

Abstract
Increased intracellular generation of reactive oxygen species [ROS] has been implicated in the pathology of metabolic [diabetes] and neurodegenerative [Alzheimer’s] diseases. Accumulating evidence suggests NADPH oxidases [Noxs] as the principal source for cellular ROS in humans. Of this class of enzymes, the phagocyte-like Nox [Nox2] has come under intense scrutiny as one of the “culprits” for the induction of cellular damage culminating in the onset of diabetes and its complications. Functional regulation of Nox2 is fairly complex due to its membranous [gp91phox, p22phox] and cytosolic [p40phox, p47phox, p67phox and Rac1] cores, which require specific post-translational modification steps [phosphorylation and lipidation] for their membrane association. Therefore, optimal efficacy of Nox2 depends upon precise regulation of these signaling steps followed by translocation of the cytosolic components to the membrane. Interestingly, numerous recent studies have reported sustained activation of Nox2, ROS-derived oxidative stress, and cellular dysfunction in in vitro and in vivo models of glucolipotoxicity and diabetes. These investigations employed a variety of cell-permeable peptides and pharmacological inhibitors to impede Nox2 holoenzyme assembly and activation in pancreatic islet β-cells, cardiomyocytes and retinal endothelial cells under conditions of glucolipotoxicity and diabetes. Herein, we highlight the existing evidence to implicate Nox2 as the “trigger” of cellular damage, and identify critical gaps in our current understanding that need to be addressed to further affirm the roles of Nox2 as a potential therapeutic target for the treatment of diabetes and other metabolic disorders.
Keywords: Nox2, Rac1, Diabetes, Complications, Cell death
1. Introduction
Diabetes is a serious medical condition resulting from failure of insulin action and/or insufficient insulin secretion from the pancreatic islet β-cells. Indeed, the rate of incidence of diabetes is reaching epidemic proportions. According to recent estimates by the International Diabetes Federation [IDF], the incidence of diabetes soured to an all-time high of 382 million in 2013 compared to 371 million in 2012. The IDF predicts that by 2035 the number of individuals afflicted with this disease will increase to 592 million. Recent estimates indicate that diabetes already accounts for annual healthcare spending of $548 billion, and this is likely to rise to $627 billion in 2035. In addition, alarmingly high number of individuals [~175 million] are as yet undiagnosed, presumedly a large proportion of these people are progressing toward developing this disease [1]. Therefore, efforts to understand the pathophysiology of diabetes and its complications are highly relevant to future developments in care and therapeutics of this disease.
It is well established in both in vitro and in vivo models that chronic exposure of cells to elevated glucose [glucotoxicity], fatty acids [lipotoxicity] or both [glucolipotoxicity] leads to impairment of cellular dysfunction and demise [2,3]. Several lines of evidence have implicated mitochondrial dysregulation as one of the hallmarks of cell death under the duress of chronic exposure to high glucose, lipids and diabetes [2–6]. Mitochondrial damage leads to release of proapoptotic factors [cytochrome C] into the soluble compartment [cytosol] culminating in the activation of a variety of caspases [caspsase-3, caspase-6 and other caspases] and degradation and inactivation of proteins [nuclear lamins A and B], which are requisite for optimal cell function [6]. A growing body of evidence in multiple cell types implicates increased intracellular [cytosolic as well as mitochondrial] reactive oxygen species [ROS] and the associated oxidative stress as causal factors in the sequence of events leading to cell dysfunction under the duress of hyperglycemia, hyperlipidemia and diabetes [2–6].
2. The phagocyte-like NADPH oxidase
NADPH oxidases represent the principal source of cellular ROS. Of this class of enzymes, the Nox2 holoenzyme [also referred to as the respiratory burst] has been implicated in phagocytosis by professional phagocytic cells including neutrophils, eosinophils, monocytes and macrophages. The Nox2 is a highly regulated membrane-associated protein complex that catalyzes the one-electron reduction of oxygen to superoxide anion involving oxidation of cytosolic NADPH, thereby generating large quantities of intracellular ROS, which, in turn, promote killing of phagocytized microorganisms, including fungi and a variety of bacteria [7]. It has been shown that defective activation of Nox2 leads to the pathology of disease states such as the chronic granulomatous disease in humans [7 and references therein]. Several plausible mechanisms have been put-forth for the generation of ROS and associated oxidative stress in a variety of non-phagocytic cell types, including the islet β-cell [2–5]. Considerable research has been conducted in numerous laboratories on the functional regulation of Nox2 activity in non-phagocytes [8–10 for recent reviews]. Briefly, the Nox2 is a multicomponent system comprised of membrane-associated as well as cytosolic components. The membrane-associated catalytic core is comprised of gp91phox, p22phox and the flavochrome b558. The cytosolic regulatory components include p40phox, p47phox, p67phox and the small G-protein Rac [9]. Under stimulatory conditions, the cytosolic components translocate to the membrane fraction for association with the catalytic core for Nox2 holoenzyme assembly and catalytic activation. Several mechanisms have been described for the translocation of the cytosolic core of Nox2 to the membrane for association with the membranous core. For example, phosphorylation of p40phox, p47phox and p67phox is considered to be an important signaling step [9]. In addition, Rac1 activation [GTP-bound conformation] leads to its association with p67phox triggering the translocation of Rac1-p67phox dimer to the membrane. Recent evidence suggests novel regulatory roles for specific guanine nucleotide exchange factors [GEFs] for Rac1 [Tiam1] in attaining its GTP-bound active configuration [11]. In the following sections, we will overview the existing body evidence that implicates Nox2-derived ROS generation and oxidative stress as pivotal for cellular dysfunction in in vitro and in vivo models of metabolic stress, diabetes and its associated complications. We will then highlight recent developments in the area of design and development of small molecule inhibitors for Nox2, and potential utility of those inhibitors in the validation of Nox2 as a therapeutic target for the prevention and treatment of diabetes and diabetic complications.
3. Roles for Nox2 in the onset of cellular [dys]function in glucolipotoxic and diabetic conditions
3.1. Studies in normal pancreatic β-cell and in models of impaired insulin secretion
Recent experimental evidence from several laboratories including our own has provided novel clues with regards to the regulatory roles of Nox2 not only in physiological insulin secretion, but also in the dysfunction of the islet β-cell under conditions of severe metabolic stress [glucolipotoxicity, exposure to ceramide (Cer) and proinflammatory cytokines]. We have recently demonstrated that glucose or a mixture of mitochondrial fuels [monomethyl succinate and α-ketoisocaproic acid] significantly increased ROS levels in INS-1 832/13 cells, which was markedly reduced by known inhibitors of Nox2 [apocynin and diphenyleneiodonium; DPI] and siRNA-p47phox [12,13]. Furthermore, specific inhibitors of protein prenylation [FTI-277 or GGTI-2147] significantly reduced nutrient-induced ROS generation implicating novel roles of protein prenylation in this signaling cascade [12]. In addition, mycophenolic acid, which selectively depletes endogenous GTP pools, significantly reduced nutrient-induced ROS generation and Rac1 activation [12]. Together, these observations have led us to suggest requisite roles for small G-proteins and their prenylation in nutrient-mediated Nox2 activation following short-term [acute] exposure conditions. These findings further suggested that a tonic increase in Nox2-derived ROS may be necessary for insulin secretion induced by metabolic fuels [12,13].
