

ABSTRACT
Antibiotic-resistant bacteria and antibiotic resistance gene (ARGs) loads dissipate through sewage treatment plants to receiving aquatic environments, but the mechanisms that mitigate the spread of these ARGs are not well understood due to the complexity of full-scale systems and the difficulty of source tracking in downstream environments. To overcome this problem, we targeted a controlled experimental system comprising a semicommercial membrane-aerated bioreactor (MABR), whose effluents fed a 4,500-L polypropylene basin that mimicked effluent stabilization reservoirs and receiving aquatic ecosystems. We analyzed a large set of physicochemical measurements, concomitant with the cultivation of total and cefotaxime-resistant Escherichia coli, microbial community analyses, and quantitative PCR (qPCR)/digital droplet PCR (ddPCR) quantification of selected ARGs and mobile genetic elements (MGEs). The MABR removed most of the sewage-derived organic carbon and nitrogen, and simultaneously, E. coli, ARG, and MGE levels dropped by approximately 1.5- and 1.0-log unit mL−1, respectively. Similar levels of E. coli, ARGs, and MGEs were removed in the reservoir, but interestingly, unlike in the MABR, the relative abundance (normalized to 16S rRNA gene-inferred total bacterial abundance) of these genes also decreased. Microbial community analyses revealed the substantial shifts in bacterial and eukaryotic community composition in the reservoir relative to the MABR. Collectively, our observations lead us to conclude that the removal of ARGs in the MABR is mainly a consequence of treatment-facilitated biomass removal, whereas in the stabilization reservoir, mitigation is linked to natural attenuation associated with ecosystem functioning, which includes abiotic parameters, and the development of native microbiomes that prevent the establishment of wastewater-derived bacteria and associated ARGs.
IMPORTANCE Wastewater treatment plants are sources of antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARGs), which can contaminate receiving aquatic environments and contribute to antibiotic resistance. We focused on a controlled experimental system comprising a semicommercial membrane-aerated bioreactor (MABR) that treated raw sewage, whose effluents fed a 4,500-L polypropylene basin that mimicked effluent stabilization reservoirs. We evaluated ARB and ARG dynamics across the raw-sewage–MABR–effluent trajectory, concomitant with evaluation of microbial community composition and physicochemical parameters, in an attempt to identify mechanisms associated with ARB and ARG dissipation. We found that removal of ARB and ARGs in the MABR was primarily associated with bacterial death or sludge removal, whereas in the reservoir it was attributed to the inability of ARBs and associated ARGs to colonize the reservoir due to a dynamic and persistent microbial community. The study demonstrates the importance of ecosystem functioning in removing microbial contaminants from wastewater.
KEYWORDS: wastewater treatment, ecological barriers, community shifts, antibiotic-resistant bacteria, antibiotic resistance genes, mobile genetic elements, microbiome, qPCR, ddPCR
INTRODUCTION
Raw sewage is a mirror of the gut microbiota of the served population (1) and is consequently a source of feces-derived antibiotic-resistant bacteria (ARB) and antibiotic resistance genes (ARGs), whose abundance and diversity vary as a function of geography and socioeconomic conditions (2, 3). Although ARGs are minor components of the sewage metagenome (0.03%), the sewage resistome constitutes a vast array of genetic determinants that confer resistance to the entire spectrum of antibiotic classes, including ARGs associated with emerging clinical threats (2, 4). Conventional wastewater treatment processes based on activated sludge generally remove 1 to 3 log units mL−1 (by volume) of total and antibiotic-resistant fecal bacteria (5) and associated ARGs (6). This removal can be augmented by disinfection (6) and membrane-based processes (7). Decentralized wastewater treatment modules are gaining popularity in developing regions that lack sewage infrastructure. These systems remove a large fraction of fecal pathogens but are generally less efficient than conventional wastewater treatment plants (WWTPs) (8). WWTP effluents frequently contain substantial ARB and ARG loads (9–14), and discharge of these determinants to receiving aquatic ecosystems or irrigated soils can result in their dissemination through the water cycle and/or the food chain, potentially expanding the global scope of antibiotic resistance (15–17).
Fecal bacterial indicators that are routinely targeted for water quality assessment do not provide insights regarding antibiotic resistance. This can be overcome by monitoring fecal bacterial indicators resistant to next-generation antibiotics concomitant with determining total counts (5), as proposed in a recent review (18). Source tracking of ARGs using quantitative PCR (qPCR)-based methods provide a rapid and accurate means of quantification that sheds light on antibiotic resistance levels in WWTPs and receiving environments, but there are close to 3,000 documented antibiotic resistance determinants (19), and currently there are no established ARG standards for assessing water quality. In an epidemiological context, ARGs are interesting to monitor only if they can be horizontally transferred to other bacteria and/or if they are associated with pathogens. With this in mind, Zhang et al. proposed ranking ARGs according to the associated level of risk for human health, where rank I refers to ARGs that are associated with mobile genetic elements (MGEs) and are present in ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) (20). The limitations of both cultivation-based and culture-independent molecular analyses underline the fact that combining the two is imperative for a holistic understanding of antibiotic resistance in WWTP effluents and downstream environments.
In warm, water-depleted countries (e.g., Israel) that reuse wastewater for irrigation, effluent storage (or stabilization) reservoirs with capacities reaching 5 million m3 enable modulation between relatively constant sewage production and generally irregular (seasonal) demand for irrigation water. These hypertrophic aquatic systems, which operate under non-steady-state conditions (21), have the capacity to improve effluent quality (i.e., reduce loads of fecal pathogen indicators and recalcitrant organic compounds), with sufficient retention times (22). Various studies have investigated the fate of WWTP effluent-derived ARB and ARGs in receiving aquatic ecosystems (14, 23–25). While certain studies indicate that ARGs can persist in receiving water and sediment (23), others suggest that they are either diluted or actively removed (14). These studies highlight the fact that mitigation of sewage-derived ARB and ARGs in these receiving aquatic ecosystems is associated with ecological interactions (24), but the scope and nature of these interactions are still not well understood due to environmental complexity and difficulty of source tracking (25).
The goal of this study was to identify factors that facilitate the removal of antibiotic resistance in WWTPs and effluent-receiving storage reservoirs. We targeted a pilot system containing a semicommercial decentralized membrane bioreactor (MABR) for secondary treatment of municipal sewage, which fed a 4,500-L pilot reservoir (Fig. 1). We specifically evaluated the temporal dynamics (over a 6-month period) of ARB and ARGs across the raw-sewage–MABR–reservoir trajectory and the impact of residence time (5 to 21 days) on reservoir performance. ARB and ARG levels were evaluated as a function of selected physicochemical parameters and microbial (bacterial and eukaryotic) community structure, in order to pinpoint factors potentially responsible for their removal. The controlled nature of the system and the coupling of isolation, culture-independent ARG quantification, and analysis of microbial community composition underlined mechanisms potentially responsible for mitigating antibiotic resistance in WWTPs and receiving reservoirs.
FIG 1.

Overview of the experimental beta-site. (A) Schematic diagram of the raw-sewage–MABR–reservoir continuum at the beta site. Sampling points include SWG (raw sewage), MABR, RT (reservoir top; faucet situated at the top 10 cm of the reservoir), and RB (reservoir bottom; faucet situated at the bottom 10 cm of the reservoir). (B) Profile (left) and aerial photos of the MABR (middle) and reservoir (right) at the beta site.
