

Abstract
Purpose:
O6-methylguanine DNA methyltransferase (MGMT)-silenced tumors reveal sensitivity to temozolomide (TMZ), which may be enhanced by PARP inhibitors. Approximately 40% of colorectal cancer has MGMT silencing and we aimed to measure antitumoral and immunomodulatory effects from TMZ and olaparib in colorectal cancer.
Experimental Design:
Patients with advanced colorectal cancer were screened for MGMT promoter hypermethylation using methylation-specific PCR of archival tumor. Eligible patients received TMZ 75 mg/m2 days 1–7 with olaparib 150 mg twice daily every 21 days. Pretreatment tumor biopsies were collected for whole-exome sequencing (WES), and multiplex quantitative immunofluorescence (QIF) of MGMT protein expression and immune markers.
Results:
MGMT promoter hypermethylation was detected in 18/51 (35%) patients, 9 received study treatment with no objective responses, 5/9 had stable disease (SD) and 4/9 had progressive disease as best response. Three patients had clinical benefit: carcinoembryonic antigen reduction, radiographic tumor regression, and prolonged SD. MGMT expression by multiplex QIF revealed prominent tumor MGMT protein from 6/9 patients without benefit, while MGMT protein was lower in 3/9 with benefit. Moreover, benefitting patients had higher baseline CD8+ tumor-infiltrating lymphocytes. WES revealed 8/9 patients with MAP kinase variants (7 KRAS and 1 ERBB2). Flow cytometry identified peripheral expansion of effector T cells.
Conclusions:
Our results indicate discordance between MGMT promoter hypermethylation and MGMT protein expression. Antitumor activity seen in patients with low MGMT protein expression, supports MGMT protein as a predictor of alkylator sensitivity. Increased CD8+ TILs and peripheral activated T cells, suggest a role for immunostimulatory combinations.
Significance:
TMZ and PARP inhibitors synergize in vitro and in vivo in tumors with MGMT silencing. Up to 40% of colorectal cancer is MGMT promoter hypermethylated, and we investigated whether TMZ and olaparib are effective in this population. We also measured MGMT by QIF and observed efficacy only in patients with low MGMT, suggesting quantitative MGMT biomarkers more accurately predict benefit to alkylator combinations.
Introduction
Colorectal cancer remains highly prevalent with few available biomarker-guided therapies. However, promoter hypermethylation of O6-methylguanine DNA methyltransferase (MGMT) is identified in approximately 40% of metastatic colorectal cancer (1, 2). Hypermethylation of the MGMT promoter is expected to result in decreased MGMT mRNA/protein expression, which reduces the capacity of tumor cells to repair the lethal O6-methylguanine lesions produced by alkylating agents and renders these tumors more susceptible to these agents, including temozolomide (TMZ; refs. 3–5). Moreover, the base excision repair pathway is a parallel process of alkylator repair and preliminary studies suggest that TMZ in combination with PARP inhibitors (PARPi) may enhance tumor cell death in MGMT-silenced tumors (6–8). The increased sensitivity of tumors to TMZ with a PARPi is multifactorial and includes the presence of stalled replication forks typically overcome by homologous recombination (HR) DNA repair, and PARPi can delay the initiation of this HR-mediated recovery (7). Furthermore, “PARP trapping” onto DNA is important for PARPi + TMZ sensitivity, which points to olaparib as an ideal candidate to use in combination with TMZ (7–10).
These preclinical findings suggesting enhanced TMZ sensitivity with PARPi have led to several clinical trials in different tumor types. One example, a prior clinical trial of TMZ with veliparib in patients with advanced colorectal cancer reported a disease control rate (DCR) of 24% after two cycles (11). This study did not select patients with MGMT promoter hypermethylated tumors and used a PARPi with limited PARP trapping effect. The biological determinants for treatment sensitivity and resistance in this setting remain poorly explored. Here, we report the results of a single-arm, investigator-initiated, phase II clinical trial of TMZ in combination with olaparib in patients with MGMT promoter hypermethylated advanced colorectal cancer. We evaluated the clinical activity and explored biological determinants using genomic and protein-based biomarkers.