In addition to its roles in physiologic insulin secretion, Nox2 has been shown to play key functional roles in the damage and demise of the islet β-cell under the conditions of metabolic stress and diabetes. For example, Syed and associates have reported marked increase in Rac1 activation, generation of superoxides and lipid peroxides in INS-1 832/13 cells following exposure to palmitate (PA; [14]). Potential involvement of Nox2 in this signaling cascade was confirmed via the use of DPI, a selective inhibitor of Nox2. Fumonisin B1, a known inhibitor of de novo synthesis of Cer, significantly inhibited PA effects, implicating roles for Cer in PA-induced effects. A requisite role for Rac1 [a member of Nox2 holoenzyme] activation in PA- and Cer-mediated effects was established by employing NSC23766, a specific inhibitor of Rac1 activation (see below; [14,15]). Together, these observations led to the hypothesis that metabolic dysfunction of the islet β-cell induced by PA/Cer requires the intermediacy of Rac1-Nox2 signaling pathway. Subsequent studies along these lines by Subasinghe and associates [16] have demonstrated contributory roles for Nox2 in cytokine-induced metabolic dysfunction of the islet β-cell. Findings from these studies showed a significant reduction in proinflammatory cytokine [IL-1β, TNFα and IFNγ]-induced Rac1 activation, Nox2-mediated ROS generation by inhibitors of Rac1 prenylation [GGTI-2147] and activation [NSC23766]. More importantly, cytokine-induced loss in mitochondrial membrane potential was also attenuated by NSC23766 > GGTI-2147 [16]. Together, these in vitro observations highlighted regulatory roles of Nox2 in the onset of metabolic dysfunction induced by PA, Cer and cytokines.
In further support of data from in vitro model systems described in the above section, recent studies have also suggested key regulatory roles for Nox2 in metabolic dysfunction of the islet in models of impaired insulin secretion and diabetes. For example, Yuan and associates noted high levels ROS generation and decreased insulin content in islets from Sprague-Dawley rats following a 24-week high fat feeding [17]. They also reported increased ROS generation and reduction in insulin content following exposure of insulin-secreting NIT-1 cells to high glucose concentrations in vitro. More importantly, high glucose-induced loss in insulin expression and impaired insulin secretion were prevented by transfecting these cells with siRNA-Nox2. These data further validate the hypothesis that Nox2 plays damaging roles in the induction of metabolic dysfunction in models of glucotoxicity [17]. In further support of this model, we recently reported significant increase in the subunit expression and activation of Nox2 in islets derived from the Zucker Diabetic Fatty [ZDF] rat, a model for type 2 diabetes [T2DM] and also in islets from T2DM human donors [18]. In addition, significantly high levels of active [GTP-bound] Rac1 were also seen in the ZDF islets compared to those from their lean counterparts. We were able to demonstrate marked increase in the stress kinase [JNK1/2] activation in INS-1 832/13 cells exposed to hyperglycemic conditions. Lastly, in a manner akin to the ZDF diabetic rat islets, Rac1 expression, JNK1/2, and caspase-3 activation were also elevated in diabetic human islets [18]. In summary, observations in pancreatic β-cells suggest increased activation of Rac1, Nox-2 mediated ROS generation, stress kinase activation and mitochondrial dysfunction leading to caspase activation in in vitro and in vivo models of metabolic stress and diabetes.
3.2. Studies in models of diabetic cardiomyopathy
Potential involvement of Nox2 in cellular dysfunction under hyperglycemic conditions and diabetes has been demonstrated in other target cells as well. For example, Shen and associates reported [19] a significant upregulation of Rac1 and Nox2 activation, ROS generation, and cellular apoptosis in the heart from the streptozotocin [Stz]-diabetic mice. Furthermore, Rac1 deficiency [Rac1-knockout model] or apocynin treatment markedly attenuated high glucose-induced ROS generation in the heart. In further support of these findings, these authors also demonstrated activation of Rac1 and Nox2 in myocytes cultured in the presence of high glucose in vitro; such effects were nullified following overexpression of inactive mutant of Rac1, knockdown of gp91phox or p47phox. Lastly, administration of NSC23766, a Tiam1-Rac1 inhibitor, to the diabetic db/db mice significantly reduced Nox2 activity, cell apoptosis resulting in partial restoration of myocardial function [19]. Additional studies from this group have conclusively demonstrated inhibition of Stz-diabetes-induced Nox2 activation, endoplasmic reticulum stress [ER stress] and myocardial remodeling in Rac1-knockout mouse model further supporting their original observations and conclusions [20]. More recent investigations by Yu et al. [21] have demonstrated contributory roles for oxidative stress [Rac1-Nox2 cascade] and inflammation in Stz-induced diabetic cardiomyopathy. Interestingly, curcumin, a known antioxidant, markedly inhibited these signal transduction pathways, and alleviated cardiomyopathy in these animals. Tawfik and coworkers have reported, among other abnormalities, a significant increases in ROS and superoxide formation in rat coronary endothelial cells exposed to hyperglycemic conditions in vitro [22]. It is noteworthy that simvastatin, an inhibitor of cholesterol biosynthesis, or apocynin treatment [Nox2 inhibition] significantly reduced glucose-induced ROS generation and superoxide formation implicating novel roles for Nox2 in the onset of these lesions [22]. Even though it remains to be verified experimentally, it is likely that the effects of simvastatin might involve inhibition of prenylation and activation of Rac1, the associated inhibition of Nox2 holoenzyme since statins also inhibit biosynthesis of prenyl pyrophosphates, which are substrates for prenyltransferases [23]. Together, these findings underscore the roles of Tiam1-Rac1-Nox2 in diabetes-induced dysfunction and apoptosis of cardiac myocytes culminating in the onset of cardiac complications associated with this disease.
3.3. Studies in models of diabetic retinopathy
Emerging evidence from several laboratories, including our own, affords further support to implicate Nox2 signaling pathway in the induction of hyperglycemia-mediated retinal endothelial dysfunction and the development of retinopathy. Studies by Al-Shabrawey and associates demonstrated novel regulatory roles for Nox2 in retinal vascular inflammation in animal models of Stz-induced diabetes [24]. They reported that apocynin treatment or genetic deletion of Nox2 prevented diabetes-induced ICAM-1 expression, leukostasis and blood-retinal barrier breakdown thus implicating critical roles for Nox2 in early signaling steps leading to the development of diabetic retinopathy [24]. More recent investigations by these researchers have suggested novel roles for 12/15 lipoxygenase signaling cascade in Nox2-mediated alterations in vascular hyperpermeability in diabetic retinopathy. They provided compelling evidence to indicate that baicalein, a known inhibitor of 12/15 lipoxygenase, reduced the levels of ICAM-1, 12- and 15-hydroxyeicosatetreanoic acids, ROS generation and Nox2 expression in the diabetic retina further implicating key roles for Nox2 in the onset of vascular hyperpermeability during diabetic retinopathy [25]. He and associates reported involvement of Nox2 in increased expression of retinal heme oxygenase-1 in the diabetic retina [26]. They observed significant increases in Nox2 activation in the retina from 8-, 12-, and 20-week db/db mice. More importantly, Nox2 inhibitors [apocynin and DPI] attenuated high glucose-induced expression of heme oxygenase in retinal explants, thus affirming roles for retinal Nox2 in the expression on this enzyme in the diabetic retinopathy. More recently, using pharmacological and gene knock-out approaches, Du and associates suggested significant regulatory roles for Nox2 and mitochondria for superoxide generation in the diabetic mouse retina [27].
Recent observations from our laboratory have provided a more direct evidence for a regulatory role for Nox2 in the onset of hyperglycemia-induced mitochondrial dysfunction and demise of retinal capillary endothelial cells [28]. Using in vitro and in vivo (retina from the Stz-diabetic mice) models of diabetic retinopathy, we observed that hyperglycemic milieu promotes activation and translocation of cytosolic components of Nox2 [p47phox and Rac1] to the membrane. We also noted a significant increase in Nox2 activity and ROS generation under those conditions. Our findings further suggested that Rac1 activation is mediated by Tiam1, a GEF for Rac1, since NSC23766, a specific inhibitor of Tiam1-Rac1 signaling, markedly attenuated Nox2 activation and ROS generation. NSC23766 also prevented glucose-induced mitochondrial dysfunction and cellular apoptosis. Based on our in vitro and in vivo data, we hypothesized that activation of cytosolic Nox2 triggers mitochondrial dysfunction and retinal dysfunction in diabetes [28].
