

Abstract
Introduction
Pituitary adenomas (PAs) are common, usually benign tumors of the anterior pituitary gland which, for the most part, have no known genetic cause. PAs are associated with major clinical effects due to hormonal dysregulation and tumoral impingement on vital brain structures. PAM encodes a multifunctional protein responsible for the essential C-terminal amidation of secreted peptides.
Methods
Following the identification of a loss-of-function variant (p.Arg703Gln) in the peptidylglycine a-amidating monooxygenase (PAM) gene in a family with pituitary gigantism, we investigated 299 individuals with sporadic PAs and 17 familial isolated PA kindreds for PAM variants. Genetic screening was performed by germline and tumor sequencing and germline copy number variation (CNV) analysis.
Results
In germline DNA, we detected seven heterozygous, likely pathogenic missense, truncating, and regulatory SNVs. These SNVs were found in sporadic subjects with growth hormone excess (p.Gly552Arg and p.Phe759Ser), pediatric Cushing disease (c.-133T>C and p.His778fs), or different types of PAs (c.-361G>A, p.Ser539Trp, and p.Asp563Gly). The SNVs were functionally tested in vitro for protein expression and trafficking by Western blotting, splicing by minigene assays, and amidation activity in cell lysates and serum samples. These analyses confirmed a deleterious effect on protein expression and/or function. By interrogating 200,000 exomes from the UK Biobank, we confirmed a significant association of the PAM gene and rare PAM SNVs with diagnoses linked to pituitary gland hyperfunction.
Conclusion
The identification of PAM as a candidate gene associated with pituitary hypersecretion opens the possibility of developing novel therapeutics based on altering PAM function.
Keywords: peptidylglycine α-amidating monooxygenase, amidation, gigantism, acromegaly, Cushing disease, pituitary tumors
Introduction
The anterior pituitary gland plays a critical role in the dynamic control of major hormonal systems, including growth, fertility, and stress responses. Anterior pituitary adenomas (PAs), also termed pituitary neuroendocrine tumors (PitNETs) (1), can be comprised of any of the secretory cell subtypes, such as lactotropes that secrete prolactin, somatotropes [growth hormone (GH)], corticotropes [adrenocorticotropic hormone (ACTH)], gonadotropes [follicle-stimulating hormone (FSH) and luteinizing hormone (LH)], and thyrotropes [thyroid-stimulating hormone (TSH)] (2). Although they are usually benign lesions, PAs can have a major clinical impact through hormonal dysregulation and direct mass effects on or the invasion of brain structures (optic chiasm and cavernous sinus). Epidemiologically, PAs are one of the most common intracranial tumor types and lead to clinically apparent disease with a frequency of about 1 per 1,000 in the general population (3, 4). Approximately 95% of PAs occur sporadically. Hereditary PAs are often distinguished by a more severe clinical presentation, such as an earlier age at onset, aggressive growth, larger size, and greater resistance to treatment, and they can coexist with other syndromic components. Therefore, the identification of germline genetic causes can have implications for earlier identification and treatment of affected individuals via genetic and clinical screening (5).
Germline changes like single-nucleotide variants (SNVs) and/or copy number variations (CNVs) in several genes have been implicated in familial isolated PAs—FIPA (AIP and GPR101 genes) (6, 7), familial syndromic PAs (MEN1, CDKN1B, PRKAR1A, PRKACB, SDHx, MAX, NF1, DICER1, and TSC2, among others), and sporadic PAs (AIP, GPR101, and CABLES1). Recurrent somatic SNVs are found most frequently in GNAS and USP8, in sporadic GH and ACTH-secreting PAs, respectively, and rarely in other genes (8). Despite these advances in establishing gene–disease links, the etiology of the overwhelming majority of inherited and sporadic PAs remains unknown. Hence, the identification and characterization of novel pathological genetic and genomic variants in PA cohorts is medically significant.
In a large, international cohort of individuals with PAs, we identified germline loss-of-function (LOF) variants of PAM (peptidylglycine α-amidating monooxygenase, MIM: 170270) in subjects with familial and sporadic GH and ACTH hypersecretion. PAM is a highly conserved, multifunctional protein that is increasingly recognized as an important regulator of peptide amidation and secretion, among many other functions, in health and disease (9). We functionally evaluated 36 SNVs, demonstrating a deleterious effect on PAM function/expression in eight variants. We also report a statistically significant association of rare PAM SNVs with the diagnoses of hyperfunction or tumors of the anterior pituitary gland. These results suggest that pathogenic PAM variants can predispose and/or contribute to the development of pathological pituitary hypersecretion.
Results
Identification of a pathogenic single-nucleotide variant in the PAM gene in a family with pituitary gigantism
The discovery cohort consisted of a three-member, non-consanguineous FIPA family that was enrolled in the Undiagnosed Diseases Program (UDP) of the National Institutes of Health Clinical Center (NIHCC). The kindred consisted of monozygotic twin brothers (individuals II-2 and II-3) and the eldest child of individual II-2, all of whom were affected by childhood-onset pituitary gigantism ( Table 1 ; Figure 1A ).
Table 1.
Summary of characteristics of seven cases with rare pathogenic peptidylglycine α-amidating monooxygenase (PAM) variants.
| ID | Diagnosis | Gender | Pathogenic PAM variant(s) | Ethnicity | Age at disease onset (years) | Age at PA diagnosis (years) | Tumor size (mm) | Treatment | Other clinical diagnoses |
|---|---|---|---|---|---|---|---|---|---|
| II-2* | GH excess (gigantism) | M | c.2108G>A (p.Arg703Gln) | Caucasian | <10 | 37 | 4 | PegV | Hypogonadism, adrenal nodules, pleural masses, severe osteopenia, muscular atrophy, diverticulosis |
| II-3* | GH excess (gigantism) | M | c.2108G>A (p.Arg703Gln) | Caucasian | <10 | 37 | 5 × 6 | TSS, SSA, PV | Hypogonadism, pancreatic and colon adenocarcinoma |
| III-3* | GH excess (gigantism) | M | c.2108G>A (p.Arg703Gln) | Caucasian | 1 | 3 | n.a. | SSA, PegV | Hypotonia |
| NIH26 | Pediatric CD | F | c.2332-2A>T (p.His778fs) | Caucasian | 12 | 14 | 3 | TSS | Scleroderma |
| NIH36 | Pediatric CD | F | c.-133T>C | African-American | 12 | 13 | 10 | TSS (×3) | None |
| Belgium128 | GH excess (gigantism) | M | c.2276T>C (p.Phe759Ser) | Latino | 18 | 20 | Macro | TSS, SRL | None |
| Belgium197 | GH excess (acromegaly) | M | c.1654G>A (p.Gly552Arg), c.1688A>G (p.Asp563Gly) | Caucasian | 40 | 41 | 40 | TSS, SRL, RTx | None |
F, female; M, male; n.a., not applicable; PA, pituitary adenoma; PegV, pegvisomant; RTx, radiotherapy; SRL, somatostatin receptor ligand; TSS, transsphenoidal surgery.
*Member of the index FIPA kindred.
Figure 1.

Clinical findings in the index family with pituitary gigantism. (A) Pedigree. Generation numbers are represented by Roman numerals, and individual numbers are in Arabic numerals. The proband is II-2, indicated by the black arrow. Open square/circle, unaffected male/female; filled square, affected male. The peptidylglycine α-amidating monooxygenase (PAM) mutational status is shown under each screened individual. In (B), the white arrowheads point to a possible 4 mm lesion (left panel, sagittal plane; right panel, coronal plane) seen in II-2. (C) Growth chart for individual III-3 before and after medical interventions. (D) A sagittal T2-weighted magnetic resonance imaging of individual III-3 that was performed before medical therapy began did not reveal a pituitary lesion. (E) Time course of the effects of treatment modalities on the growth hormone (GH; left axis) and IGF-1 (right axis; squares) in individual III-3. The rapid decrease of IGF-1 after switching from lanreotide (LAN) to pegvisomant (PegV) is evident.
The monozygotic twins were born at gestational week 31; their birth weights and lengths were 1.96 kg and 48.3 cm (II-2) and 2.3 kg and 53.3 cm (II-3), respectively. They had two normally sized siblings. Their father was 190 cm in height. Their mother was very tall (187 cm; +4.1 SDS using Centers for Disease Control charts) but had no known history of pituitary disease or established medical diagnosis of overgrowth; she died of colorectal cancer at the age of 73 years. The precise onset of the twins’ excessive growth is uncertain, but it began during childhood, such that by the age of 12, they both measured >200 cm in height (>+6.2 SDS). They were formally diagnosed with pituitary gigantism as adults (37 years), at which time their final height was 231 cm (+7.8 SDS). At diagnosis, they had clinical signs of acromegaly including coarse facial features, frontal bossing, and enlarged extremities. Elevated random GH (26 and 23.1 ng/ml) and insulin-like growth factor 1 (IGF-1) (603 and 1,130 ng/ml) levels were noted, but prolactin levels were below normal levels. Only minor abnormalities were noted on 3 Tesla pituitary magnetic resonance imaging (MRI), with possible small pituitary microadenomas, 4 and 5 mm in maximum diameter ( Figure 1B ). Individual II-3 underwent transsphenoidal surgery, but no somatotropinoma tissue was identified by histopathology. Both brothers required medical therapy with the GH-receptor antagonist, pegvisomant, to reduce excess IGF-1. Colorectal and pancreatic adenocarcinomas were diagnosed in patient II-3, and he subsequently died of the latter tumor at 48 years of age.
The third affected member was III-3, a male (son of II-2) who was of normal length and weight at birth but then developed marked overgrowth by the age of 12 months ( Figure 1C ). His siblings are dizygotic twins with normal height. Subject III-3 was diagnosed with GH and IGF-1 excess (random GH 30 ng/ml and IGF-1 389 ng/ml) before 5 years of age. An MRI demonstrated a normal pituitary gland ( Figure 1D ). To control the excessive growth, the patient was started on the long-acting somatostatin receptor ligand, lanreotide autogel, at a dose of 90–120 mg/month subcutaneously. Despite this adult-level dosing, the GH remained uncontrolled and the IGF-1 rose to 487 ng/ml. During the lanreotide autogel treatment period, the growth velocity remained accelerated, at 15.9–20.0 cm/year. Similar to II-2 and II-3, his condition markedly improved on switching to pegvisomant (10–15 mg/day subcutaneously): IGF-1 rapidly fell from 500 to 232 ng/ml, and growth velocity decreased to 3.6–5.6 cm/year during approximately 5 years of follow-up ( Figure 1E ). His growth curves for height and weight remain above the 99th centile but are no longer diverging.
We conducted WES in peripheral blood–derived DNA isolated from the index cases (see the Methods section for details). We applied a variant prioritization strategy ( Figure S1 ; Table S3 ) and selected for further analysis a heterozygous missense SNV in PAM (c.2108G>A, p.Arg703Gln), which was shared by both twins from generation II ( Figure 1A ). The DNA of their deceased mother was unavailable; thus, we could not confirm whether they inherited the PAM variant from her or if it arose de novo; the father was wild type (WT). The p.Arg703Gln variant was present in not only III-3, the child with gigantism, but also III-1, his younger brother ( Figure 1A ). Endocrine and growth studies showed that III-1 had no evidence of growth excess or hormonal dysregulation at the time of study (aged 8 years). All other family members studied were WT for PAM and had normal growth. Overall, these results suggest incomplete penetrance. No other potentially pathogenic alterations in genes were shared among the three affected members.
The PAM variant was notable among the 27 candidate genes due to its strong expression in neuroendocrine tissues and its function. As reported in The Human Protein Atlas, the pituitary is among the tissues displaying the strongest PAM mRNA and protein expression signal in humans and other mammals. PAM is highly expressed in all pituitary secretory cell types and in folliculostellate cells ( Figure S2A ), where it plays a crucial function in posttranslational hormone processing and secretion (10, 11). Staining for PAM and GM130, a cis-Golgi marker, in normal adult human pituitary cells showed diffuse cytoplasmic expression for PAM that partially overlapped the Golgi ( Figure S2B ), reflecting the established trafficking of PAM within the secretory pathway observed in rat pituitary cells.
PAM is a multifunctional protein that contains two enzymatic domains, peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine α-amidating lyase (PAL); acting sequentially; the two domains generate C-terminally amidated peptides (10). The p.Arg703Gln variant—located in the PAL domain ( Figure 2A )—has an extremely low minor allele frequency in controls (MAF: 0.0013%) and no homozygous variant entries in gnomAD and is bioinformatically predicted to be pathogenic ( Table 2 ).
Figure 2.

Location and evolutionary conservation of the missense and frameshift PAM SNVs that were functionally tested. (A) Schematic representation of the PAM gene (GenBank: NM_000919.3, PAM-1, 25 exons) and encoded protein (NP_000910.2, 974 amino acids), including functional domains, with 15 missense and one frameshift SNVs. Gene and protein structures were drawn with the Gene Structure Display Server (GSDS ver. 2.0) (12). Variants found to have deleterious effects on PAM function/expression (p < 0.01) are shown in yellow lettering, while those without major effects are shown in white lettering. Brackets identify the non-catalytic regions that precede PHMcc (N-terminus) and follow PALcc (C-terminus); CD, cytosolic domain; CDS, coding sequence; PALcc, catalytic core of peptidyl-α-hydroxyglycine α-amidating lyase; PHMcc, catalytic core of peptidylglycine α-hydroxylating monooxygenase; TMD, transmembrane domain; UTR, untranslated region. (B) Protein sequence alignments for five of the variants with deleterious effects. Conserved affected residues are shown in yellow. PAL activities, indicated in red, refer to functional experiments in PEAKrapid cells. (C) The crystal structure of rat PALcc (PDB entry 3FW0) was used to contextualize the missense variants categorized as likely pathogenic based on in silico analyses; the WT residue is shown on the left and the mutant residue on the right. PAL folds as a β-propeller, with six blades (numbered 1–6) positioned around a central cavity. The calcium and mercury ions are depicted as yellow and orange spheres, respectively. The mercury ion was used instead of zinc to capture the binding of a non-peptide substrate, α-hydroxyhippuric acid, depicted in blue. The affected residues are highlighted in purple (Ser or Trp 539), cyan (Gly or Arg 552), red (Asp or Gly 563), green (Arg or Gln 703), and orange (Phe or Ser 759), along the ribbon visualization of WT rat PALcc in gray. Arg703 is positioned at the active site and participates in substrate binding. Interestingly, p.Gly552Arg and p.Asp563Gly are located on the same face of the β-propeller. C, C-terminus; N, N-terminus.
