

Abstract
Per- and polyfluoroalkyl substances (PFAS) are synthetic chemicals; the vast majority are environmentally and biologically persistent, and some have demonstrated toxicity, including cancer, effects on metabolism, endocrine disruption, and immune dysfunction. Suppression of T-cell-dependent antibody responses (TDAR) has been observed in numerous studies of PFAS but mechanisms remain elusive. Evidence from our work suggests that B cells and how they use energy are impacted by PFAS exposure. We hypothesize that a well-studied and immunotoxic PFAS, perfluorooctanoic acid (PFOA), alters B-cell subclasses and markers of their metabolism. Adult male and female C57BL/6 mice were given PFOA (0 or 7.5 mg/kg) via gavage for 15 days, a duration and dose sufficient to suppress the TDAR. After dosing and immunization of subgroups, spleens were prepared to quantify B-cell subsets. Flow cytometric analysis revealed decreased numbers of plasmablasts, follicular, naïve, and overall B-cell subclasses in female PFOA-exposed groups. Male PFOA-exposed groups had a significant increase in follicular B cells and other subsets had decreases, including in the overall number of B cells. Twenty-four hours after naïve B-cell isolation and ex vivo activation, metabolic measurements revealed a 5-fold increase in metabolic markers in response to stimulation in PFOA-exposed groups compared with controls. These findings suggest that B-cell development and survival may be hindered by PFOA exposure, but that activation of the remaining B cells was not. Based on these findings, PFOA-mediated suppression of the primary IgM antibody response results changes to specific subsets of B cells.
Keywords: PFAS, immunotoxicity, PFOA, immunometabolism, B cells
Numerous studies have demonstrated the toxicity of several synthetic chemicals known as per- and polyfluoroalkyl substances (PFAS) and their other characteristics of concern, including environmental and biological persistence, mobility, bioaccumulation potential, and toxicity (EPA, 2022a). PFAS are used in over 60 different product use categories, including nonstick cookware, medical garments, food packaging, firefighting foams, and stain-repellent textiles (Glüge et al., 2020). A contributing source of exposure to PFAS contamination in human blood is drinking water (Sunderland et al., 2019). PFAS are also multisystem toxicants and adverse health effects associated with these chemicals include liver damage, cancer, high cholesterol, endocrine disruption, effects on reproduction and development, and immune disruption (ATSDR, 2021).
In 2022, the U.S. Environmental Protection Agency (EPA) issued an update on lifetime drinking water health advisories for 2 PFAS, perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS). These 2 PFAS, often referred to as “legacy” PFAS, have been produced and used since at least the 1940s. The critical effect underlying the health advisories is PFOA/PFOS-induced suppression of antigen-specific antibody responses in a population of children living in the Faroe Islands (EPA, 2022b). The selection of this critical effect for protection of public health is in line with a systematic review by the U.S. National Toxicology Program to classify PFOA and PFOS as presumed immune hazards to humans based on data from experimental models and epidemiological studies (NTP, 2016). Despite the acknowledged sensitivity of the immune system to PFOA and PFOS, the mechanism(s) by which these PFAS suppress antibody production remains elusive. Prior work with PFOA in our laboratory indicates that PFOA exposure in experimental animals targets B cells and we hypothesize that while overall numbers of B cells are not reduced, deficits in numbers of B-cell subsets or in B-cell functions are associated with reductions in antibody production.
In recent years, there also has been an emerging interest in immunometabolism to define alterations in metabolic pathways associated with immune function (O’Neill et al., 2016). Researchers have found that endocrine disruptors have immunomodulatory metabolic effects when in vitro models are exposed (Bansal, 2018). Previous research has shown that at high concentrations, PFOA exposure leads to disruptions in the mitochondrial membrane in rodent livers, similar to what is seen when using membrane detergents (Starkov and Wallace, 2018). These alterations to the integrity of mitochondrial membranes could affect the function of immune cells. Additionally, as PFAS like PFOA are known to be peroxisome proliferators, they have the potential to disrupt basal metabolic functions across a variety of cell types. Thus, one of our hypotheses is that PFOA affects antibody responses by altering B-cell metabolism and inhibiting the ability of B cells to appropriately transition from naïve B cells to antibody-secreting plasma cells. Therefore, the current studies were done to determine if PFOA-induced suppression of the primary IgM antibody response results from reductions in numbers of specific B-cell subsets and/or from alterations in immunometabolic functions in the first step of B development, activation of naïve B cells.
The appearance of other in use PFAS have been found in drinking water sources in coastal North Carolina, hexafluoropropylene oxide dimer acid, HFPO-DA (also known colloquially as “GenX”), and other understudied PFAS. The appearance of these toxicants has raised concerns and questions about their impact on community health, including immunization responses. These concerns are valid because although PFOA is no longer in use, due to its biopersistence, it is still present in the environment and in the serum of most individuals, thus making understanding the mechanism of PFAS-induced immune suppression even more critical. We also used a dose of PFOA that was not systemically toxic and that was based on previous research in our laboratory demonstrating suppression of the IgM TDAR. This work will help to elucidate potential molecular mechanisms by which PFOA and other PFAS may suppress antigen-specific antibody responses.
