

Abstract
Specific inhibition of a single kinase isoform is a challenging task due to the highly conserved nature of ATP binding sites. Casein kinase 1 (CK1) δ and ε share 97% sequence identity in their catalytic domains. From a comparison of the X-ray crystal structures of CK1δ and CK1ε, we developed a potent and highly CK1ε-isoform selective inhibitor (SR-4133). The X-ray co-crystal structure of the CK1δ - SR-4133 complex reveals that the electrostatic surface between the naphthyl unit of SR-4133 and CK1δ is mismatched, destabilizing the interaction of SR-4133 with CK1δ. Conversely, the hydrophobic surface area resulting from the DFG-out conformation of CK1ε stabilizes the binding of SR-4133 in the ATP binding pocket of CK1ε, leading to the selective inhibition of CK1ε. The potent CK1ε-selective agents display nanomolar growth inhibition of bladder cancer cells and inhibit the phosphorylation of 4E-BP1 in T24 cells, which is a direct downstream effector of CK1ε.
Keywords: Casein kinase 1ε, DFG-in/-out conformation, isoform specific inhibitor, structure-based design
Graphical Abstract
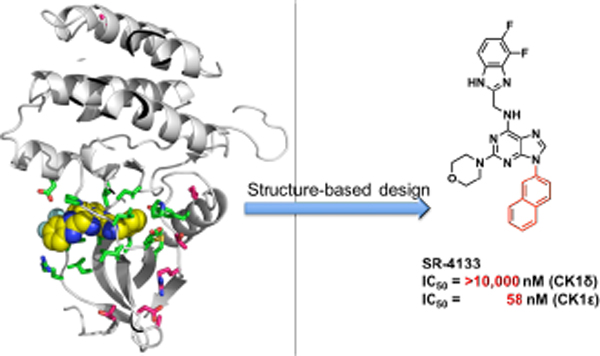
INTRODUCTION
Protein kinases are essential regulatory components of cellular function and development. Deregulation of kinases can cause inappropriate cellular responses, which can ultimately result in disease. In many instances, constitutively activated kinases are involved in the development of human cancer, and inhibition of hyperactive protein kinases is an important approach for the development of cancer-targeted therapies.1 Specific inhibition of an individual kinase is a demanding task due to the highly conserved ATP binding sites found across the kinome (>500 kinases in humans). In addition, achieving selectivity within families of kinase isoforms is challenging due to their remarkably similar three-dimensional structures and highly conserved ATP binding sites. In some cases, isoform-selective kinase inhibitors have been discovered by screening large chemical libraries followed by extensive medicinal chemistry efforts. However, a clear understanding of the differential inhibition mechanism(s) within groups of kinase isoforms is still lacking. Many attempts have been made to rationalize selective inhibition of kinase isoforms by assessing gatekeeper threonine/methionine residues,2–3 active DFG-in/inactive DFG-out conformations,4 P-loop,5 and activation loop6 differences, just to name a few.
Casein kinase 1 (CK1) belongs to a family of serine/threonine protein kinases and regulates various cellular signal transduction pathways.7–9 CK1 comprises seven members in mammals, known as the α, β, γ1, γ2, γ3, δ, and ε isoforms. Most of the CK1 isoforms are ubiquitous in tissues and cell lines, and hundreds of CK1 substrates are known.10–11 CK1 isoforms regulate various cellular signal transduction pathways, including Wnt/β-catenin signaling, circadian rhythm regulation, DNA damage-induced signal transduction, centrosome-associated functions, Hedgehog signaling, and Hippo signaling pathways.12 Among the CK1 isoforms, CK1δ and CK1ε have the highest sequence identity in their catalytic domains and have been reported to possess redundant roles in regulating the circadian clock.13 In addition, CK1δ/ε are known to behave as vital regulators in tumor formation, diabetes, neuro-degenerative diseases, and pain.13–14 However, due to the non-selective nature of known CK1 inhibitors within closely related isoforms, uncertainty remains regarding which isoform regulates specific pathophysiological functions in different cell signaling pathways. For example, nearly all CK1 isoforms are associated with Wnt signaling, and it is difficult to clearly understand the specific function of individual CK1 isoforms within this pathway. Several highly potent and CK1-selective inhibitors have been developed,15–16 and some have been used to characterize the pharmacological effects of CK1 inhibition in animal models.7, 17–18 However, most of these agents are ATP-competitive type I inhibitors, and most lack specificity for individual CK1 isoforms. Recently, a sphingosine analog, GSD0054, was identified as a highly selective CK1ε inhibitor with antiproliferative activity from the phenotypic screening of small library of enantiopure anti-β-amino alcohols and KINOME profiling assay.19–20 However, its IC50 values and modes of inhibition in the target enzyme were not reported. In addition, the high selectivity of GSD0054 to CK1ε has not been clearly understood.
All known CK1δ-inhibiting agents also inhibit CK1ε (Fig 1A) presumably due to the extremely high sequence identity in the inhibitor binding sites. Notably, we have shown that SR-3029 is highly selective among the kinome with high affinity to CK1δ/ε versus other CK1 isoforms. Furthermore, we have demonstrated that SR-3029 has potent cell-based activity and provokes tumor regression in pre-clinical efficacy models of triple negative breast cancer.17 Herein, we report the development of potent and highly isoform-selective CK1ε inhibitors developed by using structure-based design with the CK1δ - SR-3029 co-crystal structure and known CK1ε structures. Most of these new inhibitors show low inhibition potency against CK1δ with >10 μM IC50’s, while they show moderate to high inhibition of CK1ε with ca. 1 μM or lower IC50’s. The absolute binding free energy calculation21–22 shows that SR-4133 has a lower binding affinity to CK1δ than SR-3029, resulting in its high selectivity for CK1ε. SR-4133 shows high nanomolar cytotoxicity to bladder cancer cells in which CK1ε is highly expressed, and inhibits the phosphorylation of 4E-BP1, which is a downstream target of CK1ε.23
Figure 1.
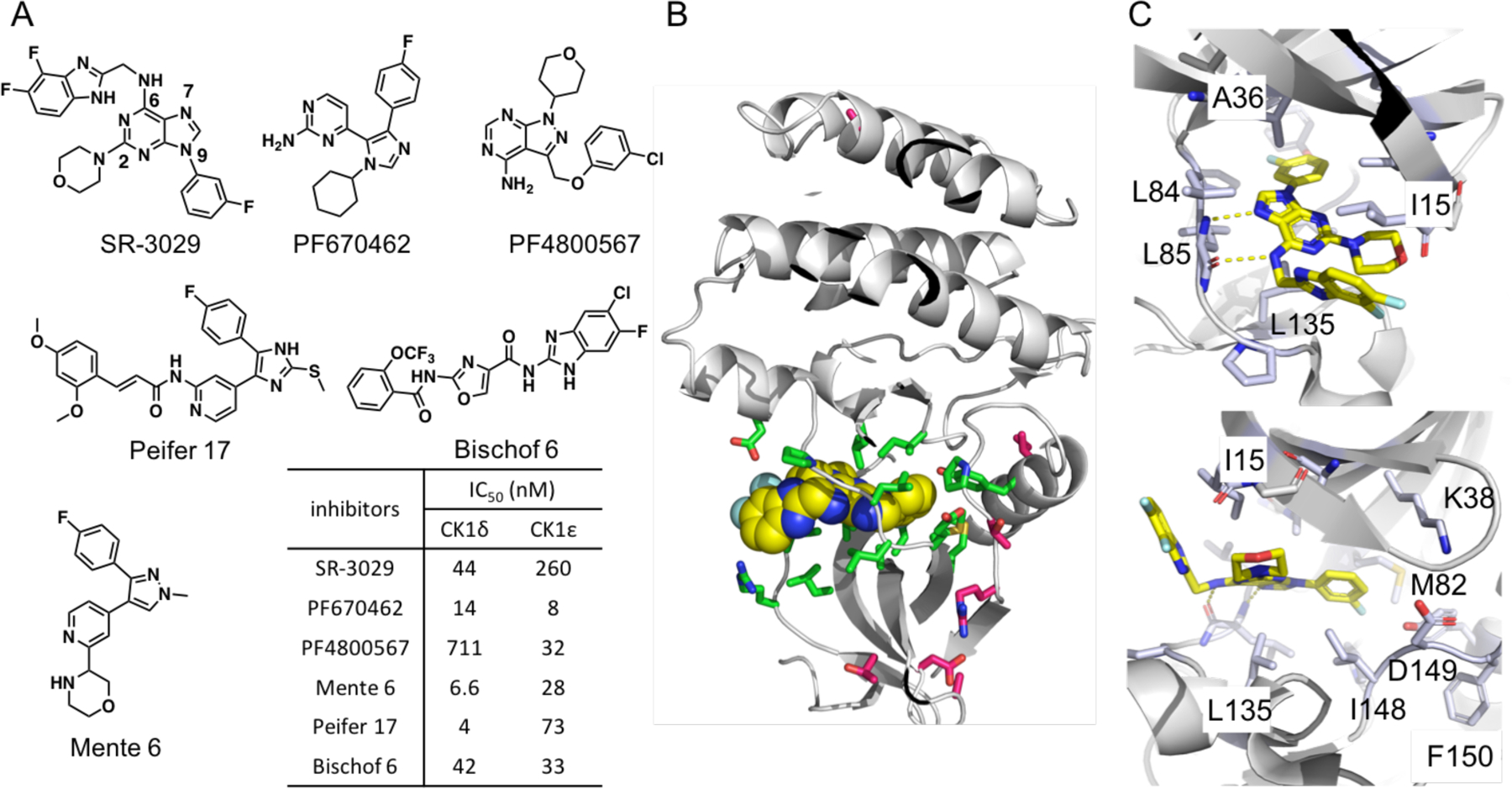
(A) Representative CK1δ/ε dual inhibitors and IC50’s, (B) The X-ray co-crystal structure of CK1δ – SR-3029 (PDB code: 6RCG). SR-3029 is shown as yellow spheres. Seven unique amino acids of CK1δ vs. CK1ε are shown as magenta sticks, and amino acids that have direct contacts with SR-3029 are shown as green sticks. (C) The binding pose of SR-3029 in the ATP pocket of CK1δ: view to the hinge region (top) and to the p-loop (bottom).
RESULTS AND DISCUSSIONS
The X-ray co-crystal Structure of CK1δ - SR-3029 and Design of Selective CK1ε inhibitors.
The X-ray co-crystal structure of CK1δ - SR-3029 has been solved and is used here for structure-based molecular design (PDB code: 6RCG). This co-crystal structure verified the binding orientation of SR-3029 in the ATP binding site predicted from docking studies using the published CK1δ - PF670462 co-crystal structure.24 The NH at the 6-position of the purine ring as well as N7 form hydrogen bond interactions with the amide backbone of L85 in the hinge region, and the 3-fluorophenyl unit at N9 of the purine occupies a hydrophobic area near the gatekeeper residue M82 (Fig. 1C). The morpholine ring and the benzimidazole unit are oriented to the solvent accessible area close to the P-loop, and I15 forms hydrophobic interactions with the surfaces of the morpholine and benzimidazole groups (Fig. 1C).
CK1δ and CK1ε share 97% sequence identity, with only 7 different amino acids in their catalytic domains: T27, D28, A31, I55, T67, R69, and A249 in CK1δ vs. A27, N28, S31, F55, S67, K69, and S249 in CK1ε (Fig. S1, sequence alignment of CK1δ and CK1ε). Furthermore, these seven amino acids (depicted as magenta sticks in Fig. 1B) are positioned far from the ATP binding site (depicted as green sticks). Therefore, most CK1δ inhibitors possess relatively similar (≤20 fold) inhibition potency against CK1ε, and vice versa, as represented by SR-3029, PF670462, PF4800567, Peifer 17, Bischof 6, Mente 6, and numerous other CK1δ inhibitors, some of which also have additional off-target activities (Fig. 1A and Table 3 in ref. 25).7 At present, 24 CK1δ X-ray co-crystal structures are available in the Protein Database Bank (PDB), while there are only two CK1ε structures, specifically of apo-CK1ε and CK1ε with bound PF4800567. Interestingly, the CK1ε structure in complex with PF4800567 revealed a ‘DFG-out’ conformation, while the apo-CK1ε structure has a ‘DFG-in’ conformation (Fig. 2A).26 The 3-chlorophenyl unit of PF4800567 forms hydrophobic contacts with M82 and the carbon chain of K38 on opposite faces of the chlorophenyl ring, but its interaction with F150 of the DFG unit is not likely significant due to the absence of π-π or π-edge interactions. Overlay of SR-3029 (docked) and PF4800567 displays that two structures, particularly the morpholine and 3-fluorophenyl units of SR-3029 and the 4-chlorophenyl ring of PF4800567, are well aligned. This analysis further suggested that the 3-fluorophenyl unit of SR-3029 could be extended to generate hydrophobic interactions with F150 of CK1ε (Fig. 2B).
Table 3.
Comparison of ABFE’s and the electrostatic and Lennard-Jones components of the transfer free energy of SR-3029 and SR-4133 to CK1δ
| SR-3029 | SR-4133 | difference | |
|---|---|---|---|
| 14.97a (1.08)b | 11.15 (1.71) | 3.82 | |
| −ΔGelec | 2.23 (0.38) | −0.07 (0.51) | 2.30 |
| −ΔGLJ | 26.47 (0.58) | 26.18 (1.18) | 0.29 |
Computed with the SDR method and integrated with the Thermodynamic Integration and the Gaussian Quadrature (TI-GQ) approach
Estimated standard errors of the mean are given in parentheses.
Figure 2.
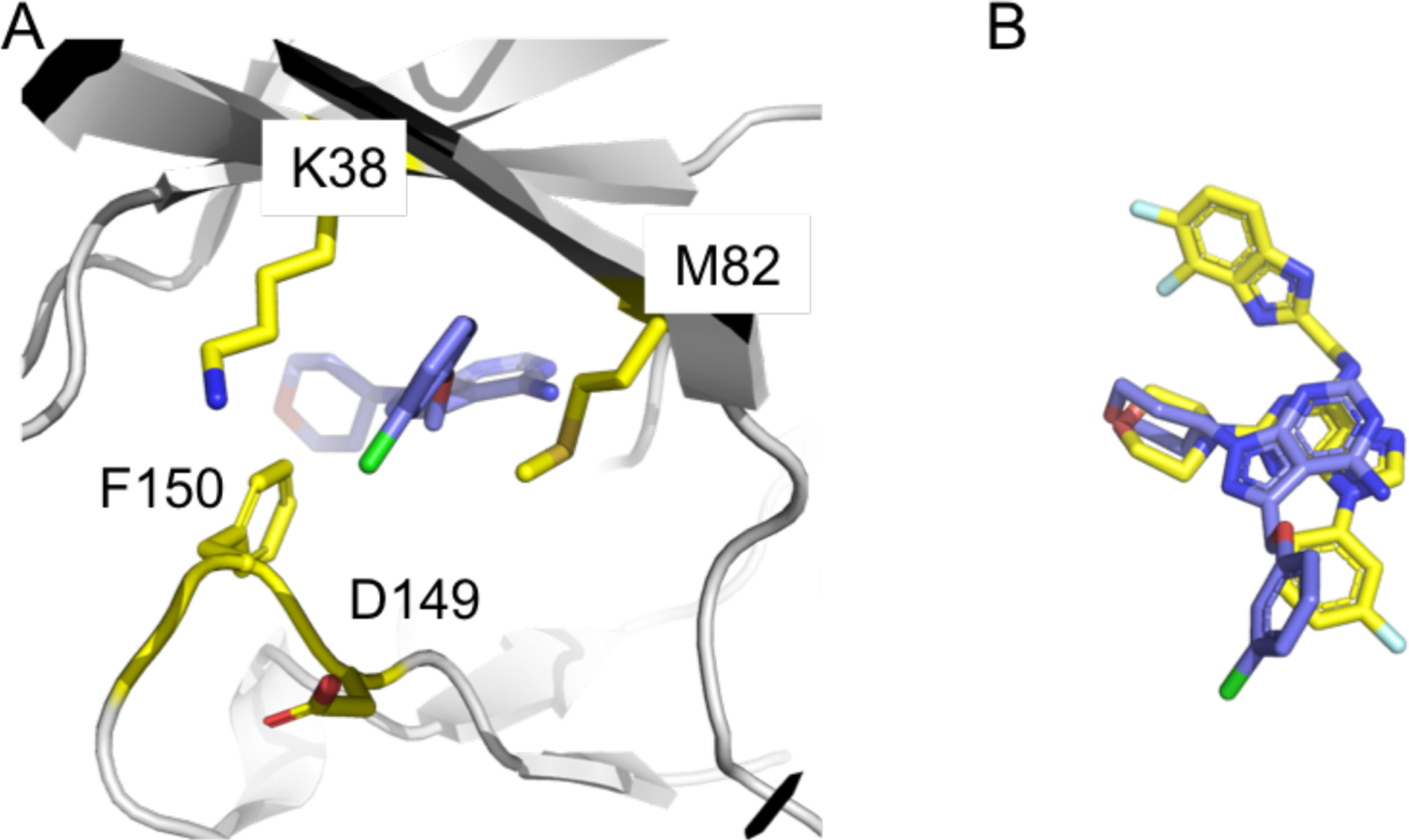
(A) The binding pose of PF-4800567 in the DFG-out conformation of CK1ε. For clarity, only amino acids near to the 3-chlorophenyl unit are present. (B) Overlay of SR-3029 and PF-4800567 based on the binding poses to CK1δ and CK1ε, respectively. SR-3029 is yellow, and PF-4800567 is purple.