Materials and Methods
Study Design and Participants
This study was a single-arm, open-label, phase II clinical trial performed at the Yale Cancer Center. Eligible patients had histopathologic confirmation of stage IV microsatellite stable (MSS) colorectal cancer that progressed after 5-fluorouracil, oxaliplatin, irinotecan ,and appropriate biologic therapy. Promoter hypermethylation of MGMT was an integral biomarker for selection/enrollment and patients were prescreened using archival tumor tissue samples. The MGMT promoter hypermethylation was measured by methylation-specific PCR (MS-PCR) in the Clinical Laboratory Improvement Amendments (CLIA)-certified Yale Molecular Diagnostics Laboratory. As an integral biomarker for enrollment, MS-PCR was selected over other methods of MGMT testing such as pyrosequencing and methyl-BEAMing given the validation of MS-PCR for clinical trial samples in our institution. These alternative methods of MGMT testing are likely only semiquantitative given the rough estimates of tumor percentage. Furthermore, while they may offer advantages to predict response to TMZ monotherapy or TMZ/cytotoxic combinations (2, 12, 13) their predictive power is uncertain for TMZ + PARPi, which may work at different MGMT levels (14, 15). A full list of eligibility criteria is available as Supplementary Materials and Methods S1. All patients provided written informed consent as a condition of participation and the Yale University Institutional Review Board approved the study, which adheres to Good Clinical Practice Guidelines. The study was conducted in accordance with the Declaration of Helsinki and followed the Consolidated Standards of Reporting Trials. The ClinicalTrials.gov Identifier is NCT04166435.
Procedures
Patients received a starting dose of TMZ 75 mg/m2 days 1–7 with continuous olaparib 150 mg twice daily during 21-day cycles. The standard olaparib dose of 300 mg twice daily was not used to due to the enhanced myelosuppression with PARPi-chemotherapy combinations. The TMZ was selected on the basis of prior experience with TMZ and olaparib by Farago and colleagues and we aimed to use continuous PARP inhibition for continuous PARP trapping and thus a slightly lower dose of olaparib 150 mg twice daily was selected (16). Participants received TMZ and olaparib until progression of disease, unacceptable toxicity, death or withdrawal. Tumor RECIST measurements were performed at baseline and every 6 weeks on study. The study mandated a pretreatment tumor biopsy along with an optional progression biopsy. Blood and buffy coat were collected at screening, during treatment, and on-progression.
For whole-exome sequencing (WES), DNA from the pretreatment tissue samples and patient-matched normal DNA extracted from buffy coats were sequenced. The WES was performed using Illumina NovaSeq 6000 at the Yale Center for Genome Analysis for tumor/normal pairs. A multiplex quantitative immunofluorescence (QIF) panel was standardized for simultaneous and spatially resolved measurement of DAPI (all nuclei), cytokeratin (CK; epithelial or tumor cells), MGMT protein, γH2AX, and CD8+ tumor-infiltrating lymphocytes (TIL) in the whole tissue sections from tumor biopsies. Antibodies for MGMT, γH2AX, and CD8 were validated for specificity and reproducibility and multiplex QIF was performed as reported previously (17, 18). A control tissue microarray sample containing positive and negative control samples was used for assay validation and stained alongside the trial samples for reproducibility assessment. Imaging was acquired on a Vectra Polaris instrument. For isolation of peripheral blood mononuclear cells (PBMC), whole blood was subjugated to density gradient separation using Ficoll-Paque PLUS. Cells were frozen in liquid nitrogen until all samples from the time course had been collected.
Outcomes
The primary endpoint of the clinical trial was the objective response rate (ORR), defined as the proportion of patients with complete or partial response by RECIST. Secondary endpoints included progression-free survival (PFS), overall survival (OS), DCR, and safety and tolerability of the combination. Exploratory endpoints included MGMT protein expression by QIF, changes in peripheral immune cell populations, and changes in tumor mutational burden.
Statistical Analysis
A null hypothesis of a 5% ORR was used with alternative hypothesis ORR of ≥25%. An early stopping rule with a Bayesian analysis plan for the ORR was planned for after the first 9 patients with ≥1 response required to continue enrollment to a total sample size of 30 patients. Four responses in a full cohort of 30 patients were required to meet the primary endpoint. Survival functions were compared using Kaplan–Meier graphical analysis and the log-rank test. Comparisons between continuous QIF scores across groups were conducted using the Mann–Whitney test. Patient characteristics were compared using the Student t test for continuous variables and χ2 test for categorical variables. The statistical analysis and graphical representation was performed in Graphpad Prism v7.01 for windows (GraphPad Software, Inc). All two-tailed P values ≤0.05 were considered as statistically significant.
Data Availability Statement
The genomic data generated in this study are publicly available in Sequence Read Archive BioProject ID PRJNA956444. Non-genomic data will be made available based on reasonable request to the corresponding author.
