

Abstract
The degradation tag (dTAG) system for target protein degradation can remove proteins from biological systems without the drawbacks of some genetic methods, such as slow kinetics, lack of reversibility, low specificity, and the inability to titrate dosage. These drawbacks can make it difficult to compare toxicity resulting from genetic and pharmacological interventions, especially in vivo. Because the dTAG system has not been studied extensively in vivo, we explored the use of this system to study the physiological sequalae resulting from CDK2 or CDK5 degradation in adult mice. Mice with homozygous knock-in of the dTAG sequence onto CDK2 and CDK5 were born at Mendelian ratios despite decreased CDK2 or CDK5 protein levels in comparison with wild-type mice. In bone marrow cells and duodenum organoids derived from these mice, treatment with the dTAG degrader dTAG-13 resulted in rapid and robust protein degradation but caused no appreciable change in viability or the transcriptome. Repeated delivery of dTAG-13 in vivo for toxicity studies proved challenging; we explored multiple formulations in an effort to maximize degradation while minimizing formulation-related toxicity. Degradation of CDK2 or CDK5 in all organs except the brain, where dTAG-13 likely did not cross the blood brain barrier, only caused microscopic changes in the testis of CDK2dTAG mice. These findings were corroborated with conditional CDK2 knockout in adult mice. Our results suggest that the dTAG system can provide robust protein degradation in vivo and that loss of CDK2 or CDK5 in adult mice causes no previously unknown phenotypes.
Keywords: CDK2, CDK5, degradation, PROTAC, toxicity, mice
In the realm of drug discovery, knowledge of a target protein’s function and potential toxicity liabilities after modulation is advantageous in assessing target safety, lead molecules, and therapeutic index. Inhibiting or removing a protein from a biological system can reveal the function of that protein and has been a staple in understanding gene function. Because genetic methods that remove proteins tend to be more specific than chemical methods that inhibit proteins, phenotypes resulting from chemicals are frequently compared with genetic techniques. Multiple genetic techniques to remove proteins from adult animals, which queries a protein’s function during homeostasis, have been developed and implemented such as conditional knockouts (cKOs) (Gu et al., 1994), RNA interference (Rubinson et al., 2003), and chemical genetic approaches (Banaszynski et al., 2008; Buckley et al., 2015; Chung et al., 2015; Yesbolatova et al., 2020). Although these techniques have yielded numerous scientific insights, RNA interference and cKOs tend to suffer from a lack of titratability, lack of reversibility, and slow kinetics (Gu et al., 1994; Rubinson et al., 2003). Numerous chemical genetic approaches have emerged for targeted protein degradation and their utility has been compared in vitro (Bondeson et al., 2022), but data surrounding in vivo compatibility is scarce.
The recently described degradation tag (dTAG) system for target protein degradation (Nabet et al., 2018) is an appealing system to remove proteins from adult organisms to study target function, safety, and efficacy. In brief, the dTAG, which is the FBKP12 protein with an F36V mutation that affords selective ligandability, is fused to a target protein, and treatment with the dTAG-targeted peptide-based proteolysis targeting chimeric molecule (PROTAC) dTAG-13 causes degradation of the target protein. This degradation is rapid, potent, titratable, and reversible. Although the dTAG system has been used in vivo (Abuhashem et al., 2022; Nabet et al., 2018), these studies did not employ repeat dosing and did not thoroughly examine potential toxicities resulting from target protein degradation or use of the dTAG system in general.
CDK2 may be an attractive therapeutic target in cancers that have amplified CDK2’s binding partner CCNE (Natrajan et al., 2012; Teng et al., 2020; Yang et al., 2015) or in breast cancers that upregulate CCNE1 after acquired resistance to CDK4/6 inhibitors (Pandey et al., 2020; Turner et al., 2019). CDK2 is highly homologous to CDK5, a protein with well-known roles in the brain (Su and Tsai, 2011). However, CDK5 has also been implicated in regulating the immune response, insulin levels, angiogenesis (Sharma and Sicinski, 2020), and lymphangiogenesis (Liebl et al., 2015). Most CDK2 inhibitors (Wells et al., 2020) or degraders (Teng et al., 2020) tend to inhibit or degrade CDK5. Given the purported physiological roles of CDK2 and CDK5, it is unknown whether on-target CDK2 or off-target CDK5 perturbation with CDK2 targeted agents will cause significant toxicity.
The dTAG system is ideal for studying CDKs as their signaling can be rapid, dynamic, and redundant (Santamaría et al., 2007), necessitating fast kinetics with high specificity. Here we generate CDK2dTAG and CDK5dTAG knock-in mice, treat these adult homozygous (HOM) mice with dTAG-13, and determine holistically if any toxicity results from use of the dTAG system itself or degradation of CDK2 or CDK5 in adult mice. Although repeated delivery of dTAG-13 safely and effectively in vivo proved challenging, we identified 2 formulations with useful but contrasting properties: one formulation that affords potent protein degradation while causing moderate toxicity due to formulation-related skin lesions, and one formulation that affords more modest protein degradation with minimal formulation-related toxicity. Using these formulations, we found that degrading CDK2 or CDK5 for 14 or 26 days in organs except the brain, where degradation did not occur potentially due to the large size of dTAG-13, results in no phenotypic changes other than microscopic findings in the testis following CDK2 degradation, which was previously found utilizing germline CDK2 KO mice (Ortega et al., 2003). We corroborated these results for CDK2 in adult mice by creating and utilizing a CDK2 cKO mouse. Our results suggest that CDK2 or CDK5 loss in adult mice in organs other than the brain only causes a phenotypic change in the testis and demonstrates the utility of the dTAG system for in vivo targeted protein degradation to facilitate target safety derisking and exploration.
Materials and methods
Test articles reagents
4-hydroxytamoxifen (4-OHT) was purchased from Selleck (catalog no. 68392-35-8) and dissolved in DMSO at 10 mM. Tamoxifen was purchased from Sigma (catalog no. T5648-5g) and prepared at 7.5 mg/ml by dissolving in sunflower seed oil (Sigma, catalog no. S5007-250ml) while stirring for about 2 h protected from light. dTAG-13 was synthesized by WuXi AppTec. For in vitro experiments, 4-OHT was used instead of taxmoxifen because 4-OHT is the active metabolite of tamoxifen in vivo (Felker et al., 2016).
Housing of mice
All procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by Pfizer’s Institutional Animal Care and Usage Committee. Mice were housed in individually ventilated cages with Bed-o’Cobs (The Andersons, Inc.) bedding at 68°F–79°F and 30%–70% humidity with 12 h light, 12 h dark cycles. Air exchange was performed 12–16 times per hour with 100% fresh filtered air. Mice were provided with municipal drinking water that is further purified by reverse osmosis and certified irradiated rodent diet 2916C (Envio Teklad Global Diet) ad libitum. Mice were generally 7–10 weeks old unless otherwise noted, and for almost all studies, both male and female mice were included for histopathological analysis.
Generation of CDK2-dTAG and CDK5-dTAG mice
To perform the knock-in, donor females were superovulated by intraperitoneal (IP) injection of pregnant mare serum gonadotropin (ProSpec, catalog no. HOR-272, 5 units/mouse) on day 0 and human chorionic gonadotropin (ProSpec, catalog no. HOR-250, 5 units/mouse) 44–48 h after pregnant mare serum gonadotropin injection. Females were individually housed with a stud male to obtain fertilized eggs, which were collected in M2 Media (CytoSpring, catalog no. M2114), transferred into KSOM media (CytoSpring, catalog no. K0104) and stored in a 37°C incubator. To prepare gRNAs, PCR was performed using PrimeStar GXL DNA polymerase (Takara Bio, catalog no. R050B) using Primer1 and Primer2 (sequences can be found in Supplementary Material in the Supplementary Sequences section). These PCR products were purified using the Qiagen PCR Purification Kit (Qiagen, catalog no. 28104) and then 200–400 ng was used for in vitro transcription using the HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs, catalog no. E2050S). This product was treated with 1 µl of DNase (from the HiScribe T7 kit) and purified using the Zymo RNA Clean and Concentrator Kit-5 (Zymo Research, catalog no. R1014). Ovaries were microinjected in M2 media on glass slides using an Eppindorf FemtoJet4i and 1 µM Cas9 protein (IDT, catalog no. 1081058), 1 µM in vitro transcribed sgRNA and 2.5 ng double-stranded DNA donor. DNA donor sequences can be found in Supplementary Material in the Supplementary Sequences section. After microinjection, embryos were implanted into pseudo-pregnant recipient females on the same or next day.