3.4. Studies in models of insulin resistance
Recent studies by Sukumar and coworkers have probed into potential regulatory roles of Nox2 in models of human insulin resistance [29]. They observed significantly high levels of superoxides in insulin-resistant endothelial cells. Pharmacological inhibition using gp91ds-tat or siRNA-mediated knockdown of Nox2 significantly attenuated superoxide generation in these cells. In further support of a role for Nox2 in the onset of these metabolic defects, these authors reported that a double transgenic mouse model [i.e., with endothelial-specific insulin resistance and deletion of Nox2] exhibited attenuated superoxide production and improved vascular function. Based on these findings, these investigators suggested Nox2 activation step as a regulatory mechanism in the signaling events leading to insulin resistance-mediated oxidative stress and vascular dysfunction [29].
In conclusion, available evidence suggests that the activation of Nox2 in hyperglycemic conditions plays key regulatory roles in generation of ROS and formation of superoxides in multiple cell types including the pancreatic islet β-cells, cardiomyocytes, and retinal endothelial cells. It should be noted however that the findings discussed above are only representative of a plethora of reports in this area. It is noteworthy that there are numerous reports implicating other NADPH oxidases [Nox1, Nox4] in cellular dysfunction and demise under various pathological conditions [30,31 for recent reviews].
3.5. A working model
Based on the above discussion, we propose a model for potential regulatory roles of Nox2 as a “trigger” for cellular dysfunction under glucolipotoxic conditions and diabetes (Fig. 1). Chronic exposure of cells to glucolipotoxic or diabetic conditions results in sustained activation of Rac1, a small G-protein, which is a key member of the Nox2 holoenzyme assembly. However, precise mechanisms underlying activation of Rac1 under these conditions remain to be defined further. Pharmacological evidence accrued from the use of inhibitors of protein geranylgeranylation [GGTI-2147] and protein palmitoylation [2-BP] suggests that both palmitoylation and geranylgeranylation of Rac1 are necessary of these signaling steps to occur. Furthermore, findings from studies involving specific inhibitors of Tiam1-Rac1 signaling [NSC23766] indicate that this pathway is critical for downstream signaling steps including Nox2 holoenzyme assembly, activation and associated ROS generation. We propose that glucolipotoxic and diabetic conditions promote activation of Rac1, and phosphorylation of cytosolic core components of Nox2 [p40phox, p47phox and p67phox] leading to their translocation and association with the membranous core for Nox2 activation and ROS generation. This is further supported by observations from several laboratories demonstrating attenuation of oxidative stress by inhibitors of Nox2 [apocynin, DPI and gp91ds-tat peptide]. Our model also predicts that induction of Nox2-derived oxidative stress results in the activation of stress kinases [p38 kinase, JNK1/2 and p53 kinases] to trigger mitochondrial dysfunction, including loss in membrane potential and cytochrome-C release and subsequent activation of proapoptotic signaling steps including activation of caspases [caspase 3]. We have recently demonstrated that activation of caspase 3 under glucotoxic and ER stress conditions leads to degradation and mislocalization of nuclear lamins in isolated β-cells, thus leading to loss in metabolic cell viability and onset of cellular demise [7]. Not highlighted in this model are recent observations from our laboratory suggesting significant protection against the induction of metabolic defects in pancreatic by inhibitors of Rac1 activation [GGTI-2147, 2-BP and NSC23766] in pancreatic β-cells exposed to proinflammatory cytokines [IL-1β, TNFα and IFNγ; 16] and Cer, a biologically active sphingolipid, which induces cell dysfunction in the islet β-cell [14,15].
Fig. 1.
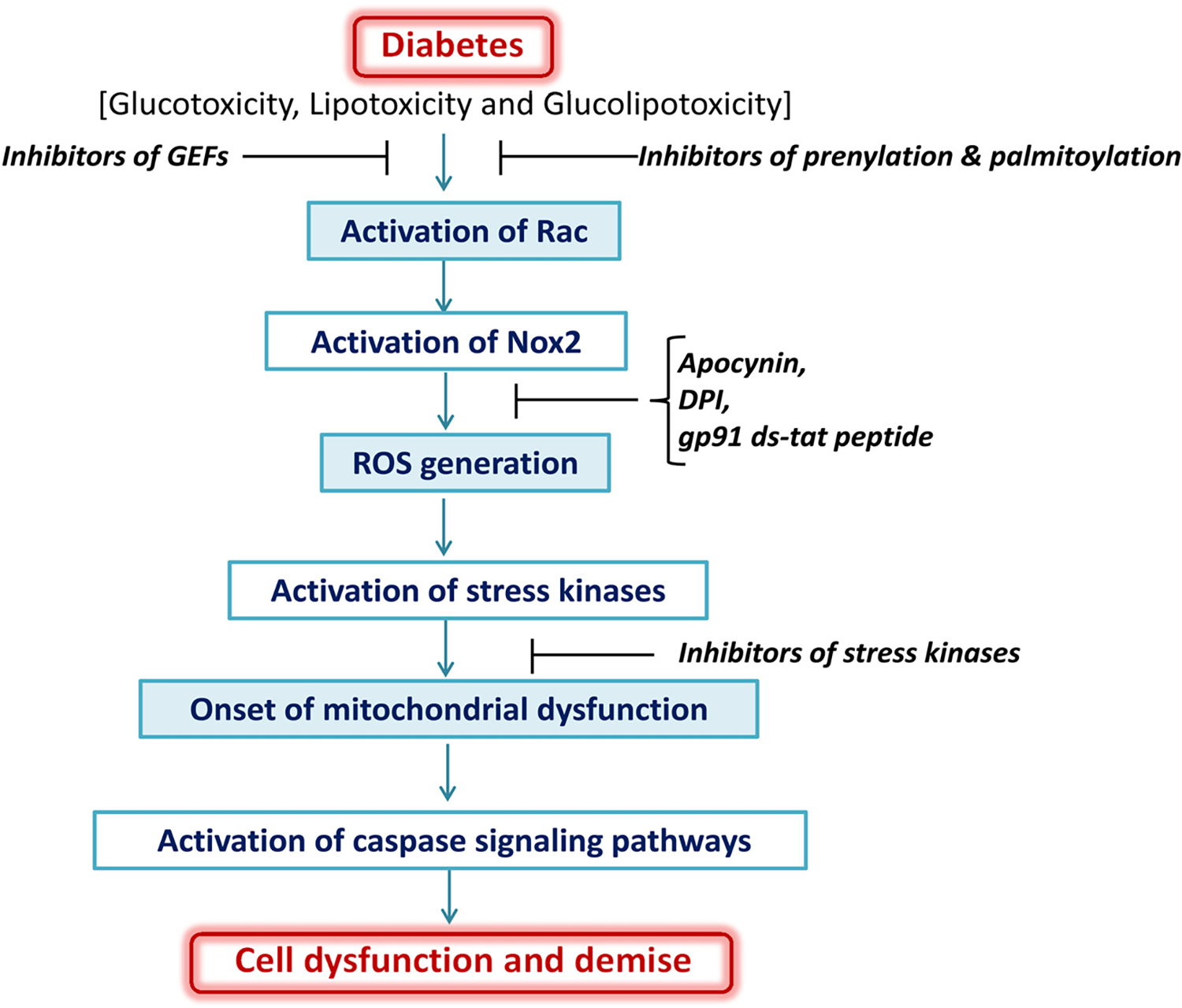
Proposed model highlighting contributory roles of Nox2 in cellular dysfunction induced by glucolipotoxic conditions and diabetes. Chronic exposure of cells to glucotoxic, lipotoxic, glucolipotoxic or diabetic conditions culminate in the generation of Nox2-mediated ROS, which, in turn, leads to activation of stress kinases and onset of mitochondrial dysregulation. Alterations in mitochondrial function including leakage of proapoptotic factors [cytochrome-C] into the soluble compartment leads to activation of caspases, which is turn, catalyze the degradation and mislocalization of key structural proteins including nuclear lamins A and B, and resulting in eventual collapse of nuclear ultrastructure. As stated in the narrative, this model is simplistic, and involves interlay and cross-talk between other signaling pathways, which are activated under these conditions. Pharmacological and molecular biological evidence strongly favors the concept that Tiam1-Rac1-Nox2 axis represents one of the areas for the development of novel therapeutics to impede metabolic defects under glucolipotoxic and diabetic conditions [see narrative for additional details].