Table 2.
Prioritized heterozygous missense, truncating, and regulatory PAM variants that were functionally analyzed.
| DNA change | Protein change | SNP ID | Location in gene | Location in protein | MAF in our cohort (%) | Global control MAF (%) | Highest control population MAF (%) | # of homozygotes in gnomAD | P-value | In silico prediction | Individuals harboring the variant |
|---|---|---|---|---|---|---|---|---|---|---|---|
| c.-607A>G † | n.a. | rs76396377 | promoter | n.a. | 0.4 | 1.479 | 2.943 (African) | 24 | n.s. | 1 TF replaced | 1 from 1 FIPA (PRL) |
| c.-550G>T ‡ | n.a. | rs1007572715 | promoter | n.a. | 0.4 | 0.0072 | 0.0415 (South Asian) | 0 | <0.05 | 2 TFs lost | 1 acro |
| c.-361G>A | n.a. | rs143617515 | 5’UTR (exon 1) | n.a. | 1.21 | 0.6275 | 3.647 (Finnish) | 12 | n.s. | 2 TFs lost, 4 TFs created | 2 gigantism, 1 NFPA |
| c.-133T>C ¥ | n.a. | rs1413721196 | 5’UTR (exon 1) | n.a. | 0.25 | 0.0007 | 0.0015 (European) | 0 | <0.05 | 1 TF lost | 1 pediatric CD |
| c.-109G>C | n.a. | Novel | 5’UTR (exon 1) | n.a. | 0.25 | n.a. | n.a. | n.a. | n.a. | 1 TF replaced | 1 acro |
| c.-35C>G | n.a. | rs201016377 | 5’UTR (exon 1) | n.a. | 0.15 | 0.0749 | 0.9040 (Finnish) | 1 (Finnish) | n.s. | n.a. | 1 gigantism |
| c.79G>A ** | p.Val27Ile | rs199856250 | exon 1 | proregion | 0.16 | 0.0004 | 0.0008 (European) | 0 | <0.05 | VUS (1, 6, 12) | 1 acro |
| c.92T>A | p.Phe31Tyr | rs114014768 | exon 2 | linker region | 0.63 | 0.8654 | 2.814 (African) | 19 | n.s. | likely benign (0, 1, 16) | 2 acro, 1 gigantism, 1 pediatric CD |
| c.145G>C | p.Val49Leu | rs2230458 | exon 2 | PHMcc | 0.94 | 0.9278 | 1.803 (Finnish) | 5 (European) | n.s. | likely benign (0, 2, 17) | 2 gigantism, 3 pediatric CD, 1 PRL |
| c.239G>A | p.Arg80Gln | rs753307443 | exon 3 | PHMcc | 0.16 | 0.0026 | 0.0066 (Latino) | 0 | <0.05 | likely benign (1, 5, 13) | 1 from 1 FIPA (GH excess) |
| c.718C>T †† | p.His240Tyr | rs761898981 | exon 9 | PHMcc | 0.15 | 0.0039 | 0.0478 (Other) | 0 | <0.05 | VUS (0, 11, 7) | 1 gigantism |
| c.731T>C ¥¥ | p.Val244Ala | rs201009674 | exon 10 | PHMcc | 0.16 | 0.0533 | 0.6632 (Ashkenazi Jewish) | 0 | n.s. | likely pathogenic (6, 12, 0) | 1 pediatric CD |
| c.1235C>T ** | p.Pro412Leu | rs761619241 | exon 14 | linker region | 0.16 | 0.0016 | 0.0029 (Latino) | 0 | <0.05 | likely pathogenic (6, 9, 4) | 2 from 1 FIPA (heterogeneous) |
| c.1354A>G | p.Ile452Val | rs145710876 | exon 14 | linker region | 0.16 | 0.0151 | 0.0555 (African) | 0 | n.s. | likely benign (0, 0, 18) | 1 gigantism |
| c.1473A>C | p.Glu491Asp | rs61736661 | exon 14 | linker region | 0.16 | 0.6662 | 1.241 (European) | 8 (European) | n.s. | likely benign (0, 1, 16) | 1 from 1 FIPA (PRL) |
| c.1616C>G | p.Ser539Trp | rs78408340 | exon 16 | PALcc | 0.15 | 0.4169 | 0.7250 (European) | 2 (European) | n.s. | likely pathogenic (10, 6, 2) | 1 acro |
| c.1654G>A ‡‡ | p.Gly552Arg | rs201249509 | exon 16 | PALcc | 0.15 | 0.0053 | 0.0103 (European) | 0 | <0.05 | likely pathogenic (10, 6, 2) | 1 acro |
| c.1688A>G | p.Asp563Gly | rs35658696 | exon 16 | PALcc | 4.00 | 3.109 | 5.657 (European) | 108 | n.s. | likely pathogenic (4, 11, 1) | 13 acro, 5 gigantism, 4 PRL, 4 CD (3 pediatric, 1 silent) |
| c.1921G>C | p.Gly641Arg | rs775364358 | exon 18 | PALcc | 0.15 | 0.0033 | 0.0197 (Latino) | 0 | <0.05 | likely benign (0, 3, 16) | 1 PRL |
| c.2108G>A | p.Arg703Gln | rs761130902 | exon 19 | PALcc | 0.15 | 0.0013 | 0.0193 (East Asian) | 0 | <0.05 | likely pathogenic (10, 9, 0) | 4 (3 affected) from 1 FIPA (gigantism) |
| c.2276T>C †† | p.Phe759Ser | rs375364507 | exon 20 | PALcc | 0.15 | 0.0092 | 0.0206 (European) | 0 | n.s. | likely pathogenic (7, 11, 0) | 1 gigantism |
| c.2332-2A>T § | p.His778fs | rs1006675725 | intron 20 | PALcc | 0.15 | 0.0007 | 0.0015 (European) | 0 | <0.05 | pathogenic (disrupts canonical SA) | 2 (1 pediatric CD and unaffected mother) |
| c.*1455C>T ¥¥¥ | n.a. | rs146343559 | 3’UTR (exon 25) | n.a. | 0.67 | 0.0362 | 0.1047 (Latino) | 0 | n.s. | creates new miRNA binding sites | 1 pediatric CD |
Variants were annotated using the NC_000005.9(NM_000919.3) reference sequence. All variants were observed in heterozygosis. Unless specified, MAFs were retrieved from the gnomAD database ver. 3.1.2. “European” excludes the Finnish population; “African” includes African-American; and “Latino” includes Admixed American. For in silico predictions, the computational verdict was based on the combined outputs of 19 softwares available in Varsome (within parentheses, the first number represents the pathogenic verdicts, the second number represents the uncertain verdicts, and the third number represents the benign verdicts) for all missense variants. For the truncating and 3’UTR variants, we used Alamut predictions; for the regulatory variants, we used Genomatix MatInspector. Individual variant allele frequencies in the study population were compared with the global frequencies reported in gnomAD using the Fisher’s exact test or chi-square test, as appropriate.
acro, acromegaly; CD, Cushing disease; FIPA, familial isolated pituitary adenoma; MAF, minor allele frequency; n.a., not available/applicable; NFPA, non-functioning pituitary adenoma; n.s., not significant; PALcc, catalytic core of peptidyl-α-hydroxyglycine α-amidating lyase; PHMcc, catalytic core of peptidylglycine α-hydroxylating monooxygenase; PRL, prolactinoma; SA, splice acceptor site; TF, transcription factor; VUS, variant of uncertain significance.
†This individual harbors the PAM p.Glu491Asp variant in exon 14.
‡This individual harbors the PAM p.Val49Leu variant in exon 2.
¥This individual harbors the PAM p.Phe31Tyr variant in exon 2 and a somatic heterozygous p.Ser718del variant in the USP8 gene (13).
††This individual is compound heterozygote for p.[His240Tyr];p.[Phe759Ser].
‡‡This individual is compound heterozygote for p.[Gly552Arg];p.[AspD563Gly].
¥¥This individual harbors a somatic heterozygous p.Ser718Pro variant in the USP8 gene (13).
¥¥¥This individual harbors a somatic heterozygous p.Ser718del variant in the USP8 gene (13).
§Somatic hotspot variants in the USP8, BRAF, and USP48 genes were ruled out in this case.
**Allele frequency obtained from gnomAD ver. 2.1.1.
Arg703 is conserved throughout evolution ( Figure 2B ). Along with Tyr651, crystallographic studies identified Arg703 as part of the PALcc catalytic dyad; the catalytic activity of soluble recombinant rat PALcc, in which Arg703 was replaced by Ala or Gln, was greatly reduced (14), a result consistent with its location at the active site ( Figure 2C ). Altogether, these data suggested that a LOF PAM variant could be associated with pituitary gigantism, prompting us to pursue this lead.
PAM screening in a validation cohort revealed multiple likely pathogenic single-nucleotide variants by in silico prediction
We next determined whether predicted pathogenic PAM variants are associated with other types of PAs, both hereditary and sporadic. To do so, we screened a diverse group of 326 germline and 60 tumor DNAs from PA patients for PAM SNVs (see the Methods section and Table S1 for details). Sequencing covered the PAM CDS and exon–intron junctions. Germline CNVs at the PAM locus were analyzed by ddPCR in a cohort of 137 individuals with PAs (16 with gigantism and 121 with different types of PAs). Germline DNA sequencing detected 51 SNVs ( Tables 2 , S4 ). There were no germline CNVs or rare/pathogenic somatic DNA SNVs (data not shown). A subset of the germline SNVs (15 of 51) was prioritized for further screening together with the p.Arg703Gln variant ( Table 2 ). This selection was based on the following non-mutually exclusive criteria: MAF < 1%, in silico analysis with multiple algorithms predicting a deleterious effect on PAM function or splicing, and published functional data indicating pathogenicity (15). To complement these data, we used Clustal and structure-based analyses to identify variants that could impact protein structure/function ( Figures 2B, C , S3 ). The remaining, non-characterized germline SNVs are listed in Table S4 .
Of the 16 heterozygous non-synonymous and splice site-affecting SNVs prioritized for functional studies, 4 were in PHMcc and 7 were in PALcc; the remainder were in the non-catalytic regions of PAM ( Figure 2A ). Based on in silico predictions, one of the four SNVs in PHMcc and six of the seven in PALcc were predicted to be pathogenic ( Table 2 ). The prioritized variants were not significantly spatially clustered when we considered the geometric mean distance between all pairs of variants normalized to complementary DNA (cDNA) length (p = 0.35). However, when we classified the variants based on their location within the N-terminus, PHMcc, linker, PALcc, or C-terminus ( Figure 2A ), the N-terminus harbored more variants than expected by random chance (p = 0.02, Table S5 ).
Expression of prioritized PAM single-nucleotide variants revealed deficits in protein expression, enzymatic activity, and glycosylation
To understand the impact of the prioritized PAM variants on the function of this membrane enzyme, we transiently expressed each PAM-1 variant in PEAKrapid (HEK-293 derivative) cells. To facilitate an accurate assessment of the PHM and PAL activity of each PAM variant, a non-ionic detergent (TX-100) was used to solubilize membrane proteins from a crude particulate fraction ( Figure 3A ). PAM expression levels and protein integrity were evaluated by Western blot analysis ( Figure 3B ), and PAM trafficking was assessed by the analysis of N- and O-linked oligosaccharide maturation ( Figures 3C, D ).
Figure 3.

Enzymatic activity, protein expression, and glycosylation pattern of PAM variants. (A) PHM and PAL activity. As described in the Methods, TMT-solubilized particulate fractions prepared from transiently transfected PEAKrapid cells were assayed for PHM and PAL activity. The data for the level of expression of WT PAM and each full-length variant were determined by quantifying the FLAG-tag signal. The levels of p.His778fs were assessed as described in the Methods. NT, not transfected; **p < 0.0001. (B) PAM protein expression. SNV expression was assessed using a FLAG tag antibody and an antibody to a peptide contained in PHMcc (JH246). The lines separating WT and p.His240Tyr from p.Arg703Gln and p.Phe759Ser indicate that the data for two intervening samples were removed. Molecular weight standards are indicated. (C) PAM protein diagram, indicating the location of the JH246 epitope (red horizontal line), the FLAG tag (gray diamond), and the expected location of N- and O-glycans (blue and yellow freeform shapes, respectively). (D) Glycosylation is altered in a subset of PAM variants. Cell lysates were treated with PNGase F or neuraminidase as described in the Methods. Proteins were visualized using the FLAG antibody. The samples treated with neuraminidase were analyzed on two separate gels, with the p.Arg703Gln samples appearing in part on both gels. Con, control; In, input; N-Gly, PNGase; Neur, neuraminidase; NT, not transfected.
To establish the efficacy of our expression system, we compared the properties of WT PAM to those of missense PAM variants designed to inactivate PHMcc (p.Thr189Ile) or PALcc (p.His529Arg and p.Gly796Glu, Figure S4 ). PHM and PAL activity measurements for WT PAM and for each engineered control were normalized to the expression of that PAM protein [(PHM or PAL activity)/PAM protein], providing a measure of its specific activity; the PHM and PAL specific activities of each engineered control were then compared to the PHM- and PAL-specific activities of WT PAM. As expected, the PHM activity of the p.Thr189Ile mutant and the PAL activity of the p.His529Arg and p.Gly796Glu mutants were reduced more than 10-fold compared to WT PAM ( Figure S4 ; Table 3 ).
Table 3.
