

Abstract
The hypoxic milieu is a critical modulator of aerobic glycolysis, yet the regulatory mechanisms between the key glycolytic enzymes in hypoxic cancer cells are largely unchartered. In particular, the M2 isoform of pyruvate kinase (PKM2), the rate-limiting enzyme of glycolysis, is known to confer adaptive advantages under hypoxia. Herein, we report that non-canonical PKM2 mediates HIF-1α and p300 enrichment at PFKFB3 hypoxia-responsive elements (HREs), causing its upregulation. Consequently, the absence of PKM2 activates an opportunistic occupancy of HIF-2α, along with acquisition of a poised state by PFKFB3 HREs-associated chromatin. This poised nature restricts HIF-2α from inducing PFKFB3 while permitting the maintenance of its basal-level expression by harboring multiple histone modifications. In addition, the clinical relevance of the study has been investigated by demonstrating that Shikonin blocks the nuclear translocation of PKM2 to suppress PFKFB3 expression. Furthermore, TNBC patient-derived organoids and MCF7 cells-derived xenograft tumors in mice exhibited substantial growth inhibition upon shikonin treatment, highlighting the vitality of targeting PKM2. Conclusively, this work provides novel insights into the contributions of PKM2 in modulating hypoxic transcriptome and a previously unreported poised epigenetic strategy exhibited by the hypoxic breast cancer cells for ensuring the maintenance of PFKFB3 expression.
Graphical Abstract
Graphical Abstract.
INTRODUCTION
Glycolysis, the central pathway of cellular metabolism, is highly responsive to various environmental cues. Numerous reports published in the last few years have revealed that cancer cells are extensively dependent on glycolysis even under optimal oxygen condition, a phenomenon referred to as the Warburg effect (1,2). Moreover, frequently observed tumor-associated microenvironmental niche of hypoxia has been well-studied to prompt the genesis of drastic metabolic alterations, further enhancing the glycolytic potential of cancer cells (3,4). The adaptative strategies exhibited by hypoxic cancer cells are primarily manifested by the activity of a family of transcription factors known as hypoxia-inducible factors (HIFs). The HIFs are heterodimeric proteins comprising an oxygen-sensitive α subunit and an oxygen-insensitive β subunit. Under optimal O2 concentration, the HIF-1α subunit undergoes prolyl hydroxylation (by prolyl hydroxylases, i.e. PHDs) at the residues Pro 402 and 564 (5), targeting it for ubiquitin-dependent proteasomal degradation (6). However, under hypoxia, PHDs are functionally less active, due to which the prolyl hydroxylation of HIF-1α is substantially blocked. Consequently, the stabilized transcription factor translocates to the nucleus and pairs with its constitutive partner, HIF-1β. This functional heterodimer drives the expression of a plethora of genes by binding to the consensus sequence (5'RCGTG3') known as hypoxia-responsive element (HRE) to govern various cellular pathways (7–9). One of the most striking effects of hypoxia can be seen on the metabolism of cancer cells. In this context, glycolysis has extensively been investigated to undergo hypoxia-mediated alterations, as numerous glycolytic genes are HIF1 targets (4).
Interestingly, PKM2 exhibits an incredible capacity to perform various non-canonical functions dictated by diverse microenvironmental cues (10). One of the most imperative non-canonical features of PKM2 is its ability to translocate into the nucleus to act as a transcriptional co-activator of HIF-1α (11). Although the role of PKM2 as a transcriptional co-activator of HIF-1α has been previously reported, there is a dearth of global transcriptomic data which can provide an estimate regarding the extent of PKM2’s influence in shaping hypoxic transcriptome. Of note, this imperative aspect is still largely elusive due to the lack of appropriate model systems selectively knockout for PKM2 and not PKM1 in the context of cancer.
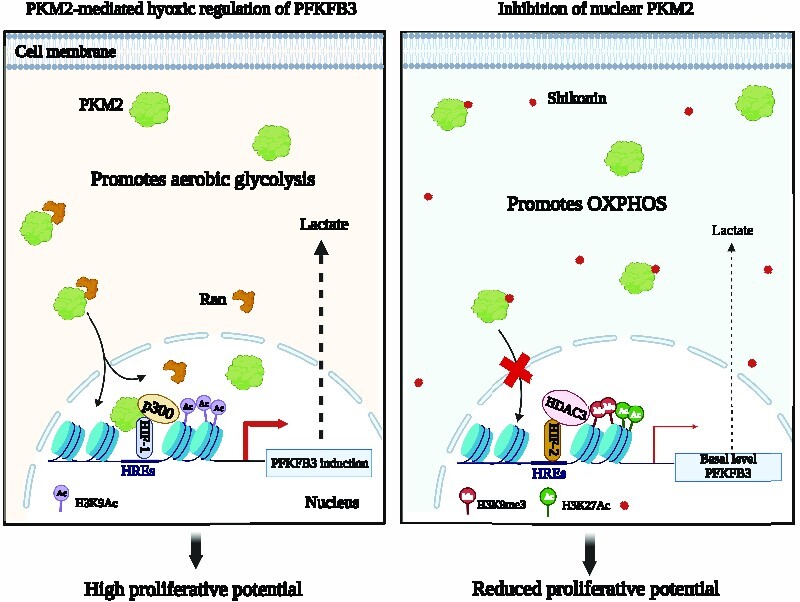
As the BORIS-binding site (BBS) present in exon 10 of the pyruvate kinase (PKM) gene has a dictatorial influence in promoting biased expression of PKM2 over PKM1 (12), we mutated the BBS to generate PKM2 knockout breast cancer cell lines. Therefore, if PKM2 acts as a transcriptional activator for a gene, its hypoxic induction will be affected in the BBS mutants (BBS Mut) compared to their wild-type BBS (WT BBS) counterpart cells. HIF-1α ChIP-seq paired with transcriptome array analysis revealed PFKFB3 as a previously unidentified target of the PKM2-HIF-1α axis. Moreover, PKM2 enhanced the binding of p300 to transcriptionally upregulation of PFKFB3. Interestingly, HIF-2α was identified as a non-canonical regulator, which maintains only basal-level expression of PFKFB3 in the absence of the master regulators HIF-1α and PKM2. Of note, we report that PFKFB3 promoter harbors a poised chromatin state, which is the fundamental basis underlying its fine-tuned hypoxic regulation. Notably, this adaptive switch significantly enhanced the mitochondrial oxidative phosphorylation (OXPHOS) dependency, ultimately compromising the proliferative potential of hypoxic breast cancer cells.
We also systematically delineate that Shikonin, a pharmacological inhibitor of PKM2, interacts with R399/400 residues to block its nuclear translocation, thus leading to the disruption of the PKM2–HIFs–PFKFB3 axis. Moreover, experimental investigations performed on TNBC patient-derived organoid, and in vivo findings clearly demonstrate that shikonin hampers the growth of breast cancer cells expressing PKM2. We envisage that targeting this axis has therapeutic potential to inhibit the proliferation of hypoxic breast cancer cells by substantially reducing the glycolytic rate.
MATERIALS AND METHODS
Cell culture
Human breast cancer cell lines MCF7 and HCC1806 were obtained from American Type Culture Collection (ATCC). MCF7 and HEK293T were cultured at 37ºC, 5% CO2 in DMEM (Invitrogen, 12800017, lot no. 2248833). HCC1806 was cultured in RPMI-1640 (Invitrogen, 23400021, lot no. 2144859). The culture media for all the cell lines was supplemented with 10% fetal bovine serum (FBS; Sigma, F7524, lot no. BCBX8466), 100 units/ml of penicillin and streptomycin (Invitrogen, 15140122, lot no. 2321120), and 2 mM/l l-glutamine (Invitrogen, 25030081, lot no. 1917006). The BBS Mut MCF7 cell line was generated as previously mentioned (12). The media and cell culture conditions for various CRISPR/Cas9 mutants were identical to the wild-type cells. Cell lines were subjected to hypoxia treatment of 1% oxygen in a Ruskinn INVIVO2 400 hypoxia chamber. The cell lines used in the study were routinely tested for mycoplasma contamination using PCR based method and were authenticated from a national cell repository facility by short tandem repeats (STR) profiling.
Plasmids
The mCherry-tagged PKM2 overexpression construct was generated as previously mentioned (12). The PKM2 overexpression lentiviral plasmid was constructed by cloning the PKM2 insert between the NotI and EcoRI sites of the plasmid pSCALPS_ZSgreen kindly gifted by A. Chande (IISER Bhopal). The PFKFB3 overexpression construct was generated by cloning the insert between NotI and EcoRI sites of the plasmid pCMV-3tag-1a (Agilent, 240195). The primers used for generating the inserts are mentioned in Supplementary Table S2.
Quantitative RT-PCR
Total RNA was extracted using TRIZol reagent (Ambion, 15596018, lot no. 260712) as per the manufacturer's instructions. The concentration was determined using Eppendorf BioSpectrometer, and 1 μg of total RNA was used to synthesize cDNA using SuperScript® III First-Strand Synthesis System (Invitrogen, 18080-051, lot no. 2291381). Amplifications were performed in duplicates using GO taq QPCR master mix (Promega, A6002, lot no. 0000385100) and light cycler 480 II (Roche) according to the manufacturer's protocol. The primers were designed using the IDT PrimerQuest tool (https://www.idtdna.com) and are enlisted in Supplementary Table S2. The average cycle thresholds from three independent biological replicate samples were calculated and normalized to housekeeping control gene RPS16 using the formula 2(Ct_control − Ct_target). Student's t-test was used to compare gene/exon expression between two different groups. P < 0.05 was considered statistically significant.
Immunoblotting
The cells were lysed using urea lysis buffer (8 M urea, 2 M thiourea, 2% CHAPS, 1% DTT) supplemented with 1× protease inhibitor cocktail (PIC; leupeptin 10–100 μM, pepstatin 1 μM, EDTA 1–10 mM, AEBSF < 1 mM) at 4°C for 30 min and spun at maximum speed (16 900 × g) in a 4°C centrifuge for 2 h. The supernatant was separated and quantified, and an equal concentration of protein samples was loaded for every experiment. The well-separated proteins were electro-transferred on an activated PVDF membrane. After transfer, the blots were incubated with recommended dilutions of primary antibodies overnight at 4°C, followed by 1 h incubation with secondary antibody. The blots were scanned using Odyssey membrane Scanning system. Quantification of the bands was performed using GelQuant software (version 1.8.2). The technical details of the antibodies used are enlisted in Supplementary Table S3.