Synthesis and Biochemical Study of CK1ε-selective Inhibitors.
As rationalized from the structure analysis and overlaid structures, several larger groups such as naphthyl rings, arylmethyls, and arylethyls (see Table 1), were attached to the N9-position of the purine scaffold of SR-3029 in an attempt to improve binding affinity to CK1ε by targeting its DFG-out conformation, particularly by enhancing hydrophobic contacts with F150. These inhibitors were synthesized by following Schemes 1 and 2 shown below.24, 27 Briefly, diaryliodonium tetrafluoroborate salts (2) were prepared with 2-iodo-1,3,5-triisopropylbenzene diacetate and the corresponding aryl boronic acids (1). Copper (I) mediated coupling of dichloropurine with the diaryliodonium salts produced N9-aryl dichloropurine (4), which was subject to the one-pot reaction with 2-(difluorobenzimidazole) methylamine followed by morpholine via microwave irradiation to produce SR-4133. Other N9-substituted purine analogs were synthesized from arylbromides and arylethyl alcohols to produce the corresponding benzylpurine (5) and phenethylpurine (6), respectively. For the synthesis of other N9-aryl derivatives, several compounds such as SR-4931, SR-4932, and SR-4933 were not accessible using the methods described in Scheme 1. This was due either to low yields (< 10%) in the initial Chan-Lam coupling step, or the inability to make the aryl iodide (2). For such cases, an alternative synthetic route was pursued, as shown in Scheme 2. First, 2,6-dichloropurine (3) was protected as a THP derivative, then two SNAr reactions led to amide 8, which was subsequently Boc protected. Selective THP deprotection with p-TSA gave key intermediate 9. Copper mediated coupling28 of 9 with an aryl bromide resulted in 10. Saponification of the ester and subsequent amide coupling with aniline derivatives gave 11. A final two-step sequence involving Boc-deprotection using 4 M HCl, followed by cyclization via heating the intermediate in acetic acid yielded the benzimidazole substituent in the desired product.
Table 1.
Inhibition of CK1δ and CK1ε by SR-3029 analogs with bulky hydrophobic groups at the N9-position.
|
IC50 (μM) | Fold selectivity for CK1ε | ||
|---|---|---|---|---|
| R | CK1δ | CK1ε | CK1δ/CK1ε | |
| SR-3029a |
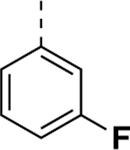
|
0.044 | 0.260 | 0.17 |
| SR-4132 |
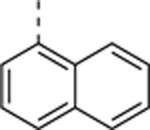
|
1.1 ± 0.017 | 0.18 ± 0.073 (n = 2)b |
6.1 |
| SR-4133 |
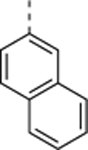
|
>10 (n = 4)b |
0.058 ± 0.013 (n = 5)b |
>172 |
| SR-4310 |
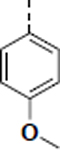
|
1.3 ± 1.1 (n = 3)b |
0.11 ± 0.045 (n = 6)b |
12 |
| SR-19839 |
|
>10 | 0.39 ± 0.04 | >26 |
| SR-19840 |
|
>10 (N.I.D.)c | 0.91 ± 0.06 | >11 |
| SR-19841 |
|
>10 | 0.25 ± 0.03 | >40 |
| SR-4932 |
|
>10 | 0.23 ± 0.15 | >43 |
| SR-4471 |
|
1.2 ± 0.9 | 0.22 ± 0.14 | 5.4 |
| SR-4931 |
|
3.2 ± 0.2 | 0.18 ± 0.17 | 18 |
| SR-4933 |
|
0.40 ± 0.03 | 0.036 ±0.014 | 11 |
| SR-3448 |
|
>10 | 1.8 ± 0.077 | >5.6 |
| SR-3449 |
|
>10 | 0.69 ± 0.091 | >14 |
| SR-3450 |
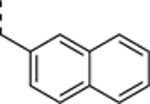
|
>10 | 4.6 ± 1.1 | >2.2 |
| SR-3451 |
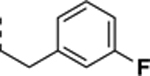
|
>10 | 0.34 ± 0.029 | >29 |
| SR-4290 |
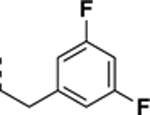
|
>10 | 0.74 ± 0.044 | >14 |
| SR-3452 |
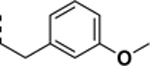
|
- | 1.4 ± 0.2 | - |
| SR-3454 |
|
>10 | 0.22 ± 0.13 | >45 |
| SR-4114 |
|
>10 | 0.46 ± 0.005 | >22 |
| SR-4116 |
|
>10 (N.I.D)c | 1.2 ± 0.002 | >8.3 |
| SR-4134 |
|
>10 | 0.57 ± 0.04 | >18 |
| SR-4152 |
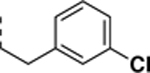
|
>10 (N.I.D)c | 0.20 ± 0.02 | >50 |
| SR-4051 |
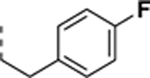
|
0.44 ± 0.26 | 0.10 ± 0.01 | 4.4 |
| SR-4052 |
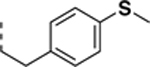
|
1.7 ± 0.4 | 0.24 ± 0.04 | 7.1 |
| SR-4053 |
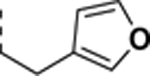
|
0.20 ± 0.02 | 0.088 ± 0.007 | 2.3 |
| SR-4054 |
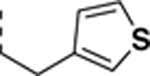
|
0.42 ± 0.04 | 0.11 ± 0.001 | 3.8 |
| SR-4055 |
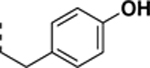
|
0.018 ± 0.001 | 0.030 ± 0.001 | 0.33 |
IC50 values are obtained from references 24 and 27
Experiments were performed (n) times in duplicates.
N.I.D. denotes compounds where no inhibition was detected at 10 μM.
Scheme 1.
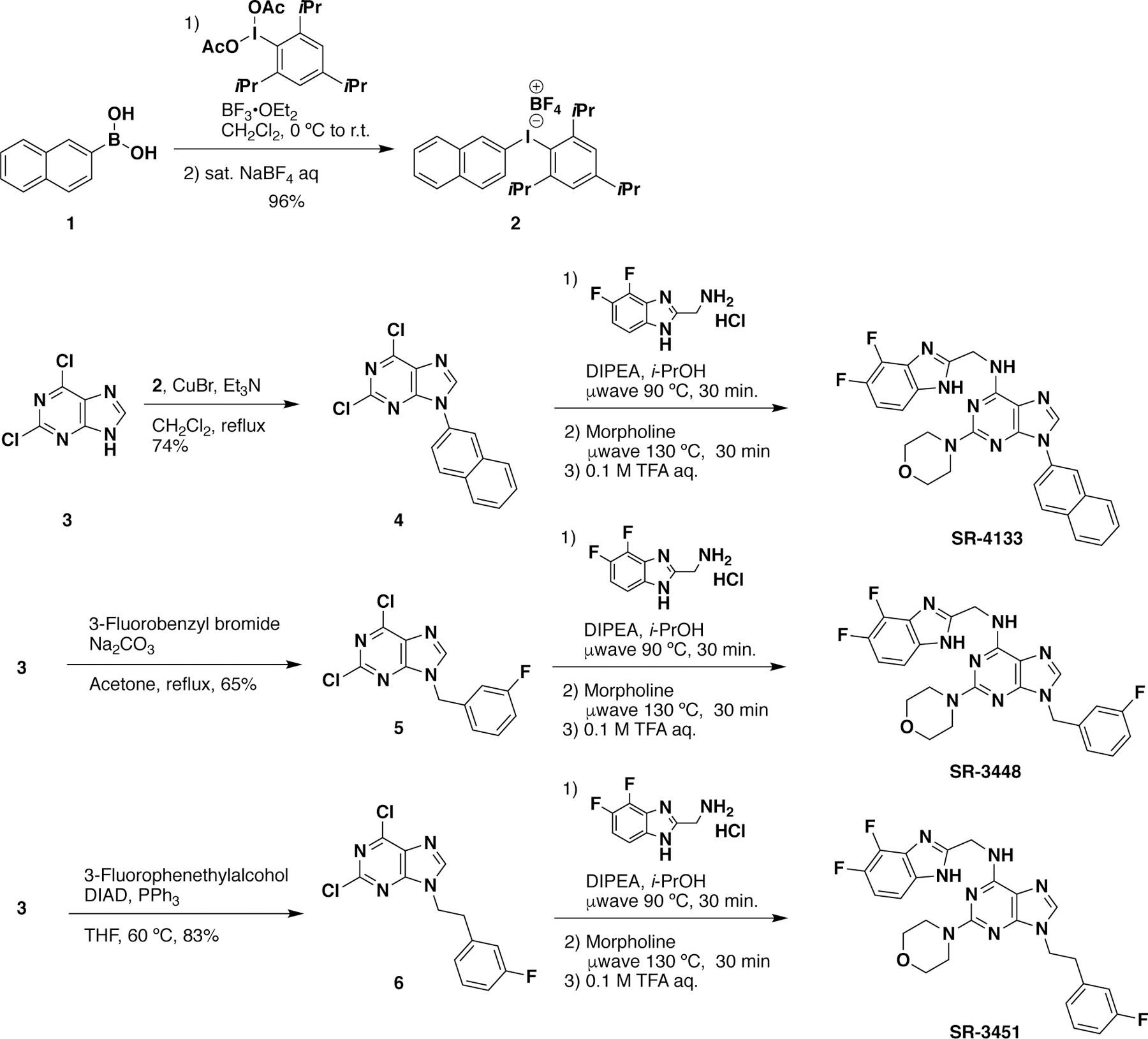
Synthesis of representative CK1ε-selective inhibitors
Scheme 2.
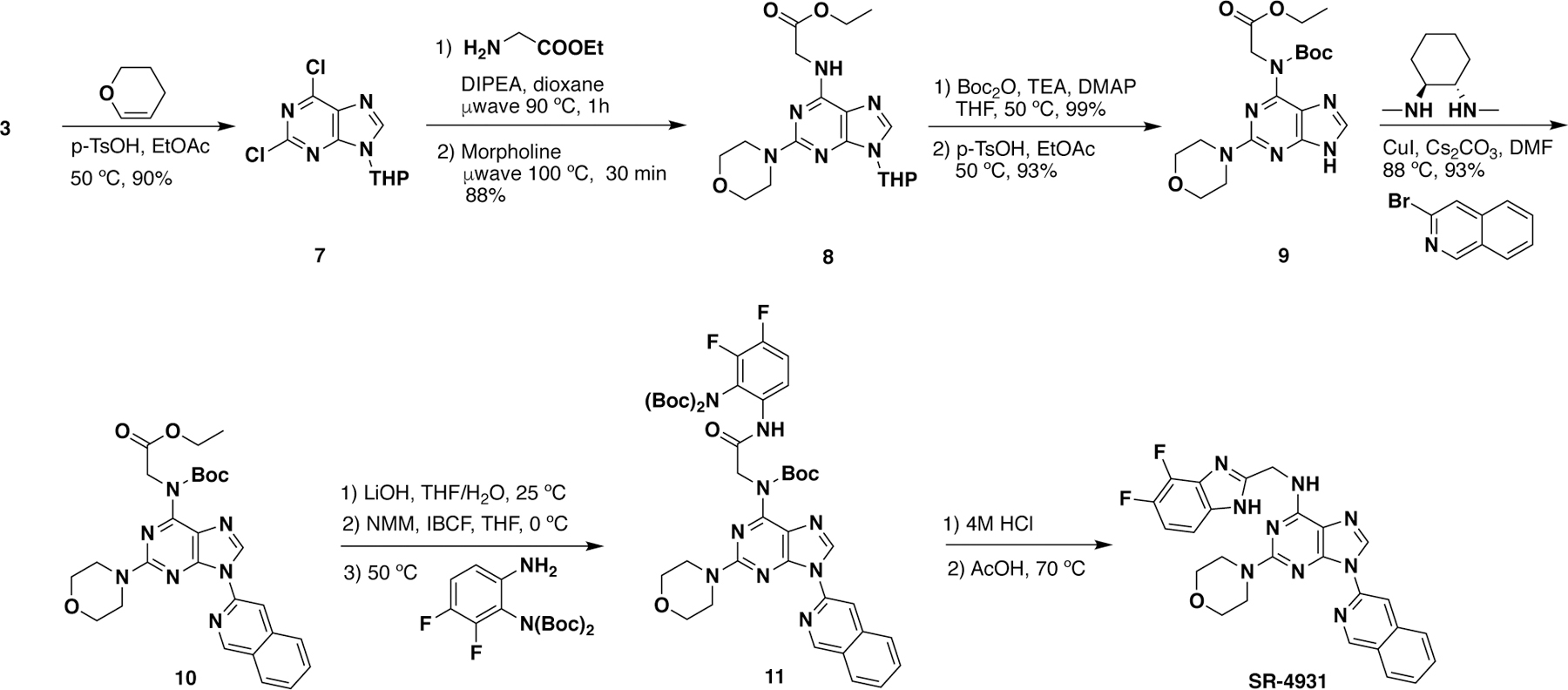
An alternative route for the synthesis of SR-4931, SR-4932, and SR-4933
All newly synthesized SR-3029 analogs were tested in a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. Inhibitors having bulkier or extended hydrophobic aryl rings such as SR-4133, SR-19841, SR-3449, and SR-4152 displayed >10 μM IC50 values against CK1δ, while having nanomolar inhibition of CK1ε. In particular, SR-4133 has a 58 nM IC50 against CK1ε and >10 μM IC50 against CK1δ. SR-4132 and SR-4310, which possess R groups such as 1-naphthyl and p-methoxyphenyl, respectively, show dual inhibition of CK1δ and CK1ε, as does SR-3029 with a m-fluorophenyl group at this position. SR-4471 and SR-4931 have quinoline and isoquinoline groups at the purine N9-position, and interestingly, these compounds show increased inhibition of CK1δ, compared to SR-4133, although their molecular sizes and shapes are very similar. Inhibitors with arylmethyl units at the N9-position such as SR-3448 and SR-3449, and SR-3450 show >10 μM IC50 values against CK1δ, and their inhibition activities vs CK1ε are decreased compared to SR-4133. Arylethyl moieties (Ar = phenyl derivatives) at the purine N9-position negatively affect inhibitor binding to CK1δ, resulting in >10 μM IC50, but compounds with such moieties inhibit CK1ε with good to moderate inhibition potency (30 – 1400 nM IC50’s). Interestingly, SR-4051 and SR-4055 which possess 4-fluorophenylethyl and 4-hydroxyphenylethyl units retain inhibition potency vs CK1δ and are non-selective CK1δ/CK1ε dual inhibitors. In addition, SR-4053 and SR-4054 with 3-furanylethyl and 3-thiophenylethyl groups show similar high nanomolar inhibition to CK1δ and CK1ε.
SR-4133 and SR-4310 were further studied for specificity among CK1 isoforms, and they do not inhibit other CK1 isoforms as expected due to the high specificity of the reference inhibitor SR-3029 against other human kinases (Fig. S2). KINOMEscan profiling of SR-4133 shows that it has a weak binding affinity only to CK1δ, PDGFRB, and TRKA out of 97 kinases examined (Fig. S3).29 Thus, from the analysis of X-ray co-crystal structures of CK1δ – SR-3029, apo-CK1ε, and CK1ε – PF4800567, we developed SR-4133 as a potent and highly isoform selective inhibitor of CK1ε. Specifically, based on our modeling, the bulky hydrophobic aryl rings extending from the N9-position of the purine ring negatively affect binding to CK1δ, leading to enhanced selectivity for CK1ε inhibition.
Cellular Activities of SR-4133 and SR-4310.