Materials and methods
Animals
C57BL/6 mice, 32 of each sex, were purchased from Charles River Laboratories and housed in the East Carolina University Brody School of Medicine animal facility (accredited by the Association for Assessment and Accreditation of Laboratory Animal Care). All procedures in this study were approved in advance by the ECU Institutional Animal Care and Use Committee and were conducted in accordance with the Guiding Principles in the Use of Animals in Toxicology. The C57BL/6 mouse strain was utilized for consistency with previously published findings as well as their sensitivity to the immunotoxic effects of PFAS (DeWitt et al., 2016). Upon arrival, animals were distributed by sex into polycarbonate cages of 3 animals/cage and weighed. Body weights were evaluated statistically by analysis of variance (ANOVA) and if statistical differences (p < .05) were detected, animals were rearranged to evenly distribute body weights across all cages within sex. Animals were acclimated to the facility for 5 days before any further manipulation, were housed in temperature-controlled conditions (23 ± 3°C) with a 12:12 h light:dark cycle with 50 ± 20% relative humidity, and were given access to food and water ad libitum.
Dosing
Fresh dosing solutions of PFOA (lot no. 1349401; Sigma) at a concentration 0.75 mg/ml were prepared weekly in sterile water with 0.5% Tween-20 as a vehicle. Vehicle control mice received sterile water with 0.5% Tween-20. Dosing solutions were stored at 4°C when not in use. Each day the dosing solutions were checked to ensure that they were free from observable signs of visible contamination. Mice were weighed daily, and dosing solution was administered via oral gavage to simulate drinking water exposure for 15 consecutive days at 0.1 ml/10 g of body weight. Dosing solutions were prepared so that this volume would result in the appropriate mg/kg of body weight dosage. The administered dose of PFOA was based on previous research in the lab demonstrating suppression of IgM TDAR at 7.5 mg/kg in the absence of overt systemic toxicity when given via gavage. This dose is one doubling above the lowest observed adverse effect level or 3.75 mg/kg reported for suppression of antigen-specific IgM antibody responses (DeWitt et al., 2008).
Serum and tissue collection
One day after the final dose was administered, mice were anesthetized with inhaled Isoflurane and blood was collected via neck vein transection into microcentrifuge tubes with a clot activator (Becton, Dickinson and Company, Franklin Lakes, New Jersey). Serum was separated from clotted blood at 4°C by centrifugation at then frozen at −80°C until further analysis. Fresh spleens were excised and weighed following terminal blood collections and immediately placed into RPMI 1640-medium (supplemented with 1% fetal bovine serum [FBS] and kept on ice for immunophenotyping or B-cell isolation and ex vivo analysis). The liver was removed and weighed and frozen at −80°C for later analyses.
Measurement of the T-cell-dependent antibody response (TDAR)
On the 11th day of dosing, mice in selected groups were immunized with 4 × 107 SRBCs (sheep red blood cells) in 0.2 ml of sterile saline by i.v. injection. The TDAR was assessed as described previously (Woodlief et al., 2021) using a commercially available mouse anti-SRBC IgM ELISA kit (Life Diagnostics, Inc., West Chester, Pennsylvania). In brief, serum samples and anti-SRBC IgM standards were diluted 50-fold and added to 96-well ELISA plates in duplicate. Plates were incubated at room temperature for 45 min on an orbital shaker. Wells were washed 5× in 1× washing solution, tapped dry, and enzyme conjugate reagent was then added to each well. Plates were incubated at room temperature for 20 min on an orbital shaker. The wells were again washed 5× in 1× wash solution and tapped dry, then TMB reagent was added to each well and plates were incubated at room temperature for 20 min. Stop solution (1 N HCL) was added to each well and the optical density at 450 nm was immediately measured on a microplate reader. The concentration of anti-SRBC IgM (units/ml) was calculated by fitting each sample onto a standard curve that was based on the absorbance of the anti-SRBC IgM standards.
B-cell phenotyping
Spleens in RPMI were mashed with the hard side of plunger from a 3-ml syringe and then passed through 70 µm filters to make single-cell suspensions. After lysis of red blood cells, suspended spleen cells were counted and viability was determined on a Nexcelon Bioscience Cellometer Auto 2000 cell counter (Nexcelom Bioscience, Lawrence, Massachusetts) and then adjusted to 2 × 107 cells/ml. Spleen cell suspensions were aliquoted and prepared for analysis by flow cytometry. Monoclonal antibodies coupled to fluorochrome-specific markers were used as follows: Spark NIR 685 CD19, PE CD43, BUV805 CD1d, PE-Cy7 CD21/CD35, eFluor 450 CD23, Super Bright 436 CD27, PE-Cy5 CD38, BV785 CD138, BUV395 CD45, and Live/Dead Blue (see Supplementary Table 1 for details on antibodies used). Each experiment contained 2 replicates of unstained cells as negative staining controls and 9 single-stained samples to establish fluorescent reference controls. Flow cytometric analysis was performed using a Cytek Aurora 5-laser full spectrum flow cytometer (Cytek Biosciences, Fremont, California) with 50 000 events collected per sample. Dead cells and debris were excluded from analysis by using Live/Dead Blue and gating the viable splenic lymphocyte populations. Unstained cells, single color reference controls, and fluorescence minus one controls were used to distinguish the negative populations from the positive populations for B cells. Leukocytes were gated based on CD45 expression. Cells were then gated for CD138, CD19, CD21/CD35, CD38, CD43, CD1D, CD27, and CD23 (see Supplementary Figure 2). B-cell subsets were classified as naïve (CD19+CD27−CD43+CD23lo/−), marginal zone (CD19+CD27−CD23/CD35hiCD1Dhi), follicular (CD19+CD23+CD21/CD35int), or plasmasblasts (CD19+CD138+CD38+). Mature B cells were identified as the sum of follicular and marginal zone cells. The total number of each cell type was determined from spleen cellularity.