PFK assay
WT BBS and BBS Mut cells subjected to normoxic or hypoxic treatment were lysed using assay buffer provided in the Phosphofructokinase (PFK) Activity Colorimetric Assay Kit (Sigma, MAK093, lot no. 6E31K07760). An equal quantity of protein lysates was used to determine the total PFK activity as per the manufacturer's instructions. The readings were measured using a microplate reader (BioTek Eon, 11-120-611) set at 37 °C and an optical density of 450 nm. The values are represented as mean ± SD of duplicates from a representative experiment.
Lactate assay
Post-treatment, an equal number of WT BBS and BBS Mut MCF7 and HCC1806 cells were lysed using ice-cold lactate assay buffer provided in the Lactate assay kit (Sigma, MAK064-1, lot no. 3F09K06270). The lactate quantification was performed using the deproteinized lysates by following the manufacturer's instructions. The readings were measured at room temperature using a microplate reader set at an optical density of 450 nm.
α-ketoglutarate assay
WT and BBS Mut cells of MCF7 and HCC1806 cells subjected to 24 h of hypoxia were processed for α-ketoglutarate estimation. Briefly, equal number of cells were lysed using assay buffer provided in the alpha KG assay kit (Abcam, ab83431, lot no. GR3410863-1). The lysates were then deproteinized using perchloric acid and neutralized using 2 M KOH as per manufacturer's instructions. Finally, an equal amount of sample was taken, and the reaction was incubated at 37°C for 30 min. The absorbance was measured using a microplate reader set at an optical density of 450 nm.
Extracellular flux assays
A seahorse XF HS mini analyzer was used to determine the oxygen consumption rate (OCR), extracellular acidification rate (ECAR) and ATP production rate. Briefly, 5 × 103 cells per well were seeded into 8-well seahorse XFp mini cell culture plate and allowed to attach overnight. Post subjecting the cells to 24 h of hypoxia, cells were washed and incubated with XF assay medium supplemented with 1 mM pyruvate (Sigma, S8636, lot no. RNBJ2351), 2 mM l-glutamine (Invitrogen, 25030081, lot no. 1917006), 10mM glucose (Gibco, A24940-01, lot no. 1969830), and 150 μM cobalt chloride (Alfa Aesar, B22031, lot no. 10194277) for 1 h at 37°C in CO2-free incubator. OCR, ECAR and ATP production rate estimation was performed as per manufacturer's instructions. OCR was assessed in response to oligomycin (1.5 μM), FCCP (0.5 μM), and rotenone/antimycin A (0.5 μM). The ECAR was assessed in response to rotenone/antimycin A (0.5 μM), and 2-deoxy-d-glucose (2-DG; 50 mM) and ATP rate assay was estimated in response to oligomycin (1.5 μM) and rotenone/antimycin A (0.5 μM). Finally, the readings were normalized to the respective protein concentrations.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed as previously described (13). Briefly, about 10 million cells were crosslinked and scraped in PBS, followed by lysis and sonication. About 25 μg of sheared chromatin (approx length 250–500 bp) was immunoprecipitated with an antibody of interest following overnight incubation at 4°C. Immunoprecipitated protein–DNA complexes and 5% input were analyzed by qRT-PCR using GO taq QPCR master mix (Promega, A6002, lot no. 0000385100) in triplicate using primers specific for PFKFB3 HREs. Additionally, each experiment was performed at least thrice. Normalization was performed using the formula [2(Ct input – Ct immunoprecipitation)]. The obtained values were further normalized to the relative rabbit IgG and control IP values. The primers were designed using the IDT PrimerQuest tool (https://www.idtdna.com) and are enlisted in Supplementary Table S2. Significance between the two groups was calculated using Student's t-test, with a value of <0.05 considered statistically significant. HIF-1α ChIP-seq data was analyzed as previously described (14). Over-representation analyses (ORA) for GO Biological processes were generated using ShinyGo 0.76.2 (15). False discovery rate (FDR)-value of <0.05 was considered significant while obtaining enriched GO terms. The top 20 enriched terms were represented using a dot plot and ranked according to the fold enrichment value. Using the online tool Morpheus (https://software.broadinstitute.org/morpheus), a heat map was constructed for the 44 common genes between HIF-1α ChIP-seq and the DEGs obtained from BBS Mut versus WT BBS Hypoxia condition.
RNA interference
The cell lines used throughout this study were plated at a seeding density of 3 × 105 cells per well of a six-well culture plate and allowed to attach for 24 h. The lentivirus containing small hairpin RNA (shRNA) (Sigma, Mission Human Genome shRNA Library) against the target gene was inoculated in the presence of 8 μg/ml polybrene (Sigma, H9268, lot no. SLBH5907V) containing media. Cells were selected for 72 h using 1 μg/ml puromycin (Sigma, P9620, lot no. 034M4008V) and subsequently used for various experiments. The sequence of shRNAs used in this study is provided in Supplementary Table S1.
Human transcriptome array 2.0
Total RNA was isolated from WT BBS and BBS mutant MCF7 cells using TRIZol (Ambion, 15596018, lot no. 260712) reagent and PureLink RNA Mini Kit (Invitrogen, 12183025, lot no. 1862249) as per manufacturer's instructions. The concentration was determined using Eppendorf BioSpectrometer, and 100 ng of total RNA was used for biotinylated cDNA synthesis using GeneChip™ WT Plus Reagent Kit (Invitrogen, 902281, lot no. 01059768) as per manufacturers protocol. Following amplification, cDNA fragmentation was performed, and 5.5 μg of fragmented cDNA was hybridized on Affymetrix GeneChip™ Human Transcriptome Array 2.0 (HTA2.0) chips (Invitrogen, 902162, lot no. 4364589) for 16 h at 45°C. The chips were washed and stained using the Affymetrix Fluidics Station 450. Post hybridization, the fluorescence intensity of the arrays was scanned using the Affymetrix Scanner 7G. The raw files generated in CEL format after the scan were further used for analysis.
Human transcriptome array 2.0 data analysis
The CEL files were analyzed using Transcriptome Array Console 4.0 (Invitrogen, version 4.0.2.15) using the gene + exon–SST-RMA method of summarization. Genes with thresholds of absolute fold-change >2, P < 0.05, and false discovery rates (FDRs) <0.05 were selected as differentially expressed genes (DEGs). Venn diagram was generated using Venn Diagram Plotter software developed by Pacific Northwest National Laboratory (https://omics.pnl.gov/software/venn-diagram-plotter). The volcano plot for DEGs was plotted using GraphPad Prism 8.
Immunofluorescence
Cells were plated at a seeding density of 3 × 104 on a coverslip in a 12-well plate. After treatment, the cells were washed thrice with ice-cold PBS, followed by 4% formaldehyde fixation and subsequent permeabilization with 0.1% Triton X-100 solution. Post blocking for 1hr at RT using 2% BSA solution, cells were incubated with appropriate primary antibodies overnight at 4°C. Cells were then washed thrice with PBS and incubated with Alexa-Flour 555 anti-rabbit IgG secondary antibody for 1hr at RT. Subsequently, the cells were counterstained with DAPI (Invitrogen, D1306, lot no. 1673432) and mounted using fluoroshield (Sigma, F6182, lot no. MKCN2676). For Ki67 staining, primary antibody incubation was directly followed by nuclei counterstaining using Hoechst 33342 (Invitrogen, HI399, lot no. 1932847). Imaging was performed using Olympus FV3000 confocal laser scanning microscope with a 60× Plan Apo N objective (oil, 1.42 NA), and image analysis was performed using Image J software.
Breast cancer sample collection
Prior to utilizing tumor specimens for research purposes, informed consent was obtained from all the patients. Paraffin-embedded breast tumor tissue sections affixed on poly-l-lysine-coated slides were collected from Bansal Hospital, Bhopal, India. The study was approved by the Institute Ethics Committee of the Indian Institute of Science Education and Research Bhopal, India.
Establishment of patient-derived cell lines
Primary cultures were established from tumor tissues obtained from breast cancer patients undergoing surgery at Bansal hospital, Bhopal, India. The tumor tissues were collected in transport media (DMEM/F12 (1:1) supplemented with 50μg/mL gentamycin sulfate (TCI, G0383, lot no. RTLNF-FJ), 200 units/ml of penicillin and streptomycin (Invitrogen, 15140122, lot no. 2321120), 0.01% antibiotic-antimycotic solution (Invitrogen, 15240062, lot no. 2441813), and 20% CELLect fetal bovine serum, gold, US origin (MP Biomedicals, 092916754, lot no. L12128014). Briefly, the transport media was discarded and the tissue was minced and subjected to enzymatic disaggregation by incubating with 5ml fresh transport medium containing 1mg collagenase (Sigma, C2674-1, lot no. 0000131473) and 120 U hyaluronidase (TCI, H064, lot no. HQV5C- II) for 45 min–1 h at 37°C with rigorous pipetting at 15 min intervals. Post incubation, the digested tissue was passed through 70 μm cell strainer (Falcon, 352350, lot no. 8362528). The single cell suspension thus obtained was subjected to centrifugation at 1500 rpm for 5 min. The resultant cell pellet was resuspended in 1× TAC buffer (1:9 ratio of 170 mM Tris [pH 7.4] and 150 mM NH4Cl [pH 7.4] respectively) and incubated at 37°C for 10 min to eliminate the RBCs. The cell suspension was centrifuged at 1500 rpm for 5 min to obtain cell pellet devoid of RBCs. Finally, the cells were resuspended in complete primary growth media (DMEM/F12 (1:1) supplemented with 50 μg/ml gentamycin sulfate, 200 U/ml penicillin and streptomycin, 0.01% antibiotic-antimycotic solution and 10% CELLect fetal bovine serum, gold, US origin. The cells were cultured in incubator set at 37°C with humidified atmosphere of 5% CO2. The cell lines were gradually cultured in DMEM supplemented with 10% FBS (Sigma, F7524, lot. no BCBX8466), 100 units/ml of penicillin and streptomycin, and 2 mM/l l-glutamine.
Immunohistochemistry
Breast tumor tissues of 5 microns size were obtained from Bansal hospital, Bhopal, India for immunohistochemical analysis. Briefly, the sections were deparaffinized at 65°C followed by xylene treatment and subsequently rehydrated using ethanol gradients. Antigen retrieval was then performed by boiling the slides at 98°C in citrate buffer (pH 6.0) for 10 min. Endogenous peroxidase was quenched using hydrogen peroxide solution for 45min at RT, followed by blocking using 5% BSA for 1 h at RT. Primary antibodies against the proteins of interest were incubated overnight at 4°C. Subsequently, secondary antibody detection and DAB staining were performed using Biogenex super sensitive™ polymer HRP IHC detection kit (QD430-XAKE, lot no. QD4300919) as per manufacturer's instructions. The details of patient samples are enlisted in Supplementary Table S4.
