

Abstract
AIM
To summarize quality of life (QoL) and its determinants, including disease severity, in individuals with developmental and epileptic encephalopathies (DEEs) through a tailored questionnaire.
METHOD
A questionnaire containing 89 items addressing demographic characteristics, genetic diagnosis, clinical features, and QoL was distributed to primary caregivers of individuals with DEEs through patient advocacy organizations. Composite scores were generated from the mean values of QoL items, grouped into domain scores.
RESULTS
Out of 176 received responses, the most common genetic diagnoses reported were SCN2A (n = 42/173, 24%), SLC6A1 (n = 28/173, 16%), SCN1A (n = 22/173, 13%), and KCNQ2 (n = 21/173, 12%). Composite QoL scores centered around a mean score of 61.67 of 100 (SD 17.10). QoL scores were strongly associated with the number of days minimally disrupted by seizures, medication side effects, genetic diagnosis, and community type. The mean QoL scores for individuals with DEEs was significantly lower than for individuals with Rett syndrome, cerebral palsy, autism spectrum disorder, and Down syndrome.
INTERPRETATION
QoL in DEEs can be assessed through a standardized instrument. QoL only partially overlaps with objective measurements of disease severity and may represent an independent outcome measure in precision medicine trials.
Graphical Abstract
This survey-based, cross-sectional study characterizes disease burden, quality-of-life, and the relationship between the two, in children with genetic developmental and epileptic encephalopathies.
The developmental and epileptic encephalopathies (DEEs) are severe epilepsies frequently starting in childhood resulting in significant morbidity. Many DEEs result in therapy-resistant seizures in addition to other neurological and non-neurological symptoms.1,2 A genetic diagnosis can be identified in up to 40% of individuals with non-lesional DEEs.3,4 While genetic etiologies in DEEs are heterogeneous, pathogenic variants in SCN1A, KCNQ2, SCN2A, and STXBP1 are amongst the most common genetic causes of DEEs. These conditions represent active targets of drug development.5–7 In some cases, there are already existing precision medicine trials that aim to address the underlying genetic etiology.8,9
Clinical trials typically rely on quantifiable outcome measures to assess disease severity. In parallel to established paradigms in clinical trials, seizure burden represents the main outcome measure.10,11 However, given the significant degree of neurological and non-neurological comorbidity, seizures likely do not represent the only parameter affecting overall quality of life (QoL) in individuals with DEEs. While seizures are a key clinical feature of the DEEs, the holistic relationship of overall disease burden, including seizure frequency, and QoL in individuals with DEEs remains poorly understood and is an emerging area of research with novel assessment tools.12–15
The development and implementation of QoL measurement tools is a priority of DEE-related projects such as the Rare Epilepsy Network and the Rare Epilepsy Landscape Analysis, with the ultimate goal of creating targeted therapies that can improve QoL.16 QoL measurements have been developed for trisomy 21 (Down syndrome) and more recently as disease-specific measures for conditions such as CDKL5 deficiency disorders through a modified Delphi method, indicating increasing interest in systematically assessing QoL in the DEEs.17–22 One such tool, the Quality of Life Inventory-Disability (QI-Disability) which we utilize in our study, was created for and with individuals with comparable neurodevelopmental disorders.17–20 However, these assessments have not been applied to larger patient populations with genetic DEEs. Accordingly, the relationship of QoL and objective measurements of disease burden in the DEEs remains unknown.
