

Case Presentation:
A 33 year-old with a diagnosis of high grade mixed germ cell tumor including immature teratoma and yolk sac tumor presented for a second opinion in the setting of growing abdominal and pelvic masses despite chemotherapy. She was originally seen 6 months prior by a local gynecologist in the setting of bladder pain and on ultrasound was found to have a 15 cm complex mass arising from the right adnexa. She then underwent a minimally invasive right salpingo-oophorectomy at an outside institution that was complicated by intraoperative spillage and final pathology demonstrated a mixed germ cell tumor. Tumor markers postoperatively were notable for an elevated AFP (581.2 ng/mL) and CA-125 (74.3 U/mL). After oocyte cryopreservation she received 4 cycles of bleomycin, etoposide and cisplatin (BEP) and two cycles of etoposide and cisplatin without bleomycin in order to receive 1 cycle past normalization of her AFP. Her adjuvant chemotherapy course was complicated by the development of pulmonary fibrosis due to the bleomycin. Prior to her cancer diagnosis, the patient was healthy with no known medical problems and no prior surgeries. Her family history was only notable for her father’s diagnosis of non-Hodgkin’s lymphoma. On exam at our institution, she was found to have a right lower quadrant mass palpated at her prior laparoscopic incision site and her abdomen was distended. Her AFP had normalized but CA-125 and CEA were increasing. She underwent a CT chest abdomen pelvis and a pelvic MRI. A biopsy of an abdominal mass was obtained.
Dr. Bhosale
A CT scan with and without contrast showed overall increasing burden of disease compared to an outside hospital study from 5 months prior. Figure 1a is a coronal post contrast image that demonstrates the low attenuation calcified implants in the pelvis. The uterus can be visualized in Figure 1b. It is superiorly displaced within the lower abdomen secondary to confluent disease within the pelvis. The pelvic