Subcellular fractionation
Subcellular fractionation was performed as described previously (16). Briefly, cells were scraped and collected in PBS, followed by centrifugation at 3000rpm for 5min. The pelleted cells were resuspended in cytoplasmic lysis buffer (10 mM HEPES [pH 8.0], 1.5 mM MgCl2, 10 mM KCl, 0.5% NP-40) supplemented with 1 × PIC and incubated on ice for 15min. The resultant mixture was pelleted by centrifugation at 3000 rpm, 4°C for 10 min. The supernatant was collected as cytoplasmic fraction and stored at –80°C until further use. The pellet was washed with PBS followed by resuspension in nuclear lysis buffer (10 mM HEPES [pH 8.0], 1.5 mM MgCl2, 400 mM NaCl, 0.1 mM EDTA, 20% Glycerol) supplemented with 1× PIC. The mixture was vortexed vigorously and was incubated at 4°C for 30 min. The mixture was then centrifuged at maximum speed (16 900 × g) for 30 min at 4°C. The supernatant was collected as nuclear fraction.
Establishment of CRISPR/cas9 knockout cells
The CRISPR/Cas9-mediated HIF-1α knockout of HCC1806 was generated as previously mentioned (13). For generating HIF-2α knockouts, exon 2 of EPAS1 (gene coding for HIF-2α) targeting sgRNA was designed using the GPP sgRNA Designer tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). The sgRNA was cloned using the BbsI site of lentiviral vector pLentiCRISPR-E (Addgene, 78852) as described (https://media.addgene.org/cms/filer_public/6d/d8/6dd83407-3b07-47db-8adb-4fada30bde8a/zhang-lab-general-cloning-protocol-target-sequencing_1.pdf). The sequence of the obtained construct was verified using Sanger sequencing. Lentivirus containing sgRNA cloned plasmid was generated in HEK293T. Post transduction, cells were subjected to puromycin (1 μg/ml) selection for 5–6 days. Subsequently, single-cell suspension was performed using a serial-dilution method and seeded in a 96-well plate. The positive knockout clones were identified using immunoblotting and the sequencing analysis of the region targeted by sgRNA. A similar protocol was followed for generating BBS Mut HCC1806 cells. The sequence of the sgRNAs used is provided in Supplementary Table S1.
Co-immunoprecipitation
The hypoxia-treated MCF7 and HCC1806 cells were lysed using cell lysis buffer (10mM Tris [pH 7.5], 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) supplemented with 1 × PIC for 30 min. The lysates were then centrifuged at the highest speed for 20 min. The supernatants were collected, quantified and incubated with anti-PKM2, anti-HIF-1α and Normal Rabbit IgG antibodies overnight at 4°C. 25 μl Protein G Dynabeads™ (Invitrogen, 10004D, lot no. 01013573) were pre-washed thrice with cell lysis buffer, added to the immunoprecipitated lysate, and further incubated for 2 h at 4°C. The immunoprecipitated complex was then isolated by magnetizing the beads and washed with cell lysis buffer four times. Post washing, the beads were boiled with 2 × Laemmli buffer for 5 min. The beads were then separated, and the eluted proteins were analyzed with immunoblotting.
Luciferase reporter assay
103, 89 and 75 bp fragments spanning PFKFB3 HRE1, HRE2 and HRE3 were individually amplified using MCF7 genomic DNA as a template and cloned between KpnI and XhoI sites of the pGL3 basic expression vector (Promega, E1751). The primers used to amplify the fragments are enlisted in Supplementary Table S2. Cells were seeded in a 24-well plate and allowed to attach. The wells were co-transfected with different PFKFB3-HRE luciferase constructs along with pRL-TK renilla luciferase plasmid (Promega, E2231). 12 h post-transfection, the cells were subjected to appropriate treatments for 12 h. Subsequently, the cells were lysed, and the luciferase activity was determined using GloMax- Multi Detection System (Promega) by normalizing to the Renilla luciferase activities.
Cell proliferation assay
The cell lines were seeded at a density of 85 × 103 cells per well of a 12-well plate. Post completion of time points, the dye exclusion method of viable cell counting using trypan blue dye (Himedia, TC193, lot no. 0000190691) and hemocytometer was employed.
MitoTracker staining
MitoTracker™ Deep Red (Invitrogen, M22426, lot no. 2112250) stock solution was prepared as per the manufacturer's instructions. 1 × 105 cells were seeded in a well of 12-well plate and allowed to attach for 24 h. 70 nM working solution of MitoTracker made using phenol red-free DMEM (Gibco, 21063-029, lot no. 1967601) or phenol red-free RPMI (Gibco, 11835-030, lot no. 1951096) was added to the cells. After 45 min of incubation in the dark at 37°C, the cells were washed with PBS and stained for nucleus using Hoechst 33342. Subsequently, the cells were trypsinized, washed twice with PBS, finally resuspended in PBS, and subjected to flow cytometry using fluorescence-activated cell sorting (FACS) Aria III by Becton Dickinson. Analysis of the data was performed using FlowJo software (version 10.7.1). For fluorescence imaging, MitoTracker was added to the spheroid culture at a final concentration of 150 nM, followed by incubation in the dark at 37°C. The staining was then visualized using Olympus FV3000 confocal laser scanning microscope with a 60× Plan Apo N objective (oil, 1.42 NA).
Mitochondria isolation from cell lines
Mitochondria were isolated from the breast cancer cells as mentioned previously (17). Briefly, cells were washed once with ice-cold PBS. Subsequently, the cells were scraped in PBS and centrifuged at 600g at 4°C for 10min. After discarding the supernatant, the pellet was resuspended in 2–3 ml of ice-cold incubation buffer (Ibc; 10 mM Tris–MOPS, 10 mM EGTA/Tris, 200 mM sucrose [pH 7.4]). The cells in resuspension were partitioned in multiple microcentrifuge tubes for homogenization using a hand-operated micro pestle, and the resultant homogenate was spun at 600g for 10 min at 4°C. The supernatant was collected and centrifuged at 7000g for 10 min at 4°C. The obtained mitochondrial pellet was washed with 200 μl of ice-cold Ibc and finally resuspended in the minimal buffer volume that remained after discarding the supernatant.
Mitochondrial complex I assay
The isolated mitochondrial lysate was quantified, and an equal amount of the preparation was used to estimate the mitochondrial complex I activity as per the manufacturer's protocol (Sigma, MAK359, lot no. 6E01K09680). The readings were measured at an optical density of 600nm using a microplate reader set at RT. The values represent the mean ± SD of technical duplicates from a representative experiment.
IC50 calculations
3 × 103 cells were seeded in each well of a 96-well cell culture plate. The cells were allowed to attach for 24 h. Appropriate serial dilutions of Shikonin (Cayman chemicals, 14751, lot no. 0566727-12) stocks were made in cell culture media and added to the wells. 24 h post-treatment, a 10 μl aliquot of 2 mg/ml tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Alfa Aesar, L11939, lot no. 10196058) dissolved in culture medium was added to each well and incubated for 2–3 h at 37°C. The formazan crystals were then solubilized using 100 μl DMSO, and the absorbance was measured at 570 nm using a microplate reader. The IC50 value was determined using GraphPad Prism 8.
Molecular docking studies
Global docking analysis for shikonin with PKM2 was performed using AutoDock Vina (18). The protein structure of PKM2 (19) (PDB ID: 1T5A) was downloaded from the RCSB Protein Data Bank (20), cleaned and processed in PyMOL 2.5 (21). The chemical structure of shikonin was downloaded from PubChem (PubChem ID: 479503), and energy optimization was performed using Chem3D 20.1 software. The protein and ligand files were prepared using AutoDockTools (ADT). Polar hydrogens, Kollman charges, and water molecule deletion were assigned using ADT and saved in PDBQT format. The grid size was set to 86 × 80 × 87 along the axes with a spacing of 1.000 Å, and the grid center was designated at X = 70.806, Y = –2.988 and Z = 90.970 to cover the entire protein surface. The docking parameters of exhaustiveness, number of modes, and energy range were set at 8, 20 and 4 kcal/mol, respectively. Rigid docking of PKM2 and shikonin was scored, and binding modes were generated using AutoDock Vina. The different binding modes of the docking were energy-optimized using the YASARA Energy Minimization server (22). The optimized poses were finally visualized and analyzed using PyMOL 2.5, Discovery Studio Visualizer v21.1.0, and Protein–Ligand Interaction profiler (PLIP) (23).
Generation of spheroid and organoid cultures
The spheroids and organoids were generated using an overlay method as previously described (24,25) with few modifications. 50 μl of Growth Factor Reduced (GFR) Basement Membrane Matrix (Corning, 356230, lot no. 9343006) was spread uniformly in a well of 96-well cell culture plate and allowed to solidify at 37°C. 7 × 103 cells were resuspended in 100 μl of appropriate cell-culture media containing 1 μg/ml hydrocortisone (Sigma, H0888, lot no. SLBG4963V) and 5 μg/ml insulin (Sigma, I1882, lot no. SLBR1114V) and added over the solidified matrix. Media containing 10% GFR matrigel was then subsequently overlayed as the topmost layer and incubated at 37°C in the presence of 5% CO2. Hormones containing media was replaced after every 72 h.
Hypoxia detection in spheroid culture
Hypoxic regions in the spheroids were detected using Hypoxyprobe™-1 (HP-1000, lot no. 111318) as described previously (26). Briefly, pimonidazole hydrochloride was added to the cell culture media at a final concentration of 100 μM and incubated at 37°C for 2 h. The samples were fixed using 4% formaldehyde and subjected to immunofluorescence protocol. The samples were finally incubated with an anti-pimonidazole mouse IgG1 monoclonal antibody that binds to protein, peptide, and amino acid adducts of pimonidazole in hypoxic cells. The hypoxic regions were visualized by using Alexa-Flour 488 anti-mouse IgG.
Animal experiments
The mice used in this study were cared for in accordance with the procedures described in the Guide for the Care and Use of Laboratory Animals. All animal experiments were conducted under the approval of the Animal Care and Use Committee (ACUC), NCI-Frederick, with a designated protocol number (21-471) specifically assigned for the experiments conducted in this study.