With potent and isoform selective CK1ε inhibitors in hand, we assessed their effects on cancer cells. According to the Cancer Atlas database (proteinatlas.org), CK1ε is highly expressed in most colorectal, breast, prostate, and urothelial cancer tissues, while lymphoma melanoma and testis cancer show less expression of CK1ε.10 Compared to bladder cancer extracts, CK1ε is overexpressed in several human bladder cancer cell lines (Fig. S4). Furthermore, knockdown of CK1ε alone impairs the growth of J82 cells in a clonogenicity assay (Fig. S5). Thus, we assessed the antiproliferative potency of SR-4133 and SR-4310 in three bladder cancer cell lines and found that both CK1ε-specific agents block the growth of bladder cancer cells with nanomolar EC50 values (Fig. 3A). Furthermore, CK1ε was also reported to play a critical role in mRNA translation and cell proliferation by blocking the phosphorylation of 4E-BP1, a negative regulator of protein synthesis.23 Our CK1ε-selective inhibitor, SR-4133, blocks the phosphorylation of T37/46, S65, and T70 of 4E-BP1 in T24 cells, as shown in Fig. 3B. The CK1δ/CK1ε dual inhibitor, SR-4310, which is 2.5-times less potent against CK1ε than SR-4133 in the enzyme assay, shows similar inhibition for the phosphorylation of 4E-BP1 in T24 cells. Thus, CK1δ also appears to play a role in the phosphorylation of T37/46 and T70 of 4E-BP1 in T24 cells, although its effect on S65 of 4E-BP1 is negligible, as shown in the previous report.23
Figure 3. Cellular activity of SR-4310 and SR-4133.
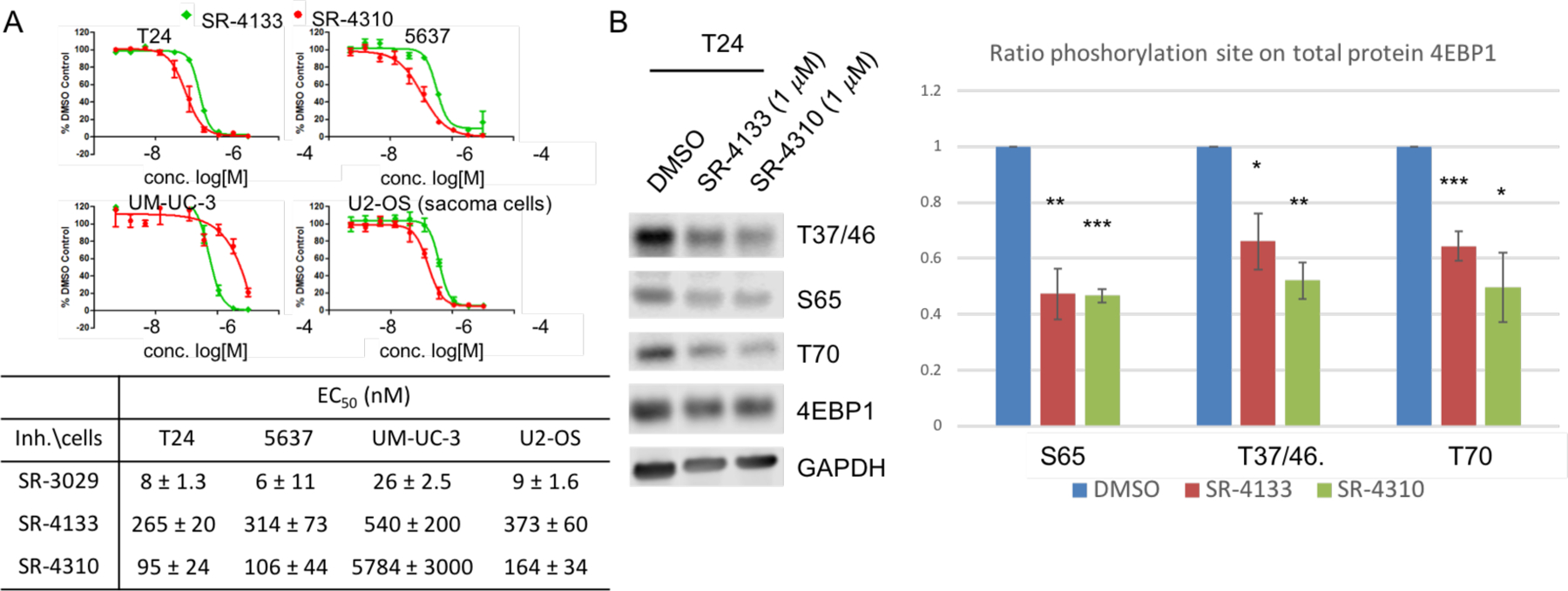
(A) Antiproliferative potency of SR-4133 and SR-4310 in the indicated bladder cancer cell lines and U2-OS cells. Cellular proliferation after 72 hours of treatment with SR-4133 and SR-4310 was analyzed by CellTiter-Glo Assay on selected bladder cancer cell and compared to DMSO control. Data shown represent the average of three replicates ± SD. (B) Inhibitory potency of SR-4133 and SR-4310 on the phosphorylation of 4E-BP1 in T24 cells. Phosphorylation of 4E-BP1 was quantified by western blot after 24h treatment with 1 μM of compound. Ratio phosphorylation had been calculated depending on respective GAPDH and the DMSO control from 3 independent experiments (* (N=3); statistical significance was calculated by two-tailed Student’s t test (* p < 0.05, ** p < 0.01, *** p < 0.001)
The binding orientation of SR-4133 in the ATP-binding site of CK1ε determined by MD simulations is similar to that in the X-ray co-crystal structure of CK1δ – SR-4133.
Because the X-ray co-crystal structure of CK1ε - SR-4133 was not available, molecular docking and molecular dynamics simulations were conducted to determine the binding orientation of SR-4133 in the ATP-binding site of CK1ε. AutoDock-GPU was used for docking of SR-4133 to CK1ε (PDB code: 4HNI),30 and 11 unique conformations (or poses in Fig. S6) were selected for MD simulations. The 11 SR-4133 conformers were merged into CK1ε to create enzyme - ligand complex systems, which were solvated in solvent boxes and simulated for 100 ns each. The last 1000 snapshots of each conformation, corresponding to 80 – 100 ns simulations, were extracted and used for the calculation of the binding affinity by MM/GBSA (Table S3). The pose 2 and 11 showed the highest binding affinities of SR-4133 to CK1ε with ΔGbinding of −45.24 kcal/mol and −47.02 kcal/mol, respectively, and they have similar binding orientations to that in the X-ray co-crystal structure of CK1δ - SR-4133 (PDB code: 6RCH; Fig. S7A, S7B, and S7C). The binding orientations of SR-4133 in pose 9 and 10 were flipped, compared to that in the X-ray co-crystal structure (Fig. S7D and S7E), and they had the lowest binding affinities to CK1ε with ΔGbinding of −35.60 kcal/mol and −26.89 kcal/mol, respectively (Table S3). Therefore, due to the high binding affinities of pose 2 and 11, SR-4133 is expected to bind to the ATP-binding site of CK1ε in a similar way as to CK1δ. The systems for the pose 2 and 11 were further simulated up to 1 μs each, and the structure of the CK1ε - SR-4133 complex having the lowest energy was extracted from trajectories (200,000 snapshots), which was overlayed to the CK1δ - SR-4133 co-crystal structure (Fig. 4A). The binding orientations of SR-4133 in the ATP-binding sites of CK1ε and CK1δ are similar, but SR-4133 of CK1ε is further moved to the pocket of the enzyme than that of CK1δ. The average structure of 20,000 snapshots from the 800 ns – 1 μs time period was also obtained, and it showed that there were no significant conformational changes of SR-4133 and residues in its binding site in this time period (Fig. S8A). Only the morpholine ring of SR-4133, the phenyl rings of F20 and F150, and side chain residues of L135, I148, R13, and E52 were often rotated, as presented as physically unmeaningful geometries (Fig. S8A), implying the system of the CK1ε - SR-4133 complex was well sustained in the 1 μs simulations. The averaged structure was overlayed with the structure having the lowest energy in the 1 μs simulations without significant difference, particularly residues in the naphthyl ring binding site (Fig. S8B). Thus, we employed the structure with the lowest energy as a representative model for analysis.
Figure 4. The binding poses of SR-4133 and SR-3029.
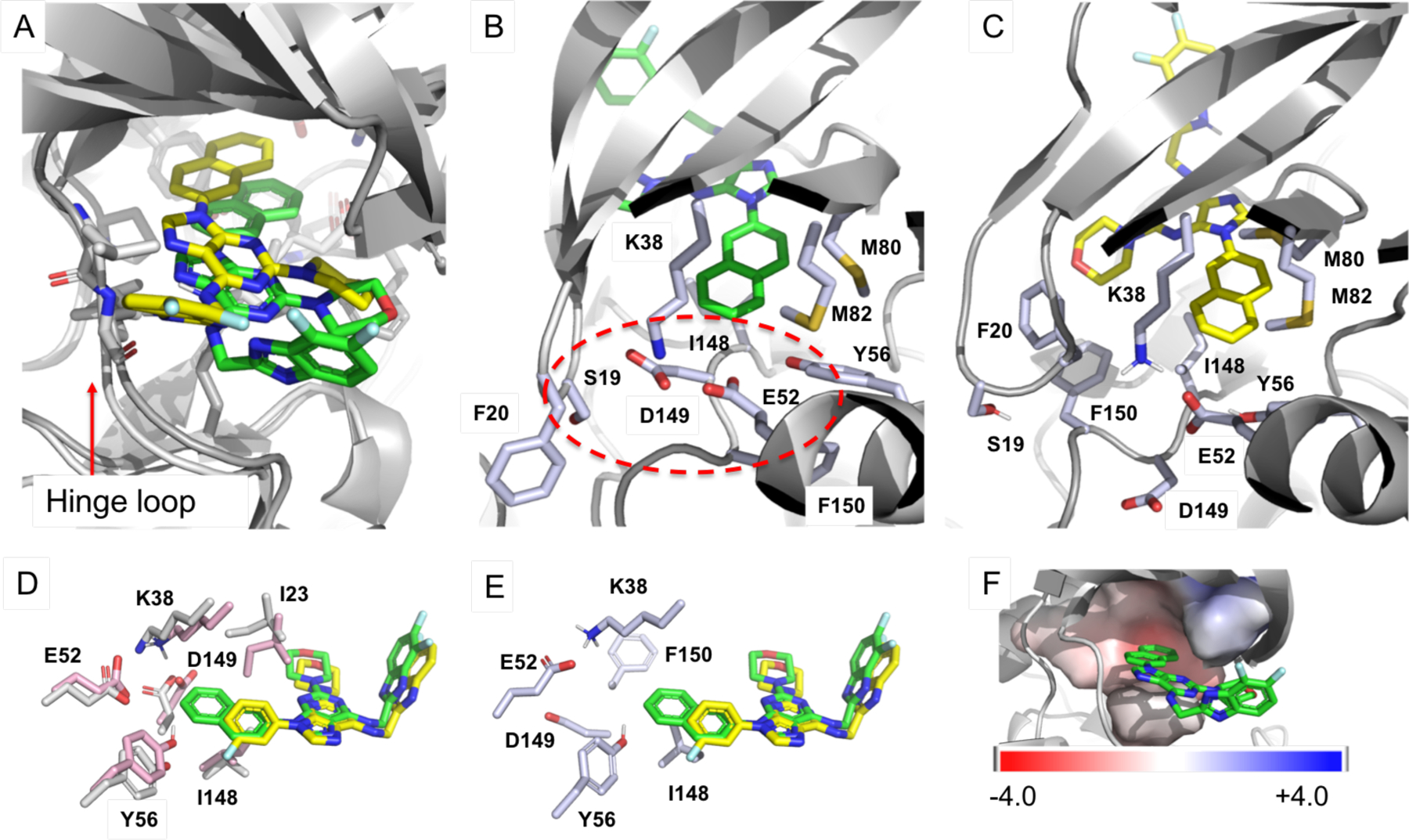
(A) The binding orientation of SR-4133 complexed with CK1ε obtained from 3.1 μs MD simulations (yellow carbon) and its overlay to CK1δ - SR-4133 X-ray co-crystal structure (green carbon, PDB code: 6RCH); view from the hinge region and same camera angle to Fig. 1C. (B) X-ray co-crystal structure of CK1δ in complex with SR-4133. D149-mediated salt bridge and hydrogen bond interactions are labelled with a red circle. (C) The MD model structure of CK1ε in complex with SR-4133. (D) Overlay of the CK1δ – SR-3029 co-crystal structure (gray carbon) to a representative structure of CK1δ (pink carbon) obtained from MD simulations without the inhibitor. SR-3029 (yellow carbon) is overlayed to SR-4133 (green carbon) based on their complex form with CK1δ co-crystal structures (PDB codes: 6RCG and 6RCH, respectively). (E) A representative structure of CK1ε obtained from MD simulation without the inhibitor (X-ray co-crystal structure of CK1ε (PDB code: 4HNI) was used for the 100 ns MD simulation. (F) The electrostatic potential map of CK1δ – SR-4133 co-crystal structure. Note: We attempted to produce crystals of CK1ε in complex with SR-4133, 4132, or 3448, but these experiments were unsuccessful. The representative structures of apo-CK1δ and apo-CK1ε were obtained from 100 ns MD simulations followed by clustering 10000 snapshots.
Rationale for the low binding affinity of SR-4133 to CK1δ, compared with SR-3029.
We solved the X-ray co-crystal structure of CK1δ in complex with SR-4133 (Fig. 4B, PDB code: 6RCH), and it was compared with the CK1δ - SR-3029 structure (Fig. 1B) and structures from MD simulations31–32 (Fig. 4C, 4D, and 4E). The CK1δ – SR-4133 co-crystal structure possesses the DFG-in conformation, with the polar D149 residue occupying the inhibitor or ATP binding site. As a result, S19, K38, D149, and E52 of CK1δ are in a position to form a salt bridge and hydrogen bond (H-bond) interactions (Fig. 4B). The hydrophobic naphthyl ring of SR-4133 is oriented into and it is mismatched with the negative electrostatic surface area composed by S19, K38, D149, and E52 of CK1δ, obtained by Adaptive Poisson-Boltzmann Solver (APBS) electrostatics calculations (Fig. 4F).33 The hydrogen bond analysis of the MD simulation of CK1δ without the inhibitor showed that S19, K38, Q48, E52, Y56, and D149 formed a strong salt bridge/H-bond network throughout the simulation (Table 2): Fraction 0.5687 implies 5687 out of 10000 frames possess the salt bridge interaction between D149 and K38 during the simulation.34–35 Particularly, D149 interacted with K38 and S19 more significantly in CK1δ than in CK1ε. Thus, the negative electrostatic potentials are sustained in the pocket. Due to the incongruous electrostatic surface, bound SR-4133 with the 2-naphthyl ring at the purine N9-position becomes unstable in the active site of CK1δ and can be easily released. In the CK1δ – SR-4133 co-crystal structure, the distance between C7 of the naphthyl ring and N of K38, O of E54, or O of D149 is 4.2 Å, 4.2 Å, or 3.9 Å, respectively. However, during the MD simulation of apo-CK1δ, these residues moved into the space where the naphthyl ring of SR-4133 was positioned in the X-ray co-crystal structure (pink carbon (from MD) vs. gray carbon residues in Fig. 4D). Thus, the apo-CK1δ structure appears to have a relatively smaller pocket with negative electrostatic surface potentials, which might hinder the binding of SR-4133, leading to diminished inhibition to CK1δ when compared to SR-3029: the 3-fluorophenyl ring of SR-3029 could not reach the area with the negative electrostatic potential (Fig. 4D. yellow carbon of SR-3029). However, in the case of CK1ε, due to the DFG-out conformation, the salt bridge interaction between only K38 and E52 can be formed and located away from the naphthyl binding site (Fig. 4C and 4E). Furthermore, since F150 of CK1ε has the out-conformation (DFG-out), F20 in the p-loop could form π - π stacking interaction with F150, making the pocket more hydrophobic (Fig. 4C). However, F20 of CK1δ has moved away from and S19 is moved into the pocket due to the H-bond and salt bridge interactions (Fig 4B).
Table 2.
Hydrogen bond analysis of the MD simulations of CK1δ and CK1ε
| Acceptora | DonorHb | Fractionc | ||
|---|---|---|---|---|
| CK1δ (6RCG) | CK1e (4HOK) | CK1e (4HNI) | ||
| D149@OD | K38@HZ | 0.5687 | 0.3357 | 0.0009 |
| D149@OD | S19@H | 0.6673 | 0.0304 | N/A |
| D149@OD | S19@HG | 0.4637 | 0.0046 | N/A |
| S19@O | K38@HZ | 0.8735 | N/A | N/A |
| E52@OE | Q48@HE | 0.8235 | 0.0778 | 0.3430 |
| E52@O | Y56@H | 0.4051 | 0.4204 | 0.0107 |
| E52@OE1 | K38@HZ | 0.5395 | 0.4604 | 0.3734 |
| E52@OE2 | K38@HZ | 0.4729 | 0.4948 | 0.4354 |
| Q48@O | E52@H | 0.5554 | 0.4463 | 0.5079 |
| Y56@OH | D149@H | 0.2868 | 0.1060 | 0.3584 |
“OD”, “OE”, and “OH” stand for acceptor oxygen in the residue, and “O” for acceptor oxygen in the amide backbone. OD1 and OD2 are combined to OD for counting frames possessing hydrogen bonds.