Results
Between February 19, 2020 and June 22, 2021, 62 patients with metastatic MSS colorectal cancer were screened for MGMT promoter hypermethylation, of which 51 had adequate tumor tissue for clinical grade MGMT testing by bisulfite deamination and MS-PCR. We identified MGMT promoter hypermethylation in 18/51 (35%) of patient samples (Fig. 1). Nine patients received TMZ with olaparib due to the predefined stopping rule and their baseline characteristics are outlined in Table 1. No patients had a confirmed partial or complete response, 5/9 (56%) had a best response of stable disease (SD) and 4/9 (44%) had progressive disease, and the study closed early per the predefined stopping rule. For those patients with SD by RECIST, 3 patients had reductions in carcinoembryonic antigen (CEA) of ≥25% (Fig. 2A) and ≥10% tumor regression by RECIST (Fig. 2B). The median PFS was 3.0 months (95% CI: 2.1–not reached) and median OS was 9.4 months (95% CI: 6.7–not reached; Supplementary Fig. S1A and S1B). Treatment-related adverse events are summarized in Table 2. Five patients underwent dose modifications for treatment-related adverse events, 3 for grade 3 neutrophil count decrease, 1 for grade 3 platelet count decrease, and 1 for grade 3 mucositis. For analysis of correlative studies, the cases were divided in two clinical groups including patients who had clinical benefit as defined by CEA reduction, minor radiographic tumor regression by RECIST v1.1 (>10%) and prolonged SD by RECIST v1.1 (≥100 days), and those who did not experience clinical benefit (e.g., progressive disease). A study representativeness table is provided by Supplementary Table S1.
FIGURE 1.
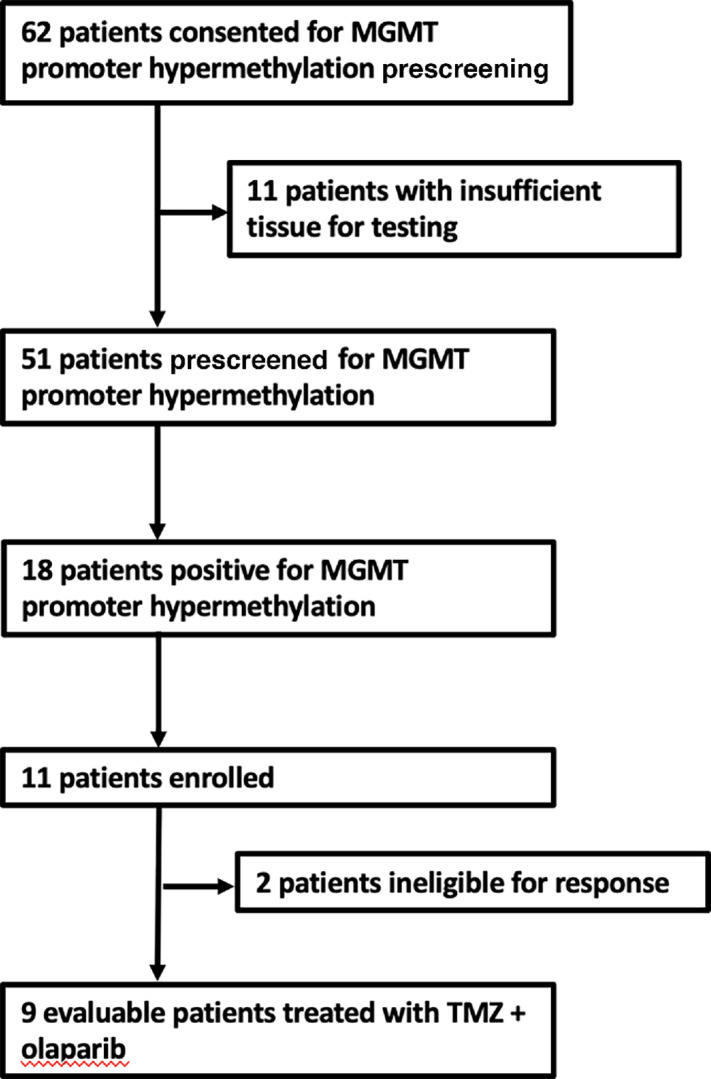
Trial profile. CONSORT diagram for enrolled participants.
TABLE 1.
Baseline characteristics
| Characteristic | TMZ + Olaparib (N = 9) |
|---|---|
| Age | |
| Median | 59 |
| Range | 45–78 |
| Sex – no. (%) | |
| Male | 4 (44) |
| Female | 5 (56) |
| Race – no. (%) | |
| White | 8 (89) |
| Black | 1 (11) |
| ECOG performance status – no. (%) | |
| 0 | 3 (33) |
| 1 | 6 (67) |
| Side of primary tumora – no. (%) | |
| Left | 4 (44) |
| Right | 5 (56) |
| Tumor grade – no. (%) | |
| Poorly differentiated | 3 (33) |
| Moderately differentiated | 6 (67) |
| Molecular results – no. (%) | |
| KRAS mutated | 7 (78) |
| KRAS/RAF wildtype | 2 (22) |
| Microsatellite stable | 9 (100) |
| Number of prior therapies – no. (%) | |
| 2 | 2 (22) |
| ≥3 | 7 (78) |
aRight side defined as tumors primary to splenic flexure.