6 × 106 cells (WT BBS and BBS Mut MCF7 cells) in 100μL of PBS was injected s.c. into the flanks of 6-week-old female athymic nude mice. One week before injection of cells, estradiol cypionate (Depo-Estradiol Covetrus NA, 074962) in cottonseed oil was administered s.c. at 1mg/kg between the shoulder blades using 25G needle. Estradiol injections was given once every week. Once the tumors reached a size of 100 mm3, mice were randomly assigned to four groups. The resource equation method was used to calculate the group size, and no selection criteria was used to assign groups. Shikonin (Sigma, 074962) dissolved in DMSO or DMSO only was injected in mice i.p. either 1.5 mg/kg of Shikonin in 20 μl DMSO or 20 μl DMSO only every other day for 2 weeks. Tumor volume was measured every other day for 5 weeks. Tumors were harvested before they reach the maximal size (2000 mm3) permitted by NCI-Frederick ACUC. Tumor sizes were measured by digital Vernier Caliper and tumor volume (in mm3) was calculated using the formula (length × width2)/2. The tumor volumes of individual mouse are provided in Supplementary Table S6. Area under the curve (AUC) was calculated using the formula 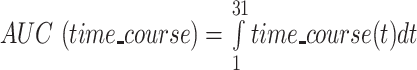 , where, time_course (t) is a function of the time variable, t, that varies between day 1 and day 31. The aforementioned formula was approximated using trapezoidal rule. Tumor growth inhibition for day 31 was calculated using the formula TGI % = (1 – [mean volume of treated tumors at day 31/mean volume of control tumors at day 31]) × 100. Mice were maintained as per ACUC guidelines.
, where, time_course (t) is a function of the time variable, t, that varies between day 1 and day 31. The aforementioned formula was approximated using trapezoidal rule. Tumor growth inhibition for day 31 was calculated using the formula TGI % = (1 – [mean volume of treated tumors at day 31/mean volume of control tumors at day 31]) × 100. Mice were maintained as per ACUC guidelines.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 or RStudio (version 1.3.1073). Unless otherwise mentioned, all data are represented as mean ± SD analyzed using a two-tailed Student's t-test. The statistical methods for each analysis are described within the figure legends or under respective sections in materials and methods. P-value less than 0.05 was considered significant. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, ns = not significant.
RESULTS
Nuclear PKM2 regulates the expression of metabolism-related genes under hypoxia
One of the most intriguing non-canonical features of PKM2 is its ability to undergo nuclear translocation to function as a co-activator of transcription factors (11,27). To explore the outcomes and vitality of non-canonical nuclear PKM2 signaling in different subtypes of breast cancer, we performed our experiments using cell lines that represent two of the commonly detected and widely studied types of breast cancer- luminal A (MCF7) and basal stubtype (HCC1806). Therefore, to understand the non-canonical effects of PKM2, we first verified its nuclear presence by performing immunofluorescence imaging and immunoblot analysis upon subjecting MCF7 and HCC1806 to 1% O2 for 24 h (Supplementary Figure S1A–C). We also identified that Ran- a GTPase enhances the nuclear translocation of PKM2 in hypoxic breast cancer cells as its knockdown led to a substantial reduction of nuclear PKM2 (Supplementary Figure S1D, E) (27). Furthermore, we also validated the physical interaction between PKM2 and HIF-1α in hypoxic MCF7 cells by performing co-immunoprecipitation assays (Supplementary Figure S1F, G).
We have previously demonstrated the crucial involvement of epigenetically-acting transcription factor BORIS in dictating the alternative splicing of PKM (12). Upon introducing mutations in the BORIS-binding site (BBS) present in the exon 10 region of PKM using CRISPR/Cas9, we generated PKM2 knockout cell lines of MCF7 and HCC1806, which expresses only PKM1 isoform (Figure 1A, B). Next, to examine the global transcriptomic alterations induced by PKM2 depletion, we performed Human Transcriptome Array 2.0 (HTA 2.0) analysis in wild-type BBS (WT BBS) and BBS mutant (BBS Mut) MCF7 cells subjected to 24 h of hypoxia. Data analysis performed using Transcriptome Analysis Console (TAC) revealed that 3190 and 1581 genes were differentially expressed in WT BBS hypoxia vs. WT BBS normoxia (condition 1), and BBS Mut hypoxia vs. BBS Mut normoxia (condition 2), respectively. Moreover, 855 genes were observed to be common between condition 1 and condition 2 (Figure 1C). Notably, 2399 genes were identified to be differentially expressed in BBS Mut hypoxia versus WT BBS hypoxia (condition 3), from which 1430 genes were upregulated, and 969 genes were downregulated. The significant events of differentially expressed genes (DEGs) in BBS Mut hypoxia versus WT BBS hypoxia with Fold Change > 2 and P < 0.05 are marked in Figure 1D, with PKM2 being one of the most significantly downregulated transcripts, indicating its successful elimination (Supplementary Figure S1H). The GO terms for biological processes enrichment analysis of DEGs in condition 3 revealed that the metabolism-related genes were transcriptionally affected, underscoring the paramount influence of PKM2 in modulating the metabolic landscape of hypoxic breast cancer cells (Figure 1E).
Figure 1.

Nuclear PKM2 affects transcriptome of hypoxic breast cancer cells. (A) Schematic representation of strategy employed for generating CRISPR/Cas9-mediated PKM2 knockout cell lines. (B) Immunoblot analysis of PKM2 and PKM1 in WT BBS and BBS Mut cell lines of MCF7 and HCC1806. (C) Venn diagram depicting the DEGs under the conditions of WT BBS MCF7 hypoxia vs. normoxia, BBS Mut MCF7 hypoxia versus normoxia, and BBS Mut MCF7 hypoxia versus WT BBS MCF7 hypoxia. (D) Volcano plot of HTA 2.0 analysis showing the DEGs in BBS Mut MCF7 hypoxia versus WT BBS MCF7 hypoxia. The significantly downregulated and upregulated genes are indicated with green and red points respectively (P < 0.05 and Fold Change > 2). The top targets have been highlighted. (E) Dot plot representing top 20 enriched terms for biological processes for the DEGs obtained from BBS Mut hypoxia vs WT BBS hypoxia HTA 2.0 analysis. (F) Venn diagram depicting the common genes between HIF-1α ChIP-seq and BBS Mut hypoxia vs. WT BBS hypoxia HTA 2.0 analysis. A total of 44 HIF-1α target genes were differentially expressed due to absence of PKM2. (G) Dot plot representing the top 20 enriched terms for GO Biological Process (FDR < 0.05) in the common genes identified from HIF-1α ChIP-Seq and DEGs from BBS Mut hypoxia vs WT BBS hypoxia HTA 2.0 analysis. (H) Heat map representation of 44 common genes from HIF-1α ChIP-Seq and DEGs from BBS Mut MCF7 hypoxia vs. WT BBS MCF7 hypoxia microarray analysis. For figure (B) representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified).
As per our microarray data, 2399 genes were differentially expressed in BBS Mut MCF7 hypoxia versus WT BBS MCF7 hypoxia (condition 3) from which 1263 genes were coding and 1136 genes were non-coding. Therefore, to identify the HIF-1α target genes that are regulated by PKM2, we overlapped HIF-1α ChIP-seq data with the microarray data obtained for coding genes from condition 3. 44 genes were common between the 323 HIF-1α target genes and the 1263 coding DEGs from condition 3 (Figure 1F). In addition, the GO terms for biological processes such as hexose metabolic process and glucose metabolic process were distinctly enriched in the 44 common genes (Figure 1G, Supplementary Table S5). Figure 1H represents a heat map of the fold change of the 44 target genes common between the DEGs in condition 3 of microarray data and HIF-1α ChIP-seq data. The fold change values are obtained from the microarray data analysis of condition 3.
Conclusively, our data indicates the vitality of the non-canonical nuclear function of PKM2 in promoting global transcriptomic alterations. Moreover, our data reveals the dynamism of PKM2-mediated regulation of the glucose metabolism-associated genes under hypoxia.
PKM2 is required for the hypoxic induction of PFKFB3
To validate the potential novel targets of the HIF-1α-PKM2 axis obtained from HTA 2.0 analysis, we performed a qRT-PCR screen of 15 glycolytic genes that are reported HIF-1α targets (Supplementary Figure S1I). Interestingly, our results suggested that 6-phosphofructo-2-kinase/fructose-2,6-biphosphatases 3 (PFKFB3) requires PKM2 for its hypoxic induction. PFKFB3 is a glycolytic enzyme which catalyzes the reversible reaction of converting fructose 6-phosphate (Fru 6 P) to fructose 2,6-bisphosphate (Fru 2,6 BP) (28,29), the latter being a potential regulator of phosphofructokinase-1 (PFK-1) activity. As PFK-1 catalyzes the committed step of glycolysis, its constitutive activation results in an elevated glycolytic rate (Figure 2A). In addition to possessing the highest kinase: bisphosphatase activity compared to other family members (30), PFKFB3 is positively associated with cancer progression and aggression (31–33).
Figure 2.

PKM2 regulates PFKFB3 expression under hypoxia. (A) Schematic representation of the role of PFKFB3 in glycolysis. PFKFB3 catalyzes the reversible reaction of converting fru 6 P to fru 2,6 BP to regulate the activity of PFK-1. (B) Immunoblot analysis depicting the hampered hypoxic induction of PFKFB3 in BBS Mut cell lines of MCF7 and HCC1806. (C) PFK assay performed in WT BBS and BBS Mut cell lines of MCF7 and HCC1806 exposed to normoxia or 24 h hypoxia. (D) Schematic representation of the location of the three HREs in the PFKFB3 promoter. The arrows indicate the primers used in ChIP-qRT PCR and for amplifying respective HREs for luciferase construct generation. (E) Luciferase assay performed in WT BBS and BBS Mut cell lines of MCF7 and HCC1806 showing the dampened activity of HRE1 and HRE2 in the BBS Mut cells (F) PFK assay results depicting enhanced PFK activity of the BBS Mut cells upon re-introduction of PKM2 under hypoxia. (G) PKM2 overexpression followed by luciferase assay in BBS Mut cell lines of MCF7 and HCC1806 cells. For figures (B), (C) and (F), representative images are provided. The error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Immunoblot analysis validated that hypoxic induction of PFKFB3 was hampered in the BBS Mut cell lines compared to their respective WT BBS cells (Figure 2B). Consequently, the BBS Mut MCF7 cell lines exhibited a decreased phosphofructokinase (PFK) activity under hypoxia as well (Figure 2C). As Ran aids the nuclear translocation of PKM2 under hypoxia, its knockdown led to substantial reduction of PFKFB3 induction in both the WT BBS cell lines used in our study (Supplementary Figure S2A, B). Further, to identify the regulatory region driving PFKFB3 expression in a PKM2-HIF-1α-dependent manner, we explored the functional contribution of each HREs located at the positions –107 (HRE1), –1279/–1288 (HRE2) and –1902 (HRE3) relative to the transcriptional start site (TSS) (Figure 2D) (34). The three fragments of the length 103, 89 and 75 bp spanning HRE1, HRE2 and HRE3 respectively were individually cloned upstream of a firefly luciferase (FLuc) coding sequence in a pGL3 basic vector. The reporter constructs were co-transfected with pRL-TK vector (containing renilla luciferase expression controlled by the SV-40 promoter) to the cell lines, followed by exposure to 1% O2 for 12 h. The FLuc activity was significantly decreased in the BBS Mut cell lines of MCF7 and HCC1806. Conclusively, HRE1 and HRE2 were identified as the functional PFKFB3 HREs from our luciferase assay data (Figure 2E).