“HZ”, “HG”, and “HH” stand for donor hydrogen in the residue, and “H” for donor hydrogen in the amide backbone. HZ1, HZ2, and HZ3 are combined to HZ for counting frames possessing hydrogen bonds.
Fraction means the number of frames possessing hydrogen bond interactions divided by the total number of frames (10000 in this 100 ns MD simulation)
This trend was also observed with SR-3448, SR-3449, and SR-3450, which possess hydrophobic aryl groups with a methylene spacer unit such as 3-fluorobenzyl, 1-naphthalenylmethyl, and 2-naphthalenylmethyl respectively. In addition, inhibitors with two methylene spacer units such as SR-3451, SR-4290, SR-3454, SR-4114, SR-4116, SR-4134, and SR-4152 showed similar tendencies toward SR-4133, having low inhibition to CK1δ. However, SR-3029, SR-4132, or SR-4310 with 3-fluorophenyl, 1-naphthyl, or 4-methoxyphenyl group showed nanomolar IC50 to CK1δ since these shorter/smaller moieties were unable to interact and clash with the negative electrostatic surface of CK1δ. In addition, the 4-methoxy of SR-4310 formed a hydrogen bond interaction with Y56 and D149, leading to its improved binding to CK1δ.
The absolute binding free energies (ABFE) of SR-3029 and SR-4133 were calculated to examine the decreased binding affinity of SR-4133 (vs. SR-3029) to CK1δ.22, 36–38 The Binding Affinity Tool (BAT.py), designed to automate ABFE calculation procedures, was used to build the MD simulation boxes, set up parameters and simulation windows, and analyze ABFE calculation results.21 The simultaneous decoupling and recoupling (SDR) method,39 which extracts the ligand from the binding site in nonphysical alchemical pathways, was used to calculate ABFE’s of SR-3029 and SR-4133 to CK1δ. Each ligand was located 40 Å away from its binding site of the enzyme, which was far enough for negligible interactions. The ABFE’s of SR-3029 and SR-4133 to CK1δ are 14.97 kcal/mol and 11.15 kcal/mol, respectively, and SR-4133 has a decreased binding affinity to CK1δ in the ABFE calculation with the SDR method (Table 3). In addition, the difference in electrostatic interaction energy term between SR-3029 and SR-4133 is more significant than Lennard-Jones potential energy term . Therefore, the MD-based ABFE calculation also shows the lower binding affinity of SR-4133 to CK1δ when compared to SR-3029, leading to the decreased inhibition to CK1δ. The energy component analysis also shows that the mismatched electrostatic interactions between SR-4133 and CK1δ could be a major contributor to the decreased binding affinity and inhibition of SR-4133, compared to SR-3029.
The CK1ε structure adopts the DFG-out conformation when complexed with type-II inhibitors, while CK1δ - SR-4133 complex has the DFG-in conformation.
Currently, only two X-ray crystal structures of CK1ε, specifically apo-CK1ε and CK1ε – PF-4800567 complex, are publicly available,26 while there are 24 structures of CK1δ complexed with various inhibitors.26, 40–50 Interestingly, the apo-CK1ε structure possesses the DFG-in conformation (Fig. 5B), and the CK1ε – PF-4800567 complex structure has the DFG-out conformation (Fig. 5C), while all CK1δ structures have the DFG-in conformation (a representative structure shown in Fig. 5A). Further analysis of these structures indicates that I55 and F55 are located close to the DFG units of CK1δ and CK1ε, respectively (Fig. 5), and that this unique pair of residues could affect the propensity of these proteins to adopt DFG conformations differently. Specifically, CK1δ may only be able to form a stable DFG-in conformation due to I55, while CK1ε can form stable DFG-in and DFG-out conformations due to F55, as witnessed with the current sets of CK1δ and CK1ε structures.26 In the DFG-in conformation of CK1δ and CK1ε, F150 occupies the hydrophobic area surrounded by M59, I65, I119, and I147, and D149 is oriented towards the solvent accessible area. The conversion of DFG-in to DFG-out conformations requires the loss of hydrophobic contacts between F150 and the aforementioned hydrophobic amino acids since F150 needs to swing out from the hydrophobic area in the DFG-out conformation. Interestingly, in the DFG-out conformation of CK1ε, the hydrophobic area where F150 was originally located is occupied by F55, which compensates for the loss of hydrophobic interactions caused by the conversion of DFG-in to DFG-out conformations (Fig. 5C). However, CK1δ has I55 at the equivalent position (Fig. 5A), and in the DFG-out conformation of CK1δ, I55 cannot swing into the area where F150 was originally located due to the smaller size of the Isoleucine residue, resulting in the unstable DFG-out conformation of CK1δ. This analysis confirms the distinct tendency of DFG conformations in CK1δ and CK1ε caused by the unique I55 and F55, which is supported by the current pool of CK1δ and CK1ε X-ray crystal structures, and this uniqueness could be applied to the design of selective CK1δ or CK1ε inhibitors.
Figure 5. The tendency of DFG conformation of CK1δ and CK1ε.
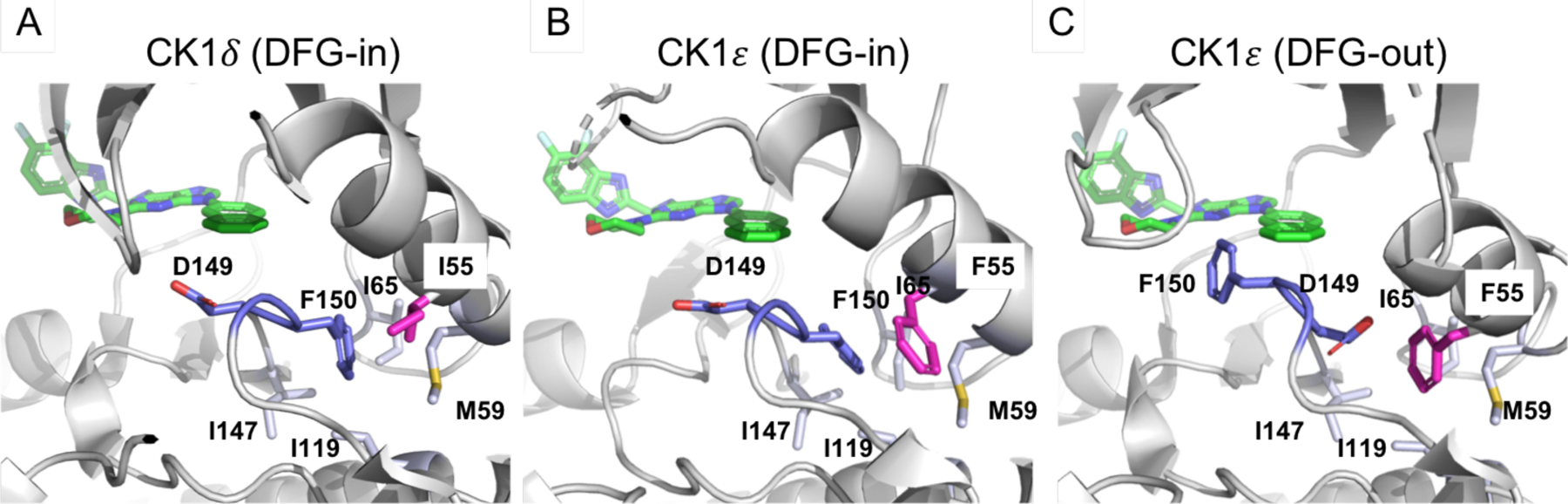
(A) DFG-in conformation of CK1δ – SR-4133 (6RCH). (B) DFG-in conformation of apo-CK1ε (4HOK) and imaginary SR-4133. (C) DFG-out conformation of CK1ε (4HNI) and imaginary SR-4133. DFG units are purple, and the unique I55 and F55 are magenta. Amino acids in the hydrophobic pocket where F150 occupies in the DFG-in conformation are labeled and shown in light purple.
The ‘in’ to ‘out’ conversion of the DFG unit in CK1ε forms the F150-mediated hydrophobic area near the p-loop where the 2-naphthyl ring of SR-4133 could occupy.
In the DFG-out of CK1ε, the hydrophobic residue F150 is in a position to form hydrophobic contacts with F20 (Fig. 4C). Due to the repositioning of F150, the salt bridge/H-bond network in the back pocket almost disappeared, and only a small fraction of salt bridge interactions remained between the conserved residues K38 and E52 in the MD simulation of apo-CK1ε (Fig 4C and 4E, and Table 2). Furthermore, this salt bridge interaction was oriented away from the binding site of the naphthyl unit, and the hydrophobic methylene unit of K38 was facing this site (Fig. 4C and 4E). Thus, the 2-naphthyl ring of SR-4133 most likely maximizes the hydrophobic contacts with I148, F150, and K38 (methylene units) in the ATP binding site of CK1ε (Fig. 4C). Two methionine residues (M80 and M82) can form a hydrophobic surface from the top face of the naphthyl ring (Fig. 4C). Thus, the inhibitor and enzyme complex can be mutually stabilized to block the enzyme activity of CK1ε. In the same way, SR-3029 analogs with the extended hydrophobic rings at the N9 position, such as SR-3451, SR-4290, and SR-3454, have higher inhibition potency versus CK1ε than CK1δ. Due to the absence of an X-ray co-crystal structure, we do not have definitive evidence that SR-4133 is a type-II inhibitor and binds to the DFG-out conformation of CK1ε. However, its potent inhibition of CK1ε with high selectivity against CK1δ supports the hypothesis that SR-4133 binds to different modes of DFG conformation in these two enzymes. In addition, maximized hydrophobic contacts, mediated by F150 (DFG-out), the 2-naphthyl ring (inhibitor), and F20 (p-loop), could be a major contributor to the potent inhibition of CK1ε.
We also conducted molecular docking studies of SR-4133 to CK1ε with the DFG-in conformation (PDB code: 4HOK), and 8 binding orientations were generated, of which complex forms with CK1ε (DFG-in) were subjected to 100 ns MD simulations and MM/GBSA calculations. The binding affinities of SR-4133 to CK1ε (DFG-in) were in the range of −41.96 kcal/mol to −21.92 kcal/mol, which is much less than that of pose 11 with SR-4133 bound to CK1ε (DFG-out, −47.02 kcal/mol). Therefore, these computational studies support our conclusion that SR-4133 is a type-II inhibitor of CK1ε.
Rationale for decreased selectivity of other SR-4133 and SR-3451 analogs.
SR-4471 and SR-4931 possess 3-quinoline and 3-isoquinoline in the N9-position of purine, respectively. Although these inhibitors were expected to show potent inhibition of CK1ε due to the similar size and shape of these substituents to the naphthyl group of SR-4133, they displayed low micromolar IC50 inhibition of CK1δ (improved relative to SR-4133) but also decreased (high nanomolar IC50) inhibition of CK1ε, resulting in only around 10-fold selectivity. Density Functional Theory (DFT) calculations using Gaussian 0951 and analysis of electrostatic potentials of SR-4133, SR-3451, and SR-4471 show that more positive electrostatic potentials are developed in the outer ring of the quinoline/isoquinoline substituents due to the high electronegativity of the nitrogen atom (Fig. S9). Therefore, the increased positive electrostatic potential may provide favorable interactions with the salt bridge/H-bond network of CK1δ, but unfavorable interactions with the hydrophobic area of CK1ε, resulting in increased or decreased inhibition to CK1δ or CK1ε, respectively.
SR-3451 displays weak inhibition (>10 μM IC50) of CK1δ, but its analogs, such as SR-4051 and SR-4055 with 4-fluorophenylethyl and 4-hydroxyphenylethyl in the purine N9-position, respectively, show enhanced inhibition potency of CK1δ. Specifically, SR-4055 shows 18 nM IC50 vs. CK1δ and 30 nM IC50 against CK1ε. This elevated inhibition potency vs. CK1δ can be explained by the -F or -OH unit’s participation at the salt bridge/H-bond network in CK1δ. Although CK1ε has a hydrophobic area at the equivalent position, K38 and E52, or D149 are still present and can participate in hydrogen bond interactions with the -F or -OH unit, leading to significant inhibition of SR-4133 (Fig. 4C). SR-4052 has a 4-methylthiophenylethyl in the purine N9-position, of which sulfur has a weak ability to participate in hydrogen bond interactions, and SR-4052 has 4-fold less inhibition potency for CK1δ compared to SR-4051. Lastly, SR-4053 and SR-4054, having 3-furanyl and 3-thiophenyl units at the N9 position, have 200 and 420 nM IC50’s against CK1δ and 88 and 110 nM IC50’s vs. CK1ε, respectively. The heterocyclic ring’s involvement in the salt bridge/H-bond network of CK1δ in addition to their hydrophobic contacts with F150 and F20 of CK1ε could explain their ability to display significant inhibition of both CK1δ and CK1ε.
CONCLUSION
From the analysis of CK1δ - SR-3029, apo-CK1ε, and CK1ε – PF4800567 crystal structures, we developed potent and highly isoform selective CK1ε inhibitors, SR-4133 and its analogs, starting from the dual CK1δ/CK1ε inhibitor (SR-3029). The electrostatic discrepancy between the naphthyl unit of SR-4133 and the polar surface area of CK1δ resulted in dramatically decreased inhibition of CK1δ, as supported by the X-ray co-crystal structure of the CK1δ - SR-4133 complex. In addition, MD simulations of apo-CK1δ displayed a strong salt bridge/H-bond interaction network and a reduced pocket area where the naphthyl unit was placed in the CK1δ - SR-4133 co-crystal structure. However, the inhibition potency of SR-4133 to CK1ε was elevated due to the favorable van der Waals binding interaction of the naphthyl unit of SR-4133 with the hydrophobic surface area generated from the DFG-out conformation. ABFE calculations of SR-3029 and SR-4133 bound to CK1δ clearly showed less binding affinity of SR-4133 to CK1δ than SR-3029 mainly due to the less favorable electrostatic energy term. The analysis of all available X-ray structures of CK1δ and CK1ε suggests that CK1δ and CK1ε, although they share 97% sequence identity in their kinase domains, have different propensities for adopting DFG conformations due to their unique I55 and F55 residues. This dissimilar tendency leads to the salt bridge/H-bond network of CK1δ or a hydrophobic pocket in CK1ε, which can be utilized for the design of selective inhibitors.
A dual phosphatidylinositol 3 kinase (PI3K)δ and CK1ε inhibitor, umbralisib, is in clinical studies for lymphocytic leukemia (CLL) and non-Hodgkin lymphomas (NHL) patients.52 In addition, the dual inhibition of CK1ε and CDK4/6 is known to arrest cell cycle progression in vitro and suppress breast tumor growth in vivo.53 Due to its cellular activity and high isoform selectivity, we envisage that SR-4133 and its analogs can be used as selective chemical probes to investigate the function(s) of CK1ε compared to CK1δ in cells and, potentially, to assess the role of CK1ε in cancer and other diseases in which CK1ε has been implicated.
EXPERIMENTAL SECTION
Biochemical assays.
CK1δ inhibitor IC50 values were measured by using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. Briefly, final assay concentrations for CK1δ (Signal Chem), Ulight peptide substrate (ULight-Topo-IIa(Thr1342) peptide, Perkin Elmer) and ATP were 2 nM, 200 nM and 50 μM respectively. The reaction was performed at room temperature in a 10 μL final volume (384-well low volume plate, Greiner) containing: 50 mM HEPES, pH 7.5, 5 mM MgCl2, 0.1 mg/ml bovine serum albumin, 1 mM dl-dithiothreitol, 0.01% Triton X-100 and 5% DMSO (Sigma-Aldrich). After 10 min, the reaction was terminated by addition of 10 μL of 4 nM Eu-anti-p-Topo-IIa (Cat:TRF-0218, PerkinElmer) in Lance Detection Buffer (Cat: CR97–100, PerkinElmer). The fluorescent signal was detected using an EnVision plate reader (PerkinElmer). Ten-point dose–response curves with 3–10-fold dilutions starting from 10 μM for each compound was generated in duplicate and data fit to a four parameter logistic (GraphPad Prism 5).
Cell culture.