We aimed to assess the relationship of QoL measurements and disease severity in the genetic DEEs through a dedicated online questionnaire that was distributed through participating patient advocacy organizations. We hypothesized that seizure frequency may account for a significant portion of QoL in patients with DEEs based on prior literature in this area.14,23–26
METHOD
Questionnaire design
The questionnaire was created via Research Electronic Data Capture (REDCap) electronic data capture tools hosted at the Children’s Hospital of Philadelphia.27 REDCap is a secure, web-based software platform designed to support data capture for research studies. The questionnaire is comprised of three sections: demographics, medical history, and QoL. Items from this questionnaire were from tools validated in similar populations including the Severity Assessment in CDKL5 Deficiency Disorder and QI-Disability as well as the Epilepsy Learning Health System and Pediatric Epilepsy Learning Health System.17,21,28 This tool was piloted to take 15 to 20 minutes to complete by individuals without medical training or medical education. Medical diagnoses were accompanied by definitions for clarity. As branching logic was used in the survey design, not all questionnaire participants completed every item of the questionnaire. Most relevant questions including QoL items were answered by most or all participants. A freely available online example of the questionnaire without branching logic can be found in Appendix S1.
Questionnaire distribution
This study was conducted through the University of Pennsylvania between 21st September 2020 and 17th December 2020. Once this period concluded, we considered those who had answered the survey to be part of our cohort. The study protocol was approved by the Institutional Review Board at the University of Pennsylvania. Written informed consent was obtained at the beginning of the questionnaire. Adult caregivers of individuals with DEEs at the time of questionnaire completion were considered for inclusion. Participants received the questionnaire through distribution by the following patient advocacy organizations: FamilieSCN2A Foundation, Dravet Syndrome Foundation, Lennox-Gastaut Syndrome Foundation, KCNQ2 Cure Alliance, STXBP1 Foundation, SLC6A1 Connect, CureGRIN Foundation, Wishes for Elliott, and PCDH19 Alliance. Participants were offered the opportunity to enter their email address for a randomized drawing for ten $20 Amazon gift cards.
Composite QoL measures
Composite QoL score was calculated for each participant using previously established methods.17 Initial responses for each of the 32 items of the QI-Disability (section 3 of the survey) were scored from 1 to 5 corresponding to the responses ‘never’, ‘rarely’, ‘sometimes’, ‘often’, or ‘very often’. Negative items were reverse coded. Once recorded, each item was scaled up to a 100-point range. QoL score for each of the five domains included in the questionnaire tool was calculated by taking the mean value of all items within a given domain. Total composite QoL scores were created by determining the mean value of these five domain scores for each participant. Normality of the composite QoL measure was assessed using the Kolmogorov–Smirnov test.
Statistical analysis
All statistical analyses were completed using the R analysis framework (R Foundation for Statistical Computing, Vienna, Austria), and accompanying figures were produced using the package ggplot2. Several analyses were performed, including Wilcoxon rank-sum test and Fisher’s exact test in order to compare groups of participants’ QoL composite scores against metrics including community type, seizure frequency, seizure freedom, ‘minimally disrupted days’, side effects, and genetic diagnosis. Statistical comparisons were not drawn between individuals with different genetic diagnoses.
RESULTS
Questionnaire responses reflect a range of genetic epilepsies
We received 176 responses to our questionnaire. Two questionnaire responses were excluded as the respondents indicated they were not a caregiver of an individual with a DEE, leaving 174 questionnaire responses for analysis. Questionnaire responses surveyed a range of genetic etiologies that reflect the range of genetic etiologies in childhood DEEs. SCN2A (n = 42/173), SLC6A1 (n = 28/173), and SCN1A (n = 22/173), represented the most common genetic etiologies named by participants (Figure S1). The average age of genetic diagnosis was 5 years 5 months (SD 6 years 3 months), and the average amount of time since the genetic diagnosis at the time of filling out the survey was 4 years 7 months (SD 7 years 6 months). A small subset of individuals did not indicate a genetic diagnosis (n = 14/173, 8%). The demographic characteristics of questionnaire participants show a predominance of families self-identifying as White with higher-than-average income with 49% of participants living in suburban areas (TableS1). A total of 99% (173/174) of individuals reported an affiliation with one of the patient advocacy organizations participating in the questionnaire.