Notably, the re-introduction of PKM2 led to its hypoxia-specific nuclear translocation (Supplementary Figure S2C) paired with a heightened induction of PFKFB3 in the BBS Mut cell lines (Supplementary Figure S2D). The rescue effect observed in PFKFB3 expression was also accompanied by a rise in hypoxic PFK activity (Supplementary Figure S2E, Figure 2F). Moreover, using luciferase assay, we further verified that reintroduction of PKM2 enhances the activity of HRE1 and HRE2 (Figure 2G). Altogether, our data conclusively suggests that HRE1 and HRE2 are the PKM2-responsive sites in PFKFB3 promoter.
PKM2 is an essential interacting partner of HIF-1α required for transcriptional upregulation of PFKFB3
To further verify luciferase assay results, ChIP assay was performed with HIF-1α and PKM2 antibody in the WT BBS and BBS Mut MCF7 cell lines exposed to 24hrs of 1% O2. As evident from the prominent enrichment of the aforementioned factors, we concluded that HRE1 and HRE2 are functionally active sites for PKM2-HIF-1α interactive binding (Figure 3A). Additionally, to investigate if HIF-1α is an obligate interacting partner of PKM2, we generated a stable HIF-1α knockout cell line of HCC1806 (H1AKO). PFKFB3 expression and PFK activity were observed to be dampened under hypoxia in H1AKO cells (Supplementary Figure S3A–C). Furthermore, the ChIP assay corroborated a complete lack of PKM2 occupancy in H1AKO cells confirming that PKM2 itself is unable to bind PFKFB3 HREs under hypoxia (Figure 3B). Additionally, the FLuc activity of HRE1 and HRE2 was also drastically reduced in H1AKO cells (Supplementary Figure S3D). To understand the mechanism underlying PKM2-mediated PFKFB3 induction, we performed ChIP assay of the histone acetyltransferase p300, primarily involved in transactivating HIF-1α target genes (35). Both p300 occupancy and the histone modification it catalyzes- H3 lysine-9 acetylation (H3K9Ac) was drastically reduced in BBS Mut cells showing that PKM2 induces epigenetic alteration, making PFKFB3 HREs accessible for transcriptional initiation (Figure 3C).
Figure 3.

Hypoxic expression of PFKFB3 requires HIF-1α and PKM2. (A) ChIP assay of HIF-1α and PKM2 performed in WT BBS and BBS Mut MCF7 cells. (B) ChIP assay of HIF-1α and PKM2 in WT BBS, BBS Mut HCC1806 and HIAKO cells. (C) ChIP assay of p300 and H3K9Ac in WT BBS and BBS Mut MCF7 cells under hypoxia. Luciferase assay performed in WT BBS and BBS Mut cell lines of (D) MCF7 and (E) HCC1806 shows the reduction in the FLuc activity of HRE1 and HRE2 only in the WT BBS cell lines upon performing HIF-1α knockdown. Immunoblot analysis demonstrating the effect of HIF-1α knockdown on PFKFB3 expression in WT BBS and BBS Mut cell lines of (F) MCF7 and (G) HCC1806. For figures (F), and (G) representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Interestingly, we observed compromised HIF-1α enrichment in BBS Mut MCF7 cells under hypoxia (Figure 3A). To investigate if in the absence of PKM2, PFKFB3 regulation exhibits diminished dependency on HIF-1α, we performed HIF-1α knockdown. Results demonstrated a hampered FLuc activity only in the WT BBS cell lines (Figure 3D, E) paired with an insignificant decrease of PFKFB3 expression in the BBS Mut cell lines (Figure 3F, G). Conclusively, our observations evidently prove that PKM2 enhances binding of HIF-1α and p300 at PFKFB3 HREs. Moreover, PKM2 is also an essential interacting partner of HIF-1α, without which the transcription factor has reduced affinity towards PFKFB3 HREs. Notably, PFKFB3 is regulated by yet unreported transcription factors other than HIF-1α in the absence of PKM2, which maintains its basal-level expression.
HIF-2α regulates PFKFB3 in the absence of PKM2 or HIF-1α
HIF-1α and HIF-2α are known to recognize an identical DNA motif (36–38). However, HIF1 and HIF2 complexes are known to regulate distinct genes, and only a few shared targets have been identified (36,39). To investigate the intriguing possibility if the reduced occupancy of HIF-1α rendered PFKFB3 HREs available for HIF-2α binding, we performed HIF-2α ChIP assay. Interestingly, a prominent enrichment of HIF-2α on HRE1 and HRE2 in the BBS Mut MCF7 cells was observed (Figure 4A). Further analysis revealed no variation of HIF-2α expression across all the WT and CRISPR/Cas9 mutants used throughout our study (Supplementary Figure S4A). These findings suggest that HIF-2α is stabilized under hypoxia and its differential occupancy on PFKFB3 HREs occurs due to reduced enrichment of HIF-1α triggered by the absence of PKM2. To further investigate this intriguing experimental observation, HIF-2α knockdown studies were performed. Functional validation assessed by performing luciferase assay indicated that HIF-2α downregulation significantly decreased the FLuc activity corresponding to HRE1 and HRE2 only in the BBS Mut cell lines (Figure 4B, C). Consequently, immunoblot analysis further corroborated our findings to reveal that PFKFB3 expression is hampered upon eliminating HIF-2α in BBS Mut cell lines (Figure 4D, E).
Figure 4.

PFKFB3 is a novel non-canonical target of HIF-2α. (A) ChIP qRT-PCR analysis indicating a predominant occupancy of HIF-2α on PFKFB3 HREs under hypoxia in BBS Mut MCF7 cells. Luciferase activity of PFKFB3 HREs upon performing HIF-2α knockdown in WT BBS and BBS Mut cell lines of (B) MCF7 and (C) HCC1806 under hypoxia. Immunoblot analysis of HIF-2α knockdown performed in WT BBS and BBS Mut cell lines of (D) MCF7 and (E) HCC1806. (F) ChIP assay showing enrichment of HIF-2α on PFKFB3 HREs in BBS Mut HCC1806 and H1AKO cells. (G) Immunoblot analysis of PFKFB3 upon performing HIF-2α knockdown in H1AKO cells. (H) ChIP assay of H3K9Ac, HDAC3 and H3K9me3 in HCC1806, H1AKO and H2AKO cells under hypoxia. (I) ChIP assay of HDAC3, H3K9me3 and H3K27Ac in WT BBS and BBS Mut MCF7 cells under hypoxia. For figures (D), (E) and (G), representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Subsequently, the PKM2-independent function of HIF-2α was further verified using H1AKO cells as well. Prominent HIF-2α occupancy was also observed on PFKFB3 HREs in BBS Mut HCC1806 and H1AKO cells as indicated by ChIP assay results, highlighting a previously unreported opportunistic regulatory mechanism exhibited by HIF-2α (Figure 4F). Furthermore, immunoblotting results revealed that PFKFB3 expression was affected upon downregulating HIF-2α in H1AKO cells (Figure 4G). As HIF-2α also interacts with PFKFB3 HREs, the FLuc activity of these sites were further dampened along with a significant reduction of PFK activity upon eliminating HIF-2α in H1AKO cells (Supplementary Figure S4B, C). We also established CRISPR/Cas9-mediated HIF-2α knockout (H2AKO) cells of HCC1806 (Supplementary Figure S4D). Lentivirus-mediated knockdown of PKM2 achieved in H2AKO cells caused decreased PFKFB3 induction, indicating that hypoxic cancer cells employ HIF-2α-mediated PFKFB3 regulation as a back-up to ensure continuous expression of the essential glycolytic enzyme (Supplementary Figure S4E).
The acquired poised nature of HREs-associated chromatin facilitates basal-level expression of PFKFB3
Remarkably, our study shows that unlike HIF-1α, HIF-2α does not cause extensive hypoxic induction of PFKFB3 and only maintains its basal-level expression. To discern the underlying mechanism, we performed ChIP assays to explore the epigenetic composition of the PFKFB3 HREs-associated chromatin. H3K9Ac ChIP assay using hypoxic HCC1806, H1AKO, and H2AKO cells revealed a loss of the activatory mark only in H1AKO cells (Figure 4H). As histone deacetylase 3 (HDAC3) is involved in deacetylating H3K9Ac modifications, we performed HDAC3 ChIP and observed its enrichment in H1AKO cells (Figure 4H). Interestingly, H1AKO cells also exhibited enhanced H3K9me3 histone marks (associated with gene repression) on PFKFB3 HREs (Figure 4H). Moreover, despite decreased p300 (Supplementary Figure S4F) and H3K9Ac status paired with enhanced H3K9me3 marks, PFKFB3 expression was basally maintained, which can possibly occur due to a cumulative effect of the simultaneous existence of multiple (activatory and inhibitory) histone modifications.
The bivalent or the poised configuration of the chromatin at the promoter regions is recognized to participate in maintaining gene expression at low levels and is an elusive aspect of cancer epigenetics (40,41). To further explore this intriguing observation of PFKFB3 HREs-associated chromatin acquiring a hypoxia-specific poised state, which is dictated by the absence PKM2, we also performed HDAC3, H3K9me3 and H3K27Ac ChIP assay in WT BBS and BBS Mut MCF7 cells. H3K27Ac (associated with gene activation) was highly present in the BBS Mut cells to underscore the paramount role played by the bivalent nature of PFKFB3 HREs (Figure 4I). Taken together, we demonstrate that the PFKFB3 HREs-associated chromatin acquires a poised nature in the absence of PKM2, which prevents HIF-2α from inducing PFKFB3 and sustains its basal-level expression.