HT-1376, J82, TCCSUP, 5637, T24 and UM-UC-3 bladder cancer cell lines, U2OS osteosarcoma epithelial cells, MDA-MB-231 breast cancer cells, as well as immortal MCF10A breast epithelial cells, were purchased from the American Type Culture Collection and culture in DMEM (Life Technologies) with 10% Fetal bovine Serum and 1% penicillin/ streptomycin in an incubator at 37 °C and 5% CO2 except MCF10 which was cultured in the medium MEGM (Lonza). Normal bladder protein extracts were purchased from Novus Biologicals Incorporation.
Immunoblotting.
SDS-PAGE gel electrophoresis was performed using NuPAGE 4–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes by semi-dry transfer using trans-blot transfer medium (Biorad). Membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences) and incubated overnight at 4 °C with primary antibodies. After repeated washes with TBST (20 mM Tris, pH 7.6, 140 mM NaCl and 0.1% Tween-20) blots were incubated with the appropriate IRDye-conjugated secondary antibody (LI-COR Biosciences) and imaged using the LI-COR Odyssey. Bands were quantified using the Odyssey software (LI-COR Biosciences). The following antibodies from cell signaling were used in this study: CK1δ, CK1ε, GAPDH, p4EBP1 (T37/46), p4EBP1 (T65), p4EBP1 (T70) and 4EBP1 total.
Lentiviral transduction.
For CK1ε knockdown, J82 cell lines were established to stably express specific shRNA against CK1ε. shRNA oligonucleotides as described below were first cloned into the Tet-pLKO-Puro vector using the recommended protocol,3 and lentiviruses were generated using the MISSION Packaging System (Sigma). J82 cells were transduced with optimized titers of lentiviruses, and infected cells were selected in puromycin (1 µg/mL) containing medium to expand stably infected pools. Knockdown of CK1ε by immunoblot was ensured after 2 days treatment of 1 µg/mL doxycycline compare to DMSO treatment and a scrambled sequence of the shRNA (Sc).
shRNA sequences:
Sh Scramble CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT
Sh CK1e #1 CCGGGGCTCTGAGTTATAAATTACTCTCGAGAGTAATTTATAACTCAGAGCCTTTTT
Sh CK1e #2 CCGGGATTAACCCATCCTTCCTAGTCTCGAGACTAGGAAGGATGGGTTAATCTTTTT
Cell proliferation and clonogenicity assays.
To evaluate the anti-proliferative activity of newly synthesized CK1ε inhibitors against T24, 5637, UM-UC-3, and U2-OS, cells were plated into a 384-well plate at a density of 500 cells per well. 10-point dose-response curves with 3-fold dilutions starting from 10 μM for each compound was generated in triplicate including DMSO control. Cell proliferation was measured 72 hours after SR-4133 or SR-4310 or vehicle treatment using CellTiter-Glo (Promega) according to the manufacturer’s instructions. EC50 values were determined by nonlinear regression and a four-parameter algorithm (GraphPad Prism 5). For clonogenic assays of CK1ε knockdown J82, cells were plated in six-wells dishes in triplicate at a density of 1000 cells per well. After overnight incubation, 1 µg/mL doxycycline or vehicle (DMSO) was added to the medium for 72 hours, and cells were allowed to grow out for 7 to 10 days, during which medium was changed every 2 to 3 days without adding compound. Colonies were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS), stained with 0.5% methylene blue in 50% ethanol for 1 hour at room temperature, and destained with water. Colonies with more than 50 cells were counted using a low-magnification light microscope.
Structural biology.
Recombinant N-terminally-His-tagged CK1δ kinase domain co-expressed with lambda phosphatase in E. coli was initially purified by Ni2+-affinity chromatography and was treated with Tobacco Etch Virus (TEV). The cleaved protein was further purified by size exclusion chromatography in buffer 20 mM Tris, pH 7.5, 200 mM NaCl and 0.5 mM Tris(2-carboxyethyl)phosphine (TCEP), and was concentrated to 6–8 mg/mL. The protein was mixed with the inhibitor at 1 mM, and the complexes were subjected to crystallization using sitting drop vapor diffusion method at 4 °C using either conditions containing 20% PEG3350, 0.1 M succinic acid for SR-3029 or 25% PEG3350, 0.2 M sodium sulfate, 0.1 M citrate pH 5.9 for SR-4133. Diffraction data collected at Diamond Light Source were processed and scaled using XDS54 and SCALA55, respectively. Structures were solved by molecular replacement using Phaser56 and the coordinate of CK1δ (PDB code: 4HNF)26. Model rebuilding alternated with refinement were performed in COOT57 and REFMAC558, respectively.
Molecular modeling and MD simulations; Protein preparation.
The X-ray co-crystal structures of CK1δ - SR-3029, CK1δ - SR-4133, and CK1ε - PF4800567 were retrieved from the Protein Data Bank (PDB codes: 6RCG, 6RCH, and 4HNI, respectively), and they were refined with Protein Preparation Wizard implemented in Maestro 12. The protein structure was imported into workspace and preprocessed to assign bond orders, add hydrogen atoms, create zero-order bonds to metals, create disulfide bonds, and delete water molecules beyond 5 Å from hetero groups. In addition, missing atoms in residues and missing loops were added using Prime to generate a complete protein structure. The protein structure was further refined via automated H-bond assignment and restrained minimization with OPLS 2005 force field by converging heavy atoms to 0.3 Å RMSD. For the MD simulations of apo-CK1δ and apo-CK1ε, 6RCG and 4HNI structures (chain A) were used by removing ligand structures. The Ser-1 residue in the 6RCG was deleted, and its C-terminus was capped with NME. Met-1 and Glu-2 were added to the N-terminus of 4HNI, and its C-terminus was capped with NME. Based on the protein preparation of the 6RCG structure in Maestro 12, His46, 162, 164, and 278 were protonated at the delta position, and His50, 12, 126, and 185 were protonated at the epsilon position. In the same way, 4HNI structure had His50, 162, 164, and 278 as delta protonated forms and His46, 120, 126, and 185 as epsilon protonated forms. Both 6RCG and 4HNI structures including Lys232 had a neutral form due to a neighboring His185. Glu52 had a neutral form in the 4HNI structure due to the presence of an inhibitor, while it is a charged form in the 6RCG structure. Thus, the charged form for Glu52 was used in both 4HNI and 6RCG structures. No disulfide bond or selenomethionine residue was identified in both 4HNI and 6RCG structures. The 6RCG and 4HNI structures were cleaned according to the protonation states and prepared for building protein systems in explicit solvent.
System buildup and MD simulations of apo-CK1δ and apo-CK1ε using Amber 20.
The cleaned apo-CK1δ and apo-CK1ε structures were solvated with the Amber ff14SB force filed and TIP3P explicit water model in a periodic box using a buffer distance of 12.0 Å containing 150 mM NaCl in 14,064 and 14,248 water molecules, respectively. The solvated protein structures were treated to 5,000 steps of steepest descent and 5,000 steps of conjugate minimizations. After the minimization, the systems were heated from 0 K to 303.15 K for 400 ps and equilibrated at 303.15 K for 100 ps under constant volume, with a timestep of 2 fs, and with SHAKE algorithm employed to restrain the calculation of forces of bonds containing hydrogen atoms. The system was further equilibrated at 303.15 K for 5 ns under constant pressure with a Berendsen barostat and Langevin thermostat. After the equilibration, the system was simulated in the production step at 303.15 K for 100 ns under the same conditions.
Analysis of MD simulation results.
The simulation data of apo-CK1δ and apo-CK1ε were analyzed with CPPTRAJ by reading and combining multiple trajectory files obtained from the MD simulations.34–35 The protein structures in the trajectories were re-oriented by using the autoimage command, and water molecules, Na, and Cl ions were striped. RMS fit of all residues were performed, and a new NetCDF trajectory was saved for further analysis. For the rmsd analysis, the rms command was used, and the amide backbones, Cα, and Cβ without hydrogen of CK1δ and CK1ε amino acids were selected for the analyses of rmsd to the first frame during the simulations. The potential energies of the system were recorded at every 10 ps and plotted for analysis. The representative conformations of apo-CK1δ and apo-CK1ε structures during the 100 ns simulations were extracted from clustering 10,000 snapshots obtained from MD simulations. Hierarchical agglomerative (bottom-up) approach was used with the options of “epsilon 2.0 clusters 10 averagelinkage” to finish clustering when a minimum distance between clusters is greater than 2 or when 10 clusters remain. The average-linkage option that uses the average distance between members of two clusters was selected. The representative structures from 100 ns MD simulations and the 6RCH structure (CK1δ - SR-4133 complex) were aligned to the 6RCG structure (CK1δ - SR-3029 complex) for the analysis of the binding pockets (Fig. 4D and 4E). Residues within 4 Å from the outer ring of the SR-4133 naphthyl unit were selected and nonpolar hydrogen atoms were removed for the calculation of APBS electrostatics implemented in Pymol. A range of +/− 4.0 was used for the projection of the electrostatic potential onto the molecular surface (Fig. 4F). Hydrogen bond analysis was performed with 10,000 frames (or structures) generated from 100 ns MD simulations. CPPTRAJ trajectory analysis software, particularly hbond command, was used to analyze hydrogen bonds in 10,000 frames. The number of hydrogen bonds within solute (solute – solute), between solute and solvent (solute – solvent), and in bridges (solute – solvent – solute) was identified and counted (Table 2).
Molecular Docking.
AutoDock-GPU was used to get the docked structures of SR-4133 to CK1ε.30 The cleaned CK1ε structure (pdb code: 4HNI) and the SR-4133 structure (pdb code: K0B) from CK1δ - SR-4133 complex (pdb code: 6RCH) were imported into AutoDockTools (ADT) to generate the coordinates (pdbqt files) of the receptor and ligand for docking studies. The grid box was centered on the binding pocket of the receptor, and it was resized by increasing the number of grid points in xyz (46/46/46) to generate a gpf file (grid parameter file). The affinity map files were generated by using autogrid4 with the gpf file. Then AutoDock-GPU was run with default options to perform docking of SR-4133, and only one unique binding pose was obtained after clustering 20 docking poses. The docking was repeated by increasing the number of runs to 50, and after clustering 50 docking poses, only two were selected as unique poses. Thus, additional 8 poses were selected manually, and 11 docking poses were used for MD simulations. In the case of docking of SR-4133 to CK1ε (DFG-in, pdb code: 4HOK), seven unique poses were generated from clustering 20 docking poses, and 8 poses (including the original pose of SR-4133 from CK1δ) were used for MD simulations.
MD simulations and MM/GBSA calculations of CK1ε - SR-4133 complexes.
The parameter modification (frcmod) files for the ligand (SR-4133) were generated by using antechamber (GAFF2 force field and AM1-BCC charges) and parmchk2 commands. The tleap command was used to form the receptor – ligand complex and solvate the system with the Amber ff14SB force field and TIP3P explicit water model in the periodic box using a buffer distance of 12Å containing 150 mM NaCl in water molecules. After the topology and coordinate files were generated by tleap, the structure, particularly the binding orientation of SR-4133, was visually inspected in VMD.59
The minimization of the system was conducted by three consecutive steps such as 1000 steps of steepest descent and 1000 steps of conjugate gradient minimization of water molecules only, same minimization steps for solute only, and 2500 steps of steepest descent and 2500 steps of conjugate gradient minimization for the whole system, respectively. After the minimization, the system was heated from 0 K to 298.15 K for 150 ps and equilibrated at the same temperature for 50 ps under constant volume, with a timestep of 2 fs, and with SHAKE algorithm employed to restrain of the calculation of forces of bonds containing hydrogen atoms. The system was further equilibrated at 298.15 K for 50 ps (5 times of 10 ps run) under constant pressure with a Berendsen barostat and Langevin thermostat. After the equilibration, the system was simulated in the production step at 298.15 K for 100 ns (10 times of 10 ns run) under the NPT condition.
All of the MM/GBSA energies were calculated based on snapshots (frames) extracted from MD trajectories obtained from the 100 ns simulations. Around 1000 snapshots in the last 10 ns simulation of each ligand were used for calculations, and the saltcon (the concentration of mobile counterions in solution) was set to 0.150. The ΔGTotal for each ligand was obtained from the result data file for the analysis.
After MM/GBSA energy calculations, two systems (pose 2 and pose 11) were subjected to extended MD runs up to 1 μs each. The simulation data were analyzed with CPPTRAJ by reading and combining multiple trajectory files obtained from the MD simulations. The protein structures in the trajectories were re-oriented by using the autoimage command, and water molecules, Na, and Cl ions were striped. RMS fit of all residues were performed, and a new NetCDF trajectory was saved for further analysis. For the Root Mean Square Distance (rmsd) analysis, the rms command was used, and the amide backbones, Cα, and Cβ without hydrogen of all CK1ε amino acids were selected for the analyses of rmsd. The CK1ε - SR-4133 complex structure with the lowest potential energy was selected from 200,000 snapshots for further analysis.
Absolute Binding Free Energy Calculations.
The BAT.py v2.1 script compatible with Amber20 was used to build the system for MD simulations and calculate the absolute binding free energies of SR-3029 and SR-4133 to CK1δ. The simultaneous decoupling/recoupling (SDR) method was applied to pull out the inhibitor from its binding site of CK1δ via nonphysical alchemical pathways. System buildup for the MD simulations with the SDR method. X-ray co-crystal structures of SR-3029 (pdb code: 6RCG) and SR-4133 (pdb code: 6RCH) were used for MD simulations. and ligand structures were extracted and refined with Protein Preparation Wizard implemented in Maestro 12. These ligand structures with hydrogen atoms were used for system building to keep the ligand protonation by adding the retain_lig_prot option. The pdb files for target enzymes were cleaned by deleting all description text, ions, ligands, and water molecules, and the 6RCG was set as a reference structure after the alignment to its principal axes using VMD. The carbon atoms of the amide backbones of Lys38, Ile51, and Met58 were assigned to the protein anchors, P1, P2, and P3, respectively, and C10 (the pdb atom type of the ligand) was assigned as L1 by setting up the ligand anchor search definitions: l1_x = 0.01, l1_y = 7.15, l1_z = 7.63, and l1_range = 2.50. L2 and L3 ligand anchor atoms were automatically assigned by the BAT.py software. Protein backbone dihedral restraints were applied between residues 37 and 59. Amber Force Field, ff14SB, was used to parametrize the protein atoms, and ligand parameters were assigned with the AM1-BCC charge model and General Amber Force Field (GAFF2). TIP3P water model was used to solvate the protein-ligand complex, and the system was neutralized with Na+ and Cl− ions with 0.15 M salt concentration. The z-distance between the last and the initial positions of the bound ligand in the SDR method was set to 40 Å, which is sufficient to provide negligible interactions between the ligand and the protein.
Simulation procedures for equilibration.
Each solvated protein – ligand complex was minimized with the protein restraints. The system was simulated at 10 K for 2 ps and then heated to 298.15 K over 100 ps using a Langevin thermostat with a collision frequency of 1.0 ps−1 at constant volume. The system was then pressurized to 1 bar with the Monte Carlo barostat by multiple short NPT simulations (5 X 14 ps). A series of molecular dynamics simulations (8 X 20 ns) was run with a sequential decrease of translational and rotational (TR) ligand restraints but without its conformational restraints. The final simulation for equilibration was conducted without any ligand restraints for 60 ns. During the above simulations for equilibration, the protein TR restraints and the distance restraints among the P1, P2, and P3 anchors remained. Once the equilibration step was complete, a new set of ligand restraints were reassigned based on the final equilibrated structure, and the simulation box was rebuilt for MD simulations for free energy calculation.
Simulation procedures for free energy calculation.
The new system obtained from the equilibration step was heated from 50 K to 298.15 K over 50 ps and equilibrated for 14 ps (5X), and the production simulation was performed for 4 ns to collect data for free energy calculation. The heating, equilibration, and production steps were simulated in 12 windows of each component to calculate the simultaneous dec/recoupling of ligand charge interactions and simultaneous dec/recoupling of ligand Lennard-Jones (LJ) interactions . Attachments of receptor conformational restraints , ligand conformation restraints , and ligand TR restraints were calculated from the 4 ns production simulations of the complex system in 10 windows for simultaneous decoupling and recoupling method. In addition, releases of ligand conformational restraint and receptor conformational restraints were calculated from the 4 ns production simulations of the complex system in 10 windows. The final absolute binding free energy was calculated by the analysis of the simulations using the Thermodynamic Integration with Gaussian Quadrature (TI-GQ), which is defined as a sum of all components:. The total simulation time for the production steps was 176 ns for each ligand, and it took 83.16 wall clock hours with a single Nvidia GTX 1080Ti GPU.
Chemistry, General Experimental Details.