Seizure phenotypes differ among participants
Participants reported a wide range of seizure and epilepsy features (Table S2). Epilepsy features were reported in 142 participants with percentages reported out of the total cohort of 174 participants. Average onset of the first seizure was at 1 year 7 months (SD 2 years 4 months, range 0–12 years). Epilepsy classifications reported by participants included generalized (33%), focal (7%), and mixed generalized and focal (26%). The most reported epilepsy syndromes included Lennox–Gastaut syndrome (14%, n = 25) and Dravet syndrome (13%, n = 23). The most common seizure types reported included tonic-clonic (50%, n = 87), myoclonic (41%, n = 72), and tonic (33%, n = 57). Most participants (69%, n = 97) either never had had a prolonged seizure or did not have a prolonged seizure in at least 6 months before questionnaire completion.
Self-reported seizure frequency was assessed with a semiquantitative measure aligned with the emerging format to assess seizure frequency in the Epilepsy Learning Health System and Pediatric Epilepsy Learning Health System (Figure 1a).19 In addition, participants were asked to report the longest period of seizure freedom (Figure 1b) and the average number of ‘minimally disrupted days’ (e.g. the average number of days not significantly affected by seizures) (Figure 1c).
Figure 1:

Multiple assessments of cumulative seizure frequency. Seizure frequency was assessed cumulatively as how often the individual has seizures. Seizure freedom was assessed cumulatively as the longest period the individual has not had a seizure. Minimally disrupted days were assessed as days that were minimally disrupted by seizures or where the individual was engaged, interactive, and able to finish therapies throughout the day. Two categories of responses were merged to create the ‘almost none/none’ group for the analysis. Frequency of a given response is shown as a percentage of total participants.
Questionnaire respondents report a range of non-seizure diagnoses
Questionnaire participants reported 36 diagnoses across six different domains of non-seizure diagnoses, including developmental delays (98% participants, 768 recorded diagnoses, 4.52 average diagnoses per participant), neurological diagnoses (98%), visual diagnoses (48%), behavioral diagnoses (38%), speech diagnoses (38%), and motor diagnoses (17%). Development delays included diagnoses such as speech/language delay (84%), motor delay (78%), and global developmental delay (78%). The most common neurological diagnosis other than seizures included intellectual disability (60%), hypotonia (60%), and autism spectrum disorder (33%). The most frequently reported diagnoses affecting vision included strabismus and/or esotropia (24%), cortical visual impairment (22%), and hyperopia (11%). The most common behavioral diagnoses were attention-deficit/hyperactivity disorder (18%), bruxism (11%), and self-injurious behavior (10%). The most reported speech diagnosis was motor speech disorder (38%), while other speech disorders, like speech/language delay, were integrated into non-seizure symptom categories. The most frequent motor diagnoses were myoclonus (9%), ataxia (8%), and dystonia and tremors, each at 7% (Figure S2). Of the 176 participants, only 18 individuals reported identical combinations of diagnoses. There was no non-seizure diagnosis shared by all participants.