4. Design, discovery and development of inhibitors of Nox2
Extant studies from several laboratories have yielded novel clues on potential roles of Nox2 as a therapeutic target for reducing or halting the excessive generation of intracellular ROS. Some of the early investigations employed selective inhibitors of Nox2 activation [apocynin and DPI]. Data from recent studies have led to the design and development of more specific small molecule inhibitors of Nox2 function; these include inhibitors of Rac1 activation and function, and inhibitors of interaction between various subunits of the Nox2 holoenzyme. The reader is referred to Cifuentes-Pagano et al. [32], El-Benna and associates [33,34], Williams and Griendling [35], and Bid et al. [36] for in depth reviews highlighting advancements in the area of development of specific inhibitors for Rac1 and Nox2. In the following section, we will briefly highlight the advances in the development of Rac1 inhibitors and their applicability in halting/preventing the activation of Nox2 under various pathological conditions, including glucolipotoxicity and diabetes. In this context, we will review the evidence implicating roles of prenylation, palmitoylation and phosphorylation of Rac1 in the regulation of its activation–deactivation cycles [GTP- or GDP-bound conformations]. The utility of specific inhibitors of GEFs for Rac1 activation and prevention of cellular dysfunction in diabetes is also discussed below. Structures of various classes of inhibitors [i.e., inhibitors of protein prenylation, protein palmitoylation, Rac1 activation and Nox2 activation] employed in the studies reviewed in this commentary are provided in Fig. 2.
Fig. 2.
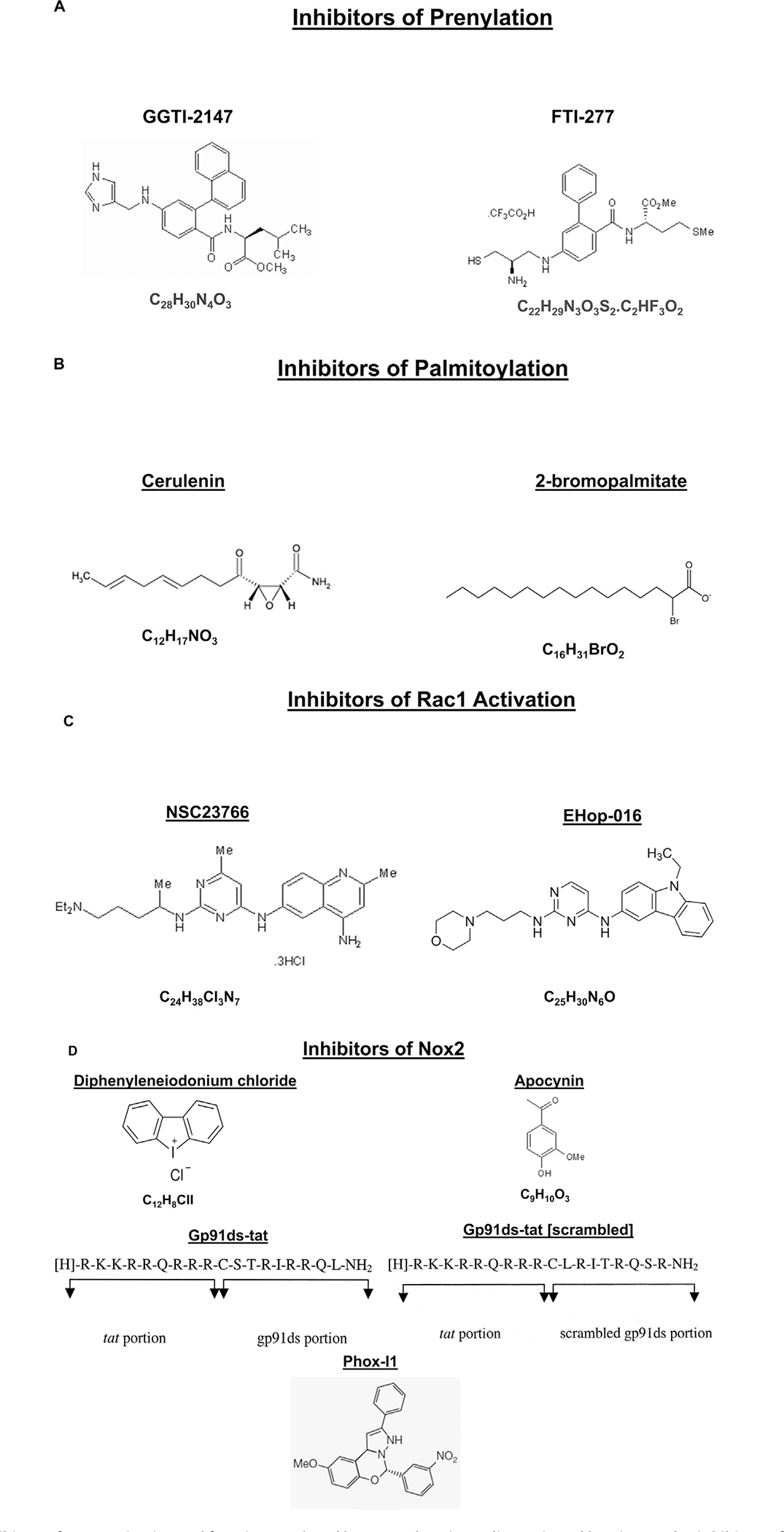
Structures of various inhibitors of Nox2 activation and function employed by researchers in studies reviewed herein. Panel A: inhibitors of protein prenylation; Panel B: inhibitors of protein palmitoylation; Panel C: inhibitors of GEF-mediated activation of Rac1; and Panel D: inhibitors of Nox2 including inhibitors of Nox2 subunit interaction and holoenzyme assembly.
4.1. Inhibitors of prenylation of Rac1
It is well established that the majority of small G-proteins and the γ-subunits of heterotrimeric G-proteins undergo post-translational modifications at their C-terminal cysteine residue. Briefly, the first of a four-step modification sequence involves incorporation of a mevalonic acid-derived 15-carbon [farnesyl] or 20-carbon [geranylgeranyl] isoprenoid moiety onto a cysteine residue toward the C-terminus of the candidate G-proteins [referred to as the CAAX motif]. Examples of farnesylated proteins include H-Ras, nuclear lamins A and B. Small G-proteins belonging to the Rho subfamily [e.g., Rho, Cdc42 and Rac1] undergo geranylgeranylation [9,37]. Several recent studies in the pancreatic islet β-cell have utilized inhibitors of prenylation of Rac1 to study its role in islet health, glucolipotoxicity and diabetes [9,15 for reviews]. In brief, geranylgeranylation of Rac1 is catalyzed by geranylgeranyltransferse-1 [GGTase-1], which is comprised of the structural asubunit [GGTase-α] and the regulatory β-subunit [GGTase-β]. Using a catalytically inactive mutant of GGTase-α, we have recently demonstrated critical regulatory roles for geranylgeranylation of Rac1 in glucose-stimulate insulin secretion [GSIS] from the pancreatic islet β-cells [38,39]. In addition, siRNA-Rac1 inhibited GSIS [15]. Moreover, GGTI-2147, a specific inhibitor of GGTase-1 markedly attenuated GSIS [38]. Together, these findings suggested requisite roles for geranylgeranylation of Rac1 in GSIS. Recent studies have shown significant inhibition, by GGTI-2147, of Nox2-mediated metabolic dysfunction of the islet β-cell under the duress of long-term exposure to proinflammatory cytokines [15]. They also reported attenuation in cytokine-induced Rac1 activation and loss in mitochondrial membrane permeability pore transition in INS-1 832/13 cells, suggesting novel regulatory roles for protein geranylgeranylation in these signaling steps [15]. Additional studies are needed to determine the potential utility of inhibitors of protein prenylation to prevent metabolic dysfunction of cells induced by glucolipotoxic conditions.