Generating CDK2 cKO mice
A cKO model was created by Pfizer’s in-house transgenic facility utilizing CRISPR/Cas9 technology (Wang et al., 2013; Yang et al., 2013) by inserting 2 LoxP sites flanking the exons 2 and 3 of the Cdk2 gene. One LoxP was positioned at 228 bp upstream of the exon 2 and the other at 106 bp downstream of the exon 3. A donor plasmid for repair consisted of the floxed genomic sequence flanked by 0.5 kb 5′ homology arm and 0.8 kb 3′ homology arm. Two sgRNAs targeting the upstream and downstream inserting sites, 5′- AAGAAAGGTAATAGGGGAGT and 5′-GACTAATGAGTAGGAGAAGG, respectively, were synthesized by in vitro transcription. For genome editing in mouse zygotes, Cas9 protein (Integrated DNA technologies, catalog no. 1081058), the 2 sgRNAs, and the donor plasmid were injected into 1-cell embryos harvested from superovulated C57BL/6J (JAX, catalog no. 000664) donors. After microinjection, embryos were transferred to pseudo-pregnant females to generate live pups. To identify founders with insertion of the 2 LoxP sites into the targeted locus, genomic PCR was performed with primers external to the 5′ and 3′ homology arms (5′-AAGGTGGAGAAGATTGGAGAG and 5′-GTCAGAGGACAATTTGCAGA) and the amplicon was sequenced to confirm correct editing. Founders were bred to wild-type (WT) C57BL/6J mice for germline transmission of the cKO allele. Resulting heterozygotes of a selected founder line were crossed with ROSA26-CreERT2 line (JAX no. 008463).
Isolation of bone marrow-derived cells
Per genotype, 3–4 male or female C57BL/6J mice (WT, CDK2dTAG, or CDK5dTAG) were humanely euthanized and femurs were removed and cleaned, scraping off as much muscle as possible. Femurs were placed into 500 µl Eppendorf tube with a hole punctured in the bottom by an 18-gauge needle, and this entire 500 µl Eppendorf tube was then placed into a 1.5 ml Eppendorf tube and spun at 8000 rpm for 15 s. Cell pellets were resuspended in 1 ml of fetal bovine serum (FBS; ThermoFisher, catalog no. 26140095), passed through a 70-micron cell strainer (Fisher, catalog no. 08-771-2) and spun down at 1600 rpm for 10 min. Supernatant was removed and cells were resuspended in 4 ml of PBS. The cell suspension was placed on top of 4 ml of Ficoll Paque Plus (Sigma, catalog no. GE17-1440-02) in a 15-ml tube and spun at 1600 rpm for 30 min with no brake. After spinning, the interphase layer of cells was removed, transferred to a new tube, PBS was added and mixed, and cells were spun down. On average, this method yields 0.5–2 million bone marrow-derived cells (BMDCs) per mouse, and bone marrow from mice with the same genotype was combined, regardless of sex. An additional PBS wash was performed, and cells were either immediately used or cryopreserved for later use.
Viability and HiBit assays with bone marrow-derived cells
Per well, 6000 BMDCs in 15 µl of media containing Stemline II media (Sigma, catalog no. S0192-6X500ML), 10% FBS (ThermoFisher, catalog no. 26140095) penicillin-streptomycin (Fisher, catalog no. MT30002CI), and cytokines (50 ng/ml SCF, RnD Systems, catalog no. 455-MC-050; 20 ng/ml G-CSF, RnD Systems, catalog no. 414-CS-025; 20 ng/ml GM-CSF, RnD Systems, catalog no. 415-ML-020; 50 ng/ml FLT3, RnD Systems, catalog no. 768-F3-050; 20 ng/ml IL-6, RnD Systems, catalog no. 406-ML-200; 10 ng/ml IL-3, RnD Systems, catalog no. 403-ML-050; 30 ng/ml TPO, RnD Systems, catalog no. 488-TO-200; 1.5 ng/ml EPO, RnD Systems, catalog no. 959-ME-010) were placed into a black-walled 384-well plate (Corning, catalog no. 3764) and left overnight. Next day, 15 µl of 2× drug-containing solution was added to each well. For HiBit assays, 6 h later the HiBit Lytic reagent was added following the manufacturer’s protocols (Promega, catalog no. N3030) and plates were read using a Biotek H1 Synergy H1 Hybrid plate reader. For viability assays, 4 days after compound addition, the CellTiter-Glo reagent was added following the manufacturer’s protocols (Promega, catalog no. G7573) and plates were read using a Biotek H1 Synergy H1 Hybrid plate reader.
Isolation of mouse duodenum organoids
Two C57BL/6J mice per genotype (WT, CDK2dTAG, CDK5dTAG, Cdk2fl/fl; Rosa26tm(Cre/ERT2)+/−, and Cdk2fl/fl; Rosa26tm(Cre/ERT2−/−)), 1 male and 1 female, were sacrificed and 10 cm of small intestine proximal to the stomach (primarily duodenum) was cleaned and removed. One male and 1 female mouse were used to establish organoids because organoids from individual mice did not show significant variations (not shown). The intestine was opened lengthwise using small scissors and shaken vigorously in ice-cold PBS in a 10-cm dish until debris no longer fell off the intestine, with PBS being changed occasionally. The cleaned, opened intestine was then cut into small pieces (approx. 2 mm) and collected in a 50-ml conical containing ice-cold PBS. These pieces were pipetted up and down 3 times, allowed to settle via gravity, supernatant was aspirated, 10 ml of new PBS was added, and this process was repeated 20 times. The tissue fragments were resuspended in 25 ml of room temperature Gentle Cell Dissociation Reagent (StemCell, catalog no. 100-0485), and incubated on a rocker at room temperature for 15 min. Tissue fragments were settled by gravity centrifugation and supernatant was aspirated. Tissue fragments were resuspended in 10 ml of ice-cold PBS with 0.1% BSA (Fisher, catalog no. BP9703100), pipetted up and down 3 times, and left to settle via gravity. Supernatant was passed through a 70-micron cell strainer (Fisher, catalog no. 08-771-2) and labeled as “Fraction 1.” This process of adding ice-cold PBS with 0.1% BSA, pipetting, collecting supernatant and filtering was performed 3 additional times to generate a total of 4 fractions. These 4 fractions were centrifuged at 290 g for 5 min at 4°C, supernatant was discarded, and the fractions were washed in ice-cold PBS with 0.1% BSA. Fractions that had a high degree of crypts were kept and/or combined, crypts from 1 male and 1 female mice were combined, and then cells were spun down, resuspended in 50:50 Matrigel (Fisher, catalog no. CB-40230C): Mouse IntestiCult OGM (StemCell, catalog no. 6005), and plated as 25 µl domes into a 10-cm dish. Domes were placed at 37°C to harden for 15 min, then 12 ml of Mouse IntestiCult OGM (StemCell, catalog no. 6005) was added to each 10 cm dish.
Plating mouse duodenum organoids into 384-well plates for HiBit or viability assays
Starting from organoids grown as 25 µl domes of 50:50 Matrigel (Fisher, catalog no. CB-40230C): Mouse IntestiCult OGM (StemCell, catalog no. 6005) in a 10-cm dish, 10 ml of ice-cold Gentle Cell Dissociation Reagent (StemCell, catalog no. 100-0485) was added for 1 min then cells were scraped using a cell lifter (Fisher, catalog no. 08-100-240). Cells were rocked for 10 min at room temperature, spun at 290 g for 5 min at 4°C, media was aspirated, cells were resuspended in 12 ml DMEM/F12 (Sigma, catalog no. D6421-500ML), spun at 200 g for 5 min at 4°C and media was aspirated. To make a single cell solution, cells were resuspended in 3 ml of TrypeLE Select (Fisher, catalog no. A1285901), and pipetted up and down 10 times using a P1000 every 5 min until a single cell solution was formed, generally within 15 min. Once a single cell solution was formed, 6 ml of DMEM/F12 was added and cells were passed through a 70-micron cell strainer (Fisher, catalog no. 08-771-2) and number of viable cells were counted by trypan blue staining using a hemocytometer. For each well, 20 000 cells were resuspended in 15 µl of 40% Matrigel 60% StemCell Mouse IntestiCult OGM and plated into the center of a black walled 384 well plate (Corning, catalog no. 3764). Plates were placed at 37°C for 20 min, then 25 µl of Mouse IntestiCult OGM was added to each well. After 2 days, when organoids begin to form and bud, inhibitors/drugs were added. For viability-based assays, compounds were left on organoids for 4 days, and viability was readout using CellTiter-Glo 3D (Promega, catalog no. G9681) following the manufacturer’s protocols.