MRI (Figure 1c) demonstrated the large confluent implant within the pelvis which was noted to closely approximate the right S2 and S3 nerve roots. This implant also exerted mass effect on the rectum and sigmoid colon. Additional peritoneal metastases were noted extending into the anterior abdominal wall.
Figure 1:
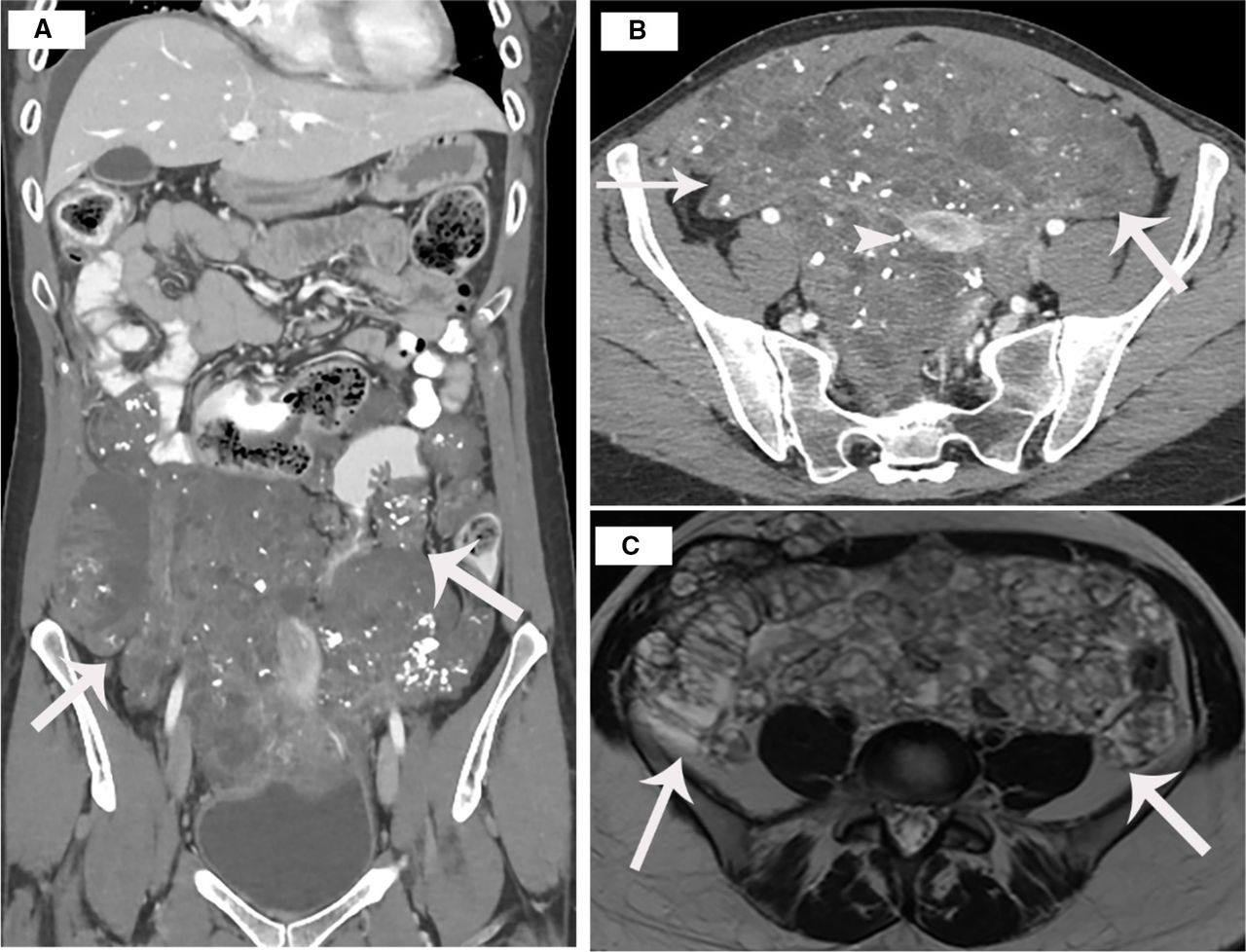
a. CT scan coronal postcontrast images demonstrate low attenuation implants in the pelvis, which have calcifications
b. Axial post-contrast images show the uterus (arrowhead) surrounded by low attenuation mass containing calcifications
c. MRI Axial T2 weighted images show high signal intensity implants in the pelvis
Dr. Hameed
Sections from the right ovarian mass from the original surgery showed a mixed germ cell tumor, composed of high-grade immature teratoma and yolk sac tumor. The immature teratoma component is composed of immature neuroepithelium arranged in rosettes with hyperchromatic nuclei and mitotically active cells (Figure 2a). The current grading system of immature teratoma is a two-tier: low and high-grade. In our case, there is immature neuroepithelium filling more than one low power (x40) microscopic field, consistent with high-grade immature teratoma. The yolk sac tumor component exhibits a combination of two patterns: the endometrioid pattern which is characterized by a glandular formation with columnar cells that can show sub- and/or supra-nuclear vacuoles (Figure 2b) and the reticular pattern which consists of a loose meshwork of interconnecting spaces lined by primitive tumor cells with clear or eosinophilic cytoplasm (Figure 2c).
Figure 2:
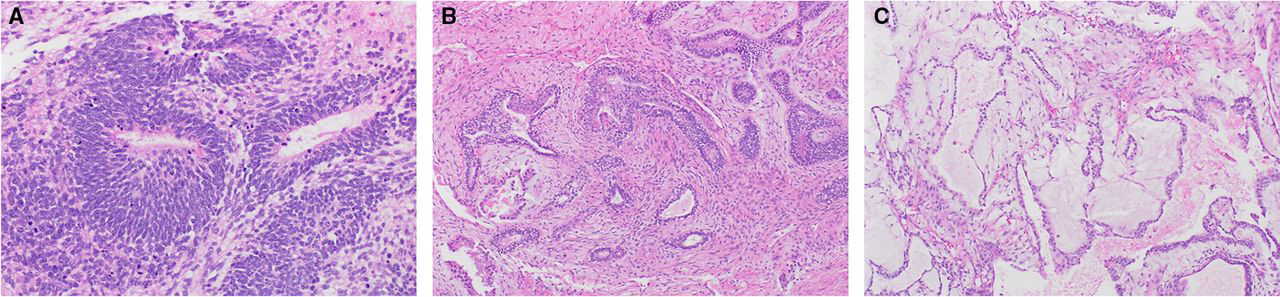
a. Immature teratoma, immature neuroepithelium
b. Yolk sac tumor, glandular pattern with endometrioid features
c. Yolk sac tumor, reticular pattern
The post-chemotherapy abdominal biopsy obtained at our institution, however, did not show evidence of this original tumor and instead showed mature teratoma (Figure 3).