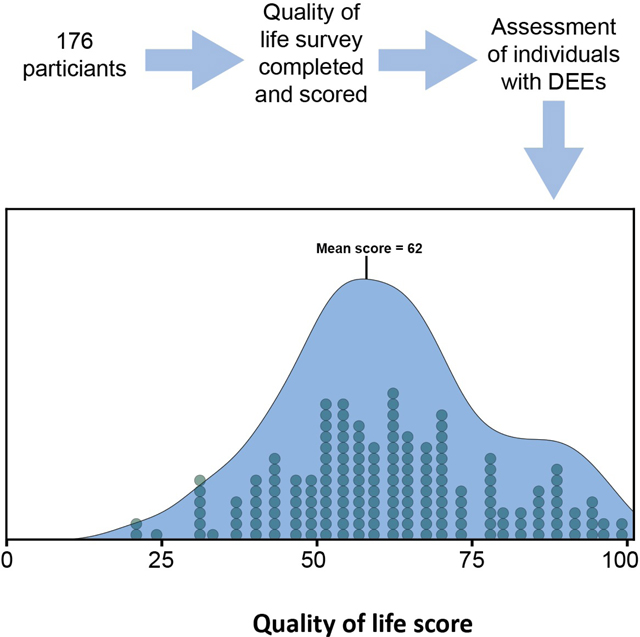
QoL shows a range of values across respondents
Across all four domains, we developed a composite measure for QoL (Figure 2). This composite measure was generated for the 173 participants who responded to QoL items of the questionnaire. The composite QoL measure was approximately normally distributed (Kolmogorov–Smirnov test, p = 0.63, mean 61.67, SD 17.10) as were most of the domain scores. The domain scores for ‘Health and wellbeing’ (p = 0.03, median 62.50, interquartile range 56.25–81.25), ‘Feelings and emotions’ (p = 0.77, mean 65.02, SD 15.88), ‘Family and friends’ (p = 0.27, mean 64.62, SD 23.16), ‘Daily life’ (p = 0.10, mean 49.34, SD 29.22), and ‘Activities and outdoors’ (p=0.13, mean 62.17, SD 24.67) had unique distributions, reflecting the variability of participant responses across these four domains.
Figure 2:

Distribution of overall and domain quality of life (QoL) scores. The distribution of QoL scores in each domain is shown below and beside the overall QoL score distribution. The number of participants with a given score is shown below the distribution curve where one circle represents one response.
QoL correlates with community factors but not other demographic variables
The only demographic variable correlated with QoL score was that participants living in rural areas have lower mean QoL (56.02, SD 15.08) than those in urban areas (67.99, SD 16.66) (estimated QoL score difference 12.25, p = 0.001, median difference 95% confidence interval [CI] 5.36–19.44). There were no other significant correlations between other demographic factors and QoL.
QoL correlates with average minimally disrupted days, but not with seizure frequency or period of longest seizure freedom
Next, how the QoL score correlated with the various assessments of seizure burden was assessed. We found that the QoL score did not correlate with reported seizure frequency (Figure 3b). Of the 141 participants with QoL scores and seizure frequency responses, there was no significant difference in QoL scores between individuals with seizures (QoL score 59.73, SD 16.45) and those who have been seizure free for at least a year (QoL score 66.47, SD 15.57) (6.39 estimated QoL score difference, p = 0.14, 95% CI –2.17 to 14.88). There was no significant difference in QoL score between participants indicating they never had seizures (70.14, SD 18.31) and participants who had a history of seizures with at least 1 day of seizure freedom (61.42, SD 16.70) (–8.71 estimated QoL score difference, p = 0.41, 95% CI –31.22 to 13.30). Overall, we did not observe a correlation with the reported longest period of seizure freedom and QoL score (Figure 3c).
Figure 3:

Relationship between quality of life (QoL) and number of minimally disrupted days, seizure frequency, and longest period of seizure freedom. Each dot represents a single participant’s response within the distribution of responses while the width of a given plot represents the probability of a given QoL score response using kernel density estimation.
The total number of average days minimally disrupted by seizures, however, was strongly correlated with QoL (Figure 3a). There was a significant difference in QoL score between participants experiencing minimally disrupted days for the majority of the month (67.12, SD 17.02), compared with those experiencing minimally disrupted days for the minority of the month (54.03, SD 11.79) (12.56 estimated QoL score difference, p < 0.001, 95% CI 7.39–18.28). Furthermore, compared against all other participants, those reporting almost always having minimally disrupted days (n = 58/173, 34%) were much less likely to have below average QoL scores (p < 0.001, odds ratio [OR] 0.15, 95% CI 0.07–0.34). Participants who report almost always having days minimally disrupted by seizures were 6.67 times more likely to have above average QoL scores. All assessments of seizure burden were significantly correlated with each other (p < 0.05).