Summary of effects on enzymatic activity and glycosylation for all PAM variants that were functionally tested.
| PAM variant | PHM activity (% WT) | PAL activity (% WT) | PNGaseF (N-sugars) |
Neuraminidase (sialic acid) |
|---|---|---|---|---|
| WT | 100 ± 2 | 100 ± 3 | yes | yes |
| Val27Ile | 79 ± 6 | 103 ± 15 | (yes) | (yes) |
| Phe31Tyr | 76 ± 16 | 84 ± 1 | (yes) | (yes) |
| Val49Leu | 80 ± 26 | 93 ± 1 | (yes) | (yes) |
| Arg80Gln | 75 ± 6 | 87 ± 9 | (yes) | (yes) |
| Thr189Ile † | 6 ± 3** | 23 ± 5** | yes | no |
| His240Tyr | 111 ± 24 | 98 ± 22 | yes | no |
| Val244Ala | 113 ± 32 | 80 ± 10 | (yes) | (yes) |
| Pro412Leu | 81 ± 32 | 85 ± 28 | (yes) | (yes) |
| Ile452Val | 112 ± 45 | 113 ± 30 | (yes) | (yes) |
| Glu491Asp | 133 ± 75 | 138 ± 55 | (yes) | (yes) |
| His529Arg † | 41 ± 13 | 7 ± 1** | n.a. | n.a. |
| Ser539Trp ‡ | 49 ± 7** | 0 ± 1** | yes | no |
| Gly552Arg | 41 ± 6** | 6 ± 2** | yes | no |
| Asp563Gly | 44 ± 8** | 14 ± 3** | yes | no |
| Gly641Arg | 104 ± 0 | 116 ± 10 | (yes) | (yes) |
| Arg703Gln | 92 ± 17 | 0 ± 1** | yes | yes |
| Phe759Ser | 110 ± 25 | 12 ± 3** | yes | no |
| His778fs | 23 ± 3** | 0 ± 1** | n.a. | n.a. |
| Gly796Glu † | 55 ± 8 | 4 ± 1** | (yes) | (yes) |
Values are presented as mean ± SEM. “yes” and “no” refer to the presence and absence, respectively, of N-linked sugars or sialic acid in the variants tested for glycosylation. “(yes)” was given when the effect on glycosylation was based only on SDS-PAGE profile.
n.a., not assessed.
†Engineered inactive mutants.
‡We prepared two independent plasmid constructs and verified by sequencing that they were correct. The p.Ser539Trp variant disrupted the normal trafficking of membrane PAM; when present in the soluble protein, it may inhibit its secretion (15).
** p < 0.0001.
The structure of bifunctional PAM has not yet been experimentally determined, but for all three engineered variants, inactivating mutations placed into one domain resulted in a modest decrease in the activity of the other domain.
The activity and Western blot data for all studied SNVs are shown in Figures 3A, B , respectively. No significant decrease in PHM activity was seen for SNVs p.Val27Ile, p.Phe31Tyr, p.Val49Leu, p.Arg80Gln, p.His240Tyr, or p.Val244Ala. The three non-catalytic linker domain SNVs tested exhibited no significant change in PHM or PAL activity and were not extensively assayed. Six of the seven SNVs located in PALcc exhibited a dramatic decrease in PAL activity. While some PALcc variants reduced PHM activity, others did not. Although the PAL activity of p.Arg703Gln was undetectable and the PAL activity of p.Phe759Ser was 12% of WT, their PHM activities were equal to that of WT. In contrast, the dramatic reductions in PAL activity observed for p.Ser539Trp, p.Gly552Arg, p.Asp563Gly, and p.His778fs were accompanied by at least a twofold decrease in PHM activity. Normal levels of both PHM and PAL activity were detected for p.Gly641Arg.
The maturation of N-linked oligosaccharides is generally completed only when the newly synthesized protein exits the Golgi complex. The sialylation of both the N- and O-linked oligosaccharides attached to rat PAM-1 occurs just before it exits the Golgi complex. The stepwise manner in which the maturation of N- and O-linked glycans occurs often results in glycoprotein heterogeneity, contributing to the diffuse bands observed for WT PAM and many of the prioritized SNVs ( Figure 3B , FLAG Ab and PHM Ab). In contrast, we consistently observed a slightly smaller, more compact band for p.Thr189Ile (engineered variant), p.His240Tyr, p.Ser539Trp, p.Gly552Arg, p.Asp563Gly, and p.Phe759Ser.
Since altered access to the enzymes responsible for the maturation of N- and O-linked glycans can reflect altered protein trafficking, we utilized PNGase F to remove N-linked oligosaccharides and neuraminidase to remove sialic acid ( Figure 3D ). The 2–3 kDa reduction in apparent molecular mass caused by PNGase F treatment demonstrates that the engineered control lacking PHM activity (Thr189Ile) and each of these variants had been N-glycosylated. As expected, neuraminidase treatment brought about a slight decrease in the apparent molecular mass of WT PAM. Strikingly, neuraminidase treatment failed to reduce the mass of any of these six SNVs, indicating that their glycans had not undergone normal sialylation. Altered trafficking in the secretory and/or endocytic pathway could limit sialylation.
A summary of the effect of each prioritized and engineered SNV on PAM expression, enzymatic activity, and oligosaccharide maturation is presented in Table 3 .
Three SNVs with diminished PAL activity were found in subjects with GH excess leading to sporadic acromegaly (p.Ser539Trp and p.Gly552Arg) or gigantism (p.Phe759Ser). The relatively common p.Asp563Gly variant (3.11% global MAF) was observed in persons with different types of PAs ( Table 2 ). Interestingly, two males with GH excess—one with acromegaly and one with gigantism—were compound heterozygotes for p.[Gly552Arg];[Asp563Gly] and p.[His240Tyr];[Phe759Ser], respectively ( Figure S5 ; Table 1 ). Although neither the PHM nor PAL activity of p.His240Tyr (located in PHMcc) differed from the WT, its heterogeneous band pattern and lack of sensitivity to neuraminidase suggest structural alterations sufficient to alter its trafficking ( Figures 3A, B, D ); its significantly higher prevalence in the cohort of subjects with PAs vs. controls, and the absence of homozygous individuals in gnomAD, argue for its classification as a variant of uncertain significance (VUS). Further studies are necessary to establish or disprove pathogenicity.
There were 3 of the 15 SNVs that were identified in individuals from FIPA families (one variant in each family, Table 2 ). SNVs p.Arg80Gln and p.Pro412Leu, which are very rare (MAF: 0.002%), are significantly more prevalent in our cohort vs. controls and are without homozygous variant entries in gnomAD. The p.Arg80Gln SNV is located within PHMcc, while the p.Pro412Leu and p.Glu491Asp SNVs are within the linker region. The p.Arg80Gln did not segregate with the phenotype in the FIPA kindred, and functional studies showed no significant effect of p.Arg80Gln on PHM enzymatic activity. In contrast, the p.Pro412Leu variant segregated with the FIPA phenotype and was seen in both the proband (acromegaly) and the daughter (prolactinoma). Neither of the p.Pro412Leu or p.Glu491Asp variants in the non-enzymatic linker region had a functional effect on PHM or PAL activity ( Figure 3A ; Table 3 ). Altogether, combining allele frequencies in our cohort vs. controls, in silico predictions, functional data, and segregation analysis, our results indicate that the p.Arg80Gln and p.Glu491Asp variants should be classified as likely benign, while p.Pro412Leu linker region change is a VUS.
Functional evaluation of a truncating variant associated with Cushing disease
A variant affecting a canonical splice acceptor site (c.2332-2A>T) was present in a pediatric female (NIH26) with ACTH excess leading to Cushing disease ( Table 1 ). Her mother also carried the variant and had evidence of disrupted circadian cortisol rhythm, based on elevated midnight serum cortisol, but did not present clinically overt Cushing disease. The variant was predicted to cause the skipping of exon 21, generating a frameshift that introduces a premature stop codon and eliminating the C-terminal region of PALcc (p.His778fs, Table 2 ; Figure S6 ). The skipping of exon 21 was confirmed by testing both the affected subject and mother’s blood-extracted RNA ( Figures 4A, B ) and by using a minigene splicing assay ( Figure 4C ). The transient expression of p.His778fs in PEAKrapid cells demonstrated, as expected, that the truncated protein lacked detectable PAL activity; in addition, significantly reduced levels of PHM activity were also observed ( Figure 3A ; Table 3 ). Western blotting indicated that the mutant protein is produced but at lower levels than those observed for WT. Moreover, the observed band was smaller than predicted (75 vs. 90 kDa, respectively), suggesting that the mutant might lack some posttranslational modifications and/or is cleaved at a pair of basic residues introduced by the frameshift ( Figure 4D ).
Figure 4.

Functional studies of the c.2332-2A>T (p.His778fs)-truncating variant. (A) RT-PCR analysis of PAM exon 21 splicing was performed using blood-extracted RNA from two family members carrying the c.2332-2A>T variant (NIH26 and her mother) and four WT control complementary DNAs (cDNAs). Primers were designed on exons 17 and 22. Both carriers and controls showed a normally spliced transcript (713 bp, upper band), while only the carriers showed an additional alternatively spliced transcript lacking exon 21 (the 613 bp band, identified by the arrow). MW, molecular weight marker. (B) The identity of the PCR products (panel A) was confirmed by Sanger sequencing. The arrows point to the variant-specific peaks present only in the carriers. MUT, mutated; WT, wild type. (C) Minigene assay. After transfection into HEK-293AD cells, mRNA synthesis from the plasmids using the cells’ own transcription and splicing machinery led to mRNA products containing (WT) or lacking (variant) exon 21 of PAM flanked by two exons from the pSPL3 vector. The RT-PCR analysis of the minigene transcripts was conducted using vector-specific primers. MOCK, cDNA from empty vector-transfected cells consisting of a 260 bp band made up of fragments of pSPL3 exons; –, negative control (RT-PCR without cDNA). The lines separating MOCK and negative control from WT and c.2332-2A>T indicate that the data for intervening samples were removed. (D) An expression vector lacking exon 21 of human PAM-1 (H778fs) was transiently expressed in PEAKrapid cells. Proteins were visualized using an antibody to PHMcc (JH246 PHM Ab). The expression of WT PAM produces a major band at 114 ± 1 kDa and a minor one at 105 kDa. The only band visible in the cells expressing p.His778fs migrated at 75 kDa; after signal peptide removal, the mass predicted for this protein—which includes only the first 777 residues of WT PAM-1 but extends 45 residues beyond residue 777 before reaching a stop codon—is 90.31 kDa. NT, not transfected.
Screening for PAM single-nucleotide variants located in regulatory regions identified two variants that reduce transcription
Next, we extended our screening to the regulatory regions of PAM, namely, the promoter and untranslated regions (UTRs), to investigate whether there are SNVs with the potential to impact PAM expression. We identified multiple variants and prioritized seven—two in the promoter, four in the 5’UTR, and one in the 3’UTR—for functional studies ( Table 2 ; Figure S7 ). All SNVs in regulatory regions were observed in the heterozygous state. The selection criteria we applied for prioritization were analogous to those used for non-synonymous variants. In vitro evaluation in HEK-293AD cells using reporter assays, in which we cloned either the promoter-5’UTR upstream or the 3’UTR downstream of the luciferase coding sequence, identified two 5’UTR variants, c.-361G>A and c.-133T>C, that significantly reduced luciferase activity when compared to the WT sequence ( Figure 5 ). In contrast, the 3’UTR variant c.*1455C>T, which was predicted to create new miRNA binding sites, did not produce any effect on luciferase activity when expressed alongside the identified miRNAs ( Figure S8 ), indicating that it does not impair PAM mRNA stability and expression. The c.-361G>A variant was identified in two patients with gigantism and in one individual with a non-functioning PA, while the c.-133T>C variant was present in a pediatric subject who also harbored the p.Phe31Tyr variant and was affected by CD (NIH36, Table 1 ). This genotype will be identified as c.-133T>C();p.Phe31Tyr. Based on in vitro enzyme assays, the p.Phe31Tyr missense SNV diminished neither PHM nor PAL activity to a significant extent ( Figure 3A ; Table 3 ).
Figure 5.

WT and mutant PAM promoter activity. A 5 kb PAM promoter-5’UTR WT sequence was cloned upstream of a luciferase reporter. Six mutant constructs were created by site-directed mutagenesis. The constructs were transiently transfected into HEK-293AD cells together with a Renilla luciferase reporter for normalization. Firefly and Renilla luciferase activities were measured 24 h post-transfection. Two SNVs, c.-361G>A and c.-133T>C, have significantly lower transcriptional activity than the WT hybrid transcript. Mock, empty pRMT-Luc vector. Differences between experimental groups were analyzed by one-way ANOVA with Dunnett’s post hoc test, using WT as the control group. RLU, relative luciferase activity. *p = 0.0443; **p = 0.0019; ***p = 0.0005.
Quantification of peptidylglycine α-hydroxylating monooxygenase and peptidyl-α-hydroxyglycine α-amidating lyase catalytic activities in serum
The endoproteolytic cleavage of PAM-1 can occur within the regulated secretory pathway and after the endocytic retrieval of PAM-1 from the cell surface (16–18). In addition, several PAM splice variants encode soluble bifunctional PAM proteins that are secreted along with the peptide hormones stored in secretory granules (10). As a result, both PHM and PAL activity are readily detected in human serum (19, 20). The anticoagulants added during the collection of plasma make the accurate assessment of PHM activity in plasma very difficult. As a result, we could only assess PHM and PAL activity in the small number of serum samples collected from our cohort. As shown in Figure 6 , we measured PHM and PAL activity and calculated the ratio of PAL to PHM activity for 13 subjects (11 affected individuals/carriers and two WT controls). To control for the fact that the serum samples assayed had been stored for varying periods of time, we evaluated the statistical significance of changes in activity by normalizing PAL activity to PHM activity.
Figure 6.

PHM and PAL activity in human sera. Assays for PHM and PAL activity were carried out on sera from individuals harboring variants showing deleterious effects in in vitro assays (p.Asp563Gly, p.Arg703Gln, and p.His778fs), subjects with more benign variants (p.Val27Ile and p.Val49Leu), and controls (WT). In NIH36, the p.Phe31Tyr variant occurs along with the 5’UTR c.-133T>C variant. The dashed line at 0.5 indicates the activity level expected with one completely inactive allele.