RESULTS
Physicochemical fluctuations along the sewage-MABR-stabilization reservoir trajectory.
We evaluated physicochemical parameters along the sewage-MABR-stabilization reservoir trajectory between July and December 2020, at 17 time points. Water temperatures ranged from 35 to 17°C. In the November and December profiles, reservoir temperatures were approximately 5°C lower than that of the raw sewage (Fig. 2A), indicating that they are more strongly impacted by ambient temperatures. The pH (Fig. 2B) and dissolved oxygen levels (Fig. 2C) were relatively stable in raw sewage and the MABR (except for 22 July and 8 September 2020, when oxygen in the MABR was low due to a system malfunction) but varied more in the stabilization reservoir, seemingly due to photosynthetic activity. Turbidity (see Fig. S2A in the supplemental material) was almost completely alleviated in the MABR (with the exception of 1 and 8 September 2020, when malfunctions occurred), correlating with a significant reduction in total organic carbon (Fig. 2D). Likewise, over 80% of total nitrogen was removed in the MABR (Fig. 2E), corresponding to the removal of most of the ammonia (Fig. 2F). Mass balance of all the analyzed species indicated that most of the carbon and nitrogen in the system was either gasified (to CO2, N2, or N2O) or removed as settled sludge biomass, considering the fact that nitrate N (Fig. 2H) and nitrite N (Fig. 2G) concentrations in the MABR effluent were 1 to 2 orders of magnitude lower than the influent ammonia N concentration. Phosphate levels (Fig. S2B) significantly dropped between raw sewage and MABR, suggesting accumulation of polyphosphate in sludge biomass, and sulfate (Fig. S2C) increased between raw sewage and MABR, implying oxidation of reduced sulfur compounds such as H2S.
FIG 2.

Physicochemical analyses in the raw sewage, MABR, and reservoir. (A) Temperature; (B) pH; (C) dissolved oxygen; (D) total organic carbon: (E) total nitrogen; (F) ammonia; (G) nitrate; (H) nitrite. Swg, raw sewage; Res, reservoir.
Fecal coliform dynamics along the sewage-MABR-stabilization reservoir trajectory.
We evaluated the abundance of total and cefotaxime-resistant Escherichia coli (Fig. 3A) and fecal coliforms (Fig. S3) in the targeted compartments. On average, the abundance of these fecal bacterial indicators decreased by approximately 1.5 log units mL−1 in both the MABR and stabilization reservoir, although fluctuations in removal capacity in both modules were observed at different time points (Fig. S4). However, normalizing to the 16S rRNA gene levels measured by qPCR analyses (Fig. 3B) revealed that the abundance of E. coli relative to the total bacterial community decreased more in the stabilization reservoir than in the MABR. E. coli values measured in the raw sewage and MABR were similar to levels of the E. coli marker gene uidA (see below), supporting the culture-based analyses. In contrast, in the reservoir uidA levels were slightly higher than cultivated-E. coli levels, suggesting the presence of free DNA or nonviable bacteria.
FIG 3.

Absolute (A) and relative (B) abundances of total and cefotaxime-resistant E. coli and of 12 ARG/MGE markers in the sewage, MABR, and reservoir. Each data point represents aggregated data collected from six different sampling times and four biological replicates. One-way ANOVA followed by pairwise two sample t test highlights significant differences. Box colors represent MGE (blue), ARG (green), and bacterial and CrAssphage (orange) indicators. Superscripts above each graph in panel A indicate culture (c)-, qPCR (q)-, and ddPCR (d)-based analyses. The boxes indicate the range between the first and third quartile. The top and bottom whiskers of the boxes represent maximum and the minimum values, respectively. The median line divides the box into interquartile ranges, and the cross represents the mean. Each box represents the spread of time point averages (four biological replicates per time point). Swg, raw sewage; Res, reservoir.
ARG and MGE dynamics along the sewage-MABR-stabilization reservoir trajectory.
The abundance of the nine targeted ARG and MGE markers was monitored by qPCR/digital droplet PCR (ddPCR) in the raw sewage, MABR, and stabilization reservoir on six sampling dates (19 and 25 August, 15 September, 24 November, and 1 and 5 December 2020) (Fig. 3A). In parallel, 16S rRNA, uidA and CrAssphage genes were monitored to estimate total bacteria, E. coli and Bacteroides phages, respectively. The low standard deviations observed for most of the targeted genes indicate that the abundance of these markers is relatively steady over time in each module. On average, bacterial loads (estimated by 16S rRNA gene abundance) dropped from 8.2 log units mL−1 in sewage to 7.1 log units mL−1 after MABR treatment and to 6.7 log units mL−1 in the stabilization reservoir. Apart from blaVIM-2, all the gene markers followed the same trend with an approximate 2-order-of-magnitude reduction across the trajectory (Fig. 3A and Fig. S5). However, excluding blaVIM-2, the reduction in normalized ARG and MGE abundance (Fig. 3B) was higher in the stabilization reservoir than in the MABR, similar to the trend observed for E. coli described above. Most notable were SXT/R391, class 1 integrons, incP plasmids, sul-1, ermF, and blaCTX-M-1, whose reduction in relative abundance was significant (P < 0.05) only in the stabilization reservoir.
Microbial diversity along the sewage-WWTP-stabilization reservoir trajectory.
Rarefaction curves (Fig. S6) and diversity indexes of bacterial communities varied between samples (Table S4) and sampling times. Average Shannon and phylogenetic diversity indices were higher in the raw sewage and MABR than in the reservoir, but the variance in the diversity indices was substantially higher in the stabilization reservoir, highlighting the dynamic nature of this ecosystem.
Principal-component analysis (PCA) revealed a strong distinction between raw sewage, MABR, and reservoir bacterial communities (Fig. 4A). Raw sewage bacterial communities appeared to be highly stable for the duration (August to December 2020) of the analysis. In contrast, the composition of MABR bacterial communities fluctuated more, but these shifts were not completely season dependent. The bacterial communities in the stabilization reservoir displayed the strongest fluctuations, principally dictated by seasonality. In contrast, the eukaryotic community dynamics (Fig. 4B) were substantially different from those of the bacteria and clustered into three primary clades. The first clade encompassed all of the samples from all three modules of the August and September profiles. The second clade contained raw sewage and MABR samples from the November and December samples, and the third clade contained all of the reservoir samples from November and December.
FIG 4.