4.2. Inhibitors of palmitoylation of Rac1
In addition to geranylgeranylation, small G-proteins [Ras and Rac1] undergo additional post-translational modifications involving the covalent addition of a long-chain fatty acid, typically palmitate, at cysteine residues, which are upstream to the CAAX motif [9,40,41]. Existing body of evidence clearly suggests novel regulatory roles for protein palmitoylation in cellular function [42–44]. The protein palmitoyltransferases [PATs] catalyze transfer of PA into the cysteine residues of proteins containing DHHC [Asp–His–His–Cys] cysteine-rich domains via a thioester linkage. Typically, palmitoylation occurs at the cytoplasmic face of membranes in the secretory pathway [e.g., ER and Golgi] and the plasma membrane. A distinct class of palmitoyl thioesterases [protein palmitoyl thioesterases 1 and 2 and acyl palmitoylesterase 1] mediate depalmitoylation via hydrolysis of the ester bonds to complete the palmitoylation–depalmitoylation [i.e., activation–inactivation] cycle [42–44].
Advancement in the area of understanding the regulatory roles of protein palmitoylation in cellular function, including activation of Nox2, has been slow, which is, in part, due to the unavailability of specific inhibitors for PATs. Previous investigations have utilized cerulenin [CER], an antifungal antibiotic, to decipher the roles of protein palmitoylation in cellular function in many cell types including the islet β-cell [23,40,43–46]. However, use of CER as inhibitor of protein palmitoylation may be limited because it has been shown to exert non-specific effects including inhibition of fatty acid and steroid biosynthesis as well as lipoprotein lipase [47]. Other investigations have utilized tunicamycin as an inhibitor to study roles of protein palmitoylation in cell function, but again, as in the case of CER, tunicamycin inhibits fatty acid synthesis and N-glycosylation of proteins [48], and induces ER stress. It is noteworthy that several extant studies have utilized 2-BP to study roles of protein palmitoylation in cellular function [44 for a review]. More recent studies by Jennings and coworkers [49] compared inhibitory effects of 2-BP and Compound V [2-(2-hydroxy-5-nitrobenzylidene)-benzo[b]thiophen-3-one] on DHHC-mediated palmitoylation in vitro. Their findings suggested that the inhibitory effects of Compound V on palmitoylation are reversible in contrast to the irreversible inhibitory effects of 2-BP.
Limited information is available, however, on the utility of 2-BP as inhibitor of metabolic dysfunction of cells under conditions of glucolipotoxicity and diabetes. In this context, we provided the first evidence to indicate that 2-BP prevents noxious effects of proinflammatory IL-1β on insulin secreting cells. These studies identified H-Ras as one of the palmitoylated proteins in the cascade of events leading to the demise of the islet β-cell [40]. More recently, we reported a time-dependent phosphorylation of p47phox by a mixture of proinflammatory cytokines [IL-1β, TNFα, and IFNγ] in insulin-secreting INS-1 832/13 cells [50]. Further, 2-BP significantly inhibited cytokine-induced, Nox2-mediated ROS generation and iNOS-mediated nitric oxide [NO] generation. Based on these findings, we proposed PAT as a target for inhibition of cytomix-induced oxidative and nitrosative stress in the pancreatic β-cell.
Besides cytokine-induced dysfunction, recent evidence appears to support key roles for protein palmitoylation in free fatty acid [FFA]-induced dysregulation [lipotoxicity] of the islet β-cell. Using insulin-secreting RINm5F cells and normal rat islets, Baldwin et al. have proposed a role for palmitoylation in FFA-induced ER stress and β-cell death [51]. Data from their studies suggested a concentration-dependent loss in β-cell viability by palmitate, which was attenuated by 2-BP. The later also inhibited palmitoylation [incorporation of (3H)PA] of RINm5F cell proteins. Lastly, 2-BP also reduced palmitate-induced inhibition of insulin secretion, ER stress and caspase activation. Based on this evidence the authors postulated key roles for protein palmitoylation in FFA-induced ER stress activation and loss of β-cell viability.
In conclusion, it appears that palmitoylation signaling step is requisite for cytokine- and FFA-induced dysregulation of the islet β-cell. Additional studies, including the development of novel inhibitors of PATs, are necessary to further decipher the roles of protein palmitoylation in cellular function, including activation of Rac1, Nox2 and their downstream signaling events [stress kinase activation] leading to cell demise under conditions of glucolipotoxicity and diabetes (Fig. 1).
4.3. Phosphorylation of Rac1
One of the emerging areas of regulation of Rac1 function is via phosphorylation–dephosphorylation. Unlike geranylgeranylation and palmitoylation, which are required for optimal activation and effector regulation of Rac1, the phosphorylation of Rac1 appears to inhibit its activation. For example, studies by Tong et al. have demonstrated phosphorylation of Rac1 [at Thr-108] by ERK1/2 in COS-7 cells culminating in inhibition of cell function [52]. Kwon et al. have reported Akt-mediated phosphorylation of Rac1 at Ser-71 in human melanoma cells resulting in inhibition of GTP-binding to Rac1 [53]. Likewise, phosphorylation of Rac1 at Ser-71 residue was reported in HEK293 cells [54], CaCo-2 monolayers [55], and HepG2 cells [56] resulting in reduction in GTP binding and altered cellular function. Lastly, studies by Chang and coworkers in mouse murine fibroblasts have provided evidence to suggest that phosphorylation of Rac1 at Tyr-64 residue results in defective cell spreading [57]. While it is clearly established that the members of the cytosolic core of Nox2 [p40phox, p47phox and p67phox] require phosphorylation for their translocation to the membrane and priming of Nox2 [58–60], very little is known about alterations in the phosphorylation status of Rac1, if any, in cellular models of glucolipotoxicity and diabetes. More definitive studies are needed therefore to conclusively demonstrate putative regulatory roles of these signaling steps in the cellular activation of Nox2 and the onset of oxidative stress in models of glucolipotoxicity and diabetes.
4.4. Other known inhibitors of Rac1 activation and function
As stated in the above sections, in addition to inhibitors of post-translational modifications, several recent studies have explored potential utility of inhibitors of specific GEFs for Rac1 as modulators of its activation under the duress of glucolipotoxicity, proinflammatory cytokine exposure and diabetes. It is well established that activation of Rac1 is mediated by a variety of GEFs including Tiam1, Vav2 and Trio [9]. In 2004, Gao and associates discovered NSC23766, a small molecule compound, that specifically inhibited Tiam1 or Trio-induced activation of Rac1 [61]. Activation of other small G-proteins [Cdc42 and Rho] remained unaffected by NSC23766. Furthermore, NSC23766 significantly attenuated Rac1-mediated proliferation, anchorage-independent growth and invasion phenotypes in human prostrate cancer PC-3 cells [60]. Since then, nearly 200 studies [a recent Medline search] employed NSC23766 as inhibitor of Rac1 activation. In the context of islet β-cell, we have utilized NSC23766 to further investigate regulatory roles of Tiam1-Rac1 signaling cascade in islet function. Compatible with original findings of Gao et al., we observed that NSC23766 significantly inhibited only Rac1, but not Cdc42 or Rho activation [11]. In addition, we suggested roles for Tiam1-Rac1 in the metabolic dysfunction on insulin-secreting cells under conditions of glucolipotoxicity, and exposure to cytotoxic proinflammatory cytokines and Cer [14–16]. Further, as mentioned above, our observations in retinal endothelial cells [28] and by others in cardiomyocytes [18,19] further affirm the involvement of Tiam1-Rac1 signaling pathway in the activation of Nox2 under glucotoxic and diabetic conditions. Importantly, NSC23766 appears to be well tolerated in diabetic animal models [19,28]. Therefore, it will be worthwhile to focus future investigations on the development novel analogs of these compounds as tools to further understand roles of Tiam1-Rac1-Nox2 signaling module (Fig. 2) in the onset of mitochondrial dysfunction and cell death in diabetes. In addition, potential involvement of other GEFs [Vav2 and others] in the sustained activation of Rac1 under these pathological conditions remains to be investigated further. Recent studies by Montalvo-Ortiz have characterized Ehop-016, a small molecule inhibitor of Rac1 activation, which is mediated by Vav2 [62]. A recent review by Bid and associates provides additional insights into Rac1 as a therapeutic target for the treatment of diseases such as cancer [36]. Indeed, this article overviewed biological properties and functional effects of a variety of Rac1 inhibitors [NSC23766, EHT 1864, Ehop-016, Phox-I1, OSU 03012, IPA-3, Compound 3, and OS2] that can be tested as inhibitors of Nox2 activation in other pathological states as well.