Several serum samples were available from patients with two SNVs that resulted in the almost total loss of the PAL activity of PAM-1, p.Arg703Gln (three samples), and p.His778fs (two samples). Consistent with our in vitro activity assays of transiently expressed PAM-1 and the total elimination of one allele, serum PAL activity fell by a factor of two for both variants. Also consistent with the data shown in Table 3 , serum PHM activity was unaffected in the p.Arg703Gln variant and was reduced substantially in the p.His778fs variant. Three of the serum samples available came from subjects carrying SNVs located in PHMcc, but none significantly reduced the PHM or PAL activity of transiently expressed PAM-1. Strikingly, both PHM and PAL activities were reduced in the serum sample from NIH36, who harbors an expression-inhibiting 5’UTR SNV (c.133T>C) in addition to the p.Phe31Tyr SNV. Taken together, our data indicate that the observed reduction in serum PHM and PAL activity can be attributed to the 5’UTR SNV, thus confirming the in vitro finding. Sera from three different subjects harboring the p.Asp563Gly variant showed substantial, but variable, reductions in both PHM and PAL activity. A better interpretation of serum PHM and PAL activity requires further insight into its multiple potential sources. Altogether, the analysis of the limited set of serum samples available emphasizes the utility of using a wide variety of in vitro systems to evaluate the functional effects of PAM SNVs.
Loss of heterozygosity studies
Next, we explored whether PAM acts as a tumor suppressor gene and requires a second somatic hit affecting the WT allele, as would be expected by the Knudson two-hit hypothesis (21). Therefore, we extracted genomic DNA from the PAs surgically removed from individuals harboring variants with demonstrated deleterious effects and studied LOH by Sanger sequencing. The available tumors that we analyzed harbored the following variants: c.-133T>C();p.Phe31Tyr, p.Ser539Trp, p.[Gly552Arg];[Asp563Gly], and p.Asp563Gly (n = 1 tested for each SNV). We did not identify the loss of the WT allele, suggesting that PAM does not behave like a typical tumor suppressor but may rather be a haploinsufficient gene. Unexpectedly, in the individual compound heterozygous for p.[Gly552Arg];[Asp563Gly], we observed the loss of the mutant allele causing the change at codon 552 ( Figure S9 ). This likely represents an instance of in vivo reversion to normal of an inherited variant (revertant mosaicism) (22).
PAM single-nucleotide variants are significantly associated with a hyperfunctioning pituitary gland
Having detected multiple SNVs with deleterious effects on PAM protein function in our cohort, we decided to test whether SNVs within the PAM locus are statistically associated with a pituitary disease phenotype in a larger and more heterogeneous cohort. To this end, we interrogated the UK Biobank (UKBB), a powerful resource for genetic association analyses that contains the exome sequencing data of 200,643 adult individuals with different and well-annotated pathologies. First, we performed a gene-based association study using the sequence kernel association test (SKAT) (23, 24). For this analysis, we considered ICD-10 codes associated with sellar lesions (D352, E220, E221, E222, E229, E237, and E240, covering the following conditions: benign neoplasm of pituitary gland; acromegaly and pituitary gigantism; hyperprolactinemia; syndrome of inappropriate secretion of antidiuretic hormone; hyperfunction of pituitary gland, unspecified; disorder of pituitary gland, unspecified; and pituitary-dependent Cushing’s disease, respectively) and SNVs at the PAM locus for which we assigned a weight based on their detrimental effect on protein (see the Methods for details). We set a MAF cutoff of 0.1% for rare variants. PAM was identified as significantly associated (exome-wide) with a diagnosis of hyperfunction of the pituitary gland (E229, p = 1.71 × 10-77, Figure 7A ). Next, we performed single-variant analyses restricted to rare (MAF < 1%) PAM variants with a predicted deleterious effect (CADD_PHRED score ≥15). We identified nine missense heterozygous variants ( Figures 7B , S10 ; Table S6 ), four in PHMcc (p.Ser97Arg, p.Arg190His, p.Pro192Arg, and p.Val351Met), four in PALcc (p.Gly531Cys, p.Ile554Val, p.Val597Met, and p.Pro693Leu), and one in the C-terminal non-catalytic region (p.Leu856Pro). The p.Ser97Arg variant is located in the loop connecting the β-strand containing the vicinal His residues that bind CuH to the preceding β-strand, and the p.Arg190His and p.Pro192Arg variants are both in the short linker that connects the two homologous domains of PHMcc. PHMcc terminates with Val351; although the final three C-terminal amino acids are not identified in the crystal structure, the removal of four C-terminal residues produces a protein lacking PHM activity. Two of the PAL variants (p.Gly531Cys and p.Ile554Val) are situated in or near the long loop that forms its hydrophobic substrate-binding pocket and connects β-strands 2 and 3 of Blade 1. The other two PAL variants (p.Val597Met and p.Pro693Leu) are contained within the structural repeats that form Blades 2 and 4. A study aimed at understanding the effects of luminal pH on the trafficking of rat PAM identified the juxtamembrane region that encompasses p.Leu856Pro as a key determinant (25).
Figure 7.

Gene- and variant-based association analyses for PAM in the UK Biobank (UKBB). (A) Results of the sequence kernel association test (SKAT) analysis of PAM variants for the diagnoses of hyperfunction of the pituitary gland (identified from UKBB fields 41270 and 41204—primary and secondary ICD-10 diagnoses from hospitalization records—and 20002—self-reported diagnosis) in 200,000 UKBB participants. The SKAT CommonRare algorithm was used for analysis. (B) Significant enrichment for PAM pathological missense PAM variants in subjects diagnosed with hyperfunction of the pituitary gland.
To complement the analyses conducted on the UKBB, we examined the outputs of a gene-specific metric, the gene damage index (GDI) (26). The GDI score predicts the liability of a gene to contain disease-causing mutations by considering the influences of selection and genetic drift (27). For this analysis, we considered 20 genes that harbor germline or somatic variants known to predispose to PAs in order to estimate a GDI cutoff above which a gene is unlikely to cause a pituitary disease, i.e., it is considered a false positive. PAM was reported to have a Phred-scaled GDI score of 3.98, below the calculated cutoff of 4.84 ( Table S7 ). Therefore, our analysis indicates an intolerance to mutational changes for PAM in large population samples, supporting the notion that the observed LOF variants are more likely to contribute to disease.
Functional analysis of splicing in other rare variants
Among the rare SNVs identified in the UKBB, an intronic SNV (c.2746 + 3A>T) was predicted to disrupt the canonical splice donor site of intron 24 ( Figure S11A ; Table S6 ); this SNV was associated with a diagnosis of acromegaly and pituitary gigantism (E220). Our splicing analysis ascertained that this SNV caused the skipping of exon 24 in most transcript molecules; the expected outcome is the in-frame deletion of 19 residues located within the cytoplasmic domain [p.(Ala897_Arg915del)]. While the absence of exon 24 does not alter the PHM or PAL activity of PAM, previous studies indicate that tissue-specific splicing at this site occurs in the rat and could affect the trafficking of membrane PAM (28–30). PAM mRNA expression levels for the mutant allele were about half of WT levels in in vitro studies ( Figure S11B ). Therefore, an individual harboring this variant is predicted to make 25% less biologically active PAM protein than healthy controls; if trafficked correctly, a reduced level of this variant might support normal PAM functions.
The finding of two non-coding SNVs affecting PAM splicing (albeit with different outcomes) and recent studies underscoring the importance of evaluating splicing defects as a disease mechanism for both conditions of hyper- and hypopituitarism (31, 32) prompted us to extend the corresponding functional studies to selected synonymous and intronic variants found in our cohort ( Figure S12A ; Table S8 ). The selection criteria were analogous to those applied for other prioritized variants. This analysis did not reveal any splicing impairment. We observed only a possible reduction in mRNA expression by the c.195G>T allele ( Figure S12B ), but this was not subsequently confirmed by qPCR studies ( Figure S12C ).
Taken together, these findings indicate that the impairment of PAM function or expression via abnormal splicing is not frequently associated with PAs.
Discussion
Studies over the last 30 years have identified a small number of germline and somatic genetic defects associated with the development of different types of PAs. However, the majority of tumors still do not have a known genetic cause (8). Here, we report that several rare variants in the PAM gene are associated with pituitary gland hypersecretion and that most have a deleterious effect on protein function.
We initially identified a deleterious missense PAM variant (p.Arg703Gln) in a FIPA family with a severe form of childhood-onset pituitary gigantism in which all other established genetic causes had been ruled out (33, 34). Among the genes filtered from the WES screening, the high level of PAM expression in the pituitary gland and its known functions in the pituitary and other endocrine cells made PAM the prioritized candidate. A bifunctional enzyme that is essential for the biosynthesis of multiple pituitary and hypothalamic peptide hormones, PAM affects regulated secretion and is involved in the biogenesis of secretory granules (10, 11, 35, 36). Based on site-directed mutagenesis and crystallographic studies of rat PALcc, this Arg residue (Arg 706 in rat PAM) is located at the active site and participates in substrate binding; the site-directed mutagenesis of this residue to Gln reduced the activity of PALcc by a factor of four, and mutagenesis to Ala decreased activity by 97% versus WT (14). Functional studies on bifunctional human PAM with the Arg703 to Gln mutation revealed an even greater decrease in its PAL activity (1% relative to WT human PAM) while retaining fully normal PHM activity.
No other likely deleterious PAM SNVs were observed in the familial cases examined, making the estimated frequency of PAM LOF variants in our FIPA kindreds 5.56% (1/18). A very rare VUS, p.Pro412Leu, located within the linker region connecting the two enzymatic domains was identified in a heterogeneous FIPA kindred.
We detected six germline SNVs with deleterious effects on protein function/expression in sporadic PA cases. Two variants (p.Ser539Trp and p.Asp563Gly) are relatively common in the general population (global MAF range: 0.42%–3.11%). The remaining four variants are very rare (MAF < 0.01%), were found in one subject each, and were significantly enriched in our cohort compared to the general population. Two of these variants (p.Gly552Arg and p.Phe759Ser) were identified in subjects with GH excess (2/173, frequency: 1.16%) and two (c.-133T>C and p.His778fs) in cases with pediatric CD (2/81, frequency: 2.47%).
The p.Ser539Trp and p.Asp563Gly mutants were previously implicated as major risk factors for type 2 diabetes (37, 38), and an important question is whether these proteins are enzymatically active. In our study, the PAM-1 p.Ser539Trp variant had 50% of normal PHM activity, lacked detectable PAL activity, and was not normally sialylated, suggesting its abnormal intracellular trafficking. When expressed as a soluble protein containing both PHMcc and PALcc, the secretion of the p.Ser539Trp protein was not detected, precluding the assessment of its catalytic activity (15). Using an assay to quantify PAM activity, Thomsen et al. reported that the p.Asp563Gly mutation was approximately 50% active (15). Our assays, which assess PHM and PAL activity separately, showed that the PHM activity of p.Asp563Gly was slightly less than half that of WT PAM, while its PAL activity was only one-seventh that of WT PAM.
Our in vitro enzyme assays for PAM-1 expressed in HEK-293 cells indicated an impairment in enzymatic activity for five missense SNVs and a splicing variant (in PAL). PAM expression was decreased for two 5’UTR variants and the COOH-terminal region splicing variant. In addition, PAM-1 glycosylation was abnormal for four of the five missense SNVs. To understand the importance of N- and O-glycosylation defects in pituitary pathology, PAM variants will need to be analyzed in pituitary cells in future studies. Experimental truncations used to define the C-terminus of rat PALcc are consistent with the lack of PAL activity observed for p.His778fs, with the same inability of the truncated rat protein to fold correctly, thereby preventing its secretion (39). Our functional data indicate a similar behavior for the human variant, along with a significant reduction in PHM activity.
We also showed that the measurement of PHM and PAL activity in the serum of subjects harboring PAM variants correlates well with the corresponding values for each single variant determined using our in vitro enzyme assay. The CD case that harbored a regulatory variant reducing PAM expression was instructive in this regard. In that case, both PHM and PAL activities were lowered to the same extent, with the PAL-to-PHM ratio showing a normal value, suggesting that enzymatic activity was not affected. These observations indicate that it is valuable to explore whether the assays of PHM and PAL activity in sera could be used as a biomarker of pituitary disease. The recent development of an efficient high-throughput amidation assay (20) could aid in this endeavor.
The association of loss-of-function PAM SNVs with PAs was further explored by screening the UKBB. Supporting our findings, PAM was significantly associated with a diagnosis of pituitary hyperfunction and nine rare missense variants, all predicted to be pathogenic, were associated with diagnoses of sellar lesions. It has to be noted that, although the sample size of 200,000 exome-sequenced individuals is considerable, the power to discover extremely rare variants is still limited due to the fact that the UKBB is a healthy volunteer cohort and the participants are mostly middle-aged individuals of European ancestry. A larger, more genetically diverse population-based study including individuals of all ages would bypass the inherent healthy volunteer bias and could also unveil additional rare variants of clinical relevance in other ethnic groups. Moreover, disease annotation may be self-reported in some cases or only after hospital admission in others, thereby excluding mild or undiagnosed pituitary hypersecretion diagnoses, which would otherwise be revealed with direct pituitary hormone measurements or more specific clinical markers of pituitary function. Regardless of the aforementioned limitations, the unbiased validation of our original findings related to PAM variants in a large independent cohort more representative of the general population strengthens the observed associations. The SKAT analysis provides a gene-level association of pituitary hyperfunction diagnoses and PAM variants that are rare and have a strong detrimental effect on the protein. This analysis takes into consideration the cumulative effect of all PAM rare variants detected in the population and calculates the strength of their association with pituitary diagnoses. Based on previous studies, each of the nine PAM variants identified in the UKBB through single-variant analysis is expected to disrupt enzymatic activity or the trafficking of membrane PAM bearing these mutations.
We did not observe LOH for PAM variants. This suggests that PAM does not behave as a classical tumor suppressor gene, leading us to propose that pathogenic LOF PAM variants act as haploinsufficient alleles. This hypothesis is supported by in silico predictions (40, 41) and by previous observations that Pam haploinsufficiency impairs several interdependent organ systems in PAM+/- mice. Specifically, PAM+/- mice developed increased adiposity and mild glucose intolerance with age (42), were deficient in their ability to maintain body temperature in the cold (43), and showed an increase in anxiety-like behavior (44). These findings are consistent with defects in β-cell function, the vascular system, and neuronal/neuroendocrine function, respectively. The production of the few amidated peptides tested was not greatly altered by PAM heterozygosity (42, 45, 46), but the production of amidated TRH in the hypothalamus was compromised and the mice were deficient in their pituitary response to hypothyroidism (43). Moreover, the pituitary levels of POMC-derived amidated peptides were significantly increased in PAM+/- mice (46). These data, together with our observations, suggest that the pituitary gland is particularly sensitive to PAM haploinsufficiency. Moreover, they indicate that changes in gene expression associated with PAM heterozygosity are tissue-specific. The intracellular pathways altered by PAM haploinsufficiency in the pituitary remain to be identified (see below).