QoL is significantly impacted by reported medication side effects and cumulative disease burden
We then assessed non-seizure features correlated with composite QoL score. Presence of medication side effects showed a strong negative correlation with QoL scores (Table 1). Participants reporting medication side effects (n = 141) had lower QoL scores (n = 89, mean 57.98, SD 14.57) compared to individuals who did not report medication side effects (n = 35, mean 68.39, SD 17.84) (10.88 estimated QoL score difference, p = 0.003, 95% CI 3.53–18.06).
Table 1:
Quality of life (QoL), medication side effect, and cumulative disease burden
| Medication side effect (n) | Mean QoL (IQR, SD) | ||
|---|---|---|---|
| Present (89) | 57.98 (50.44–67.39, 14.57) | ||
| Absent (35) | 68.39 (54.83–84.14) | ||
|
| |||
| Number of reported symptoms (n) | Mean QoL (IQR, SD) | ||
|
| |||
| <5 (23) | 72.31 (64.03–89.52, 18.02) | ||
| 5–10 (68) | 66.93 (54.83–78.31, 16.35) | ||
| 10–15 (72) | 55.17 (47.63–64.24, 14.65) | ||
| 15–19 (11) | 55.36 (47.91–58.50, 12.19) | ||
| >19 (2) | 36.33 (29.19–43.48, 20.21) | ||
|
| |||
| Symptoms with significant negative impact on QoL (p < 0.05) | Mean QoL: symptom present (SD) | Mean QoL: symptom absent (SD) | Median difference (95% CI) |
|
| |||
| Motor delay | 59.54 (16.69) | 69.48 (16.52) | –16.73 to –3.94 |
| Global developmental delay | 59.10 (16.10) | 71.09 (17.59) | –18.56 to –6.84 |
| Intellectual disability | 58.50 (16.94) | 66.44 (16.33) | –13.40 to –3.13 |
| Spasticity | 49.88 (13.55) | 64.14 (16.77) | –19.90 to –7.57 |
| Strabismus and/or esotropia | 55.30 (16.00) | 63.64 (17.00) | –14.19 to –3.32 |
| Cortical visual impairment | 54.25 (14.89) | 63.64 (17.00) | –15.19 to –3.95 |
| Dystonia | 48.40 (14.08) | 62.66 (16.93) | –23.34 to 4.83 |
| Bruxism | 53.04 (17.29) | 62.66 (16.93) | –18.11 to –1.48 |
| Self-injurious behavior | 53.45 (17.91) | 62.62 (16.81) | –18.26 to –1.19 |
Abbreviations: IQR, interquartile range; CI, confidence interval.
QoL score was also negatively correlated with total number of reported diagnoses, including steep declines in QoL score as the number of diagnoses increased above 10. Individuals with the following self-reported diagnoses had significantly lower QoL composite scores than those without: motor delay, global developmental delay, intellectual disability, spasticity, strabismus and/or esotropia, cortical visual impairment, dystonia, bruxism, and self-injurious behavior (Table 1). Additionally, there is a significant difference in QoL scores among genetic diagnoses. For example, individuals with an SCN2A-related disorder are more likely to have a below average QoL score (OR 2.47, p = 0.20, 95% CI 1.13–5.66) compared to other genetic diagnoses.
The number of individual items with the normalized QoL scores are shown in Table 1. As the number of reported diagnoses increased, an individual was more likely to be in a lower quartile than an individual with fewer diagnoses. For example, those with fewer than nine diagnoses, the average number of diagnoses for this cohort, were more likely to have a higher QoL score (70.00, SD 17.02) than those with nine or more diagnoses (55.73, SD 14.55) (14.03 estimated QoL score difference p < 0.001, 95% CI 9.24–19.10).