4.5. Inhibitors of p47phox and gp91phox interaction
Seminal contributions from the laboratory of Pagano have identified a chimeric 18 amino acid peptide [gp91ds-tat], which interferes with NoX2 holoenzyme assembly and activation [63]. These investigations demonstrated that nine of the amino acids of the peptide, which mimic the gp91phox interacting domain with p47phox, prevent the interaction between gp91phox and p47phox. Several recent studies have demonstrated the specificity of gp91ds-tat [using its scrambled peptide as negative control], in inhibiting the agonist-induced activation of Nox2 in both in vitro and in vivo systems [35]. Preliminary evidence from our laboratory suggests that gp91ds-tat peptide, but not its scrambled peptide, significantly attenuated glucose-induced Nox2 activation and associated ROS generation in INS-1 832/13 cells [Veluthakal and Kowluru; unpublished]. Clearly, additional studies are need to further assess the beneficial effects of this peptide in preventing high glucose-induced, NoX2-mediated mitochondrial dysfunction, cytochrome C release and induction of cell death (Fig. 1).
4.6. Inhibitors of p67phox and Rac1 interaction
More recently, Bosco and associates identified a novel class of inhibitors, which inhibited Rac1-p67phox interaction [64]. Through the use of rational design and in silico screen approaches, they identified a small molecule compound, Phox-I1, that effectively inhibited [in sub-mM concentrations] the interaction between p67phox and Rac1. They also demonstrated inhibition of NOX2-mediated superoxide production in human and murine neutrophils under these conditions.
4.7. Other inhibitors of Nox2 activation
As described above, numerous previous studies have utilized apocynin and DPI to assess the roles of Nox2 activation in cellular dysfunction. While these inhibitors are extremely useful in understanding the roles of Nox2, the use of these has been under scrutiny by researchers in the field due to their non-specific and/or pleotropic effects [e.g., antioxidant properties of apocynin]. Recent investigations were aimed at developing more specific inhibitors of Nox2. For example, de Faria et al. synthesized and tested novel esters of protocatechuic acid as inhibitors of Noxs [65]. They reported that an alkyl ester with a 7 carbon chain length was ~10-fold more efficient as Nox2 inhibitor than apocynin. Based on this and other supporting evidence, these authors proposed that the esters of protocatechuic acid are promising drugs for treatment of chronic inflammatory diseases. Macias-Perez and coworkers synthesized novel ether and ester derivatives of apocynin and assessed their roles as Nox inhibitors [66]. Their studies yielded several compounds with Nox2 inhibitory activity. However, it was noted that neither apocynin nor its derivatives exhibited free radical scavenging properties. Data from in silico approaches suggested that apocynin and its derivatives were recognized by the polybasic SH3A and SH3B domains, which are regions of p47phox that interact with p22phox. Based on these observations these researchers concluded that the novel derivatives of apocynin could mediate inhibition of Nox2 activation by preventing the complex formation between p47phox and p22phox, which is a requisite for translocation of these cytosolic core of Nox2 for holoenzyme assembly and activation [66]. Methodical investigations are necessary to further validate the utility of these novel class of inhibitors on ROS generation and superoxide formation in other model systems.
5. Potential caveats and non-specific or pleotropic effects of pharmacological inhibitors of Nox2
It is noteworthy that recent reports indicate pleotropic effects of inhibitors of Tiam1-Rac1 signaling pathway. For example, studies by Suzuki and associates [67] have demonstrated that both apocynin and NSC23766 inhibited cell proliferation and phosphorylation of Rac1, NF-kB and cyclin D1 in prostate cancer cells. Furthermore, NSC23766 has been shown to act as a competitive antagonist of muscarinic acetyl choline receptors [68]. In these studies, Levay et al. demonstrated inhibition of M2 muscarinic acetylcholine-induced Rac1 activation by NSC23766 in neonatal rat cardiac myocytes. Interestingly, NSC23766 also inhibited carbachol induced RhoA activation and muscarinic acetylcholine-induced inotropic response in isolated neonatal rats requiring the activation of Rho-dependent kinases. Lastly, a considerable amount of work is still needed in the development of specific inhibitors of PATs since 2-BP, the most widely used inhibitor of protein palmitoylation, is only a selective, but not specific inhibitor of palmitoylation [44]. Furthermore, 2-BP has been shown to inhibit protein deacylation by inhibiting acyl-protein thioesterases as well [69]. Therefore, data accrued from pharmacological experiments will need to be confirmed further using molecular biological approaches [inactive mutants and/or siRNAs], and in knockout animal models prior to drawing conclusions on these targets.
6. Conclusions and future directions
Increased intracellular generation of ROS has been implicated in the pathology of metabolic and neurodegenerative diseases. As reviewed above, substantial amount of evidence implicates Noxs as the principal source for cellular ROS. More importantly, recent evidencesuggeststhat Nox2 plays contributory roles in theinduction of cellular damage culminating in the onset of diabetes [β-cell dysregulation and demise] and its complications, including cardiomyopathy and retinopathy. Although not discussed herein, evidence is also accumulating on roles of NADPH oxidases in the pathogenesis of metabolic defects in the kidney and nerve in diabetes [30,70,71]. Original studies involving the use of selective inhibitors of Nox2 [apocynin and DPI] paved way for further investigations in the development of more specific inhibitors for Nox2. Despite the complexity of individual steps needed for translocation of the cytosolic core, priming and activation of Nox2, recent years have witnessed tremendous progress in the design and development of novel cell-permeable molecules with high degree of specificity to inhibit activation of Rac1 and interaction among subunits. Evidence from multiple laboratories indicates that these inhibitors significantly prevent metabolic dysfunction of cells under the duress of chronic exposure to glucose, PA, Cer and proinflammatory cytokines. NSC23766 and gp91ds-tat are effective in preventing diabetes-induced lesions in the heart and retina in animal models of diabetes. These findings raise the potential that Nox2 is indeed a “druggable” target. It is hoped that these findings would serve as “stepping stones” in our quest to develop more specific inhibitors for the prevention of metabolic and neuronal disorders.
Based on the above discussion it is evident that Nox2 plays important roles in cellular function including hormone [insulin] secretion under acute regulatory conditions, and that chronic cellular Nox2 activation under the duress of glucolipotoxicity, Cer and cytokine exposure conditions leads to oxidative damage and mitochondrial dysfunction in target cells. Therefore, due to multi-faceted functions of the Nox2 complex in various tissues and cells, we are presented with a rather narrow window of opportunity to test various inhibitors of Nox2 function [inhibitors of protein prenylation, GEFs of Rac1 and Nox2 subunit interaction] for halting/inhibiting Nox2 activation thereby limiting the amount of intracellular ROS generation to divert cellular metabolism toward the normal survival mode rather than triggering the cellular dysfunction pathways. As discussed above, some of these inhibitors appear to work well in animal models. Additional studies are needed, however, to further validate this model animal model before they can be extended to investigations in humans.
We also presented a working model (Fig. 1) to highlightindividual signaling steps involved in the onset of cellular dysfunction in diabetes. Clearly, this is based on available data implicating Nox2 as the “trigger” for metabolic defects. We realize that there are other metabolic pathways that might run in parallel and/or in concert with the steps described in this model [2]. In this context, we envision that post-translational modifications [prenylation, palmitoylation and phosphorylation] of individual core components of Nox2 play requisite roles for their translocation and association with the membranous core of Nox2. Future investigations must focus not only in understanding individual metabolic/signaling steps involved in Nox2 activation, but also in the development of specific inhibitors for these signaling steps to halt Nox2 activation and cellular defects/damage in diabetes and other disorders.