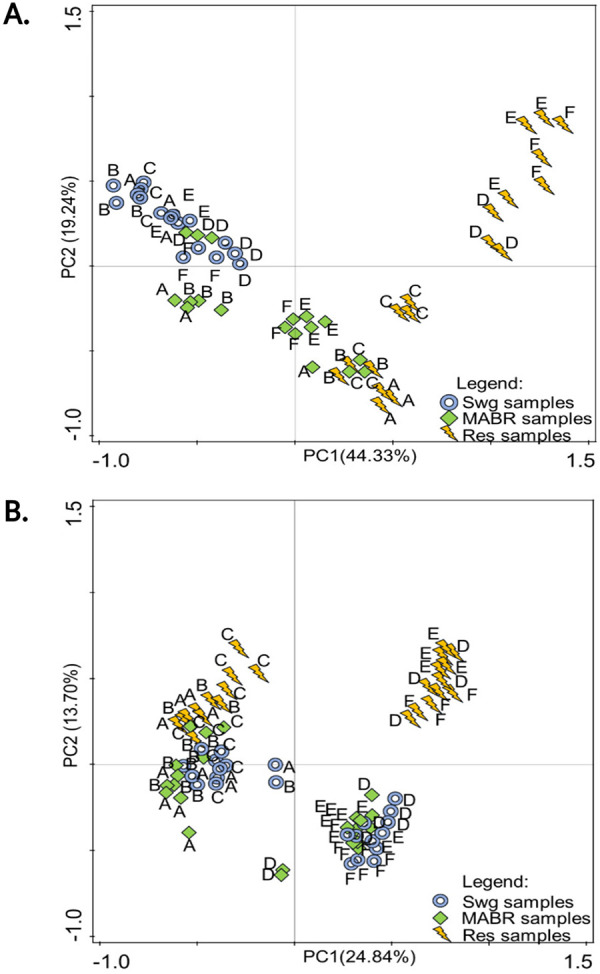
PCA showing temporal dynamics in bacterial (A) and eukaryal (B) community composition in sewage, MABR, and reservoir. Sampling dates: A, 19 August; B, 25 August; C, 15 September; D, 24 November; E, 1 December; F, 15 December. Swg, raw sewage; Res, reservoir.
Microbial community composition along the sewage-WWTP-stabilization reservoir trajectory.
The phylum-level evaluation of raw sewage, MABR, and reservoir samples revealed distinct bacterial community profiles (Fig. S7A). The reservoir samples collected in November and December were richer in Cyanobacteria than in the previous sampling times. Family-level analysis suggested that Cyanobacteria were predominantly chloroplasts (Fig. 5A), corresponding to eukaryotic algae also predominant in these microbial community profiles. Members of the phyla Pseudomonadota (31.9% in raw sewage [SWG], 37.7% in the MABR, and 34.1% in the reservoir [RES]), Bacteroidota (11.0% in SWG, 14.0% in the MABR, and 15.4% in the RES), and Actinobacteriota (6.7% in SWG, 10.9% in the MABR, and 11.9% in the RES) were among the most represented in all samples, with the relative abundances of Bacillota and Campylobacterota sharply decreasing from raw sewage to the reservoir (13.1% to 4.7% and 26.9% to 3.6%, respectively). In contrast, the relative abundance of other groups increased in the reservoir along the different sampling times, most notably, Cyanobacteria (ranging from 2.6% to 43.7% in December), Pseudomonadota (ranging from 27.6% to 41.3 in August), and Verrucomicrobiota (ranging from 0.4% to 11.9 in September), in a pattern that was sampling date dependent. Analysis of the eukaryotic community (Fig. 5B; Fig. S7B) revealed that for the duration of the analysis, the MABR was dominated by the bacterivorous protists Ciliophora (13.4 to 25.8% relative abundance), with the exception of a brief period when Euglenazoa (up to 32.9% ± 9.1% on 25 August 2020) and Ochrophyta (up to 62.8% ± 9.3% on 15 September 2020) became dominant, corresponding to the above-described MABR malfunction. Initially, the reservoir was dominated by Ciliophora (38.8% ± 2.0%), the dominant eukaryote in the MABR, and by Proteoalveolata (22.5% ± 2.3%), which was less abundant in the MABR. However, the relative abundance of both groups significantly decreased with time (P = 0.02; n = 24; nonparametric test for association based on Spearman's rho), with Proteoalveolata dropping below detection levels and the relative abundance of Ciliophora dropping to 5.2% ± 0.6%. Conversely, the relative abundance of Chlorophyta (green algae) significantly increased over time (P < 0.001; n = 24; analysis of variance [ANOVA]), and it became the dominant phylum from November onward, accounting for more than 75% of the relative abundance of eukaryotes in the reservoir. This taxon strongly correlated with the increase in the relative abundance of chloroplasts observed in the bacterial community analysis. This increase in Chlorophyta abundance was identified as the primary driver of the observed eukaryotic community composition shift (P < 0.01; n = 24; analysis of molecular variance [AMOVA]) relative to the three early profiles, in which blooms of this group of green algae were not observed in the reservoir.
FIG 5.

Dominant bacterial (A) and eukaryotic (B) families in sewage, MABR, and reservoir. Bacterial and eukaryotic analyses are based on 16S rRNA (V3-V4) and 18S rRNA (V9) gene amplicon sequencing, respectively. Additional information and statistical analyses are provided in Table S9.
We used STAMP software (26) to identify bacterial and eukaryote classes and families that are differentially abundant in the MABR versus the stabilization reservoir (Fig. 6). Arcobacteraceae, Bacteroidaceae, Aeromonadaceae, “Candidatus Competibacteraceae,” Rhodocyclaceae, and Leptotrichiaceae were the primary MABR-enriched bacterial families, and the ciliate Oligohymenophorea was the primary eukaryotic family. In contrast, Chitinophagaceae, Burkholderiaceae, unclassified PeM15 group (Actinobacteria) and the cyanobacteria Microcystaceae were the dominant bacteria and the alga family Chlorophyceae was the dominant eukaryote in the stabilization reservoir. Collectively, all of the families that were differentially more abundant in the MABR are well established in wastewater treatment systems and specifically in activated sludge, whereas those that were more abundant in the stabilization reservoir are more characteristic of freshwater ecosystems.
FIG 6.

Taxa showing significant variations in relative abundances between MABR and reservoir samples. Graphs show prokaryotic (A and B) and eukaryotic communities (C and D), at the class (A and C) and family (B and D) taxonomic levels. The comparisons of the MABR and RES samples at the class and family levels were performed using the two-sided Welch’s t test. Only the 10 most significant variances among samples are represented here.
Assessing correlations between microbial, physicochemical, and environmental parameters.
Redundancy analysis (RDA) was conducted to investigate possible statistically significant correlations (P < 0.01) between physicochemical parameters, ARGs, and MGEs and the microbial community composition in the raw sewage, MABR, and reservoir (Fig. 7; Fig. S8 and S9). Since bacterial, ARG, and MGE marker removal in the MABR was at least partially associated with biomass removal, we focused on trends that occurred in the MABR and the reservoir. While correlation does not necessarily indicate causation, RDA can potentially identify biotic (i.e., microbial populations) or abiotic factors linked to fluctuations in bacterial, ARG, or MGE abundance. The reduction of total bacteria observed in the reservoir (as measured by 16S rRNA gene abundance), total coliforms, total E. coli (including E. coli carrying uidA), and all the measured ARGs (except blaVIM-2) and MGEs were negatively correlated (P < 0.01) with pH and positively correlated with electric conductivity, total nitrogen, and dissolved oxygen. The photosynthetic eukaryotic taxa Dinoflagellata and Chlorophyta were strongly correlated (P < 0.01) with the reservoir, as were the bacterial phyla Pseudomonadota and Verrucomicrobiota. In contrast, the phyla Campylobacterota, Bacillota, and Desulfobacterota were significantly associated (P < 0.01) with the MABR.
FIG 7.