QoL scores in the population with DEE are lower than other populations with neurodevelopmental disorders
In the validation study of QI-Disability, QoL composite scores were applied to individuals with Rett syndrome, cerebral palsy, autism spectrum disorder, and Down syndrome which yielded mean QoL scores of 65.6, 64.9, 66.6, and 76.1 respectively.17 The mean QoL score of 61.7 (SD 17.1, CI 95% 59.1–64.3) in our study using the same tool was lower than each of these populations.
DISCUSSION
We assessed QoL and disease burden in genetic DEEs assessed through a de-identified questionnaire distributed through participating patient advocacy organizations. This QoL composite score allowed us to assess correlation of QoL with various disease aspects and measures of disease burden. To our knowledge, our study is the first attempt in a larger, heterogeneous group of DEEs to assess QoL and disease burden.
We found that QoL measurements were not related to self-reported parameters like seizure frequency that approximate objective measures of disease severity. However, composite QoL scores show a strong correlation to the perception of average days minimally disrupted by seizures. This finding suggests that subjective measures of seizure burden may have a greater impact on QoL than objective measures of seizure burden such as seizure frequency and longest period of seizure freedom. We expect that measurements such as ‘minimally disrupted days’ will be refined for use in future studies and potentially emerge as proxy measures to communicate perceived disease burden between families and care teams.
Overall disease burden was further assessed by the number of reported diagnoses, which was also significantly correlated to composite QoL score. We identified individual diagnoses that, when present, were associated with decreased QoL. However, it is difficult to clearly determine the effect of a single diagnosis on QoL because most individuals reported multiple diagnoses.
Other associations with QoL included community type, genetic diagnosis, and presence of medication side effects. The presence of medication side effects affecting QoL is especially essential to consider in this population as targeted therapies are developed.
Limitations
Our study had several shortcomings that will need to be addressed in future studies. First, while we attempted to reflect the populations of individuals, including a wide range of disease foundations in our questionnaire, the responses to our questionnaire only reflect a subset of the estimated >100 genetic etiologies causing DEE. Given related efforts in CDKL5 deficiency disorders where some of the QoL measures used in our study were initially developed, we limited ourselves to disease foundations representing individuals with DEEs that had not been assessed so far or where we were not aware of related ongoing studies.14 Likewise, referring to ongoing related projects, several disease foundations declined participation in our study. We expect that once information from ongoing studies is made available, they will complement our results and provide a more comprehensive picture of QoL in the DEEs. Furthermore, we chose a retrospective study format where a prospective gathering of QoL may alter or add to our findings.
Additional limitations of our study relate to the survey tool and methodology. Although the Severity Assessment in CDKL5 Deficiency Disorder and QI-Disability as well as the Epilepsy Learning Health System and Pediatric Epilepsy Learning Health System have been designed for children with epilepsy and DEE, the use of these tools together has not been specifically validated or shown to be reliable in our cohort. In addition, our study relied on de-identified responses from caregivers which may represent more limited medical information than alternate methods such as medical chart review and may introduce caregiver bias. While we asked participants to submit information on genetic etiology and variant, we could not validate these findings. In our study, none of the self-reported genetic variants was found to be a population variant or a genetic change that we interpreted as non-contributory.
Finally, the demographics of the families contributing to our study do not fully reflect the DEE population at large. Participants were largely White, non-Hispanic/Latino, and had above average income to a much higher degree than the general US population, indicating a lack of diversity in the participants of our study. Recruiting participants through disease foundations and distributing the questionnaire in English only may have contributed to this disparity. Currently, the degree of diversity in disease foundations is poorly understood. We did not find significant differences in QoL and reported diagnoses in individuals from the small group of underrepresented minorities in our study.
Conclusion
In summary, we provide evidence that QoL in DEEs can be comprehensively assessed through a detailed questionnaire that integrates self-reported diagnoses with QoL composite scores. The lack of association of QoL with reported seizure frequency emphasizes the need to address disease severity in DEEs holistically to inform patient care and outcome measures in future precision medicine trials.
