

SUMMARY
In this review, we consider the regulatory strategies of aquatic oligotrophs, microbial cells that are adapted to thrive under low-nutrient concentrations in oceans, lakes, and other aquatic ecosystems. Many reports have concluded that oligotrophs use less transcriptional regulation than copiotrophic cells, which are adapted to high nutrient concentrations and are far more common subjects for laboratory investigations of regulation. It is theorized that oligotrophs have retained alternate mechanisms of regulation, such as riboswitches, that provide shorter response times and smaller amplitude responses and require fewer cellular resources. We examine the accumulated evidence for distinctive regulatory strategies in oligotrophs. We explore differences in the selective pressures copiotrophs and oligotrophs encounter and ask why, although evolutionary history gives copiotrophs and oligotrophs access to the same regulatory mechanisms, they might exhibit distinctly different patterns in how these mechanisms are used. We discuss the implications of these findings for understanding broad patterns in the evolution of microbial regulatory networks and their relationships to environmental niche and life history strategy. We ask whether these observations, which have emerged from a decade of increased investigation of the cell biology of oligotrophs, might be relevant to recent discoveries of many microbial cell lineages in nature that share with oligotrophs the property of reduced genome size.
KEYWORDS: marine microbiology, metabolic regulation, transcriptional regulation
INTRODUCTION
Microbial cells monitor intracellular and extracellular variables and respond to detected changes using regulation. Regulation governs intercellular interactions (e.g., quorum sensing), exploits or protects the cell from environmental conditions (e.g., catabolic operons and stress responses), and modulates internal processes to achieve homeostasis across growth cycles (e.g., the control of cellular growth and division). Regulatory mechanisms of microbial cells have been explored at length, with the consensus that bacteria generally respond to environmental stimuli via transcriptional regulation (expressing or repressing genes). However, there are many additional layers of regulation in bacterial cells: regulation mediated by structural RNAs (e.g., riboswitches), mechanisms that modulate mRNA translation (e.g., DEAD box ATPases), covalent and allosteric posttranslational modifications of proteins (e.g., acetylation of proteins), and internal regulation of cellular metabolism based on the kinetic properties of enzymes (e.g., partitioning of glucose and galactose metabolism). Most of what we know about microbial regulation has been derived from the study of cells that can easily be cultured, although significant work has been done on regulation in extremophiles and the broader uncultured microbial diversity.
The study of microbial regulation is relevant beyond the study of cell physiology. On an ecological scale, regulation in response to environmental changes and fluctuations in cell physiology determines which genes are turned on and off at a given point in time, for example impacting which organic compounds are oxidized. Thus, the mere presence of a gene in the environment does not mean that the protein coded by that gene is produced. By grasping how cells in the ocean regulate protein production, we might increase our ability to predict carbon oxidation functions vital to understanding the global carbon cycle. Understanding regulatory responses of microbes to fluctuating environments is also critical in a variety of other fields, including in plant-microbe interactions, the study of virulent bacteria, and biotechnological settings, as summarized in a recent article (1).
Prokaryotes are generally divided into two categories based on lifestyle strategy: oligotroph or copiotroph, with a broad spectrum between the two opposites. These two strategies are also relative measures of lifestyle based on the magnitude of nutrient fluxes and concentrations in environments; for instance, even the highest concentrations of nutrients in the ocean are lower than concentrations regularly experienced by gut bacteria. Oligotrophic microorganisms are adapted to thrive in environments with very low nutrient fluxes (<0.1 mg of C/L per day) and achieve maximal growth rates under these conditions (2); high-nutrient concentrations are known to inhibit growth of oligotrophs (3, 4). Copiotrophs are opportunists, only experiencing maximal growth rates at high nutrient concentrations, but are capable of surviving at low nutrient conditions for long periods of time, essentially in a state of starvation (5–8). The oligotroph/copiotroph dichotomy is a simplistic classification of microbes: many microorganisms fall somewhere in between, with varied combinations of characteristics associated with the two extremes (9). Because of this, various other frameworks have been proposed, such as the competitive/stress tolerator/ruderals triangle (C-S-R) (9–11), among others (12, 13), which provide useful insights into microbial ecosystem functioning. Despite this, the oligotroph/copiotroph dichotomy benefits from its simplicity, providing simple categories that broadly assess the niches and physiologies of microbes and ease communication among researchers.
Overall, there are more oligotrophic bacteria in the world than copiotrophic bacteria, primarily because the majority of the world is covered by oligotrophic ocean environments (14, 15). The two most abundant clades of bacteria in the world are also its most well-studied oligotrophs: SAR11 and Prochlorococcus (16, 17). Prochlorococcus is a group of very small (cell volume, ~0.14 μm3) photosynthetic cyanobacteria that are ubiquitous in the world’s oceans between 40°S and 40°N latitudes and have a minimal genome (18–20). They are the most abundant type of phytoplankton in the world, with an estimated 2.9 ± 0.1 × 1027 cells worldwide (21). There are several reviews of Prochlorococcus, including a recent, highly relevant review of regulation in Prochlorococcus (18, 19, 22). SAR11 is a clade of heterotrophic alphaproteobacteria that are very small (cell volume, 0.01 μm3), ubiquitous in the world’s oceans at high densities (especially at the surface), are nonmotile, and have one of the smallest genomes of any free-living organism (17, 23). Globally, it is estimated that there are 2.4 × 1028 SAR11 cells in the world (17). As with Prochlorococcus, there are several helpful SAR11 reviews, including a 2017 general review of SAR11 and a review of genome streamlining in SAR11 (24, 25).
The oligotroph/copiotroph life history strategies are theorized to be associated with a variety of physiological and genomic adaptations, with oligotrophs generally thought of as being small in cell size, free-living, nonmotile, and slow-growing and having streamlined genomes with low numbers of rRNA gene operons and low GC content (2, 14, 15, 23, 24, 26–30). Other studies have found a lack of correlation between assigned lifestyle strategy and proposed phenotypic traits across a range of environments (31, 32). In uncovering broad associations between phenotypic traits and lifestyle strategy, there are two difficulties: first, most cultured microbes are copiotrophs, due to the ease of isolating and culturing them (15, 27, 33, 34); second, microbes are often misclassified as oligotrophs based solely on their presence in oligotrophic environments. For instance, Caulobacter has long been studied as a model aquatic oligotroph (see, for example, reference 35); however, a recent study has shown that Caulobacter microbes are primarily found in high-nutrient soils and use aquatic systems mainly for dispersal (36), casting doubt on their reputed character as aquatic oligotrophs.
In this review, we examine one of the most salient examples of a theorized broad association between regulatory strategies and life history strategies. The comparative study of microbial regulation is not a “field” in the sense that you will find symposia devoted to the topic. Rather, we tend to see each cell type as an amalgam of regulatory functions that suits its needs, and we do not often consider whether cellular regulatory strategies can be divided into broad categories that fit modalities in lifestyle. For each type of regulation known to operate in prokaryotic organisms (transcriptional, posttranscriptional, posttranslational, and kinetic), we first briefly introduce its utility to cells, describe its mode of action, review the evidence (if any) for different uses between copiotrophs and oligotrophs, and finally provide examples of each type of regulation in aquatic oligotrophs. We show that the evidence indicates a reduction in transcriptional regulation in oligotrophs, resulting in constitutive expression of many genes (Fig. 1). Many posttranscriptional levels of regulation seem to be present at similar levels in oligotrophs as in copiotrophs, but they take on added importance to the functioning of cells in the absence of transcriptional regulation.
FIG 1.

Illustration of the primary hypothesis explored in this article. Aquatic oligotrophic microorganisms are depleted in transcriptional regulation compared to copiotrophs. This results in constitutive expression of most of their genes, no matter the current nutrient regime they are experiencing. This constitutive expression may further be modified by posttranscriptional, posttranslational, or kinetic/metabolic regulation, but the result remains that a majority of proteins from oligotrophic genomes are expressed most of the time. “Metabolic activity” here refers to all levels of regulation, from transcriptional to posttranslational.
Finally, we ask why copiotrophs and oligotrophs might use a common set of regulatory mechanisms with very different preferences on how, and how frequently, they are employed. We ask whether these differences are part of a multivariate continuum of cell properties, such that theorizing mainly serves a heuristic role, or whether there are trends in evolution that can lead different lineages to converge on a set of cell regulatory properties associated with oligotrophy that are distinctly identifiable and uniquely different from those associated with copiotrophy. We ask how creating such categories might help us understand the ecology and physiology of cells and whether it makes sense to develop broad rules that could be used to define these categories.
REGULATION AT THE TRANSCRIPTIONAL LEVEL
The central dogma of molecular biology states that genes, encoded in DNA, are transcribed into mRNA and then translated into protein. Cells can regulate the amount of a given protein in the cell by modulating all parts of this process. Transcriptional regulation has the benefit of saving on cellular resources when a nutrient is unavailable, with no excess production of mRNA or protein. The downside is that it can be relatively slow (on the order of minutes) to respond to a stimulus (37) and requires extra regulatory machinery. In Escherichia coli, it has been shown that the majority of protein expression changes are attributable to transcriptional regulation (38).
Introduction to Transcriptional Regulation
Regulation at the transcriptional level modulates the amount of a given gene being expressed at any given time. Generally, aquatic microbes are thought to respond to environmental changes via a sense-response system, where an environmental stimulus initiates a transcriptional response in the cell (39). These transcriptional responses can take multiple forms.
Suites of genes that are regulated together in response to a stimulus are called regulons; regulons are generally regulated by a combination of transcription factors (40), proteins that either enhance or repress the binding of RNA polymerase to the promoter of a gene, and sigma factors (σ-factors) (41). σ-Factors are transcription initiation factors that are required for the binding of RNA polymerase to gene promoters. By having different σ-factors that bind to different gene promoters, cells can change expression of large sets of genes by simply changing which σ-factor is expressed (42). σ-Factors are usually used to modulate responses to large environmental changes, such as starvation, heat stress, etc. (43). E. coli cells have seven σ-factors, while some Verrucomicrobia have more than 30 σ-factors.
Transcriptional regulation can also happen in a more targeted way, through two-component regulatory systems, which translate environmental stimuli into a targeted regulatory response using phosphotransferase systems (44). Two-component systems are comprised of a histidine kinase, which senses the environmental stimulus, and a response regulator protein, which receives the signal from the kinase and activates the transcriptional response (45). The breadth of possible transcriptional regulation in bacteria, such as the types of transcription factors, σ-factors, and two-component systems, is extensive and has been reviewed before (40, 44, 46–48).
Relevance of Transcriptional Regulation to Oligotrophs
Oligotrophs have less transcriptional regulation than would be expected from their genome sizes. This has been demonstrated both bioinformatically by mining oligotroph genomes for genes associated with regulation, as well as experimentally, looking at changes in transcription in response to environmental changes.
A reduction in the number of mechanisms for transcriptional regulation is evident at the genome level in the most-studied representative of the SAR11 clade, “Candidatus Pelagibacter ubique” strain HTCC1062, which only has two σ-factors and four two-component regulatory systems (23). In a comparison of σ-factors in bacteria to genome size, a variety of oligotrophs, including SAR11, Prochlorococcus, and the abundant oligotrophic gammaproteobacterial clades SAR86 and SAR92, were found to have fewer σ-factors than would be expected based on genome size (24). In a survey of two-component systems in marine bacteria, it was discovered that oligotrophy was associated with a reduced number of two-component regulatory systems, with oligotrophs having, on average, 0.2 histidine kinases per 100 genes, while copiotrophs had 1.4 (45). Similarly, the oligotrophic clade II of the globally abundant cyanobacteria Synechococcus, such as Prochlorococcus, has limited numbers of histidine kinases and response regulators (7 and 12, respectively) compared to the abundant freshwater, more copiotrophic Synechocystis cyanobacteria (47 and 42, respectively) (49). Within the Dadabacteria phylum, the marine pelagic subclade that exhibits genome streamlining has fewer genes associated with two-component systems and motility (50).
In an analysis of the genomic features that differentiate oligotrophs and copiotrophs, it was discovered that, compared to copiotrophs, oligotroph genomes were depleted in genes in all categories relating to regulation (14). These categories included σ-factors, transcriptional regulators (COG0583), AraC-type DNA-binding domain-containing proteins (COG2207), DNA-binding winged-helix–turn–helix (HTH) domains (COG3710), regulators with FOG:GGDEF domains (COG2199), and regulators with FOG:EAL domains (COG2200) (14). Another, more recent paper found a similar result (i.e., depletion of genes in the transcriptional regulation category in oligotrophs) when categorizing oligotroph/copiotroph based on maximal growth rates as predicted by codon usage bias (27). This definition of oligotroph/copiotroph is based on one cell property but has the advantage of being quantitative in nature. However, this functional analysis used genomes from the RefSeq database, which spans a variety of habitats. We recently repeated this analysis using a large collection of marine metagenome-assembled genomes (51) (Fig. 2). We first found that the genomes of the oligotrophic microbes in this data set generally conform to predicted genome characteristics of oligotrophs: smaller genomes and lower GC content (Fig. 2A and B). Next, as found in the previous analysis that used RefSeq genomes, copiotrophs were significantly (P < 0.05, Mann-Whitney test with Benjamini-Hochberg correction) enriched in genes for transcriptional regulators compared to oligotrophs (Fig. 2C), again confirming that oligotrophs are depleted in transcriptional regulation at the genomic level. Finally, a recent review compared traits of marine pelagic microbes (generally oligotrophs) to freshwater sedimentary microbes (generally copiotrophs) again found that the marine pelagic microbes tended to have smaller genomes, lower GC content, fewer σ-factors, and fewer two-component systems than the freshwater sediment microbes (52).
FIG 2.

Analysis of marine copiotroph/oligotroph metagenome assembled genomes (MAGs) from Tully et al. (51), with lifestyle strategy defined by predicted maximal growth rate based on codon usage patterns, as in Weissman et al. (27). (A and B) Comparison of genome characteristics of the two lifestyle categories from this data set. (C) Enrichment of different COG categories in copiotrophs or oligotrophs. (For full methods for this analysis, see reference 76.) Category T was split between COG categories having “chemotaxis” in their description and all others. Category N was split between categories with “flagella” in their description and all others. Category K was split between categories with “transcriptional regulator” in their description and all others. Category O was split between categories representing common posttranslational modifications (acetylation, phosphorylation [kinases], ubiquitination, methylation, glycosylation, adenylation, and peptidases) and all others. (The figure was adapted from reference 76.)