Shikonin inhibits the nuclear translocation of PKM2 to hamper hypoxic induction of PFKFB3
PKM2 is associated with various non-canonical functions that drive cancer progression and aggression; therefore, drugs targeting PKM2 present as desirable candidates for anti-cancer therapy. To investigate the effects of PKM2 inhibition under hypoxia, we employed the use of shikonin- a compound well-characterized to inhibit only PKM2 and not PKM1 (42). Molecular docking studies revealed that shikonin interacts with PKM2 with a best binding score of –8.1 kcal/mol. Additionally, the energy-optimized structure demonstrated that the inhibitor exclusively interacts with protein region coded by exon 10 (present only in PKM2) (Supplementary Figure S5A). R399/400 residue has been previously reported to be involved in forming a putative nuclear localization signal (NLS) of PKM2 (43). Interestingly, the optimized poses of the protein and ligand interaction as analyzed by PyMOL 2.5 and subsequently visualized by PLIP indicated that R399/400 can form hydrogen bonds with shikonin (Supplementary Figure S5B). Therefore, we suspected that subjecting hypoxic breast cancer cells to shikonin-mediated PKM2 inhibition will block its NLS and suppress non-canonical nuclear functions.
To test this hypothesis, we treated WT BBS cell lines with IC50 concentration of shikonin and exposed the cells to 24 h of hypoxia (Supplementary Figure S5C, D). Immunofluorescence imaging performed to visualize the sub-cellular localization of PKM2 revealed its significantly reduced nuclear pool in the drug-treated cells (Figure 5A, B). Furthermore, upon subjecting the cells to shikonin, only the WT BBS cell lines exhibited reduced hypoxic induction of PFKFB3, to indicate that shikonin inhibits the nuclear translocation of PKM2 (Figure 5C, D) without altering its expression (Supplementary Figure S5E). Additionally, dampened activity of PFKFB3 HREs assessed by luciferase assay further confirmed that shikonin blocks the nuclear non-canonical function of PKM2. Since PFKFB3 is maintained in a PKM2-independent manner by HIF-2α, no significant difference was observed in the FLuc activity of the PFKFB3 HREs in the BBS Mut cell lines (Figure 5E, F). Consequently, the restricted PFKFB3 induction in the shikonin-treated WT BBS cell lines also resulted in a decreased PFK activity, indicating that the rate of the committed step of glycolysis catalyzed by PFK-1 is affected (Figure 5G).
Figure 5.

Shikonin treatment affects PFKFB3 expression in hypoxic breast cancer cells. Immunofluorescence imaging analysis and quantification showing blocked PKM2 nuclear translocation upon shikonin treatment in WT BBS (A) MCF7 and (B) HCC1806 cells. Magnification used: 60×. Immunoblot analysis of PFKFB3 expression upon treating WT BBS and BBS Mut cell lines of (C) MCF7 and (D) HCC1806 with shikonin for 24 h under hypoxia. Luciferase assay performed upon subjecting WT BBS and BBS Mut cells of (E) MCF7 and (F) HCC1806 to shikonin treatment under hypoxia. (G) PFK assay performed in WT BBS MCF7 and HCC1806 cells showing the reduced PFK activity upon shikonin treatment in hypoxia. For figures (A), (B), (C), (D) and (G), representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Conclusively, our results evidently proves that PKM2-NLS interacting drugs can effectively restrict its nuclear translocation under hypoxia to inhibit PFKFB3 expression.
The PKM2-HIFs-PFKFB3 axis determines the glycolytic fate of hypoxic breast cancer cells
As PFKFB3 is directly involved in altering the dynamics of the committed step of glycolysis, we further explored the functional impact of restricted PFKFB3 expression on the metabolic landscape of hypoxic breast cancer cells. As PFKFB3 promotes aerobic glycolysis (29), we investigated if conditions leading to PFKFB3 downregulation (which naturally occurs in the BBS Mut cells) lead to enhanced dependency on OXPHOS for meeting the ATP requirement. As tricarboxylic acid (TCA) cycle is the main producer of reduced equivalents fueling OXPHOS, we estimated α-ketoglutarate (α-KGA) level. The PKM2 knockout cells exhibited significantly elevated α-KGA level (Figure 7A). Next, mitochondrial complex I assay was performed using mitochondria isolated from hypoxic WT BBS and BBS Mut MCF7 cells. The purity of the mitochondrial isolate was verified by checking for cytoplasmic and nuclear protein markers (Supplementary Figure S6C). Our results indicated a rapid drop in the absorbance by the BBS Mut cells, demonstrating the existence of highly active complex I under hypoxia (Supplementary Figure S6D). A direct evidence of enhanced OXPHOS rate was also obtained by utilizing MitoTracker, which is up-taken in mitochondrial membrane potential (ΔΨ) dependent manner (44,45). Upon treatment with rotenone under hypoxia, the ΔΨ of the BBS Mut cells was found to be significantly decreased denoting a higher dependency on OXPHOS even under hypoxic condition (Supplementary Figure S6E). Moreover, the BBS Mut cells lines exhibited significantly elevated OCR under hypoxia as well (Figure 6B, C, Supplementary Figure S6A,B). Finally, mitochondrial ATP rate assay confirmed the functionality of the enhanced mitochondrial activity in the BBS Mut cell lines (Supplementary Figure S6F).
Figure 7.

PFKFB3 promotes the proliferation of hypoxic breast cancer cells. (A) Ki67 immunofluorescence analysis showing decreased proliferation of BBS Mut cell lines under hypoxia. Magnification used: 10×. (B) Ki67 immunofluorescence analysis performed upon PFKFB3 overexpression showing rescued proliferation of BBS Mut cell lines under hypoxia. Magnification used: 10×. (C) BBS Mut HCC1806 cells-derived spheroids exhibit significantly less spheroid diameter compared to the WT counterpart cells. Magnification used: 40×. (D) Shikonin treatment performed in WT BBS HCC1806-derived spheroids (post 5 days of growth). (E) Shikonin treatment performed in BBS Mut HCC1806-derived spheroids (post 5 days of growth). Shikonin did not alter the proliferation of the BBS Mut cells. For all the figures, representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Figure 6.

Abrogating PFKFB3 expression enhances OXPHOS dependency under hypoxia. (A) α-KGA assay indicating elevated α-KGA levels in the BBS Mut cell lines of MCF7 and HCC1806 under hypoxia. (B) Real-time analysis of the OCR in the hypoxic WT BBS and BBS Mut MCF7 cells. (C) Quantification of the basal ΔOCR in hypoxic WT BBS and BBS Mut MCF7 cells. (D) Real-time analysis of the ECAR in WT BBS and BBS Mut MCF7 cells under hypoxia. (E) Quantification of the basal ΔECAR in hypoxic WT BBS and BBS Mut MCF7 cells. (F) Immunoblot analysis depicting PFKFB3 overexpression in BBS Mut cell lines. (G) Intracellular lactate production upon PFKFB3 overexpression in the BBS Mut cell lines estimated by lactate assay. (H) Immunoblot analysis of PFKFB3 knockdown performed in the WT BBS cell lines under hypoxia. (I) MitoTracker FACS analysis depicting an increased ΔΨ of PFKFB3 knockdown WT BBS MCF7 and HCC1806 cells. For figures (F), (H) and (I), representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
As glycolysis primarily causes lactate accumulation, which in turn influences a plethora of oncogenic properties (46–48), we assessed the glycolytic rate and the lactate producing capability of the cell lines used in our study. Lactate assay and ECAR estimation revealed reduced intracellular lactate level which resulted in its decreased extracellular secretion in the BBS Mut cell lines (Figure 6D, E, Supplementary Figure S6G–I). To investigate if the observed metabolic alterations are triggered due to modulations of the PFKFB3 expression, we performed PFKFB3 overexpression studies in the BBS Mut cell lines. Immunoblotting and PFK assay was used to verify successful PFKFB3 overexpression under hypoxia (Figure 6F, Supplementary Figure S6J). Our results clearly demonstrate that PFKFB3 overexpression significantly increased the intracellular lactate level in the BBS Mut cell lines under hypoxia (Figure 6G). Moreover, PFKFB3 downregulation was also found to suppress glycolytic rate to promote enhance OXPHOS dependency (Figure 6H, I).
Disrupting the PKM2-HIFs-PFKFB3 axis using shikonin impedes the proliferation of breast cancer cells
The metabolic state of a cancer cell is known to influence its replicative potential. A shift from a glycolysis-dependent state to an OXPHOS-dependent state is known to decrease the proliferation of cancer cells (49). We observed that the proliferation of BBS Mut cells was significantly reduced (Supplementary Figure S7A–C, Figure 7A). However, to dissect the role of hypoxia-specific metabolic alterations mediated by the restricted expression of PFKFB3, we overexpressed the glycolytic enzyme and assessed the proliferation of the BBS Mut cells. As indicated by the higher % of Ki67 positive cells, it is evident that nuclear PKM2 regulates PFKFB3 expression to stimulate the proliferation of hypoxic breast cancer cells (Figure 7B).
Furthermore, to investigate the effects of blocking the PKM2-HIFs-PFKFB3 axis in an in vitro model mimicking physiological conditions, we performed experiments using spheroid culture. We evaluated the presence of low O2 niches in the spheroids by utilizing Hypoxyprobe, and as shown in Supplementary Figure S6D, multiple hypoxic foci were detected only in the dense spheroids formed by WT BBS HCC1806 cells. The spheroid size plateaued post 7 days of seeding with an average diameter of 62 ± 8.9 μm for WT BBS HCC1806; whereas the BBS Mut HCC1806 formed remarkably smaller spheroids with a diameter size of 38.96 ± 5.86 μm to indicate a loss of proliferative potential (Figure 7C). Additionally, to understand the vitality of inhibiting the nuclear functions of PKM2 under hypoxia, we treated the spheroid cultures (post 5th day of seeding) with 2.2 μM of shikonin for 48 h. Microscopic analysis clearly indicated a significant decrease in the spheroid size (Figure 7D) paired with enhanced OXPHOS rate (Supplementary Figure S7E) of the WT BBS HCC1806-derived spheroids. On the contrary, shikonin did not alter the growth kinetics of BBS Mut HCC1806-derived spheroids (Figure 7E).