Commercially available reagents were used without further purification. Tetrahydrofuran and dichloromethane were purified by passing through a solvent column composed of activated A-1 alumina. Anhydrous acetone was purchased from Aldrich Chemical Company. Triethylamine and diisopropylethylamine were purified by distillation from calcium hydride. Unless indicated otherwise, all reactions were conducted under an atmosphere of argon using flame-dried or oven-dried (140 °C) glassware. The term “concentrated under reduced pressure” refers to the removal of solvents and other volatile materials using a rotary evaporator with the water bath temperature below 40 °C, followed by the removal of residual solvent at high vacuum (< 0.2 mbar). Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a commercial instrument at 400 and 600 MHz. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded at 100 MHz, 150 MHz, and 175 MHz. The proton signal for residual non-deuterated solvent (δ 7.26 for CHCl3, 2.50 for dimethyl sulfoxide) was used as an internal reference for 1H NMR spectra. For 13C NMR spectra, chemical shifts are reported relative to the δ 77.1 resonance of CHCl3 and δ 39.52 resonance of dimethyl sulfoxide. Coupling constants are reported in Hz. Infrared (IR) spectra were recorded as films on a commercial FTIR instrument. High resolution mass spectra were recorded on a commercial high resolution mass spectrometer. Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. The TLC plates were visualized with UV light and/or by staining with KMnO4. Column chromatography was generally performed using Kieselgel 60 (230–400 mesh) silica gel, typically using a 50–100:1 weight ratio of silica gel to crude product. All compounds are >95% pure by HPLC analysis.
General procedure A for Preparation of iodonium tetrafluoroborate
To a solution of the 1-naphthalene boronic acid (6.28 mmol) in CH2Cl2 (100 mL) was added BF3•OEt2 (6.93 mmol) at 0 °C. The reaction mixture stirred for 10 min before addition of a solution of 2-iodo-1,3,5-triisopropylbenzene diacetate (6.71 mmol) in CH2Cl2 (20 mL) dropwise over 10 minutes. The reaction was allowed to warm to 25 °C over the course of 2 h and then 100 mL saturated aq. NaBF4 solution was added with rapid stirring and the stirring continued for 30 minutes. After this time the phases were separated, the aqueous layer extracted twice with CH2Cl2. The combined organics dried over NaSO4 and passed through a short silica pad (30 g), eluted with CH2Cl2 (300 mL) to remove byproduct, followed by CH2Cl2/MeOH (300 mL, 20:1), to elute the product. The iodonium tetrafluoroborate was precipitated from Et2O. The solid was filtered, washed with Et2O and dried under vacuum.
General procedure B – Chan-Lam Boronic Acid Coupling
To a solution of 2,6-dichloropurine (3, 1 eq.) in CH2Cl2 (0.25 M) was added arylboronic acid (2 eq.), triethylamine (3 eq.), and copper acetate (2 eq.). The reaction mixture was refluxed at 50 °C for 1 h, then cooled to 25 °C and filtered over celite washing 3 times with CH2Cl2. The filtrate was dry-loaded onto a silica column and purified by flash chromatography to give the coupled product.
General procedure C for the Preparation of final compounds.
A 2–5 mL Biotage microwave vial was charged with 2,6-dichloropurine analogs (1.0 mmol), 2-(methylamino)-4,5-difluorobenzimidazole • HCl (1.1 mmol), N,N-diisopropylethylamine (5.0 mmol,), and isopropanol (2.5 mL). The vial was resealed and the reaction was heated to 90 °C for 30 minutes in the microwave unit. The cooled vial was then placed on a rotary evaporator and the reaction mixture was concentrated. Morpholine (2.5 mL) was added to the vial containing the concentrated crude mixture. The vial was resealed and the reaction was heated to 130 °C for 30 minutes in the microwave unit. The cooled reaction mixture was concentrated on a rotary evaporator and then the crude product was purified by flash chromatography on silica gel to give the desired compound. Then, 0.1 M TFA aq. (1.0 mmol) was added to the product (1.0 mmol) and sonicated for 30 min. Followed by freeze under –78 °C, dried using a lyophilizer.
General procedure D for Preparation of the final compounds
A Biotage microwave vial was charged with N9 substituted 2,6-dichloropurine (1 eq.), 2-(methylamino)-4,5-difluorobenzimidazole • HCl (1.1 eq.), N,N-diisopropylethylamine (5 eq), and isopropanol (0.4 M). The vial was sealed and the reaction was heated at 90 °C for 30 minutes in the microwave unit. The cooled vial was then placed on a rotary evaporator and the reaction mixture was concentrated. Morpholine (0.4 M) was added to a microwave vial containing the concentrated crude mixture. The vial was sealed and the reaction was heated at 130 °C for 30 min in the microwave unit. The cooled reaction mixture was concentrated on a rotary evaporator and then the crude product was purified by flash chromatography or HPLC to give the final product.
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(naphthalen-1-yl)-9Hpurin-6-amine (SR-4132).
Synthesized according to general procedure from 2,6-dichloro-9-(naphthlen-1-yl)-9H-purine in 53% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.37 (brs, 1H), 8.13 (s, 1H), 8.11 (dd, J = 14.0, 8.2 Hz), 7.69 (app t, J = 7.6 Hz), 7.64–7.60 (m, 2H), 7.57–7.53 m, 1H), 7.39 (d, J = 8.4 Hz), 7.31–7.20 (m, 2H), 4.89 (s, 2H), 3.34 (s, 4H), 3.29 (s, 4H); 13C NMR (175 MHz, DMSO-d6, δ): 158.6, 158.3, (q, J = 36.4 Hz), 156.1, 154.1, 152.5. 146.0 (d, J = 9.5 Hz), 144.6 (q, J = 9.3 Hz), 139.6 (d, J = 15.2 Hz), 139.2, 138.2 (d, J = 15.2 Hz), 133.8, 133.5, 131.2, 129.3 (d, J = 7.4 Hz), 128.3, 127.2, 126.8, 125.7, 125.4, 122.7, 115.7 (q, J = 289.6 Hz), 112.9, 111.5 (d, J = 20.8 Hz), 108.4, 65.7 (x2), 44.3 (x2).
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(naphthalen-2-yl)-9Hpurin-6-amine (SR-4133).
Synthesized according to general procedure from 2,6-dichloro-9-(naphthlen-2-yl)-9H-purine in 74% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.48 (s, 1H), 8.46 (s, 1H), 8.33 (brs,1H), 8.13–8.06 (m, 2H), 8.00–7.99 (m, 2H), 7.62–7.55 (m, 2H), 7.29–7.19 (m, 2H), 4.87 (s, 2H), 3.52 (s, 4H), 3.49 (s, 4H); 13C NMR (175 MHz, DMSO-d6, δ): 158.6, 158.3 (q, J = 35.2 Hz), 156.0, 154.1, 150.9, 146.0 (d, J = 9.5 Hz), 144.6 (d, J = 9.3 Hz), 139.5 (d, J = 14.4 Hz), 138.1 (d, J = 14.4 Hz), 137.2, 133.4, 133.1, 133.0, 131.4, 129.2, 127.9, 127.7, 127.0, 126.4, 121.1, 119.8, 115.6 (q, J = 289.4 Hz), 113.9, 111.7 (d, J = 21.0 Hz), 108.4, 65.9, 44.5; HRMS (ESI-TOF) calculated for C27H23N8OF2 (M+H)+ 513.1963, found 513.1971.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(4-methoxyphenyl)-2-morpholino-9H-purin-6-amine (SR-4310).
Synthesized according to general procedure from 2,6-dichloro-9-(4-methoxyphenyl)-9H-purine in 72% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ) 8.28 (s, 1H), 7.84–7.65 (m, 2H), 7.35–7.17 (m, 2H), 7.13–7.05 (m, 2H), 4.87 (s, 2H), 3.46 (m, 8H). HRMS (ESI-TOF) calculated for C24H23N8O2F2 (M+H)+ 493.1912, found 493.1930.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(4-ethoxyphenyl)-2-morpholino-9H-purin-6-amine (SR-19839).
Synthesized according to the general procedure B followed by the general procedure D (100 °C, oil bath heating, for 18 h in step 2). The product was isolated as a white solid (45 mg, 70% yield from 4c, 12% yield from 3 to 4c). 1H NMR (400 MHz, DMSO-d6, δ): 8.25 (s, 2H), 7.85 – 7.63 (m, 2H), 7.37 – 7.18 (m, 2H), 7.15 – 7.04 (m, 2H), 4.88 (s, 2H), 4.07 (q, J = 7.0 Hz, 2H), 3.47 (s, 8H), 1.34 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, DMSO-d6, δ): 158.56, 157.32, 156.01, 154.07, 150.76, 144.16, 143.93, 137.20, 133.72, 128.31, 123.94, 115.03, 113.57, 111.41, 111.20, 65.88, 63.40, 44.51, 14.64; HRMS (ESI-TOF) calculated for C25H25N8O2F2 (M+H)+ 507.2069, found 507.2091.
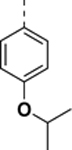
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(4-isopropoxyphenyl)-2-morpholino-9H-purin-6-amine (SR-19840).
Synthesized according to the general procedure B followed by the general procedure D (100 °C, oil bath heating, for 18 h in step 2). The product was isolated as a white solid (63 mg, 77% yield from 4d, 11% yield from 3 to 4d). 1H NMR (400 MHz, DMSO-d6, δ): 8.28 (d, J = 14.3 Hz, 2H), 7.76 – 7.64 (m, 2H), 7.34 – 7.15 (m, 2H), 7.12 – 7.00 (m, 2H), 4.90 (s, 2H), 4.66 (hept, J = 6.0 Hz, 1H), 3.47 (s, 8H), 1.28 (d, J = 6.0 Hz, 6H); 13C NMR (151 MHz, DMSO-d6, δ): 158.55, 156.27, 156.00, 154.06, 150.68, 145.18 (dd, J = 234.8, 9.3 Hz), 139.00 (dd, J = 250.4, 14.9 Hz), 137.19, 133.74, 129.47, 128.15, 123.98, 116.12, 113.54, 111.28 (d, J = 21.2 Hz), 108.19, 69.54, 65.87, 44.48, 38.41, 21.77; HRMS (ESI-TOF) calculated for C26H27N8O2F2 (M+H)+ 521.2225, found 521.2206.
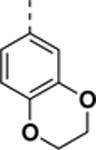
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-morpholino-9H-purin-6-amine (SR-19841).
Synthesized according to the general procedure B followed by the general procedure D (100 °C, oil bath heating, for 18 h in step 2). 1H NMR (400 MHz, DMSO-d6, δ): 8.29 (s, 2H), 7.42 (d, J = 2.6 Hz, 1H), 7.36 – 7.21 (m, 3H), 7.01 (d, J = 8.7 Hz, 1H), 4.89 (s, 2H), 4.29 (s, 4H), 3.46 (s, 8H); 13C NMR (151 MHz, DMSO-d6, δ): 170.04, 158.98, 156.47, 154.49, 151.05, 150.19, 146.83 – 144.78 (m), 144.01, 143.78, 143.14, 142.78, 137.61, 133.63, 131.01, 130.11, 129.35, 127.75, 127.26, 124.31, 123.40, 117.98, 115.69, 114.08, 111.84, 108.77, 66.32, 64.70, 64.55, 44.95, 38.84; HRMS (ESI-TOF) calculated for C25H23N8O3F2 (M+H)+ 521.1861, found 521.1852.
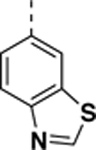
9-(benzo[d]thiazol-6-yl)-N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9H-purin-6-amine (SR-4932).
Synthesized according to the general procedure E with 6-bromobenzo[d]thiazole. 1H NMR (600 MHz, DMSO-d6, δ): 9.51 (s, 1H), 8.64 (d, J = 2.1 Hz, 1H), 8.47 (s, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.32 – 8.24 (m, 1H), 8.06 (dd, J = 8.7, 2.1 Hz, 1H), 7.25 (dd, J = 8.9, 3.8 Hz, 1H), 7.19 (ddd, J = 11.4, 8.7, 7.1 Hz, 1H), 4.95 – 4.79 (m, 2H), 3.50 (d, J = 26.7 Hz, 8H); 13C NMR (151 MHz, DMSO-d6, δ): 174.27, 159.13, 158.78, 156.43, 155.14, 154.79, 154.01, 151.27, 145.50 (dd, J = 234.2, 9.8 Hz), 137.69, 134.57, 132.31, 130.08, 123.81, 120.51, 116.73, 114.57, 111.35 (d, J = 21.3 Hz), 108.29 (d, J = 23.5 Hz), 66.35, 44.99, 39.06; HRMS (ESI-TOF) calculated for C24H20N9OF2S (M+H)+ 520.1480, found 520.1479.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(quinolin-3-yl)-9H-purin-6-amine (SR-4471).
Synthesized according to general procedure C from 3-(2,6-dichloro-9H-purin-9-yl)quinoline (white solid, 120 mg, 67% yield). 1H NMR (400 MHz, DMSO-d6, δ): 9.49 (d, J = 2.5 Hz, 1H), 8.87 (d, J = 2.5 Hz, 1H), 8.54 (s, 1H), 8.40 (s, 1H), 8.09 (td, J = 7.4, 6.6, 1.2 Hz, 2H), 7.83 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.71 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H), 7.32 – 7.17 (m, 2H), 4.88 (s, 2H), 3.50 (d, J = 16.9 Hz, 8H); 13C NMR (151 MHz, DMSO-d, δ): 159.25, 159.14, 159.01, 158.77, 156.43, 154.65, 151.33, 146.53 (d, J = 9.5 Hz), 146.10, 145.73, 139.36 (d, J = 251.3 Hz), 137.25, 133.89, 130.32, 129.13, 128.75, 128.17, 127.89, 117.19, 115.26, 114.27, 112.07 (d, J = 20.3 Hz), 108.83, 66.32, 44.92, 38.81; HRMS (ESI-TOF) calculated for C26H22N9OF2 (M+H)+ 514.1915, found 514.1898.
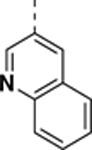
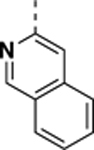
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(isoquinolin-3-yl)-2-morpholino-9H-purin-6-amine (SR-4931).
Synthesized according to the general procedure E with 3-bromoisoquinoline. 1H NMR (600 MHz, DMSO-d6, δ): 9.35 (s, 1H), 8.90 (s, 1H), 8.75 (s, 1H), 8.34 (s, 1H), 8.20 (d, J = 8.2 Hz, 1H), 8.18 – 8.09 (m, 1H), 7.88 – 7.81 (m, 1H), 7.69 (dd, J = 8.2, 6.9 Hz, 1H), 7.26 (dd, J = 8.8, 3.8 Hz, 1H), 7.20 (ddd, J = 11.1, 8.7, 7.0 Hz, 1H), 4.98 – 4.75 (m, 2H), 3.62 (s, 8H); 13C NMR (151 MHz, DMSO-d6, δ): 159.17, 158.95, 158.72, 158.48, 156.39, 154.72, 152.60, 152.16, 150.78, 145.56 (dd, J = 234.0, 9.6 Hz), 144.16, 137.35, 136.05, 132.05, 131.51, 128.42, 127.69, 127.47, 126.52, 115.10, 111.44, 109.45, 108.43, 107.01, 66.46, 45.11, 39.02; HRMS (ESI-TOF) calculated for C26H22N9OF2 (M+H)+ 514.1915, found 514.1916.
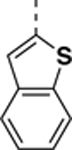
9-(benzo[b]thiophen-2-yl)-N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9H-purin-6-amine (SR-4933).
Synthesized according to the general procedure E with 2-bromobenzo[b]thiophene. 1H NMR (400 MHz, DMSO-d6, δ): 8.53 (d, J = 9.5 Hz, 1H), 8.42 (s, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.83 (d, J = 6.7 Hz, 2H), 7.38 (dt, J = 23.7, 7.4 Hz, 2H), 7.24 (ddt, J = 20.0, 11.3, 6.2 Hz, 2H), 4.89 (s, 2H), 3.55 (d, J = 32.1 Hz, 8H); 13C NMR (151 MHz, DMSO-d6, δ): 159.10, 156.32, 154.69, 150.58, 145.61 (dd, J = 234.3, 9.8 Hz), 139.57 (dd, J = 249.1, 13.9 Hz), 137.66, 136.60, 136.40, 136.31, 134.38, 130.27, 125.54, 124.86 (d, J = 2.9 Hz), 123.77, 122.87, 117.28, 115.34, 113.69 (d, J = 22.6 Hz), 112.24, 111.65 (d, J = 20.9 Hz), 108.60, 66.36, 45.01, 38.99; HRMS (ESI-TOF) calculated for C25H21N8OF2S (M+H)+ 519.1527, found 519.1526.
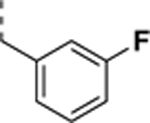
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(3-fluorobenzyl)-2-morpholino-9Hpurin-6-amine trifluoroacetate salt (SR-3448).