Several experimental studies have reported a global reduction of transcriptional regulation in oligotrophs. In a comparison of rRNA/rDNA ratios in two oligotrophic bacteria, SAR11 and SAR92, and two copiotrophic bacteria, Roseobacter and Flavobacteria, Lankiewicz et al. found that the two oligotrophs did not modulate their ribosome number in response to growth rate, indicating a lack of transcriptional regulation of rRNA in the oligotrophs compared to copiotrophs (53). In a subsequent paper, the same four organisms were compared for their global transcriptional changes in response to transitioning from exponential to stationary growth phases (54). In the two oligotrophs, the change in transcript abundances between exponential and stationary phases was extraordinarily minimal, with almost all changes falling below a log2-fold change of 2 (54). In the copiotrophs, on the other hand, transcript abundances varied widely between the two growth stages, with log2-fold changes reaching 10 (54). The low amplitude changes in SAR11 likely reflect the absence of the stationary-phase sigma factor σS in these cells, which results in a muted proteomic response to entering stationary phase (55). Instead of wholesale proteome remodeling, modest increases in the abundance of proteins involved in maintaining cellular protein pools, such as chaperones, signal transducing proteins, and amino acid synthesis enzymes (especially methionine and cysteine) were reported in SAR11 cells entering stationary phase (55). Sowell et al. argued that the muted proteomic response of “Ca. Pelagibacter ubique” allows them to cope with short periods of nutrient deprivation and resume growth quickly, making a comprehensive global stationary-phase response unnecessary (55).
Studies of Prochlorococcus using cultures, metatranscriptomics, and pigment analyses point to control of chlorophyll production by protein-level, not transcriptional, regulation (56, 57). In a proteomic analysis of the oligotrophic alphaproteobacterium Sphingopyxis alaskensis in glucose-limited chemostats, only 12 of over 1,000 resolved proteins were found to be significantly different in abundance compared to nutrient-replete conditions (58). Similarly, the oligotroph Sphingomonas sp. strain RB2256 showed consistent growth rates across low and high nutrient conditions, which was hypothesized to be due to constitutive expression of protein-synthesizing machinery (59). In a mesocosm experiment where surface marine microbial communities were exposed to deep-see water, the highly abundant oligotrophs (SAR11 and Prochlorococcus) showed little to no transcriptional response, compared to the copiotrophic Alteromonas species detected (60).
There is additional experimental evidence indicating that oligotrophs have limited transcriptional regulation at the global level. Much of this evidence comes from studies that compare data from transcriptomic (total RNA from cells that is sequenced [61]) and proteomic (total proteins from cells that are analyzed via mass spectrometry [62]) studies. By simultaneously comparing changes in transcript and protein abundance for genes, the regulatory control of that gene can be examined (63). If a gene is under transcriptional control, one would expect changes in mRNA and protein abundance to be correlated for that gene, while genes under posttranscriptional regulation would, in principle, have decoupled protein and transcript abundance. However, differences in the stability of mRNA and proteins in cells can also skew correlations (63). As an example, in E. coli, the half-life of mRNA molecules is only minutes long, while proteins have an average half-life of 20 h, resulting in a lack of correlation between transcripts and proteins on a single-cell basis (64). However, in general, when transcript and protein abundances are averaged over a population, there is a linear correlation, with at least 40% of variation in protein abundance being explained by transcript abundance (38, 65). While these findings point to posttranscriptional regulation being widespread across all domains of life, they also indicate that, in general, protein and transcript abundances are correlated (66). In three papers examining transcriptional and proteomic responses in SAR11 to iron, sulfur, and nitrogen limitation, there was no correlation between transcript and protein abundances, except for a few genes, indicating a lack of transcriptional regulation at a global level in SAR11 (Fig. 3A) (67–69). Similarly, in an examination of diel oscillations in Prochlorococcus transcripts and proteins, transcript abundances were found to vary much more widely than protein abundance for genes, signifying heavy posttranscriptional regulation (56). In several of these papers, the possibility of either mRNA degradation or missed pulses of mRNA skewing transcript abundances was tested, but no evidence was found to support these possibilities (56, 68). On the environmental level, one study that paired metagenomic and metaproteomic data from oligotrophic ocean samples found that the most abundant members of the microbial community (SAR11, Prochlorococcus, Synechococcus, and SAR116) had strong coupling between the presence of a gene in the genome and the corresponding protein being expressed, while the opposite was true for the rarer, copiotrophic members of the community (70). This suggests that oligotrophs were expressing most of their genes in the samples studied, while copiotrophs were not.
FIG 3.

(A) The prevalence of posttranscriptional regulation in SAR11 cells is apparent when comparing transcript and protein abundances (log2-fold) in SAR11 cells under a variety of nutrient limitations. The dashed line indicates a 1:1 correlation between transcript and protein abundance, which the data clearly does not fit. Data were digitized from previous publications (67–69) using WebPlotDigitizer v4.6 (189). (B and C) Comparison of log2-fold changes in transcript abundance in copiotrophs or oligotrophs in response to a variety of environmental challenges. Aggregated data are shown in panel B and are broken out by nutrient limitation state in panel C. The following species are represented for iron limitation: for copiotrophs, Pseudomonas fluorescens (190), Listeria monocytogenes (191), Alteromonas macleodii (192), Campylobacter jejuni (193), Synechocystis (194), Pasteurella multocida (195), and Synechococcus sp. strain PCC 7002 (196); for oligotrophs, Prochlorococcus (141) and “Ca. Pelagibacter ubique” (67). The following species are represented for nitrogen limitation: for copiotrophs, Pseudomonas putida (197), Synechococcus elongatus PCC7942 (198), Mycobacterium smegmatis (199), and E. coli (77); and for oligotrophs, Dehalococcoides ethenogenes (200), Prochlorococcus (201), and “Ca. Pelagibacter ubique” (68). The following species are represented for phosphate limitation: for copiotrophs, E. coli (78), Rhodobacter sphaeroides (202), and Synechococcus sp. strain PCC 7002 (196); and for oligotrophs, “Ca. Pelagibacter ubique” (94). The following species are represented for sulfur limitation: for copiotrophs, Pseudomonas aeruginosa (203), Synechococcus sp. strain PCC 7002 (196); and for oligotrophs, Emiliania huxleyi (204), Arthrospira (205), “Ca. Pelagibacter ubique” (69). The following species are represented for other environmental challenges: for copiotrophs, Bacillus subtilis (superoxide stress) (206), Marinobacter hydrocarbonoclasticus (hydrocarbon exposure) (207, 208), Alcanivorax borkumensis (hydrocarbon exposure) (209), Alteromonas naphthalenivorans (contaminated seawater exposure) (210), and Synechococcus sp. strain PCC 7002 (CO2 limitation) (196); and for oligotrophs, OM43 (variety of nutrient exposures) (86), Prochlorococcus (coculture with heterotroph [211]; CO2 limitation [212]), and “Ca. Pelagibacter ubique” (DMSP versus methionine exposure [74]). A threshold log2-fold change of 2 and −2 was used to reduce the amount of data; for “Ca. Pelagibacter ubique” nitrogen limitation and DMSP exposure, this threshold removed all transcripts. For species with a murky lifestyle strategy, their maximal growth rate reported in literature was used for classification as described previously (27). Not all data from these papers was included for ease of visualization, but the largest changes in transcript abundances were selected.
When examining a trove of published and unpublished proteomic data collected on HTCC1062 cells over a variety of nutrient limitation conditions, it becomes clear that these cells constitutively express the majority of their genes. Rarefaction curves of the proteomic coverage of HTCC1062 shows almost 80% coverage due to the constitutive expression of most genes (Fig. 4). In contrast, the nonoligotrophic cyanobacterium Synechocystis PCC68803 proteome only shows around 50% coverage, indicating that only about half of the genes in this organism are being expressed under a given set of conditions (Fig. 4). This has been borne out in other oligotrophs as well, including a cultured representative of the ammonia-oxidizing Thaumarchaeota phylum, Nitrosopumilus maritimus SCM1, which is ubiquitous in oligotrophic waters and has a small genome (71). The vast majority of genes in these cells were also constitutively expressed in exponentially growing cells (72).
FIG 4.
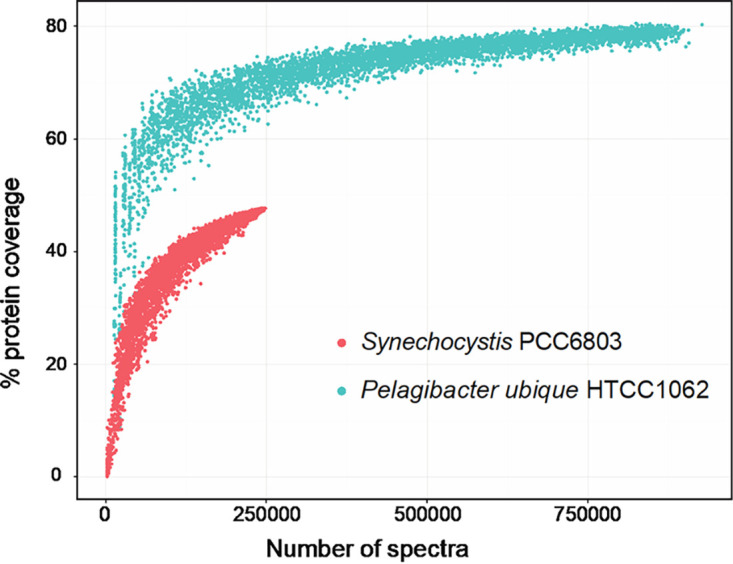
Rarefaction plots for proteomic coverage for “Ca. Pelagibacter ubique” HTCC1062 (oligotroph) compared to that of the copiotrophic Synechocystis PCC6803. For HTCC1062, protein expression data from a range of lab experiments across a variety of conditions (nutrient depletions, stationary phase, etc.) were uploaded to the Proteomics Identifications database (PRIDE). The coverage of the predicted cellular proteins in HTCC1062, and the numbers of spectra measured for these peptide fragments were compared to a copiotrophic organism (Synechocystis) that had undergone lab-based experiments from a similar set of conditions and whose proteomics measurements had been done on a similar type of mass spectrometer, to allow for direct comparisons. The reduction in transcriptional regulation found in HTCC1062 is correlated with a highly conserved (and thus well-covered) proteome, with the majority of genomic proteins having a high percentage of coverage at high peptide spectra levels.
In many studies of SAR11 and other heterotrophic oligotrophs, the data have pointed to a lack of transcriptional regulation of genes involved in carbon metabolism. One of the easiest ways to test for this is to compare the uptake rates and/or metabolic rates of carbon compounds in naive cells (i.e., cells grown in the absence of a compound of interest) and preconditioned cells (i.e., cells grown with the compound of interest for a reasonable amount of time). If naive and preconditioned cells have the same uptake and/or metabolic rates, then the reasonable conclusion is that the genes required for uptake and/or metabolism of that compound are constitutively expressed at the same level in the presence and absence of that compound. In SAR11 for dimethylsulfoniopropionate (DMSP), dimethyl arsenate (DMA), and l-alanine, naive and preconditions cells were found to have the same uptake and/or metabolic rates (73–76). Conversely, if there is a significantly larger uptake and/or metabolic rate of the compound in preconditioned cells, one can conclude that the cell has the genes required for uptake and/or metabolism under transcriptional regulation. In a study comparing regulation of l-alanine metabolism in SAR11 and a marine copiotroph, Alteromonas macleodii, the copiotroph was found to have a significantly larger metabolic rate of l-alanine in preconditioned cells compared to naive cells (76).
Examples of Transcriptional Regulation in Oligotrophs
Oligotrophic bacteria do use transcriptional regulation to respond to some environmental stimuli, but many of these responses (as described below) are significantly smaller in magnitude than commonly observed transcriptional responses in copiotrophs. For instance, the maximum log2-fold change in transcripts found in nitrogen-starved Pelagibacter cells was 1.5 compared to 15 in nitrogen-starved E. coli (77); the maximum log2-fold change in transcripts in phosphate-limited Pelagibacter cells was 30 compared to 131 in phosphate-limited E. coli (78). This is illustrated on a broad scale in Fig. 3B and C, which shows that, in response to the same types of nutrient limitation, oligotrophs have a highly muted transcriptional response compared to copiotrophs. Poindexter theorized that oligotrophs would have low-magnitude regulatory responses, since a large regulatory response would be out of proportion to the minimal nutrient concentrations they are regularly exposed to (79). A common observation in oligotroph biology is that growth rates rarely accelerate in response to positive environmental changes. This factor might mute the apparent intensity of positive transcriptional changes when comparisons are made to copiotrophs.
In a study of diel transcript abundances in the oligotrophic open ocean, Ottesen et al. found significant diel changes in transcripts in a variety of oligotrophic bacteria, including Prochlorococcus, SAR11, SAR86, and SAR116 (80). However, the number of transcripts showing diel changes and magnitude of those changes were lower in the heterotrophic oligotrophic groups of organisms than the copiotrophs (80). Some of the changes in transcript levels may also be due to growth dilution, i.e., as cells grow or shrink, all cellular components must increase or decrease to some extent with changes in biomass, as observed in E. coli (38). Prochlorococcus, as a photoautotroph, showed strong, diel transcriptional responses that matched light levels (80). In a similar study of transcriptional changes in coastal and open-ocean microbes, several heterotrophic oligotrophs, including SAR11 and SAR86, were found to have significant transcriptional changes on a diel cycle (81). In contrast, a similar examination of diel transcriptomes in a coastal region did not find significant diel changes in gene expression in the heterotrophic oligotrophic groups, including SAR11 and SAR86 (82). There were, however, pathway-level, covariant changes in transcript abundances in these heterotrophic oligotrophic groups; as an example, in SAR11, ribosomal proteins and oxidative phosphorylation had a strong positive correlation with each other and a negative correlation with several transport gene transcripts (82). Interestingly, in two separate experiments, there were found to be diel, synchronous changes in transcript abundance between SAR11 and SAR86, which could either be due to the two groups responding to similar environmental signals or could be indicative of interspecies interactions (81, 82). Upon addition of dissolved organic matter (DOM) derived either from Prochlorococcus cultures or high molecular-weight DOM from surface waters to a surface microbial community, a swift (within 2 h) transcriptional response was observed in both SAR11 and Prochlorococcus cells (83). In both oligotrophs, genes involved in assimilating organic N compounds were upregulated, in addition to protein biosynthesis genes (83). Similarly, in response to polyamine addition to surface water mesocosms, SAR11 transcripts for genes involved in polyamine metabolism increased in abundance within hours of polyamine addition (84).
In cultures of oligotrophs, transcriptional responses to changing growth conditions or nutrient limitation have been observed. In cells from the oligotrophic methylotrophic clade OM43, which have highly streamlined genomes (85), strong changes in transcript abundance were observed when cells transitioned from exponential to stationary growth phase and in response to addition of specific nutrients (86). Genes involved in C metabolism showed the strongest response, including ribulose monophosphate, proteorhodopsin, and methanol dehydrogenase (86). Nitrosopumilus maritimus SCM1 cells have been found to downregulate genes involved in ammonia transport and metabolism (amoA, amoB, nirK, and amtB) upon ammonia limitation, while the hsp20 gene for the molecular chaperone protein was upregulated (72, 87). This finding has been observed in other cell types undergoing stress, including SAR11 (67–69, 72, 88). Overall, however, a large proportion of genes are constitutively expressed by these Thaumarchaeota and the transcriptional response to ammonia limitation is relatively muted (71, 72, 87, 89).
