna® C18 column (5μM, 250mm x 10 mm) (Phenomenex) with product eluting from a mixture of solvent A (water, 0.1 % TFA) and solvent B (acetonitrile, 0.1% TFA). TLC analysis was performed on aluminum backed 60 F254 silica sheets from Sigma Aldrich. NMR spectra were recorded on a Bruker Ascend 700 MHz, Bruker Avance III 500 MHz, or Bruker Nanobay 400 MHz spectrometers and referenced to deuterated solvent peak. NMR spectra were processed using Mestrelab Research’s Mnova software (Santiago de Compostela, Spain). ESI-MS was performed on an Aligent 6110 single quad mass spectrometer, HRMS was performed on a Bruker Impact II QTOF mass spectrometer, and MALDI mass spectrometry was performed on a Bruker Microflex MALDI-TOF instrument. All final compounds were purified to >95% as assessed by HPLC with UV detection at λ= 213 and 353 nm.
Experimental details for Schemes 1, 2, 3, and 4.
tert-butyl(2,4-dibromo-3-methylphenoxy)dimethylsilane (4)
Tert-Butyldimethylsilyl chloride (3.4 g, 22.6 mmol, 1.2 equiv), imidazole (3.84 g, 56.4 mmol, 3 equiv), and 4-(dimethylamino)pyridine (230 mg, 1.88 mmol, 0.1 equiv) were added to a solution of 2,4-dibromo-3-methylphenol (5 g, 18.8 mmol, 1.0 equiv) in CH2Cl2 (40mL). After 2 h, TLC showed reaction completion. The mixture was washed with 1 M HCl (2 x 50 mL) and brine (50 mL). The organic layer was then dried over MgSO4, filtered, and concentrated. The crude oil was purified using flash chromatography, eluting with hexanes to yield 3 as a clear oil (5.97g, 84%). 1H NMR (700 MHz, CDCl3) δ 7.34 (d, J = 8.7 Hz, 1H), 6.61 (d, J = 8.7, 1H), 2.57 (s, 3H), 1.04 (s, 9H), 0.24 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 131.30, 118.38, 25.89, 24.55, 18.53, −4.07.
tert-butyl(3,5-dibromo-4-methylphenoxy)dimethylsilane (4-p)
Starting from 3,5-dibromo-4-methylphenol, 4-p was synthesized following same procedure as 4 to yield 4-p as a clear oil (1.34g, 94%). 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 2H), 2.55 (s, 3H), 1.04 (s, 9H), 0.27 (s, 6H). 13C NMR (176 MHz, CDCl3) δ 154.29, 130.14, 129.94, 124.78, 123.82, 25.71, 22.79, 18.30.
tert-butyl(2,4-dibromo-3-(bromomethyl)phenoxy)dimethylsilane (5)
N-bromo-succinimide (4.49 g, 25.25 mmol, 1.5 equiv) and benzoyl peroxide (163 mg, 0.5 mmol, 0.03 equiv) were added to a solution of 4 (6.4 g, 16.83 mmol, 1.0 equiv) in CCl4 (60 mL) and heated at reflux for 4 h. The reaction was quenched with sodium thiosulfate (60 mL). Layers were separated, and the aqueous layer was washed with CH2Cl2 (2 x 50 mL). The combined organic layers were washed with water, dried over MgSO4, filtered, and concentrated. The crude oil was then purified using flash chromatography, eluting with hexanes to yield 5 as a clear oil (5.89 g, 76%). 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.7 Hz, 1H), 6.71 (d, J = 8.8 Hz, 1H), 4.84 (s, 2H), 1.53 (s, 1H), 1.35 – 1.22 (m, 1H), 1.04 (s, 7H), 1.08 – 1.01 (m, 1H), 0.25 (s, 5H). 13C NMR (101 MHz, CDCl3) δ 152.99, 137.53, 132.23, 120.89, 119.54, 116.31, 35.00, 25.82, 18.52, −4.08.
tert-butyl(3,5-dibromo-4-(bromomethyl)phenoxy)dimethylsilane (5-p)
Starting from 4-p, 5-p was synthesized following the same procedure as 5 to yield 5-p as a clear oil (1.36g, 84%). 1H NMR (400 MHz, CDCl3) δ 7.07 (s, 2H), 4.83 (s, 2H), 1.00 (s, 9H), 0.25 (s, 6H). 13C NMR (176 MHz, CDCl3) δ 156.69, 128.99, 125.58, 124.45, 77.34, 77.16, 76.98, 34.42, 25.61, 18.26, −4.34.
(2,6-dibromo-3-hydroxyphenyl)acetonitrile (6)
Sodium cyanide (2.71 g, 61.58 mmol, 1.5 equiv) was added to a solution of 5 (18.85 g, 41 mmol, 1.0 equiv) in DMF (23 mL) and water (3 mL), and the mixture stirred for 1.5 h. The reaction was concentrated then diluted with water and washed with ethyl acetate (3 x 100 mL). The combined organic layers were dried over MgSO4, filtered, then concentrated. The crude oil was then purified using flash chromatography, eluting with 2:8 ethyl acetate:toluene yielding 6 as a white solid (9.43g, 79%). 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 5.64 (s, 1H), 4.09 (s, 2H), 1.55 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 133.21, 133.17, 132.47, 118.20, 117.71, 115.57, 72.39, 51.05, 26.40, 26.33. ESI-MS for C8H5Br2NO (m/z) 289.9 [M-H]−.
(2,6-dibromo-4-hydroxyphenyl)acetonitrile (6-p)
Starting from 5-p, 6-p was synthesized following the same procedure as 6 to yield 6-p as a white solid (295 mg, 34%). 1H NMR (400 MHz, CD3OD) δ 7.11 (s, 2H), 4.09 (s, 2H). 13C NMR (176 MHz, MeOD) δ 158.89, 124.46, 120.52, 119.46, 116.11, 23.98.
General Procedure for Synthesis of Phenols 8-n
K2CO3 (4 equiv) and 6 (1 equiv) was added to a solution of 6-iodo-1-hexyne (1 equiv) or tosylated alkynes (7a-f) (1 equiv) in DMF (0.5 M). The mixture was heated to 80°C and stirred for 1 h. The mixture was filtered then concentrated. The crude oil was then purified using flash chromatography (9.6:0.4 CH2Cl2:MeOH) to yield 8-n as a brown oil.
The following compounds were prepared following the general procedure above.
(2,6-dibromo-3-(hex-5-yn-1-yloxy)phenyl)acetonitrile (8a)
Starting from 6-iodo-hexyne (363 mg, 1.75 mmol) and 6 (635 mg, 1.75 mmol), the general procedure gave 8a as a brown oil (381 mg, 59%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.9 Hz, 1H), 4.13 (s, 2H), 4.05 (s, 2H), 2.31 (td, J = 7.0, 2.7 Hz, 2H), 2.02 – 1.94 (m, 3H), 1.82 – 1.73 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 155.58, 132.36, 131.22, 115.98, 115.72, 115.13, 114.06, 84.01, 69.21, 68.96, 28.04, 26.31, 25.04, 18.20. ESI-MS for C14H13Br2NO (m/z) 371.9 [M+H]+.
(2,6-dibromo-3-(2-(2-(2-(hex-5-yn-1-yloxy)ethoxy)ethoxy)ethoxy)phenyl)acetonitrile (8b)
Starting from 6 (399 mg, 1.37 mmol) and 7a (527 mg, 1.37 mmol), the general procedure gave 8b as a brown oil (595 mg, 86%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.9 Hz, 1H), 6.84 (d, J = 8.9 Hz, 1H), 4.21 – 4.16 (m, 2H), 4.13 (s, 2H), 3.92 (dd, J = 5.4, 4.3 Hz, 2H), 3.80 – 3.74 (m, 2H), 3.70 – 3.62 (m, 4H), 3.62 – 3.55 (m, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.93 (t, J = 2.7 Hz, 1H), 1.76 – 1.65 (m, 2H), 1.65 – 1.54 (m, 2H), 1.26 (d, J = 1.8 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 162.16, 155.17, 131.99, 130.93, 115.46, 115.29, 114.94, 114.29, 70.72, 70.29, 70.24, 69.72, 69.29, 69.01, 68.21, 68.10, 36.11, 30.98, 28.27, 25.82, 24.80, 17.81. ESI-MS for C20H25Br2NO4 (m/z) 504.0 [M+H]+.
(2,6-dibromo-3-(3,6,9,12,15,18-hexaoxatetracos-23-yn-1-yloxy)phenyl)acetonitrile (8c)
Starting from 6 (181 mg, 0.62 mmol) and 7b (214 mg, 0.414 mmol), the general procedure gave 8c as a brown oil (163 mg, 62%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.8 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 4.19 (t, J = 4.8 Hz, 2H), 4.13 (s, 2H), 3.94 – 3.89 (m, 2H), 3.79 – 3.74 (m, 2H), 3.69 – 3.61 (m, 16H), 3.61 – 3.54 (m, 2H), 3.48 (td, J = 6.4, 2.3 Hz, 2H), 2.21 (td, J = 7.1, 2.7 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.74 – 1.65 (m, 2H), 1.65 – 1.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 155.59, 132.37, 132.35, 131.21, 116.05, 115.65, 115.47, 114.63, 84.43, 71.21, 70.80, 70.75, 70.69, 70.68, 70.65, 70.17, 69.70, 69.46, 68.52, 28.71, 26.29, 25.22, 18.28. ESI-MS for C26H37Br2NO7 (m/z) 636.1 [M+H]+.
(2,6-dibromo-4-(3,6,9,12,15,18-hexaoxatetracos-23-yn-1-yloxy)phenyl)acetonitrile (8c-p)
Starting from 6-p (70 mg, 0.24 mmol) and 7b (125 mg, 0.24 mmol), the general procedure gave 8c-p as a brown oil (122 mg, 80%). 1H NMR (700 MHz, CDCl3) δ 7.17 (s, 2H), 4.12 – 4.09 (m, 2H), 4.02 (s, 2H), 3.85 – 3.81 (m, 2H), 3.70 (td, J = 4.0, 1.0 Hz, 2H), 3.70 – 3.61 (m, 16H), 3.57 (dd, J = 5.9, 3.8 Hz, 2H), 3.47 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.1, 2.6 Hz, 2H), 1.95 – 1.93 (m, 1H), 1.72 – 1.66 (m, 2H), 1.59 (dq, J = 9.9, 7.1 Hz, 2H). 13C NMR (176 MHz, CDCl3) δ 162.67, 159.52, 125.22, 122.21, 119.25, 116.04, 84.49, 71.05, 70.87, 70.23, 69.49, 68.41, 28.76, 25.15, 18.34. ESI-MS for C26H37Br2NO7 (m/z) 636.1 [M+H]+.
(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27-nonaoxatritriacont-32-yn-1-yloxy)phenyl)acetonitrile (8d)
Starting from 6 (56 mg, 0.193 mmol) and 7c (125 mg, 0.193 mmol), the general procedure gave 8d as a brown oil (117 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.9 Hz, 1H), 6.85 (d, J = 8.9 Hz, 1H), 4.19 (t, J = 4.8 Hz, 2H), 4.13 (s, 2H), 3.94 – 3.88 (m, 2H), 3.79 – 3.74 (m, 2H), 3.70 – 3.61 (m, 27H), 3.58 (dd, J = 5.8, 3.6 Hz, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.70 (dq, J = 8.6, 6.2 Hz, 2H), 1.65 – 1.55 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 162.66, 155.66, 132.43, 131.29, 116.16, 115.71, 115.56, 114.69, 114.65, 84.50, 72.57, 71.36, 71.29, 70.88, 70.83, 70.76, 70.72, 70.64, 70.25, 69.77, 69.54, 68.55, 61.95, 36.62, 31.57, 28.78, 26.36, 25.29, 18.35. ESI-MS for C32H49Br2NO10 (m/z) 768.2 [M+H]+.