Synthesized according to general procedure from 2,6-dichloro-9-(3-fluorobenzyl)-9H-purine in 49% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.37 (d, J = 9.2 Hz, 1H), 8.28 (brs, 1H), 8.20 (brs, 1H), 7.97 (m, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.57 (m, 2H), 7.49 (app t, J = 7.4 Hz, 1H), 7.38 (d, J = 6.8 Hz, 1H), 7.30–7.21 (m, 2H), 5.76 (s, 2H), 4.86 (brs, 2H), 3.51 (brs, 4H), 3.46 (brs, 4H); 13C NMR (175 MHz, DMSO-d6, δ): 162.1 (d, J = 242.2 Hz), 158.5, 158.4 (q, J = 36.9 Hz), 155.7, 153.0, 150.7, 146.1 (d, J = 8.8 Hz), 144.8 (d, J = 9.3 Hz), 139.3, 138.0, 133.1, 130.7 (d, J = 8.6 Hz), 128.1, 124.1 (d, J = 2.1 Hz), 115.9 (q, J = 288.7 Hz), 114.9 (d, J = 21.7 Hz), 114.7 (d, J = 21.0 Hz), 112.1 (d, J = 21.0 Hz), 110.4, 108.7, 65.8 (x2), 45.8, 44.5 (x2), 38.3.
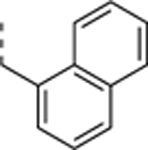
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(naphthalen-1-ylmethyl)-9Hpurin-6-amine trifluoroacetate salt (SR-3449).
Synthesized according to general procedure from 2,6-dichloro-9-(naphthalen-1-ylmethyl)-9H-purine in 57% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.30 (brs, 1H), 8.25 (s, 1H), 7.39 (m, 2H), 7.31–7.10 (m, 7H), 5.29 (s, 2H), 4.86 (s, 2H), 3.49 (brs, 4H), 3.46 (brs, 4H); 13C NMR (175 MHz, DMSO-d6, δ): 158.5, 158.4 (q, J = 36.9 Hz), 155.7, 153.8, 150.9, 146.1 (d, J = 9.5 Hz), 144.7 (d, J = 9.5 Hz), 139.5 (d, J = 15.3 Hz), 138.3, 138.0 (d, J = 15.2 Hz), 133.3, 132.1, 130.5, 128.6 (d, J = 4.2 Hz), 128.5, 126.7 (d, J = 5.1 Hz), 126.1, 125.5, 116.0 (q, J = 288.9 Hz), 111.9 (d, J = 20.8 Hz), 110.7, 108.6, 65.8 (x2), 44.5 (x2), 44.0, 38.3.
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(naphthalen-2-ylmethyl)-9Hpurin-6-amine trifluoroacetate salt (SR-3450).
Synthesized according to general procedure from 2,6-dichloro-9-(naphthalen-2-ylmethyl)-9H-purine in 58% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.23 (brs, 2H), 7.90–7.85 (m, 4H), 7.53–7.48 (m, 3H), 7.27–7.18 (m, 2H), 5.43 (s, 2H), 4.84 (s, 2H), 3.52 (brs, 4H), 3.46 (brs, 4H); 13C NMR (175 MHz, DMSO-d6, δ): 158.5, 158.4 (q, J = 36.9 Hz), 155.7, 153.2, 150.9, 145.9 (d, J = 9.5 Hz), 144.6 (d, J = 9.5 Hz), 139.6 (d, J = 13.7 Hz), 138.2, 134.2, 133.5, 132.7, 132.4, 128.3, 127.7, 127.6, 126.8, 126.5, 126.3, 126.0, 115.7 (q, J = 289.6 Hz), 111.5 (d, J = 20.8 Hz), 111.1, 108.4, 65.8 (x2), 46.5, 44.5 (x2), 38.4.
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(3-fluorophenethyl)-2-morpholino-9Hpurin-6-amine trifluoroacetate salt (SR-3451).
Synthesized according to general procedure from 2,6-dichloro-9-(3-fluorophenethyl)-9H-purine in 72% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.26 (brs, 1H), 8.07 (s, 1H), 7.32–7.21 (m, 3H), 7.08–7.00 (m, 2H), 6.96 (d, J = 7.6 Hz, 1H), 4.86 (s, 2H), 4.35 (t, J = 7.0 Hz, 2H), 3.52 (s, 4H), 3.50 (s, 4H), 3.15 (t, J = 7.0 Hz, 2H); 13C NMR (175 MHz, DMSO-d6, δ): 162.6 (d, J = 241.3 Hz), 158.9, 158.8 (q, J = 36.4 Hz), 156.0, 153.0, 151.1, 146.4 (d, J = 9.5 Hz), 145.1 (d, J = 9.3 Hz), 141.2 (d, J = 7.2 Hz), 140.0 (d, J = 15.2 Hz), 138.6 (d, J = 13.7 Hz), 138.4, 134.0, 130.7 (d, J = 8.6 Hz), 129.3, 125.3, 116.0 (q, J = 289.4 Hz), 115.9 (d, J = 20.8 Hz), 113.8 (d, J = 20.3 Hz), 112.1 (d, J = 20.1 Hz), 110.0, 109.0, 66.3 (x2), 44.9 (x2), 44.6, 38.8, 34.8; HRMS (ESI-TOF) calculated for C25H24N8OF3 (M+H)+ 509.2025, found 509.2022.
N-((4,5-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(3,5-difluorophenethyl)-2-morpholino-9Hpurin-6-amine (SR-4290).
Synthesized according to general procedure from 2,6-dichloro-9-(3,5-difluorophenethyl)-9H-purine in 74% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.15 (brs, 1H), 7.91 (s, 1H), 7.28–7.18 (m, 2H), 7.08–7.03 (m, 1H), 6.95–6.91 (m, 1H), 4.83 (s, 2H), 4.33 (t, J = 7.0 Hz), 3.51 (brs, 8H), 3.15 (t, J = 7.0 Hz); 13C NMR (100 MHz, DMSO-d6, δ): 162.3 (d, J = 244.3 Hz), 162.2 (t, J = 244.3 Hz), 158.5 (q, J = 35.4 Hz), 158.4, 155.7, 153.0, 150.8, 145.9 (d, J = 9.5 Hz),.144.5 (d, J = 9.5 Hz), 142.6 (t, J = 9.0 Hz), 139.7 (d, J = 15.2 Hz), 138.3 (d, J = 15.2 Hz), 138.1, 133.8, 129.4, 115.9 (d, J = 290.4 Hz), 112.0 (d, J = 4.4 Hz), 111.9 (d, J = 4.4 Hz), 110.8, 108.3, 102.0 (t, J = 25.3 Hz), 65.9 (x2), 44.5 (x2), 43.5, 38.4.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(3-methoxyphenethyl)-2-morpholino-9H-purin-6-amine (SR-3452).
Synthesized according to general procedure D from 6b (heating in oil bath for 60 min) in 70% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.20 (s, 1H), 7.97 (s, 1H), 7.32 – 7.11 (m, 3H), 6.80 – 6.67 (m, 3H), 4.84 (s, 2H), 4.31 (t, J = 7.1 Hz, 2H), 3.51 (m, 8H), 3.69 (s, 3H), 3.09 (t, J = 7.1 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 159.76, 158.84 (d, J = 36.0 Hz), 156.14, 152.29 (d, J = 327.7 Hz), 145.68 (d, J = 234.8 Hz), 140.38, 139.93, 138.55, 134.30, 129.95, 121.36, 117.27, 115.33, 114.71, 112.46, 111.83 (d, J = 20.9 Hz), 108.80, 66.33, 55.33, 44.95, 44.67, 38.90, 35.17; HRMS (ESI-TOF) calculated for C26H27N8O2F2 (M+H)+ 521.2225, found 521.2244.
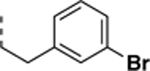
9-(3-bromophenethyl)-N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9H-purin-6-amine (SR-3454).
Synthesized according to general procedure D from 6c (heating in oil bath for 60 min) in 63% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.20 (s, 1H), 7.99 (s, 1H), 7.46 (t, J = 1.8 Hz, 1H), 7.39 (ddd, J = 7.9, 2.1, 1.1 Hz, 1H), 7.32 – 7.16 (m, 3H), 7.11 (dt, J = 7.6, 1.3 Hz, 1H), 4.84 (s, 2H), 4.32 (t, J = 7.0 Hz, 2H), 3.50 (s, 8H), 3.12 (t, J = 7.0 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 159.76, 159.20, 158.96, 158.72, 156.14, 152.29 (d, J = 327.7 Hz), 145.68 (d, J = 234.8 Hz), 140.38, 139.93, 138.55, 134.30, 129.95, 121.36, 117.27, 115.33, 114.71, 112.46, 111.83 (d, J = 20.9 Hz), 108.80, 66.33, 55.33, 44.95, 44.67, 38.90, 35.17; HRMS (ESI-TOF) calculated for C25H24N8OF2Br (M+H)+ 569.1225, found 569.1239.
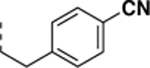
4-(2-(6-(((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)amino)-2-morpholino-9H-purin-9-yl)ethyl)benzonitrile (SR-4114).
Synthesized according to general procedure D from 6d in 60% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.16 (s, 1H), 7.91 (s, 1H), 7.72 (d, J = 8.3 Hz, 2H), 7.38 – 7.31 (m, 2H), 7.31 – 7.16 (m, 2H), 4.83 (s, 2H), 4.34 (t, J = 6.8 Hz, 2H), 3.49 (s, 8H), 3.21 (t, J = 6.9 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 158.74, 158.50, 158.30 (d, J = 9.9 Hz), 158.03, 155.76, 153.09, 150.85, 145.18 (dd, J = 234.4, 9.8 Hz), 144.18, 138.11, 133.84, 132.29, 129.90, 118.87, 116.86, 114.92, 111.32 (d, J = 20.8 Hz), 109.39, 108.28, 65.89, 44.46, 43.65, 38.42, 34.92; HRMS (ESI-TOF) calculated for C26H24N9OF2 (M+H)+ 516.2072, found 516.2065.
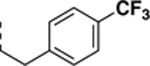
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(4-(trifluoromethyl)phenethyl)-9H-purin-6-amine (SR-4116).
Synthesized according to general procedure D from 6e in 75% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.15 (s, 1H), 7.90 (s, 1H), 7.62 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.31 – 7.15 (m, 2H), 4.82 (s, 2H), 4.34 (t, J = 7.0 Hz, 2H), 3.49 (s, 8H), 3.22 (t, J = 7.0 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 158.66, 158.43, 155.78, 153.10, 150.86, 145.18 (dd, J = 234.6, 9.4 Hz), 143.05, 138.15, 133.86, 129.61, 127.61 – 126.76 (m), 125.24 (d, J = 3.7 Hz), 123.45, 121.65, 117.03, 115.09, 114.30, 113.15, 111.29 (d, J = 21.1 Hz), 108.27, 65.88, 44.48, 43.85, 38.45, 34.65; HRMS (ESI-TOF) calculated for C26H24N8OF5 (M+H)+ 559.1993, found 559.2005.
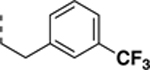
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(3-(trifluoromethyl)phenethyl)-9H-purin-6-amine (SR-4134).
Synthesized according to general procedure D from 6f in 72% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.13 (s, 1H), 7.90 (s, 1H), 7.56 (d, J = 10.8 Hz, 2H), 7.52 – 7.39 (m, 2H), 7.30 – 7.15 (m, 2H), 4.82 (s, 2H), 4.34 (t, J = 7.0 Hz, 2H), 3.49 (s, 8H), 3.22 (t, J = 7.0 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 158.41, 155.80, 153.08, 146.00, 145.93, 144.44, 144.38, 139.3, 139.54, 138.24, 133.85, 132.97, 129.46, 129.39, 129.25, 129.04, 128.84, 126.94, 125.36, 125.32, 125.13, 123.32, 123.28, 121.52, 111.40, 111.26, 108.31, 65.87, 63.32, 44.46, 43.85, 42.84, 40.03, 38.44, 34.59; HRMS (ESI-TOF) calculated for C26H24N8OF5 (M+H)+ 559.1993, found 559.1992.
9-(3-chlorophenethyl)-N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9H-purin-6-amine (SR-4152).
Synthesized according to general procedure D from 6g in 63% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.17 (s, 1H), 7.94 (s, 1H), 7.34 – 7.13 (m, 5H), 7.13 – 7.04 (m, 1H), 4.83 (s, 2H), 4.32 (t, J = 7.0 Hz, 2H), 3.50 (s, 8H), 3.13 (t, J = 7.0 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 158.96, 158.72, 158.49, 158.25, 155.72, 152.92, 150.77, 145.22 (dd, J = 234.8, 9.4 Hz), 140.57, 139.85 (d, J = 15.5 Hz), 138.07, 133.77, 133.05, 130.22, 129.46, 128.63, 128.49, 127.50, 126.56, 111.40 (d, J = 21.5 Hz), 110.54, 108.33, 65.90, 44.48, 43.91, 38.43, 34.43; HRMS (ESI-TOF) calculated for C25H24N8OF2Cl (M+H)+ 525.1730, found 525.1755.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(4-fluorophenethyl)-2-morpholino-9H-purin-6-amine (SR-4051).
Synthesized according to general procedure D from 6h in 80% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.18 (s, 1H), 7.92 (s, 1H), 7.31 – 7.13 (m, 4H), 7.13 – 7.03 (m, 2H), 4.83 (s, 2H), 4.29 (t, J = 7.0 Hz, 2H), 3.51 (s, 9H), 3.10 (t, J = 7.1 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 162.30, 160.69, 159.40, 159.17, 158.93, 158.70, 156.13, 153.29, 151.17, 145.67 (dd, J = 234.8, 9.7 Hz), 139.43 (d, J = 274.6 Hz), 134.54, 134.20, 131.02 (d, J = 8.4 Hz), 129.86, 117.38, 115.63 (d, J = 20.9 Hz), 111.86 (d, J = 21.0 Hz), 110.73, 108.79, 66.33, 44.92, 38.88, 34.37; HRMS (ESI-TOF) calculated for C25H24N8OF3 (M+H)+ 509.2025, found 509.2022.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(4-(methylthio)phenethyl)-2-morpholino-9H-purin-6-amine (SR-4052).
Synthesized according to general procedure D from 6i in 60% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.16 (s, 1H), 7.91 (s, 1H), 7.31 – 7.17 (m, 2H), 7.20 – 7.05 (m, 4H), 4.83 (s, 2H), 4.28 (t, J = 6.9 Hz, 2H), 3.51 (s, 8H), 3.07 (t, J = 7.1 Hz, 2H), 2.42 (s, 3H); 13C 13C NMR (151 MHz, DMSO-d6, δ): 159.42, 159.18, 158.94, 158.70, 156.08, 153.14, 151.11, 145.70 (dd, J = 235.2, 9.8 Hz), 138.48, 136.53, 134.93, 134.16, 129.78, 126.56, 117.33, 115.40, 111.94 (d, J = 21.4 Hz), 110.27, 108.85, 66.32, 44.91, 38.87, 34.58, 15.20; HRMS (ESI-TOF) calculated for C26H27N8OF2S (M+H)+ 537.1997, found 537.2012.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-9-(2-(furan-3-yl)ethyl)-2-morpholino-9H-purin-6-amine (SR-4053).
Synthesized according to general procedure D from 6j in 80% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.19 (s, 1H), 8.00 (s, 1H), 7.57 (t, J = 1.7 Hz, 1H), 7.45 – 7.40 (m, 1H), 7.31 – 7.15 (m, 2H), 6.36 (dd, J = 1.8, 0.9 Hz, 1H), 4.84 (s, 2H), 4.26 (t, J = 7.0 Hz, 2H), 3.51 (m, 8H), 2.94 (t, J = 6.9 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 159.42, 159.18, 158.94, 158.70, 156.08, 153.14, 151.11, 145.70 (dd, J = 235.2, 9.8 Hz), 138.48, 136.53, 134.93, 134.16, 129.78, 126.56, 117.33, 115.40, 111.94 (d, J = 21.4 Hz), 110.27, 108.85, 66.32, 44.91, 38.87, 34.58, 15.20; HRMS (ESI-TOF) calculated for C23H23N8O2F2 (M+H)+ 481.1912, found 481.1898.
N-((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)-2-morpholino-9-(2-(thiophen-3-yl)ethyl)-9H-purin-6-amine (SR-4054).
Synthesized according to general procedure D from 6k in 70% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 8.19 (s, 1H), 7.97 (s, 1H), 7.47 (dd, J = 4.9, 2.9 Hz, 1H), 7.32 – 7.15 (m, 3H), 6.97 (dd, J = 4.9, 1.3 Hz, 1H), 4.84 (s, 2H), 4.32 (t, J = 7.1 Hz, 2H), 3.51 (m, 8H), 3.14 (t, J = 7.1 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 158.97, 158.73, 158.49, 158.25, 155.65, 152.70, 150.65, 145.28 (dd, J = 235.1, 9.6 Hz), 138.08, 133.72, 129.26, 128.25, 126.37, 122.06, 118.80, 116.86, 114.93, 112.99, 111.54 (d, J = 21.3 Hz), 109.73, 108.43, 65.89, 44.49, 43.79, 38.44, 29.38; HRMS (ESI-TOF) calculated for C23H23N8OF2S (M+H)+ 497.1684, found 497.1684.