In SAR11 cells, the regulatory response to inorganic nitrogen limitation was found to rely on a much simpler system for ammonium assimilation than the PII signal-transducing system that is found in most proteobacteria, with five fewer genes than other free-living alphaproteobacteria. This system in SAR11 cells is under transcriptional control of the two-component system NtrX/NtrY; transporters for organic nitrogen compounds, such as amino acids, opines, and taurine, were also upregulated in N-depleted SAR11 cells (68). Smith et al. speculated that the absence of the PII system in SAR11 contributes to genome streamlining at the cost of making these cells vulnerable to metabolic disruptions caused by competition for the metabolic intermediate 2-oxoglutarate, which is an intermediate both in ammonium uptake and in energy metabolism (68). All Prochlorococcus strains appear to have active versions of the PII regulatory system, in addition to the global nitrogen regulatory protein NtcA (22, 90). However, these proteins show unique regulatory responses to N in Prochlorococcus: the PII system may not be fully phosphorylated, the urease enzyme is constitutively expressed, and the response of NtcA to N limitation is not as strong as in other cyanobacteria (91). Methylotrophic cells from OM43 also encode the PII regulatory system; under nitrogen limitation, these cells were found to have large transcriptional responses (log2-fold changes above 10 in transcript levels) (86).
When SAR11 cells were iron limited, the ABC transporter for iron, SfuABC, was found to be highly upregulated, both in transcript and protein abundance, but other transcriptionally upregulated genes involved in iron metabolism were not translated (67). The abundance of the RNA chaperone CspL increased dramatically in the iron limited Pelagibacter cultures, whereas the paralog CspE declined, leading Smith et al. to speculate that Pelagibacter utilizes RNA chaperones (also know as DEAD-box ATPases) to control a global posttranscriptional regulatory response to iron limitation (67). RNA chaperones are found in all cell types and have been implicated in a variety of processes involving RNA, including RNA sequestration into subcellular structures (92), a process that can involve the recognition of RNA sequences by RNA chaperones. Smith et al. postulated that CspL might function by inhibiting the translation of nonessential transcripts until iron became available, although this hypothesis was not tested (67). However, RNA chaperones recently were implicated in the regulation of iron metabolism in E. coli (93).
Similar to iron limitation, when SAR11 cells experienced phosphate limitation, they vastly upregulated (30-fold) transporters for phosphorous-containing compounds (94), although this regulatory response was much lower than that observed in phosphate-limited E. coli cells (maximum fold change of 130) (78). However, there were large differences observed between the coastal (“Ca. Pelagibacter ubique” strain HTCC1062) and open ocean (“Ca. Pelagibacter” strain HTCC7211) strains of SAR11 studied, with the coastal strain upregulating its phosphate transporter and the open ocean strain upregulating its phosphonate (organic phosphate) transporter (94). HTCC7211 also upregulated genes for a C-P lyase (phnGHIJKLM), which cleaves phosphonate compounds into inorganic phosphate and methane (94). The transcriptional response in the coastal strain, HTCC1062, appears to be due to the upregulation of general stress-response genes such as recA, lexA, and umuD, but the transcriptional regulatory mechanism in the open ocean strain, HTCC7211, is not fully understood (94). In cells from the OM43 clade, phosphate limitation resulted in a decrease in the phosphate-specific transport system gene, but the response was relatively muted (only changes of 2- to 5-fold) (86).
In SAR11 cells exposed to light and starved for carbon, 9.7% of coding sequences showed differential expression compared to cells in the dark, including upregulation of genes involved in oxidative phosphorylation (95). Three of the upregulated genes were found to be transcriptional regulators, with one being a putative ferric uptake regulator protein (Fur), known to bind to specific sequences (Fur-boxes) upstream of regulated genes (95). A search for relaxed fits to Fur-boxes showed potential binding sequences upstream of several of the genes upregulated in the light, implicating this Fur protein in the transcriptional regulatory response to light and C starvation in SAR11 (95). The presence of light-dependent proteorhodopsin pumps in SAR11, combined with the lack of large-scale transcriptional changes in SAR11 in response to light availability, indicates that SAR11 cells use their proteorhodopsin pumps to produce enough energy to survive until more organic carbon nutrients are available, instead of undergoing proteome remodeling leading to a dormant state (95, 96). SAR11 cells seem to focus on small cellular changes to promote homeostasis and survive periods without nutrients, allowing them to quickly start up metabolism and growth when they encounter nutrient patches (25).
REGULATION BY NONCODING RNA
Gene regulation via the use of noncoding RNA molecules mainly differs from transcriptional regulation in that it generally regulates genes after transcription of the target gene has occurred and is carried out by an RNA molecule, not a protein (97). RNA-based regulation provides a faster response to environmental stimuli that comes at a lower genomic cost than protein-based regulation but usually does not result in changes in protein abundance that are as large of a magnitude as transcriptional regulation (24, 98). RNA-based regulation encompasses riboswitches, small RNA molecules, and CRISPR RNAs, the last of which will not be discussed here, since oligotrophs rarely have CRISPR arrays (99).
Introduction to Riboswitches and Small Noncoding RNAs
Riboswitches are structural mRNA elements, usually located in the 5′ untranslated region of the RNA, that act in a cis-regulatory way, causing the early termination of transcription which either prevents translation of the gene it is present in (type 1) or causes mRNA degradation (type 2) (100). There are numerous reviews on riboswitches (100–103), but some relevant highlights are presented below. In both cases, the binding of a regulator molecule to the riboswitch changes the riboswitch conformation, which results in the regulatory outcome (101, 104). Another method of riboswitch action involves modulating the stability of the mRNA molecule, with ligand binding exposing an RNase-sensitive site (105, 106). Riboswitches are comprised of two domains, an aptamer domain and an expression platform (107). The regulatory ligand (aptamer) binds to the aptamer domain, which is highly conserved within classes of riboswitches (107). The expression platform is much more variable in sequence and is the portion that changes structure in response to the binding of the molecule to the aptamer domain (107). Many riboswitches act more as “dimmers” than on/off switches, with a gradient of responses to physiochemical factors, such as pH and temperature, as the stability of the riboswitch domain changes at a given physiochemical state (108, 109). There are currently at least 40 known classes of riboswitches, with many more likely to be discovered (102, 110, 111).
Small RNA regulators (sRNAs), or antisense RNAs, play critical regulatory roles in bacteria. For reviews of sRNA categories, functions, strategies for detection, etc. (112–116). The three primary categories of sRNAs are: cis-sRNAs, trans-sRNAs, and sRNAs that regulate proteins (117, 118). cis-sRNAs are located on the complementary strand from their regulatory target and carry out their regulation by binding directly to the mRNA produced from the gene and inhibiting translation (119). These cis-sRNAs usually regulate genes whose product is toxic to the cell at high concentrations (120). trans-sRNAs, in contrast, have limited complementation to the mRNA that they regulate, giving them a broader range of regulatory capacity, and are usually found at different genomic sites than the genes they regulate (114). Their mechanisms of action are also much broader, similar to riboswitches; binding of a trans-sRNA to a target mRNA can inhibit translation from occurring by occluding the ribosome binding site (Shine-Dalgarno sequence), which also often marks the mRNA for degradation (121, 122). They can also have positive effects on the target mRNA by relieving a secondary RNA structure in the mRNA, exposing the ribosome binding site (118). These sRNAs are often expressed during a specific physiological state and coordinate a cellular response to environmental stimuli (118). The binding of these trans-sRNA molecules to their target is often mediated by the ribosomal S1 protein, the Sm-like Hfq protein, or other RNA-binding proteins (123, 124). sRNAs that regulate proteins do so by mimicking RNA or DNA targets of the regulated protein, as in the case of the 6S RNA in E. coli that inhibits RNA polymerase activity (125).
Relevance of RNA-Based Regulation to Oligotrophs
Our primary argument here is that oligotrophs are generally reduced in transcriptional regulation compared to copiotrophs, while retaining other forms of regulation, including RNA-based regulation. This does not necessitate that oligotrophs have a greater abundance of RNA-based regulators on a per-kilobase basis, although this is a distinct possibility as discussed below. Rather, at the very least, the general lack of transcriptional regulation in oligotrophs means that other forms of regulation take on added importance in these cell types (24, 126). To our knowledge, there has not been any direct comparison of the prevalence of RNA-based regulation between aquatic oligotrophs and copiotrophs, which offers an interesting further area of research. However, there are two pieces of evidence that suggest that marine oligotrophs may have larger numbers of RNA regulators than copiotrophs, although this will require further work.
The first piece of evidence comes from Prochlorococcus, where an impressively large number of RNA-based regulators has been reported (126), with over 73% of gene expression being linked to RNA regulators over a diel cycle (56). In addition, one of the two glutamine riboswitches (glutamine type 1) found in Prochlorococcus is unique in that it controls the expression of four genes, in contrast to the one gene generally regulated by glutamine type 1 riboswitches in other, more copiotrophic marine phytoplankton, hinting at an expanded role of this and, potentially, other riboswitches in Prochlorococcus (22). Finally, a high proportion (44%) of the primary transcriptome of two strains of Prochlorococcus, MED4 and MIT9313, are devoted to cis-sRNAs, much more than any other type of transcript, with trans-sRNA only comprising 5 to 9% of the transcriptome (127). Other, nonoligotrophic phytoplankton have much lower proportions of cis-sRNA (15 to 30%) (128).
The second piece of evidence comes from the vast amount of RNA molecules measured in marine metatranscriptomic studies from oligotrophic waters that are involved in regulation. Early studies of metatranscriptomic data found that riboswitches in particular are common and diverse in the environment, both in the oligotrophic open ocean and soil samples (129). At the Hawaii Oceanic Time Series (HOT), many measured transcripts (~16%) were small RNA molecules, many of which were either known to be regulatory or were putatively involved in regulation (130, 131). At multiple other oligotrophic, open ocean sites, a large proportion (~19%) of measured transcripts were found to be unannotated, >75-bp RNA transcripts, most likely regulatory RNA (132). In addition, when some of the most abundant transcripts measured at various sites in the open ocean were examined closely, they were found to be regulatory RNAs such as riboswitches (133).
Examples of Riboswitches in Oligotrophs
SAR11 cells are known to have riboswitches from a variety of riboswitch classes, some of which appear to be unique to SAR11 cells and are of unknown functions (134). One of the best-studied examples is a pair of glycine riboswitches in “Ca. Pelagibacter ubique” strain HTCC1062. These cells are conditionally auxotrophic for glycine and serine as a consequence of gene losses that appear to have been driven by selection for small genome size. They elegantly regulate glycine metabolism with riboswitches to avoid glycine starvation, while channeling excess glycine and glycine precursor molecules to energy metabolism (4, 135). When intracellular glycine is low, the two independent, cis-acting glycine riboswitches repress transcription/translation of GlcB (malate synthase) and the aminomethyltransferase GcvT. As intracellular glycine concentrations rise, GlcB is activated by its riboswitch, channeling excess glyoxylate (a glycine precursor) into the TCA cycle. Higher intracellular glycine concentrations activate the second riboswitch, causing transcription/translation GcvT, which cleaves glycine to produce ATP, NH3, and CO2 (136). This unusual configuration, as seen also in the absence of the PII regulated system for nitrogen uptake in SAR11, appears to replace a common, vital, and complex regulated system with a system composed of fewer genes.
Two other instances of riboswitches being involved in essential metabolic pathways in SAR11 have also been uncovered. The response of Pelagibacter cells to sulfur limitation is largely mediated by riboswitches (69). The four most upregulated genes by Pelagibacter cells under sulfur limitation are all located downstream from S-adenosylmethionine riboswitches, which inhibit the translation of mRNA into protein (69). One of the most upregulated genes, ordL, was found to be located downstream of a conserved motif suggestive of a novel riboswitch (69). The response of Pelagibacter cells to sulfur limitation involves re-allocating sulfur to methionine instead of increasing expression of organosulfur transporter proteins, as was found to be the case for nitrogen and phosphate limitation (69). This was postulated to be due to Pelagibacter cells being adapted to a marine environment where organosulfur compounds are rarely limiting (69). In addition, riboswitches are also thought to be involved in vitamin B1 synthesis in Pelagibacter cells (137). Pelagibacter cells require thiamine, a vitamin B1 precursor, for growth; the putative transporter for thiamine in these cells, ThiV, is located next to a riboswitch that binds thiamine-diphosphate (ThPP; the active form of vitamin B1) (137). The authors of that study theorized that, when ThPP is present in sufficient quantities in the cell, it binds the riboswitch and prevents the transcription and/or translation of ThiV, preventing excess production of thiamine transporters (137).
As in SAR11, oligotrophic cyanobacteria such as Prochlorococcus have multiple instances of riboswitches, many of which are related to vitamin B12 or B1 metabolism, in addition to two types of glutamine riboswitches, as mentioned above (138). The breadth of riboswitches in Prochlorococcus has recently been reviewed at length (22).
Examples of Small Noncoding RNAs in Oligotrophs
In oligotrophic cyanobacteria, there are a plethora of well-characterized sRNAs, many of which are involved in core processes such as photosynthesis and nitrogen metabolism (139). The recent review of Prochlorococcus regulation has in-depth discussion on sRNAs in Prochlorococcus (22). In general, Prochlorococcus tends to primarily utilize cis-sRNAs in regulation, as noted above. The sRNAs in Prochlorococcus have been shown to be involved in light adaptation, nitrogen limitation, and iron limitation, with a range of different sRNAs showing differential expression under different light regimes (126), nitrogen limitation (140), and iron limitation (141). cis-sRNAs are also involved in phage infection by protecting a wide set of mRNAs from degradation by RNase E, which is upregulated during phage infection (142).
In heterotrophic oligotrophic bacteria, there are several instances of sRNAs playing an important regulatory role. In the genomes of SAR86 cells, genome regions containing carbon assimilation pathways were found to be heavily populated with putative sRNAs (143). In the oligotrophic Sphingopyxis granuli strain TFA, isolated from river sediment, more than 90 putative sRNAs have been identified, with the trans-sRNA suhB playing an important role in the regulation of the degradation of tetralin, an environmentally contaminating hydrocarbon (144).
POSTTRANSLATIONAL REGULATION
At the posttranslational level, enzyme activity can be modulated through either covalent modifications (often called posttranslational modifications [PTMs]) or allosteric regulation. This allows for rapid (seconds to minutes) responses to environmental stimuli that, in many cases, are reversible and spare the cell from the machinery needed for a transcriptional response (145). Posttranslational regulation has similar advantages to RNA-based regulation, with the added benefit of a potentially shorter time scale response, since translation is usually the slowest part of gene expression (146).
Introduction to Covalent and Allosteric Posttranslational Regulation
Covalent posttranslational regulation (PTMs) involves the modification of proteins via the addition or removal of certain groups (e.g., phosphate, acetyl, methyl, etc.) or other modifications to amino acid residues (e.g., disulfide-bond formation or cleavage) (147). These modifications result in changes to enzyme function, the formation of protein complexes, etc. (148). There are numerous different types of PTMs, with acetylation and phosphorylation being two of the most widespread mechanisms (148, 149). The usage of PTMs as a regulatory mechanism in bacterial cells is only beginning to be understood but appears to be broadly used across bacterial taxa in diverse processes such as metabolism, protein synthesis, cell cycle, etc. (147–150), although modifications happen at a lower rate in bacteria than eukaryotes and at low stoichiometric levels, making detection challenging (147). In addition, different enrichment steps are needed for measuring different types of modifications, making it challenging to encompass all PTMs in one study. It was long thought that PTMs were only used in eukaryotes, since bacteria were too “simple” to use PTMs (151). Thus, it is only in recent years that global studies of PTMs in a time-resolved manner in response to environmental stimuli have been conducted (150).