Performing HiBit assay on mouse duodenum organoids grown in Matrigel in 384-well plates
Starting from organoids grown in 40:60 Matrigel: Mouse IntestiCult OGM (protocol to plate in 384-well plates shown above), the entire 384-well plate was moved to a 4°C room and placed on ice for 15 min. After 15 min, the entire cell solution was pipetted up and down 6 times on ice, then 40 µl HiBit reagent (Promega, catalog no. N3030) that was prewarmed to 37°C was added, the plate was wrapped in aluminum foil, and shaken at room temperature vigorously for 20 min. After 20 min, luminescence was read using a Biotek H1 Synergy H1 Hybrid plate reader.
Western blotting
For cell lines and organoids, proteins were isolated by resuspending cells and rotating at 4° for 2 h in ice-cold RIPA (Fisher, catalog no. 89900) containing 2 mM MgCl2 (Sigma, catalog no. M1028), Halt Phosphatase/Protease inhibitors (Fisher, catalog no. 78440) and 1 unit/µl benzonase (Sigma, catalog no. E1014-25KU). For tissues, an NP-40-based lysis buffer was used with the same amount of MgCl2, protease and phosphatase inhibitors. Additionally, tissues were homogenized using a Qiagen TissueLyser II at 20 Hz for 2 min, changing tube placement, and running an additional 20 Hz for 2 min. After homogenization (tissues) or addition of lysis buffer (organoids and cell lines) 1 unit/µl benzonase was added (Sigma, catalog no. E1014-25KU) and samples were rotated at 4°C for 2 h. Lysates were clarified by spinning at 4°C and 18 000 g for 15 min. Supernatant from this spin was quantified by BCA assay (Fisher, catalog no. 23225) following the manufacturer’s protocols, mixed with Laemmli’s 2× sample loading buffer (BioRad, catalog no. 1610737EDU) and boiled for 5 min. Twenty micrograms of protein were loaded on a 4%–20% Criterion TGX Precast Midi Protein Gel (BioRad, catalog no. 5671094), ran at 200 V in Tris/Glycine/SDS buffer (BioRad, catalog no. 1610732), and transferred onto a nitrocellulose membrane using a BioRad Trans-Blot Turbo and a Trans-Blot Turbo Midi .2 µm Nitrocellulose Transfer Pack (BioRad, catalog no. 1704159). Membranes were blocked for 1 h using LiCor Intercept TBS Blocking Buffer (Fisher, catalog no. NC1863051) and primary antibodies were added overnight at 4°C in LiCor Intercept T20 Antibody Diluent (Fisher, catalog no. NC1703226) on a rocker. The following primary antibodies were used in this study at the indicated dilutions: CDK2 1:1000 (Abcam, ab32147), CDK5 1:2000 (Abcam, catalog no. ab40773), Vinculin 1:4000 (Cell Signaling, catalog no. 13901), and GAPDH 1:4000 (Cell Signaling, catalog no. 97166). The next morning, membranes were washed 3 times for 5 min using TBST (Cell Signaling, catalog no. 9997), then secondary antibodies were diluted into LiCor Intercept T20 Antibody Diluent (Fisher, catalog no. NC1703226) and added to membranes for 1 h at room temperature on a rocker. The following secondary antibodies were used in this study at the indicated dilutions: Donkey anti-Rabbit 800CW, 1:10 000 (Fisher, catalog no. NC9523609), Donkey anti-Mouse 680RD, 1:10 000 (Fisher, catalog no. NC0250903). After secondary antibody incubation, membranes were washed 4 times for 5 min with TBST (Cell Signaling, catalog no. 9997), once with TBS (Cell Signaling, catalog no. 12498) for 5 min, and then imaged on a LiCor Odyssey CLx. When necessary, membranes were stripped using LiCor NewBlot Nitro Stripping Buffer (LiCor, catalog no. 928-40030) following the manufacturer’s suggestions.
Far-western for HiBit
For far-western HiBit assays, western blotting was performed as described above. After probing with all antibodies, the Nano-Glo HiBit Blotting System (Promega, catalog no. N2410) was used to visualize HiBit expression. In brief, following the manufacturer’s recommendations on volumes and timing, membranes were probed overnight with the LgBit protein diluted in 1× Nano-Glo HiBit Blotting Reagent on a rocker at 4°C. Next day, this was allowed to equilibrate to room temperature, then Nano-Glo Luciferase Assay Substrate was added, placed on a rocker for 5 min, and imaged using a BioRad Chemidoc MP.
Wes
Wes was used instead of western to analyze protein lysates in Supplementary Figure 1. Protein lysates were generated as shown in the “Western blotting” methods section and after determination of protein concentration via the BCA assay, lysates were diluted to 1 mg/ml using the minimal amount of RIPA and protein lysate necessary, with the rest of the sample volume being composed of Wes Sample Buffer (Protein Simple catalog no. 042-195). Samples were loaded and ran on a Wes by Protein Simple using the 12–230 kDa Separation Module, 8 × 25 capillary cartridges (Protein Simple catalog no. SM-W004) and the anti-rabbit detection module (Protein Simple catalog no. DM-001) under default settings. The following primary antibodies were used at the indication dilutions: CDK2 (Abcam catalog no. ab32147) at 1:500 and Vinculin (Cell Signaling Technologies catalog no. 13901) at 1:2000.
Sperm analysis
Sperm analyses (phenotypic and morphological) were performed by Charles Rivers Laboratories. In brief, mice were euthanized and sperm was isolated from the caudal epididymis and vas deferens. Sperm was chilled overnight at 4°C, then analyzed by an IVOS machine (Hamilton Thorne). Sperm from 2 pairs of mice (4 total) were pooled and ran individually on the IVOS.
Hematology and clinical chemistry
For clinical pathology assessments, mice were euthanized with isoflurane and exsanguination and blood was collected from the inferior vena cava. Standard hematology and clinical chemistry parameters were measured using a Siemens Advia 2120 Hematology analyzer (Siemens Healthcare Diagnostics, Tarrytown, New York) and a Siemens Advia 1800 Chemistry analyzer (Siemens Healthcare Diagnostics, Tarrytown, New York), respectively.
Descriptive statistics were generated for each parameter and group at each scheduled sampling time or each time interval. Statistical tests were conducted at the 5% and 1% significance levels. A nonparametric (rank-transform) 1-way analysis of variance on all groups was conducted, with 2-sided trend tests and 2-sided pairwise comparisons of each group to the reference group using Dunnett’s test. Statistical tests were not done if n was less than 3 animals. Clinical pathology results were reported as ratio of the dTAG-13-related finding relative to concurrent control group mean values. Conclusions regarding relationship of clinical pathology results to dTAG-13 were made using a weight-of-evidence approach as described previously (Aulbach et al., 2019).
Animals from asynchronous studies were not cross-analyzed to avoid any study-specific confounders. Thus, results from WT mice were not compared directly to results from CDK2dTAG or CDK5dTAG mice; rather comparisons between vehicle versus dTAG-13 treatment groups for CDK2dTAG and CDK5dTAG mice were performed.
The following parameters were captured during hematological assessment: red blood cell count (cells/µl), hemoglobin (g/dl), hematocrit (%), mean cell volume (fl), mean cell hemoglobin (pg), mean cell hemoglobin concentration (g/dl), red cell distribution width (%), reticulocytes (cells/µl), platelets (cells/µl), mean platelet volume (fl), white blood cells (cells/µl) neutrophils (cells/µl), lymphocytes (cells/µl), monocytes (cells/µl), eosinophils (cells/µl), basophils (cells/µl), and large unstained cells (cells/µl).
The following parameters were captured during assessment of clinical chemistry: alanine aminotransferase (µ/l), aspartate aminotransferase (µ/l), alkaline phosphatase (µ/l), total cholesterol (mg/dl), triglycerides (mg/dl), glucose (mg/dl), total protein (g/dl), albumin (g/dl), globulin (g/dl), blood urea nitrogen (mg/dl), creatinine (mg/dl), phosphorus (mg/dl), calcium (mg/dl), sodium (mmol/l), potassium (mmol/l), and chloride (mmol/l).