Ex vivo spleen B-cell preparation and activation
Spleens were processed as described previously in the phenotyping section, with a few modifications. Spleen single-cell suspensions were counted and assessed for viability as described previously. B-cell fractions were isolated using Dynabeads (mouse CD43: Invitrogen/ThermoFisher Scientific, Waltham, Massachusetts) by negative selection. Live cell counts were used to determine a cell to bead ratio of 5 × 107 cells/ml to 50 ml CD43 beads + 2 ml cell medium (RPMI-1640 medium + 1% FBS). Cell and bead mixtures were incubated at room temperature for 20 min on an orbital shaker, and then placed on a cell separation magnet for 2–3 min to allow for beads to bind to the magnet. The resulting cell suspension was then transferred to a clean tube and allowed to sit for 2–3 min to remove residual beads; this step was performed twice. Samples were then centrifuged at 400 × g for 5 min at 4°C, resuspended in 1 ml of complete B-cell medium (RPMI-1640 + 10% FBS + 50 µM β-mercaptoethanol, 1% PenStrep, 1% Glutamax, 1% nonessential ammino acids, 1% sodium pyruvate) and counted on a Nexcelon Bioscience Cellometer Auto 2000 cell counter. Enriched B-cell fractions were combined from spleens of 2 mice of the same dose and sex and were then plated into 6-well cell culture plates (5 × 106 cells/well) and incubated for 24 h in the absence or presence of anti-CD40 (1 µg/ml) and IL-4 (20 ng/1 ml). Cells were harvested after 24 h, centrifuged at 400 × g at 4°C, resuspended in 500–1 ml of B-cell medium, counted, and then placed on ice for phenotyping or bioenergetic analysis.
Ex vivo B-cell phenotyping
Cells from the ex vivo spleen B-cell preparation and activation were added to microcentrifuge tubes at a concentration of 2 × 107 cells/ml. Activated B cells were identified with CD86-PE, a monoclonal antibody (eBiosciences, San Diego, California; see Supplementary Table 1 for details on antibodies used). Each experiment contained one tube of unstained unstimulated cells and one unstained tube of stimulated cells to provide the autofluorescence of each condition. Flow cytometric analysis was performed using a Cytek Aurora 5-laser full spectrum flow cytometer with 50 000 events collected per sample. Dead cells and debris were excluded from analysis by gating for lymphocytes using SSC-A and FSC-A. A singlet gate was then created using FSC-H and FSC-A to exclude doublets. After this gate was established, the cells were then gated for CD86. The total number of specific cells was determined from spleen cellularity.
Ex vivo B-cell mitochondrial markers
Oxygen consumption rate (OCR) was evaluated in enriched B cells 24 h after incubation in the presence or absence of anti-CD40 and IL-4. After incubation, cells were resuspended in respiration buffer (Seahorse RPMI, 20 nM glutamine, 200 nM glucose, 200 mM pyruvate) and plated at 5 × 105 cells/180 µl on a 96-well Seahorse plate (Agilent, Santa Clara, California). Prior to plating cells, Seahorse plates were coated with poly-d-lysine (20 µl/well) as per manufacturer’s instructions. OCR was determined using the Mitochondrial Stress Test kit (Agilent) as per the manufacturer’s instructions with the following inhibitors: Port A: Oligomycin (0.5 µM); Port B: FCCP (2.0 µM); Port C: FCCP (2.0 µM); Port D: Rot/Ana (0.5 µM). Basal, maximal (FCCP induced), and reserve capacity (maximal-basal) were calculated by OCR/5 × 105 cells. Metabolic parameters were acquired from XF Wave software (Agilent, Santa Clara, California) and calculated using Microsoft Excel.
Statistical analysis
Statistical analyses were performed using the Statistical Analysis System (SAS, Cary, North Carolina). Daily body weights within sex were analyzed by a 2-way repeated measures ANOVA with the between-subjects variable being dose and the within-subjects variable being day. Within each sex a 1-way ANOVA by dose was used to analyze liver weights, anti-SRBC IgM levels, spleen cellularity, and B-cell phenotype. A 2-way ANOVA was used for ex vivo B-cell activation and B-cell bioenergetics with the between-subjects variable being dose and the within-subjects variable being stimulation. Post hoc 1-way ANOVAs and pairwise t-tests were used when F-statistics from 2-way or 1-way ANOVAs indicated statistical significance. Statistical significance was determined when the p-value was less than .05. A paired t-test was used to examine the difference between the control unstimulated and control stimulated groups as well as the PFOA unstimulated and PFOA stimulated groups for B-cell bioenergetics data.
Results
Organ weights and cellularity
PFOA exposure resulted in a significant reduction in spleen weights in both female (30.4%; p < .05) and male (27.7%; p < .05) cohorts, compared with sex-matched vehicle controls (Table 1). In addition to reduced splenic weight, dosed female and male mice also had a significant decrease in spleen cellularity, measured as cells/mg tissue weight; in female mice cellularity was decreased by 27.1% and in male mice cellularity was decreased by 20.5% (Table 1). Oppositely, liver weights from the PFOA-dosed mice in both male and female groups increased statistically by 144%, on average, compared with sex-matched vehicle controls (Table 1).