Allosteric posttranslational regulation involves the binding of a regulatory molecule to an enzyme at a noncompetitive (or allosteric) site, changing the conformation of the enzyme and thus its activity (152). Allosteric regulation differs from PTMs in that allosteric regulation is generally reversible, while PTMs are generally not reversible. Also, allosteric regulation generally does not require an effector enzyme to carry out the posttranslational regulation, as PTMs do. Similar to PTMs, allosteric regulation of enzyme activity has been understudied to date on a broad scale (153), although it is likely to be widespread (153, 154).
Relevance of Posttranslational Regulation to Oligotrophs
In theory, as with RNA-based regulation, posttranslational regulation might be especially relevant for oligotrophs, especially given the ability to modulate enzyme activity quickly, since translation generally is the longest part of gene expression, as noted before. However, as noted with RNA-based regulation, this does not necessitate that oligotrophs have a greater abundance of posttranslational regulators than copiotrophs, although this is possible. Between the two types of posttranslational regulation, allosteric is the most likely to be enriched in oligotrophs, since PTMs come with the added cost of needing an enzyme to carry out the PTM, which takes up genomic space, as well as the energy cost necessary to carry out the PTM. In addition, the most common type of PTM, phosphorylation, requires the use of phosphate, which is a limiting nutrient in the oligotrophic ocean. Allosteric regulation does not take up any genomic space, does not require energy input, and is readily reversible. Thus, if oligotrophs do have an enrichment in posttranslational regulation compared to copiotrophs, we would predict that allosteric regulation would be the most likely mechanism.
However, the hypothesis that oligotrophs have a greater reliance on allosteric regulation than copiotrophs will be challenging to fully examine on a whole-cell basis (sometimes called the “allosterome”). In fact, there are very few studies of the allosterome of a bacterial species, and the few that have been conducted are not comprehensive of all metabolic pathways in the cell (154–156). The challenges behind studying the allosteromes of cells, especially oligotrophs, are several: (i) the number of potential regulatory interactions in a cell is vast, given the large number of active enzymes in a cell and the large diversity of potentially regulatory small molecules in cells (variable by cell type and growth status [157]); (ii) allosteric regulation usually involves weak interactions, which are difficult to measure (156); and (iii) past large-scale computational studies identifying whole-cell allosteric interactions rely on decades of metabolic studies, something that is not currently available for most oligotrophs (155). Most likely, a whole-cell allosterome study comparing an oligotrophic and copiotrophic species is not plausible in the near future. Instead, as a start, focusing on one or more well-studied metabolic pathways and characterizing the allosteric interactions present in a model marine oligotroph and copiotroph would be a more achievable start, using some of the techniques already employed in model organisms (154–156).
We used our previously discussed functional comparison of marine oligotrophs and copiotrophs (Fig. 2) to address the question of whether genes involved in PTM are more common in oligotrophs than copiotrophs, with our prediction being that oligotrophs would have similar numbers of PTM genes as copiotrophs. We separated genes for known PTMs (acetylation, phosphorylation [kinases], ubiquitination, methylation, glycosylation, adenylation, and peptidases) into a separate category (O:PTM) from other category O (translation) genes. O:PTM genes were found to be at similar genomic proportions in both lifestyle categories, while the remaining category O genes were significantly enriched in oligotrophs (Fig. 2C). This could indicate that regulation via these known PTMs is used at similar rates within both copiotrophs and oligotrophs, in contrast to transcriptional regulation. One difficulty with this approach is that the PTMs used in this analysis have all been discovered and studied almost exclusively in copiotrophs. Further studies are needed to assess the relative importance of regulation at the PTM level in oligotrophs compared to copiotrophs, both at the physiological and the genomic level.
Examples of Posttranslational Regulation in Oligotrophs
Reported instances of posttranslational regulation in oligotrophs are rare, but a few have been reported. Several oligotrophic species from the Sphingomonadales order were found to have numerous instances of PTMs, both under normal growth conditions and in response to starvation, but the identity of the proteins modified and the nature of the modifications are unclear, as these experiments were conducted using two-dimensional electrophoresis methods (158–160). In oligotrophic Nitrosopumilus maritimus SCM1, 6% of the detected proteins were found to be modified by N-terminal acetylation under normal growth (72), which is a lower proportion than found in other archaea (~29%) (161). A recent study of l-alanine metabolism in SAR11 also found evidence for posttranslational regulation of the l-alanine transporter, perhaps via allosteric regulation, although this was not experimentally confirmed (76).
KINETIC REGULATION
All of the types of regulation discussed so far have been part of hierarchical regulation, which modulate protein abundance/activity via the abundance of transcripts or enzymes. Kinetic regulation occurs outside of hierarchical regulation, where enzyme activities are modulated by metabolite-enzyme interactions, completely separate from the abundances of transcripts or enzymes (162). Thus, kinetic regulation is instantaneous and does not require extra genomic space or energy costs.
Introduction to Kinetic Regulation
Kinetic regulation relies on different enzymes having different affinities for the same metabolites. Thus, at low levels of a given metabolite, it might be processed by a high-affinity enzyme, which sends it through one metabolic pathway. At higher concentrations, however, the same metabolite might be processed by an enzyme with lower affinity for the metabolite, but which has a higher activity, resulting in the metabolite being shunted down a different metabolic pathway. This form of regulation is widespread in cells and coordinates the flux of nutrients through cellular metabolism with hierarchical regulation, making it challenging to dissect on a case-by-case basis. Thus, many studies of kinetic regulation have been carried out in model organisms for central pathways on a large scale using modeling approaches. Examples include whole-cell modeling and use of 13C flux analysis in E. coli, which found that kinetic regulation occurs at the level of individual intracellular metabolite concentrations, as well as a whole-cell coordination of metabolite uptake and growth rate (162–164).
Relevance of Kinetic Regulation to Oligotrophs and an Example
As noted previously, forms of regulation that do not require excess genomic space or extra energetic/nutrient costs and are able to respond quickly are likely the most useful to aquatic oligotrophs. Since all of these are true of kinetic regulation, we would predict that kinetic regulation would be especially prevalent in oligotrophs. This was also the prediction of Button, who predicted that oligotrophs, due to the relatively constant nutrient concentrations that they are exposed to, might emphasize kinetic control more than hierarchical regulation (26). This hypothesis, however, will be challenging to study on a global scale, as noted with allosteric regulation, given the difficulties posed with studying kinetic regulation (noted above). However, the one reported instance of kinetic regulation in an aquatic oligotroph (regulation of dimethylsulfoniopropionate, DMSP, and metabolism in SAR11), provides support for our hypothesis that kinetic regulation is more prevalent in oligotrophs than copiotrophs. This regulation is illustrated in Fig. 5 in comparison to the regulation of DMSP metabolism in many copiotrophic bacteria.
FIG 5.

Illustration of the differences between a common copiotrophic strategy of regulating dimethylsulfoniopropionate (DMSP) metabolism and the kinetic regulation of DMSP metabolism found in SAR11 cells. DMS, dimethyl sulfide; MeSH, methanethiol.
DMSP is primarily metabolized in marine bacteria through either a cleavage pathway, which results in the gas dimethylsulfide (DMS) being released, or the demethylation pathway, which results in the incorporation of the sulfur (S) from DMSP into biomass and release of methanethiol gas (73). In copiotrophic bacteria such as Roseobacter species, either DMSP itself or its catabolic products act as inducers of the DMSP lyase or demethylation genes (73). In the SAR11 strain “Ca. Pelagibacter ubique” HTCC1062, DMSP metabolism is under kinetic control instead of being transcriptionally regulated. No transcriptional response was observed in HTCC1062 cells in response to the addition of DMSP, and metabolism of DMSP did not increase upon preconditioning with DMSP (74). Instead, both the demethylation (mediated by DmdA) and cleavage (mediated by DddK) pathways are operational and constitutively expressed, resulting in the two pathways competing for metabolism of DMSP (74). However, DmdA has a higher affinity for DMSP than DddK, resulting in DMSP metabolism primarily being shunted into absorption of S from DMSP until cellular demands for S have been met, when increasing amounts of DMSP are shunted to the cleavage pathway for release as DMS (74). It seems that one of the adaptations of SAR11 to a life of oligotrophy is the replacement of transcriptional regulation of DMSP metabolism with kinetic regulation, reducing the amount of genomic coding and proteome complexity needed and increasing the speed of response.
WHY DO OLIGOTROPHS USE LESS TRANSCRIPTIONAL REGULATION?
One of the primary questions that has yet to be definitively answered is why there is an evolutionary trend in oligotrophs to less transcriptional regulation; below, we propose three potential factors that may have played a role in this evolutionary trend, illustrated in Fig. 6.
FIG 6.

Illustration of three potential factors that may have played a role in driving aquatic oligotrophs to a reduction in transcriptional regulation. See the text for a full discussion.
One viable explanation is that transcriptional regulation has diminished fitness value in many of the niches found in nutrient-limited environments because of the elemental and energetic costs of transcriptional regulation. Cellular resources required for transcriptional regulation include the costs of coding sequences and transcribing and translating regulatory proteins (165). Most oligotrophs have streamlined genomes, with reduced intragenic regions, few pseudogenes and paralogs, an enrichment in nucleotides and amino acids with lower nitrogen content, and few regulatory genes (24). These adaptations are thought to be a strategy to reduce the cellular requirement for essential nutrients such as nitrogen and phosphorous that are limiting in oligotrophic environments (24). The reduction in genome complexity that accompanies the loss of transcriptional regulatory systems would, in theory, result in cells that need fewer macronutrients and less energy to reproduce (166, 167). However, it is an open question of whether the loss of transcriptional regulatory systems results in a large enough conservation of macronutrients and energy to explain the absence of transcriptional regulation in oligotrophs.
Another potential explanation for diminished regulation in oligotrophs lies in the nonmotile lifestyle of many oligotrophic cells. Cells in the ocean inhabit a patchy environment where they encounter ephemeral patches of high nutrient concentrations, interspersed within larger regions of low nutrient concentrations (168). Cells with transcriptional regulation must be able to turn on the genes needed to metabolize nutrients either before the patch dissipates, which can happen within 10 min of the patch forming (169, 170), or before they leave the nutrient patch. This is perhaps one reason why copiotrophs tend to be motile (14, 52), which allows them to locate and occupy nutrient patches and attach to particles, providing enough time to switch on metabolic enzymes. Oligotrophs, on the other hand, are generally nonmotile and/or nonchemotactic (Fig. 2) (14), possibly because their small cell size renders motility unfeasible due to Brownian effects, or because motility, like all genome complexity, seems to lose some of its fitness value in niches where resources are expensive (171, 172). Thus, nonmotile oligotrophs have no means to direct their drift through the water, resulting in a reduced amount of time spent in nutrient patches and an absence of time spent attached to particles (76, 169). If nonmotile oligotrophs relied on transcriptional regulation to turn on the necessary metabolic enzymes for nutrients encountered in patches, the time required for these systems to initiate would preclude them from being able to make full use of the nutrients they encounter, as they would exit the patch before their metabolic systems are fully activated (76). The constitutive expression of most genes in oligotrophs, especially those related to carbon metabolism, means that the oligotrophs are able to instantly utilize whatever nutrients they encounter in a patch before they exit it, which reduces the cost of missed opportunities that nonmotile oligotrophs would otherwise incur.
On the other hand, the strategy of constitutively expressing genes for carbon metabolism incurs the costs of synthesizing proteins that have no use when the corresponding nutrient is absent. This strategy would not make much sense for a heterotrophic cell in a niche where the composition of the nutrient field varied qualitatively on time scales much larger than transcriptional response times. However, this strategy would make sense for cells that specialize in harvesting organic compounds that are not special, i.e., the small molecules and polymers common to all cell types. Enzyme multifunctionality has been reported in streamlined oligotrophs, such as SAR11 (173–175), which in principle broadens substrate range at no regulatory cost and potentially could be a factor offsetting the costs of deregulation strategies. This may be why heterotrophic oligotrophs are especially lacking in transcriptional regulation of carbon oxidation functions: there is a much wider diversity of carbon compounds available than inorganic nutrients. This strategy of constitutively expressing metabolic genes has been observed previously in E. coli cells that become adapted to a fluctuating environment as a memory mechanism or adaptive plasticity and has also been observed in thermal priming of dinoflagellates (39, 176). But instead of a short-term, epigenetic plasticity, oligotrophs seem to have undergone genomic adaptations to deal with the consistently low-nutrient, fluctuating ocean environment they experience.
Another potential reason for the lack of transcriptional regulation in oligotrophs revolves around the low ambient nutrient concentrations in the open ocean. If fluctuations in ambient nutrient concentrations tended to be below the threshold concentrations necessary to turn on regulatory systems, then, even if there was a potential advantage, regulation would not be effective. The threshold concentration above which motility is induced in marine bacteria has been found to be 0.001% (wt/vol) tryptic soy broth (177). A similar result was found in E. coli, where chemotaxis was not induced under a threshold concentration of a variety of compounds, and was only induced upon increases in nutrient concentrations that followed the Weber law (178). In marine diatoms, chemotaxis toward silicate particles was only induced when the silicate concentration was above a certain threshold (179). It may be that motility, with its large energy expenditure, is more tightly controlled based on nutrient concentrations than metabolic enzymes to diminish the potential for large costs to cells without benefits. This would argue for a second option, that cells can sense even low concentrations of nutrients, but do not expend energy to acquire them because of the risks (180).
The discussion above highlights some of the complexities in understanding why selection has favored different regulatory strategies depending on the pathway, the organism, and the niche. While some trends, for example the lesser amount of transcriptional regulation in oligotrophs, are clear, there are only a few examples from the cell biology of oligotrophs to illustrate other key concepts, and therefore our understanding of these issues will have to await investigations that find ways to probe these questions across a larger selection of different cell types.
IMPLICATIONS OF THE OLIGOTROPHIC REGULATORY STRATEGY AND FUTURE PROSPECTS
Much of this review has focused on marine oligotrophs, since the majority of experimental studies of regulation in oligotrophs have been carried out in marine oligotrophs. Freshwater oligotrophs with streamlined features have been reported, as with the freshwater clade of SAR11 (181), but regulation in these organisms has been less studied. We would expect to see similar patterns of regulation in freshwater oligotrophs as well, although this requires further confirmation. Similarly, in soil, it was long thought that large genomes were the norm, until the discovery of a highly abundant soil microbe with hallmarks of a streamlined genome (182). The adaptive pressures of soil environments are very different from aquatic environments, so it remains to be seen whether the same patterns of adaptation and genome evolution reported in marine oligotrophs will be repeated in soil oligotrophs. However, a previously described study that examined functional enrichment by lifestyle category across all environments (animal-associated, soil, marine, etc.) found a very strong depletion of genes involved in transcriptional regulation in oligotrophs (27). Thus, we predict that the pattern of regulation described here is generalizable to all oligotrophs across all environments. Nonetheless, it is clear that oligotrophs use transcriptional regulation when it is selectively advantageous, as described previously, and that different oligotrophs have different nuances of this general regulatory strategy.