Figure 3:
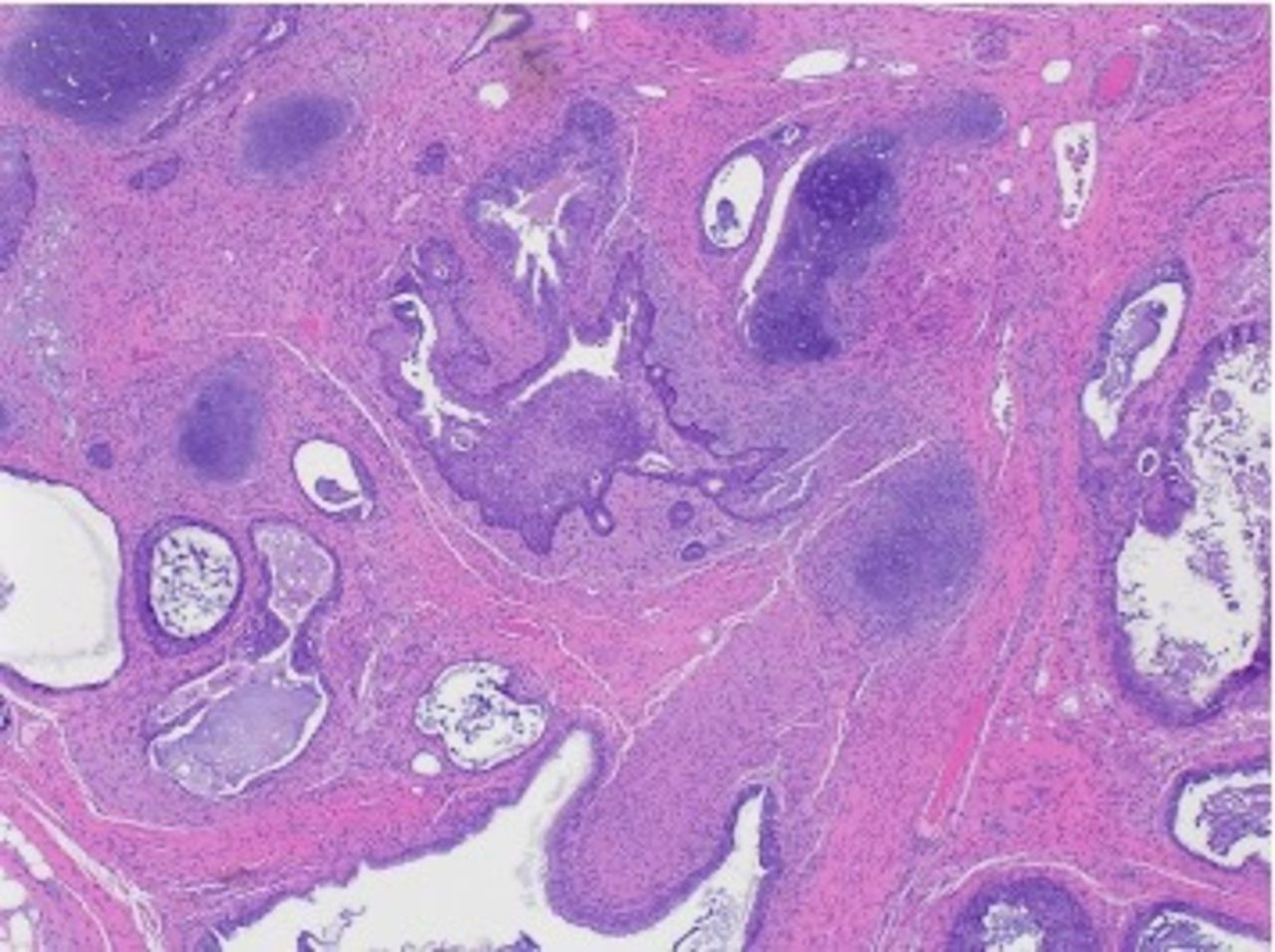
Mature teratoma with cartilage, and various types of epithelia including respiratory epithelium.
Dr. Shafer: Based on these findings, describe the suspected diagnosis
The present scenario is consistent with an entity known as “growing teratoma syndrome”, a term that was coined by Logothetis in 1982[1]. It was first described, however, by DiSaia and colleagues in 1977 based on their observation of 3 female patients with ovarian immature teratoma who received chemotherapy and then underwent “chemotherapeutic retroconversion”, or the conversion of immature elements to mature elements after exposure to chemotherapy[2]. The mechanisms that DiSaia proposed are still considered the leading hypotheses used to explain how the growing teratoma syndrome develops, namely that chemotherapy promotes conversion (or retroconversion) at the cellular level or that chemotherapy eradicates all immature chemo-sensitive elements, leaving the mature chemo-resistant elements to propagate. The growing teratoma syndrome has 3 main components: presence of clinical enlargement of tumors during or after chemotherapy administered for non seminomatous germ cell tumor (in men), normalization of previously elevated tumor markers, and presence of only mature elements upon histologic examination of the tumor[1]. We were suspicious of growing teratoma syndrome in this patient as she met all 3 criteria—she had clear radiologic and clinical evidence of growing tumors after chemotherapy for an immature teratoma despite normalization of AFP and histologic review of the abdominal biopsy showed mature teratoma.