(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27,30,33,36-dodecaoxadotetracont-41-yn-1-yloxy)phenyl)acetonitrile (8e)
Starting from 6 (138 mg, 0.47 mmol) and 7d (370 mg, 0.47 mmol), the general procedure gave 8e as a brown oil (306 mg, 72%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.9 Hz, 1H), 6.85 (d, J = 9.0 Hz, 1H), 4.21 – 4.16 (m, 2H), 4.13 (s, 2H), 3.94 – 3.88 (m, 2H), 3.80 – 3.74 (m, 2H), 3.69 – 3.61 (m, 40H), 3.61 – 3.55 (m, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.1, 2.7 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.74 – 1.65 (m, 2H), 1.65 – 1.55 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 162.66, 155.66, 132.43, 131.30, 116.14, 115.71, 115.56, 114.69, 84.49, 71.29, 70.87, 70.83, 70.76, 70.72, 70.25, 69.78, 69.54, 68.55, 36.62, 31.57, 28.78, 26.35, 18.35. ESI-MS for C38H61Br2NO13 (m/z) 900.3 [M+H]+.
(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45-pentadecaoxahenpentacont-50-yn-1-yloxy)phenyl)acetonitrile (8f)
Starting from 6 (41 mg, 0.14 mmol) and 7e (130 mg, 0.14 mmol), the general procedure gave 8f as a brown oil (127 mg, 86%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.9 Hz, 1H), 6.85 (d, J = 8.9 Hz, 1H), 4.22 – 4.11 (m, 3H), 3.97 – 3.86 (m, 2H), 3.76 (dd, J = 5.9, 3.6 Hz, 2H), 3.65 (d, J = 5.0 Hz, 53H), 3.61 – 3.55 (m, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.7 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.70 (dq, J = 8.5, 6.5 Hz, 2H), 1.59 (dq, J = 9.5, 6.9 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 155.67, 132.43, 131.30, 116.15, 115.71, 115.57, 114.70, 84.50, 71.29, 70.88, 70.84, 70.77, 70.73, 70.72, 70.26, 69.78, 69.54, 68.55, 28.78, 26.36, 25.29, 18.35. ESI-MS for C44H73Br2NO16 (m/z) 1032.4 [M+H]+.
(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54-octadecaoxahexacont-59-yn-1-yloxy)phenyl)acetonitrile (8g)
Starting from 6 (51 mg, 0.17 mmol) and 7f (182 mg, 0.17 mmol), the general procedure gave 8g as a brown oil (184 mg, 90%). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 8.9 Hz, 1H), 6.84 (d, J = 8.9 Hz, 1H), 4.18 (t, J = 4.8 Hz, 2H), 4.12 (s, 2H), 3.91 (t, J = 4.8 Hz, 2H), 3.79 – 3.73 (m, 2H), 3.71 – 3.60 (m, 64H), 3.60 – 3.54 (m, 2H), 3.47 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.74 – 1.64 (m, 2H), 1.62 – 1.57 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 132.43, 131.30, 114.70, 84.50, 71.30, 70.88, 70.84, 70.77, 70.72, 70.26, 69.78, 69.54, 68.55, 36.61, 28.78, 26.36, 25.29, 18.35. ESI-MS for C50H85Br2NO19 (m/z) 1164.4 [M+H]+.
General Procedure for Pyridopyrimidine Alkynes
60% NaH in oil (0.4 equiv) was added to a solution of 8-n in 2-ethoxyethanol in an ice bath. The mixture was allowed to warm to r.t., then 2,4-diaminopyrimidine-5-carbaldehyde was added. The mixture was heated to reflux for 4 h. The mixture was then concentrated and taken directly to purification by flash chromatography (9:1 CH2Cl2:MeOH) yielding (9-n) as an orange oil. For in vitro assessment and MIC, oil was purified by RP-HPLC resulting 9-n as an off white solid.
The following compounds were prepared following the general procedure above.
6-(2,6-dibromo-3-(hex-5-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9a)
Starting from 8a (290 mg, 0.709 mmol), the general procedure gave 9a as an orange oil (189 mg, 54%) that was purified by RP-HPLC (isocratic 45% B, retention time = 15.2 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.48 (s, 1H), 7.12 (d, J = 9.0 Hz, 1H), 6.70 (s, 2H), 6.50 (s, 2H), 4.11 (td, J = 6.3, 2.8 Hz, 1H), 3.31 (s, 1H), 2.79 (t, J = 2.7 Hz, 1H), 2.26 (td, J = 7.1, 2.6 Hz, 2H), 1.85 (h, J = 6.4 Hz, 2H), 1.65 (p, J = 7.2 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.77, 161.18, 160.04, 155.03, 138.49, 136.93, 132.25, 120.60, 115.33, 115.06, 114.79, 108.07, 84.29, 71.49, 68.57, 27.67, 24.66, 17.41. HRMS (ESI) calculated for C19H17Br2N5O [M+H]+, 489.9873; found, 489.9871.
6-(2,6-dibromo-3-(2-(2-(2-(hex-5-yn-1-yloxy)ethoxy)ethoxy)ethoxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9b)
Starting from 8b (545 mg, 1.08 mmol), the general procedure gave 9b as an orange oil (239 mg, 35%) that was purified by RP-HPLC (isocratic 40% B, retention time = 22.5 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.48 (s, 1H), 7.14 (d, J = 8.9 Hz, 1H), 6.69 (s, 2H), 6.49 (s, 2H), 4.21 (q, J = 4.2 Hz, 2H), 3.80 (t, J = 4.6 Hz, 2H), 3.64 (dd, J = 6.0, 3.7 Hz, 2H), 3.53 (ddd, J = 12.0, 5.4, 3.2 Hz, 4H), 3.46 (dd, J = 5.8, 3.6 Hz, 2H), 3.37 (t, J = 6.3 Hz, 2H), 2.74 (t, J = 2.7 Hz, 1H), 2.15 (td, J = 7.0, 2.7 Hz, 2H), 1.60 – 1.51 (m, 2H), 1.46 (p, J = 7.0 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.77, 161.18, 160.21, 160.03, 155.06, 138.55, 136.88, 132.24, 120.60, 115.36, 115.30, 115.01, 108.07, 84.44, 71.25, 70.18, 69.87, 69.81, 69.68, 69.48, 69.13, 68.76, 28.23, 24.79, 17.47. HRMS (ESI) calculated for C25H29Br2N5O4 [M+H]+, 622.0659; found, 622.0661.
6-(2,6-dibromo-3-(3,6,9,12,15,18-hexaoxatetracos-23-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9c)
Starting from 8c (112 mg, 0.177 mmol), the general procedure gave 9c as an orange oil (45 mg, 34%) that was purified by RP-HPLC (isocratic 40% B, retention time = 19.4 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.48 (s, 1H), 7.14 (d, J = 9.0 Hz, 1H), 6.70 (s, 2H), 6.49 (s, 2H), 4.21 (q, J = 4.1 Hz, 2H), 3.85 – 3.76 (m, 2H), 3.64 (dd, J = 5.8, 3.7 Hz, 2H), 3.57 – 3.43 (m, 18H), 3.37 (t, J = 6.3 Hz, 2H), 2.74 (t, J = 2.6 Hz, 1H), 2.15 (td, J = 7.1, 2.7 Hz, 2H), 1.55 (ddd, J = 13.5, 7.4, 4.7 Hz, 2H), 1.46 (dtd, J = 10.5, 8.3, 7.6, 5.8 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.77, 161.18, 160.19, 160.02, 155.06, 138.55, 136.87, 132.23, 120.60, 115.35, 115.29, 114.99, 108.07, 84.43, 71.23, 70.18, 69.84, 69.81, 69.80, 69.78, 69.67, 69.45, 69.12, 68.75, 28.23, 24.79, 17.47. HRMS (ESI) calculated for C31H41Br2N5O7 [M+H]+, 754.1446; found, 754.1442.
6-(2,6-dibromo-4-(3,6,9,12,15,18-hexaoxatetracos-23-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9c-p)
Starting from 8c-p, 9c-p was synthesized following the same procedure as 9c to yield 9c-p as a yellow solid (59 mg, 48%). 1H NMR (700 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.50 (s, 1H), 7.38 (s, 2H), 6.72 (s, 1H), 6.66 (s, 2H), 6.45 (s, 2H), 4.20 – 4.16 (m, 2H), 3.77 – 3.72 (m, 2H), 3.59 (dd, J = 5.9, 3.5 Hz, 4H), 3.50 (d, J = 4.7 Hz, 11H), 3.48 – 3.39 (m, 4H), 3.37 (t, J = 6.4 Hz, 2H), 2.71 (t, J = 2.7 Hz, 1H), 2.14 (td, J = 7.1, 2.7 Hz, 2H), 1.55 (dq, J = 8.6, 6.5 Hz, 2H), 1.49 – 1.41 (m, 2H). ). 13C NMR (176 MHz, DMSO-d6) δ 163.85, 161.25, 160.83, 160.46, 159.47, 137.82, 129.77, 125.67, 120.32, 118.76, 108.31, 84.72, 71.39, 70.12, 70.01, 69.99, 69.97, 69.95, 69.87, 69.64, 68.86, 68.42, 28.40, 24.96, 17.65. HRMS (ESI) calculated for C31H41Br2N5O7 [M+H]+, 754.1446; found, 754.1438.
6-(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27-nonaoxatritriacont-32-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9d)
Starting from 8d (132 mg, 0.172 mmol), the general procedure gave 9d as an orange oil (45 mg, 29%) that was purified by RP-HPLC (isocratic 40% B, retention time = 17.2 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.48 (s, 1H), 7.14 (d, J = 9.0 Hz, 1H), 6.69 (s, 2H), 6.49 (s, 2H), 4.21 (q, J = 4.1 Hz, 2H), 3.80 (t, J = 4.7 Hz, 2H), 3.64 (dd, J = 5.8, 3.8 Hz, 2H), 3.57 – 3.43 (m, 30H), 3.38 (t, J = 6.3 Hz, 2H), 2.74 (t, J = 2.7 Hz, 1H), 2.16 (td, J = 7.0, 2.7 Hz, 2H), 1.56 (dq, J = 8.6, 6.5 Hz, 2H), 1.51 – 1.42 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.77, 161.18, 160.18, 160.02, 155.06, 138.55, 136.87, 132.23, 120.59, 115.35, 115.29, 114.99, 108.07, 84.43, 71.23, 70.18, 69.84, 69.80, 69.77, 69.67, 69.46, 69.12, 68.75, 28.24, 24.80, 17.47. HRMS (ESI) calculated for C37H53Br2N5O10 [M+H]+, 886.2232; found, 886.2229.