In addition, much of our review has focused on two oligotrophic groups, SAR11 and Prochlorococcus, since most of the available information about regulation in aquatic oligotrophs has come from studies of these important model organisms, which are the most abundant microbial lineages in aquatic environments worldwide. As noted in the introduction, however, the oligotrophy/copiotrophy concept idealizes a spectrum of ecological strategies, so we would not predict that aquatic microbes are strictly dichotomous with respect to the alternate strategies we describe. Rather, the study of SAR11 and Prochlorococcus, which are the most successful oligotrophs in the world, offers broadly generalizable conclusions about the oligotrophic lifestyle that will likely vary based on a given oligotroph’s niche.
One intriguing consequence of reduced transcriptional regulation in marine oligotrophs is that they may have less control over influxes of nutrients and growth rate, which is not a problem as long as their environment remains oligotrophic but could lead to metabolite imbalances in artificial environments. Increases in intracellular metabolites to toxic levels has been proposed as a cause of growth inhibition in oligotrophs exposed to high nutrient concentrations (175, 183). On the other hand, several instances have been reported in which very high intracellular accumulations of transported metabolites are metabolized later, providing these cells a means of exploiting transient nutrient surfeits. This “excess uptake” strategy has been observed in bulk marine microbial communities for glycine betaine (184), for DMSP in cultured SAR11 (74), and has been proposed to cause increases in polyamine storage and cell size, presumably from changes in turgor pressure, in SAR11 cells exposed to high polyamine levels (175).
The expansion of microbial diversity by metagenomics has revealed many new uncultivated lineages, including major new groups of abundant organisms that have highly reduced genomes such as the Patescibacteria and DPANN superphyla. Interestingly, although evidence about regulatory strategies in these two large groups is minimal, the genomic evidence available for Patescibacteria indicates many hallmarks of the oligotrophic strategy described above, with severe reductions in transcriptional regulatory proteins, two-component systems, and chemotaxis having been reported (185). In addition, the majority of reported Patescibacteria and DPANN genomes contain fewer σ-factors than expected per genome size (mean σ-factors/kb of 0.0066 and 0.043 for Patescibacteria and DPANN, respectively; one would expect closer to 0.01 for these genome sizes) based on a previously reported model fit for σ-factors and genome size (24, 186). In a number of cases, it has been established that Patescibacteria and DPANN superphyla are involved in symbioses (187, 188), providing a ready explanation for reductive genome evolution.
The evidence reviewed above indicates a dichotomy in paths to microbial success, wherein one path leads to versatile cells that can exploit environmental fluctuations, and incidentally grow well in labs, whereas the other path, seemingly more common, leads to success when relative stability in nutrient production and sustained competition combine to hold nutrient fluctuations in narrow ranges. Generally, when microbiologists observe very small genomes in nature, they suspect either mutualistic or parasitic symbioses. The patterns of gene regulation we report suggest an alternate explanation: if simplicity/efficiency and complexity/versatility represent polar strategies for success, is it possible that some cells might be prospering in nature that are even more simple than the global oligotrophs that have become model organisms? By recognizing that the imprint of oligotrophy on regulation is not simply diminished transcriptional regulation but instead fine network tuning by selection to achieve success in a different niche, we might be able to recognize more extreme examples of reductive evolution that are due to environmental adaptation rather than coevolution.
CONCLUSIONS
Like many aspects of biology, regulatory strategies of microorganisms are more complicated than was originally thought. With the increased awareness of the importance of oligotrophic microbes to environmental processes, more and more oligotrophs are being brought into culture, where the nuances of their regulatory strategies can be studied more closely. The realization that many abundant oligotrophs have reduced transcriptional regulation and constitutively express most of their genes for carbon oxidation has led to the testable hypothesis that oligotrophs instead rely primarily on posttranscriptional regulation. As has been noted above, many types of posttranscriptional regulation have only begun to be explored, especially in oligotrophs. For example, the breadth of potential riboswitches in microbes is still being uncovered and offers a promising avenue for further discoveries of posttranscriptional regulation. Both posttranslational and metabolic regulation are other vast, underexplored fields, especially in oligotrophs. We hope this review has communicated to readers that there are broad trends in regulation that distinguish oligotrophs from copiotrophs, even if as yet we do not fully understand the selective pressures that drive these trends or the integration of different types of regulation in relation to these two very different trophic strategies.
ACKNOWLEDGMENTS
We thank Chris Suffridge and Holger Buchholz for their helpful comments that improved the manuscript. This research was partially supported by the Simons Foundation International BIOS-SCOPE program.
This study was funded by the National Science Foundation grant IOS-1838445.
Biographies

Stephen E. Noell is a Postdoctoral Fellow at the University of Waikato in Hamilton, New Zealand. Dr. Noell was born and raised in Oklahoma and received his B.S. from Geneva College in Pennsylvania. His undergraduate research involved culturing novel psycrophilic bacteria from a creek influenced by acid-mine drainage. He earned his PhD in 2021 with Dr. Giovannoni at Oregon State University, studying the physiology and metabolism of the highly abundant and ubiquitous marine bacterial group SAR11. His current postdoctoral research uses microbial community analyses and novel culturing methods to study the microbes that inhabit geothermal areas, especially in Antarctica.

Ferdi L. Hellweger was educated at Northeastern University, the University of Texas and Columbia University. He previously served as Project Manager at the water quality modeling consultant firm HydroQual, and as an Assistant and Associate Professor at Northeastern University. He is presently Professor at the Technical University of Berlin. Dr. Hellweger’s research and teaching is in the area of surface water quality, biogeochemistry, and the microbial ecology of lakes, rivers, and marine systems. He specializes in the development and application of mathematical models, and since his PhD research two decades ago, his work has focused on bringing modern biology knowledge into operational models that are used for research and management.

Ben Temperton is an associate professor of Microbiology at the University of Exeter. He received his PhD from Queens University Belfast in marine microbiology before completing a postdoctoral position at Oregon State University with Prof. Steve Giovannoni. His current research investigates how host-virus interactions shape global biogeochemical cycles, as well as the application of clinical phage therapy to treat patients with antibiotic-resistant infections.

Stephen J. Giovannoni is a Distinguished Professor and the Head of the Department of Microbiology at Oregon State University. Dr. Giovannoni was born and raised in San Francisco, did undergraduate work at UC San Diego, and received a master’s degree from Boston University with Lynn Margulis. He received his Ph.D. in 1984 with Richard Castenholz for isolating and describing novel thermophilic bacteria from Yellowstone. He left Oregon to join Norman Pace’s group at Indiana University to work on the development of molecular techniques for studying microbial ecology. Since leaving Indiana for Oregon State University, his research has focused on ocean microbiology and the carbon cycle.
REFERENCES
- 1.Moreno R, Rojo F. 2023. The importance of understanding the regulation of bacterial metabolism. Environ Microbiol 25:54–58. 10.1111/1462-2920.16123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poindexter J. 1981. Oligotrophy: fast and famine existence. Adv Microb Ecol 5:63–89. [Google Scholar]
- 3.Moore LR, Coe A, Zinser ER, Saito MA, Sullivan MB, Lindell D, Frois-Moniz K, Waterbury J, Chisholm SW. 2007. Culturing the marine cyanobacterium Prochlorococcus: Prochlorococcus culturing. Limnol Oceanogr Methods 5:353–362. 10.4319/lom.2007.5.353. [DOI] [Google Scholar]
- 4.Carini P, Steindler L, Beszteri S, Giovannoni SJ. 2013. Nutrient requirements for growth of the extreme oligotroph “Candidatus Pelagibacter ubique” HTCC1062 on a defined medium. ISME J 7:592–602. 10.1038/ismej.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novitsky JA, Morita RY. 1978. Possible strategy for the survival of marine bacteria under starvation conditions. Mar Biol 48:289–295. 10.1007/BF00397156. [DOI] [Google Scholar]
- 6.Morita RY. 1982. Starvation-survival of heterotrophs in the marine environment, p 171–198. In Marshall KC (ed), Advances in microbial ecology. Springer US, Boston, MA. [Google Scholar]
- 7.Moriarty DJW, Bell RT. 1993. Bacterial growth and starvation in aquatic environments, p 25–53. In Kjelleberg S (ed), Starvation in bacteria. Springer US, Boston, MA. [Google Scholar]
- 8.Lever MA, Rogers KL, Lloyd KG, Overmann J, Schink B, Thauer RK, Hoehler TM, Jørgensen BB. 2015. Life under extreme energy limitation: a synthesis of laboratory- and field-based investigations. FEMS Microbiol Rev 39:688–728. 10.1093/femsre/fuv020. [DOI] [PubMed] [Google Scholar]
- 9.Ho A, Di Lonardo DP, Bodelier PLE. 2017. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiol Ecol 93. 10.1093/femsec/fix006. [DOI] [PubMed] [Google Scholar]
- 10.Grime JP, Pierce S. 2012. The evolutionary strategies that shape ecosystems. John Wiley & Sons, Inc, New York, NY. https://books.google.co.nz/books?id=kDnGU-elg3kC. [Google Scholar]
- 11.Santillan E, Seshan H, Constancias F, Wuertz S. 2019. Trait-based life-history strategies explain succession scenario for complex bacterial communities under varying disturbance. Environ Microbiol 21:3751–3764. 10.1111/1462-2920.14725. [DOI] [PubMed] [Google Scholar]
- 12.Romillac N, Santorufo L. 2021. Transferring concepts from plant to microbial ecology: a framework proposal to identify relevant bacterial functional traits. Soil Biol Biochem 162:108415. 10.1016/j.soilbio.2021.108415. [DOI] [Google Scholar]
- 13.Westoby M, Gillings MR, Madin JS, Nielsen DA, Paulsen IT, Tetu SG. 2021. Trait dimensions in bacteria and archaea compared to vascular plants. Ecol Lett 24:1487–1504. 10.1111/ele.13742. [DOI] [PubMed] [Google Scholar]
- 14.Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S, DeMaere MZ, Ting L, Ertan H, Johnson J, Ferriera S, Lapidus A, Anderson I, Kyrpides N, Munk AC, Detter C, Han CS, Brown MV, Robb FT, Kjelleberg S, Cavicchioli R. 2009. The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA 106:15527–15533. 10.1073/pnas.0903507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, Gonzalez JM, Luo H, Wright JJ, Landry ZC, Hanson NW, Thompson BP, Poulton NJ, Schwientek P, Acinas SG, Giovannoni SJ, Moran MA, Hallam SJ, Cavicchioli R, Woyke T, Stepanauskas R. 2013. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc Natl Acad Sci USA 110:11463–11468. 10.1073/pnas.1304246110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goericke R, Welschmeyer NA. 1993. The marine prochlorophyte Prochlorococcus contributes significantly to phytoplankton biomass and primary production in the Sargasso Sea. Deep Sea Res Part I Oceanogr Res Pap 40:2283–2294. 10.1016/0967-0637(93)90104-B. [DOI] [Google Scholar]
- 17.Morris RM, Rappé MS, Connon SA, Vergin KL, Siebold WA, Carlson CA, Giovannoni SJ. 2002. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 420:806–810. [DOI] [PubMed] [Google Scholar]
- 18.Partensky F, Hess WR, Vaulot D. 1999. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol Mol Biol Rev 63:106–127. 10.1128/MMBR.63.1.106-127.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Partensky F, Garczarek L. 2010. Prochlorococcus: advantages and limits of minimalism. Annu Rev Mar Sci 2:305–331. 10.1146/annurev-marine-120308-081034. [DOI] [PubMed] [Google Scholar]
- 20.Heldal M, Scanlan DJ, Norland S, Thingstad F, Mann NH. 2003. Elemental composition of single cells of various strains of marine Prochlorococcus and Synechococcus using X-ray microanalysis. Limnol Oceanogr 48:1732–1743. 10.4319/lo.2003.48.5.1732. [DOI] [Google Scholar]
- 21.Flombaum P, Gallegos JL, Gordillo RA, Rincon J, Zabala LL, Jiao N, Karl DM, Li WKW, Lomas MW, Veneziano D, Vera CS, Vrugt JA, Martiny AC. 2013. Present and future global distributions of the marine cyanobacteria Prochlorococcus and Synechococcus. Proc Natl Acad Sci USA 110:9824–9829. 10.1073/pnas.1307701110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambrecht SJ, Steglich C, Hess WR. 2020. A minimum set of regulators to thrive in the ocean. FEMS Microbiol Rev 44:232–252. 10.1093/femsre/fuaa005. [DOI] [PubMed] [Google Scholar]
- 23.Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappé MS, Short JM, Carrington JC, Mathur EJ. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245. 10.1126/science.1114057. [DOI] [PubMed] [Google Scholar]
- 24.Giovannoni SJ, Cameron Thrash J, Temperton B. 2014. Implications of streamlining theory for microbial ecology. ISME J 8:1553–1565. 10.1038/ismej.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giovannoni SJ. 2017. SAR11 bacteria: the most abundant plankton in the oceans. Annu Rev Mar Sci 9:231–255. 10.1146/annurev-marine-010814-015934. [DOI] [PubMed] [Google Scholar]
- 26.Button DK. 2004. Life in extremely dilute environments: the major role of oligobacteria, p 160–168. In Bull AT (ed), Microbial diversity and bioprospecting. American Society of Microbiology, Washington, DC. [Google Scholar]