Much of what is known about growing teratoma syndrome was described in male patients with non-seminomatous germ cell tumors, whereas the literature in female patients is composed of scant case reports and small series and therefore is quite limited. While this syndrome may occur after treatment for other ovarian germ cell tumors[3], such as a high grade mixed tumor in this case, it primarily arises after systemic treatment for ovarian immature teratoma[4]. It is important to distinguish growing teratoma syndrome, which can contain tissue from any of the three embryologic lineages, from gliomatosis peritonei, a rare entity composed only of mature glial tissue. While both entities are benign and may develop in association with an immature teratoma, gliomatosis peritonei may arise in the absence of chemotherapy, whereas growing teratoma syndrome is, by definition, an outcome of systemic treatment [5–7].
One of the larger case series by Bentivegna and colleagues demonstrated that of 196 cases of immature teratoma, 19% developed growing teratoma syndrome[8], consistent with a series by Wang and colleagues[3]. The median age at diagnosis is in the second decade, though it may be diagnosed in premenstrual females as well[3,8]. The median time of onset is on the order of 7–18 months in most series, though this syndrome may develop during treatment, or years after initial diagnosis[3,8,9]. While some case series suggest that the risk of growing teratoma syndrome is independent of disease remaining at the end of initial surgery[9], the more robust series demonstrated that residual disease and stage at initial surgery were significantly associated with growing teratoma syndrome [3,10]. In our case, for example, surgical spillage likely contributed to the development of growing teratomas. The presence of mature elements upon initial surgery is not necessary to the development of this syndrome, though presence of gliomatosis peritonei at initial surgery may predict it[3].
Dr. Bhosale
Preoperative images which were obtained after 6 cycles of chemotherapy demonstrated slightly increased extensive supracolic and infracolic peritoneal carcinomatosis with extensive omental caking with punctate calcifications occupying the lower abdomen and pelvis. Large carcinomatosis involving abdomen and pelvis approximately measuring 22.5×10.6 compared to 19.7×9.9 previously (Figure 4). Implants involving the right abdominal wall muscles and subcutaneous soft tissues have increased. Segmental bowel wall thickening secondary to serosal implants for example a left descending colonic serosal implant is visualized. There was stable mild bilateral hydronephrosis. The rectum was displaced to the left secondary to extensive carcinomatosis within cul de sac.