For both hematology and clinical chemistry, only conclusions deemed statistically significant and robust based on a weight-of-evidence approach are shown in Supplementary Table 1. In cases where hematology and/or clinical chemistry was captured but not reported, no changes were deemed statistically significant and robust.
UPLC-MS/MS analysis of dTAG-13 in plasma
Serial blood samples of 35 µl were collected from individual animals at 1, 3, 7, and 24 h post dose. A standard curve containing dTAG-13 was prepared in control mouse plasma before extraction by protein precipitation. To an individual polypropylene tube 5 µl of unknown sample or standard was added followed by 50 µl of acetonitrile containing internal standard (Indomethacin) at 150 ng/ml. Samples were vortexed for 1 min, and then centrifuged at 3000 rpm for 5 min. Approximately 35 µl of supernatant was removed and transferred to a clean 96-well block, and 100 µl of 0.1% formic acid in water was added to the supernatant. Sample block was then vortexed briefly and 15 µl was injected on the LC-MS for analysis.
Chromatography was performed on a Waters Acquity UPLC System (Milford, Massachusetts). The autosampler and column were kept at 9°C and 40°C, respectively. Separation was achieved with a Waters Acquity UPLC HSS T3 (2.1*50 mm, 1.8 µm) using a gradient of 0.1% formic acid in water (Mobile Phase A) and 0.1% Formic Acid in acetonitrile (Mobile Phase B) at a flow rate of 0.600 ml/min. An initial mobile phase composition of 5% B was ramped to 95% in 2.3 min, held at 95% for 0.3 min, and then returned to initial 5% B for 0.3 min re-equilibration. The total analysis time for each sample was 3 min.
Data were collected on an AB Sciex API5500 (QTRAP) mass spectrometer (Foster City, California) using positive Turbo IonSpray electrospray ionization and multiple reaction monitoring mode. Key source parameters including source temperature, gas1, gas2, and curtain gas were set at 500°C, 40, 40, and 10, respectively. Transitions for dTAG-13 and Indomethacin were m/z 1049.4 → 684.2 and m/z 358.3 → 139.2, respectively. Data acquisition and processing was carried out with Analyst software version 1.7 (Applied Biosystems/MDS Sciex, Canada).
The unbound concentration of dTAG-13 was determined using previously published methods (Riccardi et al., 2015). Fu was estimated as 0.008 in mouse plasma.
Tissue processing, H&E, and pathology readings
Histopathology was performed using standard methods. Tissues were collected, fixed in 10% neutral buffered formalin, the sternum was decalcified using Cal-Rite (Epredia 5501), and all tissues were processed to paraffin blocks (FFPE) following standard histology protocols. Tissue sectioning was performed on a Leica microtome (Autocut) at 4 microns, sectioned onto glass slides and hematoxylin and eosin (H&E) stained on an automated Sakura Prisma H&E stainer & glass coverslipper. After processing, all slides for all mice were evaluated by a board-certified veterinary anatomic pathologist.
In vivo studies using wild-type and transgenic mice
All animal studies were conducted in a facility accredited by the Association for Assessment of Laboratory Animal Care under Institutional Animal Care and Use Committee guidelines and with appropriate animal research approval. Initially, single-dose pharmacokinetic (PK) studies were conducted in WT mice with dTAG-13 to assess the effects of different routes of administration and dose on exposure. Then, multiple formulations for dTAG-13 were tested in a 26-day repeat dose study to determine PKs of each formulation and repeat dose toleration. To further assess exposure, tolerability, protein degradation, and the effects of CDK2 or CDK5 loss following repeated doses of dTAG-13, 14-day study and 24-day repeat dose studies were conducted using 2 different formulations for dTAG-13 in WT, CDK2dTAG and CDK5dTAG mice. The 14-day study (utilizing Formulation no. 4) provided the strongest protein degradation, but its results were complicated by formulation-related toxicity. The 24-day study (utilizing Formulation no. 1) provided less protein degradation but minimal formulation-related toxicity. Finally, in a separate study, reproductive tissues from aged WT, CDK2dTAG and CDK5dTAG mice were evaluated by standard histopathology. Detailed study designs are described in the following sections.
dTAG-13 toxicokinetic/pharmacokinetic studies
For the first study (Figure 3A), the objective was to investigate the tolerability and PKs of a single-dose of dTAG-13 following the subcutaneous (SC), IP, intravenous (IV) route of administration in 7–9-week-old C57BL/6J WT mice at a single dose level. This study utilized 2 male and 2 female WT C57BL/6J mice and tested dTAG-13 delivered IV in an IV formulation (DMSO: PEG400: Water [10:50:40] [v: v: v]) at 1 mg/kg, dTAG-13 delivered IP in Formulation no. 1 (Solutol: DMSO: Saline [20:5:75] [v:v:v]) (Nabet et al., 2018) at 1 mg/kg, and dTAG-13 delivered SC in Formulation no. 2 (ethanol: benzyl bBenzoate: PEG400: Tween80 [10:30:40:20] [v:v:v: v] at 10 mg/kg). The parameters evaluated in this study included clinical observations, blood collection at 1, 3, 7, 24, and 48 h post dose for systemic exposure, and tissue collection (brain, bone marrow, stomach, ileum, colon, heart, kidney and liver) at 24 and 48 h post dose for tissue exposures. Tissue exposures are not shown due to high mouse-to-mouse variability. Details on how the concentration of dTAG-13 was determined in the plasma can be found in the section entitled “UPLC-MS/MS analysis of dTAG-13 in plasma.”
Figure 3.

Delivering dTAG-13 in vivo for toxicity studies. A, Determining the optimal route to deliver dTAG-13 in vivo. A single dose of dTAG-13 was given intravenously (IV) dissolved in an IV formulation, intraperitoneally (IP) dissolved in Formulation no. 1, or subcutaneously (SC) dissolved in Formulation no. 2, and levels of unbound dTAG-13 in plasma were quantified over time. n = 4 mice per group (2 males, 2 females); values shown are averages of the estimated unbound plasma levels of dTAG-13 ± SEM. B, Linear dose-exposure relationship for dTAG-13 delivered SC using Formulation no. 2. Pharmacokinetics of various doses of dTAG-13 dissolved in Formulation no. 2 and delivered SC. n = 6 male mice per group. Values shown are averages of the estimated unbound plasma levels of dTAG-13 ± SEM. C–E, Testing multiple dTAG-13 formulations in WT mice to identify a formulation suitable for repeat dose toxicity studies. (C) study outline for treating 3 male WT mice with dTAG-13 delivered in 3 different formulations. (D) histopath results of the injection site after 24 days of dosing. H&E slides from all mice in all groups were evaluated by a board-certified veterinary anatomic pathologist. Findings deemed secondary to injection site toxicity are displayed in Supplementary Figure 3. E, Estimated unbound plasma levels of dTAG-13 after 1 day or 9 days of dosing. Values are displayed as averages ± SEM and n = 3. q.d., once daily. Formulation composition can be found in Materials and Methods.
The objective of the second study (Figure 3B) was to determine if increasing doses of dTAG-13 delivered SC would yield increasing plasma levels of dTAG-13. The SC route of administration was chosen over IP and IV because it tends to result in less damage to surrounding organs than IP and is more convenient than IV. Six C57BL/6J WT male mice aged 7–9 weeks old were administered dTAG-13 dissolved in Formulation no. 2 (ethanol: benzyl benzoate: PEG400: Tween80 [10:30:40:20] [v:v:v:v]) delivered SC at 3, 30 or 300 mg/kg. The parameters evaluated in this study included clinical observations, blood collection at 1, 3, 7, 24, and 48 h post dose for systemic exposure, and tissue collection (brain, bone marrow, stomach, ileum, colon, heart, kidney and liver) at 24 and 48 h post dose for tissue exposures. Tissue exposures are not shown due to high mouse-to-mouse variability. Details on how the concentration of dTAG-13 was determined in the plasma can be found in the section entitled “UPLC-MS/MS analysis of dTAG-13 in plasma.”