RDA showing correlations between ARGs, MGEs, and prokaryotic and eukaryotic populations, physicochemical conditions, and system compartments. (A) Correlation between ARG, MGE, and uidA abundance and selected physicochemical parameters in the MABR and reservoir (Res). (B) Correlation between ARG, MGE, and uidA abundance and specific prokaryotic and eukaryotic phyla. (C) Correlation between selected taxa and system compartments (MABR and reservoir). (D) Correlation between selected taxa and ARG, MGE, and uidA abundance. The test variables (ARGs, MGEs, and uidA) are represented in black, prokaryotes are in blue, and eukaryotes are in green. The test variables are portrayed in black and the explanatory variables in blue. The explanatory variables were associated with 74.4% of the observed variation among the test variables. Additional information and statistical analyses are provided in Table S9.
DISCUSSION
The recent proposal for a directive of the European Parliament and of the council concerning urban wastewater treatment (https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52022PC0541), which dictates that member states regularly monitor antimicrobial resistance (AMR) determinants in the outlets of urban wastewater treatment plants, underlines the potential contribution of WWTPs to AMR in downstream environments. However, understanding mechanisms associated with AMR dissemination and mitigation in WWTPs and receiving environments require comprehensive analysis and cannot be addressed by periodic effluent monitoring alone.
Our results demonstrated that stabilization reservoirs used for effluent storage prior to irrigation remove both antibiotic-resistant coliforms and ARGs, corroborating previous results (14). Maximal ARB and ARG removal capacity approached levels comparable to those of more sophisticated disinfection processes, such as chlorination (27) and UV irradiation (28), suggesting that reservoirs can potentially provide a simple and environmentally sustainable means of alleviating AMR determinants in wastewater effluents if properly managed. The comprehensive evaluation of large-scale stabilization reservoirs has indicated that long retention times (exceeding 3 months) facilitate optimal fecal pathogen indicator removal (21), and we foresee that such retention times can also reduce AMR indicators in reservoirs. Optimally, disinfection processes such as chlorination or UV irradiation should be applied to reservoir effluents to ensure water quality; however, properly managed reservoirs can potentially provide a stand-alone solution in certain decentralized wastewater treatment configurations, or in developing regions where sophisticated disinfection platforms are not available.
The decrease in relative abundance of E. coli, ARG, and MGE markers (normalized to 16S rRNA genes), coupled with the strong shift in the microbial community composition observed in the reservoir, underlines the fact that fecal and AMR marker decay in the reservoir is at least partially attributed to its autochthonous microbiota (29, 30). Compared to the MABR, the reservoir microbiome was enriched in bacterial and eukaryotic families (i.e., unclassified PeM15 group, Microcystaceae, and Chlorophyceae) characteristic of aquatic ecosystems (31–33). In contrast, the bacterial community composition of the MABR effluent was closer to that of the raw sewage than to that of the reservoir, with enrichment of bacterial families (i.e., Arcobacteraceae, Bacteroidaceae, Aeromonadaceae, “Candidatus Competibacteraceae,” Rhodocyclaceae, and Leptotrichiaceae) known to be profuse in raw sewage and wastewater treatment plants (34–39). The green algal family Chlorophyceae was ubiquitous in the reservoir microbiome but became dominant in the last three profiles (November and December 2020) analyzed. Similar algal blooms were also observed in a previous study that evaluated plankton community changes in a freshwater reservoir amended with treated wastewater effluents (40). It is unclear whether these green blooms were stimulated by temperature or other physicochemical parameters in the reservoir or whether it simply took time for these algae to mature in the reservoir. Algal and cyanobacterial blooms can significantly affect water quality and operation of effluent stabilization reservoirs (21, 41, 42), and thus, future studies need to identify specific parameters that induce their proliferation.
Biotic interactions between influent-derived fecal indicators (i.e., ARB and ARGs) and the indigenous microbiota (including predation and/or competition) are undoubtedly a significant factor in mitigating ARB and ARG markers in reservoirs and other receiving aquatic ecosystems (43). Nonetheless, abiotic factors, such as UV light exposure, temperature fluctuations, salinity, and nutrient availability, undoubtedly play a major role as well (43). Determining the relative contribution of specific biotic and abiotic factors to pathogen/ARG indicator decay rates is extremely complex, and small-scale models do not always properly predict large-scale scenarios (43). This is underlined by the fact that reducing retention time in the reservoir from 21 to 5 days in this study did not significantly impede its capacity to remove fecal bacteria and ARGs, contrary to previous reports that evaluated longer retention times and showed strong correlations between retention time and fecal pathogen indicator removal in large-scale stabilization reservoirs (21). The negligible effect of residence time on AMR marker mitigation observed in the pilot reservoir is surprising, but it may be explained by the fact that ARB and associated resistance genes may decline rapidly upon entering the reservoir but later acclimate or enter a more resilient state, thereby reducing the impact of retention time (44). Alternatively, the longer retention times in the pilot reservoir may not have been maintained long enough to reach steady-state conditions and therefore did not show enhanced performance compared to the shorter times.
Further understanding of processes responsible for mitigating fecal pathogens and AMR in reservoirs requires long-term temporal and spatial monitoring of specific ARB and ARG markers in commercial-scale stabilization reservoir influents and effluents, using standardized protocols as recently suggested (45). To determine the relative contribution of specific biotic and abiotic factors that most strongly facilitate marker decline, generated data should be comprehensively evaluated concomitant with the evaluation of bacterial and eukaryotic community composition and selected physicochemical parameters (i.e., temperature, light intensity, pH, organic matter, and nutrients) using multivariate statistics and correlation analyses (46). Quantitative (cultivation or qPCR-based) evaluation of predicted competitors and predators (selected based on microbial community analyses) can be added to this evaluation to provide even higher resolution.
Conclusions.
Stabilization reservoirs provide a simple means of removing ARB and ARGs from secondary effluents. Based on our results, we stipulate that removal of these constituents in the reservoir is facilitated by interactions with a robust, albeit dynamic, microbial community, coupled with abiotic stress. Further understanding the mechanism in which reservoirs mitigate fecal pathogens and ARGs necessitates research that implements harmonized protocols to elucidate the interplay between ARB and ARG markers, reservoir microbial communities, and physicochemical conditions.
MATERIALS AND METHODS
Description of the experimental system.
An experimental wastewater treatment beta site (Fig. 1), situated within the Maayn Zvi municipal wastewater treatment plant (32.59684, 34.92975) in Israel, was operated between July and December 2020. The system consisted of an Aspiral L3 (www.fluencecorp.com/wp-content/uploads/2018/05/Aspiral-Product-Brochure.pdf) membrane-aerated biofilm reactor (MABR) connected to a cylindrical polypropylene reservoir (4,500-L working volume), designed to mimic operational reservoirs commonly used for effluent storage prior to irrigation. The passive aeration by diffusion of oxygen through MABR membranes supports an aerobic nitrifying biofilm that develops on their surface, while suspended solids are held in the mixed liquor, enabling simultaneous nitrification and denitrification. To date, approximately 300 commercial decentralized MABR units have been installed at various sites in Asia, Africa, and North America. The feed flow rate of primary effluent to the MABR was approximately 5 m3/h, with slight variations due to occasional equipment failures (i.e., clogged feed pump or ruptured diffuser), power failures (4 overall), and excess sludge removal, which reduced the desired effluent quality. Mixing frequency and duration, return activated sludge (RAS), sludge wasting (WAS), and aeration regimens were modulated to maintain bioreactor performance. The reservoir retention time was initially 21 days (from system initiation to 14 October 2020), after which it was reduced to 10 days (14 October 2020 to 4 November 2020) and later to 5 days (4 to 25 November 2020) in order to evaluate the potential impact on the capacity of the reservoir to remove fecal bacteria and ARGs.