Table 1.
Average absolute and relative organ weights and splenic cellularity in female and male C57BL/6 mice given PFOA by gavage for 15 days
| Females |
Males |
|||
|---|---|---|---|---|
| 0 mg/kg | 7.5 mg/kg | 0 mg/kg | 7.5 mg/kg | |
| Body weight (g) | 19.65 ± 0.86 | 20.78 ± 0.58 | 25.45 ± 0.60 | 20.78 ± 0.58 |
| Absolute organ weight | ||||
| Liver (mg) | 910.0 ± 0.02 | 2260 ± 0.09* | 1280 ± 0.04 | 3140 ± 0.13* |
| Spleen (mg) | 116.3 ± 12.11 | 81.0 ± 4.43* | 90.0 ± 0.00 | 65.0 ± 0.00* |
| Relative (organ weight/body weight) organ weight | ||||
| Liver | 46.9 ± 2.37 | 108.4 ± 2.14* | 50.2 ± 1.32 | 152.1 ± 7.14* |
| Spleen | 6.2 ± 1.06 | 3.8 ± 0.13 | 3.6 ± 0.12 | 2.9 ± 0.23 |
| Splenic cellularity | ||||
| Cell counts | 7.55 ± 6.18 × 107 | 5.50 ± 4.83 × 107* | 6.04 ± 3.87 × 107 | 4.80 ± 2.73 × 107* |
A statistical (p < .05) difference from match-sex control group.
N = 8 animals/group.
The TDAR
As measured by anti-SRBC soluble IgM in serum, both female and male mice dosed with PFOA had a statistical reduction in the TDAR; 79% and 89.9%, respectively (p < .05; Figure 1).
Figure 1.
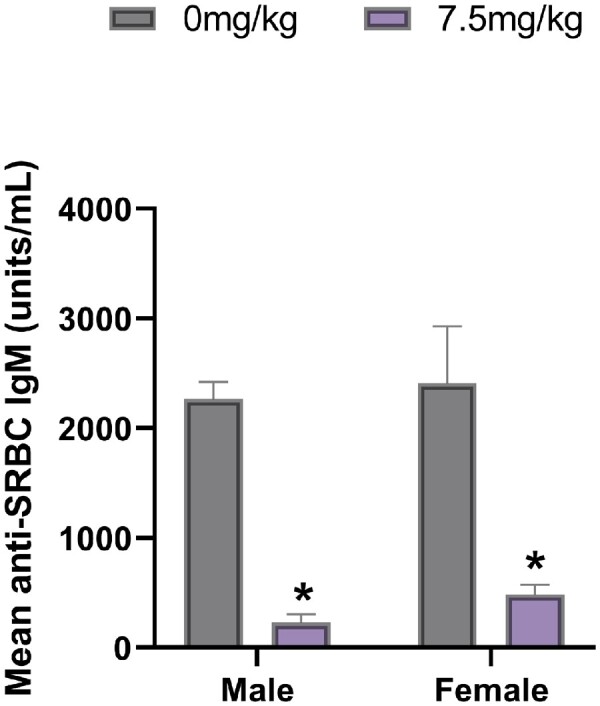
Mean (± SD) SRBC-specific IgM antibody concentrations in male and female C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. *p < .05 compared with sex-matched control group. N = 8 animals/sex/group.
B-cell phenotype
Exposure to PFOA resulted in a nonsignificant decrease in the total number of overall B cells by 1.45% (Supplementary Figure 1) and in the numbers of naïve, follicular, plasmablasts, and memory B cells (Figure 2A) in the spleens of female mice, compared with vehicle controls. Alternatively, male mice displayed a more robust response to PFOA exposure. The number of overall B cells was statistically reduced by 15%. The number of follicular cells was statistically increased by 9 ± 1.15% and the number of plasmablasts was statistically reduced from vehicle controls by 55 ± 0.08%. Although naïve, marginal zone, memory cells were reduced in dosed male mice, these reductions were not statistically significant (Figure 2B). Additionally, female and male mice exhibited a biologically based sex difference, with female mice having higher overall numbers within follicular, plasmablast, and memory B cells. Numbers of naïve and marginal B cells were approximately equal between female and male mice. PFOA exposure did not change the direction of this sex difference.
Figure 2.

A, Mean (± SD) number of B cells counted in spleens of female C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. The CD19+ marker was used to define the following populations: Memory [CD19 +CD27], Plasmablasts [CD138 +CD38], Marginal [CD1D], Naive [CD43], and Follicular [CD23]. No statistical differences were detected among the dose groups. N= 8 animals/dose. B, Mean (± SD) number of B cells counted in spleens from male C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. The CD19+ marker was used to define the following populations: Memory [CD19 +CD27], Plasmablasts [CD138 +CD38], Marginal [CD1D], Naive [CD43], and Follicular [CD23] *p < .05 compared with control group. N = 8 animals/dose.