Figure 4:
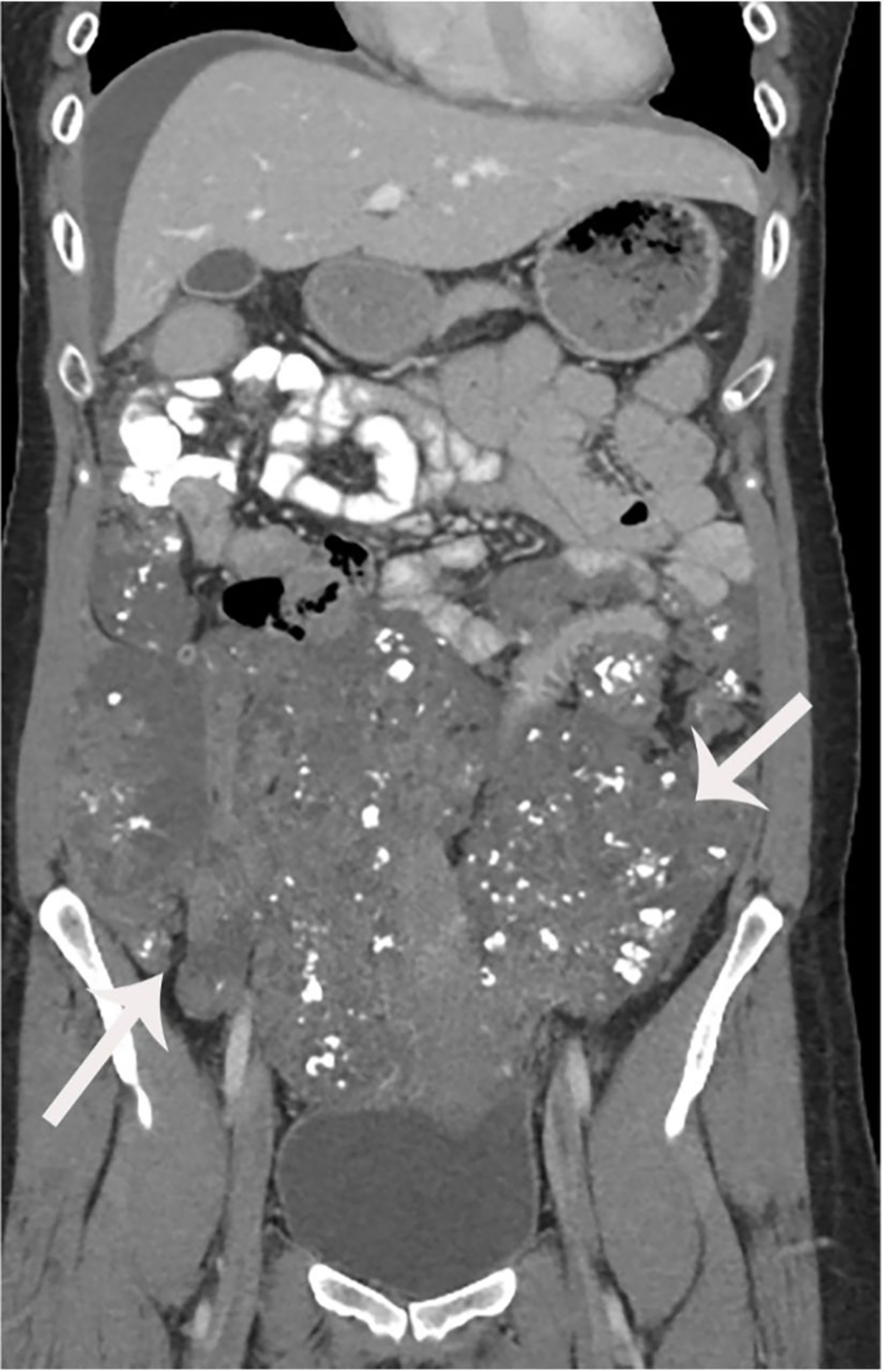
Coronal post-contrast images show low attenuation implants which have slightly increased following chemotherapy.
Dr. Shafer: Please comment on your decision to pursue surgery and the multidisciplinary team that you assembled.
The cornerstone of management of this chemo-resistant, benign entity, is surgery. The growing masses can provoke compression of surrounding organs leading to discomfort, and importantly dysfunction of surrounding organs manifesting as, for example, bowel perforations, ischemia, pulmonary embolism and so forth[8]. Resection also allows for histologic evaluation of all the tumors, which is necessary to exclude recurrence of the malignant germ cell tumor. For this reason and the increased risk of recurrence of growing teratoma syndrome when not completely resected [3], an important goal is to cytoreduce all visible disease. In addition, though rare, malignant transformation primarily to sarcoma, carcinoid tumor, and primitive neuroectodermal tumor, has been reported in 3%–5% of cases of growing teratoma syndrome[3,8,11], which again, makes surgical management (when possible) the management strategy of choice.
Given the extent of disease we assembled a multidisciplinary team to maximally cytoreduce the disease and therefore ensure the best possible outcome for the patient. Pre-operatively the patient was referred to hepatobiliary surgical oncology team given perihepatic involvement with sizable tumor behind the liver and abutting the right hemidiaphragm. She was also seen by the plastic surgery team to plan for abdominal wall reconstruction as on her abdominal exam she had a 15×4cm area in the right lower quadrant consistent with tumor implants surrounding a 4cm scar from the prior incision. She was counseled on the possibility of flaps in the event that primary closure of the skin was not possible as well as bridge underlay mesh reconstruction of her abdominal wall should primary skin and subcutaneous tissue closure remain a viable possibility. Finally, she was seen by the colorectal surgery team given the possibility of extensive colon resection, as the tumor was seen in close proximity to the descending colon above the pelvic brim, it appeared to at the very least displace the rectum, and we suspected there was invasion of it as well. Finally, she was seen by the urology team to plan for intraoperative ureteral stent placement.
Dr. Shafer: What were the key operative findings?