4-(2-(6-(((6,7-difluoro-1H-benzo[d]imidazol-2-yl)methyl)amino)-2-morpholino-9H-purin-9-yl)ethyl)phenol (SR-4055).
Synthesized according to general procedure D from 6l in 55% yield as a white solid. 1H NMR (400 MHz, DMSO-d6, δ): 9.22 (s, 1H), 8.19 (s, 1H), 7.95 (s, 1H), 7.31 – 7.16 (m, 2H), 6.98 – 6.90 (m, 2H), 6.69 – 6.60 (m, 2H), 4.84 (s, 2H), 4.24 (t, J = 7.1 Hz, 2H), 3.51 (m, 8H), 2.98 (t, J = 7.2 Hz, 2H); 13C NMR (151 MHz, DMSO-d6, δ): 159.36, 159.12, 158.89, 158.66, 156.45, 156.06, 153.13, 151.06, 145.66 (dd, J = 234.7, 9.6 Hz), 140.57 – 137.50 (m), 134.27, 130.07, 128.22, 117.41, 115.72, 115.47, 111.84 (d, J = 20.7 Hz), 110.17, 108.79, 66.33, 45.29, 44.92, 38.91, 34.37; HRMS (ESI-TOF) calculated for C25H25N8O2F2 (M+H)+ 507.2069, found 507.2067.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH NCI grants CA175094 and CA223823, Queens College start-up funds, and PSC-CUNY Enhanced Award (J.Y.C). J.Y.C was supported by NIH SC2 award (SC2GM130470). S. K. and A. C. are grateful for support by the Structural Genomics Consortium (SGC), a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer and Takeda.
ABBREVIATIONS USED
- APBS
Adaptive Poisson-Boltzmann Solver
- ATP
adenosine triphosphate
- CK1
Casein kinase 1
- MD
molecular dynamics
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures, spectroscopic data, dose-response curves in biochemical and cell-based assays, x-ray data collection and refinement statistics, sequence alignment, and Molecular Formula Strings are included in the electronic supplement. The Supporting Information is available free of charge on the ACS Publications website.
Accession Numbers
The atomic coordinates and structure factors of CK1δ - SR-3029 (PDB code: 6RCG) and CK1δ - SR-4133 (PDB code: 6RCH) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The authors declare no competing financial interest.
REFERENCES
- 1.Zhang J; Yang PL; Gray NS, Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009, 9 (1), 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang D; Zhou T; Lafleur K; Nevado C; Caflisch A, Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 2010, 26 (2), 198–204. [DOI] [PubMed] [Google Scholar]
- 3.Azam M; Seeliger MA; Gray NS; Kuriyan J; Daley GQ, Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol 2008, 15 (10), 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Treiber DK; Shah NP, Ins and outs of kinase DFG motifs. Chem Biol 2013, 20 (6), 745–746. [DOI] [PubMed] [Google Scholar]
- 5.Guimaraes CR; Rai BK; Munchhof MJ; Liu S; Wang J; Bhattacharya SK; Buckbinder L, Understanding the impact of the P-loop conformation on kinase selectivity. J Chem Inf Model 2011, 51 (6), 1199–1204. [DOI] [PubMed] [Google Scholar]
- 6.Steichen JM; Kuchinskas M; Keshwani MM; Yang J; Adams JA; Taylor SS, Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J Biol Chem 2012, 287 (18), 14672–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knippschild U; Kruger M; Richter J; Xu P; Garcia-Reyes B; Peifer C; Halekotte J; Bakulev V; Bischof J, The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front Oncol 2014, 4, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheong JK; Virshup DM, Casein kinase 1: Complexity in the family. Int J Biochem Cell Biol 2011, 43 (4), 465–469. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg LH; Cleveland JL; Roush WR; Duckett DR, CK1delta: an exploitable vulnerability in breast cancer. Ann Transl Med 2016, 4 (23), 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schittek B; Sinnberg T, Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer 2014, 13, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu P; Ianes C; Gartner F; Liu C; Burster T; Bakulev V; Rachidi N; Knippschild U; Bischof J, Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 2019, 715, 144005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fulcher LJ; Sapkota GP, Functions and regulation of the serine/threonine protein kinase CK1 family: moving beyond promiscuity. Biochem J 2020, 477 (23), 4603–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y; Xu T; Zhang Y; Qin X, Molecular basis for the regulation of the circadian clock kinases CK1delta and CK1epsilon. Cell Signal 2017, 31, 58–65. [DOI] [PubMed] [Google Scholar]
- 14.Roth A; Gihring A; Bischof J; Pan L; Oswald F; Knippschild U, CK1 Is a Druggable Regulator of Microtubule Dynamics and Microtubule-Associated Processes. Cancers (Basel) 2022, 14 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunkari YK; Meijer L; Flajolet M, The protein kinase CK1: Inhibition, activation, and possible allosteric modulation. Front Mol Biosci 2022, 9, 916232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li SS; Dong YH; Liu ZP, Recent Advances in the Development of Casein Kinase 1 Inhibitors. Curr Med Chem 2021, 28 (8), 1585–1604. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg LH; Lafitte M; Quereda V; Grant W; Chen W; Bibian M; Noguchi Y; Fallahi M; Yang C; Chang JC; Roush WR; Cleveland JL; Duckett DR, Therapeutic targeting of casein kinase 1delta in breast cancer. Sci Transl Med 2015, 7 (318), 318ra202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richter J; Ullah K; Xu P; Alscher V; Blatz A; Peifer C; Halekotte J; Leban J; Vitt D; Holzmann K; Bakulev V; Pinna LA; Henne-Bruns D; Hillenbrand A; Kornmann M; Leithauser F; Bischof J; Knippschild U, Effects of altered expression and activity levels of CK1delta and varepsilon on tumor growth and survival of colorectal cancer patients. Int J Cancer 2015, 136 (12), 2799–2810. [DOI] [PubMed] [Google Scholar]
- 19.Lagunes I; Martin-Batista E; Silveira-Dorta G; Fernandes MX; Padron JM, Differential mechanism of action of the CK1ε inhibitor GSD0054. J Mol Clin Med 2018, 1 (2), 77–84. [Google Scholar]
- 20.Silveira-Dorta G; Sousa IJ; Fernandes MX; Martin VS; Padron JM, Synthesis and identification of unprecedented selective inhibitors of CK1epsilon. Eur J Med Chem 2015, 96, 308–17. [DOI] [PubMed] [Google Scholar]
- 21.Heinzelmann G; Gilson MK, Automation of absolute protein-ligand binding free energy calculations for docking refinement and compound evaluation. Sci Rep 2021, 11 (1), 1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo HJ; Roux B, Calculation of absolute protein-ligand binding free energy from computer simulations. Proc Natl Acad Sci U S A 2005, 102 (19), 6825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin S; Wolgamott L; Roux PP; Yoon SO, Casein kinase 1epsilon promotes cell proliferation by regulating mRNA translation. Cancer Res 2014, 74 (1), 201–211. [DOI] [PubMed] [Google Scholar]
- 24.Bibian M; Rahaim RJ; Choi JY; Noguchi Y; Schurer S; Chen W; Nakanishi S; Licht K; Rosenberg LH; Li L; Feng Y; Cameron MD; Duckett DR; Cleveland JL; Roush WR, Development of highly selective casein kinase 1delta/1epsilon (CK1delta/epsilon) inhibitors with potent antiproliferative properties. Bioorg Med Chem Lett 2013, 23 (15), 4374–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao Y; Chen T; Yang H; Chen Y; Lin H; Qu W; Feng F; Liu W; Guo Q; Liu Z; Sun H, Small molecule modulators targeting protein kinase CK1 and CK2. Eur J Med Chem 2019, 181, 111581. [DOI] [PubMed] [Google Scholar]
- 26.Long AM; Zhao H; Huang X, Structural basis for the potent and selective inhibition of casein kinase 1 epsilon. J Med Chem 2012, 55 (22), 10307–10311. [DOI] [PubMed] [Google Scholar]
- 27.Monastyrskyi A; Nilchan N; Quereda V; Noguchi Y; Ruiz C; Grant W; Cameron M; Duckett D; Roush W, Development of dual casein kinase 1delta/1epsilon (CK1delta/epsilon) inhibitors for treatment of breast cancer. Bioorg Med Chem 2018, 26 (3), 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antilla JC; Baskin JM; Barder TE; Buchwald SL, Copper−Diamine-Catalyzed N-Arylation of Pyrroles, Pyrazoles, Indazoles, Imidazoles, and Triazoles. The Journal of Organic Chemistry 2004, 69 (19), 6514–6514. [DOI] [PubMed] [Google Scholar]
- 29.Fabian MA; Biggs WH 3rd; Treiber DK; Atteridge CE; Azimioara MD; Benedetti MG; Carter TA; Ciceri P; Edeen PT; Floyd M; Ford JM; Galvin M; Gerlach JL; Grotzfeld RM; Herrgard S; Insko DE; Insko MA; Lai AG; Lelias JM; Mehta SA; Milanov ZV; Velasco AM; Wodicka LM; Patel HK; Zarrinkar PP; Lockhart DJ, A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005, 23 (3), 329–36. [DOI] [PubMed] [Google Scholar]
- 30.Santos-Martins D; Solis-Vasquez L; Tillack AF; Sanner MF; Koch A; Forli S, Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J Chem Theory Comput 2021, 17 (2), 1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber biomolecular simulation programs. J Comput Chem 2005, 26 (16), 1668–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salomon-Ferrer R; Gotz AW; Poole D; Le Grand S; Walker RC, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput 2013, 9 (9), 3878–88. [DOI] [PubMed] [Google Scholar]
- 33.Dolinsky TJ; Nielsen JE; McCammon JA; Baker NA, PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 2004, 32 (Web Server issue), W665–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roe DR; Cheatham TE 3rd, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput 2013, 9 (7), 3084–95. [DOI] [PubMed] [Google Scholar]
- 35.Roe DR; Cheatham TE 3rd, Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. J Comput Chem 2018, 39 (25), 2110–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aldeghi M; Bluck JP; Biggin PC, Absolute Alchemical Free Energy Calculations for Ligand Binding: A Beginner’s Guide. Methods Mol Biol 2018, 1762, 199–232. [DOI] [PubMed] [Google Scholar]
- 37.Aldeghi M; Heifetz A; Bodkin MJ; Knapp S; Biggin PC, Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations. J Am Chem Soc 2017, 139 (2), 946–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilson MK; Given JA; Bush BL; McCammon JA, The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J 1997, 72 (3), 1047–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinzelmann G; Chen PC; Kuyucak S, Computation of standard binding free energies of polar and charged ligands to the glutamate receptor GluA2. J Phys Chem B 2014, 118 (7), 1813–24. [DOI] [PubMed] [Google Scholar]
- 40.Cullati SN; Chaikuad A; Chen JS; Gebel J; Tesmer L; Zhubi R; Navarrete-Perea J; Guillen RX; Gygi SP; Hummer G; Dotsch V; Knapp S; Gould KL, Kinase domain autophosphorylation rewires the activity and substrate specificity of CK1 enzymes. Mol Cell 2022, 82 (11), 2006–2020 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gebel J; Tuppi M; Chaikuad A; Hotte K; Schroder M; Schulz L; Lohr F; Gutfreund N; Finke F; Henrich E; Mezhyrova J; Lehnert R; Pampaloni F; Hummer G; Stelzer EHK; Knapp S; Dotsch V, p63 uses a switch-like mechanism to set the threshold for induction of apoptosis. Nat Chem Biol 2020, 16 (10), 1078–1086. [DOI] [PubMed] [Google Scholar]
- 42.Minzel W; Venkatachalam A; Fink A; Hung E; Brachya G; Burstain I; Shaham M; Rivlin A; Omer I; Zinger A; Elias S; Winter E; Erdman PE; Sullivan RW; Fung L; Mercurio F; Li D; Vacca J; Kaushansky N; Shlush L; Oren M; Levine R; Pikarsky E; Snir-Alkalay I; Ben-Neriah Y, Small Molecules Co-targeting CKIalpha and the Transcriptional Kinases CDK7/9 Control AML in Preclinical Models. Cell 2018, 175 (1), 171–185 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Reyes B; Witt L; Jansen B; Karasu E; Gehring T; Leban J; Henne-Bruns D; Pichlo C; Brunstein E; Baumann U; Wesseler F; Rathmer B; Schade D; Peifer C; Knippschild U, Discovery of Inhibitor of Wnt Production 2 (IWP-2) and Related Compounds As Selective ATP-Competitive Inhibitors of Casein Kinase 1 (CK1) delta/epsilon. J Med Chem 2018, 61 (9), 4087–4102. [DOI] [PubMed] [Google Scholar]
- 44.Shinohara Y; Koyama YM; Ukai-Tadenuma M; Hirokawa T; Kikuchi M; Yamada RG; Ukai H; Fujishima H; Umehara T; Tainaka K; Ueda HR, Temperature-Sensitive Substrate and Product Binding Underlie Temperature-Compensated Phosphorylation in the Clock. Mol Cell 2017, 67 (5), 783–798 e20. [DOI] [PubMed] [Google Scholar]
- 45.Halekotte J; Witt L; Ianes C; Kruger M; Buhrmann M; Rauh D; Pichlo C; Brunstein E; Luxenburger A; Baumann U; Knippschild U; Bischof J; Peifer C, Optimized 4,5-Diarylimidazoles as Potent/Selective Inhibitors of Protein Kinase CK1delta and Their Structural Relation to p38alpha MAPK. Molecules 2017, 22 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mente S; Arnold E; Butler T; Chakrapani S; Chandrasekaran R; Cherry K; DiRico K; Doran A; Fisher K; Galatsis P; Green M; Hayward M; Humphrey J; Knafels J; Li J; Liu S; Marconi M; McDonald S; Ohren J; Paradis V; Sneed B; Walton K; Wager T, Ligand-protein interactions of selective casein kinase 1delta inhibitors. J Med Chem 2013, 56 (17), 6819–6828. [DOI] [PubMed] [Google Scholar]
- 47.Bischof J; Leban J; Zaja M; Grothey A; Radunsky B; Othersen O; Strobl S; Vitt D; Knippschild U, 2-Benzamido-N-(1H-benzo[d]imidazol-2-yl)thiazole-4-carboxamide derivatives as potent inhibitors of CK1delta/epsilon. Amino Acids 2012, 43 (4), 1577–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeringo NA; Murphy L; McCloskey EA; Rohal L; Bellizzi JJ 3rd, A monoclinic crystal form of casein kinase 1 delta. Acta Crystallogr Sect F Struct Biol Cryst Commun 2013, 69 (Pt 10), 1077–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang H; Acquaviva L; Berry V; Bregman H; Chakka N; O’Connor A; DiMauro EF; Dovey J; Epstein O; Grubinska B; Goldstein J; Gunaydin H; Hua Z; Huang X; Huang L; Human J; Long A; Newcomb J; Patel VF; Saffran D; Serafino R; Schneider S; Strathdee C; Tang J; Turci S; White R; Yu V; Zhao H; Wilson C; Martin MW, Structure-Based Design of Potent and Selective CK1gamma Inhibitors. ACS Med Chem Lett 2012, 3 (12), 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long A; Zhao H; Huang X, Structural basis for the interaction between casein kinase 1 delta and a potent and selective inhibitor. J Med Chem 2012, 55 (2), 956–960. [DOI] [PubMed] [Google Scholar]
- 51.Gaussian 09, R. A, Gaussian, Inc., Wallingford CT. 2016. [Google Scholar]
- 52.Janovska P; Normant E; Miskin H; Bryja V, Targeting Casein Kinase 1 (CK1) in Hematological Cancers. Int J Mol Sci 2020, 21 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dang F; Nie L; Zhou J; Shimizu K; Chu C; Wu Z; Fassl A; Ke S; Wang Y; Zhang J; Zhang T; Tu Z; Inuzuka H; Sicinski P; Bass AJ; Wei W, Inhibition of CK1epsilon potentiates the therapeutic efficacy of CDK4/6 inhibitor in breast cancer. Nat Commun 2021, 12 (1), 5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans P, Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 2006, 62 (Pt 1), 72–82. [DOI] [PubMed] [Google Scholar]
- 56.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J Appl Crystallogr 2007, 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA, REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humphrey W; Dalke A; Schulten K, VMD: visual molecular dynamics. J Mol Graph 1996, 14 (1), 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.