Supplementary Material
Figure S1: Genetic diagnosis of affected individuals.
Table S1: Demographics of probands and caregivers participating in questionnaire.
Table S2: Epilepsy features.
Figure S2: Frequency of reported symptoms.
Appendix S1: Non-seizure symptoms & quality of life in epileptic encephalopathies.
What this paper adds.
Quality of life (QoL) in individuals with genetic developmental and epileptic encephalopathies (DEEs) can be assessed through standardized instruments.
Seizure frequency is unrelated to QoL in individuals with genetic DEEs.
Individuals who rarely or never have days disrupted by seizures have higher QoL scores.
Individuals with DEEs have lower QoL than individuals with other neurodevelopmental disorders.
Acknowledgements
The authors would like to especially thank the patient advocacy organizations and families who participated in this study. Full funding for this project was provided by the National Society of Genetic Counselors Neurogenetics Special Interest Group.
The data that supports the findings of this study are available on request from the corresponding author. The data are not publicly available because of privacy or ethical restrictions.
DMCN 15187 was supported by NIH funding.
U24 NS120854
ABBREVIATIONS
- DEE
Developmental and epileptic encephalopathy
- QI-Disability
Quality of Life Inventory-Disability
- QoL
Quality of life
REFERENCES
- 1.McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol2016;15(3):304–16. [DOI] [PubMed] [Google Scholar]
- 2.Ellis CA, Petrovski S, Berkovic SF. Epilepsy genetics: clinical impacts and biological insights. Lancet Neurol 2020;19(1):93–100. [DOI] [PubMed] [Google Scholar]
- 3.Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med 2016;18(9):898–905. [DOI] [PubMed] [Google Scholar]
- 4.Weber YG, Biskup S, Helbig KL, Von Spiczak S, Lerche H. The role of genetic testing in epilepsy diagnosis and management. Expert Rev Mol Diagn 2017;17(8):739–50. [DOI] [PubMed] [Google Scholar]
- 5.Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol Rev 2018;70(1):142–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maljevic S, Reid CA, Petrou S. Models for discovery of targeted therapy in genetic epileptic encephalopathies. J Neurochem 2017;143(1):30–48. [DOI] [PubMed] [Google Scholar]
- 7.Hussain S Developing a PPI inhibitor-based therapy for STXBP1 haploinsufficiency-associated epileptic disorders. Front Mol Neurosci 2014;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med 2020;12(558). [DOI] [PubMed] [Google Scholar]
- 9.Lenk GM, Jafar-Nejad P, Hill SF, Huffman LD, Smolen CE, Wagnon JL, et al. Scn8a Antisense Oligonucleotide Is Protective in Mouse Models of SCN8A Encephalopathy and Dravet Syndrome. Ann Neurol 2020;87(3):339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet 2019;394(10216):2243–54. [DOI] [PubMed] [Google Scholar]
- 11.Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. N Engl J Med 2017;376(21):2011–20. [DOI] [PubMed] [Google Scholar]
- 12.Brunklaus A, Dorris L, Zuberi SM. Comorbidities and predictors of health-related quality of life in Dravet syndrome. Epilepsia 2011;52(8):1476–82. [DOI] [PubMed] [Google Scholar]
- 13.Lagae L, Brambilla I, Mingorance A, Gibson E, Battersby A. Quality of life and comorbidities associated with Dravet syndrome severity: a multinational cohort survey. Dev Med Child Neurol2018;60(1):63–72. [DOI] [PubMed] [Google Scholar]
- 14.Leonard H, Junaid M, Wong K, Demarest S, Downs J. Exploring quality of life in individuals with a severe developmental and epileptic encephalopathy, CDKL5 Deficiency Disorder. Epilepsy Res 2021;169:106521. [DOI] [PubMed] [Google Scholar]