Ex vivo B-cell activation
B-cell functionality was assessed by upregulation of the co-stimulatory marker CD86 in response to anti-CD40 and IL-4 stimulation. At baseline, without stimulation, there was no difference in the number of CD86+ B cells between vehicle and PFOA exposed mice, for either female or male cohorts. In vitro stimulation with anti-CD40 and IL-4 significantly increased the number of CD86+ B cells in all groups, when compared with their appropriate unstimulated controls (Figure 3A and 3B). There were no observed statistical differences between vehicle and PFOA exposed stimulated B cells in either female or male cohorts, suggesting that PFOA did not affect the ability of B cells to respond to stimulation.
Figure 3.

A, Mean (± SD) number of B cells isolated from spleens of female C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. Isolated naïve B cells were stimulated ex vivo with IL-4 and anti-CD40 and counted following a 24-h incubation. *p < .05 compared with respective unstimulated group. N = 3 animals/dose. B, Mean (± SD) number of B cells isolated from spleens of male C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. Isolated naïve B cells were stimulated ex vivo with IL-4 and anti-CD40 and counted following a 24-h incubation. *p < .05 compared with control group. N = 3 animals/dose.
B-cell mitochondrial markers
Mitochondrial function was determined by measuring oxygen consumption in response to ex vivo anti-CD40 and IL-4 stimulation. The measurements included basal, maximal, and spare respiratory capacity. Basal respiration was captured to provide a baseline of the cellular OCR under normal conditions. Maximal respiration provided values of maximum cellular oxygen consumption under the influence of increased energetic demand. Spare respiratory capacity reflected the cellular capability to respond to changes in energetic demand by evaluating the difference between basal and maximal respiration. These measurements were made 24 h after the 15-day exposure to PFOA. In the overall model, changes in mitochondrial markers were statistically significant for both male and female groups, indicating differences due to stimulation and/or PFOA exposure. Within control groups for both sexes, pmol/min increased following stimulation for all mitochondrial markers, but these increases were not statistically significant. In cells from female animals, basal respiration, maximum respiration, and spare capacity increased by 46.7%, 33.6%, and 29.5%, respectively, following stimulation (Figure 4A). In cells from male animals, basal respiration, maximum respiration, and spare capacity increased by 98%, 44% and 34%, respectively, following stimulation (Figure 4B). In comparison, cells from animals exposed to PFOA had statistically significant increases in all mitochondrial markers. In cells from female animals, basal respiration, maximum respiration, and spare capacity increased by 264%, 172% and 153%, respectively, following stimulation (Figure 4A). Although not statistically significant, responses due to stimulation in cells from the PFOA-exposed group represented a 5-fold increase across all measurements when compared with stimulation in the control group. In cells from male animals, basal respiration, maximum respiration, and spare capacity increased by 200%, 252% and 266%, respectively, following stimulation (Figure 4B). Additionally, following stimulation, maximum respiration and spare capacity differed between cells from control animals and cells from PFOA-exposed animals, with PFOA-exposed cells having about a 130% increase, on average, compared with the control cells. Responses due to stimulation in cells from the PFOA-exposed group represented a 2-fold increase in basal respiration, a 7-fold increase in maximum respiration, and a 5-fold increase in spare capacity (Figure 4A and 4B).
Figure 4.

A, Mean (± SD) measures of mitochondrial function in B cells isolated from spleens of female C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. Isolated naïve B cells were stimulated ex vivo with IL-4 and anti-CD40 and were evaluated with a mitochondrial stress test following a 24-h incubation. The overall 2-way repeated measures ANOVA was statistically significant (p < .05). Basal respiration, maximum capacity, and spare capacity differed statistically regardless of PFOA exposure or stimulation, demonstrating that these 3 mitochondrial measures differed from one another. Within control groups, stimulation did not statistically increase responses from unstimulated cells and within PFOA-treated groups, stimulation did statistically increase responses from unstimulated cells (*p < .05). N = 3 animals per dose. B, Mean (± SD) measures of mitochondrial function in B cells isolated from spleens of male C57BL/6 mice exposed to PFOA or a vehicle control via gavage for 15 days. Isolated naïve B cells were stimulated ex vivo with IL-4 and anti-CD40 and were evaluated with a mitochondrial stress test following a 24-h incubation. The overall 2-way repeated measures ANOVA was statistically significant. Basal respiration, maximum capacity, and spare capacity differed statistically regardless of PFOA exposure or stimulation, demonstrating that these 3 mitochondrial measures differed from one another. Within the control groups, ex vivo stimulation did not statistically alter the pmol/min within basal respiration, maximum capacity, or spare capacity. Within the PFOA-exposed groups, ex vivo stimulation did statistically alter the pmol/min within basal respiration, maximum respiration, and spare capacity (*p < .05). There was not a statistical difference between control and PFOA exposed groups for basal respiration regardless of stimulation, but there was a difference between control and PFOA-exposed groups for both maximum capacity and spare capacity in stimulated cells (*p < .05). N = 3 animals per dose.
DISCUSSION
Studies with animal models and of exposed human populations have reported that exposure to PFAS diminishes the ability of the immune system to fight infections or respond to vaccines (Fenton et al., 2021). According to current projections, up to 69 million deaths will be prevented between 2000 and 2030 due to vaccinations and more than 21 million hospitalizations and 732 000 deaths among children born in the last 20 years will be prevented (CDC, 2014). An effective immune system is critical in reaching these projections and exposure to agents, such as PFAS, that compromise the immune system are detrimental to public health.