The objective of the third study (Figs. 3C–E) was to determine the optimal formulation to deliver dTAG-13 SC; an optimal formulation would provide high exposure and minimal toxicity. Based on dTAG-13 solubility and prior knowledge, 3 formulations were tested: Formulation no. 1 (Solutol: DMSO: Saline [20:5:75] [v: v: v]), Formulation no. 2 (ethanol: benzyl benzoate: PEG400: Tween80 [10:30:40:20] [v:v:v:v]) and Formulation no. 3 (10% DMSO, 25% Miglyol 812: Kolliphor EL: Capmul MCM [30:40:30] [w:w:w]), 65% deionized water). These formulations alone (vehicle) or dTAG-13 in these formulations (3 mg/kg and 15 mg/kg for Formulation no. 1 and no. 3, or 15 mg/kg and 60 mg/kg for Formulation no. 2—more dTAG-13 could be used in Formulation no. 2 due to better solubility in this formulation) was delivered to 7–9-week-old male WT C57BL/6J mice (n = 3 per group) SC daily for 26-days. After the first (day 1) or ninth (day 9) dose, blood was collected for PK analysis 1, 3, 7, and 24 h after dosing. Details on how the concentration of dTAG-13 was determined in the plasma can be found in the section entitled “UPLC-MS/MS analysis of dTAG-13 in plasma.” Throughout the study, mice were monitored for clinical signs and bodyweight loss and there were no significant signs (other than injection site inflammation/scarring as noted in the manuscript) or changes. After 26 days, animals were sacrificed and the following tissues were harvested and processed for histopathological analyses: bone marrow (sternum), injection sites, duodenum, spleen, small intestine and stomach. After processing, all tissues were evaluated by a board-certified veterinary anatomic pathologist.
Repeat-dose toxicity and protein degradation studies in wild-type, CDK2dTAG and CDK5dTAG mice
Two repeat-dose toxicity studies with dTAG-13 were performed to assess tolerability, degree of protein degradation (pharmacodynamics), and the effect of CDK2 or CDK5 degradation on the physiology of the adult mouse.
In the first study (Figs. 4A–C), 7–9-week-old HOM C57BL/6J WT (n = 24 males), CDK2dTAG (n = 6 males, 6 females), or CDK5dTAG (n = 6 males, 6 females) mice were administered dTAG-13 in Formulation no. 4 (ethanol: PEG400: Tween80 [20:60:20] [v: v: v: v]) or this vehicle alone. Formulation no. 4 is the same as Formulation no. 3 but it lacks benzyl benzoate, which was removed from Formulation no. 4 in a futile attempt to mitigate injection site toxicity stemming from Formulation no. 3. For WT mice, 6 male mice were administered vehicle alone, 30 mg/kg dTAG-13, 100 mg/kg dTAG-13, or 300 mg/kg dTAG-13. For CDK2dTAG and CDK5dTAG mice, 4 mice per genotype were administered vehicle (2 males, 2 females) and 8 mice per genotype (4 males, 4 females) were administered 300 mg/kg dTAG-13. Clinical observations were performed daily, and bodyweights were captured throughout the study but did not change significantly (Supplementary Figure 3). Four hours after the second dose (on day 2), 8 CDK2dTAG and 8 CDK5dTAG mice (4 vehicle, 4 dTAG-13 treated; 2 males and 2 females per group) were sacrificed and the following tissues were snap frozen in liquid nitrogen to assess the degree of CDK2 or CDK5 degradation by Western blot and HiBit assays (Figure 4C): bone marrow, brain, heart, ileum, kidney, liver, lung, pancreas, spleen, stomach, testis, thymus, and ovaries. The remaining animals (all 24 WT, 8 CDK2dTAG, and 8 CDK5dTAG) were sacrificed after 14 days. Blood was collected for hematology and clinical chemistry, and the following tissues were collected for histopathological analysis: bone marrow, brain, epididymis, esophagus, heart, kidney, liver, lung, draining lymph node from the injection site, mesentery, pancreas, injection site, duodenum, ileum, jejunum, spleen, stomach, testis, and thymus. After processing, all tissues were evaluated by a board-certified veterinary anatomic pathologist.
Figure 4.

Degradation of CDK2 or CDK5 outside the brain in adult mice only causes a morphological change in the testis of CDK2dTAG mice. A–C, Potent degradation of CDK2 or CDK5 for 14 days using dTAG-13 delivered in Formulation no. 4 only causes a morphological change in the testis. A, Study outline for treating HOM WT, CDK2dTAG or CDK5dTAG mice with dTAG-13 delivered in Formulation no. 4 for 14 days. B, CDK2-specific and injection site histopathology results following 14 days of dTAG-13 treatment using Formulation no. 4. For most findings, H&E slides from 5 to 8 mice were evaluated by a board-certified veterinary anatomic pathologist. Results from male and female mice were combined due to a lack of gender specific differences. Findings deemed secondary to injection site toxicity are displayed in Supplementary Figure 5. C, Levels of CDK2 or CDK5 via a plate-based HiBit assay in HOM CDK2dTAG or CDK5dTAG mice after 2 days of dosing dTAG-13 or vehicle in vivo using Formulation no. 4. Data points are from individual mice and are relative to the average HiBit signal from vehicle-treated mice. A dashed line separates results from CDK2dTAG and CDK5dTAG mice. For most tissues, results from 2 male and 2 female mice are shown. M, male; F, female. ND, not determined. D–F, Modest degradation of CDK2 or CDK5 for 26 days using dTAG-13 delivered in Formulation no. 1 only causes a morphological change in the testis. D, Study outline for treating HOM CDK2dTAG or CDK5dTAG mice with dTAG-13 in Formulation no. 1 for 26 days. E, Summary of all histopathologic findings. For most findings, H&E slides from at least 4 mice per gender were evaluated by a board-certified veterinary anatomic pathologist. Results from male and female mice were combined due to lack of gender differences. Degen, degeneration, regen, regeneration. F, Levels of CDK2 or CDK5 via a plate-based HiBit assay in HOM CDK2dTAG or CDK5dTAG mice after 2 days of dosing dTAG-13 or vehicle in vivo using Formulation no. 1. Values are from individual mice and are relative to the average HiBit signal from vehicle treated mice. A dashed line separates results from CDK2dTAG and CDK5dTAG mice. For most tissues, results from 1 male and 1 female mouse are shown.
In the second study (Figs. 4D–F), 7–9-week-old HOM C57BL/6J WT (n = 4 males, 4 females), CDK2dTAG (n = 5 males, 5 females), or CDK5dTAG (n = 5 males, 5 females) mice were administered dTAG-13 SC daily in Formulation no. 1 (Solutol: DMSO: Saline [20:5:75] [v: v: v]) or this vehicle only. CDK2dTAG (n = 4 males, 4 females), CDK5dTAG (n = 4 males, 4 females), and all WT mice received 15 mg/kg dTAG-13, whereas 1 male and 1 female mouse from the CDK2dTAG and CDK5dTAG genotypes received vehicle only. Clinical observations were performed daily, and bodyweights were captured throughout the study but did not change After 2 days, 4 h after the second dose, 4 CDK2dTAG and 4 CDK5dTAG mice (2 vehicle and 2 dTAG-13 treated; 1 male and 1 female per group) were sacrificed and the following tissues were snap frozen in liquid nitrogen to assess the degree of CDK2 or CDK5 degradation via Western blot and HiBit assay (Figure 4E): bone marrow, ileum, liver, kidney, heart, lung, spleen, testis, and pancreas. The remaining animals were sacrificed after 26 days. Blood was collected for hematology and clinical chemistry, and the following tissues were collected for histopathological analysis: bone marrow, brain, epididymis, esophagus, heart, kidney, liver, lung, draining lymph node from the injection site, mesentery, pancreas, injection site, duodenum, ileum, jejunum, spleen, stomach, testis, ovaries, and thymus. After processing, all tissues were evaluated by a board-certified veterinary anatomic pathologist.