Limitations to our current study include the fact that only a few subjects with sporadic non-functioning tumors and families with multiple endocrine neoplasia were screened. To better estimate the prevalence of PAM variants and their possible contribution to pituitary tumorigenesis, the search for PAM variants needs to be extended to those subtypes as well as cohorts with hyperprolactinemia (tumoral or non-tumoral), the most common form of pituitary hypersecretion. In particular, it would be interesting to better assess the risk that the relatively common p.Asp563Gly variant confers to the development of pituitary hyperfunction, particularly in the homozygous state. The variable PAL activity measured in PA subjects that were heterozygous for this variant suggests that other factors (e.g., genetic, biochemical, or dietary) (19), contribute to the regulation of PAM expression and/or activity and, hence, predisposition to pituitary dysfunction and potentially tumorigenesis. Second, our LOH and serum activity analyses were limited to the small number of available PAs and serum samples, respectively. It will be important to screen more tumors harboring deleterious PAM variants to definitively exclude the absence of LOH. Likewise, assessing circulating PHM and PAL activity in more PAM variant carriers will better define their risk profile. Third, the question of how disordered PAM function leads to increased hormonal secretion and/or pituitary tumor development remains to be determined. PAM is a complex, multifunctional protein involved in several pathways related to the regulation of secretion; the disruption of one or more of these pathways could play a role in hormonal hypersecretion ( Figure 8 ). The lack of PAM did not affect neuronal cell proliferation (42). Similarly, we found that PAM silencing does not affect the proliferation of pituitary tumor cells ( Figure S13 ).
Figure 8.
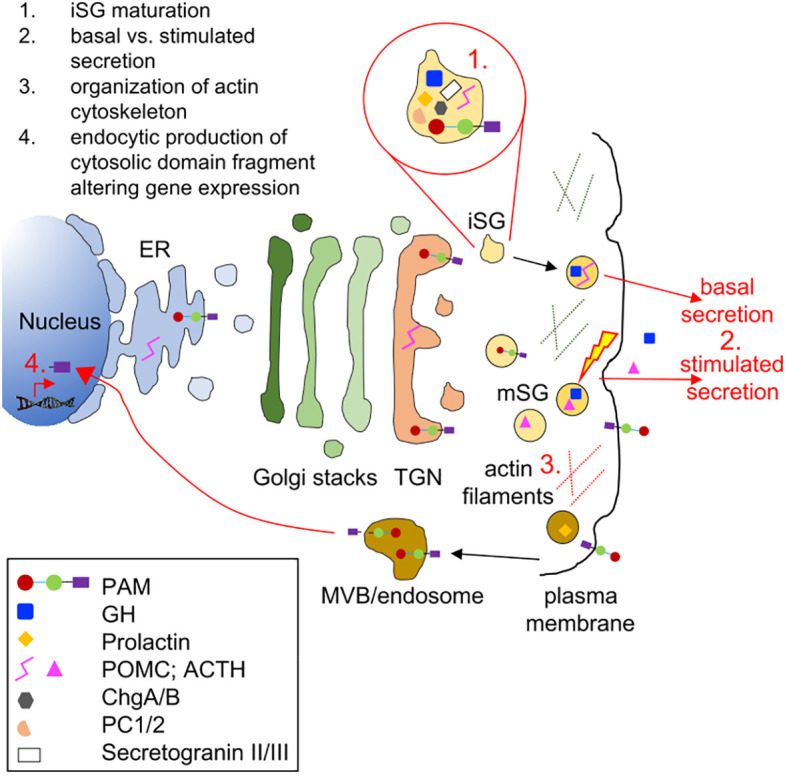
Biological processes and cellular components that might be affected by PAM haploinsufficiency in pituitary hormone-secreting cells. The trafficking of integral membrane PAM and pituitary hormones through the regulated secretory pathway is depicted. Immature secretory granules (iSGs) budding from the trans-Golgi network (TGN) contain prohormones and processing enzymes like the prohormone convertases (PCs), granins [chromogranins A and B (ChgA/B) and secretogranins II and II], and PAM. Mature secretory granules (mSGs) release their soluble content during regulated exocytosis (the yellow lightning bolt represents the external stimulus triggering secretion). Although PAM appears on the cell surface during exocytosis, its rapid endocytosis means that very little PAM remains on the plasma membrane at the steady state. Cleavage by secretase-like enzymes can generate a soluble, cytosolic fragment of PAM that can enter the nucleus, where it relays information about the status of the secretory granule pool. Several in vitro studies have shown that PAM misexpression can affect a variety of steps in the regulated secretory pathway; these are highlighted in red in the cartoon. ER, endoplasmic reticulum; GH, growth hormone; MVB, multivesicular body.
In our study, pathological PAM variants were associated with the hypersecretion of both amidated and non-amidated pituitary hormones, which suggests that the altered PAM function may affect a different target than the hormones themselves. Granin proteins, including chromogranin A and B, are crucial factors in hormonal secretion. Chromogranin A-derived peptides such as pancreastatin are themselves targets for amidation by PAM (47), such that changes in PHM and PAL function could alter the packaging and release of hormones contained in secretory granules. Separately, the linker region connecting the PHM and PAL enzymes is O-glycosylated and pH sensitive and is known to influence PAM function and its effects on vesicular trafficking (48).
In atrial myocytes, proANP storage is a highly PAM-dependent process even though neither proANP nor ANP itself are amidated proteins. In the absence of PAM, proANP storage granules failed to form; the reintroduction of PAM facilitated proANP storage. Crucially, enzymatically inactive PAM also supported proANP storage (49, 50), leading to the suggestion that PAM acts as a cargo receptor that directly binds to and protects newly synthesized proANP, facilitating its delivery to granules (9, 49). A potential role for PAM as a cargo receptor in pituitary cells remains to be explored. Taken together, these data underline the multiple roles that PAM can play in the secretion of hormones.
In conclusion, PAM was identified as a candidate gene associated with pituitary hypersecretion. Our comprehensive analysis of the impact of multiple PAM variants on protein function can also improve our understanding of other endocrine disorders recently linked to PAM defects, such as type 2 diabetes. Together, these new results raise the prospect of therapeutically targeting PAM function to influence disease in the pituitary and elsewhere across the endocrine system.
Materials and methods
The study population consisted of index cases and a validation cohort.
The index cases were part of a three-member FIPA acrogigantism family referred to the National Institutes of Health Clinical Center (NIHCC) as part of the Undiagnosed Diseases Program (UDP) (51–53). The presence of germline SNVs/CNVs in genes known to predispose to pituitary tumors (AIP, GPR101, CDKN1B, MEN1, PRKAR1A, GNAS, and DICER1) had been previously ruled out in all affected individuals.
The validation cohort consisted of 359 individuals with PAs ( Table S1 ). Germline DNA was studied in 299 unrelated individuals with sporadic PAs, in 17 FIPA families (including 19 individuals), and in three families with syndromic presentations of PAs and other endocrine tumors (MEN-like syndromes) (7, 54). The presence of germline SNVs in genes known to predispose to pituitary tumors had been previously ruled out. Among the sporadic cases, 173 had somatotroph adenomas (40 had pituitary gigantism), 84 had Cushing disease associated with corticotroph adenomas (CD, 81 were pediatric—age < 18 years at diagnosis—and one had Nelson syndrome), four had silent corticotroph adenomas, 25 had lactotroph adenomas (six were pediatric; one was a carcinoma), and 13 had clinically non-functioning or silent adenomas (one was pediatric). Somatic PA DNA was available from additional 38 subjects with sporadic acromegaly; 5 of them had matched germline DNA (7, 55). There were 22 of the pediatric CD cases (1 familial and 21 sporadic) that had matched tumor DNA available (54). Three somatic heterozygous USP8 pathogenic variants were previously detected in this cohort ( Table S1 ) (13). Subjects for the study were recruited from the Centre Hospitalier Universitaire (CHU), University of Liège (231), the NIH (90), and the Bicêtre Hospital and Foch Hospital Paris (38).
UDP patients provided written informed consent under clinical protocol 76-HG-0238, “Diagnosis and Treatment of Patients with Inborn Errors of Metabolism and Other Genetic Disorders,” approved by the NHGRI Institutional Review Board. Other individuals from the NIH were recruited between 1997 and 2017 under research protocol 97-CH-0076 (ClinicalTrials.gov: NCT00001595). The study was approved by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Institutional Review Board and by the Ethics Committee of the University of Liège, Belgium. Affected subjects or their relatives signed informed consent or assent forms approved by the local ethics committee. The parents and siblings of the affected individuals were also recruited, when appropriate and available.
DNA sequencing
Index cases
DNA was extracted from peripheral whole blood samples of the twin probands affected with gigantism from generation II, their father, two unaffected siblings, and the three individuals from generation III (sons of II-2). Whole exome sequencing (WES) was performed by a commercial vendor (Axeq Technologies, Rockville, MD) using the TruSeq Human Exome 62 Mb capture kit for library preparation for individuals from generation I and II. Exome sequencing was completed using the Illumina HiSeq2000 platform resulting in raw sequence FASTQ files. WES in individuals from generation III (and in II-2 as control for kit-specific artifacts) was performed using the NimbleGen SeqCap EZ Exome Library v3.0 kit (Roche) for library preparation. Exome sequencing was completed using the Illumina NovaSeq 6000 platform. Alignments were made to human reference assembly GRCh38 using BWA-MEM (56). Variant calling and joint genotyping were performed using GATK Best Practices (GATK ver. 4.2.3.0, HTSJDK ver. 2.24.1, Picard ver. 2.25.4) (57). The VCF files were annotated with population data from the gnomAD database (ver. 3.1.2). Functional and predictive annotations were attached using Ensembl Variant Effect Predictor (ver. 102) (58) and the dbNSFP (ver. 4.3a) (59) database. Pathogenicity predictions used in filtering came from SIFT, ClinPred, PolyPhen-2, FATHMM, and Combined Annotation Dependent Depletion (CADD) score. No known disease-causing or damaging biallelic/hemizygous variants segregating to the affected status were identified. Slivar (60) was used to filter the variants and apply pedigree analysis. We prioritized rare heterozygous damaging variants with non-zero pituitary median expression from GTEx that appear in affected individuals for downstream validation and analysis, excluding variants with a population minor allele frequency (MAF) > 1%. Variants reported in the ClinVar and/or the UniProt databases as benign or likely benign were also excluded. PCR (GoTaq Green Master Mix, M7123, Promega) and Sanger sequencing (BigDye Terminator 3.1 Cycle Sequencing Kit, 4337456, ThermoFisher Scientific) were used to confirm the PAM variant identified by WES. Sequences were aligned and analyzed using the SeqMan Pro 11.1.0 (DNASTAR Lasergene) software. Sequence chromatograms were visualized using SnapGene Viewer ver. 5.0.4.
Validation cohort
WES data on germline and tumor DNA samples from subjects with CD were generated as described previously (54). These WES datasets were manually assessed using the Integrative Genomics Viewer ver. 2.12.3 platform (Broad Institute) (61). For all other subjects in the validation cohort, genomic DNA was extracted from peripheral blood and screened for PAM variants by Sanger sequencing. Whenever a PAM pathogenic/likely pathogenic variant was identified and family members were available for analysis, cascade screening was initiated. This led to two instances of genotype-positive family members. In 38 individuals with sporadic acromegaly, tumor DNA was available for analysis. However, due to the limited amount of DNA available, Sanger sequencing was targeted to specific regions of PAM where the majority of likely pathogenic variants were previously observed ( Figure 2 ), i.e., the proximal promoter and exons 9, 16, 18, 19, 20, and 21. The primers included in Table S2 were used to amplify the promoter and 5’UTR regions, the coding regions, and exon–intron junctions by PCR and for Sanger sequencing. In the available adenomas, DNA was extracted from unstained sections using the Pinpoint Slide DNA Isolation System (D3001, Zymo Research) and loss of heterozygosity (LOH) was investigated by Sanger sequencing. The NC_000005.9(NM_000919.3) reference sequence was used to annotate PAM variants.
Single-cell RNA sequencing analysis
Figure S2A was derived from data published in the NCBI Gene Expression Omnibus (GEO) Series (accession number GSE132224) using methods described in detail (62). Briefly, scRNA-seq data were derived from freshly dispersed rat anterior pituitary cells from postpubertal males (3,562 cells) and diestrus females (3,334 cells). Cell type clusters were identified using known genetic markers for secretory cell types, folliculostellate cells, erythrocytes, leukocytes, and endothelial cells. Here, we examined the expression levels of the Pam gene and the percentage of cells of each type expressing this gene, plotted as a tSNE map (63) and percent-expressing heatmaps using Matlab (R2018b).
Copy number variation analysis
There were 16 pituitary gigantism patients and 121 other individuals with different subtypes of PAs who were tested for germline PAM deletions/duplications by droplet digital PCR (ddPCR) using five FAM-labeled TaqMan CNV assays spanning the entire PAM gene (Hs03560663_cn in intron 2, Hs03041534_cn in intron 5, Hs06720527_cn in intron 13, Hs06071620_cn in intron 16, and Hs06028580_cn in exon 25, all ThermoFisher Scientific). The VIC-labeled RPP30 (Rnase P) assay (4403326, ThermoFisher Scientific) was used as an internal control. All PCR reactions were prepared using the ddPCR SuperMix for Probes (no dUTP, 1863024, Bio-Rad) and the HindIII restriction enzyme in a QX200 Droplet Digital PCR System (Bio-Rad). Results were analyzed with Quanta Soft software ver. 1.7.4.0917 (Bio-Rad).