This study observed that PFOA exposure reduced antibody responses and reduced numbers of splenic B cells, which may be linked to modulation of B-cell immunometatoblic functions. Evaluation of the TDAR is essential for detecting immunotoxicity and immunosuppression (DeWitt et al., 2012, 2019). Previous studies in our lab have demonstrated a reduction in the TDAR following PFOA exposure and our current study is in agreement. PFOA (7.5 mg/kg) given orally to male and female mice for 15 days suppressed the TDAR (Figure 1). Similar to past findings, no reductions in body weight (Table 1) or signs of systemic toxicity over the duration of exposure were observed (DeWitt et al., 2008, 2016). Liver weights also were statistically increased in PFOA-exposed mice, consistent with previous studies of PFOA (Fenton et al., 2021).
Although this immune suppression has been linked to B-cell function, it has not previously been linked to reductions in total numbers of splenic B cells (DeWitt et al., 2016). However, to date, no studies have reported enumeration of B-cell subsets following PFOA exposure, which is why the current study was conducted. In a study with PFOS, the total number of splenic B cells did not change but there was a reduced percentage of mature B cells (Torres et al., 2021). In an immunotoxicity study of PFOA, De Guise and Levin (2021) exposed B6C3F1 female mice to 0, 1.88, or 7.5 mg/kg of PFOA in drinking water for 4 weeks and evaluated the TDAR using keyhole limpet hemocyanin as an antigen. PFOA exposure reduced the TDAR and suppressed Th2 cytokines, which they interpreted as mechanistically relevant to suppression of the TDAR. Although our study differed in exposure duration (15 days vs 4 weeks) and in administration (gavage vs drinking water), we did not evaluate serum cytokine levels in the current study and De Guise and Levin (2021) did not enumerate B- or T-cell populations. We agree that PFOA can modify cytokine levels and have effects on T cells (Son et al., 2009). However, PFOA and PFOS exposure in mice can reduce antibody production when immunized with T cell-independent antigens, indicating that deficiencies in B cells is a more likely mechanistic explanation (DeWitt et al., 2016; Peden-Adams et al., 2008).
We report here reductions in total number of B cells in PFOA-exposed female (not statistically significant) and male mice (Supplemental Figure 1), which has not been consistently observed in previous studies of adult mice. In DeWitt et al. (2016), adult female C57BL/6 mice had no alterations in total B-cell numbers after oral exposure to 0, 7.5, or 30 mg/kg of PFOA for 15 days. Regarding other PFAS, in a study of sulfluramid, an insecticide that degrades to PFOS, no changes in total B-cell numbers in adult female B6C3F1 mice were observed (Peden-Adams et al., 2007). Keil et al. (2008) exposed male and female B6C3F1 mice to PFOS during gestation and observed a 20% reduction in total B-cell numbers in female offspring exposed to the highest dose (5 mg/kg). Although these studies had slight differences in exposure duration and strain of mice used, they were largely in agreement that total numbers of B cells were not robustly diminished by exposure to PFOS. However, as antibody suppression has been reported across experimental animal and epidemiological studies, a logical first step in uncovering potential mechanisms of this suppression was enumeration of B-cell subsets.
B-cell development is a complex sequence of events dependent on the microenvironment in which cells exist. If conditions are favorable, this allows for progression to the next step of development (Loder et al., 1999). Modifications in the microenvironment can lead to alterations that can disrupt the B-cell differentiation pathway (Loder et al., 1999). Our flow cytometric analyses revealed that exposure to PFOA decreased numbers of naïve and marginal zone B cells in male mice (although these reductions were not statistically significant). These cells provide a rapid response of IgM and IgG antibodies following infections by blood-borne viruses and encapsulated bacteria (Cerutti, 2013). Reduction of naïve B-cell numbers affects development of the B-cell life cycle because naïve B cells must become activated to ultimately differentiate into memory or plasma cells. If fewer naïve B cells are circulating, there likely will be reduced humoral responses (Schulz et al., 2021). Plasmablasts are precursors to plasma cells, which secrete antibodies; these cell numbers were decreased by PFOA exposure in male and female mice. Both male and female mice also had reductions in the TDAR, with males having a slightly greater reduction (89.9%) compared with females (79.0%). Along the B-cell development pathway, plasmablasts stem from follicular B cells (Abbas et al., 2022). We observed a nonstatistical decrease in follicular B-cell numbers in the females and a statistical increase in follicular B-cell numbers in male mice exposed to PFOA. Follicular B cells participate in TDAR and are essential for T-dependent humoral immunity (Mirzaei, 2022). The transition from activated follicular B cells into plasma cells is an intricate process influenced by cytokines, chemokines, and coreceptors of various cells (Nera et al., 2015). For example, T follicular helper cells function by activating B follicular cells, which allows them to differentiate into high-affinity B cells (De Silva and Klein, 2015). These B cells then differentiate into plasmablasts. Variation in this process leads to lower affinity B cells that may not be fit to continue (Nera et al., 2015). Decreases in male plasmablasts may be due to B cells being lower affinity cells, which can differentiate into memory B cells or return to the germinal center to undergo more rounds of somatic hypermutation (Victora and Nussenzweig, 2012). Diminishment in numbers of multiple B-cell subsets demonstrates that PFOA has effects on the B-cell development pathway leading to decreased immune function. Although a limitation of this study is a focus on B cells at the peak of IgM antibody levels, important next steps will require evaluating B cells across the trajectory of B-cell development, from stimulation to antibody production. This will improve understanding of effects of PFAS exposure on B-cell differentiation and proliferation.