Acknowledgements
This work was supported by grants from the National Institutes of Health [EY014370, EY017313 and EY022230 to RK; DK74921 and EY022230 to AK], Juvenile Diabetes Research Foundation [5-2012-313 to RK and 5-2012-257 to AK] and the Department of Veterans Affairs [1BX000469 to AK]. AK is the recipient of a Senior Research Career Scientist Award from the Department of Veterans Affairs [13S-RCS-006]. This work was also supported by unrestricted funds to the Department of Ophthalmology from Research to Prevent Blidness.We sincerely thank the former and the current members of our laboratories who have contributed to the studies highlighted in this article.
Abbreviations:
- 2-BP
2-bromopalmitate
- BRECs
bovine retinal endothelial cells
- Cer
ceramide
- CER
cerulenin
- DPI
diphenyleneiodonium
- ER stress
endoplasmic reticulum stress
- FTase
farnesyltransferase
- FTI
farnesyltransferase inhibitor
- GEFs
guanine nucleotide exchange factors
- GGTase
geranylgeranyltransferase
- GGTI
geranylgeranyltransferase inhibitor
- GSIS
glucose-stimulated insulin secretion
- IFNγ
interferon-γ
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- Nox2
phagocyte-like NADPH oxidase
- NSC23766
(N(6)-[2-[[4-(diethylamino)-1-methylbutyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl-4,6-quinolinediamine trihydrochloride
- PA
palmitic acid
- PAT
palmitoyltransferase
- ROS
reactive oxygen species
- Stz
streptozotocin
- T2DM
type 2 diabetes mellitus
- Tiam1
T-lymphoma invasion and metastasis inducing factor 1
- TNFα
tumor necrosis factora
- ZDF rat
Zucker diabetic fatty rat
References
- [1].IDF. International Diabetes Federation – Diabetes Atlas. 6th ed. 2013;p. 1–160. [PubMed]
- [2].Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 2008;29:351–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Blake R, Trounce IA. Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta 2013. 10.1016/j.bbagen.2013.11.007. [DOI] [PubMed] [Google Scholar]
- [4].Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic beta-cells. Trends Endocrinol Metab 2012;23:477–87. [DOI] [PubMed] [Google Scholar]
- [5].Newsholme P, Gaidel C, Krause M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv Exp Med Biol 2012;942:235–47. [DOI] [PubMed] [Google Scholar]
- [6].Syeda K, Mohammed AM, Arora DK, Kowluru A. Glucotoxic conditions induce endoplasmic reticulum stress to cause caspase 3 mediated lamin B degradation in pancreatic β-cells: protection by nifedipine. Biochem Pharmacol 2013;86:1338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kuijpers T, Lutter R. Inflammation and repeated infections in CGID: two sides of a coin. Cell Mol Life Sci 2012;69:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal 2009;11:791–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kowluru A Small G proteins in islet beta-cell function. Endocr Rev 2010;31:52–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Newsholme P, Haber EP, Hirabara SM, Rebelato ELO, Procopio J, Morgan D, et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and nonmitochondrial ROS production and activity. J Physiol 2007;583:9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Veluthakal R, Madathilperambil SV, McDonald P, Olson LK, Kowluru A. Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem Pharmacol 2009;77:101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Syed I, Kyathanahalli CN, Kowluru A. Phagocyte-like NADPH oxidase generates ROS in INS 832/13 cells and rat islets: role of protein prenylation. Am J Physiol Regul Integr Comp Physiol 2011;300:R756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Matti A, Kyathanahalli C, Kowluru A. Protein farnesylation is requisite for mitochondrial fuel-induced insulin release: further evidence to link reactive oxygen species generation to insulin secretion in pancreatic beta-cells. Islets 2012;4:74–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Syed I, Jayaram B, Subasinghe W, Kowluru A. Tiam1/Rac1 signaling pathway mediates palmitate-induced, ceramide-sensitive generation of superoxides and lipid peroxides and the loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol 2010;80:874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kowluru A Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol 2011;81:965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. Am J Physiol Regul Integr Comp Physiol 2011;300:R12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yuan H, Lu Y, Huang X, He Q, Man Y, Zhou Y, et al. Suppression of NADPH oxidase 2 substantially restores glucose-induced dysfunction of pancreatic NIT-1 cells. FEBS J 2010;277:5061–71. [DOI] [PubMed] [Google Scholar]
- [18].Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, et al. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 2011;60:2843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen E, Li Y, Li Y, Shan L, Zhu H, Feng Q, et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes 2009;58:2386–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibitsendoplasmic reticulum stress, andreducesmyocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010;59: 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D, et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLOS ONE 2012;7:E52013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tawfik HE, El-Remessy AB, Matragoon S, Ma G, Caldwell RB, Caldwell RW. Simvastatin improves diabetes-induced coronary endothelial dysfunction. J Pharmacol Exp Ther 2006;319:386–95. [DOI] [PubMed] [Google Scholar]
- [23].Metz SA, Rabaglia ME, Stock JB, Kowluru A. Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GTP-binding proteins. Biochem J 1993;295:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Al-Shabrawey M, Rojas M, Sanders T, Behzadian A, El-Remessy A, Bartoli M, et al. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci 2008;49:3239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Othman A, Ahmad S, Megyerdi S, Mussell R, Choksi K, Maddipati KR, et al. 12/15-lipoxygenase-derived lipid metabolites induce retinal endothelial barrier dysfunction: contribution of NADPH oxidase. PLOS ONE 2013;8(2):e57254. 10.1371/journal.pone.0057254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].He M, Pan H, Xiao C, Pu M. Roles for redox signaling by NADPH oxidase in hyperglycemia-induced heme oxygenase-1 expression in the diabetic retina. Invest Ophthalmol Vis Sci 2013;54:4092–101. [DOI] [PubMed] [Google Scholar]
- [27].Du Y, Veenstra A, Palczewski K, Kern TS. Photoreceptor cells are major contributors to oxidative stress and local inflammation in the retina. Proc Natl Acad Sci U S A 2013;110:16586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kowluru R, Kowluru A, Mohammad G, Syed I, Santos JM, Veluthakal R. Tiam1-Rac1 signaling axis mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014. [in press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sukumar P, Viswambharan H, Imrie H, Cubbon RM, Yuldasheva N, Gage M, et al. Nox2 NADPH oxidase has a critical role in insulin resistance-related endothelial cell dysfunction. Diabetes 2013;62:2130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gorin Y, Block K. Nox as a target for diabetic complications. Clin Sci (Lond) 2013;125:361–82. [DOI] [PubMed] [Google Scholar]
- [31].Taylor-Fishwick DA. NOX, NOX who is there? The contribution of NADPH oxidase one to beta cell dysfunction. Front Endocrinol (Lausanne) 2013;4:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cifuentes-Pagano E, Meijles DN, Pagano PJ. Sly as a Nox: the challenges, triumphs and pitfalls of selective NADPH Oxidase inhibition. Antioxid Redox Signal 2013. 10.1089/ars.2013.5620 [ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].El-Benna J, Dang PM, Périanin A. Towards specific NADPH oxidase inhibition by small synthetic peptides. Cell Mol Life Sci 2012;69:2307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].El-Benna J, Dang PM, Périanin A. Peptide-based inhibitors of the phagocyte NADPH oxidase. Biochem Pharmacol 2010;80:778–85. [DOI] [PubMed] [Google Scholar]