- 27.Weissman JL, Hou S, Fuhrman JA. 2021. Estimating maximal microbial growth rates from cultures, metagenomes, and single cells via codon usage patterns. Proc Natl Acad Sci USA 118:e2016810118. 10.1073/pnas.2016810118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klappenbach JA, Dunbar JM, Schmidt TM. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ Microbiol 66:1328–1333. 10.1128/AEM.66.4.1328-1333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zoccarato L, Sher D, Miki T, Segrè D, Grossart H-P. 2022. A comparative whole-genome approach identifies bacterial traits for marine microbial interactions. Commun Biol 5:276. 10.1038/s42003-022-03184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Neilson JW, Kushwaha P, Maier RM, Barberán A. 2021. Life-history strategies of soil microbial communities in an arid ecosystem. ISME J 15:649–657. 10.1038/s41396-020-00803-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westoby M, Nielsen DA, Gillings MR, Litchman E, Madin JS, Paulsen IT, Tetu SG. 2021. Cell size, genome size, and maximum growth rate are near-independent dimensions of ecological variation across bacteria and archaea. Ecol Evol 11:3956–3976. 10.1002/ece3.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schut F, Prins RA, Gottschal JC. 1997. Oligotrophy and pelagic marine bacteria: facts and fiction. Aquat Microb Ecol 12:177–202. 10.3354/ame012177. [DOI] [Google Scholar]
- 33.Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu Rev Microbiol 57:369–394. 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 34.Giovannoni S, Stingl U. 2007. The importance of culturing bacterioplankton in the “omics” age. Nat Rev Microbiol 5:820–826. 10.1038/nrmicro1752. [DOI] [PubMed] [Google Scholar]
- 35.Poindexter JS. 1964. Biological properties and classification of the Caulobacter group. Bacteriol Rev 28:231–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilhelm RC. 2018. Following the terrestrial tracks of Caulobacter: redefining the ecology of a reputed aquatic oligotroph. ISME J 12:3025–3037. 10.1038/s41396-018-0257-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pardee AB, Prestidge LS. 1961. The initial kinetics of enzyme induction. Biochim Biophys Acta 49:77–88. [DOI] [PubMed] [Google Scholar]
- 38.Balakrishnan R, Mori M, Segota I, Zhang Z, Aebersold R, Ludwig C, Hwa T. 2022. Principles of gene regulation quantitatively connect DNA to RNA and proteins in bacteria. Science 378:eabk2066. 10.1126/science.abk2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arromrak BS, Li Z, Gaitán-Espitia JD. 2022. Adaptive strategies and evolutionary responses of microbial organisms to changing oceans. Front Mar Sci 9:864797. 10.3389/fmars.2022.864797. [DOI] [Google Scholar]
- 40.Burley SK, Kamada K. 2002. Transcription factor complexes. Curr Opin Struct Biol 12:225–230. 10.1016/S0959-440X(02)00314-7. [DOI] [PubMed] [Google Scholar]
- 41.van Hijum SAFT, Medema MH, Kuipers OP. 2009. Mechanisms and evolution of control logic in prokaryotic transcriptional regulation. Microbiol Mol Biol Rev 73:481–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruber TM, Gross CA. 2003. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol 57:441–466. [DOI] [PubMed] [Google Scholar]
- 43.Sharma UK, Chatterji D. 2010. Transcriptional switching in Escherichia coli during stress and starvation by modulation of σ70 activity. FEMS Microbiol Rev 34:646–657. 10.1111/j.1574-6976.2010.00223.x. [DOI] [PubMed] [Google Scholar]
- 44.Stock AM, Robinson VL, Goudreau PN. 2000. Two-component signal transduction. Annu Rev Biochem 69:183–215. 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 45.Held NA, McIlvin MR, Moran DM, Laub MT, Saito MA. 2019. Unique patterns and biogeochemical relevance of two-component sensing in marine bacteria. mSystems 4:e00317-18. 10.1128/mSystems.00317-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goulian M. 2010. Two-component signaling circuit structure and properties. Curr Opin Microbiol 13:184–189. 10.1016/j.mib.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Österberg S, del Peso-Santos T, Shingler V. 2011. Regulation of alternative sigma factor use. Annu Rev Microbiol 65:37–55. 10.1146/annurev.micro.112408.134219. [DOI] [PubMed] [Google Scholar]
- 48.Feklístov A, Sharon BD, Darst SA, Gross CA. 2014. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol 68:357–376. [DOI] [PubMed] [Google Scholar]
- 49.Scanlan DJ, Ostrowski M, Mazard S, Dufresne A, Garczarek L, Hess WR, Post AF, Hagemann M, Paulsen I, Partensky F. 2009. Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev 73:249–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Graham ED, Tully BJ. 2021. Marine Dadabacteria exhibit genome streamlining and phototrophy-driven niche partitioning. ISME J 15:1248–1256. 10.1038/s41396-020-00834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tully BJ, Graham ED, Heidelberg JF. 2018. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data 5:170203. 10.1038/sdata.2017.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiriac M, Haber M, Salcher MM. 2023. Adaptive genetic traits in pelagic freshwater microbes. Environ Microbiol 25:606–641. 10.1111/1462-2920.16313. [DOI] [PubMed] [Google Scholar]
- 53.Lankiewicz TS, Cottrell MT, Kirchman DL. 2016. Growth rates and rRNA content of four marine bacteria in pure cultures and in the Delaware estuary. ISME J 10:823–832. 10.1038/ismej.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cottrell MT, Kirchman DL. 2016. Transcriptional control in marine copiotrophic and oligotrophic bacteria with streamlined genomes. Appl Environ Microbiol 82:6010–6018. 10.1128/AEM.01299-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sowell SM, Norbeck AD, Lipton MS, Nicora CD, Callister SJ, Smith RD, Barofsky DF, Giovannoni SJ. 2008. Proteomic analysis of stationary phase in the marine bacterium “Candidatus Pelagibacter ubique.” Appl Environ Microbiol 74:4091–4100. 10.1128/AEM.00599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waldbauer JR, Rodrigue S, Coleman ML, Chisholm SW. 2012. Transcriptome and proteome dynamics of a light-dark synchronized bacterial cell cycle. PLoS One 7:e43432. 10.1371/journal.pone.0043432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Becker KW, Harke MJ, Mende DR, Muratore D, Weitz JS, DeLong EF, Dyhrman ST, Van Mooy BAS. 2021. Combined pigment and metatranscriptomic analysis reveals highly synchronized diel patterns of phenotypic light response across domains in the open oligotrophic ocean. ISME J 15:520–533. 10.1038/s41396-020-00793-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ostrowski M, Cavicchioli R, Blaauw M, Gottschal JC. 2001. Specific growth rate plays a critical role in hydrogen peroxide resistance of the marine oligotrophic ultramicrobacterium Sphingomonas alaskensis strain RB2256. Appl Environ Microbiol 67:1292–1299. 10.1128/AEM.67.3.1292-1299.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eguchi M, Nishikawa T, Macdonald K, Cavicchioli R, Gottschal JC, Kjelleberg S. 1996. Responses to stress and nutrient availability by the marine ultramicrobacterium Sphingomonas sp. Appl Environ Microbiol 62:1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi Y, McCarren J, DeLong EF. 2012. Transcriptional responses of surface water marine microbial assemblages to deep-sea water amendment: microbial transcriptome responses to deep-water addition. Environ Microbiol 14:191–206. 10.1111/j.1462-2920.2011.02598.x. [DOI] [PubMed] [Google Scholar]
- 61.Sorek R, Cossart P. 2010. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet 11:9–16. 10.1038/nrg2695. [DOI] [PubMed] [Google Scholar]
- 62.Schweder T, Markert S, Hecker M. 2008. Proteomics of marine bacteria. Electrophoresis 29:2603–2616. 10.1002/elps.200800009. [DOI] [PubMed] [Google Scholar]
- 63.Haider S, Pal R. 2013. Integrated analysis of transcriptomic and proteomic data. CG 14:91–110. 10.2174/1389202911314020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taniguchi Y, Choi PJ, Li G-W, Chen H, Babu M, Hearn J, Emili A, Xie XS. 2010. Quantifying Escherichia coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329:533–538. 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vogel C, Marcotte EM. 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13:227–232. 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moran MA, Kujawinski EB, Stubbins A, Fatland R, Aluwihare LI, Buchan A, Crump BC, Dorrestein PC, Dyhrman ST, Hess NJ, Howe B, Longnecker K, Medeiros PM, Niggemann J, Obernosterer I, Repeta DJ, Waldbauer JR. 2016. Deciphering ocean carbon in a changing world. Proc Natl Acad Sci USA 113:3143–3151. 10.1073/pnas.1514645113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith DP, Kitner JB, Norbeck AD, Clauss TR, Lipton MS, Schwalbach MS, Steindler L, Nicora CD, Smith RD, Giovannoni SJ. 2010. Transcriptional and translational regulatory responses to iron limitation in the globally distributed marine bacterium “Candidatus Pelagibacter ubique.” PLoS One 5:e10487. 10.1371/journal.pone.0010487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith DP, Thrash JC, Nicora CD, Lipton MS, Burnum-Johnson KE, Carini P, Smith RD, Giovannoni SJ. 2013. Proteomic and transcriptomic analyses of “Candidatus Pelagibacter ubique” describe the first PII-independent response to nitrogen limitation in a free-living alphaproteobacterium. mBio 4:e00133-12. 10.1128/mBio.00133-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith DP, Nicora CD, Carini P, Lipton MS, Norbeck AD, Smith RD, Giovannoni SJ. 2016. Proteome remodeling in response to sulfur limitation in “Candidatus Pelagibacter ubique.” mSystems 1:e00068-16. 10.1128/mSystems.00068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie Z-X, Yan K-Q, Kong L-F, Gai Y-B, Jin T, He Y-B, Wang Y-Y, Chen F, Lin L, Lin Z-L, Xu H-K, Shao Z-Z, Liu S-Q, Wang D-Z. 2022. Metabolic tuning of a stable microbial community in the surface oligotrophic Indian Ocean revealed by integrated meta-omics. Mar Life Sci Technol 4:277–290. 10.1007/s42995-021-00119-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santoro AE, Dupont CL, Richter RA, Craig MT, Carini P, McIlvin MR, Yang Y, Orsi WD, Moran DM, Saito MA. 2015. Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc Natl Acad Sci USA 112:1173–1178. 10.1073/pnas.1416223112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qin W, Amin SA, Lundeen RA, Heal KR, Martens-Habbena W, Turkarslan S, Urakawa H, Costa KC, Hendrickson EL, Wang T, Beck DA, Tiquia-Arashiro SM, Taub F, Holmes AD, Vajrala N, Berube PM, Lowe TM, Moffett JW, Devol AH, Baliga NS, Arp DJ, Sayavedra-Soto LA, Hackett M, Armbrust EV, Ingalls AE, Stahl DA. 2018. Stress response of a marine ammonia-oxidizing archaeon informs physiological status of environmental populations. ISME J 12:508–519. 10.1038/ismej.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Curson ARJ, Todd JD, Sullivan MJ, Johnston AWB. 2011. Catabolism of dimethylsulphoniopropionate: microorganisms, enzymes and genes. Nat Rev Microbiol 9:849–859. 10.1038/nrmicro2653. [DOI] [PubMed] [Google Scholar]
- 74.Sun J, Todd JD, Thrash JC, Qian Y, Qian MC, Temperton B, Guo J, Fowler EK, Aldrich JT, Nicora CD, Lipton MS, Smith RD, De Leenheer P, Payne SH, Johnston AWB, Davie-Martin CL, Halsey KH, Giovannoni SJ. 2016. The abundant marine bacterium Pelagibacter simultaneously catabolizes dimethylsulfoniopropionate to the gases dimethyl sulfide and methanethiol. Nat Microbiol 1:16065. 10.1038/nmicrobiol.2016.65. [DOI] [PubMed] [Google Scholar]
- 75.Giovannoni SJ, Halsey KH, Saw J, Muslin O, Suffridge CP, Sun J, Lee C-P, Moore ER, Temperton B, Noell SE. 2019. A parasitic arsenic cycle that shuttles energy from phytoplankton to heterotrophic bacterioplankton. mBio 10:e00246-19. 10.1128/mBio.00246-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Noell SE, Brennan E, Washburn Q, Davis EW, Hellweger FL, Giovannoni SJ. 2023. Differences in the regulatory strategies of marine oligotrophs and copiotrophs reflect differences in motility. Environ Microbiol 46:1721–1728. 10.1111/1462-2920.16357. [DOI] [PubMed] [Google Scholar]
- 77.Switzer A, Brown DR, Wigneshweraraj S. 2018. New insights into the adaptive transcriptional response to nitrogen starvation in Escherichia coli. Biochem Soc Trans 46:1721–1728. 10.1042/BST20180502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chekabab SM, Jubelin G, Dozois CM, Harel J. 2014. PhoB activates Escherichia coli O157:H7 virulence factors in response to inorganic phosphate limitation. PLoS One 9:e94285. 10.1371/journal.pone.0094285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poindexter JS. 1981. Oligotrophy, p 63–89. In Alexander M (ed), Advances in microbial ecology. Springer US, Boston, MA. [Google Scholar]
- 80.Ottesen EA, Young CR, Gifford SM, Eppley JM, Marin R, Schuster SC, Scholin CA, DeLong EF. 2014. Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science 345:207–212. 10.1126/science.1252476. [DOI] [PubMed] [Google Scholar]
- 81.Aylward FO, Eppley JM, Smith JM, Chavez FP, Scholin CA, DeLong EF. 2015. Microbial community transcriptional networks are conserved in three domains at ocean basin scales. Proc Natl Acad Sci USA 112:5443–5448. 10.1073/pnas.1502883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ottesen EA, Young CR, Eppley JM, Ryan JP, Chavez FP, Scholin CA, DeLong EF. 2013. Pattern and synchrony of gene expression among sympatric marine microbial populations. Proc Natl Acad Sci USA 110:E488–E497. 10.1073/pnas.1222099110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sharma AK, Becker JW, Ottesen EA, Bryant JA, Duhamel S, Karl DM, Cordero OX, Repeta DJ, DeLong EF. 2014. Distinct dissolved organic matter sources induce rapid transcriptional responses in coexisting populations of Prochlorococcus, Pelagibacter, and the OM60 clade: metatranscriptomics of DOM perturbations. Environ Microbiol 16:2815–2830. 10.1111/1462-2920.12254. [DOI] [PubMed] [Google Scholar]
- 84.Mou X, Vila-Costa M, Sun S, Zhao W, Sharma S, Moran MA. 2011. Metatranscriptomic signature of exogenous polyamine utilization by coastal bacterioplankton: polyamine-transforming genes in marine bacterial communities. Environ Microbiol Rep 3:798–806. 10.1111/j.1758-2229.2011.00289.x. [DOI] [PubMed] [Google Scholar]
- 85.Halsey KH, Carter AE, Giovannoni SJ. 2012. Synergistic metabolism of a broad range of C1 compounds in the marine methylotrophic bacterium HTCC2181: C1 metabolism in the marine isolate HTCC2181. Environ Microbiol 14:630–640. 10.1111/j.1462-2920.2011.02605.x. [DOI] [PubMed] [Google Scholar]
- 86.Gifford SM, Becker JW, Sosa OA, Repeta DJ, DeLong EF. 2016. Quantitative transcriptomics reveals the growth- and nutrient-dependent response of a streamlined marine methylotroph to methanol and naturally occurring dissolved organic matter. mBio 7:e01279-16. 10.1128/mBio.01279-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carini P, Dupont CL, Santoro AE. 2018. Patterns of thaumarchaeal gene expression in culture and diverse marine environments. Environ Microbiol 20:2112–2124. 10.1111/1462-2920.14107. [DOI] [PubMed] [Google Scholar]