Samples for physicochemical, bacterial, and molecular analyses (Table S1) were taken from faucets situated at different points along the sewage-MABR-reservoir trajectory (Fig. 1). Faucets were opened for 30 s, and sampling vessels were washed 3 times before sampling to remove residual water in the pipes. Water samples for bacterial enumeration and community DNA extraction were either immediately filtered on site or transferred on ice to the lab at the Volcani Institute and filtered within 3 h. Samples for physicochemical analyses and bacterial quantification were taken on 22 July 2020, whereas samples for microbiome and quantitative PCR analyses were taken on 19 August 2020.
Physical and chemical analyses.
Temperature, oxygen, pH, and conductivity were measured using a model HQ40D digital two-channel multimeter (Hach, Colorado, USA) using specific electrodes for each parameter, and turbidity was measured with a 2100Q portable turbidimeter (Hach, Colorado, USA). Total organic carbon (TOC) was determined by dry combustion with a Flashea 1112 NC elemental analyzer (Thermo Fisher Scientific, Hanau, Germany). Ammonia, nitrite, nitrate, phosphorus, and sulfate were measured colorimetrically with a Quickchem 8000 Autoanalyzer (Lachat Instruments, Milwaukee, WI) using standard protocols provided by the manufacturer.
Microbial quantification, isolation, and characterization.
Cultivation-based analysis was applied to enumerate total and cefotaxime-resistant coliform and Escherichia coli in the raw sewage, MABR effluent, and reservoir, using a modified version of the standard membrane filtration method (ISO 9308-1). Briefly, 10-fold serial dilutions (10−2 to 10−5 and 10° to 10−4 for total and cefotaxime resistant coliforms, respectively) of the collected raw sewage and effluent samples were prepared in sterile saline solution (0.85% [wt/vol] NaCl), and 1 mL of diluted sample was filtered in triplicate through a 0.45-μm nitrocellulose grid membrane filter using a vacuum filtration system. Subsequently, filters were placed (grid facing upward) on Chromocult coliform agar (CCA; per ISO 9308-1 [47]) culture medium plates with or without cefotaxime (4 mg/L), which is above CLSI and EUCAST clinical breakpoints for E. coli (48), and plates were incubated at 37°C overnight. Coliform and E. coli colonies on the CCA medium were enumerated based on the color classification defined by the supplier.
To validate presumptive CCA colorimetric identifications, 108 randomly selected colonies from raw sewage, the MABR, and the reservoir were classified on a Microflex LT matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI-TOF MS) system (Bruker Daltonics GmbH, Bremen, Germany) using the Flex Control v3.4 Biotyper automation software as previously described (49). Results supported the manufacturer’s colorimetric taxonomic characterizations, as 55 blue colonies were identified as E. coli or Shigella, and 53 red colonies were identified as Klebsiella spp. or Enterobacter spp.
DNA extraction, storage, and shipment.
For DNA extraction, 10 to 100 mL of the freshly collected samples was filtered through 0.22-μm polycarbonate membranes using a filtration unit and then stored at −80°C prior to extraction. Sampling dates and specific volumes filtered for each sample are shown in Table S2. DNA was extracted from these membranes using the DNeasy PowerWater kit (catalog no. 14900-100-NF; Qiagen, USA), using the protocols provided by the manufacturer. Purified DNA was divided into four aliquots and stored at −80°C until shipping. Composite (top and bottom) reservoir samples were prepared by mixing equal volumes of DNA, after top and bottom physicochemical parameters were found to be very similar to each other. Samples for bacterial community analysis and qPCR quantification of ARGs were shipped to Universidade Católica Portuguesa (UCP) in Porto, Portugal; samples for digital droplet PCR (ddPCR) quantification of ARGs and MGEs were shipped to Université de Lorraine, LCPME, Nancy, France; and samples for eukaryotic community analysis were shipped to Technische Universität Dresden, Germany. All samples were shipped on dry ice by 2-day express delivery and stored at −80°C upon arrival.
Quantification of the 16S rRNA gene, MGEs, and ARGs.
ddPCR and qPCR were used to quantify the bacterial 16S rRNA gene, the E. coli indicator gene uidA, and the CrAssphage fecal contamination indicator, as well as five ARGs (sul1, ermF, blaVIM-2, blaKPC, and blaCTX-M-1) and four MGE families (class 1 integrons, Tn916/Tn1545 ICE family, SXT ICE family, and IncP plasmid family). The above-mentioned genes were all analyzed in the samples from 19 and 25 August, 15 September, 24 November, and 1 and 15 December 2020. The ddPCR was conducted on a one-step QX200 system (Bio-Rad Hercules, CA, USA) using Evagreen or TaqMan technology. The qPCR was performed on a StepOnePlus machine (Applied Biosystems, Life Technologies, Carlsbad, CA, USA) using Sybr technology (Table S3). Sample dilutions were adjusted to avoid saturation with 100% positive droplets (for 16S rRNA genes, for instance), and the absolute number of target copies was calculated from the proportion of positive partitions and statistically corrected with a Poisson distribution. Primers, mix reactions, and amplification conditions for ddPCR and qPCR are described in Table S3. For ddPCR, the QX Manager software (version 1.7.4, Bio-Rad) was used to assign positive/negative droplets, after manual adjustment of the thresholds, and to convert counts into copies per microliter. Negative controls (DNA- and RNA-free water) and a positive control (artificial target DNA) were used in the first ddPCR assay to confirm the proper position of the positive/negative droplets. Negative controls and a calibration curve were run with each qPCR determination. We initially compared qPCR and ddPCR quantifications of 16S rRNA and ermF genes using identical sets of DNA to determine the relationship between the two approaches (Fig. S1).
The rationale for choosing the targeted genes was as follows. The beta-glucuronidase-encoding gene uidA was chosen because it is frequently used to source track E. coli (the most common fecal bacterial indicator) in aquatic ecosystems, and the CrAssphage bacteriophage was selected because it is highly abundant in the human gut. The ARGs were targeted based on expected abundance and ubiquity and different risk levels, as described by Zhang et al. (20), included the widespread sul1 gene (rank IV), the human-enriched ermF gene (rank III/IV), and three ARGs of concern with regard to public health, blaCTX-M-1 (rank III), blaVIM-2 (rank I/III), and blaKPC-2 (rank I). Regarding the rationale for MGE selection, the Tn916/Tn1545 ICE family is abundant in Bacillota, the SXT/R391 ICE family is predominant in Gammaproteobacteria, IncP-1 conjugative plasmids are profuse in Pseudomonadota, and class 1 integrons are broadly found in Gram-negative bacteria.
Microbial community analyses.