Allele phasing
The PacBio long seq run was employed to determine the phasing of PAM variants c.718C>T (p.His240Tyr) and c.2276T>C (p.Phe759Ser), located 60 kb apart (exons 9 and 20, respectively), in a person with sporadic gigantism. His parents were not available for genetic studies. Briefly, we employed single-molecule, real-time (SMRT) Sequencing technology and a Sequel sequencer (Pacific Biosciences, Menlo Park, CA). The general strategy was to generate a whole-genome library and then perform a hybridization-based pulldown to enrich for the genomic region including the two variants. The probes were targeted against chr5:102,939,611–103,019,811 (hg38 coordinates). This encompasses the positions of the two variants plus an extra 10 kb upstream and downstream for a total of approximately 83 kb. The biotinylated xGen Lockdown Probes were designed and synthesized by IDT (Coralville, IA) with one probe approximately every 1 kb, for a total of 86 probes. The long reads enable heterozygous variant walking to determine the phasing of the variants. The library was constructed according to the Pacific Biosciences protocol given at https://www.pacb.com/wp-content/uploads/Procedure-Checklist-%E2%80%93-Multiplex-Genomic-DNA-Target-Capture-Using-IDT-xGen-Lockdown-Probes.pdf, except that SeqCap EZ reagents and the SeqCap Library SR User’s Guide (Roche, Indianapolis, IN) were used for the hybridization pulldown. Briefly, the genomic DNA was fragmented using a g-TUBE (Covaris, Woburn, MA) and Pacific Biosciences linear adapters were ligated to the ends to generate a library with an average insert length of approximately 10 kb. The region of interest was pulled down using a pool of 400 attomoles of each of the 86 probes described above. The resulting material was amplified, and SMRTbell adapters were ligated to the ends. This library was sequenced on a Sequel (Pacific Biosciences) on a SMRT Cell 1M v2 cell with Sequencing kit 2.1 and a 10 h acquisition time. The resulting reads were analyzed using the program “Targeted Phasing Consensus” from Pacific Biosciences (https://github.com/WenchaoLin/targeted-phasing-consensus) to perform the read phasing and alignment using CCS reads (circular consensus sequences from 3+ passes), basic subreads, and the hg38 genome for the target region.
A male with sporadic acromegaly harbored PAM variants c.1654G>A (p.Gly552Arg) and c.1688A>G (p.Asp563Gly), both located in exon 16. To determine the phasing of these closely located SNVs, exon 16 was PCR-amplified from his germline DNA and the PCR product cloned into the TOPO-TA vector. Transformed bacterial colonies were screened for the presence of the variants by PCR and Sanger sequencing.
Bioinformatic analyses
Variant pathogenicity and gene damage predictions
Aside from the bioinformatic tools used in the variant prioritization process, Alamut Visual ver. 2.9 software (Interactive Biosoftware) was used for annotation, for in silico prediction, and for determining the frequency in public databases of all the PAM variants identified. For non-synonymous variants, unless specified, the computational verdict was based on the combined outputs of 19 software packages available in Varsome (accessed on 11/24/2022). Five algorithms (Splice Site Finder, MaxEnt, NNSplice, GeneSplicer, and Human Site Finder) integrated in Alamut were used to evaluate candidate splicing-affecting variants. Variants were considered probably damaging or affecting splicing when most of the algorithms agreed. Otherwise, they were considered variants of uncertain significance (VUS). The gene damage index (GDI) (26) score for PAM and other genes known to predispose to pituitary tumors was retrieved from The Gene Damage Index Server.
Spatial clustering analysis
The spatial clustering of a set of variants is obtained by calculating the geometric mean distance between all pairs of variants and normalizing them to the gene’s cDNA length (2,922 bp, isoform P19021-5). An empirical p-value is calculated by randomly generating 100,000 permutations of the variants and comparing their clustering distance against the clustering of the actual variants. As input, we used the 16 prioritized missense and truncating variants found in the index cases and validation cohort and reported them in Table 2 . We then classified the number of variants located in particular domains of the protein (converted to cDNA coordinates) and counted the number of variants appearing in each domain across all the permutations to calculate an empirical p-value comparing the actual number of variants found in each domain to the number of variants in each domain of the 100,000 permutation analysis.
Promoter analysis
The computational search for transcription factor binding sites within the promoter region of PAM was executed using Genomatix. The MatInspector ver. 3.1 tool of Genomatix (Matrix Library 11.0) was used with a core similarity threshold of 0.75 and an optimized matrix similarity threshold to search for the presence of transcription factor binding sites. We limited our search to vertebrate general core promoter elements and the + strand.
Protein sequence alignment and structural analysis
Protein sequence alignments were performed using the following sequences from UniProt and Clustal Omega: human PAM-1 (P19021-5), chimpanzee PAM-1 (A0A2I3SM67-1), rat PAM-1 (P14925-1), Aplysia PAM-1 (Q9NJI4-1), Drosophila PHM (O01404-1) and PAL2 (Q9W1L5-1), and Chlamydomonas PAM (A0A0S2C767-1). The enzymatically active cores for PHM and PAL were aligned. Enzyme activity has been demonstrated for each of these proteins except Aplysia and chimpanzee (39, 64–69). We included chimpanzee as this species is evolutionarily close to human, rat because both its PHM and PAL domains have been crystalized and extensively analyzed (14, 70), Aplysia and Drosophila to cover other major families, and Chlamydomonas as the most evolutionarily distant species at the boundary of the animal/plant kingdoms. Full-length alignments are available in the Supplemental Methods . The ribbon diagrams of the crystal structures of the catalytic cores of rat PHM (PHMcc) (PDB: 1OPM) and PAL (PALcc) (PDB: 3FW0) were used to contextualize the identified variants. Three-dimensional models were generated using the PyMOL molecular graphics system (Schrödinger, LLC). Rat PHMcc and PALcc are 93% and 92% identical to human PHMcc and PALcc, respectively.
Analysis of PAM single-nucleotide variant enrichment in pituitary diagnoses from the UK Biobank cohort
UK Biobank dataset and disease annotation
The UK Biobank (UKBB) is a prospective study that recruited 502,611 participants aged 38–73 years from 22 sites across the UK with baseline measures collected between 2006 and 2010 (71). This research was conducted using the UKBB Resource (Application Number 48008). The UKBB obtained ethics approval from the North West Multi-centre Research Ethics Committee (approval number: 11/NW/0382) and obtained informed consent from all participants. The diagnoses of hyperfunctioning pituitary diseases were retrieved from hospital inpatient diagnosis fields 41270, 41202, and 41204, with International classification of diseases (ICD-10) codes E220 (acromegaly and pituitary gigantism), E221 (hyperprolactinemia), E222 (syndrome of inappropriate secretion of antidiuretic hormone), E229 (unspecified hyperfunction of the pituitary gland), E240-E248-E249 combined as E240 (pituitary-dependent Cushing disease), D352-D359 combined as D352 (benign neoplasm of pituitary gland), E237 (disorder of pituitary gland), or as self-reported medical condition with codes 1237 (disorder of pituitary gland combined with ICD-10 code E237), 1238 (PA/tumor combined with ICD-10 code D352), 1239 (Cushing’s syndrome combined with ICD-10 code E240), 1431 (hyperprolactinemia combined with ICD-10 code E221), and 1429 (acromegaly combined with ICD-10 code E220).
Sequence kernel association test analysis
Gene-based diagnosis-association analysis was performed on 200,643 individuals for whom exome sequencing data were available using the Sequence Kernel Association Test (SKAT). The R package SKAT was employed (24). We used the CommonRare algorithm for binary traits with the adaptive sum method to account for common and rare variants within the PAM gene, using a MAF cutoff of 0.1% and considering the weight of the SNVs based on their detrimental effect on protein. SNV weights were assigned as previously described (72) and adapted as follows:
weight of 5 for UTR variants, synonymous variants, and splice region variants;
weight of 10 for protein-altering variants;
weight of 20 for start lost, stop lost, in-frame deletions, and in-frame insertions;
for missense variants, weight of 20 + score from combined PolyPhen-2 and SIFT predictions: PolyPhen-2 + 10 if possibly damaging or +20 if probably damaging or +5 if unknown; SIFT +20 if deleterious, +0 if tolerated;
weight of 75 for frameshift variants, non-sense, splice acceptor variants, and splice donor variants;
weight of 100 for transcript ablation.
Statistical significance for gene-based tests was set at a Bonferroni-corrected threshold of p < 2 × 10−6 (threshold for 25,000 genes).
Analysis of single-nucleotide variants in the UK Biobank
The analysis of SNVs in UKBB individuals with pituitary diagnoses was performed with PLINK and a custom code in Python ver. 3.7. The CADD v1.660 VEP plugin was used to provide prediction scores for their detrimental effect. A CADD_PHRED score of ≥15 and an MAF < 1% were considered as pathogenicity criteria.
Functional characterization studies
Cell lines
The PEAKrapid (ATCC CRL-2828) and HEK-293AD (ATCC CRL-1573) cells are lines derived from the Human Embryonic Kidney (HEK)-293 cell line. The PEAKrapid cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)-F12, with 10% fetal bovine serum (FBS, Hyclone), pen-strep, and HEPES to net 25 mM, while the HEK-293AD cells were maintained in DMEM containing low glucose, pyruvate, 2 mM glutamine (Gibco), with 10% FBS (Gemini Bio Products), and 1% antibiotic–antimycotic (Gibco). The rat pituitary tumor GH3 cell line (ATCC CCL-82.1) was maintained in the same medium as HEK-293AD cells. All cell lines were kept in a humidified atmosphere at 37°C with 5% CO2.
Vectors and mutagenesis
All cloning reactions were performed using the In-Fusion cloning system (Clontech). Cloning primers are reported in Table S2 . Tissue-specific alternative splicing generates multiple PAM isoforms (73). The longest human isoform is a 974 amino acid integral membrane protein identified as P19021-5 in UniProt. Since a GenBank search revealed that this isoform is more prevalent than the one chosen as the canonical sequence (UniProt P19021-1/NM_001177306.2, 973 amino acid-long), we regarded it as human PAM-1, corresponding to what was previously extensively characterized as rat PAM-1, a 976 amino acid integral membrane protein. The ORF expression clone for WT human PAM-1 (NM_000919.3) cloned into pReceiver-M02 (EX-A3104-M02-GS, Genecopoeia) was subcloned into the pCMV-FLAG-C vector (635688, Clontech) using the EcoRI and XhoI restriction sites. The resulting PAM-1 protein has an in-frame C-terminal FLAG tag (DYKDDDDK) separated from its normal C-terminus by a five amino acid spacer (LEVPA). A 5,373 bp sequence including the human WT PAM promoter (5,000 bp) and 5’UTR (373 bp) was generated by de novo gene synthesis (Blue Heron Biotech) and cloned into the pRMT-Luc vector (PR100001, Origene) using the EcoRI and MluI restriction sites located upstream of Firefly luciferase. The WT and mutant (harboring the c.*1455C>T variant) 3’UTRs of human PAM (2,040 bp) were PCR-amplified from human genomic DNA and cloned into psiCHECK-2 (Promega) using the XhoI and NotI restriction sites located downstream of Renilla luciferase. WT and mutant PAM sequences consisting of seven different human PAM exons (exon 2, 6, 16, 18, 19, 20, and 21) together with their flanking intronic sequences (100/300-bp-long) were PCR-amplified from human genomic DNA and cloned into the EcoRI-digested pSPL3 vector (Invitrogen). See Supplemental Methods for more details.
Mutagenic primers (reported in Table S2 ) were used to introduce PAM SNVs using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies). Mutagenesis products were verified by Sanger sequencing. Since our findings (see the Results section) showed a discrepancy with what was previously reported by Thomsen et al. (15) for the p.Ser539Trp variant, we prepped the construct harboring this variant twice and sequence-verified it again. To introduce the deletion caused by the splice-altering c.2332-2A>T variant into the PAM-1_pCMV-FLAG-C vector, the PCR splicing technique was used. A pair of primers flanking the region where the deletion occurs (entire exon 21) and a pair of complementary primers comprising a region of −15 to +15 bp related to the junction point are reported in Table S2 .
Antibodies
The antibodies used for the various experiments included the following: an affinity-purified rabbit polyclonal antibody (Ab JH629) raised to purified bacterially expressed Exon A (exon 16 in rat Pam) (74); a rabbit polyclonal antibody (Ab JH246) raised to a synthetic peptide corresponding to human PAM(111-126), which is identical to rat PAM(116-131) (75); a mouse anti-FLAG monoclonal antibody (F1804, Sigma-Aldrich); an anti-GM130 antibody (#610822, BD Biosciences); and a mouse anti-beta actin antibody (ab6276, Abcam).
Biochemical analysis of non-synonymous and splice site–affecting single-nucleotide variants
The transient transfection of PAM-1 WT and mutant vectors was performed 24 h after plating using PEAKrapid cells and TransIT-2020 (MIR 5400, Mirus Bio) (76). Cells were harvested 24–36 h after transfection. Protein extraction and Western blotting were performed as described previously (74). After rinsing with serum-free medium, cells were scraped into serum-free medium and pelleted. Cell pellets were extracted into ice-cold 20 mM Na TES, 10 mM mannitol, 1% TX-100, pH 7.4 (TMT) containing protease inhibitors and subjected to three freeze/thaw cycles. Particulate material was removed by centrifugation, and protein concentrations were determined using the bicinchoninic acid assay with bovine serum albumin (BSA) as the standard. For enzyme assays, lysates were diluted using TMT containing 1 mg/ml BSA. For Western blot analysis, lysates were denatured using 2X or 4X Laemmli Sample Buffer (Bio-Rad).
PHM and PAL assays were performed using [125I]-Ac-Tyr-Val-Gly and [125I]-Ac-Tyr-Val-α-hydroxy-Gly with 0.5 μM unlabeled substrate, as described previously (77). Samples were assayed in triplicate, within the linear range of the assay. Data for mutants were normalized to data for human PAM-1 analyzed in parallel. Normalized data from multiple different transfections were averaged to ensure that each mutant was assessed in at least two independent transfections. Human serum samples were diluted 10-fold into 20 mM Na TES, 10 mM mannitol, 1 mg/ml bovine serum albumin (PHM diluent). Assays for PHM and PAL activity were carried out in duplicate or triplicate at pH 5.5; PHM assays included 4 μM CuSO4 and PAL assays contained 1 mM CoCl2.
The analysis of the glycosylation patterns for PAM WT and mutants was performed as described previously (39). Cells were harvested 24 h after transfection. Cell pellets were frozen and thawed in 20 mM Na TES, 10 mM mannitol, pH7.4 containing a protease inhibitor mix. Following centrifugation at 17,000 g for 20 min at 4°C, the pellets were extracted in TMT with protease inhibitors, and the supernatants were denatured using SDS and digested with PNGase F or neuraminidase as described by the manufacturer (NEB). For PAM and for each PAM variant, one aliquot remained on ice (In, input) and two aliquots were prepared for enzymatic digestion; both were incubated at 37°C for 60 min, but one received enzyme (N-Gly or Neur) and one did not (Con). After SDS-PAGE and transferring to PVDF membranes, epitope-tagged PAM and its variants were visualized using a FLAG antibody. Neuraminidase removes sialic acid from both N-linked and O-linked oligosaccharides. Samples treated with neuraminidase were analyzed on two separate gels, with the p.Arg703Gln samples appearing in part on both gels.