Repeat-dose study using cCDK2 conditional KO mice
The objective of this study was to investigate the effects of conditionally deleting Cdk2 in the adult mouse. Animals that express CreERT2 and have a floxed CDK2 allele (genotype Cdk2fl/fl; Rosa26tm(Cre/ERT2)+/−) are referred to as CDK2 cKO mice, because upon tamoxifen administration, CreERT2 is activated, resulting in deletion of the CDK2 active site, frameshift and loss of protein expression. Animals that do not express CreERT2 but contain a floxed CDK2 allele (genotype Cdk2fl/fl; Rosa26tm(Cre/ERT2)−/−) are used as comparators and are referred to as “Wild-type” because administration of tamoxifen will not activate CreERT2 because it is not expressed and will not delete CDK2. All mice received daily oral administration of 75 mg/kg/day tamoxifen for 6 days to activate CreERT2 where applicable. A cohort of animals (n = 5/sex/group) were euthanized 3 weeks post dose, whereas the remaining animals (n = 10/sex/group) were euthanized at 24 weeks post dose to evaluate both short- and long-term effects of CDK2 deletion. The parameters evaluated in this study included daily clinical observations, body weight, and blood collection for hematology measurement at 24-week necropsy. Body weight did not change significantly throughout the study. A list of tissues (bone marrow, heart, kidney, liver, lung, pancreas, ileum, jejunum, stomach, colon, ovary, uterus, testis, hemolymphoreticular tissue, spleen, and skin) were collected for microscopic evaluation at 3- and 24-week necropsy time points. After processing, all tissues were evaluated by a board-certified veterinary anatomic pathologist.
Evaluation of reproductive-related organs from aged mice
The objective of this study was to determine if reproductive organs from aged WT, CDK2dTAG or CDK5dTAG mice had findings that may explain the reduced fecundity of HOM CDK2dTAG and CDK5dTAG mice. Two separate cohorts were analyzed. One cohort consisted of HOM 20–22-week-old WT, CDK2dTAG and CDK5dTAG mice (n = 10; 5 males, 5 females). The following tissues were collected, processed, and evaluated by a board-certified veterinary anatomic pathologist: testes, epididymis, prostate, seminal vesicles, ovary, oviduct, uterus, cervix, vagina, and mammary gland. An additional cohort consisted of 15 32–39-week-old HOM CDK5dTAG female mice. The following tissues were collected, processed, and evaluated by a board-certified veterinary anatomic pathologist: ovary, uterus, and cervix. As mentioned in the manuscript, there were no findings in any of the aged CDK5dTAG mice above background (in comparison to WT). Aged CDK2dTAG mice displayed microscopic testicular findings similar to CDK2 cKO mice treated with tamoxifen and CDK2dTAG mice treated with dTAG-13.
RNA isolation
RNA isolation was performed using the Qiagen RNeasy Micro Plus Kit (Qiagen catalog t no. 74034) on the Qiacube according to the manufacturer’s instructions. Briefly, cells lysed in 350 µl of Buffer RLT Plus were vortexed before the lysate was transferred to a QIAshredder spin column (Qiagen, catalog no. 79656) placed in a 2-ml collection tube and centrifuged for 2 min at full speed. The homogenized lysate was transferred to a gDNA Eliminator spin column placed in a 2-ml collection tube and centrifuged for 30 s at ≥8000 g. This flow through was then added to 350 µl of 70% ethanol and mixed well by pipetting. Up to 700 µl of the sample was then transferred to an RNeasy spin column placed in a 2-ml collection tube and centrifuged for 15 s at ≥8000 g before discarding the flow through. Buffer RW1 (700 µl) was then added to the spin column before further centrifugation for 15 s at ≥8000 g. This was followed by an additional 500 µl buffer RPE wash for 15 s at ≥8000 g. Next, 500 µl of 80% ethanol was added for a second time to the spin column and centrifuged for 2 min at 20 000 g. Following these washes, the matrix column was dried by centrifugation at full speed for 5 min. Total RNA was eluted from the solid support matrix by applying 14 µl of RNase free water (Qiagen, catalog no. 129112) and collected in a sample tube through centrifugation at full speed for 1 min. After elution, 2 µl of each sample was evaluated for total RNA concentration using the Qubit RNA HS Assay Kit (ThermoFisher Scientific, catalog no. Q32852) on the Qubit 4 Fluorometer (ThermoFisher Scientific, catalog no. Q33238).
QuantSeq library generation/quantification
A total of 50 ng RNA was used as input for the Lexogen QuantSeq 3′ mRNA-Seq Library Preparation kit for Illumina (FWD) (Lexogen, catalog no. 015.96) and libraries were prepared according to the manufacturer’s protocol. Briefly, for first-strand cDNA synthesis, 5 µl of RNA was mixed with 5 µl FS1 and incubated for 3 min at 85°C and then cooled to 42°C. A total of 10 µl of a pre-warmed FS2/E1 mix was then added to the samples on the thermocycler at 42°C and incubated for 15 min before proceeding to RNA removal (duodenum organoid samples) or globin removal (blood samples). For RNA or globin removal, either 5 µl of RS or 5 µl of RS-GB (Lexogen, catalog no. 070.96) was added to the samples and then incubated for 10 min at 95°C before cooling to 25°C. Second-strand synthesis was then performed by adding 10 µl of SS1, incubating for 1 min at 98°C before ramping to 25°C at a ramp speed of 0.50°C/s. Samples were then incubated for 30 min at 25°C. A master mix of 5 µl of SS2/E2 was then added to each reaction and incubated for 15 min at 25°C. Following a purification process with PB/EB/PS beads/buffers, 20 µl of each sample was eluted in EB. A total of 17 µl of this elution was added to a master mix of 8 µl of PCR/E3 and 5 µl i7 primer (7001–7096) and PCR amplified using the following conditions: 98°C for 30 s, followed by 16 cycles of 98°C for 10 s, 65°C for 20 s, 72°C for 30 s, followed by 72°C for 1 min before cooling to 10°C indefinitely. Each QuantSeq library was then purified again using PB/EB/PS beads/buffers and eluted in 20 µl of EB buffer. After elution, libraries were quantified using the Qubit 4 Fluorometer (ThermoFisher Scientific, catalog no. Q33238) using the Qubit 1× dsDNA HS Assay Kit (ThermoFisher Scientific, catalog no. Q33231).
QuantSeq processing and analysis
For BMDCs, the raw reads from all samples were aligned using a Salmon-based RNA-Seq pipeline developed at Pfizer. The differential expression analyses were done in R v.3.5.0 using DESeq2 package, and differentially expressed genes were selected based on p-adjusted < .05.
For duodenum organoid samples, sequencing data was imported into CLC Genomics Workbench (Qiagen), quality-controlled and thereafter processed using that package (version 20) for alignment and gene expression analysis.
Sequencing data were deposited in GEO under accession number GSE226390. SubSeries can also be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226386 and https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226388.
Results
Generation of CDK2dTAG and CDK5dTAG mice knock-in mice
The dTAG, a hemagglutinin affinity tag, and a split luciferase (HiBiT) was appended to the C-terminus of Cdk2 or Cdk5 in the germline of C57BL/6J mice to yield CDK2dTAG or CDK5dTAG knock-in mice (Figure 1A). Crossing heterozygous (HET) CDK2dTAG or CDK5dTAG mice yielded pups at Mendelian ratios (Figure 1B), suggesting that the dTAG did not abrogate protein function because HOM CDK5 KO is perinatally lethal (Ohshima et al., 1996). Homozygous CDK2 KO mice weigh less than WT littermates and have atrophy of their testis or ovaries (Berthet et al., 2003; Ortega et al., 2003). Compared to WT or HET littermates, HOM CDK2dTAG mice tended to weigh less (Figure 1C). However, in contrast to CDK2 KO mice, we did not observe any overt phenotype in the testis of 7–9-week-old CDK2dTAG mice by H&E staining. Tissues from HOM CDK2dTAG or CDK5dTAG mice had 60%–80% less CDK2 or CDK5 protein (Figure 1D) than their WT counterparts, which may explain the decreased bodyweight of CDK2dTAG mice. Both dTAG fusion proteins were faithfully expressed in multiple tissues (Figs. 1D and 1E), with a strong correlation between CDK2 and CDK5 protein levels in knock-in mice and mRNA levels of CDK2 or CDK5 in WT mice (Figure 1F), suggesting that incorporation of the dTAG does not alter the expression pattern of these genes across various tissues.
Figure 1.