Prokaryotic communities were analyzed by targeting the V3-V4 region of the 16S rRNA gene, using the primers 341F and 806R (Table S3). Amplicons were sequenced using an Illumina paired-end platform to generate 250-bp paired-end raw reads that were merged with FLASH (V1.2.7) and quality filtered using QIIME software (V1.7.0). The chimeric sequences were removed and the reads with good quality were assigned to operational taxonomic units (OTUs; ≥97% sequence identity). OTU annotation was performed against the small-subunit (SSU)-rRNA SILVA v.138 database (http://www.arb-silva.de/) (50).
Eukaryotic communities were analyzed by targeting the 18S rRNA gene using the universal primers 1391f and EukBr (51) (Table S3), which target the V9 variable region. PCR products of the proper size were selected following validation by 2% agarose gel electrophoresis. Equimolar amounts of PCR products from each sample were pooled, end repaired, A tailed, and further ligated with Illumina adapters. Libraries were sequenced at Novogene Co. (Cambridge, UK) using the Illumina MiSeq platform generating 250-bp paired-end raw reads, which were assigned to samples based on their unique barcodes and truncated by cutting off the barcode and primer sequences. Subsequently, FLASH (52) (v1.2.11; http://ccb.jhu.edu/software/FLASH/) was applied to merge reads, and fastp (53) was sued for quality control of raw tags, to obtain high-quality clean tags. Vsearch software (54) was used to conduct a BLAST analysis of clean tags against the database, to detect and remove chimeric sequences. The deblur module in QIIME2 (55) was used to denoise, and sequences with fewer than 5 reads were filtered out to obtain the final amplicon sequence variants (ASVs) and feature table. Finally, the Classify-sklearn module in QIIME2 software was used to compare ASVs with the SILVA database (http://www.arb-silva.de/) (50) and to obtain the species annotation of each ASV.
Statistical analyses.
One-way ANOVA followed by a least-significant-difference (LSD) post hoc test was used to evaluate statistical significance between the raw sewage, MABR, and reservoir for each of the measured parameters, and one-way ANOVA followed by a Tukey-Kramer post hoc test was used to evaluate temporal variance. For all analyses, differences were considered significant when P values were below 0.05.
Potential relationships between species-level microbial community composition and structure, ARG and MGE markers, and environmental and physicochemical variables were assessed using redundancy analysis with Canoco 5.01 software (56). The relationship between species and environmental variables was assessed based on 1,000 Monte Carlo permutations, followed by forward selection with the significance criterion of a P value of <0.01.
IBM SPSS Statistics version 28 was used to statistically analyze the alpha diversity of the most abundant prokaryotic (>5%) and eukaryotic (>1%) microbial communities and to compare the qPCR and ddPCR analyses (UCP and LCPME labs, respectively) of 16S rRNA and ermF genes. For alpha diversity, ANOVA was applied with Tukey’s post hoc test, with a significance level of a P value of <0.01. For the comparison between laboratory results, a Friedman test was performed (with a significance level of a P value of <0.01), because the data did not follow a normal distribution. Statistical calculations for gene abundance and microbial community analyses are shown in Tables S5 to S8.
Data availability.
The 16S and 18S rRNA sequences were uploaded to the NCBI SRA archive under BioProject number PRJNA805207.
ACKNOWLEDGMENTS
This study was supported by the European Union's Horizon 2020 research and innovation program project DSWAP under the PRIMA program under grant agreement 1822. C.M., U.K., and T.U.B. were also supported through the ANTIVERSA project funded by the Agence Nationale de la Recherche (France) and the Bundesministerium für Bildung, und Forschung (Germany), respectively, under grant number 01LC1904A. C.M. received additional support from the LTSER-France and the Lorraine Region through the research network of Zone Atelier Moselle (ZAM). Responsibility for the information and views expressed lies entirely with the authors.
Footnotes
Supplemental material is available online only.
Contributor Information
Eddie Cytryn, Email: eddie@volcani.agri.gov.il.
Jeremy D. Semrau, University of Michigan-Ann Arbor
REFERENCES
- 1.Aarestrup FM, Woolhouse MEJ. 2020. Using sewage for surveillance of antimicrobial resistance. Science 367:630–632. doi: 10.1126/science.aba3432. [DOI] [PubMed] [Google Scholar]
- 2.Hendriksen RS, Munk P, Njage P, van Bunnik B, McNally L, Lukjancenko O, Röder T, Nieuwenhuijse D, Pedersen SK, Kjeldgaard J, Kaas RS, Clausen PTLC, Vogt JK, Leekitcharoenphon P, van de Schans MGM, Zuidema T, de Roda Husman AM, Rasmussen S, Petersen B, Bego A, Rees C, Cassar S, Coventry K, Collignon P, Allerberger F, Rahube TO, Oliveira G, Ivanov I, Vuthy Y, Sopheak T, Yost CK, Ke C, Zheng H, Baisheng L, Jiao X, Donado-Godoy P, Coulibaly KJ, Jergović M, Hrenovic J, Karpíšková R, Villacis JE, Legesse M, Eguale T, Heikinheimo A, Malania L, Nitsche A, Brinkmann A, Saba CKS, Kocsis B, Solymosi N. Global Sewage Surveillance project consortium, et al. 2019. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat Commun 10:1124. doi: 10.1038/s41467-019-08853-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pärnänen KMM, Narciso-da-Rocha C, Kneis D, Berendonk TU, Cacace D, Do TT, Elpers C, Fatta-Kassinos D, Henriques I, Jaeger T, Karkman A, Martinez JL, Michael SG, Michael-Kordatou I, O'Sullivan K, Rodriguez-Mozaz S, Schwartz T, Sheng H, Sørum H, Stedtfeld RD, Tiedje JM, Giustina SVD, Walsh F, Vaz-Moreira I, Virta M, Manaia CM. 2019. Antibiotic resistance in European wastewater treatment plants mirrors the pattern of clinical antibiotic resistance prevalence. Sci Adv 5:eaau9124. doi: 10.1126/sciadv.aau9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bürgmann H, Frigon D, Hg W, Mm C, Pruden A, Singer AC, Fs B, Zhang T. 2018. Water and sanitation: an essential battlefront in the war on antimicrobial resistance. FEMS Microbiol Ecol 94:fiy101. doi: 10.1093/femsec/fiy101. [DOI] [PubMed] [Google Scholar]
- 5.Marano RBM, Fernandes T, Manaia CM, Nunes O, Morrison D, Berendonk TU, Kreuzinger N, Tenson T, Corno G, Fatta-Kassinos D, Merlin C, Topp E, Jurkevitch E, Henn L, Scott A, Heß S, Slipko K, Laht M, Kisand V, Di Cesare A, Karaolia P, Michael SG, Petre AL, Rosal R, Pruden A, Riquelme V, Agüera A, Esteban B, Luczkiewicz A, Kalinowska A, Leonard A, Gaze WH, Adegoke AA, Stenstrom TA, Pollice A, Salerno C, Schwermer CU, Krzeminski P, Guilloteau H, Donner E, Drigo B, Libralato G, Guida M, Bürgmann H, Beck K, Garelick H, Tacão M, Henriques I, Martínez-Alcalá I, Guillén-Navarro JM, et al. 2020. A global multinational survey of cefotaxime-resistant coliforms in urban wastewater treatment plants. Environ Int 144:106035. doi: 10.1016/j.envint.2020.106035. [DOI] [PubMed] [Google Scholar]