- 15.Morris C, Janssens A, Allard A, Coon J, Shilling V, Tomlinson R, et al. Informing the NHS Outcomes Framework: evaluating meaningful health outcomes for children with neurodisability using multiple methods including systematic review, qualitative research, Delphi survey and consensus meeting. NIHR Journals Library. 2014. [PubMed] [Google Scholar]
- 16.Miller IP. Raring for change: Confluence of scientific discovery and advocate alignment warrants vital new investments in The Epilepsies. Epilepsy Behav. 2020; 111: 107276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Downs J, Jacoby P, Leonard H, Epstein A, Murphy N, Davis E, et al. Psychometric properties of the Quality of Life Inventory-Disability (QI-Disability) measure. Qual Life Res 2019;28(3):783–94. [DOI] [PubMed] [Google Scholar]
- 18.Epstein A, Williams K, Reddihough D, Murphy N, Leonard H, Whitehouse A, Jacoby P, Downs J. Content validation of the Quality of Life Inventory-Disability. Child Care Health Dev 2019;45(5):654–659. [DOI] [PubMed] [Google Scholar]
- 19.Jacoby P, Epstein A, Kim R, Murphy N, Leonard H, Williams K, Reddihough D, Whitehouse A, Downs J. Reliability of the Quality of Life Inventory-Disability Measure in Children with Intellectual Disability. J Dev Behav Pediatr 2020;41(7):534–539. [DOI] [PubMed] [Google Scholar]
- 20.Whitehouse A, Jacoby P, Reddihough D, Leonard H, Williams K, Downs J. The effect of functioning on Quality of Life Inventory-Disability measured quality of life is not mediated or moderated by parental psychological distress [DOI] [PubMed] [Google Scholar]
- 21.Demarest S, Pestana-Knight EM, Olson HE, Downs J, Marsh ED, Kaufmann WE, et al. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr Neurol 2019;97:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Humphrey A, Ploubidis GB, Yates JR, Steinberg T, Bolton PF. The Early Childhood Epilepsy Severity Scale (E-Chess). Epilepsy Res 2008;79(2–3):139–45. [DOI] [PubMed] [Google Scholar]
- 23.Fitzgerald MP, Kaufman MC, Massey SL, Frindinger S, Prelack M, Ellis C, et al. Assessing seizure burden in pediatric epilepsy using an electronic medical record-based tool through a common data element approach. Epilepsia 2021; 62(7):1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong CY, Chang WM, Kong AN, Rithauddin AM, Khoo TB, Ong LC. Quality of life in Malaysian children with epilepsy. Epilepsy Behav 2018;80:15–20. [DOI] [PubMed] [Google Scholar]
- 25.Jain P, Smith ML, Speechley K, Ferro M, Connolly M, Ramachandrannair R, Almubarak S, Andrade A, Widjaja E; Pepsqol Study Team. Seizure freedom improves health-related quality of life after epilepsy surgery in children. Dev Med Child Neurol 2020;62(5):600–608. [DOI] [PubMed] [Google Scholar]
- 26.Sadleir L, Hulihan J, Messenheimer J, Ali S, Gutterman D, Sebree T, et al. Quality of Life and Qualitative Caregiver Assessments in Children and Adolescents with Developmental and Epileptic Encephalopathies Treated with Cannabidiol Transdermal Gel: An Open-Label Clinical Trial. JAMA Netw Open. 4(9):e2123930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42(2):377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grinspan ZM, Patel AD, Shellhaas RA, Berg AT, Axeen ET, Bolton J, et al. Design and implementation of electronic health record common data elements for pediatric epilepsy: Foundations for a learning health care system. Epilepsia 2021;62(1):198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Genetic diagnosis of affected individuals.
Table S1: Demographics of probands and caregivers participating in questionnaire.
Table S2: Epilepsy features.
Figure S2: Frequency of reported symptoms.
Appendix S1: Non-seizure symptoms & quality of life in epileptic encephalopathies.