FIGURE 2.

Biochemical and radiographic response. A, Spider plot of the percent change of CEA (ng/dL) from baseline for evaluable patients. Each color represents a patient. B, Waterfall plot for all evaluable patients, representing radiographic best response of tumor change from baseline according to RECIST v1.1. The * denotes patients that had progression of non-target lesions at the time of disease progression.
TABLE 2.
Frequency of treatment-related adverse events and laboratory abnormalities
| Event | Any grade | Grade ≥3 |
|---|---|---|
| Any event – no. (%) | 9 (100) | 7 (78) |
| Any serious event – no. (%)a | 0 | 0 |
| Most common events – no. (%) | ||
| Nausea | 3 (34) | 0 |
| Fatigue | 2 (22) | 0 |
| Anorexia | 2 (22) | 0 |
| Constipation | 1 (11) | 0 |
| Mucositis | 1 (11) | 1 (11) |
| Concentration impairment | ||
| Laboratory abnormalities – no. (%) | ||
| White blood cell count decreased | 6 (67) | 3 (34) |
| Neutrophil count decrease | 5 (56) | 4 (44) |
| Platelet count decreased | 5 (56) | 2 (22) |
| Anemia | 4 (44) | 0 |
| Lymphocyte count decrease | 3 (34) | 2 (22) |
| Aspartate aminotransferase increased | 2 (22) | 0 |
| Alkaline phosphatase increased | 1 (11) | 0 |
aOne serious adverse event was reported for one patient nausea/vomiting that was related to underlying disease.
Whole-exome DNA sequencing was performed on tumor and matched non-tumor samples from all patients who received study treatments. The mean coverage was 486 reads and the mean number of nonsynonymous mutations across cases was 527 (range, 279–527). Molecular analysis revealed 8/9 (89%) had tumor-specific deleterious variants in genes of the MAP kinase pathway (7 KRAS and 1 ERBB2), and 8/9 (89%) had TP53 variants (Supplementary Fig. S2). The mean tumor mutational burden was 15 mutations/megabase (range, 6–41) as outlined in Supplementary Fig. S2A. One patient without any clinical benefit had a BRCA2 mutation, and no HR-related mutations were identified in any of the patients who experienced clinical benefit. Given the limited sample size no association was noted between patient outcomes and genomic alterations.
To obtain a quantitative and spatially resolved assessment of MGMT protein expression, DNA damage response (DDR), and TILs in pretreatment and posttreatment tumors, we studied the biopsy samples with a 5-colored multiplexed QIF panel containing the markers DAPI for all cells, cytokeratin for tumor cells, MGMT, γH2AX as a marker of DDR, and CD8 for cytotoxic T cells (representative example: Fig. 3A). The marker scores were selectively measured in tumor and non-tumor stromal cells based on their colocalization with the epithelial/tumor cell marker cytokeratin as reported previously (18). Despite showing MGMT promoter hypermethylation by MS-PCR, MGMT protein was detected in cytokeratin-positive tumor cells from six of the nine baseline biopsy samples. Repeat bisulfite deamination and MS-PCR of the MGMT promoter was conducted as confirmation on all pretreatment biopsies and all 9 patients remained MGMT promoter-hypermethylated at enrollment.
FIGURE 3.

Low tumor-specific MGMT protein expression and high CD8+ T-cell infiltration are seen in the pretreatment biopsies of patients who derived clinical benefit from the trial and trial therapy alters the PBMC population. A, Representative multicolor images of a pretreatment biopsy sample with tumor cell–selective MGMT downregulation and paired posttreatment biopsy with increased tumor-specific MGMT expression. Epithelial and tumor cells are highlighted by CK (green channel). MGMT-positive cells are shown in the red channel, γH2AX-positive cells in the yellow channel, and CD8-positive cells in the white channel. Bar = 100 μm. Mean levels of MGMT (B), γH2AX (C), and CD8 (D) in the pretreatment biopsies of patients who derived clinical benefit on trial (white bars) and who did not show any clinical benefit (gray bars). Differences between groups were compared using the nonparametric Mann–Whitney test. The P values obtained for marker comparisons are indicated within each chart. E–I, The change from baseline mean for each cell population was plotted for two timepoints (i) on-treatment (cycle 2) and (ii) progression by RECIST. The maximal positive/negative mean % change values are plotted for five different panels; (E) Basic Immunophenotype panel; (F) Memory-Naïve and Activated T Cell panel; (G) T Cell Proliferation panel; (H) Monocytes/DC/NK Cell panel; and (I) T Cell Exhaustion panel.