The patient underwent an exploratory laparotomy total abdominal hysterectomy left salpingo-oophorectomy, omentectomy, diaphragmatic stripping, rectosigmoid resection with reanastomosis, diverting loop ileostomy and abdominal wall resection with reconstruction. Upon exploratory laparotomy there was diffuse peritoneal disease primarily in the pelvis. (Figure 5a) There was tumor in the infracolic omentum and a large right hemidiaphragm plaque as well as disease involving the peritoneum overlying Gerota’s fascia. There was a large mass that was approximately 12–14 cm that encompassed the right lower quadrant intra-abdominal wall and invaded through the muscle and fascia. Resection of this mass required sacrifice of the deep inferior epigastric artery pedicle of the rectus. There was a large 18–20 cm multilobed mass that encompassed and surrounded the remaining uterus and left ovary which was densely adherent to the rectosigmoid colon as well as part of the descending colon. (Figure 5b) A low anterior resection was performed to remove the entire pelvic tumor contents en bloc. There was tumor involving the serosal surface of the bladder. At the end of the case all visible disease was removed other than some miliary disease which was ablated with cautery. The estimated blood loss was 1L and the patient was given 2 units of packed red blood cells intraoperatively.
Figure 5:
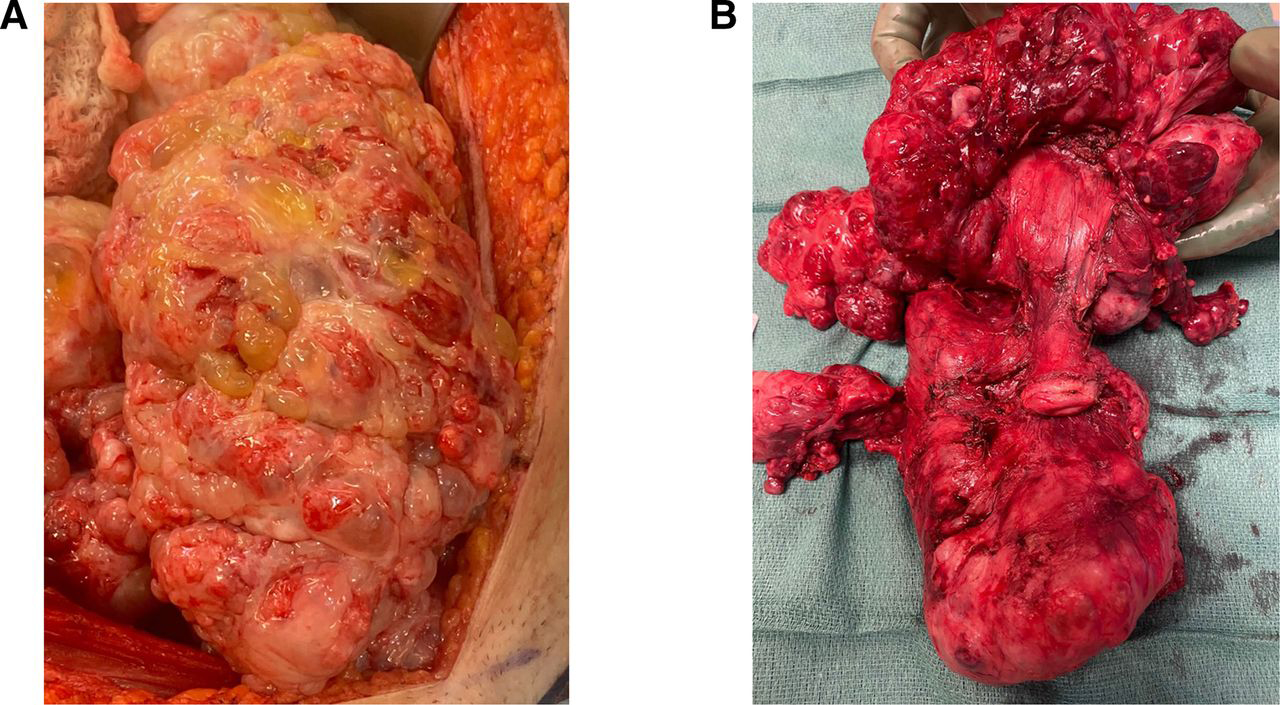
5a. The large growing teratoma in situ upon opening the abdomen. This tumor was emanating from the omentum and attached to the uterus and anterior pelvic peritoneum.
5b. En bloc resection of the uterus, left ovary and fallopian tube, posterior cul de sac tumor and rectosigmoid colon. The rectosigmoid colon is underneath the pelvic tumor
Given the complexity of the procedure the patient was admitted to the intensive care unit postoperatively, but by post-operative day 2 she transitioned to a regular floor. Her postoperative course was complicated by an ileus that required a nasogastric tube and bowel rest. On post-operative day 9 she was noted to have a fever and imaging demonstrated intra-abdominal fluid, but no organized collection and no evidence of an anastomotic leak. The interventional radiology team placed a drain, and she was discharged on oral antibiotics. Cultures and remainder of infectious work-up was ultimately unrevealing.