Derivation and validation of CDK2dTAG and CDK5dTAG knock-in mice. A, Knock-in of the dTAG onto the C-terminus of Cdk2 and Cdk5 in the germline of C57BL/6J mice. Cartoon diagram of the strategy to generate dTAG knock-in mice. A DNA sequence encoding a linker, the dTAG, an HA tag and a HiBit sequence were knocked onto the C-terminus of Cdk2 or Cdk5 in the germline of C57BL/6J mice using CRISPR/Cas9 and homology-directed repair. Dashed lines represent introns, boxes represent exons. Not drawn to scale and all exons/introns are not included for illustrative purposes. B, HET × HET crosses of CDK2dTAG or CDK5dTAG mice yield pups with Mendelian ratios. Mendelian ratio is the expected genotype frequency from crossing heterozygous mice. HET, heterozygous; HOM, homozygous. n > 17 litters for CDK2dTAG and CDK5dTAG. Data are displayed as averages ± standard deviations (SDs). C, Developing HOM CDK2dTAG mice tend to weigh less than CDK5dTAG or WT mice. Bodyweight of developing wild-type (WT), CDK2dTAG and CDK5dTAG mice. n > 3 mice for all data points, n > 6 for most data points. Lines fit using a smoothing spline with 4 knots in GraphPad prism. D, CDK2dTAG and CDK5dTAG mice have decreased expression of the dTAG fusion protein. Western blot for CDK2 and CDK5 and far western for the HiBit tag in HOM mice for the indicated genotype. Each lane represents an individual mouse. Highlighted regions are saturated and asterisks at the top right of bands represent the presumed CDK2 or CDK5 proteins (black or red for untagged or dTAG’d proteins, respectively). Mice were approximately 6–10 weeks old. Molecular weight markers are in kDa. E, Expression of CDK2dTAG and CDK5dTAG fusion proteins by the HiBit assay in various tissues. Protein levels of CDK2 and CDK5 as read out via a plate-based HiBit assay. Values are averages ± SD, n= 2. Same protein lysate as from (D). F, Strong correlation between mRNA levels of CDK2 or CDK5 in WT mice and CDK2dTAG or CDK5dTAG protein levels, respectively, in dTAG knock-in mice. Correlation between HiBit signal and mRNA levels (RNA-seq) for CDK2 and CDK5. Tissues shown are brain, ileum, kidney, liver, muscle, testis, and ovaries. For the brain, the HiBit signal is for the entire brain and mRNA level is the average transcripts per million (TPM) from RNA-seq on the cerebellum and cerebral cortex. HiBit data is from (E). G, Knock-in of loxP sites flanking exon 2 and 3 to generate Cdk2 conditional KO mice. Cartoon diagram of the strategy to delete Cdk2. Exons 2 and 3 were flanked by loxP sites, leading to deletions of exon 2 and 3 and a frameshift in CDK2 upon Cre-mediated recombination. Dashed lines represent introns, boxes represent exons. Not drawn to scale and all exons/introns are not included for illustrative purposes. H, Treatment of CDK2 cKO mice with tamoxifen causes loss of CDK2 protein expression. CDK2 cKO mice (genotype Cdk2fl/fl; Rosa26tm(Cre/ERT2)+/−) or “wild-type” mice (genotype Cdk2fl/fl; Rosa26tm(Cre/ERT2−/−)) were administered 75 mg/kg tamoxifen orally for 6 days. Twelve days later tissues were analyzed for CDK2 protein expression by immunoblot. Molecular weight markers are in kDa.
To corroborate any potential findings from our CDK2dTAG mice we generated CDK2 cKO mice in the C57BL/6J background by flanking exons 2 and 3 of Cdk2 with loxP sites (Figure 1G). These mice were then bred with Rosa26tm(Cre/ERT2) mice. Treatment of CDK2 cKO mice (Cdk2fl/fl; Rosa26tm(Cre/ERT2)+/−) with tamoxifen resulted in potent loss of CDK2 protein in multiple organs in comparison to “wild-type” controls (Cdk2fl/fl; Rosa26tm(Cre/ERT2−/−) (Figure 1H).
Degradation of CDK2 or CDK5 in bone marrow cells and duodenum organoids in vitro causes no appreciable phenotype
Gastrointestinal and bone marrow toxicities are common side effects of many therapies in oncology (Lau et al., 2004). Thus, we derived in vitro cultures of bone marrow cells and duodenum organoids from HOM WT, CDK2dTAG or CDK5dTAG mice. Treatment of duodenum organoids derived from HOM CDK2dTAG or CDK5dTAG mice with dTAG-13 resulted in dose-dependent, rapid (maximal degradation occurs within 4 h) and potent (>80% degradation for CDK2dTAG and >85% degradation for CDK5dTAG with > 100 nM dTAG-13) protein destruction (Figs. 2A and 2B). Degradation was likely time-dependent because a ternary complex must be formed, degrade the tagged protein, and repeat, with maximum degradation occurring when the rate of protein degradation and protein resynthesis reaches equilibrium. Despite this potent and rapid degradation, destruction of CDK2 or CDK5 for 4 days in duodenum organoids did not alter viability in comparison with WT controls (Figure 2C). Viability was inhibited at concentrations of dTAG-13 > 100 nM; however, because maximal degradation occurred at approximately 100 nM and viability was inhibited in all genotypes at concentrations >100 nM, even in WT cells which lack the dTAG, these viability effects are likely due to off-target effects. To holistically query whether CDK2 or CDK5 loss effects the duodenum, CDK2dTAG and CDK5dTAG organoids were treated with 250 nM dTAG-13 or DMSO for either 6 or 24 h and QuantSeq was performed. These time points and dose were chosen because they provide maximal (>80%) protein degradation (Figs. 2A and 2B) without significant toxicity in WT organoids (Figure 2C) and capture both early and late responses. Differential gene expression analysis showed that degradation of CDK2 or CDK5 causes no significant transcriptional responses (Figs. 2D–G). Processed and unprocessed QuantSeq results can be found in the accompanying GEO accession GSE226390. We confirmed this result for CDK2 using duodenum organoids derived from HOM CDK2 cKO mice, which show potent loss of CDK2 protein 24 h after treatment with 4-hydroxytamoxifen (4-OHT), the active metabolite of tamoxifen (Felker et al., 2016) (Supplementary Figure 1), without significant changes in organoid viability or transcriptome (Figs. 2H and 2I).
Figure 2.

Kinetics and consequences of CDK2 or CDK5 destruction using in vitro models of the bone marrow and small intestine. A and B, Kinetics of CDK2 or CDK5 destruction after dTAG-13 administration in duodenum organoids derived from HOM CDK2dTAG or CDK5dTAG mice. HOM CDK2dTAG (A) and HOM CDK5dTAG (B) duodenum organoids were treated as indicated and then the HiBit assay was performed to determine the amount of CDK2 (A) or CDK5 (B) protein. Values shown are normalized to DMSO treatment and are averages ± standard error of the mean (SEM) for 3 biological replicates. C, Degradation of CDK2 or CDK5 in CDK2dTAG or CDK5dTAG duodenum organoids does not affect viability. Duodenum organoids derived from WT, CDK2dTAG or CDK5dTAG HOM mice were treated with dTAG-13 for 4 days, then viability was measured by CellTiter-Glo 3D. Values shown are normalized to DMSO treatment and are averages ± SEM for 3 biological replicates. D and E, No significant changes in gene transcription after degrading CDK2 in CDK2dTAG duodenum organoids. Duodenum organoids derived from HOM CDK2dTAG mice were treated for 6 h (D) or 24 h (E) with 250 nM dTAG-13 or DMSO, QuantSeq was performed, and differential gene expression analysis was performed between the 2 groups. n = 3 biological replicates. FDR, false discovery rate. F and G, No significant changes in gene transcription after degrading CDK5 in CDK5dTAG duodenum organoids. Duodenum organoids derived from HOM CDK5dTAG mice were treated for 6 h (F) or 24 h (G) with 250 nM dTAG-13 or DMSO, QuantSeq was performed, and differential gene expression analysis was performed between the 2 groups. n = 3 biological replicates. (H and I) No significant changes in gene transcription after CDK2 KO in CDK2 cKO duodenum organoids. Duodenum organoids derived from HOM CDK2 cKO mice were treated for 24 h (H) or 48 h (I) with 1 µM 4-hydroxytamoxifen (4-OHT) or DMSO, QuantSeq was performed, and differential gene expression analysis was performed between the 2 groups. n = 3 biological replicates. J, Potent degradation of CDK2 or CDK5 in BMDCs from CDK2dTAG and CDK5dTAG mice. Bone marrow cells were derived from multiple CDK2dTAG or CDK5dTAG HOM mice, pooled, treated with dTAG-13 in vitro for 6 h at the indicated concentrations, and the HiBit assay was performed to determine levels of CDK2 or CDK5 protein. Values are normalized to DMSO treatment and are averages ± SEM for 3 technical replicates. K, No change in viability after degrading CDK2 or CDK5 in BMDCs from CDK2dTAG or CDK5dTAG mice. Bone marrow cells were derived from 3–4 WT, CDK2dTAG or CDK5dTAG HOM mice, pooled, treated with dTAG-13 in vitro for 4 days at the indicated concentrations, and cell viability measured via CellTiter-Glo. Values are normalized to DMSO treatment and are averages ± SEM for 3 technical replicates. L, No significant changes in gene transcription after degrading CDK2 in BMDCs from CDK2dTAG mice. BMDCs from HOM CDK2dTAG were treated with 250 nM dTAG-13 or DMSO for 24 h, QuantSeq was performed, and differential gene expression analysis was performed between the 2 groups. n = 3 technical replicates. BMDCs, bone marrow derived cells.