- [35].Williams HC, Griendling KK. NADPH oxidase inhibitors: new antihypertensive agents? J Cardiovasc Pharmacol 2007;50:9–16. [DOI] [PubMed] [Google Scholar]
- [36].Bid HK, Roberts RD, Manchanda PK, Houghton PJ. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther 2013;12:1925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kowluru A Protein prenylation in glucose-induced insulin secretion from the pancreatic islet beta-cell: a perspective. J Cell Mol Med 2008;12:164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Veluthakal R, Kaur H, Goalstone M, Kowluru A. Dominant negative alpha subunit of farnesyl and geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated insulin secretion in INS 832/13 cells. Diabetes 2007;56:204–10. [DOI] [PubMed] [Google Scholar]
- [39].Kowluru A, Veluthakal R, Rhodes CJ, Kamath V, Syed I, Koch BJ. Protein farnesylation-dependent Raf/extracellular signal-related kinase signaling links to cytoskeletal remodeling to facilitate glucose-induced insulin secretion in pancreatic beta-cells. Diabetes 2010;59:967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chen HQ, Tannous M, Veluthakal R, Amin R, Kowluru A. Novel roles for palmitoylation of Ras in IL-1β-induced nitric oxide release and caspase-3 activation in insulin-secreting β-cells. Biochem Pharmacol 2003;66:1681–94. [DOI] [PubMed] [Google Scholar]
- [41].Navarro-Lérida I, Sánchez-Perales S, Calvo M, Rentero C, Zheng Y, Enrich C, et al. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J 2012;31:534–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bijlmakers MJ, Marsh M. The on-off story of protein palmitoylation. Trends Cell Biol 2003;13:32–42. [DOI] [PubMed] [Google Scholar]
- [43].Blaskovic S, Blanc M, van der Goot FG. What does S-palmitoylation do to membrane proteins. FEBS J 2013;280:2766–74. [DOI] [PubMed] [Google Scholar]
- [44].Mohammed AM, Chen F, Kowluru A. The two faces of protein palmitoylation in islet beta-cell function: potential implications in the pathophysiology of islet metabolic dysregulation and diabetes. Recent Pat Endocr Metab Immune Drug Discov 2013;7:203–12. [DOI] [PubMed] [Google Scholar]
- [45].Straub SG, Yajima H, Komatsu M, Aizawa T, Sharp GW. The effects of cerulenin, an inhibitor of protein acylation, on the two phases of glucose-stimulated insulin secretion. Diabetes 2002;51(Suppl. 1):S91–5. [DOI] [PubMed] [Google Scholar]
- [46].Yajima H, Komatsu M, Yamada S, Straub SG, Kaneko T, Sato Y, et al. Cerulenin, an inhibitor of protein acylation, selectively attenuates nutrient stimulation of insulin release: a study in rat pancreatic islets. Diabetes 2000;49:712–7. [DOI] [PubMed] [Google Scholar]
- [47].Falo LD Jr, Benacerraf B, Rothstein L, Rock KL. Cerulenin is a potent inhibitor of antigen processing by antigen-presenting cells. J Immunol 1987;139:3918–23. [PubMed] [Google Scholar]
- [48].Gabius HJ, van de Wouwer M, Andre S, Villabolo A. Down regulation of epidermal growth factor receptor by altering N-glycosylation: emerging role for beta1,4-galactosyltransferases. Anti Cancer Res 2012;32:1565–72. [PubMed] [Google Scholar]
- [49].Jennings BC, Nadolski MJ, Ling Y, Baker MB, Harrison ML, Deschenes RJ, et al. 2-Bromopalmitate and 2-(2-hydroxy-5-nitro-benzylidene)-benzo[b]thiophen-3-one inhibit DHHC-mediated palmitoylation in vitro. J Lipid Res 2009;50: 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mohammed AM, Syeda K, Hadden T, Kowluru A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem Pharmacol 2013;85:109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Baldwin AC, Green CD, Olson LK, Moxley MA, Corbett JA. A role for aberrant protein palmitoylation in FFA-induced ER stress and β-cell death. Am J Physiol Endocrinol Metab 2010;302:E1390–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tong J, Li L, Ballermann B, Wang Z. Phosphorylation of Rac1 T108 by ERK in response to EGF: a novel mechanism to regulate Rac1 function. Mol Cell Biol 2013;33:4538–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits Rac1-GTP binding through phosphorylation at serine 71 of Rac1. J Biol Chem 2000;275:423–8. [DOI] [PubMed] [Google Scholar]
- [54].Schwarz J, Proff J, Hävemeier A, Ladwein M, Rottner K, Barlag B, et al. Serine-71 phosphorylation of Rac1 modulates downstream. PLOS ONE 2012;7:e44358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schoentaube J, Olling A, Tatge H, Just I, Gerhard R. Serine-71 phosphorylation of Rac1/Cdc42 diminishes the pathogenic effect of Clostridium difficile toxin A. Cell Microbiol 2009;11:1816–26. [DOI] [PubMed] [Google Scholar]
- [56].Brandes V, Schelle I, Brinkmann S, Schulz F, Schwarz J, Gerhard R, et al. Protection from Clostridium difficile toxin B-catalysed Rac1/Cdc42 glucosylation by tauroursodeoxycholic acid-induced Rac1/Cdc42 phosphorylation. Biol Chem 2012;393:77–84. [DOI] [PubMed] [Google Scholar]
- [57].Chang F, Lemmon C, Lietha D, Eck M, Romer L. Tyrosine phosphorylation of Rac1: a role in regulation of cell spreading. PLoS ONE 2011;6:e28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and Nox2 mobilization to the plasma membrane. Semi Immunopathol 2008;30:279–89. [DOI] [PubMed] [Google Scholar]
- [59].Sheppard FR, Kelher MR, Moore EE, McLaughlin NJ, Banerjee A, Silliman CC. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming. J Leukoc Biol 2005;78:1025–42. [DOI] [PubMed] [Google Scholar]
- [60].El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in disease. Exp Mol Med 2009;41:217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 2004;101:7618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, et al. Characterization of Ehop-016, a novel small molecule inhibitor of Rac GTPase. J Biol Chem 2012;287:13228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res 2001;89:408–14. [DOI] [PubMed] [Google Scholar]
- [64].Bosco EE, Kumar S, Marchioni F, Biesiada J, Kordos M, Szczur K, et al. Rational design of small molecule inhibitors targeting the Rac GTPase-p67(phox) signaling axis in inflammation. Chem Biol 2012;19:228–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].de Faria CM, Nazare AC, Petronio MS, Paracatu LC, Zeraic ML, Regasini LO, et al. Protocatechuic acid alkyl esters: hydrophobicity as a determinant factor for inhibition of NADPH oxidase. Curr Med Chem 2012;19:4885–93. [DOI] [PubMed] [Google Scholar]
- [66].Macías-Pérez ME, Martıínez-Ramos F, Padilla-Martıínez II, Correa-Basurto J, Kispert L, Mendieta-Wejebe JE, et al. Ethers and esters derived from apocynin avoid the interaction between p47phox and p22phox subunits of NADPH oxidase: evaluation in vitro and in silico. Biosci Rep 2013;33(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Suzuki S, Pitchakarn P, Sato S, Shirai T, Takahashi S. Apocynin, an NADPH oxidase inhibitor, suppresses progression of prostate cancer via Rac1 dephosphorylation. Exp Toxicol Pathol 2013;65:1035–41. [DOI] [PubMed] [Google Scholar]
- [68].Levay M, Krobert KA, Wittig K, Voigt N, Bermudez M, Wolber G, et al. NSC23766, a widely used inhibitor of Rac1 activation, additionally acts as a competitive antagonist at muscarinic acetylcholine receptors. J Pharmacol Exp Ther 2013;347:69–79. [DOI] [PubMed] [Google Scholar]
- [69].Pedro MP, Vilcaes AA, Tomatis VM, Oliveira RG, Gomez GA, et al. 2-Bromopalmitate reduces protein deacylation by inhibition of acyl-protein thioesterase enzymatic activities. PLOS ONE 2013;8:e75232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].You YH, Okada S, Ly S, Jandeleit-Dahm K, Barit D, Namikoshi T, et al. Role of Nox2 in diabetic kidney disease. Am J Physiol Renal Physiol 2013;304:F840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sedeek M, Nasrallah R, Touyz RM, Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend or foe. J Am Soc Nephrol 2013;24:1512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]