- 88.Pellitteri-Hahn MC, Halligan BD, Scalf M, Smith L, Hickey WJ. 2011. Quantitative proteomic analysis of the chemolithoautotrophic bacterium Nitrosomonas europaea: comparison of growing- and energy-starved cells. J Proteomics 74:411–419. 10.1016/j.jprot.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 89.Nakagawa T, Stahl DA. 2013. Transcriptional response of the archaeal ammonia oxidizer Nitrosopumilus maritimus to low and environmentally relevant ammonia concentrations. Appl Environ Microbiol 79:6911–6916. 10.1128/AEM.02028-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palinska KA, Laloui W, Bédu S, Loiseaux-de Goer S, Castets AM, Rippka R, Tandeau de Marsac N. 2002. The signal transducer P(II) and bicarbonate acquisition in Prochlorococcus marinus PCC 9511, a marine cyanobacterium naturally deficient in nitrate and nitrite assimilation. Microbiology 148(Pt 8):2405–2412. 10.1099/00221287-148-8-2405. [DOI] [PubMed] [Google Scholar]
- 91.García-Fernández JM, de Marsac NT, Diez J. 2004. Streamlined regulation and gene loss as adaptive mechanisms in Prochlorococcus for optimized nitrogen utilization in oligotrophic environments. Microbiol Mol Biol Rev 68:630–638. 10.1128/MMBR.68.4.630-638.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hondele M, Sachdev R, Heinrich S, Wang J, Vallotton P, Fontoura BMA, Weis K. 2019. DEAD-box ATPases are global regulators of phase-separated organelles. Nature 573:144–148. 10.1038/s41586-019-1502-y. [DOI] [PubMed] [Google Scholar]
- 93.Yair Y, Michaux C, Biran D, Bernhard J, Vogel J, Barquist L, Ron EZ. 2022. Cellular RNA targets of cold shock proteins CspC and CspE and their importance for serum resistance in septicemic Escherichia coli. mSystems 7:e00086-22. 10.1128/msystems.00086-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carini P, White AE, Campbell EO, Giovannoni SJ. 2014. Methane production by phosphate-starved SAR11 chemoheterotrophic marine bacteria. Nat Commun 5. 10.1038/ncomms5346. [DOI] [PubMed] [Google Scholar]
- 95.Steindler L, Schwalbach MS, Smith DP, Chan F, Giovannoni SJ. 2011. Energy-starved “Candidatus Pelagibacter ubique” substitutes light-mediated ATP production for endogenous carbon respiration. PLoS One 6:e19725. 10.1371/journal.pone.0019725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lennon JT, Jones SE. 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9:119–130. 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 97.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nudler E. 2004. The riboswitch control of bacterial metabolism. Trends Biochem Sci 29:11–17. 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 99.Thrash JC, Temperton B, Swan BK, Landry ZC, Woyke T, DeLong EF, Stepanauskas R, Giovannoni SJ. 2014. Single-cell enabled comparative genomics of a deep ocean SAR11 bathytype. ISME J 8:1440–1451. 10.1038/ismej.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith AM, Fuchs RT, Grundy FJ, Henkin T. 2010. Riboswitch RNAs: regulation of gene expression by direct monitoring of a physiological signal. RNA Biol 7:104–110. 10.4161/rna.7.1.10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Winkler WC, Breaker RR. 2005. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol 59:487–517. 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 102.McCown PJ, Corbino KA, Stav S, Sherlock ME, Breaker RR. 2017. Riboswitch diversity and distribution. RNA 23:995–1011. 10.1261/rna.061234.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Antunes D, Jorge NAN, Caffarena ER, Passetti F. 2018. Using RNA sequence and structure for the prediction of riboswitch aptamer: a comprehensive review of available software and tools. Front Genet 8. 10.3389/fgene.2017.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Henkin TM. 2008. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev 22:3383–3390. 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Caron M-P, Bastet L, Lussier A, Simoneau-Roy M, Masse E, Lafontaine DA. 2012. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc Natl Acad Sci USA 109:E3444–E3453. 10.1073/pnas.1214024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spinelli SV, Pontel LB, García Véscovi E, Soncini FC. 2008. Regulation of magnesium homeostasis in Salmonella: Mg2+ targets the mgtA transcript for degradation by RNase E: control of magnesium homeostasis in bacteria. FEMS Microbiol Lett 280:226–234. [DOI] [PubMed] [Google Scholar]
- 107.Mandal M, Breaker RR. 2004. Gene regulation by riboswitches. Nat Rev Mol Cell Biol 5:451–463. 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 108.Bastet L, Dubé A, Massé E, Lafontaine DA. 2011. New insights into riboswitch regulation mechanisms: new riboswitch regulation mechanisms. Mol Microbiol 80:1148–1154. 10.1111/j.1365-2958.2011.07654.x. [DOI] [PubMed] [Google Scholar]
- 109.Baird NJ, Kulshina N, Ferré-D’Amaré AR. 2010. Riboswitch function: flipping the switch or tuning the dimmer? RNA Biol 7:328–332. 10.4161/rna.7.3.11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roth A, Breaker RR. 2009. The structural and functional diversity of metabolite-binding riboswitches. Annu Rev Biochem 78:305–334. 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Serganov A, Patel DJ. 2012. Metabolite recognition principles and molecular mechanisms underlying riboswitch function. Annu Rev Biophys 41:343–370. 10.1146/annurev-biophys-101211-113224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Altuvia S. 2007. Identification of bacterial small noncoding RNAs: experimental approaches. Curr Opin Microbiol 10:257–261. 10.1016/j.mib.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 113.Pichon C, Felden B. 2008. Small RNA gene identification and mRNA target predictions in bacteria. Bioinformatics 24:2807–2813. 10.1093/bioinformatics/btn560. [DOI] [PubMed] [Google Scholar]
- 114.Repoila F, Darfeuille F. 2009. Small regulatory noncoding RNAs in bacteria: physiology and mechanistic aspects. Biol Cell 101:117–131. 10.1042/BC20070137. [DOI] [PubMed] [Google Scholar]
- 115.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891. 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Melamed S, Peer A, Faigenbaum-Romm R, Gatt YE, Reiss N, Bar A, Altuvia Y, Argaman L, Margalit H. 2016. Global mapping of small RNA-target interactions in bacteria. Mol Cell 63:884–897. 10.1016/j.molcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang H-Y, Chang H-Y, Chou C-H, Tseng C-P, Ho S-Y, Yang C-D, Ju Y-W, Huang H-D. 2009. sRNAMap: genomic maps for small noncoding RNAs, their regulators and their targets in microbial genomes. Nucleic Acids Res 37:D150–D154. 10.1093/nar/gkn852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gottesman S, Storz G. 2011. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3:a003798. 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wagner EGH, Romby P. 2015. Small RNAs in Bacteria and Archaea, p 133–208. In Advances in genetics. Elsevier, New York, NY. [DOI] [PubMed] [Google Scholar]
- 120.Gerdes K, Wagner EGH. 2007. RNA antitoxins. Curr Opin Microbiol 10:117–124. 10.1016/j.mib.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 121.Wagner EGH. 2009. Kill the messenger: bacterial antisense RNA promotes mRNA decay. Nat Struct Mol Biol 16:804–806. 10.1038/nsmb0809-804. [DOI] [PubMed] [Google Scholar]
- 122.Bandyra KJ, Said N, Pfeiffer V, Górna MW, Vogel J, Luisi BF. 2012. The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E. Mol Cell 47:943–953. 10.1016/j.molcel.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hajnsdorf E, Boni IV. 2012. Multiple activities of RNA-binding proteins S1 and Hfq. Biochimie 94:1544–1553. 10.1016/j.biochi.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 124.Van Assche E, Van Puyvelde S, Vanderleyden J, Steenackers HP. 2015. RNA-binding proteins involved in posttranscriptional regulation in bacteria. Front Microbiol 6. 10.3389/fmicb.2015.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cavanagh AT, Klocko AD, Liu X, Wassarman KM. 2008. Promoter specificity for 6S RNA regulation of transcription is determined by core promoter sequences and competition for region 4.2 of σ70. Mol Microbiol 67:1242–1256. 10.1111/j.1365-2958.2008.06117.x. [DOI] [PubMed] [Google Scholar]
- 126.Steglich C, Futschik ME, Lindell D, Voss B, Chisholm SW, Hess WR. 2008. The challenge of regulation in a minimal photoautotroph: noncoding RNAs in Prochlorococcus. PLoS Genet 4:e1000173. 10.1371/journal.pgen.1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Voigt K, Sharma CM, Mitschke J, Joke Lambrecht S, Voss B, Hess WR, Steglich C. 2014. Comparative transcriptomics of two environmentally relevant cyanobacteria reveals unexpected transcriptome diversity. ISME J 8:2056–2068. 10.1038/ismej.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Muro-Pastor AM, Hess WR. 2020. Regulatory RNA at the crossroads of carbon and nitrogen metabolism in photosynthetic cyanobacteria. Biochim Biophys Acta Gene Regul Mech 1863:194477. 10.1016/j.bbagrm.2019.194477. [DOI] [PubMed] [Google Scholar]
- 129.Kazanov MD, Vitreschak AG, Gelfand MS. 2007. Abundance and functional diversity of riboswitches in microbial communities. BMC Genomics 8:347. 10.1186/1471-2164-8-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, DeLong EF. 2008. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA 105:3805–3810. 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shi Y, Tyson GW, DeLong EF. 2009. Metatranscriptomics reveals unique microbial small RNAs in the ocean’s water column. Nature 459:266–269. 10.1038/nature08055. [DOI] [PubMed] [Google Scholar]
- 132.Hewson I, Poretsky RS, Tripp HJ, Montoya JP, Zehr JP. 2010. Spatial patterns and light-driven variation of microbial population gene expression in surface waters of the oligotrophic open ocean: comparative oceanic gene expression profiles. Environ Microbiol 12:1940–1956. 10.1111/j.1462-2920.2010.02198.x. [DOI] [PubMed] [Google Scholar]
- 133.Brown J, Hewson I. 2010. Ecophysiology of a common unannotated gene transcript in surface water microbial assemblages of the oligotrophic open ocean. Aquat Microb Ecol 60:289–297. 10.3354/ame01426. [DOI] [Google Scholar]
- 134.Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ, Breaker RR. 2009. Identification of candidate structured RNAs in the marine organism “Candidatus Pelagibacter ubique.” BMC Genomics 10:268. 10.1186/1471-2164-10-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tripp HJ, Schwalbach MS, Meyer MM, Kitner JB, Breaker RR, Giovannoni SJ. 2009. Unique glycine-activated riboswitch linked to glycine-serine auxotrophy in SAR11. Environ Microbiol 11:230–238. 10.1111/j.1462-2920.2008.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tezuka T, Ohnishi Y. 2014. Two glycine riboswitches activate the glycine cleavage system essential for glycine detoxification in Streptomyces griseus. J Bacteriol 196:1369–1376. 10.1128/JB.01480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Carini P, Campbell EO, Morré J, Sañudo-Wilhelmy SA, Thrash JC, Bennett SE, Temperton B, Begley T, Giovannoni SJ. 2014. Discovery of a SAR11 growth requirement for thiamin’s pyrimidine precursor and its distribution in the Sargasso Sea. ISME J 8:1727–1738. 10.1038/ismej.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Singh P, Kumar N, Jethva M, Yadav S, Kumari P, Thakur A, Kushwaha HR. 2018. Riboswitch regulation in cyanobacteria is independent of their habitat adaptations. Physiol Mol Biol Plants 24:315–324. 10.1007/s12298-018-0504-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kopf M, Hess WR. 2015. Regulatory RNAs in photosynthetic cyanobacteria. FEMS Microbiol Rev 39:301–315. 10.1093/femsre/fuv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lambrecht SJ, Kanesaki Y, Fuss J, Huettel B, Reinhardt R, Steglich C. 2019. Interplay and targetome of the two conserved cyanobacterial sRNAs Yfr1 and Yfr2 in Prochlorococcus MED4. Sci Rep 9. 10.1038/s41598-019-49881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thompson AW, Huang K, Saito MA, Chisholm SW. 2011. Transcriptome response of high- and low-light-adapted Prochlorococcus strains to changing iron availability. ISME J 5:1580–1594. 10.1038/ismej.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stazic D, Lindell D, Steglich C. 2011. Antisense RNA protects mRNA from RNase E degradation by RNA-RNA duplex formation during phage infection. Nucleic Acids Res 39:4890–4899. 10.1093/nar/gkr037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dupont CL, Rusch DB, Yooseph S, Lombardo M-J, Alexander Richter R, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL, Lasken RS, Nealson K, Friedman R, Craig Venter J. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. 10.1038/ismej.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.García-Romero I, Förstner KU, Santero E, Floriano B. 2018. SuhB, a small noncoding RNA involved in catabolite repression of tetralin degradation genes in Sphingopyxis granuli strain TFA: SuhB-regulated thn expression in TFA. Environ Microbiol 20:3671–3683. 10.1111/1462-2920.14360. [DOI] [PubMed] [Google Scholar]
- 145.Nussinov R, Tsai C-J, Xin F, Radivojac P. 2012. Allosteric posttranslational modification codes. Trends Biochem Sci 37:447–455. 10.1016/j.tibs.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 146.Kepes A. 1963. Kinetic of induced enzyme synthesis determination of the mean life of galactosidase-specific messenger RNA. Biochim Biophys Acta 76:293–309. 10.1016/0926-6550(63)90043-4. [DOI] [PubMed] [Google Scholar]
- 147.Macek B, Forchhammer K, Hardouin J, Weber-Ban E, Grangeasse C, Mijakovic I. 2019. Protein posttranslational modifications in bacteria. Nat Rev Microbiol 17:651–664. 10.1038/s41579-019-0243-0. [DOI] [PubMed] [Google Scholar]
- 148.Cain JA, Solis N, Cordwell SJ. 2014. Beyond gene expression: the impact of protein posttranslational modifications in bacteria. J Proteomics 97:265–286. 10.1016/j.jprot.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 149.Pisithkul T, Patel NM, Amador-Noguez D. 2015. Posttranslational modifications as key regulators of bacterial metabolic fluxes. Curr Opin Microbiol 24:29–37. 10.1016/j.mib.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 150.Soufi B, Krug K, Harst A, Macek B. 2015. Characterization of the Escherichia coli proteome and its modifications during growth and ethanol stress. Front Microbiol 6. 10.3389/fmicb.2015.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Abu-Qarn M, Eichler J, Sharon N. 2008. Not just for Eukarya anymore: protein glycosylation in Bacteria and Archaea. Curr Opin Struct Biol 18:544–550. 10.1016/j.sbi.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 152.Sanwal BD. 1970. Allosteric controls of amphilbolic pathways in bacteria. Bacteriol Rev 34:20–39. 10.1128/br.34.1.20-39.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lindsley JE, Rutter J. 2006. Whence cometh the allosterome? Proc Natl Acad Sci USA 103:10533–10535. 10.1073/pnas.0604452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chubukov V, Uhr M, Le Chat L, Kleijn RJ, Jules M, Link H, Aymerich S, Stelling J, Sauer U. 2013. Transcriptional regulation is insufficient to explain substrate-induced flux changes in Bacillus subtilis. Mol Syst Biol 9:709. 10.1038/msb.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Reznik E, Christodoulou D, Goldford JE, Briars E, Sauer U, Segrè D, Noor E. 2017. Genome-scale architecture of small molecule regulatory networks and the fundamental trade-off between regulation and enzymatic activity. Cell Rep 20:2666–2677. 10.1016/j.celrep.2017.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Orsak T, Smith TL, Eckert D, Lindsley JE, Borges CR, Rutter J. 2012. Revealing the allosterome: systematic identification of metabolite-protein interactions. Biochemistry 51:225–232. 10.1021/bi201313s. [DOI] [PubMed] [Google Scholar]