- 6.Manaia CM, Rocha J, Scaccia N, Marano R, Radu E, Biancullo F, Cerqueira F, Fortunato G, Iakovides IC, Zammit I, Kampouris I, Vaz-Moreira I, Nunes OC. 2018. Antibiotic resistance in wastewater treatment plants: tackling the black box. Environ Int 115:312–324. doi: 10.1016/j.envint.2018.03.044. [DOI] [PubMed] [Google Scholar]
- 7.Gurung K, Ncibi MC, Fontmorin J-M, Särkkä H, Sillanpää M. 2016. Incorporating submerged MBR in conventional activated sludge process for municipal wastewater treatment: a feasibility and performance assessment. J Membra Sci Technol 6. doi: 10.4172/2155-9589.1000158. [DOI] [Google Scholar]
- 8.Geetha Varma V, Jha S, Himesh Karthik Raju L, Lalith Kishore R, Ranjith V. 2022. A review on decentralized wastewater treatment systems in India. Chemosphere 300:134462. doi: 10.1016/j.chemosphere.2022.134462. [DOI] [PubMed] [Google Scholar]
- 9.Krzeminski P, Tomei MC, Karaolia P, Langenhoff A, Almeida CMR, Felis E, Gritten F, Andersen HR, Fernandes T, Manaia CM, Rizzo L, Fatta-Kassinos D. 2019. Performance of secondary wastewater treatment methods for the removal of contaminants of emerging concern implicated in crop uptake and antibiotic resistance spread: a review. Sci Total Environ 648:1052–1081. doi: 10.1016/j.scitotenv.2018.08.130. [DOI] [PubMed] [Google Scholar]
- 10.Tong J, Tang A, Wang H, Liu X, Huang Z, Wang Z, Zhang J, Wei Y, Su Y, Zhang Y. 2019. Microbial community evolution and fate of antibiotic resistance genes along six different full-scale municipal wastewater treatment processes. Bioresour Technol 272:489–500. doi: 10.1016/j.biortech.2018.10.079. [DOI] [PubMed] [Google Scholar]
- 11.Czekalski N, Sigdel R, Birtel J, Matthews B, Bürgmann H. 2015. Does human activity impact the natural antibiotic resistance background? Abundance of antibiotic resistance genes in 21 Swiss lakes. Environ Int 81:45–55. doi: 10.1016/j.envint.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Chu BTT, Petrovich ML, Chaudhary A, Wright D, Murphy B, Wells G, Poretsky R, Löffler FE. 2018. Metagenomics reveals the impact of wastewater treatment plants on the dispersal of microorganisms and genes in aquatic sediments. Appl Environ Microbiol 84:e02168-17. doi: 10.1128/AEM.02168-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cacace D, Fatta-Kassinos D, Manaia CM, Cytryn E, Kreuzinger N, Rizzo L, Karaolia P, Schwartz T, Alexander J, Merlin C, Garelick H, Schmitt H, de Vries D, Schwermer CU, Meric S, Ozkal CB, Pons M-N, Kneis D, Berendonk TU. 2019. Antibiotic resistance genes in treated wastewater and in the receiving water bodies: a pan-European survey of urban settings. Water Res 162:320–330. doi: 10.1016/j.watres.2019.06.039. [DOI] [PubMed] [Google Scholar]
- 14.Marano RBM, Zolti A, Jurkevitch E, Cytryn E. 2019. Antibiotic resistance and class 1 integron gene dynamics along effluent, reclaimed wastewater irrigated soil, crop continua: elucidating potential risks and ecological constraints. Water Res 164:114906. doi: 10.1016/j.watres.2019.114906. [DOI] [PubMed] [Google Scholar]
- 15.Galvin S, Boyle F, Hickey P, Vellinga A, Morris D, Cormican M. 2010. Enumeration and characterization of antimicrobial-resistant Escherichia coli bacteria in effluent from municipal, hospital, and secondary treatment facility sources. Appl Environ Microbiol 76:4772–4779. doi: 10.1128/AEM.02898-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karkman A, Do TT, Walsh F, Virta MPJ. 2018. Antibiotic-resistance genes in waste water. Trends Microbiol 26:220–228. doi: 10.1016/j.tim.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Berendonk TU, Manaia CM, Merlin C, Fatta-Kassinos D, Cytryn E, Walsh F, Bürgmann H, Sørum H, Norström M, Pons M-N, Kreuzinger N, Huovinen P, Stefani S, Schwartz T, Kisand V, Baquero F, Martinez JL. 2015. Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 13:310–317. doi: 10.1038/nrmicro3439. [DOI] [PubMed] [Google Scholar]
- 18.Garner E, Organiscak M, Dieter L, Shingleton C, Haddix M, Joshi S, Pruden A, Ashbolt NJ, Medema G, Hamilton KA. 2021. Towards risk assessment for antibiotic resistant pathogens in recycled water: a systematic review and summary of research needs. Environ Microbiol 23:7355–7372. doi: 10.1111/1462-2920.15804. [DOI] [PubMed] [Google Scholar]
- 19.Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen A-LV, Cheng AA, Liu S, Min SY, Miroshnichenko A, Tran H-K, Werfalli RE, Nasir JA, Oloni M, Speicher DJ, Florescu A, Singh B, Faltyn M, Hernandez-Koutoucheva A, Sharma AN, Bordeleau E, Pawlowski AC, Zubyk HL, Dooley D, Griffiths E, Maguire F, Winsor GL, Beiko RG, Brinkman FSL, Hsiao WWL, Domselaar GV, McArthur AG. 2020. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 48:D517–D525. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang A-N, Gaston JM, Dai CL, Zhao S, Poyet M, Groussin M, Yin X, Li L-G, van Loosdrecht MCM, Topp E, Gillings MR, Hanage WP, Tiedje JM, Moniz K, Alm EJ, Zhang T. 2021. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat Commun 12:4765. doi: 10.1038/s41467-021-25096-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedler E, Juanico M, Shelef G. 2003. Simulation model of wastewater stabilization reservoirs. Ecol Eng 20:121–145. doi: 10.1016/S0925-8574(03)00009-0. [DOI] [Google Scholar]
- 22.Juanico M, Shelef G. 1991. The performance of stabilization reservoirs as a function of design and operation parameters. Water Sci Technol 23:1509–1516. doi: 10.2166/wst.1991.0604. [DOI] [Google Scholar]
- 23.Czekalski N, Gascón Díez E, Bürgmann H. 2014. Wastewater as a point source of antibiotic-resistance genes in the sediment of a freshwater lake. ISME J 8:1381–1390. doi: 10.1038/ismej.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckert EM, Di Cesare A, Coci M, Corno G. 2018. Persistence of antibiotic resistance genes in large subalpine lakes: the role of anthropogenic pollution and ecological interactions. Hydrobiologia 824:93–108. doi: 10.1007/s10750-017-3480-0. [DOI] [Google Scholar]
- 25.Paulus GK, Hornstra LM, Medema G. 2020. International tempo-spatial study of antibiotic resistance genes across the Rhine river using newly developed multiplex qPCR assays. Sci Total Environ 706:135733. doi: 10.1016/j.scitotenv.2019.135733. [DOI] [PubMed] [Google Scholar]