Despite the limited number of cases, the tumor cell–specific MGMT protein expression in the pretreatment biopsies of the 3 patients who derived clinical benefit from study treatment was significantly lower compared with the pretreatment biopsies of the 6 patients who did not experience disease stabilization on study (Fig. 3B). The cases with clinical activity showed also numerically higher levels of γH2AX in tumor cells and significantly higher CD8+ TILs (Fig. 3C and D). Two patients with clinical benefit underwent biopsy at the time of disease progression with 1 patient biopsied at the same site for both pretreatment and progression and another patient with a pretreatment lymph node biopsy and progression biopsy of peritoneal metastasis. Both patients remained mismatch repair proficient on progression biopsy. The progression biopsies showed marked increased in tumoral MGMT protein levels relative to the pretreatment biopsy and a trend toward decreased γH2AX and CD8+ T-cell tumor infiltration (Supplementary Fig. S3). Together, these results suggest that MGMT protein can be expressed in a substantial fraction of MGMT promoter-methylated colorectal cancer, and low MGMT expression in tumor cells and increased local effector TILs were associated with benefit from TMZ and olaparib. Conversely, elevated tumoral-cell MGMT protein expression was associated with lack of any clinical benefit from the treatment regimen, supporting the value of this metric as a biomarker.
Peripheral blood was collected to assess the pharmacodynamic effects of the study treatment on PBMC. For each PBMC population (Fig. 3E–I) the baseline mean for all patients was calculated and the percent change from baseline mean was measured on cycle 2 day 1 (“On Treatment”) and at the time of progression for all patients. Most prominently, an expansion and proliferation of recently activated CD8+ T cells was seen with treatment, as well as increases in both naïve and memory T cells (Fig. 3E–G). There was also an expansion of tumor-associated natural killer (NK) cells (CD56bright CD16−), and an increase in multiple T-cell exhaustion markers (Fig. 3H and I). A classification of the immune cell subtype from the flow cytometry results is available in Supplementary Table S2.
Discussion
Our study represents the first clinical evaluation of TMZ plus a PARPi in MGMT promoter hypermethylated colorectal cancer. Although, no responses by RECIST v1.1 were observed, correlative studies suggest potential for further evaluation of this therapy by refining the biomarker selection strategy. Moreover, this is the first prospective assessment of the MGMT protein levels, γH2AX and TILs using spatially resolved quantitative analysis in this setting. Here we found prominent discordance between the MGMT promoter hypermethylation status and MGMT protein expression in colorectal cancers and the associated clinical outcomes. Furthermore, in patients with clinical benefit, increased MGMT protein expression at progression was noted, representing a potential resistance mechanism to study treatment. Our data suggest that localized MGMT protein measurements could be a superior biomarker than MGMT methylation by MS-PCR in this population and tumors with low MGMT protein and high CD8+ TILs derived increased clinical benefit from TMZ plus olaparib combination therapy. Flow cytometry of PBMCs also revealed an expansion in activated CD8+ T cells after treatment with TMZ plus olaparib (Supplementary Fig. S3). Our findings of an increase in CD8+ TILs and PBMCs may support the potential investigation of alkylator-immunotherapy combinations in MGMT silenced tumors.
The antitumoral activity for approved agents in the third line setting for colorectal cancer is extremely limited with response rates of approximately 1% with regorafenib and TAS-102 (19, 20). Furthermore, our patient population was heavily pretreated with 7/9 (78%) of patients receiving at least three prior therapies, likely making it more difficult to achieve a radiographic response. Thus, while we did not observe an objective response, the reductions in target lesions described in Fig. 2 are noteworthy and consistent with treatment activity in a subset of patients. It is also possible that the lower dose of olaparib may have limited the efficacy, but 300 mg twice daily olaparib is intolerable in a chemotherapy combination.