6-(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27,30,33,36-dodecaoxadotetracont-41-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9e)
Starting from 8e (292 mg, 0.325 mmol), the general procedure gave 9e as an orange oil (68 mg, 20%) that was purified by RP-HPLC (isocratic 40% B, retention time = 17 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.47 (s, 1H), 7.14 (d, J = 9.0 Hz, 1H), 6.69 (s, 2H), 6.48 (s, 2H), 4.21 (q, J = 4.1 Hz, 2H), 3.80 (t, J = 4.7 Hz, 2H), 3.64 (dd, J = 5.9, 3.7 Hz, 2H), 3.57 – 3.50 (m, 5H), 3.50 (s, 21H), 3.46 (dd, J = 5.8, 3.3 Hz, 2H), 3.38 (t, J = 6.4 Hz, 2H), 3.33 (d, J = 2.4 Hz, 14H), 2.74 (t, J = 2.7 Hz, 1H), 2.16 (td, J = 7.0, 2.7 Hz, 2H), 1.56 (dq, J = 8.5, 6.5 Hz, 2H), 1.51 – 1.42 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.77, 160.20, 155.06, 136.87, 132.23, 120.60, 115.00, 71.23, 70.19, 69.84, 69.81, 69.77, 69.68, 69.47, 69.13, 68.75, 28.24, 24.80, 17.47. HRMS (ESI) calculated for C43H65Br2N5O13 [M+H]+, 1018.3018; found, 1018.3019.
6-(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45-pentadecaoxahenpentacont-50-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9f)
Starting from 8f (117 mg, 0.113 mmol), the general procedure gave 9f as an orange oil (24 mg, 17%) that was purified by RP-HPLC (isocratic 40% B, retention time = 16.3 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.48 (s, 1H), 7.15 (d, J = 9.0 Hz, 1H), 6.69 (s, 2H), 6.49 (s, 2H), 4.21 (q, J = 4.1 Hz, 2H), 3.80 (t, J = 4.7 Hz, 2H), 3.64 (dd, J = 5.9, 3.7 Hz, 2H), 3.57 – 3.43 (m, 54H), 3.38 (t, J = 6.3 Hz, 2H), 2.74 (t, J = 2.7 Hz, 1H), 2.16 (td, J = 7.0, 2.6 Hz, 2H), 1.56 (dq, J = 8.5, 6.5 Hz, 2H), 1.47 (dtd, J = 9.3, 7.0, 5.2 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 163.75, 161.16, 160.21, 160.01, 155.06, 138.53, 136.88, 132.24, 120.60, 115.34, 115.28, 115.01, 108.05, 84.42, 71.24, 70.18, 69.76, 69.67, 69.46, 69.13, 68.75, 61.26, 28.24, 24.80, 17.47. HRMS (ESI) calculated for C49H77Br2N5O16 [M+H]+, 1150.3805; found, 1150.3809.
6-(2,6-dibromo-3-(3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54-octadecaoxahexacont-59-yn-1-yloxy)phenyl)pyrido[2,3-d]pyrimidine-2,7-diamine (9g)
Starting from 8g (174 mg, 0.149 mmol), the general procedure gave 9g as an orange oil (46 mg, 24%) that was purified by RP-HPLC (isocratic 35% B, retention time = 11.9 min). 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.47 (s, 1H), 7.14 (d, J = 9.0 Hz, 1H), 6.69 (s, 2H), 6.37 (s, 2H), 4.21 (q, J = 4.2 Hz, 2H), 3.80 (t, J = 4.7 Hz, 2H), 3.64 (dd, J = 5.9, 3.7 Hz, 2H), 3.50 (dd, J = 5.3, 1.8 Hz, 68H), 2.74 (t, J = 2.7 Hz, 1H), 2.16 (td, J = 7.1, 2.7 Hz, 2H), 1.61 – 1.41 (m, 4H). 13C NMR (126 MHz, DMSO-d6) δ 132.23, 115.35, 71.23, 70.18, 69.81, 69.77, 69.67, 69.46, 60.51, 59.67, 28.24, 24.80, 17.47. HRMS (ESI) calculated for C55H89Br2N5O19 [M+H]+, 1282.4591; found, 1282.4601.
General Procedure for Alkyne Addition
Polyethylene glycol (1.1 equiv) (n=3,6) was added to DMF (0.5 M), and the mixture was cooled to 0°C. Sodium hydride (1 equiv) was then added, and the reaction was stirred for 10 mins. 6-iodo-1-hexyne or substituted (11a-d) alkyne (1 equiv) was then added, and the reaction was removed from the ice bath. After 4 h, the TLC showed completion. The reaction was then concentrated and taken directly to purification by column chromatography eluting with 9:1 CH2Cl2:MeOH.
The following compounds were prepared following the general procedure above.
2-(2-(2-(hex-5-yn-1-yloxy)ethoxy)ethoxy)ethanol (10a)
Starting from triethylene glycol (500 mg, 3.32 mmol) and 6-iodo-hexyne (692 mg, 3.32 mmol), the general procedure gave 10a as a yellow oil (190 mg, 25%). 1H NMR (500 MHz, CDCl3) δ 8.02 (s, 1H), 3.76 – 3.56 (m, 12H), 3.49 (t, J = 6.4 Hz, 2H), 2.22 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.75 – 1.66 (m, 2H), 1.65 – 1.55 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 162.65, 84.48, 72.61, 70.90, 70.78, 70.74, 70.51, 70.20, 68.51, 61.90, 36.61, 31.56, 28.72, 25.26, 18.32. ESI-MS for C12H22O4 (m/z) 231.3 [M+H]+.
3,6,9,12,15,18-hexaoxatetracos-23-yn-1-ol (10b)
Starting from hexaethylene glycol (6.78 g, 24 mmol) and 6-iodo-hexyne (5.0 g, 24 mmol), the general procedure gave 10b as a yellow oil (3.42 g, 39%).1H NMR (500 MHz, CDCl3) δ 3.75 – 3.70 (m, 2H), 3.70 – 3.55 (m, 22H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.7 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.69 (dt, J = 8.6, 6.3 Hz, 2H), 1.65 – 1.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 171.22, 84.50, 72.65, 70.87, 70.76, 70.75, 70.72, 70.69, 70.49, 70.24, 69.27, 68.53, 63.76, 61.89, 53.56, 28.77, 25.28, 21.11, 18.34. ESI-MS for C18H34O7 (m/z) 363.3 [M+H]+.
3,6,9,12,15,18,21,24,27-nonaoxatritriacont-32-yn-1-ol (11a)
Starting from triethylene glycol (145 mg, 0.97 mmol) and 7b (500 mg, 0.97 mmol), the general procedure gave 11a as a yellow oil (153 mg, 32%).1H NMR (500 MHz, CDCl3) δ 3.75 – 3.66 (m, 2H), 3.70 – 3.63 (m, 29H), 3.62 (ddd, J = 9.1, 5.6, 3.3 Hz, 4H), 3.58 (dd, J = 5.8, 3.7 Hz, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.72 (s, 0H), 1.73 – 1.65 (m, 2H), 1.63 (s, 1H), 1.59 (dtd, J = 9.4, 7.1, 5.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 171.22, 162.69, 129.93, 128.09, 84.47, 72.74, 72.59, 70.84, 70.71, 70.66, 70.64, 70.61, 70.42, 70.39, 70.37, 70.21, 69.40, 69.24, 68.79, 68.53, 63.55, 61.86, 61.79, 36.61, 31.56, 28.74, 25.25, 21.75, 21.05, 18.31. ESI-MS for C24H46O10 (m/z) 495.4 [M+H]+.
3,6,9,12,15,18,21,24,27,30,33,36-dodecaoxadotetracont-41-yn-1-ol (11b)
Starting from hexaethylene glycol (1.3 g, 4.6 mmol) and 7b (2.4 g, 4.6 mmol), the general procedure gave 11b as a yellow oil (1.85 g, 64%).1H NMR (500 MHz, CDCl3) δ 3.75 – 3.67 (m, 2H), 3.70 – 3.61 (m, 42H), 3.62 (dd, J = 5.4, 3.7 Hz, 2H), 3.58 (dd, J = 5.8, 3.7 Hz, 2H), 3.48 (t, J = 6.4 Hz, 2H), 2.22 (td, J = 7.1, 2.7 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.69 (dt, J = 8.6, 6.4 Hz, 2H), 1.60 (dtd, J = 9.5, 7.1, 5.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 84.44, 72.73, 70.82, 70.70, 70.65, 70.62, 70.60, 70.37, 70.19, 68.53, 61.78, 53.55, 36.58, 28.72, 25.23, 18.29. ESI-MS for C30H58O13 (m/z) 627.5 [M+H]+.
3,6,9,12,15,18,21,24,27,30,33,36,39,42,45-pentadecaoxahenpentacont-50-yn-1-ol (11c)
Starting from triethylene glycol (120 mg, 0.8 mmol) and 7d (622 mg, 0.8 mmol), the general procedure gave 11c as a yellow oil (203 mg, 33%).1H NMR (500 MHz, CDCl3) δ 3.81 – 3.55 (m, 60H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.7 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.70 (tt, J = 8.7, 6.2 Hz, 2H), 1.59 (dtd, J = 9.5, 7.1, 5.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 162.64, 84.47, 72.65, 70.85, 70.74, 70.69, 70.46, 70.23, 68.53, 61.84, 36.60, 31.55, 28.76, 25.27, 18.33. ESI-MS for C36H70O16 (m/z) 759.5 [M+H]+.
3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54-octadecaoxahexacont-59-yn-1-ol (11d)
Starting from hexaethylene glycol (225 mg, 0.8 mmol) and 7d (622 mg, 0.8 mmol), the general procedure gave 11d as a yellow oil (256 mg, 36%). 1H NMR (500 MHz, CDCl3) δ 3.80 – 3.53 (m, 72H), 3.48 (t, J = 6.4 Hz, 2H), 2.21 (td, J = 7.0, 2.7 Hz, 3H), 1.94 (t, J = 2.6 Hz, 1H), 1.69 (dd, J = 10.1, 4.6 Hz, 2H), 1.60 (dtd, J = 9.5, 7.1, 5.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 84.49, 72.65, 70.87, 70.76, 70.71, 70.48, 70.25, 68.55, 61.87, 53.57, 31.07, 28.78, 25.29, 18.35. ESI-MS for C42H82O19 (m/z) 891.6 [M+H]+.
General Procedure for Tosylation
4-Toluenesulfonyl chloride (1.5 equiv) was added to a solution of 10a-b or 11a-d (1 equiv) and triethylamine (3 equiv) in dry CH2Cl2 (0.3 M) at 0°C. The mixture was allowed to warm to r.t. and stir overnight. The mixture was then washed with 2M HCl, sat. Na2CO3, and water, dried over MgSO4, filtered, and concentrated. The crude product was then purified by column chromatography eluting with 9.5:0.5 CH2Cl2:MeOH.
The following compounds were prepared following the general procedure above.
2-(2-(2-(hex-5-yn-1-yloxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (7a)
Starting from 4-toluenesulfonyl chloride (542 mg, 2.84 mmol) and 10a (803 mg, 2.84 mmol), the general procedure gave 7a as a yellow oil (793 mg, 73%).1H NMR (500 MHz, CDCl3) δ 7.83 – 7.76 (m, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.20 – 4.12 (m, 2H), 3.69 (dd, J = 5.5, 4.3 Hz, 2H), 3.64 – 3.53 (m, 8H), 3.47 (t, J = 6.4 Hz, 2H), 2.45 (s, 3H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.75 – 1.64 (m, 2H), 1.63 – 1.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 144.92, 133.17, 129.95, 128.13, 84.49, 70.91, 70.88, 70.80, 70.71, 70.22, 69.38, 68.83, 68.54, 28.76, 25.29, 21.79, 18.34. ESI-MS for C19H28O6S (m/z) 385.2 [M+H]+.