- 26.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. 2014. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narciso-da-Rocha C, Rocha J, Vaz-Moreira I, Lira F, Tamames J, Henriques I, Martinez JL, Manaia CM. 2018. Bacterial lineages putatively associated with the dissemination of antibiotic resistance genes in a full-scale urban wastewater treatment plant. Environ Int 118:179–188. doi: 10.1016/j.envint.2018.05.040. [DOI] [PubMed] [Google Scholar]
- 28.Wen Q, Yang L, Duan R, Chen Z. 2016. Monitoring and evaluation of antibiotic resistance genes in four municipal wastewater treatment plants in Harbin, Northeast China. Environ Pollut 212:34–40. doi: 10.1016/j.envpol.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 29.Nnadozie CF, Odume ON. 2019. Freshwater environments as reservoirs of antibiotic resistant bacteria and their role in the dissemination of antibiotic resistance genes. Environ Pollut 254:113067. doi: 10.1016/j.envpol.2019.113067. [DOI] [PubMed] [Google Scholar]
- 30.Ribeirinho-Soares S, Moreira NFF, Graça C, Pereira MFR, Silva AMT, Nunes OC. 2021. Overgrowth control of potentially hazardous bacteria during storage of ozone treated wastewater through natural competition. Water Res 209:117932. doi: 10.1016/j.watres.2021.117932. [DOI] [PubMed] [Google Scholar]
- 31.Tandon K, Baatar B, Chiang P-W, Dashdondog N, Oyuntsetseg B, Tang S-L. 2020. A large-scale survey of the bacterial communities in lakes of western Mongolia with varying salinity regimes. Microorganisms 8:1729. doi: 10.3390/microorganisms8111729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komárek J. 2016. Review of the cyanobacterial genera implying planktic species after recent taxonomic revisions according to polyphasic methods: state as of 2014. Hydrobiologia 764:259–270. doi: 10.1007/s10750-015-2242-0. [DOI] [Google Scholar]
- 33.Visser PM, Ibelings BW, Bormans M, Huisman J. 2016. Artificial mixing to control cyanobacterial blooms: a review. Aquat Ecol 50:423–441. doi: 10.1007/s10452-015-9537-0. [DOI] [Google Scholar]
- 34.Venâncio I, Luís Â, Domingues F, Oleastro M, Pereira L, Ferreira S. 2022. The prevalence of Arcobacteraceae in aquatic environments: a systematic review and meta-analysis. Pathogens 11:244. doi: 10.3390/pathogens11020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newton RJ, McLellan SL, Dila DK, Vineis JH, Morrison HG, Eren AM, Sogin ML. 2015. Sewage reflects the microbiomes of human populations. mBio 6:e02574-14. doi: 10.1128/mBio.02574-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML. 2010. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol 12:378–392. doi: 10.1111/j.1462-2920.2009.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shanks OC, Newton RJ, Kelty CA, Huse SM, Sogin ML, McLellan SL. 2013. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Appl Environ Microbiol 79:2906–2913. doi: 10.1128/AEM.03448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brand VR, Crosby LD, Criddle CS. 2019. Niche differentiation among three closely related Competibacteraceae clades at a full-scale activated sludge wastewater treatment plant and putative linkages to process performance. Appl Environ Microbiol 85:e02301-18. doi: 10.1128/AEM.02301-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu H, Chandran K, Stensel D. 2014. Microbial ecology of denitrification in biological wastewater treatment. Water Res 64:237–254. doi: 10.1016/j.watres.2014.06.042. [DOI] [PubMed] [Google Scholar]
- 40.Teltsch B, Azov Y, Juanico M, Shelef G. 1992. Plankton community changes due to the addition of treated effluents to a freshwater reservoir used for drip irrigation. Water Res 26:657–668. doi: 10.1016/0043-1354(92)90242-V. [DOI] [Google Scholar]
- 41.Wallace J, Champagne P, Hall G. 2016. Multivariate statistical analysis of water chemistry conditions in three wastewater stabilization ponds with algae blooms and pH fluctuations. Water Res 96:155–165. doi: 10.1016/j.watres.2016.03.046. [DOI] [PubMed] [Google Scholar]
- 42.Beran B, Kargi F. 2005. A dynamic mathematical model for wastewater stabilization ponds. Ecol Modell 181:39–57. doi: 10.1016/j.ecolmodel.2004.06.022. [DOI] [Google Scholar]
- 43.Korajkic A, Wanjugi P, Brooks L, Cao Y, Harwood VJ. 2019. Persistence and decay of fecal microbiota in aquatic habitats. Microbiol Mol Biol Rev 83:e00005-19. doi: 10.1128/MMBR.00005-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie H, Ogura Y, Suzuki Y. 2022. Persistence of antibiotic-resistant Escherichia coli strains belonging to the B2 phylogroup in municipal wastewater under aerobic conditions. Antibiotics 11:202. doi: 10.3390/antibiotics11020202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liguori K, Keenum I, Davis BC, Calarco J, Milligan E, Harwood VJ, Pruden A. 2022. Antimicrobial resistance monitoring of water environments: a framework for standardized methods and quality control. Environ Sci Technol 56:9149–9160. doi: 10.1021/acs.est.1c08918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang C, Zhu W, Strong PJ, Zhu F, Han X, Hong C, Wang W, Yao Y. 2021. Disentangling the effects of physicochemical, genetic, and microbial properties on phase-driven resistome dynamics during multiple manure composting processes. Environ Sci Technol 55:14732–14745. doi: 10.1021/acs.est.1c03933. [DOI] [PubMed] [Google Scholar]
- 47.ISO. 2014. 9308–1:2014 I. Water quality — enumeration of Escherichia coli and coliform bacteria — part 1: membrane filtration method for waters with low bacterial background flora., 3rd ed.
- 48.Rodríguez-Baño J, Picón E, Navarro MD, López-Cerero L, Pascual Á. ESBL-REIPI Group. 2012. Impact of changes in CLSI and EUCAST breakpoints for susceptibility in bloodstream infections due to extended-spectrum β-lactamase-producing Escherichia coli. Clin Microbiol Infect 18:894–900. doi: 10.1111/j.1469-0691.2011.03673.x. [DOI] [PubMed] [Google Scholar]
- 49.Kornspan D, Brendebach H, Hofreuter D, Mathur S, Blum SE, Fleker M, Bardenstein S, Al Dahouk S. 2021. Protein biomarker identification for the discrimination of Brucella melitensis field isolates from the Brucella melitensis Rev.1 vaccine strain by MALDI-TOF MS. Front Microbiol 12:712601. doi: 10.3389/fmicb.2021.712601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medlin L, Elwood HJ, Stickel S, Sogin ML. 1988. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71:491–499. doi: 10.1016/0378-1119(88)90066-2. [DOI] [PubMed] [Google Scholar]
- 52.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Šmilauer P, Lepš J. 2014. Multivariate analysis of ecological data using CANOCO 5, 2nd ed. Cambridge University Press, Cambridge, UK. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aem.00170-23-s0001.pdf, PDF file, 4.5 MB (4.5MB, pdf)
Supplemental material. Download aem.00170-23-s0002.xlsx, XLSX file, 0.1 MB (106.3KB, xlsx)
Data Availability Statement
The 16S and 18S rRNA sequences were uploaded to the NCBI SRA archive under BioProject number PRJNA805207.