To explore our study's clinical relevance, we performed immunohistochemical analyses of tissue sections obtained from breast cancer patients. We found a strong co-expression of PKM2 and PFKFB3 in the hypoxic regions of the tumor sections (Figure 8A, C). Notably, the non-hypoxic regions exhibited reduced PKM2 and PFKFB3 expression (Figure 8B, C). Interestingly, upon subjecting our established triple negative breast tumor-derived cell line BC8322 to shikonin treatment, PFKFB3 induction was hampered, thus corroborating our findings that in hypoxic regions of breast cancers, PFKFB3 is primarily driven in a PKM2-HIF-1α-dependent manner (Figure 8D). Next, we also treated BC8322-derived organoid culture with shikonin, and consistently, we observed a marked decrease as indicated by the diminished organoid diameter (Figure 8E). Moreover, to verify if shikonin can successfully hinder the proliferation of breast cancer cells in vivo, we performed experiments using xenograft tumor models in athymic nude mice. Mice carrying WT BBS or BBS Mut MCF7 tumors were treated with shikonin or DMSO and relative tumor growth was monitored over time (Figure 8F). As evident from the drastic difference in the tumor volume, shikonin treatment resulted in a marked decrease in WT BBS MCF7 tumor growth (Figure 8G, H). Importantly, shikonin treatment exhibited >∼50-fold higher growth inhibitory effect on WT BBS MCF7 tumors (TGI = 49.03%) as compared to the BBS Mut tumors (TGI = –2.53%) (Figure 8I). Similarly, area under the curve (AUC) measurements strongly corroborated with the TGI values to indicate that shikonin treatment impedes the PKM2-HIFs-PFKFB3 axis to restrict the growth of breast cancer cells (Figure 8J).
Figure 8.

Shikonin blocks the HIFs-PKM2-PFKFB3 axis to exhibit anti-tumoral activity. (A) Immunohistochemistry analysis of CAIX, PKM2 and PFKFB3. Hypoxic regions marked by the strong membranous and/or cytoplasmic immunostaining for CAIX also exhibit strong expression of PKM2 and PFKFB3. (B) Normoxic regions of tumors indicated by a negative staining for CAIX also shows relatively poor expression of PKM2 and PFKFB3. Magnification used: 40×. (C) Quantification of CAIX, PKM2 and PFKFB3 for the normoxic and hypoxic regions of 18 breast cancer patients. (D) Immunoblot analysis of PFKFB3 upon treating triple negative breast tumor-derived cell line BC8322 with shikonin. (E) BC8322-derived organoid culture depicting significantly decreased spheroid diameter on shikonin treatment. (F) Schematic representation of the in vivo experimental protocol. (G) Representative tumor images from athymic nude mice bearing WT BBS MCF7-derived tumors and receiving DMSO (n = 4) or shikonin (n = 6) treatment. (H) Representative tumor images from athymic nude mice bearing BBS Mut MCF7-derived tumors and receiving DMSO (n = 5) or shikonin (n = 7) treatment. (I) Line graph depicting relative growth rate of WT BBS and BBS Mut MCF7 xenograft tumors. Tumor growth inhibition (TGI) is indicated. WT BBS MCF7 DMSO vs WT BBS MCF7 Shikonin, P = 0.045; BBS Mut MCF7 DMSO versus BBS Mut MCF7 Shikonin P = 0.68 (as calculated using Welch's t-test). (J) Box plot depicting AUC measurements of WT BBS and BBS Mut MCF7 xenograft tumors. WT BBS MCF7 DMSO vs WT BBS MCF7 Shikonin, P = 0.006; BBS Mut MCF7 DMSO vs BBS Mut MCF7 Shikonin, P = 1.0 (as calculated using t-test, combined with the Bonferroni P-value correction for multiple comparisons). For figures (A), (B), (D) and (E), representative images are provided. Error bars show mean values ± SD (n = 3 unless otherwise specified). As calculated using two-tailed Student's t-test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Taken in concert, the cell-based data as well as the in vivo data consistently prove that inhibition of PKM2 hampers PFKFB3 induction to restrict breast cancer cell proliferation by enhancing OXPHOS dependency.
DISCUSSION
It is estimated that almost 10 million cancer patient deaths occurred in 2020, and breast cancer was identified as the most commonly diagnosed type of cancer (50). Due to the multinodular structure of breast cancers, numerous hypoxic core regions surrounded by a normoxic frame of cells can often be detected (51). This microenvironmental condition of low pO2 is known to increase aggressiveness of cancers that ultimately presents as major hurdle in anti-cancer therapy (52,53). An eminent feature of hypoxic breast cancer cells which presents as one of the paramount orchestrators of rewired cellular signalling is altered metabolism (1). Moreover, the hypoxic niches also promote ‘repurposing’ of various metabolic enzymes to achieve additional functional advantages that aid cancer cell survival (54). PKM2 is one such metabolic enzyme known to exhibit moonlighting functions under hypoxia and is also a critical contributor of the Warburg effect.
To specifically dissect the contributions of PKM2 in shaping hypoxic transcriptome, we compared the ChIP-seq analysis of HIF-1α with transcriptomic data obtained from comparing WT BBS and BBS Mut cells. Besides governing glycolytic rate, we identified for the first time, a plethora of cellular pathways that are affected by PKM2; highlighting its role in sculpting global gene transcription. Till date, there are no reports which have investigated the epigenetic regulation of PFKFB3 under hypoxia. Remarkably, this is the first instance where novel insights into the PKM2-mediated alterations in the epigenetic landscape of PFKFB3 HREs has been provided which dictates PFKFB3 expression. Further, we have not only shown a complex interplay of various epigenetic factors, but have also highlighted the functional contributions of the poised nature of the promoter-associated chromatin of PFKFB3. Collectively, our findings lay a strong foundation for future studies based on exploring the potent involvement of metabolic enzymes in orchestrating cancer-specific epigenetic alterations.
Notably, our study substantiates the PKM2-independent transcriptional regulation of PFKFB3 by HIF-2α results in a shift from glycolysis to OXPHOS under hypoxia. Mitochondrial respiration can function even at 0.5% of O2 availability (55,56). Luengo and co-workers have previously negatively correlated ΔΨ with cellular proliferation (49). Consistently, our present study also corroborated a decrease in the proliferation of the BBS Mut cell lines, which are predominantly OXPHOS-dependent. Finally, we investigate the clinical relevance of this study by employing PKM2 inhibitor- shikonin. Shikonin dampens the hypoxic induction of PFKFB3 by interacting with a putative-NLS of PKM2 and preventing its hypoxic nuclear translocation. Notably, this phenomenon was coupled with a concomitant rise in the ΔΨ, a direct indicator of OXPHOS rate as verified using 2D and 3D cell culture methods. Moreover, mice carrying WT BBS MCF7 tumors and subjected to shikonin treatment exhibited remarkably decreased tumor growth, highlighting the paramount importance of targeting PKM2 signaling to impede tumor progression.
Evidently, this body of work provides concrete evidence for a unique regulatory network that exists between two crucial glycolytic enzymes- PKM2 and PFKFB3 under hypoxia. To the best of our knowledge, our work also reports for the first time a novel phenomenon in which HIF-2α compensates for the reduced occupancy of HIF-1α to regulate PFKFB3. It will be further interesting to discern the epigenetic writer responsible for H3K9me3 enrichment at PFKFB3 HREs, whose binding is negatively regulated by PKM2. Moreover, we strongly believe that implementing PKM2-targeting drugs as a part of anti-cancer regimen can be beneficial in combating the adverse repercussions of hypoxic cancer cells by suppressing the non-canonical functions of PKM2.
DATA AVAILABILITY
The microarray data generated in this study is deposited under the GEO accession number: GSE190401. All the other data supporting the findings are included in the article and its supporting information.
Supplementary Material
ACKNOWLEDGEMENTS
Graphical representations are generated using BioRender.
Author contributions: S.S. and M.R.P. conceived and designed the study. M.R.P. performed ChIP, immunoblotting, Co-IP, luciferase, and qRT-PCR assays. Spheroid and organoid experiments, extracellular flux assays, FACS, knockdown, rescue and CRISPR/Cas9 experiments as well as generation of schematic representations for the manuscript were contributed by M.R.P. M.R.P. and S.A.M. contributed to performing HTA 2.0 array. ChIP-seq and HTA 2.0 array data was analyzed by A.R. M.R.P. and A.R. contributed in performing metabolic assays. M.R.P. and S.A.M. contributed to proliferation assays, confocal microscopy and image analyses as well as establishing breast tumor-derived cell lines. A.R contributed to molecular docking studies and cloning of all the used plasmid constructs. S.A.M. contributed to immunohistochemical analysis of patient-derived tumor sections. K.B. contributed to animal experiments. T.M. and A.M. contributed to performing statistical analysis of in vivo data. M.R.P., A.R. and S.S. analyzed the data. A.S., J.M., S.K.S. and S.S. contributed new reagents. S.S., S.K.S. and M.R.P. contributed to scientific discussions. M.R.P. wrote the manuscript with input from all authors. S.S. oversaw all the experiments, data analyses and manuscript preparation.
Notes
Present address: Adarsh Raveendran, German Cancer Research center (DKFZ), Im Neuenheimer Feld 581, 69120 Heidelberg, Germany.
Kajal Biswas, Division of Cancer Prevention, NCI, Rockville, MD 20850, USA.
Jharna Mishra, Neuberg Supratech Kotgirwar Diagnostics, Bhopal, Madhya Pradesh 462003, India.
Contributor Information
Madhura R Pandkar, Department of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal, Madhya Pradesh 462066, India.
Adarsh Raveendran, Department of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal, Madhya Pradesh 462066, India.
Kajal Biswas, Center for Cancer Research, National Cancer Institute (NCI), Frederick, MD 21702-1201, USA.
Srinivas Abhishek Mutnuru, Department of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal, Madhya Pradesh 462066, India.
Jharna Mishra, Department of Pathology, Bansal Hospital (BH), Bhopal, Madhya Pradesh 462016, India.
Atul Samaiya, Department of Surgical Oncology, BH, Bhopal, Madhya Pradesh 462016, India.
Tyler Malys, Statistical Consulting and Scientific Programming, Frederick National Laboratory for Cancer Research, National Institutes of Health, Frederick, MD 21702, USA.
Alexander Y Mitrophanov, Statistical Consulting and Scientific Programming, Frederick National Laboratory for Cancer Research, National Institutes of Health, Frederick, MD 21702, USA.