1-(4-(methylsulfonyl)phenoxy)-3,6,9,12,15,18-hexaoxatetracos-23-yne (7b)
Starting from 4-toluenesulfonyl chloride (2.7 g, 14.15 mmol) and 10b (3.42 g, 9.43 mmol), the general procedure gave 7b as a yellow oil (4.06 g, 83%). 1H NMR (500 MHz, CDCl3) δ 7.82 – 7.77 (m, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.16 (dd, J = 5.7, 4.0 Hz, 2H), 3.69 (d, J = 4.9 Hz, 2H), 3.63 (dd, J = 12.1, 2.6 Hz, 15H), 3.58 (s, 5H), 3.48 (t, J = 6.4 Hz, 2H), 2.45 (s, 3H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.6 Hz, 1H), 1.70 (dq, J = 8.6, 6.2 Hz, 2H), 1.64 – 1.56 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 144.91, 133.16, 129.95, 128.13, 84.49, 70.89, 70.87, 70.76, 70.72, 70.70, 70.66, 70.24, 69.37, 68.82, 68.53, 53.57, 28.78, 25.28, 21.78, 18.34. ESI-MS for C25H40O9S (m/z) 517.2 [M+H]+.
1-(4-(methylsulfonyl)phenoxy)-3,6,9,12,15,18,21,24,27-nonaoxatritriacont-32-yne (7c)
Starting from 4-toluenesulfonyl chloride (72 mg, 0.38 mmol) and 11a (125 mg, 0.25 mmol), the general procedure gave 7c as a yellow oil (142 mg, 86%). 1H NMR (500 MHz, CDCl3) δ 7.82 – 7.78 (m, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.18 – 4.14 (m, 2H), 3.70 – 3.55 (m, 34H), 3.48 (t, J = 6.4 Hz, 2H), 2.45 (s, 3H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.69 (dt, J = 8.6, 6.4 Hz, 2H), 1.60 (dtd, J = 9.4, 7.0, 5.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 144.97, 144.93, 133.15, 129.97, 128.14, 84.50, 70.90, 70.77, 70.72, 70.67, 70.64, 70.26, 69.39, 69.33, 68.89, 68.83, 68.55, 63.67, 28.78, 25.29, 21.79, 21.10, 18.35. ESI-MS for C31H52O12S (m/z) 649.3 [M+H]+.
1-(4-(methylsulfonyl)phenoxy)-3,6,9,12,15,18,21,24,27,30,33,36-dodecaoxadotetracont-41-yne (7d)
Starting from 4-toluenesulfonyl chloride (429 mg, 2.25 mmol) and 11b (941 mg, 1.5 mmol), the general procedure gave 7d as a yellow oil (932 mg, 79%). 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.16 (dd, J = 5.7, 4.1 Hz, 2H), 3.71 – 3.57 (m, 40H), 3.58 (s, 5H), 3.48 (t, J = 6.4 Hz, 2H), 2.45 (s, 3H), 2.21 (td, J = 7.1, 2.7 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.70 (dq, J = 8.6, 6.6 Hz, 2H), 1.64 – 1.56 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 144.92, 129.97, 128.14, 84.50, 70.90, 70.88, 70.77, 70.72, 70.67, 70.26, 69.38, 68.83, 68.55, 28.79, 25.30, 21.80, 18.36. ESI-MS for C37H64O15S (m/z) 781.4 [M+H]+.
1-(4-(methylsulfonyl)phenoxy)-3,6,9,12,15,18,21,24,27,30,33,36,39,42,45-pentadecaoxahenpentacont-50-yne (7e)
Starting from 4-toluenesulfonyl chloride (70 mg, 0.37 mmol) and 11c (187 mg, 0.25 mmol), the general procedure gave 7e as a yellow oil (140 mg, 62%). 1H NMR (500 MHz, CDCl3) δ 7.83 – 7.77 (m, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.19 – 4.13 (m, 2H), 3.76 – 3.55 (m, 58H), 3.48 (t, J = 6.4 Hz, 2H), 2.45 (s, 3H), 2.21 (td, J = 7.1, 2.7 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.70 (dq, J = 8.6, 6.7, 6.2 Hz, 2H), 1.60 (qd, J = 7.1, 3.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 144.92, 133.17, 129.97, 128.13, 84.50, 70.90, 70.88, 70.77, 70.72, 70.67, 70.26, 69.38, 68.83, 68.55, 28.78, 25.29, 21.79, 18.35. ESI-MS for C43H76O18S (m/z) 913.5 [M+H]+.
1-(4-(methylsulfonyl)phenoxy)-3,6,9,12,15,18,21,24,27,30,33,36,39,42,45,48,51,54-octadecaoxahexacont-59-yne (7f)
Starting from 4-toluenesulfonyl chloride (77 mg, 0.40 mmol) and 11d (240 mg, 0.27 mmol), the general procedure gave 7f as a yellow oil (194 mg, 69%). 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.19 – 4.11 (m, 2H), 3.61 (m, 72H), 3.48 (t, J = 6.4 Hz, 2H), 2.45 (s, 3H), 2.21 (td, J = 7.0, 2.6 Hz, 2H), 1.94 (t, J = 2.7 Hz, 1H), 1.72 – 1.67 (m, 2H), 1.63 – 1.56 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 144.92, 133.15, 129.96, 128.13, 84.49, 70.88, 70.75, 70.71, 70.70, 70.25, 69.38, 68.82, 68.56, 28.78, 25.29, 21.79, 18.35. ESI-MS for C49H88O21S (m/z) 1045.6 [M+H]+.
3-(10-carboxydecyl)triaza-1,2-dien-2-ium-1-ide (12)
Sodium azide (735 mg, 11.31 mmol, 3.0 equiv) was added to a solution of 11-bromoundecanoic acid (1.0 g, 3.77 mmol, 1.0 equiv) in DMSO (10 mL). The mixture was heated at 80°C for 5 h, then the reaction was quenched with water (10 mL). The mixture was diluted with ethyl acetate (10 mL), and aqueous layer was washed with ethyl acetate (3 x 10 mL). The organic layer was dried with MgSO4, filtered, and concentrated. The crude oil was then purified by column chromatography eluting with 3:2 hexanes:ethyl acetate to give 12 as a white solid (770 mg, 90%).
1H NMR (500 MHz, DMSO-d6) δ 11.95 (s, 1H), 3.31 (t, J = 6.8 Hz, 2H), 2.18 (t, J = 7.4 Hz, 2H), 1.50 (dp, J = 20.8, 6.9 Hz, 4H), 1.25 (s, 12H). 13C NMR (126 MHz, DMSO-d6) δ 174.49, 50.60, 33.64, 28.85, 28.80, 28.68, 28.53, 28.50, 28.22, 26.12, 24.48. ESI-MS for C11H21N3O2 (m/z) 226.2 [M-H]−.
tert-butyl((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)carbamate (13)
Precursor 13 was synthesized following the procedure for Moiramide B from Silvers et al.23 1H NMR (500 MHz, DMSO-d6) δ 11.36 (s, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.34 – 7.20 (m, 5H), 7.20 (dd, J = 6.2, 2.7 Hz, 1H), 4.96 – 4.82 (m, 1H), 4.70 (dd, J = 8.5, 5.0 Hz, 1H), 4.01 (dd, J = 20.3, 6.3 Hz, 1H), 3.33 (d, J = 2.3 Hz, 1H), 2.96 – 2.84 (m, 1H), 2.79 – 2.70 (m, 1H), 2.70 – 2.60 (m, 1H), 2.50 – 2.42 (m, 1H), 2.32 (dq, J = 13.4, 6.8 Hz, 1H), 1.33 (s, 9H), 1.15 – 1.06 (m, 2H), 0.85 (d, J = 6.7 Hz, 3H), 0.77 (d, J = 6.8 Hz, 2H). ESI-MS for C24H33N3O6 (m/z) 458.2 [M-H]−.
3-(11-(((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)amino)-11-oxoundecyl)triaza-1,2-dien-2-ium-1-ide (14)
Trifluoracetic acid (4.0 mL) was added dropwise to a solution of 13 (500 mg, 1.09 mmol, 1.0 equiv) in CH2Cl2 at 0°C and was stirred for 2 h. Toluene (20 mL) was then added to the mixture, and the reaction was concentrated. After azeotroping with toluene two times, the crude product was suspended in DMF (10 mL). 12 (297 mg, 1.3 mmol, 1.2 equiv), HATU (496 mg, 1.3 mmol, 1.2 equiv), and diisopropylethylamine (570 μL, 3.26 mmol, 3.0 equiv) was added to the solution and stirred overnight. The reaction was concentrated and partitioned between water (100 mL) and ethyl acetate (100 mL). The organic layer was washed with sat. NaHCO3 (100 mL) and brine (100 mL), dried over MgSO4, filtered, and concentrated. The reaction was purified by column chromatography eluting with 9.5:0.5 CH2Cl2:MeOH to give an off-white foam (426 mg, 69%). 1H NMR (500 MHz, DMSO-d6) δ 11.36 (s, 1H), 8.29 – 8.17 (m, 1H), 8.05 (d, J = 8.5 Hz, 1H), 7.32 – 7.23 (m, 4H), 7.23 – 7.14 (m, 0H), 7.20 (p, J = 4.3 Hz, 1H), 5.25 – 5.15 (m, 1H), 4.66 (dd, J = 8.4, 5.3 Hz, 1H), 3.95 (d, J = 5.5 Hz, 1H), 3.34 – 3.27 (m, 2H), 2.97 – 2.85 (m, 1H), 2.75 – 2.67 (m, 1H), 2.62 (dd, J = 23.2, 6.7 Hz, 1H), 2.59 – 2.52 (m, 0H), 2.31 (dq, J = 13.1, 6.7 Hz, 1H), 2.06 (tq, J = 5.6, 2.9 Hz, 2H), 1.56 – 1.39 (m, 4H), 1.33 – 1.14 (m, 14H), 1.14 – 1.03 (m, 2H), 0.89 – 0.80 (m, 2H), 0.77 (p, J = 6.5 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 179.99, 174.09, 173.73, 171.15, 169.87, 143.09, 128.16, 128.07, 128.02, 126.73, 126.60, 126.37, 126.33, 126.30, 63.21, 62.96, 58.84, 57.85, 50.61, 49.74, 49.69, 42.04, 41.85, 38.24, 35.49, 35.43, 35.38, 28.85, 28.84, 28.73, 28.59, 28.52, 28.23, 28.18, 27.09, 26.13, 25.26, 25.22, 25.20, 19.71, 19.59, 19.44, 18.93, 18.84, 17.57, 17.47, 17.18, 16.89, 14.64, 14.62, 14.53, 10.38. HRMS (ESI) calculated for C30H44N6O5 [M+H]+, 569.3446; found, 569.3443.
General Procedure for Heterobivalent Inhibitors
Copper (II) sulfate pentahydrate (1.0 equiv) and sodium ascorbate (2.0 equiv) was added to a solution of alkyne (8a-g, 1.0 equiv) and azide (13, 1.0 equiv) in 9:1 DMF:H2O (1 mL). The reaction was stirred overnight. The reaction was then filtered through celite, washed with MeOH, concentrated, and purified by RP-HPLC.
The following compounds were prepared following the general procedure above.