Dr. Hameed
Sections from the omentum and peritoneum showed mature teratoma with acellular mucin dissecting its stroma (Figure 6a). Acellular mucin is also noted on the peritoneal surface (Figure 6b). No immature elements were identified.
Figure 6:
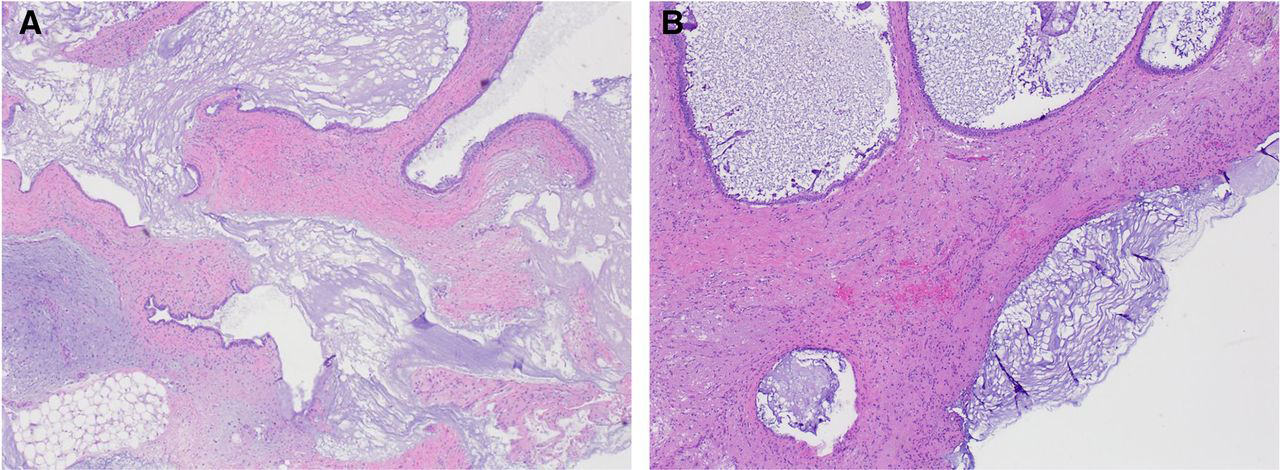
a. Mature teratoma with acellular mucin dissecting its stroma
b. Acellular mucin on the peritoneal surface
Dr. Shafer: What does surveillance entail?
After surgery, surveillance for patients with growing teratoma syndrome involves mostly clinical exam and imaging. While there are no consensus guidelines on surveillance after surgery for growing teratoma syndrome, at the very least, follow-up should be every 3 months for the first 6–12 months. This should include, physical exam, imaging (preferably CT chest, abdomen, pelvis), and tumor markers. Tumor markers should include AFP, beta hCG, and CA-125 (if elevated at diagnosis). Tumor markers are most helpful if they were elevated at initial diagnosis for monitoring for recurrence of immature elements or other mixed elements such as yolk sac tumor. If imaging is stable after 6–12 months with either no evidence of disease or no growth of lesions, then one can consider spacing out imaging to every 6 months. While the goal of surgery for growing teratoma syndrome is resection of all visible disease, if there is small volume disease remaining, it may remain stable. If there is growth of a lesion, then repeat surgery should be considered, especially if there are symptoms. However, if small lesions remain stable and the patient is asymptomatic, continued surveillance is appropriate. Patients with disease on imaging, even if stable, should continue to be followed as these lesions can begin to grow even years later. For patients with completely resected disease, recurrences can occur but these are usually within the first 2 years after surgery[3,12]. When recurrences are found after previously negative imaging, it is important to get histologic documentation of mature teratoma as if immature or mixed elements are present, then the patients will often require chemotherapy in addition to surgery. Our patient has now undergone her first surveillance imaging which showed no evidence of disease. Additionally, her CA-125 has normalized and her AFP remains normal as well.
Closing Summary:
Growing teratoma syndrome is a rare entity that can occur in women who have been treated for ovarian immature teratoma or mixed germ cell tumors. While our case is certainly a dramatic example, this patient demonstrates a number of important risk factors and clinical characteristics of this disease. She was relatively old at time of diagnosis for ovarian germ cell tumor (33 years old), had intra-operative rupture of her tumor at her initial surgery, received adjuvant chemotherapy, and had normalization of her germ call tumor markers (beta hCG and AFP). Biopsy to rule out persistent or recurrent immature teratoma is important as those patients often require further systemic therapy. Surgery is the cornerstone of treatment of growing teratoma syndrome and the goal of surgery should be complete gross resection. In patients with a large burden of disease, a multi-disciplinary may be necessary and should be engaged if it maximizes the chance of complete gross resection.