Shyam K Sharan, Center for Cancer Research, National Cancer Institute (NCI), Frederick, MD 21702-1201, USA.
Sanjeev Shukla, Department of Biological Sciences, Indian Institute of Science Education and Research Bhopal, Bhopal, Madhya Pradesh 462066, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Wellcome Trust/Department of Biotechnology (DBT) India Alliance Fellowship Grant [IA/I/16/2/502719]; Science and Engineering Research Board (SERB) Grant [CRG/2021/004949, STR/2020/000093, IPA/2021/000148) to S.S.]; University Grants Commission (to M.R.P); Indian Institute of Science Education and Research Bhopal fellowship (to S.A.M.); Department of Science and Technology, Ministry of Science and Technology (to A.R).
Conflict of interest statement. None declared.
REFERENCES
- 1. Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 2. Hsu P.P., Sabatini D.M. Cancer cell metabolism: warburg and beyond. Cell. 2008; 134:703–707. [DOI] [PubMed] [Google Scholar]
- 3. Chen C., Pore N., Behrooz A., Ismail-Beigi F., Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1: interaction between H-ras and hypoxia. J. Biol. Chem. 2001; 276:9519–9525. [DOI] [PubMed] [Google Scholar]
- 4. Semenza G.L., Roth P.H., Fang H.M., Wang G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994; 269:23757–23763. [PubMed] [Google Scholar]
- 5. Kaelin W.G., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008; 30:393–402. [DOI] [PubMed] [Google Scholar]
- 6. Ohh M., Park C.W., Ivan M., Hoffman M.A., Kim T.Y., Huang L.E., Pavletich N., Chau V., Kaelin W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel - Lindau protein. Nat. Cell Biol. 2000; 2:423–427. [DOI] [PubMed] [Google Scholar]
- 7. Ikeda E., Achen M.G., Breier G., Risau W. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J. Biol. Chem. 1995; 270:19761–19766. [DOI] [PubMed] [Google Scholar]
- 8. Semenza G.L., Jiang B.H., Leung S.W., Passantino R., Concordat J.P., Maire P., Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase a gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996; 271:32529–32537. [DOI] [PubMed] [Google Scholar]
- 9. Wenger R.H., Rolfs A., Marti H.H., Bauer C., Gassmann M. Hypoxia, a novel inducer of acute phase gene expression in a human hepatoma cell line. J. Biol. Chem. 1995; 270:27865–27870. [DOI] [PubMed] [Google Scholar]
- 10. Amin S., Yang P., Li Z. Pyruvate kinase M2: a multifarious enzyme in non-canonical localization to promote cancer progression. Biochim. Biophys. Acta - Rev. Cancer. 2019; 1871:331–341. [DOI] [PubMed] [Google Scholar]
- 11. Luo W., Hu H., Chang R., Zhong J., Knabel M., O’Meally R., Cole R.N., Pandey A., Semenza G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011; 145:732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh S., Narayanan S.P., Biswas K., Gupta A., Ahuja N., Yadav S., Panday R.K., Samaiya A., Sharan S.K., Shukla S. Intragenic DNA methylation and BORIS-mediated cancer-specific splicing contribute to the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:11440–11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahuja N., Ashok C., Natua S., Pant D., Cherian A., Pandkar M.R., Yadav P., Narayanan S.S.V., Mishra J., Samaiya A. et al. Hypoxia-induced TGF-β–RBFOX2–ESRP1 axis regulates human MENA alternative splicing and promotes EMT in breast cancer. NAR Cancer. 2020; 2:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schödel J., Oikonomopoulos S., Ragoussis J., Pugh C.W., Ratcliffe P.J., Mole D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011; 117:e207–e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ge S.X., Jung D., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020; 36:2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou Z., Li M., Zhang L., Zhao H., Sahin O., Chen J., Zhao J.J., Songyang Z., Yu D. Oncogenic kinase–induced PKM2 tyrosine 105 phosphorylation converts nononcogenic PKM2 to a tumor promoter and induces cancer stem–like cells. Cancer Res. 2018; 78:2248–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured filroblasts. Nat. Protoc. 2007; 2:287–295. [DOI] [PubMed] [Google Scholar]
- 18. Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009; 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dombrauckas J.D., Santarsiero B.D., Mesecar A.D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005; 44:9417–9429. [DOI] [PubMed] [Google Scholar]
- 20. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baugh E.H., Lyskov S., Weitzner B.D., Gray J.J. Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS One. 2011; 6:e21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. Improving physical realism, stereochemistry and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009; 77:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adasme M.F., Linnemann K.L., Bolz S.N., Kaiser F., Salentin S., Haupt V.J., Schroeder M. PLIP 2021: expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021; 49:W530–W534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Debnath J., Muthuswamy S.K., Brugge J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003; 30:256–268. [DOI] [PubMed] [Google Scholar]
- 25. Vaapil M., Helczynska K., Villadsen R., Petersen O.W., Johansson E., Beckman S., Larsson C., Påhlman S., Jögi A. Hypoxic conditions induce a cancer-like phenotype in Human breast epithelial cells. PLoS One. 2012; 7:e46543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ducommun B., Lobjois V., Laurent J., Frongia C., Mondesert O., Cazales M. Abstract 4404: multicellular tumor spheroid models to evaluate drugs targeting cell cycle checkpoints in 3D. Cancer Res. 2013; 73:4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y.C., Chien M.H., Liu H.Y., Chang Y.C., Chen C.K., Lee W.J., Kuo T.C., Hsiao M., Hua K.T., Cheng T.Y. Nuclear translocation of PKM2/AMPK complex sustains cancer stem cell populations under glucose restriction stress. Cancer Lett. 2018; 421:28–40. [DOI] [PubMed] [Google Scholar]
- 28. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy. Nat. Rev. Cancer. 2016; 16:635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu L., Chen Y., Zhu Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget. 2017; 8:62793–62802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi L., Pan H., Liu Z., Xie J., Han W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017; 2:17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. La Belle Flynn A., Calhoun B.C., Sharma A., Chang J.C., Almasan A., Schiemann W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019; 10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H.M., Yang J.G., Liu Z.J., Wang W.M., Yu Z.L., Ren J.G., Chen G., Zhang W., Jia J. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017; 36:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peng F., Li Q., Sun J.Y., Luo Y., Chen M., Bao Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int. J. Oncol. 2018; 52:945–954. [DOI] [PubMed] [Google Scholar]
- 34. Obach M., Navarro-Sabaté À., Caro J., Kong X., Duran J., Gómez M., Perales J.C., Ventura F., Rosa J.L., Bartrons R. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J. Biol. Chem. 2004; 279:53562–53570. [DOI] [PubMed] [Google Scholar]
- 35. Dames S.A., Martinez-Yamout M., De Guzman R.N., Jane Dyson H., Wright P.E. Structural basis for hif-1α/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dengler V.L., Galbraith M.D., Espinosa J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014; 49:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee J.W., Ko J., Ju C., Eltzschig H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019; 51:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian H., McKnight S.L., Russell D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997; 11:72–82. [DOI] [PubMed] [Google Scholar]
- 39. Skuli N., Majmundar A.J., Krock B.L., Mesquita R.C., Mathew L.K., Quinn Z.L., Runge A., Liu L., Kim M.N., Liang J. et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Invest. 2012; 122:1427–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chaffer C.L., Marjanovic N.D., Lee T., Bell G., Kleer C.G., Reinhardt F., D’Alessio A.C., Young R.A., Weinberg R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013; 154:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Curry E., Zeller C., Masrour N., Patten D.K., Gallon J., Wilhelm-Benartzi C.S., Ghaem-Maghami S., Bowtell D.D., Brown R. Genes predisposed to DNA hypermethylation during acquired resistance to chemotherapy are identified in ovarian tumors by bivalent chromatin domains at initial diagnosis. Cancer Res. 2018; 78:1383–1391. [DOI] [PubMed] [Google Scholar]
- 42. Chen J., Xie J., Jiang Z., Wang B., Wang Y., Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011; 30:4297–4306. [DOI] [PubMed] [Google Scholar]
- 43. Yang W., Zheng Y., Xia Y., Ji H., Chen X., Guo F., Lyssiotis C.A., Aldape K., Cantley L.C., Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012; 14:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Greene A.W., Grenier K., Aguileta M.A., Muise S., Farazifard R., Haque M.E., McBride H.M., Park D.S., Fon E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012; 13:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469:221–226. [DOI] [PubMed] [Google Scholar]
- 46. Hayes C., Donohoe C.L., Davern M., Donlon N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021; 500:75–86. [DOI] [PubMed] [Google Scholar]
- 47. Yang Z., Yan C., Ma J., Peng P., Ren X., Cai S., Shen X., Wu Y., Zhang S., Wang X. et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat. Metab. 2023; 5:61–79. [DOI] [PubMed] [Google Scholar]
- 48. San-Millán I., Brooks G.A. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis. 2017; 38:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luengo A., Li Z., Gui D.Y., Sullivan L.B., Zagorulya M., Do B.T., Ferreira R., Naamati A., Ali A., Lewis C.A. et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell. 2021; 81:691–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021; 71:209–249. [DOI] [PubMed] [Google Scholar]
- 51. Pandkar M.R., Dhamdhere S.G., Shukla S. Oxygen gradient and tumor heterogeneity: the chronicle of a toxic relationship. Biochim. Biophys. Acta - Rev. Cancer. 2021; 1876:188553. [DOI] [PubMed] [Google Scholar]
- 52. Vaupel P., Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007; 26:225–239. [DOI] [PubMed] [Google Scholar]
- 53. Wilson W.R., Hay M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer. 2011; 11:393–410. [DOI] [PubMed] [Google Scholar]
- 54. Williams D., Fingleton B. Non-canonical roles for metabolic enzymes and intermediates in malignant progression and metastasis. Clin. Exp. Metastasis. 2019; 36:211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chandel N.S., Budinger G.R.S., Schumacker P.T. Molecular oxygen modulates cytochrome c oxidase function. J. Biol. Chem. 1996; 271:18672–18677. [DOI] [PubMed] [Google Scholar]
- 56. Rumsey W.L., Schlosser C., Nuutinen E.M., Robiolio M., Wilson D.F. Cellular energetics and the oxygen dependence of respiration in cardiac myocytes isolated from adult rat. J. Biol. Chem. 1990; 265:15392–15399. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data generated in this study is deposited under the GEO accession number: GSE190401. All the other data supporting the findings are included in the article and its supporting information.