11-(4-(4-(2,4-dibromo-3-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)butyl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (15)
Starting from 9a and 14 the general procedure yielded an off-white powder (5.2mg, 20%) purified by RP-HPLC (45% B held for 5 min, 48% in 25 min, 27.5 min ret time). 1H NMR (500 MHz, DMSO-d6) δ (11.37 s, 1H), 8.63 (s, 1H), 8.33 (dt, J = 19.9, 8.1 Hz, 1H), 7.85 (s, 1H), 7.68 (d, J = 8.9 Hz, 1H), 7.48 (s, 1H), 7.28 (tt, J = 10.6, 5.6 Hz, 4H), 7.18 (q, J = 5.0, 4.0 Hz, 1H), 7.11 (d, J = 9.0 Hz, 1H), 6.69 (s, 2H), 6.54 (s, 1H), 6.49 (s, 2H), 5.15 (s, 1H), 4.27 (t, J = 7.0 Hz, 2H), 4.10 (d, J = 5.6 Hz, 2H), 2.89 (q, J = 6.6 Hz, 0H), 2.68 (d, J = 6.7 Hz, 2H), 2.65 – 2.51 (m, 1H), 2.31 (dt, J = 13.8, 8.0 Hz, 1H), 2.05 (qd, J = 9.0, 6.0, 4.9 Hz, 2H), 1.78 (dt, J = 15.4, 5.4 Hz, 6H), 1.43 (d, J = 24.7 Hz, 1H), 1.43 (s, 2H), 1.26 – 0.96 (m, 18H), 1.11 (s, 1H), 0.89 – 0.73 (m, 4H), 0.68 (dt, J = 12.6, 8.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 203.91, 164.23, 161.64, 160.66, 160.50, 155.53, 146.98, 138.95, 137.38, 132.70, 128.61, 127.06, 126.77, 126.64, 126.59, 122.16, 121.08, 115.80, 115.48, 115.24, 108.55, 70.25, 69.27, 63.64, 58.27, 49.60, 30.20, 29.48, 29.29, 29.26, 29.18, 29.08, 28.84, 28.64, 28.51, 28.37, 26.32, 25.99, 25.66, 25.10. HRMS (ESI) calculated for C49H61Br2N11O6 [M+H]+, 1058.3246; found, 1058.3237.
11-(4-(4-(2-(2-(2-(2,4-dibromo-3-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)ethoxy)ethoxy)ethoxy)butyl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (16)
Starting from 9b and 14 the general procedure yielded an off-white powder (33 mg, 33.5%) purified by RP-HPLC (45% B held for 5 min, 48% in 25 min, 25.3 min ret time). 1H NMR (500 MHz, DMSO-d6) δ 11.37 (s ,1H)9.04 (s, 1H), 8.63 (s, 1H), 8.20 (d, J = 10.2 Hz, 0H), 7.81 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.47 (s, 1H), 7.34 – 7.10 (m, 6H), 6.69 (s, 2H), 6.61 (s, 1H), 6.50 (s, 2H), 5.15 – 5.06 (m, 1H), 4.30 – 4.14 (m, 3H), 3.79 (t, J = 4.6 Hz, 2H), 3.66 – 3.59 (m, 2H), 3.53 (t, J = 5.8 Hz, 2H), 3.54 – 3.43 (m, 2H), 3.44 (d, J = 4.6 Hz, 1H), 3.38 (t, J = 6.4 Hz, 2H), 2.93 (q, J = 7.3 Hz, 1H), 2.58 (t, J = 7.5 Hz, 2H), 2.31 (dd, J = 13.8, 4.6 Hz, 0H), 2.04 (tp, J = 14.1, 6.9 Hz, 2H), 1.75 (p, J = 7.2 Hz, 2H), 1.58 (tt, J = 17.5, 9.3 Hz, 2H), 1.46 (ddd, J = 27.7, 14.4, 7.3 Hz, 4H), 1.33 (s, 1H), 1.26 – 1.06 (m, 21H), 0.89 – 0.59 (m, 7H). 13C NMR (126 MHz, DMSO-d6) δ 160.05, 155.07, 146.68, 138.55, 132.24, 128.14, 126.16, 121.63, 115.37, 115.30, 115.01, 70.18, 70.01, 69.88, 69.83, 69.49, 69.13, 68.76, 62.02, 50.32, 49.12, 35.60, 29.72, 28.83, 28.81, 28.72, 28.69, 28.65, 28.38, 25.85, 25.71, 25.50, 25.13, 24.79, 20.18, 17.45, 16.31. HRMS (ESI) calculated for C55H73Br2N11O9 [M+H]+, 1190.4032; found, 1190.4028.
11-(4-(22-(2,4-dibromo-3-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)-5,8,11,14,17,20-hexaoxadocos-1-yl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (17)
Starting from 9c and 14 the general procedure yielded an off-white powder (16.1 mg, 14%) purified by RP-HPLC (45% B held for 5 min, 48% in 25 min, 23.6 min ret time). 1H NMR (500 MHz, DMSO-d6) δ 11.37 (s, 1H), 8.63 (s, 1H), 7.82 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.47 (s, 1H), 7.27 (h, J = 4.1, 3.6 Hz, 5H), 7.16 (dd, J = 21.2, 7.1 Hz, 2H), 6.69 (s, 2H), 6.49 (s, 2H), 5.26 – 5.05 (m, 1H), 4.26 (t, J = 7.1 Hz, 2H), 4.20 (q, J = 4.1 Hz, 2H), 3.80 (t, J = 4.8 Hz, 2H), 3.63 (dd, J = 5.8, 3.8 Hz, 2H), 3.56 – 3.48 (m, 5H), 3.48 (s, 12H), 3.44 (d, J = 5.1 Hz, 2H), 3.38 (t, J = 6.3 Hz, 2H), 2.59 (t, J = 7.5 Hz, 3H), 2.09 – 1.97 (m, 3H), 1.75 (q, J = 7.3 Hz, 2H), 1.64 – 1.55 (m, 2H), 1.51 (q, J = 6.9 Hz, 2H), 1.43 (s, 3H), 1.23 (s, 3H), 1.20 – 1.06 (m, 16H), 0.84 (dd, J = 12.1, 5.1 Hz, 2H), 0.76 (d, J = 6.5 Hz, 2H), 0.66 (ddd, J = 25.3, 12.7, 6.7 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 169.89, 163.77, 161.17, 160.19, 160.03, 155.06, 146.67, 138.55, 136.88, 132.23, 128.14, 126.59, 126.29, 126.15, 121.61, 120.61, 115.36, 115.29, 114.99, 108.08, 70.18, 70.00, 69.84, 69.81, 69.79, 69.77, 69.46, 69.13, 68.75, 49.72, 49.11, 29.72, 28.83, 28.81, 28.73, 28.70, 28.61, 28.37, 25.85, 25.72, 25.21, 24.80, 14.54. HRMS (ESI) calculated for C61H85Br2N11O12 [M+H]+, 1322.4819; found, 1322.4817.
11-(4-(22-(3,5-dibromo-4-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)-5,8,11,14,17,20-hexaoxadocos-1-yl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (17-p)
Starting from 9c-p and 14 the general procedure yielded an off-white powder (16 mg, 20%). 1H NMR (700 MHz, DMSO-d6) δ 11.37 (s, 1H), 8.63 (s, 1H), 7.82 (s, 1H), 7.49 (s, 1H), 7.39 (s, 2H), 7.32 – 7.22 (m, 4H), 7.18 (dq, J = 8.7, 4.8 Hz, 1H), 6.69 (s, 2H), 6.55 (s, 2H), 5.21 – 5.07 (m, 1H), 4.26 (t, J = 7.1 Hz, 2H), 4.21 – 4.17 (m, 2H), 3.77 – 3.73 (m, 2H), 3.59 (dd, J = 5.9, 3.6 Hz, 2H), 3.56 – 3.51 (m, 3H), 3.51 (dd, J = 10.9, 2.3 Hz, 14H), 3.45 (dd, J = 5.9, 3.7 Hz, 2H), 3.38 (d, J = 12.9 Hz, 1H), 2.59 (t, J = 7.6 Hz, 3H), 2.10 – 1.97 (m, 3H), 1.76 (p, J = 7.2 Hz, 2H), 1.60 (qd, J = 8.8, 7.8, 6.2 Hz, 2H), 1.54 – 1.48 (m, 2H), 1.47 – 1.41 (m, 3H), 1.24 (d, J = 5.4 Hz, 4H), 1.18 (h, J = 8.7, 8.2 Hz, 13H), 1.13 (d, J = 7.3 Hz, 1H), 1.02 (s, 3H), 0.87 – 0.78 (m, 1H), 0.75 (ddt, J = 12.5, 9.6, 4.9 Hz, 2H), 0.69 (d, J = 6.5 Hz, 1H), 0.69 – 0.60 (m, 1H). 13C NMR (176 MHz, DMSO-d6) δ 208.70, 176.71, 174.96, 171.12, 171.05, 169.71, 163.77, 161.20, 160.64, 160.17, 159.25, 146.67, 143.09, 137.53, 129.71, 128.14, 128.05, 126.65, 126.59, 126.54, 126.29, 126.18, 126.15, 125.52, 121.63, 120.04, 118.58, 108.12, 86.89, 70.01, 69.94, 69.83, 69.82, 69.80, 69.48, 69.47, 68.69, 68.22, 58.20, 49.12, 29.73, 28.85, 28.82, 28.76, 28.74, 28.71, 28.65, 28.62, 28.61, 28.59, 28.39, 25.86, 25.73, 25.71, 24.81, 19.87, 19.84, 15.98, 13.98, 7.76. HRMS (ESI) calculated for C61H85Br2N11O12 [M+H]+, 1322.4819; found, 1324.4809.
11-(4-(31-(2,4-dibromo-3-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)-5,8,11,14,17,20,23,26,29-nonaoxahentriacont-1-yl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (18)
Starting from 9d and 14 the general procedure yielded an off-white powder (21.9 mg, 17%) purified by RP-HPLC (45% B held for 10 min, 50% in 20 min, 21.8 min ret time).
1H NMR (700 MHz, DMSO-d6) δ 11.36 (s, 1H), 8.85 (s, 1H), 8.30 – 8.21 (m, 0H), 8.21 (d, J = 8.4 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.82 (s, 1H), 7.78 (s, 1H), 7.74 (d, J = 8.9 Hz, 1H), 7.33 – 7.21 (m, 4H), 7.21 – 7.14 (m, 2H), 5.25 – 5.13 (m, 1H), 4.65 (dd, J = 8.5, 5.4 Hz, 1H), 4.31 – 4.17 (m, 4H), 3.95 (d, J = 5.5 Hz, 0H), 3.80 (q, J = 7.7, 6.5 Hz, 2H), 3.63 (dd, J = 5.8, 3.8 Hz, 2H), 3.56 – 3.42 (m, 30H), 3.34 (dd, J = 52.9, 13.3 Hz, 2H), 2.96 – 2.83 (m, 1H), 2.78 – 2.67 (m, 1H), 2.66 – 2.52 (m, 3H), 2.30 (dq, J = 13.2, 6.7 Hz, 1H), 2.18 (t, J = 7.4 Hz, 0H), 2.05 (hept, J = 7.3 Hz, 2H), 1.76 (p, J = 7.2 Hz, 2H), 1.60 (ddd, J = 12.3, 8.9, 6.2 Hz, 2H), 1.55 – 1.39 (m, 5H), 1.33 – 1.21 (m, 5H), 1.18 (q, J = 5.8, 5.2 Hz, 12H), 1.15 – 1.06 (m, 2H), 0.88 – 0.72 (m, 5H), 0.68 (t, J = 7.1 Hz, 1H). 13C NMR (176 MHz, DMSO-d6) δ 204.51, 203.33, 203.31, 180.67, 180.27, 180.24, 174.50, 174.11, 173.75, 171.18, 171.14, 169.97, 169.87, 155.14, 146.69, 143.10, 142.70, 132.45, 128.17, 128.13, 128.08, 128.04, 126.80, 126.74, 126.60, 126.36, 126.32, 126.30, 121.63, 115.52, 115.26, 115.06, 70.18, 70.00, 69.84, 69.82, 69.80, 69.77, 69.47, 69.18, 68.73, 63.22, 62.97, 58.82, 57.83, 50.60, 49.74, 49.69, 49.11, 42.05, 42.03, 41.85, 35.48, 35.42, 35.38, 33.65, 29.72, 28.85, 28.84, 28.81, 28.74, 28.70, 28.60, 28.53, 28.50, 28.38, 28.34, 28.23, 28.18, 27.44, 27.09, 26.13, 25.85, 25.72, 25.26, 25.22, 25.20, 24.80, 24.48, 19.72, 19.60, 19.45, 19.17, 18.94, 18.85, 17.47, 17.19, 16.89, 14.64, 14.54, 14.52, 10.39, 9.62, 1.11. HRMS (ESI) calculated for C67H97Br2N11O15 [M+H]+, 1454.5605; found, 1454.5595.