B cells encounter paramount metabolic demands to complete the activation phase of cell division (Pearce et al., 2013). To survive, B cells must be able to handle bioenergetic requirements of an increase in cell proliferation and a changing environment (Pearce et al., 2013).Environmental changes include an increased rate of transcription and translation, increased consumption of nutrients, and changes in cell size (D’Souza and Bhattacharya, 2019). Assessing bioenergetic health of immune cells is often used as an indicator of overall bioenergetic health (Kramer et al., 2014) and an important feature of mitochondria is the ability to regulate activation, differentiation, and survival of immune cells. This leads to the interlinking of immune functions such as the TDAR with mitochondrial-based immunometabolism to understand immunosuppression. B cells from both control and PFOA-exposed animals responded to stimulation and became activated, an essential step to initiating downstream events. We also observed that only cells from male mice exposed to PFOA had exaggerated responses to stimulation. This suggests that mitochondria in B cells exposed to PFOA were fit to handle energy challenges of activation but may be hyperresponsive following PFOA exposure. This is concerning because cells may not be able to keep up with progressive energy demands of proliferation and further differentiation. These observations suggest that PFOA exposure leads to a B-cell phenotype overreactive to activation that may not be sustainable long term. If energy availability is insufficient due to over expenditure in earlier steps this could lead to B-cell subset development arrest, diminishment, or a metabolic collapse.
We observed changes in numbers of B-cell subsets as well as mitochondrial markers following PFOA exposure, indicating that the B-cell developmental trajectory may have deficiencies that affect differentiation or proliferation, leading to suppression of the TDAR. Our analysis also highlighted sex differences with males appearing more sensitive to effects of PFOA exposure than females. Although female mice had higher overall numbers within follicular, plasmablast, and memory B cells, PFOA exposure did not change the direction of this sex difference, supporting the increased sensitivity of male mice to PFOA exposure. Our findings suggest that B-cell development and survival may be hindered by PFOA exposure, but that activation of B cells was not. This work will help to further elucidate potential molecular mechanisms by which PFOA and other PFAS may suppress the TDAR.
Supplementary Material
Acknowledgments
The authors thank the ECU Department of Comparative Medicine for their humane treatment and husbandry of research animals, Matthew Wittenborn for his quality assurance assistance, and Dr Debajit Bhowmick for his assistance with flow cytometric analyses.
Contributor Information
Krystal D Taylor, Department of Pharmacology & Toxicology, Brody School of Medicine, East Carolina University, Greenville, North Carolina 27834-4300, USA.
Tracey L Woodlief, Department of Pharmacology & Toxicology, Brody School of Medicine, East Carolina University, Greenville, North Carolina 27834-4300, USA.
Aya Ahmed, Department of Pharmacology & Toxicology, Brody School of Medicine, East Carolina University, Greenville, North Carolina 27834-4300, USA.
Qing Hu, Department of Pharmacology & Toxicology, Brody School of Medicine, East Carolina University, Greenville, North Carolina 27834-4300, USA.
Patrick C Duncker, Cytek Biosciences, Mid-Atlantic Region, Fremont, California 94538-6407, USA.
Jamie C DeWitt, Department of Pharmacology & Toxicology, Brody School of Medicine, East Carolina University, Greenville, North Carolina 27834-4300, USA.
Supplementary data
Supplementary data are available at Toxicological Sciences online.
Declaration of conflicting interests
The funders had no role in the study’s design or in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results. J.C.D. declares that she currently is engaged as a plaintiff’s expert witness in several cases involving PFAS.
Funding
US National Institute of Environmental Health Sciences (1P42ES031009-01, NC State University Center for Environmental and Human Health Effects of PFAS) via a subaward to East Carolina University (ECU) through NC State University.
References
- Abbas A. K., Lichtman A. H., Pillai S. (2022). Leukocyte circulation and migration into tissues. Cell. Mol. Immunol. 10, 43–61. [Google Scholar]
- ATSDR. (2021). Toxicological profile for PFAS. Available at: https://www.atsdr.cdc.gov/toxprofiles/tp200.pdf. Accessed January 10, 2023.
- Bansal A., Henao-Mejia J., Simmons R. A. (2018). Immune system: An emerging player in mediating effects of endocrine disruptors on metabolic health. Endocrinology 159, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. (2014). Benefits from immunization during the vaccines for children program era – United States, 1994–2013. Morbidity and Mortality Weekly Report (MMWR), Centers for Disease Control and Prevention. Available at: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6316a4.htm. Accessed January 10, 2023.