Analysis of splicing by minigene assay
The transient transfection of WT and mutant pSPL3 vectors was performed using HEK-293AD cells and TurboFect (ThermoFisher Scientific). Cells were transfected 24 h after plating; 24 h after transfection, total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen). RNA (500 ng) was reverse-transcribed to cDNA using the Superscript III Kit (ThermoFisher Scientific). mRNA synthesis from the plasmids using the cells’ own transcription and splicing machinery led to mRNA products containing the tested PAM exon flanked by two exons from the pSPL3 vector. Splicing products were analyzed by RT-PCR using the vector-specific SD6 and SA2 primers ( Table S2 ). The empty pSPL3 vector was used as negative control.
Analysis of single-nucleotide variants in regulatory regions by luciferase-based reporters
SNVs located upstream (promoter and 5’UTR) and downstream (3’UTR) of PAM CDS were investigated using luciferase-based reporter assays transfected into HEK-293AD cells. The empty pRMT-Luc and psiCHECK-2 vectors were used as negative controls. For the 3’UTR experiments, four microRNA (miRNA) mimics (hsa-miR-138-2-3p, MC12814; hsa-miR-192-3p, MC12893; hsa-miR-3143, MC17293; and hsa-miR-556-3p, MC12806; all ThermoFisher Scientific) were transfected alongside the WT and mutant reporter vectors. Cells were lysed 24 h after transfection, and Firefly and/or Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s protocol. When appropriate, the ratios of Firefly/Renilla luminescence signals, serving as a measure for reporter activity normalized for transfection efficiency, were determined using a FLUOstar Omega microplate reader (BMG Labtech).
Pam silencing by small interfering RNA–mediated knockdown
Double-stranded RNA targeting the rat Pam gene (NM_013000) was purchased from Dharmacon (Lafayette, CO). Four siRNA duplexes were mixed in ON-TARGETplus SMARTpool (L-089153-02-0005). Reverse transfection using TurboFect was carried out in GH3 cells using a 25 nM final concentration of siRNA. Non-targeting siRNA (D-001810-10-05) was used as negative control.
Cell proliferation assay
Cell proliferation was assessed in GH3 cells by the colorimetric measurement of bromodeoxyuridine (BrdU) uptake using the Cell Proliferation Biotrak Version 2 ELISA (GERPN250, Sigma Aldrich), according to the manufacturer’s instructions. Briefly, 72 h after Pam silencing, the transfection medium was changed. 24 h later, cells were incubated with BrdU for 2 h to allow its incorporation into newly synthesized DNA. Absorbance measurements (450 nm) were normalized with the non-specific binding control.
Immunohistochemistry
Immunofluorescence analysis was performed on histological sections from non-pathological pituitary cells (control). The full protocol has been detailed elsewhere (78). Briefly, all slides were blocked with 10% normal donkey serum in 1X PBS for 1 h at room temperature and then incubated overnight at 4°C with affinity-purified 1:500 anti-PAM JH629 and anti-GM130. All slides were incubated for 1 h with 1:500 donkey anti-rabbit 488 and donkey anti-mouse 594 (A-11055 and A-21207, respectively, ThermoFisher Scientific). Slides were imaged at ×40 on a Keyence BZ- X710 microscope.
Statistical analysis
All graphs were plotted as mean ± standard error of the mean (SEM). Data distributions were assessed for approximate normality. Differences between experimental groups were analyzed by two-tailed Student’s t-test or one-way ANOVA with Dunnett’s post hoc test, or corresponding non-parametric tests, as appropriate. PAM SNV allele frequencies in the study population were compared with frequencies in the general population reported in the gnomAD public database using Fisher’s exact test or the chi-square test, as appropriate. Data were analyzed using GraphPad Prism (GraphPad, San Diego, CA, USA). P-values < 0.05 were considered statistically significant.
Data availability statement
The code used to conduct the spatial clustering analysis can be found at https://github.com/NICHD-BSPC/spatial_clustering. WES data for the index cases have been deposited in the National Institutes of Health dbGaP database under the accession codes phs000721.v1.p1 and phs003245.v1.p1. The original contributions presented in the study are included in the article/ Supplementary Material and at Zenodo (DOI 10.5281/zenodo.7789345). The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a article that does not adhere to our open data policies.
Ethics statement
The studies involving human participants were reviewed and approved by the NHGRI Institutional Review Board, NIH, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Institutional Review Board, NIH, and the Ethics Committee of the University of Liège, Belgium. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
GT and CS conceived the study. GT, AD, LH-R, RD, GF, AM, BE, RM, EA, and CS formulated study hypotheses and conceived and designed the experiments. GT, LH-R, GF, AM, JI, TL, CTo, EA, FF, CTa, BE, and RM performed the experiments. GT, AD, LH-R, GF, EA, FF, CTo, CTa, BE, and RM analyzed the data. GT, AD, EA, BE, and RM constructed the figures. GT wrote the original manuscript draft. All authors reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
This work was partially supported by “Ricerca Corrente” funding from Italian Ministry of Health to IRCCS Humanitas Research Hospital. The authors would like to thank Steven Coon of the NICHD’s IRP Molecular Genomics Core for conducting the phasing experiment, the late Mario Amzel (Johns Hopkins University School of Medicine) for useful comments on the impact of PAM variants on its 3D structure, Lyssikatos Charalampos and María de la Luz Sierra (NICHD/NIH) for their help with the collection and preparation of DNA and histopathological samples used in this study; Emilie Castermans and Leonor Palmeira (CHU de Liège) for genetic and bioinformatic analyses of FIPA kindreds, and Jean-Francois Bonneville (CHU de Liège, Belgium) for discussions on the neuroradiological images.
Appendix
BWA, https://github.com/lh3/bwa
CADD, https://cadd.gs.washington.edu/
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/
Clustal Omega, https://www.ebi.ac.uk/Tools/msa/clustalo/
dbNSFP (ver. 4.3a), http://database.liulab.science/dbNSFP
dbSNP, https://www.ncbi.nlm.nih.gov/snp/
Ensembl VEP (ver. 102), https://grch37.ensembl.org/info/docs/tools/vep/index.html
GATK (ver. 4.2.3.0), https://gatk.broadinstitute.org/hc/en-us/articles/4409678362139-GATK-4-2-3-0-release
Genomatix, https://mygga.genomatix.de/online_help//help_eldorado/genomatix_portal_help.html
gnomAD (ver. 3.1.2), https://gnomad.broadinstitute.org/
Gene Structure Display Server (GSDS 2.0), http://gsds.gao-lab.org/
GTEx Project, https://www.gtexportal.org/home/
HIPred score, https://github.com/HAShihab/HIPred
HTSJDK (ver. 2.24.1), https://samtools.github.io/htsjdk/
OMIM, https://www.ncbi.nlm.nih.gov/omim
PCR splicing technique, http://www.methods.info/Methods/Mutagenesis/PCR_splicing.html
Picard (ver. 2.25.4), http://broadinstitute.github.io/picard/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, https://bioinfo.ut.ee/primer3/
Protein Molecular Weight, https://www.bioinformatics.org/sms/prot_mw.html
Slivar, https://github.com/brentp/slivar
The Gene Damage Index (GDI) Server, http://pec630.rockefeller.edu:8080/GDI/
The Human Protein Atlas, https://www.proteinatlas.org/
UniProt, https://www.uniprot.org/
Varsome, https://varsome.com/
Funding Statement
The work was supported by the following funding sources: Society for Endocrinology equipment grant (to GT); Intramural Research Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH) Research projects Z1A HD008920 (to CS, supporting GT, LH-R, and FF), R01-DK032949 (to BE and RM); the Intramural Research Program of the National Human Genome Research Institute (to CT and WG); the Daniel Schwartzberg Fund (to RM and BE); Fonds d’Investissement pour la Recherche Scientifique (FIRS) of the Centre Hospitalier Universitaire de Liège (to AD and AB); Novo Nordisk Belgium Educational Grant, Belgium (to AD and AB); and the JABBS Foundation, UK (to AB). AD was supported, in part, by Action de Recherche Concertée (ARC) Grant 17/21-01 from Liège University.
Conflict of interest
AB, AD, FF, CS, and GT hold a patent on the GPR101 gene and its function US Patent No. 10,350,273, Treatment of Hormonal Disorders of Growth. CS holds patents on technologies involving PRKAR1A and related genes causing adrenal, pituitary, and other tumors. In addition, his laboratory has received research funding support by Pfizer Inc. for investigations on growth hormone–producing pituitary adenomas. CS has also consulted within the last 12 months with Lundbeck Pharmaceuticals and Sync, LLC, and is currently employed by ELPEN Pharmaceuticals. AB and AD have received research funding from Pfizer Inc. and Novo-Nordisk. M-LJ-R is part of the advisory board of Recordati Rare diseases since 2022.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1166076/full#supplementary-material
References
- 1. Figarella-Branger D, Appay R, Metais A, Tauziede-Espariat A, Colin C, Rousseau A, et al. [The 2021 WHO classification of tumours of the central nervous system]. Ann Pathol (2021) 42(5):367–82. doi: 10.1016/j.annpat.2021.11.005 [DOI] [PubMed] [Google Scholar]
- 2. Melmed S, Kaiser UB, Lopes MB, Bertherat J, Syro LV, Raverot G, et al. Clinical biology of the pituitary adenoma. Endocr Rev (2022) 43(6):1003–37. doi: 10.1210/endrev/bnac010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of liege, Belgium. J Clin Endocrinol Metab (2006) 91(12):4769–75. doi: 10.1210/jc.2006-1668 [DOI] [PubMed] [Google Scholar]
- 4. Srirangam Nadhamuni V, Korbonits M. Novel insights into pituitary tumorigenesis: genetic and epigenetic mechanisms. Endocr Rev (2020) 41(6):821–46. doi: 10.1210/endrev/bnaa006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vandeva S, Daly AF, Petrossians P, Zacharieva S, Beckers A. Somatic and germline mutations in the pathogenesis of pituitary adenomas. Eur J Endocrinol (2019) 181(6):R235–R54. doi: 10.1530/EJE-19-0602 [DOI] [PubMed] [Google Scholar]
- 6. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev (2013) 34(2):239–77. doi: 10.1210/er.2012-1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med (2014) 371(25):2363–74. doi: 10.1056/NEJMoa1408028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coopmans EC, Korbonits M. Molecular genetic testing in the management of pituitary disease. Clin Endocrinol (Oxf). (2022) 97(4):424–35. doi: 10.1111/cen.14706 [DOI] [PubMed] [Google Scholar]
- 9. Back N, Mains RE, Eipper BA. PAM: diverse roles in neuroendocrine cells, cardiomyocytes, and green algae. FEBS J (2022) 289(15):4470–96. doi: 10.1111/febs.16049 [DOI] [PubMed] [Google Scholar]
- 10. Kumar D, Mains RE, Eipper BA. 60 YEARS OF POMC: from POMC and alpha-MSH to PAM, molecular oxygen, copper, and vitamin c. J Mol Endocrinol (2016) 56(4):T63–76. doi: 10.1530/JME-15-0266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mains RE, Blaby-Haas C, Rheaume BA, Eipper BA. Changes in corticotrope gene expression upon increased expression of peptidylglycine alpha-amidating monooxygenase. Endocrinology (2018) 159(7):2621–39. doi: 10.1210/en.2018-00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu B, Jin J, Guo A-Y, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics (2015) 31(8):1296–7. doi: 10.1093/bioinformatics/btu817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernandez-Ramirez LC, Settas N, et al. Somatic USP8 gene mutations are a common cause of pediatric cushing disease. J Clin Endocrinol Metab (2017) 102(8):2836–43. doi: 10.1210/jc.2017-00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chufan EE, De M, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: the structure of the lyase domain of the amidating enzyme. Structure (2009) 17(7):965–73. doi: 10.1016/j.str.2009.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thomsen SK, Raimondo A, Hastoy B, Sengupta S, Dai XQ, Bautista A, et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic beta-cells. Nat Genet (2018) 50(8):1122–31. doi: 10.1038/s41588-018-0173-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Braas KM, Stoffers DA, Eipper BA, May V. Tissue specific expression of rat peptidylglycine alpha-amidating monooxygenase activity and mRNA. Mol Endocrinol (1989) 3(9):1387–98. doi: 10.1210/mend-3-9-1387 [DOI] [PubMed] [Google Scholar]
- 17. Ferraro F, Eipper BA, Mains RE. Retrieval and reuse of pituitary secretory granule proteins. J Biol Chem (2005) 280(27):25424–35. doi: 10.1074/jbc.M414156200 [DOI] [PubMed] [Google Scholar]
- 18. May V, Cullen EI, Braas KM, Eipper BA. Membrane-associated forms of peptidylglycine alpha-amidating monooxygenase activity in rat pituitary. tissue specificity. J Biol Chem (1988) 263(16):7550–4. doi: 10.1016/S0021-9258(18)68534-5 [DOI] [PubMed] [Google Scholar]
- 19. Gaier ED, Kleppinger A, Ralle M, Covault J, Mains RE, Kenny AM, et al. Genetic determinants of amidating enzyme activity and its relationship with metal cofactors in human serum. BMC Endocr Disord (2014) 14:58. doi: 10.1186/1472-6823-14-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaufmann P, Bergmann A, Melander O. Novel insights into peptide amidation and amidating activity in the human circulation. Sci Rep (2021) 11(1):15791. doi: 10.1038/s41598-021-95305-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knudson AG. Antioncogenes and human cancer. Proc Natl Acad Sci (1993) 90(23):10914–21. doi: 10.1073/pnas.90.23.10914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hirschhorn R. In vivo Reversion to normal of inherited mutations in humans. J Med Genet (2003) 40(10):721–8. doi: 10.1136/jmg.40.10.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet (2011) 89(1):82–93. doi: 10.1016/j.ajhg.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ionita-Laza I, Lee S, Makarov V, Buxbaum Joseph D, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet (2013) 92(6):841–53. doi: 10.1016/j.ajhg.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bell-Parikh LC, Eipper BA, Mains RE. Response of an integral granule membrane protein to changes in pH. J Biol Chem (2001) 276(32):29854–63. doi: 10.1074/jbc.M103936200 [DOI] [PubMed] [Google Scholar]