11-(4-(40-(2,4-dibromo-3-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)-5,8,11,14,17,20,23,26,29,32,35,38-dodecaoxatetracont-1-yl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (19)
Starting from 9e and 14 the general procedure yielded an off-white powder (17.3 mg, 12.4%) purified by RP-HPLC (45% B held for 10 min, 50% in 20 min, 21.2 min ret time).
1H NMR (700 MHz, DMSO-d6) δ 11.37 (s, 1H), 8.63 (s, 1H), 8.22 (dd, J = 24.0, 8.5 Hz, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.82 (s, 1H), 7.69 (d, J = 8.9 Hz, 1H), 7.47 (s, 1H), 7.33 – 7.21 (m, 5H), 7.21 – 7.12 (m, 3H), 6.70 (s, 3H), 6.55 (s, 2H), 6.50 (s, 2H), 5.25 – 5.09 (m, 1H), 4.65 (dd, J = 8.5, 5.3 Hz, 1H), 4.29 – 4.16 (m, 6H), 3.95 (d, J = 5.5 Hz, 1H), 3.84 – 3.76 (m, 3H), 3.64 (dd, J = 5.8, 3.8 Hz, 3H), 3.56 – 3.48 (m, 7H), 3.45 (dd, J = 5.9, 3.7 Hz, 3H), 3.38 (t, J = 6.5 Hz, 3H), 3.33 (s, 9H), 2.92 (qd, J = 7.4, 5.4 Hz, 1H), 2.71 (ddd, J = 14.2, 8.3, 3.1 Hz, 1H), 2.66 – 2.51 (m, 4H), 2.30 (dq, J = 13.1, 6.7 Hz, 1H), 2.11 – 1.97 (m, 3H), 1.76 (p, J = 7.2 Hz, 3H), 1.60 (p, J = 7.6 Hz, 3H), 1.57 – 1.45 (m, 3H), 1.44 (tt, J = 12.3, 6.7 Hz, 3H), 1.27 – 1.11 (m, 20H), 1.09 (d, J = 7.4 Hz, 2H), 0.88 – 0.79 (m, 2H), 0.76 (dd, J = 9.6, 6.8 Hz, 3H), 0.73 – 0.62 (m, 2H). 13C NMR (176 MHz, DMSO-d6) δ 203.34, 203.31, 180.25, 180.02, 174.11, 173.76, 171.17, 171.14, 171.04, 169.98, 169.87, 163.78, 161.19, 160.19, 160.03, 155.06, 146.67, 143.10, 142.71, 138.56, 136.87, 132.23, 128.17, 128.14, 128.12, 128.08, 128.04, 126.81, 126.74, 126.60, 126.30, 126.16, 121.62, 120.60, 115.35, 115.29, 114.98, 108.08, 70.19, 70.01, 69.82, 69.80, 69.78, 69.48, 69.12, 68.75, 63.22, 62.97, 58.83, 57.84, 49.74, 49.70, 49.11, 42.06, 41.86, 35.59, 35.49, 35.44, 35.38, 29.73, 28.84, 28.82, 28.74, 28.71, 28.65, 28.61, 28.38, 28.19, 27.09, 25.86, 25.72, 25.27, 25.21, 25.15, 24.81, 19.73, 19.60, 19.45, 17.48, 17.20, 14.65, 14.63, 14.54, 9.62, 1.12. HRMS (ESI) calculated for C73H109Br2N11O18 [M/2+H]+, 793.8232; found, 793.8224.
11-(4-(49-(2,4-dibromo-3-(2,7-diaminopyrido[2,3-d]pyrimidin-6-yl)phenoxy)-5,8,11,14,17,20,23,26,29,32,35,38,41,44,47-pentadecaoxanonatetracont-1-yl)-1H-1,2,3-triazol-1-yl)-N-((1S)-3-(((2S)-3-methyl-1-((3R,4S)-4-methyl-2,5-dioxopyrrolidin-3-yl)-1-oxobutan-2-yl)amino)-3-oxo-1-phenylpropyl)undecanamide (20)
Starting from 9f and 14 the general procedure yielded an off-white powder (23.1 mg, 15.3%) purified by RP-HPLC (43.5% B held for 10 min, 90% in 20 min, 18.0 min ret time).
1H NMR (700 MHz, DMSO-d6) δ 11.37 (s, 1H), 8.94 (s, 1H), 8.64 (s, 1H), 8.55 (s, 1H), 8.23 (dd, J = 17.8, 8.2 Hz, 1H), 8.13 (s, 1H), 8.09 (d, J = 8.5 Hz, 0H), 7.91 (s, 1H), 7.84 (s, 1H), 7.79 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.32 – 7.25 (m, 4H), 7.21 (dd, J = 19.3, 7.3 Hz, 2H), 6.67 (s, 2H), 5.19 (p, J = 8.8, 7.8 Hz, 1H), 4.67 – 4.61 (m, 0H), 4.24 (dt, J = 21.4, 5.9 Hz, 4H), 3.95 (d, J = 5.4 Hz, 0H), 3.81 (d, J = 6.0 Hz, 2H), 3.63 (t, J = 4.8 Hz, 2H), 3.54 (d, J = 4.5 Hz, 2H), 3.49 (s, 50H), 3.45 (s, 1H), 2.91 (p, J = 6.7 Hz, 1H), 2.71 (dd, J = 14.4, 8.5 Hz, 1H), 2.59 (t, J = 7.9 Hz, 3H), 2.30 (q, J = 6.6 Hz, 0H), 2.05 (hept, J = 9.5, 8.2 Hz, 2H), 1.76 (p, J = 7.0 Hz, 2H), 1.59 (q, J = 7.6 Hz, 2H), 1.51 (p, J = 6.7 Hz, 2H), 1.43 (s, 3H), 1.24 (d, J = 9.0 Hz, 2H), 1.18 (s, 13H), 1.08 (d, J = 7.4 Hz, 1H), 0.88 – 0.78 (m, 5H), 0.76 (q, J = 9.8, 8.9 Hz, 2H), 0.71 – 0.63 (m, 1H). 13C NMR (176 MHz, DMSO-d6) δ 204.46, 203.30, 203.28, 203.05, 180.64, 180.24, 180.21, 180.03, 179.98, 179.22, 179.08, 178.85, 174.60, 174.47, 174.07, 173.72, 171.15, 171.12, 170.59, 169.95, 169.85, 169.76, 165.71, 163.48, 160.21, 155.15, 152.90, 143.08, 143.00, 142.98, 142.68, 139.61, 135.97, 132.51, 128.25, 128.13, 128.10, 128.04, 128.00, 127.89, 126.77, 126.70, 126.61, 126.56, 126.35, 126.32, 126.29, 126.26, 126.04, 121.84, 115.75, 115.19, 114.92, 106.44, 103.68, 83.76, 77.00, 72.31, 70.15, 69.97, 69.77, 69.74, 69.44, 69.18, 68.69, 63.25, 63.20, 62.98, 62.88, 62.48, 60.16, 59.23, 58.78, 58.62, 57.78, 57.75, 55.09, 54.37, 50.57, 49.83, 49.74, 49.71, 49.68, 49.62, 49.10, 42.03, 41.82, 38.35, 37.17, 35.44, 35.39, 35.34, 33.62, 30.34, 29.68, 28.80, 28.78, 28.70, 28.67, 28.57, 28.50, 28.47, 28.35, 28.30, 28.20, 28.14, 27.39, 27.24, 27.05, 26.10, 25.82, 25.67, 25.27, 25.23, 25.17, 24.80, 24.45, 22.44, 19.80, 19.68, 19.56, 19.41, 19.19, 19.13, 18.91, 18.88, 18.82, 17.77, 17.53, 17.44, 17.17, 16.84, 16.46, 16.36, 14.91, 14.61, 14.59, 14.50, 14.46, 10.36, 9.58, 5.87. HRMS (ESI) calculated for C79H121Br2N11O21 [M/2+H]+, 859.8625; found, 859.8603.
Bacterial strains.
Wild-type E. coli strain K12 sub strain MG1655 was obtained from ATCC. E. coli ΔacrAB was a gift from Prof. Vincent Tam at the University of Houston, and E. coli ΔtolC was a gift from Professor Zgurskaya, University of Oklahoma.
Minimum inhibitory concentration (MIC).
Antibacterial susceptibility tests for aerobically growing bacteria were performed with the microbroth dilution assay according to the Clinical and Laboratory Standard Institute, using visual inspection of cells grown in transparent 96-well plates.50 Briefly, bacteria were grown to mid log phase (OD600 of 0.6–0.7) in cation-adjusted Mueller-Hinton (CaMH) media at 37 °C in an orbital shaker. An inoculum of 106 CFU/mL per well was added to media containing a2-fold dilutions of compounds to give final concentrations ranging from 0.012 μM to 25 μM. The MIC was defined as the minimum concentration of inhibitor at which no visible growth could be detected after 24 h of incubation at 37 °C.
Post-antibiotic effect (PAE).
Bacteria were grown to mid log phase (OD600 of 0.6–0.7) in CaMH media at 37 °C and then exposed to different concentrations of ACC inhibitors or vehicle (DMSO). After shaking for 1 h at 37 °C, cultures were diluted 1000-fold into fresh CaMH media to remove any unbound drug. Regrowth was monitored by withdrawing 0.1 mL aliquots at 1 h time intervals and plating serial dilutions on Muller-Hinton agar plates. The CFUs were determined by counting colonies after overnight incubation at 37 °C. The post-antibiotic effect (PAE) was calculated as the time required for the antibiotic-treated cell population to increase 1 log10 CFU minus the time needed for the control population to increase by 1 log10 CFU.51
Time-kill assays.
Bacteria were grown to mid log phase (OD600 of 0.6–0.7) in CaMH media at 37 °C and then exposed to 32x MIC of ACC inhibitors or vehicle (DMSO). Subsequently, 0.1 mL aliquots were taken at 1 h time intervals and plated in serial dilutions on Muller-Hinton agar plates. The CFUs were determined by counting colonies after overnight incubation at 37 °C. Bactericidal activity was defined as a reduction in CFUs of 3 log10 CFU/mL within the first 3 h.
Cloning, expression, and purification of E. coli ACC proteins.