- Cerutti A., Cols M., Puga I. (2013). Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 13, 118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Guise S., Levin M. (2021). Suppression of Th2 cytokines as a potential mechanism for reduced antibody response following PFOA exposure in female B6C3F1 mice. Toxicol. Lett. 351, 155–162. [DOI] [PubMed] [Google Scholar]
- DeWitt J. C., Copeland C., Strynar M., Luebke R. (2008). Perfluorooctanoic acid-induced immunomodulation in adult C57BL/6J or C57BL/6N female mice. Environ. Health Perspect. 116, 644–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt J. C., Williams W., Creech N. J., Luebke R. W. (2016). Suppression of antigen-specific antibody responses in mice exposed to perfluorooctanoic acid: Role of PPARα and B cell targeting. J. Immunotoxicol. 13, 38–45. [DOI] [PubMed] [Google Scholar]
- DeWitt J. C., Blossom S. J., Schaider L. (2019). Exposure to per- and polyfluoroalkyl substances leads to immunotoxicity: Epidemiological and toxicological evidence. J. Expo. Sci. Environ. Epidemiol. 29, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt J. C., , Peden-AdamsM. M., , KellerJ. M., and , Germolec D. R. (2012). Immunotoxicity of perfluorinated compounds: Recent developments. Toxicol. Pathol. 40, 300–311. [DOI] [PubMed] [Google Scholar]
- De Silva N., Klein U. (2015). Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 15, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza L., Bhattacharya D. (2019). Plasma cells: you are what you eat. Immunol. Rev. 288, 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Environmental Protection Agency. (2022a). PFAS explained. Available at: https://www.epa.gov/pfas/basic-information-pfas. Accessed January 10, 2023.
- Environmental Protection Agency (2022b). Drinking water health advisories for PFOA and PFOS. 2022 interim updated PFOA and PFOS health advisories. Available at: https://www.epa.gov/sdwa/drinking-water-health-advisories-pfoa-and-pfos. Accessed January 10, 2023.
- Fenton S., Ducatman A., Boobis A., DeWitt J., Lau C., Ng C., Smith J. S., Roberts S. M. (2021). Per‐ and polyfluoroalkyl substance toxicity and human health review: Current state of knowledge and strategies for informing future research. Environ. Toxicol. Chem. 40, 606–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glüge J., Scheringer M., Cousins I. T., DeWitt J. C., Goldenman G., Herzke D., Lohmann R., Ng C. A., Trier X., Wang Z. (2020). An overview of the uses of per- and polyfluoroalkyl substances (PFAS). Environ. Sci. Process. Impacts. 22, 2345–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil D. E., Mehlmann T., Butterworth L., Peden-Adams M. M. (2008). Gestational exposure to perfluorooctane sulfonate suppresses immune function in B6C3F1 mice. Toxicol. Sci. 103, 77–85. [DOI] [PubMed] [Google Scholar]
- Kramer P. A., Ravi S., Chacko B., Johnson M. S., Darley-Usmar V. M. (2014). A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: implications for their use as bioenergetic biomarkers. Redox Biol. 2, 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loder B. F., Mutschler B., Ray R. J., Paige C. J., Sideras P., Torres R., Lamers M. C., Carsetti R. (1999). B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor–derived signals. J. Exp. Med. 190, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei H. R. (2022). Adaptive immunity. Encycl. Infect. Immun. 1, 39–55. [Google Scholar]
- Nera K.-P., Kyläniemi M. K., Lassila O. (2015). Regulation of B cell to plasma cell transition within the follicular B cell response. Scand. J. Immunol. 82, 225–234. [DOI] [PubMed] [Google Scholar]
- O’Neill L. A. J., Kishton R. J., Rathmell J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce E. L., Poffenberger M. C., Chang C. H., Jones R. G. (2013). Fueling immunity: Insights into metabolism and lymphocyte function. Science 342, 1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden-Adams M. M., , EuDalyJ. G., , DabraS., , EuDalyA., , HeesemannL., , SmytheJ., and , Keil D. E. (2007). Suppression of humoral immunity following exposure to the perfluorinated insecticide sulfluramid. J. Toxicol. Environ. Health A. 70, 1130–1141. [DOI] [PubMed] [Google Scholar]
- Peden-Adams M. M., Keller J. M., EuDaly J. G., Berger J., Gilkeson G. S., Keil D. E. (2008). Suppression of humoral immunity in mice following exposure to perfluorooctane sulfonate. Toxicol. Sci. 104, 144–154. [DOI] [PubMed] [Google Scholar]
- Schulz E., Hodl I., Forstner P., Hatzl S., Sareban N., Moritz M., Fessler J., Dreo B., Uhl B., Url C., et al. (2021). Cd19+IgD+CD27- naïve B cells as predictors of humoral response to COVID 10 mRNA vaccination in immunocompromised patients. Front. Immunol. 12, 803742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H.-Y., Lee S., Tak E.-N., Cho H.-S., Shin H.-I., Kim S.-Y., Yang J.-H. (2009). Perfluorooctanoic acid alters T lymphocyte phenotypes and cytokine expression in mice. Environ. Toxicol. 24, 580–588. [DOI] [PubMed] [Google Scholar]
- Starkov A. A., Wallace K. B. (2002). Structural determinants of fluorochemical-induced mitochondrial dysfunction. Toxicol. Sci. 66, 244–252. [DOI] [PubMed] [Google Scholar]
- Sunderland E. M., Hu X. C., Dassuncao C., Tokranov A. K., Wagner C. C., Allen J. G. (2019). A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFAS) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 29, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres L., Redko A., Limper C., Imbiakha B., Chang S., August A. (2021). Effect of perfluorooctanesulfonic acid (PFOS) on immune cell development and function in mice. Immunol. Lett. 233, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora G. D., Nussenzweig M. C. (2012). Germinal centers. Annu. Rev. Immunol. 30, 429–457. [DOI] [PubMed] [Google Scholar]
- Woodlief T., Vance S., Hu Q., DeWitt J. (2021). Immunotoxicity of per- and polyfluoroalkyl substances: Insights into short-chain PFAS exposure. Toxics 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.