- 26. Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Vélez M, et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci (2015) 112(44):13615–20. doi: 10.1073/pnas.1518646112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alyousfi D, Baralle D, Collins A. Gene-specific metrics to facilitate identification of disease genes for molecular diagnosis in patient genomes: a systematic review. Brief Funct Genomics (2019) 18(1):23–9. doi: 10.1093/bfgp/ely033 [DOI] [PubMed] [Google Scholar]
- 28. Eipper BA, Park LP, Dickerson IM, Keutmann HT, Thiele EA, Rodriguez H, et al. Structure of the precursor to an enzyme mediating COOH-terminal amidation in peptide biosynthesis. Mol Endocrinol (1987) 1(11):777–90. doi: 10.1210/mend-1-11-777 [DOI] [PubMed] [Google Scholar]
- 29. Stoffers DA, Green CB, Eipper BA. Alternative mRNA splicing generates multiple forms of peptidyl-glycine alpha-amidating monooxygenase in rat atrium. Proc Natl Acad Sci USA (1989) 86(2):735–9. doi: 10.1073/pnas.86.2.735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ciccotosto GD, Hand TA, Mains RE, Eipper BA. Breeding stock-specific variation in peptidylglycine alpha-amidating monooxygenase messenger ribonucleic acid splicing in rat pituitary. Endocrinology (2000) 141(2):476–86. doi: 10.1210/endo.141.2.7337 [DOI] [PubMed] [Google Scholar]
- 31. Brue T, Camper SA. Novel mechanism of pituitary hormone deficiency: genetic variants shift splicing to produce a dominant negative transcription factor isoform. Eur J Endocrinol (2021) 185(6):C19–25. doi: 10.1530/EJE-21-0949 [DOI] [PubMed] [Google Scholar]
- 32. Vazquez-Borrego MC, Fuentes-Fayos AC, Venegas-Moreno E, Rivero-Cortes E, Dios E, Moreno-Moreno P, et al. Splicing machinery is dysregulated in pituitary neuroendocrine tumors and is associated with aggressiveness features. Cancers (Basel) (2019) 11(10):1439. doi: 10.3390/cancers11101439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Franke M, Daly AF, Palmeira L, Tirosh A, Stigliano A, Trifan E, et al. Duplications disrupt chromatin architecture and rewire GPR101-enhancer communication in X-linked acrogigantism. Am J Hum Genet (2022) 109(4):553–70. doi: 10.1016/j.ajhg.2022.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rostomyan L, Daly AF, Petrossians P, Nachev E, Lila AR, Lecoq AL, et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocr Relat Cancer. (2015) 22(5):745–57. doi: 10.1530/ERC-15-0320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ciccotosto GD, Schiller MR, Eipper BA, Mains RE. Induction of integral membrane PAM expression in AtT-20 cells alters the storage and trafficking of POMC and PC1. J Cell Biol (1999) 144(3):459–71. doi: 10.1083/jcb.144.3.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El Meskini R, Mains RE, Eipper BA. Cell type-specific metabolism of peptidylglycine alpha-amidating monooxygenase in anterior pituitary. Endocrinology (2000) 141(8):3020–34. doi: 10.1210/endo.141.8.7620 [DOI] [PubMed] [Google Scholar]
- 37. Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The genetic architecture of type 2 diabetes. Nature (2016) 536(7614):41–7. doi: 10.1038/nature18642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet (2014) 46(3):294–8. doi: 10.1038/ng.2882 [DOI] [PubMed] [Google Scholar]
- 39. Kolhekar AS, Quon AS, Berard CA, Mains RE, Eipper BA. Post-translational n-glycosylation of a truncated form of a peptide processing enzyme. J Biol Chem (1998) 273(36):23012–8. doi: 10.1074/jbc.273.36.23012 [DOI] [PubMed] [Google Scholar]
- 40. Shihab HA, Rogers MF, Campbell C, Gaunt TR. HIPred: an integrative approach to predicting haploinsufficient genes. Bioinformatics (2017) 33(12):1751–7. doi: 10.1093/bioinformatics/btx028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schierup MH, Huang N, Lee I, Marcotte EM, Hurles ME. Characterising and predicting haploinsufficiency in the human genome. PloS Genet (2010) 6(10):e1001154. doi: 10.1371/journal.pgen.1001154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Czyzyk TA, Ning Y, Hsu MS, Peng B, Mains RE, Eipper BA, et al. Deletion of peptide amidation enzymatic activity leads to edema and embryonic lethality in the mouse. Dev Biol (2005) 287(2):301–13. doi: 10.1016/j.ydbio.2005.09.001 [DOI] [PubMed] [Google Scholar]
- 43. Bousquet-Moore D, Ma XM, Nillni EA, Czyzyk TA, Pintar JE, Eipper BA, et al. Reversal of physiological deficits caused by diminished levels of peptidylglycine alpha-amidating monooxygenase by dietary copper. Endocrinology (2009) 150(4):1739–47. doi: 10.1210/en.2008-1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gaier ED, Rodriguiz RM, Ma XM, Sivaramakrishnan S, Bousquet-Moore D, Wetsel WC, et al. Haploinsufficiency in peptidylglycine alpha-amidating monooxygenase leads to altered synaptic transmission in the amygdala and impaired emotional responses. J Neurosci (2010) 30(41):13656–69. doi: 10.1523/JNEUROSCI.2200-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bousquet-Moore D, Prohaska JR, Nillni EA, Czyzyk T, Wetsel WC, Mains RE, et al. Interactions of peptide amidation and copper: novel biomarkers and mechanisms of neural dysfunction. Neurobiol Dis (2010) 37(1):130–40. doi: 10.1016/j.nbd.2009.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yin P, Bousquet-Moore D, Annangudi SP, Southey BR, Mains RE, Eipper BA, et al. Probing the production of amidated peptides following genetic and dietary copper manipulations. PloS One (2011) 6(12):e28679. doi: 10.1371/journal.pone.0028679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eipper BA, Stoffers DA, Mains RE. The biosynthesis of neuropeptides: peptide alpha-amidation. Annu Rev Neurosci (1992) 15:57–85. doi: 10.1146/annurev.ne.15.030192.000421 [DOI] [PubMed] [Google Scholar]
- 48. Vishwanatha K, Back N, Mains RE, Eipper BA. A histidine-rich linker region in peptidylglycine alpha-amidating monooxygenase has the properties of a pH sensor. J Biol Chem (2014) 289(18):12404–20. doi: 10.1074/jbc.M113.545947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Back N, Luxmi R, Powers KG, Mains RE, Eipper BA. Peptidylglycine alpha-amidating monooxygenase is required for atrial secretory granule formation. Proc Natl Acad Sci USA (2020) 117(30):17820–31. doi: 10.1073/pnas.2004410117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bartels ED, Gotze JP, Mains RE, Eipper BA. Commentary on: peptidylglycine alpha-amidating monooxygenase is required for atrial secretory granule formation. J Clin Cardiol (2021) 2(4):75–80. doi: 10.33696/cardiology.2.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, et al. The national institutes of health undiagnosed diseases program: insights into rare diseases. Genet Med (2012) 14(1):51–9. doi: 10.1038/gim.0b013e318232a005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, et al. The NIH undiagnosed diseases program and network: applications to modern medicine. Mol Genet Metab (2016) 117(4):393–400. doi: 10.1016/j.ymgme.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gahl WA, Tifft CJ. The NIH undiagnosed diseases program: lessons learned. JAMA (2011) 305(18):1904–5. doi: 10.1001/jama.2011.613 [DOI] [PubMed] [Google Scholar]
- 54. Hernandez-Ramirez LC, Gam R, Valdes N, Lodish MB, Pankratz N, Balsalobre A, et al. Loss-of-function mutations in the CABLES1 gene are a novel cause of cushing's disease. Endocr Relat Cancer (2017) 24(8):379–92. doi: 10.1530/ERC-17-0131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lecoq AL, Bouligand J, Hage M, Cazabat L, Salenave S, Linglart A, et al. Very low frequency of germline GPR101 genetic variation and no biallelic defects with AIP in a large cohort of patients with sporadic pituitary adenomas. Eur J Endocrinol (2016) 174(4):523–30. doi: 10.1530/EJE-15-1044 [DOI] [PubMed] [Google Scholar]
- 56. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. (2013). doi: 10.48550/arxiv.1303.3997 [DOI] [Google Scholar]
- 57. van der Auwera G, O'Connor BD. Genomics in the cloud: using docker, GATK, and WDL in Terra. O'Reilly Media, Incorporated; (2020). [Google Scholar]
- 58. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol (2016) 17(1):122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med (2020) 12(1):103. doi: 10.1186/s13073-020-00803-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pedersen BS, Brown JM, Dashnow H, Wallace AD, Velinder M, Tristani-Firouzi M, et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom Med (2021) 6(1):60. doi: 10.1038/s41525-021-00227-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol (2011) 29(1):24–6. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fletcher PA, Smiljanic K, Maso Previde R, Iben JR, Li T, Rokic MB, et al. Cell type- and sex-dependent transcriptome profiles of rat anterior pituitary cells. Front Endocrinol (Lausanne). (2019) 10:623. doi: 10.3389/fendo.2019.00623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. van der Maaten L, Hinton G. Visualizing high-dimensional data using t-SNE. J Mach Learn Res (2008) 9:2579–605. [Google Scholar]
- 64. Eipper BA, Quon AS, Mains RE, Boswell JS, Blackburn NJ. The catalytic core of peptidylglycine alpha-hydroxylating monooxygenase: investigation by site-directed mutagenesis, Cu X-ray absorption spectroscopy, and electron paramagnetic resonance. Biochemistry (1995) 34(9):2857–65. doi: 10.1021/bi00009a016 [DOI] [PubMed] [Google Scholar]
- 65. Fan X, Spijker S, Akalal DB, Nagle GT. Neuropeptide amidation: cloning of a bifunctional alpha-amidating enzyme from aplysia. Brain Res Mol Brain Res (2000) 82(1-2):25–34. doi: 10.1016/s0169-328x(00)00173-x [DOI] [PubMed] [Google Scholar]
- 66. Han M, Park D, Vanderzalm PJ, Mains RE, Eipper BA, Taghert PH. Drosophila uses two distinct neuropeptide amidating enzymes, dPAL1 and dPAL2. J Neurochem (2004) 90(1):129–41. doi: 10.1111/j.1471-4159.2004.02464.x [DOI] [PubMed] [Google Scholar]
- 67. Kolhekar AS, Keutmann HT, Mains RE, Quon ASW, Eipper BA. Peptidyl a-hydroxylating monooxygenase: active site residues, disulfide linkages and a two-domain model of the catalytic core. Biochemistry (1997) 36:10901–9. doi: 10.1021/bi9708747 [DOI] [PubMed] [Google Scholar]
- 68. Kumar D, Blaby-Haas CE, Merchant SS, Mains RE, King SM, Eipper BA. Early eukaryotic origins for cilia-associated bioactive peptide-amidating activity. J Cell Sci (2016) 129(5):943–56. doi: 10.1242/jcs.177410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luxmi R, Kumar D, Mains RE, King SM, Eipper BA. Cilia-based peptidergic signaling. PloS Biol (2019) 17(12):e3000566. doi: 10.1371/journal.pbio.3000566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Amidation of bioactive peptides: the structure of peptidylglycine alpha-hydroxylating monooxygenase. Science (1997) 278(5341):1300–5. doi: 10.1126/science.278.5341.1300 [DOI] [PubMed] [Google Scholar]
- 71. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK biobank resource with deep phenotyping and genomic data. Nature (2018) 562(7726):203–9. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Curtis D, Bakaya K, Sharma L, Bandyopadhyay S. Weighted burden analysis of exome-sequenced late-onset alzheimer's cases and controls provides further evidence for a role for PSEN1 and suggests involvement of the PI3K/Akt/GSK-3beta and WNT signalling pathways. Ann Hum Genet (2020) 84(3):291–302. doi: 10.1111/ahg.12375 [DOI] [PubMed] [Google Scholar]
- 73. Bousquet-Moore D, Mains RE, Eipper BA. Peptidylgycine alpha-amidating monooxygenase and copper: a gene-nutrient interaction critical to nervous system function. J Neurosci Res (2010) 88(12):2535–45. doi: 10.1002/jnr.22404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Powers KG, Ma XM, Eipper BA, Mains RE. Identifying roles for peptidergic signaling in mice. Proc Natl Acad Sci U S A. (2019) 116(40):20169–79. doi: 10.1073/pnas.1910495116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Husten EJ, Eipper BA. The membrane-bound bifunctional peptidylglycine alpha-amidating monooxygenase protein. exploration of its domain structure through limited proteolysis. J Biol Chem (1991) 266:17004–10. doi: 10.1016/S0021-9258(19)47332-8 [DOI] [PubMed] [Google Scholar]
- 76. Katrancha SM, Wu Y, Zhu M, Eipper BA, Koleske AJ, Mains RE. Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum Mol Genet (2017) 26(23):4728–40. doi: 10.1093/hmg/ddx355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kolhekar AS, Mains RE, Eipper BA. Peptidylglycine alpha-amidating monooxygenase: an ascorbate-requiring enzyme. Methods Enzymol (1997) 279:35–43. doi: 10.1016/s0076-6879(97)79007-4 [DOI] [PubMed] [Google Scholar]
- 78. Trivellin G, Bjelobaba I, Daly AF, Larco DO, Palmeira L, Faucz FR, et al. Characterization of GPR101 transcript structure and expression patterns. J Mol Endocrinol (2016) 57(2):97–111. doi: 10.1530/JME-16-0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used to conduct the spatial clustering analysis can be found at https://github.com/NICHD-BSPC/spatial_clustering. WES data for the index cases have been deposited in the National Institutes of Health dbGaP database under the accession codes phs000721.v1.p1 and phs003245.v1.p1. The original contributions presented in the study are included in the article/ Supplementary Material and at Zenodo (DOI 10.5281/zenodo.7789345). The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a article that does not adhere to our open data policies.