The plasmid encoding BC was constructed by amplifying the accC gene using primers accC-f and accC-r (Table 4) and plasmid pACM4-ACA (Addgene #49809) as template DNA. The polymerase chain reaction (PCR) product was digested with NdeI and BamHI and ligated into pET28a digested with the same enzymes. The plasmid encoding the CT domain was cloned as one operon in two steps after amplifying the accA and accD genes using the forward and reverse primers in Table 4 and plasmid pACM4-ACA as the template. Initially, the accA PCR product was digested with NdeI and BamHI and ligated into pET28a using the same restriction sites. Subsequently, the PCR product of the accD gene was digested with BamHI and XhoI and ligated into the pET 28a plasmid containing the accA gene using the same restriction sites. Therefore, the final order of the plasmid was constructed to a N-terminus His-tag, α subunit and β subunit, which was used to express the CT domain. Finally, a plasmid encoding BCCP was produced by amplifying accB gene using the primers in Table S2, pACM4-ACA as the template, and NdeI and BamHI to ligate into pET28a. The plasmid constructs were confirmed by DNA sequencing.
Protein expression was performed by transforming the plasmids into E. coli strain BL21 (DE3) pLysS. After transformation, one individual colony was used to inoculate 10 mL of Luria Broth (LB) media containing 30 μg/mL kanamycin in a 50-mL falcon tube that was shaken in the incubator at 37°C overnight. The overnight culture was then used to inoculate 1 L of LB media containing 30 μg/mL of kanamycin, which was shaken at 37°C until the optical density at 600 nm (O.D. 600) increased to 0.8–1.0. Protein expression was induced by adding 1 mM isopropyl-1-thio-b-D-galactopyranoside after which the temperature was reduced to 25°C for ~16 h. Cells were harvested by centrifugation at 5,000 rpm for 15 min at 4°C. The cell pellet was then resuspended in 30 mL of His binding buffer (20 mM Tris HCl, pH 7.9 containing 5 mM imidazole and 0.5 M NaCl) and lysed by sonication. Cell debris was removed by centrifugation at 40,000 rpm for 1 h at 4°C. The BC and CT domains were purified using His affinity chromatography. The supernatant was loaded onto a His-bind column (2 cm x 20 cm) containing 7 mL of Ni- NTA resin that had been charged with 10 mL of charge buffer (50 mM NiSO4) after which the column was washed first with 30 mL of His-bind buffer followed by 60 mL of wash buffer (20 mM Tris HCl, pH 7.9 containing 60 mM imidazole and 0.5 M NaCl). Subsequently, the protein was eluted using 15 mL elute buffer (20 mM Tris HCl, pH 7.9 containing 500 mM imidazole and 0.5 M NaCl). Fractions containing protein were collected, and further purification was performed on an AKTA using a size exclusion column Superdex200 prep grade (GE Healthcare) and 10 mM HEPES buffer pH 8.0 containing 0.5 M KCl. SDS PAGE was used to confirm protein purity, and 50 μL aliquots of each protein were flash frozen in liquid nitrogen and stored at −80°C.
BCCP is post translationally modified though the covalent attachment of biotin to K122,26 and E. coli BirA was used to ensure that BCCP was fully biotinylated. BCCP and BirA were separately expressed and after lysing the cell pellets, the supernatants were mixed, and 2 mM biotin, 10 mM ATP, and 1 mM DTT were added. The reaction mixture was then incubated on ice for 1 h followed by a 15 to 20 min incubation at 37°C. Since BirA precipitates in the presence of high concentrations of ATP at 37°C, the supernatant was centrifuged and filtered before loading onto the His affinity column. Pure BCCP was subsequently obtained following the wash and elution steps described above and stored at −80°C. Biotinylation of BCCP was confirmed by MALDI-TOF mass spectrometry (Figure S5).
ACC Activity Assay.
ACC activity was measured by quantifying ADP production using a Transcreener® ADP FP assay kit from BellBrooks Labs (Madison, Wisconsin. Cat #3010–1K). Initially 1 nM BC, 2 nM BCCP, and 1nM CT were incubated with inhibitors or DMSO in assay buffer (100 mM HEPES pH 8.0 containing 15 mM KHCO3 and 20 mM MgCl2) in a 384 well plate (Corning Cat #4514), and then the reaction was initiated by addition of 10 μM ATP and 200 μM acetyl Coenzyme A. Endpoint reactions were performed by allowing the 10 μL reaction mixtures to run for 45 min before being quenched with 10 μL of Stop and Detection Buffer for a final concentration of 2 nM of Alexa Fluor 633 and 11.8 μg/mL of ADP antibody. In the kinetic mode, all concentrations remained the same, but EDTA was omitted from the detection solution and the plate was read every 2 min over 1 h using a BioTek Synergy Neo2 (BioTek, Winooski, Vermont). Data was analyzed using GraphPad Prism 9 (GraphPad, San Diego, California).
Inhibitor off rate measurements.
A direct binding competition assay was used to determine the rate of inhibitor dissociation from ACC. A 1 μM solution of ACC in 100 mM HEPES pH 8.0 containing 15 mM KHCO3 and 20 mM MgCl2 was incubated for 1 h with heterobivalent inhibitor after which either 100 μM of 1 or 50 μM of 1 and 3 were added. At various time points, a 100 μL aliquot was removed from the mixture and subjected to centrifugal gel filtration chromatography using a PD SpinTrap™ G-25 spin column (Cytiva, Marlborough, MA) previously equilibrated 10 mM NH4HCO2 pH 8.0 buffer. The amount of inhibitor present in the filtrate was quantified using quantified by LC/MSMS mass spectrometry using an Agilent 1290 Infinity II UHPLC and a Poroshell 120 EC-C18 column (1.9 μm; 2.1 x 50 mm). Separation was achieved using a gradient from solvent A (0.1% formic acid in water) to solvent B (acetonitrile, 0.1% formic acid) as follows: 45% B held for 0.3 min, 50% B in 2.7 min, 100% B in 0.1 min and held for 0.3 min, 100% A in 0.1 min, and re-equilibration for 1.5 min. The flow rate was maintained at 0.2 mL/min for the duration of the run, the column was maintained at 40°C, and samples were stored at 4°C. The column eluate was infused into an Agilent 6495c triple quadruple mass spectrometer with an electrospray source for MRM analysis. The instrument was operated in positive mode and two transitions were set up for each of the desired compounds, one for quantitation and the other for qualification. Data analysis was performed using Agilent MassHunter Workstation Quantitative Analysis V 10.0. Inhibitor concentration was converted to target occupancy by using the 0 time point as 100% target occupancy and fitting the data to a single exponential decay using GraphPad Prism 9.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (GM102864 to P.J.T.). M.T.C. was supported by a National Institutes of Health Chemistry-Biology interface training grant (T32GM136572), and Genentech is acknowledged for predoctoral internship support.
Abbreviations Used
- ACC
Acetyl CoA Carboxylase
- BC
Biotin Carboxylase
- BCCP
Biotin Carboxylase Carrier Protein
- CFU
Colony Forming Units
- CT
Carboxyltransferase
- FIC
Fractional inhibitory concentration
- MIC
Minimum inhibitory concentration
- PAE
Post antibiotic effect
- PMBN
Polymyxin b nonapeptide
Footnotes
The authors declare the following competing financial interest(s): P.J.T. is the cofounder of Chronus Pharmaceuticals Inc.
Supporting Information
The Supporting Information is available free of charge at https://
Experimental methods, supplementary tables and figures, chemical synthesis schemes, analytical data, and NMR spectra (PDF)
Molecular formula strings (CSV)
Contributor Information
Matthew T. Cifone, Center for Advanced Study of Drug Action, and Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States
YongLe He, Center for Advanced Study of Drug Action, and Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States.
Rajeswari Basu, Center for Advanced Study of Drug Action and Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY11794-3400, United States.
Nan Wang, Center for Advanced Study of Drug Action, and Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States.
Shabnam Davoodi, Center for Advanced Study of Drug Action, and Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States.
Lauren A. Spagnuolo, Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States
Yuanyuan Si, Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States.
Taraneh Daryaee, Department of Chemistry, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States.