Acknowledgments
Funding National Cancer Institute (P30CA016672) National Institutes of Health T32 grant (#5T32 CA101642; RN) The funding sources were not involved in the development of the research hypothesis, study design, data analysis, or manuscript writing. Competing interests: None.
Contributor Information
Roni Nitecki, Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Nadia Hameed, Department of Pathology, University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Priya Bhosale, Department of Diagnostic Imaging, University of Texas MD Anderson Cancer Center, Houston, Texas, USA Department of Radiology, University of Texas MD Anderson Cancer Center Division of Cancer Medicine, Houston, Texas, USA.
Aaron Shafer, Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
References
- 1.Logothetis CJ, Samuels ML, Trindade A, et al. The growing teratoma syndrome. Cancer 1982;50:1629–35. doi: [DOI] [PubMed] [Google Scholar]
- 2.DiSaia PJ, Saltz A, Kagan AR, et al. Chemotherapeutic retroconversion of immature teratoma of the ovary. Obstet Gynecol 1977;49:346–50.http://www.ncbi.nlm.nih.gov/pubmed/65751 [PubMed] [Google Scholar]
- 3.Wang D, Zhu S, Jia C, et al. Diagnosis and management of growing teratoma syndrome after ovarian immature teratoma: A single center experience. Gynecol Oncol 2020;157:94–100. doi: 10.1016/j.ygyno.2019.12.042 [DOI] [PubMed] [Google Scholar]
- 4.Imran H, Siddiqui AH, Wilson F, et al. Growing Teratoma Syndrome After Chemotherapy For Ovarian Immature Teratoma. J Pediatr Hematol Oncol 2020;42:e630–3. doi: 10.1097/MPH.0000000000001525 [DOI] [PubMed] [Google Scholar]
- 5.Li S, Su N, Jia C, et al. Growing Teratoma Syndrome with Synchronous Gliomatosis Peritonei during Chemotherapy in Ovarian Immature Teratoma: A Case Report and Literature Review. Curr Oncol 2022;29:6364–72. doi: 10.3390/curroncol29090501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shukkur N, Penumadu P, Ganesan P, et al. Growing teratoma syndrome - ovarian germ cell tumor. Am J Med Sci Published Online First: November 2022. doi: 10.1016/j.amjms.2022.11.001 [DOI] [PubMed] [Google Scholar]
- 7.Liang L, Zhang Y, Malpica A, et al. Gliomatosis peritonei: a clinicopathologic and immunohistochemical study of 21 cases. Mod Pathol 2015;28:1613–20. doi: 10.1038/modpathol.2015.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bentivegna E, Azaïs H, Uzan C, et al. Surgical Outcomes After Debulking Surgery for Intraabdominal Ovarian Growing Teratoma Syndrome: Analysis of 38 Cases. Ann Surg Oncol 2015;22:964–70. doi: 10.1245/s10434-015-4608-y [DOI] [PubMed] [Google Scholar]
- 9.Zagamé L, Pautier P, Duvillard P, et al. Growing Teratoma Syndrome After Ovarian Germ Cell Tumors. Obstet Gynecol 2006;108:509–14. doi: 10.1097/01.AOG.0000231686.94924.41 [DOI] [PubMed] [Google Scholar]
- 10.Kikawa S, Todo Y, Minobe S, et al. Growing teratoma syndrome of the Ovary: A case report with FDG -PET findings. J Obstet Gynaecol Res 2011;37:926–32. doi: 10.1111/j.1447-0756.2010.01439.x [DOI] [PubMed] [Google Scholar]
- 11.André F, Fizazi K, Culine S, et al. The growing teratoma syndrome: results of therapy and long-term follow-up of 33 patients. Eur J Cancer 2000;36:1389–94. doi: 10.1016/S0959-8049(00)00137-4 [DOI] [PubMed] [Google Scholar]
- 12.Rathod PS, Singh A, Punyashree RM, et al. Growing Teratoma Syndrome a Rare Clinical Entity: Two Decades Management Experience from the Regional Cancer Institute. Indian J Surg Oncol 2021;12:31–8. doi: 10.1007/s13193-020-01224-1 [DOI] [PMC free article] [PubMed] [Google Scholar]