Prior to our clinical trial evaluating TMZ and olaparib in colorectal cancer, a previous clinical evaluated TMZ with veliparib, a PARPi (11). However, veliparib does not “trap” PARP like other PARPis, such as olaparib, and is presumed to be inferior to PARPis with the ability to PARP-“trap” when used in combination with TMZ (7–10). Moreover, this study did not restrict enrollment to MGMT promoter hypermethylated colorectal cancer as our study did, nor did it analyze MGMT expression and TILs. Recently, data from Morano and colleagues in the MAYA trial described the combination of TMZ and low-dose ipilimumab/nivolumab in patients with MSS colorectal cancer, indicating a very promising ORR and survival with the combination (21). These observations were further supported by the ARETHUSA trial, which treated patients with colorectal cancer with “TMZ priming” followed by pembrolizumab and revealed acquired MSH6 variants, a genomic signature for TMZ, and increased tumor mutational burden after the “TMZ priming” (22). While preliminary, the MAYA and ARETHUSA trials suggests immunogenicity with TMZ treatment, and support further novel strategies to use alkylators together with immunostimulatory therapies. However, in glioma, TMZ-induced hypermutation has not translated into enhanced sensitivity to immune checkpoint inhibitors, which highlights the need for additional biomarkers (23). The results from our clinical trial also support the concept that tumors with lower levels of MGMT are more immunogenic given our observations of higher CD8+ T-cell levels in MGMT low tumors and that TMZ may enhance the antitumor immune response by a pharmacodynamic effect of increased immunostimulatory PBMCs. These observations are novel and have not been described in previous TMZ single monotherapy or combination strategies, and the role that the addition of olaparib may play in the pharmacodynamic PBMCs effects will require further study. Additional exploratory descriptive analysis of the PBMC changes for patients with clinical benefit versus no benefit is outlined in Supplementary Fig. S4, which are limited by the small sample size, but do suggest an increase in multiple PBMC populations for the clinical benefit group.
Our genomic analysis revealed APC to be among the most commonly mutated genes, which is consistent with the typical molecular profile of colorectal cancer. In contrast, previous observations for MGMT promoter hypermethylation in colorectal cancer were that these tumors are more likely to arise from serrated adenomas and serrated adenocarcinomas, which are less frequently APC-mutated (24, 25). Thus, MGMT promoter hypermethylated colorectal cancer may be heterogenous in development compared with the conventional development of adenomatous polyps. The elevated tumor mutational burden of 15 mutations/megabase, may suggest these tumors have increased levels of tumor neoantigens and therefore more immunogenic, which could be due to impaired DNA repair in the setting of MGMT promoter hypermethylation. The high prevalence of MAP kinase mutations (KRAS and ERBB2) is important should future biologic therapy be added to TMZ combinations, and support the use of bevacizumab over cetuximab/panitumumab. Moreover, tumor hypoxia induced by bevacizumab may sensitize tumors to PARPis by creating homologous recombination deficiency (26, 27).
For metastatic colorectal cancer, the successful use of alkylator combinations in the future will require a careful selection of biomarkers, which should include a quantitative measure of MGMT protein expression, as supported by our findings. Comparable findings are described by other groups showing that MGMT promoter hypermethylation is necessary but not sufficient for TMZ sensitivity for colorectal cancer (21). We confirm this observation by showing a lack of correlation between these two markers in the current study and in our related presented work (28). Thus, a future direction of TMZ and PARPi combination could be in patients with specifically low tumoral MGMT protein levels.
The major limitation of our study is the small sample size of patients that received TMZ and olaparib. While we did identify MGMT promoter hypermethylation in 18/51 (35%) prescreened patients, only 9 received the study treatment due to the predefined early stopping rule. Furthermore, we assessed MGMT testing by MS-PCR which is our CLIA lab's validated MGMT test, but it is possible that alternative MGMT promoter hypermethylation testing such as methyl-BEAMing or pyrosequencing would have better enriched our cohort for response (2, 12, 13). Our correlative analysis is also limited by a lack of on-treatment or progression biopsies to study changes in the tumor microenvironment after treatment with TMZ and olaparib.
In conclusion, TMZ and olaparib was tolerable and did reveal antitumor activity in a subset of patients with MGMT promoter hypermethylated tumors that also had low MGMT protein expression and increased CD8+ effector TILs. However, given the lack of response in the overall treatment group, this did not justify continuing enrollment beyond 9 patients. Nevertheless, it was noteworthy that MGMT promoter hypermethylation was seen in 35% of prescreened patients, and treated patients had higher than expected MAPK signaling alterations. Furthermore, the treatment of TMZ plus olaparib has a noticeable impact on activated CD8+ T cells in patients with advanced colorectal cancer, and therefore novel alkylator-immunotherapy combinations may be warranted.
Supplementary Material
Supplementary Figure 1: Survival Analysis.
{kind=link}
Supplementary Figure 2: Molecular Analysis
{kind=link}
Supplementary Figure 3: Changes in MGMT expression, yH2AX and CD8+ T cells in pre-treatment and post-treatment biopsies of patients who derived clinical benefit from the trial.
{kind=link}
Supplementary Figure 4: The change in PBMCs for patients with clinical benefit (blue) and no benefit (red).
{kind=link}
Supplementary Table 1: Study representativeness table.
{kind=link}
Supplementary Table 2: A classification of the immune cell subtypes.
{kind=link}
Supplementary Methods 1: Clinical trial protocol.