- 157.Moran MA, Kujawinski EB, Schroer WF, Amin SA, Bates NR, Bertrand EM, Braakman R, Brown CT, Covert MW, Doney SC, Dyhrman ST, Edison AS, Eren AM, Levine NM, Li L, Ross AC, Saito MA, Santoro AE, Segrè D, Shade A, Sullivan MB, Vardi A. 2022. Microbial metabolites in the marine carbon cycle. Nat Microbiol 7:508–523. 10.1038/s41564-022-01090-3. [DOI] [PubMed] [Google Scholar]
- 158.Fegatella F, Ostrowski M, Cavicchioli R. 1999. An assessment of protein profiles from the marine oligotrophic ultramicrobacterium, Sphingomonas sp. strain RB2256. Electrophoresis 20:2094–2098. . [DOI] [PubMed] [Google Scholar]
- 159.Fegatella F, Cavicchioli R. 2000. Physiological responses to starvation in the marine oligotrophic ultramicrobacterium Sphingomonas sp. strain RB2256. Appl Environ Microbiol 66:2037–2044. 10.1128/AEM.66.5.2037-2044.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ostrowski M, Fegatella F, Wasinger V, Guilhaus M, Corthals GL, Cavicchioli R. 2004. Cross-species identification of proteins from proteome profiles of the marine oligotrophic ultramicrobacterium, Sphingopyxis alaskensis. Proteomics 4:1779–1788. 10.1002/pmic.200300695. [DOI] [PubMed] [Google Scholar]
- 161.Vorontsov EA, Rensen E, Prangishvili D, Krupovic M, Chamot-Rooke J. 2016. Abundant lysine methylation and N-terminal acetylation in Sulfolobus islandicus revealed by bottom-up and top-down proteomics. Mol Cell Proteomics 15:3388–3404. 10.1074/mcp.M116.058073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Millard P, Smallbone K, Mendes P. 2017. Metabolic regulation is sufficient for global and robust coordination of glucose uptake, catabolism, energy production and growth in Escherichia coli. PLoS Comput Biol 13:e1005396. 10.1371/journal.pcbi.1005396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mannan AA, Toya Y, Shimizu K, McFadden J, Kierzek AM, Rocco A. 2015. Integrating kinetic model of Escherichia coli with genome scale metabolic fluxes overcomes its open system problem and reveals bistability in central metabolism. PLoS One 10:e0139507. 10.1371/journal.pone.0139507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Haverkorn van Rijsewijk BRB, Nanchen A, Nallet S, Kleijn RJ, Sauer U. 2011. Large-scale 13C-flux analysis reveals distinct transcriptional control of respiratory and fermentative metabolism in Escherichia coli. Mol Syst Biol 7:477. 10.1038/msb.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ranea JAG, Grant A, Thornton JM, Orengo CA. 2005. Microeconomic principles explain an optimal genome size in bacteria. Trends Genet 21:21–25. 10.1016/j.tig.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 166.Conaway R, Conaway J. 1988. ATP activates transcription initiation from promoters by RNA polymerase II in a reversible step prior to RNA synthesis. J Biol Chem 263:2962–2968. 10.1016/S0021-9258(18)69162-8. [DOI] [PubMed] [Google Scholar]
- 167.Jewett MC, Miller ML, Chen Y, Swartz JR. 2009. Continued protein synthesis at low [ATP] and [GTP] enables cell adaptation during energy limitation. J Bacteriol 191:1083–1091. 10.1128/JB.00852-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Stocker R. 2012. Marine microbes see a sea of gradients. Science 338:628–633. 10.1126/science.1208929. [DOI] [PubMed] [Google Scholar]
- 169.Blackburn N, Fenchel T, Mitchell J. 1998. Microscale nutrient patches in planktonic habitats shown by chemotactic bacteria. Science 282:2254–2256. 10.1126/science.282.5397.2254. [DOI] [PubMed] [Google Scholar]
- 170.Stocker R, Seymour JR, Samadani A, Hunt DE, Polz MF. 2008. Rapid chemotactic response enables marine bacteria to exploit ephemeral microscale nutrient patches. Proc Natl Acad Sci USA 105:4209–4214. 10.1073/pnas.0709765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mitchell JG. 1991. The influence of cell size on marine bacterial motility and energetics. Microb Ecol 22:227–238. [DOI] [PubMed] [Google Scholar]
- 172.Dusenbery DB. 1997. Minimum size limit for useful locomotion by free-swimming microbes. Proc Natl Acad Sci USA 94:10949–10954. 10.1073/pnas.94.20.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kelkar YD, Ochman H. 2013. Genome reduction promotes increase in protein functional complexity in bacteria. Genetics 193:303–307. 10.1534/genetics.112.145656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Noell SE, Giovannoni SJ. 2019. SAR11 bacteria have a high affinity and multifunctional glycine betaine transporter. Environ Microbiol 21:2559–2575. 10.1111/1462-2920.14649. [DOI] [PubMed] [Google Scholar]
- 175.Noell SE, Barrell GE, Suffridge C, Morré J, Gable KP, Graff JR, VerWey BJ, Hellweger FL, Giovannoni SJ. 2021. SAR11 cells rely on enzyme multifunctionality to metabolize a range of polyamine compounds. mBio 12:e01091-21. 10.1128/mBio.01091-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lambert G, Kussell E. 2014. Memory and fitness optimization of bacteria under fluctuating environments. PLoS Genet 10:e1004556. 10.1371/journal.pgen.1004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mitchell JG, Pearson L, Bonazinga A, Dillon S, Khouri H, Paxinos R. 1995. Long lag times and high velocities in the motility of natural assemblages of marine bacteria. Appl Environ Microbiol 61:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mesibov R, Ordal GW, Adler J. 1973. The range of attractant concentrations for bacterial chemotaxis and the threshold and size of response over this range. J Gen Physiol 62:203–223. 10.1085/jgp.62.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bondoc KGV, Heuschele J, Gillard J, Vyverman W, Pohnert G. 2016. Selective silicate-directed motility in diatoms. Nat Commun 7:10540. 10.1038/ncomms10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bray D. 2002. Bacterial chemotaxis and the question of gain. Proc Natl Acad Sci USA 99:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Henson MW, Lanclos VC, Faircloth BC, Thrash JC. 2018. Cultivation and genomics of the first freshwater SAR11 (LD12) isolate. ISME J 12:1846–1860. 10.1038/s41396-018-0092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Brewer TE, Handley KM, Carini P, Gilbert JA, Fierer N. 2016. Genome reduction in an abundant and ubiquitous soil bacterium “Candidatus Udaeobacter copiosus.” Nat Microbiol 2:16198. 10.1038/nmicrobiol.2016.198. [DOI] [PubMed] [Google Scholar]
- 183.Ray JCJ, Wickersheim ML, Jalihal AP, Adeshina YO, Cooper TF, Balázsi G. 2016. Cellular growth arrest and persistence from enzyme saturation. PLoS Comput Biol 12:e1004825. 10.1371/journal.pcbi.1004825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Boysen AK, Durham BP, Kumler W, Key RS, Heal KR, Carlson LT, Groussman RD, Armbrust EV, Ingalls AE. 2022. Glycine betaine uptake and metabolism in marine microbial communities. Environ Microbiol 24:2380–2403. 10.1111/1462-2920.16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tian R, Ning D, He Z, Zhang P, Spencer SJ, Gao S, Shi W, Wu L, Zhang Y, Yang Y, Adams BG, Rocha AM, Detienne BL, Lowe KA, Joyner DC, Klingeman DM, Arkin AP, Fields MW, Hazen TC, Stahl DA, Alm EJ, Zhou J. 2020. Small and mighty: adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 8:1–15. 10.1186/s40168-020-00825-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Castelle CJ, Brown CT, Anantharaman K, Probst AJ, Huang RH, Banfield JF. 2018. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol 16:629–645. 10.1038/s41579-018-0076-2. [DOI] [PubMed] [Google Scholar]
- 187.Lemos LN, Medeiros JD, Dini-Andreote F, Fernandes GR, Varani AM, Oliveira G, Pylro VS. 2019. Genomic signatures and co-occurrence patterns of the ultra-small Saccharimonadia (phylum CPR/Patescibacteria) suggest a symbiotic lifestyle. Mol Ecol 28:4259–4271. 10.1111/mec.15208. [DOI] [PubMed] [Google Scholar]
- 188.Kuroda K, Yamamoto K, Nakai R, Hirakata Y, Kubota K, Nobu MK, Narihiro T. 2022. Symbiosis between “Candidatus Patescibacteria” and archaea discovered in wastewater-treating bioreactors. mBio 13:e01711-22. 10.1128/mbio.01711-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rohatgi A. 2022. Webplotdigitizer: version 4.6. https://automeris.io/WebPlotDigitizer/.
- 190.Lim CK, Hassan KA, Tetu SG, Loper JE, Paulsen IT. 2012. The effect of iron limitation on the transcriptome and proteome of Pseudomonas fluorescens Pf-5. PLoS One 7:e39139. 10.1371/journal.pone.0039139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ledala N, Sengupta M, Muthaiyan A, Wilkinson BJ, Jayaswal RK. 2010. Transcriptomic response of Listeria monocytogenes to iron limitation and fur mutation. Appl Environ Microbiol 76:406–416. 10.1128/AEM.01389-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Manck LE, Espinoza JL, Dupont CL, Barbeau KA. 2020. Transcriptomic study of substrate-specific transport mechanisms for iron and carbon in the marine copiotroph Alteromonas macleodii. mSystems 5:e00070-20. 10.1128/mSystems.00070-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Holmes K, Mulholland F, Pearson BM, Pin C, McNicholl-Kennedy J, Ketley JM, Wells JM. 2005. Campylobacter jejuni gene expression in response to iron limitation and the role of Fur. Microbiology (Reading) 151:243–257. [DOI] [PubMed] [Google Scholar]
- 194.Hernández-Prieto MA, Schön V, Georg J, Barreira L, Varela J, Hess WR, Futschik ME. 2012. Iron deprivation in synechocystis : inference of pathways, noncoding RNAs, and regulatory elements from comprehensive expression profiling. G3 (Bethesda) 2:1475–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Paustian ML, May BJ, Kapur V. 2001. Pasteurella multocida gene expression in response to iron limitation. Infect Immun 69:4109–4115. 10.1128/IAI.69.6.4109-4115.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ludwig M, Bryant DA. 2012. Acclimation of the global transcriptome of the cyanobacterium Synechococcus sp. Strain PCC 7002 to nutrient limitations and different nitrogen sources. Front Microbiol 3. 10.3389/fmicb.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hervás AB, Canosa I, Santero E. 2008. Transcriptome analysis of Pseudomonas putida in response to nitrogen availability. J Bacteriol 190:416–420. 10.1128/JB.01230-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Choi SY, Park B, Choi I-G, Sim SJ, Lee S-M, Um Y, Woo HM. 2016. Transcriptome landscape of Synechococcus elongatus PCC 7942 for nitrogen starvation responses using RNA-seq. Sci Rep 6:30584. 10.1038/srep30584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Petridis M, Benjak A, Cook GM. 2015. Defining the nitrogen regulated transcriptome of Mycobacterium smegmatis using continuous culture. BMC Genomics 16:821. 10.1186/s12864-015-2051-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lee PKH, Dill BD, Louie TS, Shah M, VerBerkmoes NC, Andersen GL, Zinder SH, Alvarez-Cohen L. 2012. Global transcriptomic and proteomic responses of Dehalococcoides ethenogenes strain 195 to fixed nitrogen limitation. Appl Environ Microbiol 78:1424–1436. 10.1128/AEM.06792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Read RW, Berube PM, Biller SJ, Neveux I, Cubillos-Ruiz A, Chisholm SW, Grzymski JJ. 2017. Nitrogen cost minimization is promoted by structural changes in the transcriptome of N-deprived Prochlorococcus cells. ISME J 11:2267–2278. 10.1038/ismej.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang L, Liu L, Wang K-F, Xu L, Zhou L, Wang W, Li C, Xu Z, Shi T, Chen H, Li Y, Xu H, Yang X, Zhu Z, Chen B, Li D, Zhan G, Zhang S-L, Zhang L-X, Tan G-Y. 2019. Phosphate limitation increases coenzyme Q10 production in industrial Rhodobacter sphaeroides HY01. Synth Syst Biotechnol 4:212–219. 10.1016/j.synbio.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tralau T, Vuilleumier S, Thibault C, Campbell BJ, Hart CA, Kertesz MA. 2007. Transcriptomic analysis of the sulfate starvation response of Pseudomonas aeruginosa. J Bacteriol 189:6743–6750. 10.1128/JB.00889-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bochenek M, Etherington GJ, Koprivova A, Mugford ST, Bell TG, Malin G, Kopriva S. 2013. Transcriptome analysis of the sulfate deficiency response in the marine microalga Emiliania huxleyi. New Phytol 199:650–662. 10.1111/nph.12303. [DOI] [PubMed] [Google Scholar]
- 205.Kumaresan V, Nizam F, Ravichandran G, Viswanathan K, Palanisamy R, Bhatt P, Arasu MV, Al-Dhabi NA, Mala K, Arockiaraj J. 2017. Transcriptome changes of blue-green algae, Arthrospira sp. in response to sulfate stress. Algal Res 23:96–103. 10.1016/j.algal.2017.01.012. [DOI] [Google Scholar]
- 206.Mostertz J, Scharf C, Hecker M, Homuth G. 2004. Transcriptome and proteome analysis of Bacillus subtilis gene expression in response to superoxide and peroxide stress. Microbiology 150:497–512. 10.1099/mic.0.26665-0. [DOI] [PubMed] [Google Scholar]
- 207.Mounier J, Camus A, Mitteau I, Vaysse P-J, Goulas P, Grimaud R, Sivadon P. 2014. The marine bacterium Marinobacter hydrocarbonoclasticus SP17 degrades a wide range of lipids and hydrocarbons through the formation of oleolytic biofilms with distinct gene expression profiles. FEMS Microbiol Ecol 90:816–831. 10.1111/1574-6941.12439. [DOI] [PubMed] [Google Scholar]
- 208.Gunasekera TS, Bowen LL, Radwan O, Striebich RC, Ruiz ON. 2022. Genomic and transcriptomic characterization revealed key adaptive mechanisms of Marinobacter hydrocarbonoclasticus NI9 for proliferation and degradation of jet fuel. Int Biodeterior Biodegrad 175:105502. 10.1016/j.ibiod.2022.105502. [DOI] [Google Scholar]
- 209.Naether DJ, Slawtschew S, Stasik S, Engel M, Olzog M, Wick LY, Timmis KN, Heipieper HJ. 2013. Adaptation of the hydrocarbonoclastic bacterium Alcanivorax borkumensis SK2 to alkanes and toxic organic compounds: a physiological and transcriptomic approach. Appl Environ Microbiol 79:4282–4293. 10.1128/AEM.00694-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jin HM, Jeong HI, Kim KH, Hahn Y, Madsen EL, Jeon CO. 2016. Genome-wide transcriptional responses of Alteromonas naphthalenivorans SN2 to contaminated seawater and marine tidal flat sediment. Sci Rep 6:21796. 10.1038/srep21796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Aharonovich D, Sher D. 2016. Transcriptional response of Prochlorococcus to coculture with a marine Alteromonas: differences between strains and the involvement of putative infochemicals. ISME J 10:2892–2906. 10.1038/ismej.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bagby SC, Chisholm SW. 2015. Response of Prochlorococcus to varying CO2:O2 ratios. ISME J 9:2232–2245. 10.1038/ismej.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]