Craig E. Stivala, Discovery Chemistry, Genentech Inc., 1 DNA Way, South San Francisco, CA 94080
Stephen G. Walker, Department of Oral Biology and Pathology, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States
Peter J. Tonge, Center for Advanced Study of Drug Action, and Departments of Chemistry and Radiology, John S. Toll Drive, Stony Brook University, Stony Brook, NY 11794-3400, United States
References
- (1).Copeland RA; Pompliano DL; Meek TD (2006) Drug-target residence time and its implications for lead optimization Nat Rev Drug Discov, 5, 730–739. DOI: 10.1038/nrd2082 [DOI] [PubMed] [Google Scholar]
- (2).Swinney DC (2009) The role of binding kinetics in therapeutically useful drug action Curr Opin Drug Discov Devel, 12, 31–39. DOI: [PubMed] [Google Scholar]
- (3).Lu H; Tonge PJ (2010) Drug-target residence time: critical information for lead optimization Curr Opin Chem Biol, 14, 467–474. DOI: 10.1016/j.cbpa.2010.06.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Copeland RA (2016) The drug-target residence time model: a 10-year retrospective Nat Rev Drug Discov, 15, 87–95. DOI: 10.1038/nrd.2015.18 [DOI] [PubMed] [Google Scholar]
- (5).Schuetz DA; de Witte WEA; Wong YC; Knasmueller B; Richter L; Kokh DB; Sadiq SK; Bosma R; Nederpelt I; Heitman LH; Segala E; Amaral M; Guo D; Andres D; Georgi V; Stoddart LA; Hill S; Cooke RM; De Graaf C; Leurs R; Frech M; Wade RC; de Lange ECM; A.P. I; Muller-Fahrnow A; Ecker GF (2017) Kinetics for Drug Discovery: an industry-driven effort to target drug residence time Drug Discov Today, 22, 896–911. DOI: 10.1016/j.drudis.2017.02.002 [DOI] [PubMed] [Google Scholar]
- (6).Tonge PJ (2018) Drug-target kinetics in drug discovery ACS Chem Neurosci, 9, 29–39. DOI: 10.1021/acschemneuro.7b00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Walkup GK; You Z; Ross PL; Allen EK; Daryaee F; Hale MR; O'Donnell J; Ehmann DE; Schuck VJ; Buurman ET; Choy AL; Hajec L; Murphy-Benenato K; Marone V; Patey SA; Grosser LA; Johnstone M; Walker SG; Tonge PJ; Fisher SL (2015) Translating slow-binding inhibition kinetics into cellular and in vivo effects Nat Chem Biol, 11, 416–423. DOI: 10.1038/nchembio.1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Daryaee F; Chang A; Schiebel J; Lu Y; Zhang Z; Kapilashrami K; Walker SG; Kisker C; Sotriffer CA; Fisher SL; Tonge PJ (2016) Correlating drug-target kinetics and in vivo pharmacodynamics: long residence time inhibitors of the FabI enoyl-ACP reductase Chem Sci, 7, 5945–5954. DOI: 10.1039/C6SC01000H [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Davoodi S; Daryaee F; Chang A; Walker SG; Tonge PJ (2020) Correlating drug-target residence time and post-antibiotic effect: insight into target vulnerability ACS Infect Dis, 6, 629–636. DOI: 10.1021/acsinfecdis.9b00484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Basak S; Tonge PJ In Med Chem Rev; Bronson, J. J, Ed.; Medicinal Chemistry Division of the American Chemical Society: Washington, DC., 2020; Vol. 55, p 367–380. [Google Scholar]
- (11).Basu R; Wang N; Basak S; Daryaee F; Babar M; Allen EK; Walker SG; Haley JD; Tonge PJ (2021) Impact of target turnover on the translation of drug-target residence time to time-dependent antibacterial activity ACS Infect Dis, 7, 2755–2763. DOI: 10.1021/acsinfecdis.1c00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Miller JR; Dunham S; Mochalkin I; Banotai C; Bowman M; Buist S; Dunkle B; Hanna D; Harwood HJ; Huband MD; Karnovsky A; Kuhn M; Limberakis C; Liu JY; Mehrens S; Mueller WT; Narasimhan L; Ogden A; Ohren J; Prasad JV; Shelly JA; Skerlos L; Sulavik M; Thomas VH; VanderRoest S; Wang L; Wang Z; Whitton A; Zhu T; Stover CK (2009) A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore Proc Natl Acad Sci U S A, 106, 1737–1742. DOI: 10.1073/pnas.0811275106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mochalkin I; Miller JR; Narasimhan L; Thanabal V; Erdman P; Cox PB; Prasad JV; Lightle S; Huband MD; Stover CK (2009) Discovery of antibacterial biotin carboxylase inhibitors by virtual screening and fragment-based approaches ACS Chem Biol, 4, 473–483. DOI: 10.1021/cb9000102 [DOI] [PubMed] [Google Scholar]
- (14).Freiberg C; Brunner NA; Schiffer G; Lampe T; Pohlmann J; Brands M; Raabe M; Habich D; Ziegelbauer K (2004) Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity J Biol Chem, 279, 26066–26073. DOI: 10.1074/jbc.M402989200 [DOI] [PubMed] [Google Scholar]
- (15).Guchhait RB; Polakis SE; Lane MD (1975) Biotin carboxylase component of acetyl-CoA carboxylase from Escherichia coli Methods Enzymol, 35, 25–31. DOI: 10.1016/0076-6879(75)35134-3 [DOI] [PubMed] [Google Scholar]
- (16).Choi-Rhee E; Cronan JE (2003) The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase J Biol Chem, 278, 30806–30812. DOI: 10.1074/jbc.M302507200 [DOI] [PubMed] [Google Scholar]
- (17).Guchhait RB; Polakis SE; Lane MD (1975) Carboxyltransferase component of acetyl-CoA carboxylase from Escherichia coli Methods Enzymol, 35, 32–37. DOI: 10.1016/0076-6879(75)35135-5 [DOI] [PubMed] [Google Scholar]
- (18).Broussard TC; Price AE; Laborde SM; Waldrop GL (2013) Complex Formation and Regulation of Escherichia coli Acetyl-CoA Carboxylase Biochemistry, 52, 3346–3357. DOI: 10.1021/bi4000707 [DOI] [PubMed] [Google Scholar]
- (19).Heath RJ; White SW; Rock CO (2002) Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics Appl Microbiol Biotechnol, 58, 695–703. DOI: 10.1007/s00253-001-0918-z [DOI] [PubMed] [Google Scholar]
- (20).Liu X; Fortin PD; Walsh CT (2008) Andrimid producers encode an acetyl-CoA carboxyltransferase subunit resistant to the action of the antibiotic Proc Natl Acad Sci U S A, 105, 13321–13326. DOI: 10.1073/pnas.0806873105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pohlmann J; Lampe T; Shimada M; Nell PG; Pernerstorfer J; Svenstrup N; Brunner NA; Schiffer G; Freiberg C (2005) Pyrrolidinedione derivatives as antibacterial agents with a novel mode of action Bioorg Med Chem Lett, 15, 1189–1192. DOI: 10.1016/j.bmcl.2004.12.002 [DOI] [PubMed] [Google Scholar]
- (22).Freiberg C; Pohlmann J; Nell PG; Endermann R; Schuhmacher J; Newton B; Otteneder M; Lampe T; Habich D; Ziegelbauer K (2006) Novel bacterial acetyl coenzyme A carboxylase inhibitors with antibiotic efficacy in vivo Antimicrob Agents Chemother, 50, 2707–2712. DOI: 10.1128/AAC.00012-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Silvers MA; Robertson GT; Taylor CM; Waldrop GL (2014) Design, synthesis, and antibacterial properties of dual-ligand inhibitors of acetyl-CoA carboxylase J Med Chem, 57, 8947–8959. DOI: 10.1021/jm501082n [DOI] [PubMed] [Google Scholar]
- (24).Waldrop GL; Rayment I; Holden HM (1994) Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase Biochemistry, 33, 10249–10256. DOI: 10.1021/bi00200a004 [DOI] [PubMed] [Google Scholar]
- (25).Bilder P; Lightle S; Bainbridge G; Ohren J; Finzel B; Sun F; Holley S; Al-Kassim L; Spessard C; Melnick M; Newcomer M; Waldrop GL (2006) The structure of the carboxyltransferase component of acetyl-coA carboxylase reveals a zinc-binding motif unique to the bacterial enzyme Biochemistry, 45, 1712–1722. DOI: 10.1021/bi0520479 [DOI] [PubMed] [Google Scholar]
- (26).Broussard TC; Kobe MJ; Pakhomova S; Neau DB; Price AE; Champion TS; Waldrop GL (2013) The Three-Dimensional Structure of the Biotin Carboxylase-Biotin Carboxyl Carrier Protein Complex of E. coli Acetyl-CoA Carboxylase Structure, 21, 650–657. DOI: 10.1016/j.str.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tran TH; Hsiao YS; Jo J; Chou CY; Dietrich LE; Walz T; Tong L (2015) Structure and function of a single-chain, multi-domain long-chain acyl-CoA carboxylase Nature, 518, 120–124. DOI: 10.1038/nature13912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wei J; Tong L (2015) Crystal structure of the 500-kDa yeast acetyl-CoA carboxylase holoenzyme dimer Nature, 526, 723–727. DOI: 10.1038/nature15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hunkeler M; Stuttfeld E; Hagmann A; Imseng S; Maier T (2016) The dynamic organization of fungal acetyl-CoA carboxylase Nat Commun, 7, 11196. DOI: 10.1038/ncomms11196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vauquelin G; Charlton SJ (2013) Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands Br J Pharmacol, 168, 1771–1785. DOI: 10.1111/bph.12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tornoe CW; Christensen C; Meldal M (2002) Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides J Org Chem, 67, 3057–3064. DOI: 10.1021/jo011148j [DOI] [PubMed] [Google Scholar]
- (32).Rostovtsev VV; Green LG; Fokin VV; Sharpless KB (2002) A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes Angew Chem Int Ed Engl, 41, 2596–2599. DOI: [DOI] [PubMed] [Google Scholar]
- (33).Thirumurugan P; Matosiuk D; Jozwiak K (2013) Click Chemistry for Drug Development and Diverse Chemical–Biology Applications Chem Rev, 113, 4905–4979. DOI: 10.1021/cr200409f [DOI] [PubMed] [Google Scholar]
- (34).Banerjee SS; Aher N; Patil R; Khandare J (2012) Poly(ethylene glycol)-Prodrug Conjugates: Concept, Design, and Applications J Drug Deliv, 2012, 103973–103973. DOI: 10.1155/2012/103973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Veronese FM; Pasut G (2005) PEGylation, successful approach to drug delivery Drug Discov Today, 10, 1451–1458. DOI: 10.1016/s1359-6446(05)03575-0 [DOI] [PubMed] [Google Scholar]
- (36).PyMOL. (2015) The PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC. DOI: [Google Scholar]
- (37).Andrews LD; Kane TR; Dozzo P; Haglund CM; Hilderbrandt DJ; Linsell MS; Machajewski T; McEnroe G; Serio AW; Wlasichuk KB; Neau DB; Pakhomova S; Waldrop GL; Sharp M; Pogliano J; Cirz R; Cohen F (2019) Optimization and Mechanistic Characterization of Pyridopyrimidine Inhibitors of Bacterial Biotin Carboxylase J Med Chem. DOI: 10.1021/acs.jmedchem.9b00625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Williams JW; Morrison JF (1979) The kinetics of reversible tight-binding inhibition Methods Enzymol, 63, 437–467. DOI: 10.1016/0076-6879(79)63019-7 [DOI] [PubMed] [Google Scholar]
- (39).Pankey GA; Sabath LD (2004) Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections Clin Infect Dis, 38, 864–870. DOI: 10.1086/381972 [DOI] [PubMed] [Google Scholar]
- (40).Mammen M; Choi S-K; Whitesides GM (1998) Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors Angew Chem Int Ed Engl, 37, 2754–2794. DOI: [DOI] [PubMed] [Google Scholar]
- (41).Valant C; Robert Lane J; Sexton PM; Christopoulos A (2012) The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors Annu Rev Pharmacol Toxicol, 52, 153–178. DOI: 10.1146/annurev-pharmtox-010611-134514 [DOI] [PubMed] [Google Scholar]
- (42).Peschel A; Cardoso FC; Walker AA; Durek T; Stone MRL; Braga Emidio N; Dawson PE; Muttenthaler M; King GF (2020) Two for the Price of One: Heterobivalent Ligand Design Targeting Two Binding Sites on Voltage-Gated Sodium Channels Slows Ligand Dissociation and Enhances Potency J Med Chem, 63, 12773–12785. DOI: 10.1021/acs.jmedchem.0c01107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Tran HNT; Tran P; Deuis JR; Agwa AJ; Zhang AH; Vetter I; Schroeder CI (2020) Enzymatic Ligation of a Pore Blocker Toxin and a Gating Modifier Toxin: Creating Double-Knotted Peptides with Improved Sodium Channel NaV1.7 Inhibition Bioconjug Chem, 31, 64–73. DOI: 10.1021/acs.bioconjchem.9b00744 [DOI] [PubMed] [Google Scholar]
- (44).Steinfeld T; Mammen M; Smith JA; Wilson RD; Jasper JR (2007) A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor Mol Pharmacol, 72, 291–302. DOI: 10.1124/mol.106.033746 [DOI] [PubMed] [Google Scholar]
- (45).Steinfeld T; Hughes AD; Klein U; Smith JA; Mammen M (2011) THRX-198321 is a bifunctional muscarinic receptor antagonist and beta2-adrenoceptor agonist (MABA) that binds in a bimodal and multivalent manner Mol Pharmacol, 79, 389–399. DOI: 10.1124/mol.110.069120 [DOI] [PubMed] [Google Scholar]
- (46).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings Adv Drug Deliv Rev, 46, 3–26. DOI: 10.1016/s0169-409x(00)00129-0 [DOI] [PubMed] [Google Scholar]
- (47).Vaara M (1992) Agents that increase the permeability of the outer membrane Microbiol Rev, 56, 395–411. DOI: [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lebraud H; Wright DJ; Johnson CN; Heightman TD (2016) Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras ACS Cent Sci, 2, 927–934. DOI: 10.1021/acscentsci.6b00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Blackman ML; Royzen M; Fox JM (2008) Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity J Am Chem Soc, 130, 13518–13519. DOI: 10.1021/ja8053805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).CLSI In CLSI Standard M07; 11 ed.; Clinical and Laboratory Standards Institute: Wayne, PA, 2018. [Google Scholar]
- (51).Bundtzen RW; Gerber AU; Cohn DL; Craig WA (1981) Postantibiotic suppression of bacterial growth Rev Infect Dis, 3, 28–37. DOI: 10.1093/clinids/3.1.28 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.