Acknowledgments
M. Cecchini receives support from an NCI Career Development Award (1K08CA255465-01A1). The authors would like to acknowledge all trial participants, and AstraZeneca for providing olaparib and funding the clinical trial. The authors would also like to thank Dr. Charles Fuchs for his helpful suggestions and Ashraf Khalil from the Yale University Flow Cytometry Core Facility for technical assistance.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Communications Online (https://aacrjournals.org/cancerrescommun/).
Authors’ Disclosures
M. Cecchini reports grants from AstraZeneca during the conduct of the study; personal fees from Bayer Pharmaceuticals, DAVA Oncology, Taiho, Seattle Genetics, Macrogenics, Daiichi Sankyo and other from Parthenon Therapeutics outside the submitted work. J. Sklar reports grants from NCI during the conduct of the study. S.M. Stein reports personal fees from Genentech, Merck, Abbvie, AstraZeneca, Coherus, and Imvax outside the submitted work. P. LoRusso reports personal fees from AbbVie, Agios, Five Prime, GenMab, Halozyme, Genentech, CytomX, Takeda, SOTIO, Cybrexa, Agenus, Tyme, IQVIA, TRIGR, Pfizer, ImmunoMet, Black Diamond, Glaxo-Smith Kline, QED Therapeutics, AstraZenenca, EMD Serono, Shattuck, Astellas, Salarius, Silverback, MacroGenics, Kyowa Kirin, Kineta, Zentalis, Molecular Templates, ABL Bio, SK Life Science, STCube, Bayer, I-Mab, Seagen, imCheck, Relay Therapeutics, Stemline, Compass BADX, Mekanist, Mersana, BAKX Therapeutics, Scenic Biotech, Qualigen, Roivant, NeuroTrials, and Actuate Therapeutics; other from Roche-Genentech outside the submitted work. K.A. Schalper reports personal fees from Clinica Alemana Santiago, Shattuck Labs, AstraZeneca, Takeda, Agenus, Genmab, Parthenon Therapeutics, Bristol-Myers Squibb, Sensei, Roche, Molecular Templates, Janssen, CDR life, Merck, PeerView, PER and Forefront collaborative; grants from Tesaro/GSK, Takeda, Surface Oncology, Merck, Bristol-Myers Squibb, AstraZeneca, Ribon Therapeutics, Eli Lilly, Boehringer-Ingelheim, Roche, Akoya Biosciences outside the submitted work. No disclosures were reported by the other authors.
Authors’ Contributions
M. Cecchini: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, methodology, writing-original draft. J.Y. Zhang: Conceptualization, data curation, software, formal analysis, writing-review and editing. W. Wei: Formal analysis, writing-review and editing. J. Sklar: Software, formal analysis, supervision, writing-review and editing. J. Lacy: Investigation, writing-review and editing. M. Zhong: Investigation, writing-review and editing. Y. Kong: Investigation, writing-review and editing. H. Zhao: Investigation, writing-review and editing. J. DiPalermo: Investigation, writing-review and editing. L. Devine: Investigation, writing-review and editing. S.M. Stein: Investigation, writing-review and editing. J. Kortmansky: Investigation, writing-review and editing. K.L. Johung: Writing-review and editing. R.S. Bindra: Conceptualization, writing-review and editing. P. LoRusso: Conceptualization, writing-review and editing. K.A. Schalper: Conceptualization, resources, data curation, formal analysis, supervision, validation, investigation, methodology, writing-original draft, writing-review and editing.
References
- 1. Thomas A, Tanaka M, Trepel J, Reinhold WC, Rajapakse VN, Pommier Y. Temozolomide in the era of precision medicine. Cancer Res 2017;77:823–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morano F, Corallo S, Niger M, Barault L, Milione M, Berenato R, et al. Temozolomide and irinotecan (TEMIRI regimen) as salvage treatment of irinotecan-sensitive advanced colorectal cancer patients bearing MGMT methylation. Ann Oncol 2018;29:1800–6. [DOI] [PubMed] [Google Scholar]
- 3. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003. [DOI] [PubMed] [Google Scholar]
- 4. Brandes AA, Tosoni A, Cavallo G, Reni M, Franceschi E, Bonaldi L, et al. Correlations between O6-methylguanine DNA methyltransferase promoter methylation status, 1p and 19q deletions, and response to temozolomide in anaplastic and recurrent oligodendroglioma: a prospective GICNO study. J Clin Oncol 2006;24:4746–53. [DOI] [PubMed] [Google Scholar]
- 5. Chinot OL, Barrié M, Fuentes S, Eudes N, Lancelot S, Metellus P, et al. Correlation between O6-methylguanine-DNA methyltransferase and survival in inoperable newly diagnosed glioblastoma patients treated with neoadjuvant temozolomide. J Clin Oncol 2007;25:1470–5. [DOI] [PubMed] [Google Scholar]
- 6. Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, et al. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res 2000;6:2860–7. [PubMed] [Google Scholar]
- 7. Murai J, Huang SYN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012;72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for Poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on parp trapping versus catalytic inhibition. J Pharmacol Exp Ther 2014;349:408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pommier Y, O'Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps17. [DOI] [PubMed] [Google Scholar]
- 10. Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, et al. PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res 2019;17:409–19. [DOI] [PubMed] [Google Scholar]
- 11. Pishvaian MJ, Slack RS, Jiang W, He AR, Hwang JJ, Hankin A, et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018;124:2337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sartore-Bianchi A, Pietrantonio F, Amatu A, Milione M, Cassingena A, Ghezzi S, et al. Digital PCR assessment of MGMT promoter methylation coupled with reduced protein expression optimises prediction of response to alkylating agents in metastatic colorectal cancer patients. Eur J Cancer 2017;71:43–50. [DOI] [PubMed] [Google Scholar]
- 13. Barault L, Amatu A, Bleeker FE, Moutinho C, Falcomatà C, Fiano V, et al. Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann Oncol 2015;26:1994–9. [DOI] [PubMed] [Google Scholar]
- 14. Hwang K, Lee JH, Kim SH, Go KO, Ji SY, Han JH, et al. The combination PARP inhibitor olaparib with temozolomide in an experimental glioblastoma model. In Vivo 2021;35:2015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yuan AL, Ricks CB, Bohm AK, Lun X, Maxwell L, Safdar S, et al. ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PLoS One 2018;13:e0202860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Farago AF, Yeap BY, Stanzione M, Hung YP, Heist RS, Marcoux JP, et al. Combination olaparib and temozolomide in relapsed small-cell lung cancer. Cancer Discov 2019;9:1372–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res 2019;25:4663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schalper KA, Brown J, Carvajal-Hausdorf D, McLaughlin J, Velcheti V, Syrigos KN, et al. Objective measurement and clinical significance of TILs in non–small cell lung cancer. J Natl Cancer Inst 2015;107:dju435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, et al. Randomized Trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–19. [DOI] [PubMed] [Google Scholar]
- 20. Grothey A, Cutsem EV, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:303–12. [DOI] [PubMed] [Google Scholar]
- 21. Morano F, Raimondi A, Pagani F, Lonardi S, Salvatore L, Cremolini C, et al. Temozolomide followed by combination with low-dose ipilimumab and nivolumab in patients with microsatellite-stable, O6-methylguanine–DNA methyltransferase–silenced metastatic colorectal cancer: the MAYA trial. J Clin Oncol 2022;40:1562–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crisafulli G, Sartore-Bianchi A, Lazzari L, Pietrantonio F, Amatu A, Macagno M, et al. Temozolomide treatment alters mismatch repair and boosts mutational burden in tumor and blood of colorectal cancer patients. Cancer Discov 2022;12:1656–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020;580:517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dong SM, Lee EJ, Jeon ES, Park CK, Kim K-M. Progressive methylation during the serrated neoplasia pathway of the colorectum. Mod Pathol 2005;18:170–8. [DOI] [PubMed] [Google Scholar]
- 25. Jass J, Baker K, Zlobec I, Higuchi T, Barker M, Buchanan D, et al. Advanced colorectal polyps with the molecular and morphological features of serrated polyps and adenomas: concept of a ‘fusion’ pathway to colorectal cancer. Histopathology 2006;49:121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bindra RS, Schaffer PJ, Meng A, Woo J, Måseide K, Roth ME, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol 2004;24:8504–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci U S A 2010;107:2201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang JY, Cecchini M, Desai SS, Schalper KA. Clinical significance and biomarker potential of MGMT protein measurement in colorectal cancer. J Clin Oncol 40: 4s, 2022. (suppl; abstr 136). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Survival Analysis.
Supplementary Figure 2: Molecular Analysis
Supplementary Figure 3: Changes in MGMT expression, yH2AX and CD8+ T cells in pre-treatment and post-treatment biopsies of patients who derived clinical benefit from the trial.
Supplementary Figure 4: The change in PBMCs for patients with clinical benefit (blue) and no benefit (red).
Supplementary Table 1: Study representativeness table.
Supplementary Table 2: A classification of the immune cell subtypes.
Supplementary Methods 1: Clinical trial protocol.
Data Availability Statement
The genomic data generated in this study are publicly available in Sequence Read Archive BioProject ID PRJNA956444. Non-genomic data will be made available based on reasonable request to the corresponding author.