To determine if loss of CDK2 or CDK5 affects cells within the bone marrow we isolated BMDCs from WT, HOM CDK2dTAG or HOM CDK5dTAG mice. Treatment of these BMDCs with dTAG-13 ex vivo for 6 h at various concentrations caused >95% protein degradation (Figure 2J) but did not affect viability (Figure 2K). Similar to our results in duodenum organoids, treatment of BMDCs from CDK2dTAG mice with dTAG-13 for 24 h did not induce any transcriptional changes (Figure 2L). In totality, our ex vivo results suggest that loss of CDK2 or CDK5 in cells or organoids derived from the bone marrow or duodenum, respectively, causes no significant changes in cell viability or the transcriptome.
Delivering dTAG-13 in vivo for repeat dose toxicity studies
With a molecular weight over 1000 g/mol, efficient and safe delivery of dTAG-13 in vivo could prove challenging. We first determined the optimal route to deliver dTAG-13 by administering a single dose of dTAG-13 via various routes and monitoring unbound plasma levels over time. Administration of dTAG-13 intravenously (IV), intraperitoneally (IP), or subcutaneously (SC) resulted in reasonable exposure (Figure 3A) with no differences between sexes (Supplementary Figure 2). SC dosing was chosen for subsequent studies due to reliability, ease, and a low likelihood to cause undesirable effects in nearby organs. Increasing doses of dTAG-13, up to 300 mg/kg, delivered SC resulted in increasing plasma levels of unbound dTAG-13 (Figure 3B).
Encouraged by these results we tested 3 different formulations for SC delivery of dTAG-13 in a 24-day repeat dose toxicity study. Three male WT C57BL/6J mice were treated with vehicle alone or different doses of dTAG-13 in 3 different formulations once a day SC for 24 days and signs of toxicity were evaluated by clinical observation and microscopic evaluation of a subset of tissues, including the bone marrow, injection site, small intestine, stomach, and spleen (Figure 3C). These tissues were chosen because they can report toxicities arising from SC dosing (injection site, bone marrow, and spleen) and/or because they are common sites for toxicity from anticancer drugs (small intestine, stomach, bone marrow, and spleen). All formulations caused some toxicity at the injection site (Figure 3D), with secondary responses in other organs (spleen, stomach, and bone marrow, Supplementary Figure 3). For all formulations, mice administered vehicle and dTAG-13 had similar toxicities; thus, it is likely that most toxicity stems from the formulation and SC dosing, not dTAG-13. Formulation no. 1 was the least toxic, however, Formulation no. 2 provided the highest level of exposure, especially after 9 days of repeat dosing, while being the most toxic (Figure 3E).
Degradation of CDK2 or CDK5 outside the brain in vivo causes no unexpected phenotypic changes
Given the inability to find a single formulation for dTAG-13 that provides strong exposure without multi-organ toxicity we used both Formulation no. 1 (least toxic but lower exposure) and Formulation no. 2 (more toxic but higher exposure) in separate repeat dose toxicity studies. In an attempt to mitigate toxicity from Formulation no. 2, we removed benzyl benzoate (now referred to as Formulation no. 4) and treated WT, CDK2dTAG or CDK5dTAG mice with vehicle or 300 mg/kg dTAG-13 in Formulation no. 4 subcutaneously daily for 14 days and measured a number of toxicity parameters, including hematology, clinical pathology, and microscopic evaluation of a select list of tissues (Figure 4A, see Methods for tissue list). Administration of vehicle or dTAG-13 to WT mice resulted in similar toxicities, suggestive of formulation-related toxicity, with most toxicities likely driven by lesions at the injection site that became prominent after 4–8 days of dosing. Addition of dTAG-13 increased the severity of most findings, possibly due to its poor solubility in aqueous environments. Although repeated dTAG-13 administration did not affect overall bodyweight (Supplementary Figure 4), histopathological evaluation of tissues showed that treatment of CDK2dTAG or CDK5dTAG mice with 300 mg/kg dTAG-13 caused similar toxicities as WT mice treated with 300 mg/kg dTAG-13 or vehicle. These toxicities included inflammation, ulceration, and acanthosis at the injection site (Figure 4B) and associated regenerative responses secondary to the injection site toxicity (Supplementary Figure 5). In the hematological system, treatment of WT, CDK2dTAG or CDK5dTAG mice with dTAG-13 lowered red blood cell mass and increased reticulocyte numbers, indicative of a regenerative process. Additionally, WT mice treated with dTAG-13 had lowered lymphocytes and higher neutrophils, whereas CDK2dTAG and CDK5dTAG mice treated with dTAG-13 had higher numbers of white blood cells, composed of neutrophils, lymphocytes, monocytes, eosinophils, and basophils (Supplementary Table 1). Treatment of WT, CDK2dTAG or CDK5dTAG with dTAG-13 caused no changes in the clinical chemistry parameters in these mice (not shown, parameters measured are listed in Materials and Methods section). The only toxicity that was unique, prevalent, and likely due to protein degradation was germ cell depletion in the testis and intraluminal cell debris (sloughed germ cells) in the epididymis of CDK2dTAG mice treated with dTAG-13 (Figure 4B), a finding that was previously described in the literature using germline CDK2 KO mice (Berthet et al., 2003; Ortega et al., 2003). A subset of animals were taken down following 2 days of dTAG-13 administration, and these mice showed potent degradation of CDK2 or CDK5 in nearly all organs other than the brain using a plate-based HiBit assay (Figure 4C). Given the large size of dTAG-13, it is not surprising that this molecule does not pass the blood brain barrier and promote degradation in the brain. Protein degradation was corroborated using Western blot, and degradation was observed in every tissue other than the brain, even those that did not appear to have protein degradation via the plate-based HiBit assay such as CDK5 in the liver (Supplementary Figure 6).
Although dTAG-13 delivered SC in Formulation no. 4 yielded potent protein degradation (Figure 4C), this study was complicated by formulation-related toxicity and the limited study duration (14 days) that was ethically feasible using this formulation. Thus, we decided to test the better tolerated but less effective Formulation no. 1 in a 26-day repeat-dose toxicity study. We administered 15 mg/kg dTAG-13 in Formulation no. 1 or vehicle alone to WT, CDK2dTAG or CDK5dTAG mice for 26 days and then measured several toxicity parameters, including hematology, clinical chemistry, and microscopic evaluation of a standard list of tissues (Figure 4D). This formulation caused less toxicity than Formulation no. 4, and similar to our previous results, the only unique toxicity seen across the different genotypes and treatments was seminiferous tubular degeneration in the testis with associated decreases in spermatocytes and increases in intraluminal cellular debris (sloughed germ cells) in the epididymis of CDK2dTAG mice (Figure 4E). These findings correlated with macroscopic findings of small testes. There were no changes in the hematology or clinical chemistry parameters in these mice (not shown). Although administration of 15 mg/kg dTAG-13 in Formulation no. 1 did cause multi-organ degradation of CDK2 or CDK5 protein (Figure 4F), as expected based on exposure data (Figs. 3B and 3E), this formulation provided more modest protein degradation than Formulation no. 4 delivered at 300 mg/kg (Figure 4C).
Similar to degradation of CDK2, conditional knockout of CDK2 only causes phenotypic changes in the testis
To corroborate the apparent lack of novel phenotypic changes in the CDK2dTAG mice following CDK2 degradation, we performed similar studies using the CDK2 cKO mice we generated (Figure 1G). CDK2 cKO or WT mice were treated with tamoxifen and assessed by histopathology after 3 or 24 weeks, and hematology was assessed after 24 weeks (Figure 5A). Three weeks after deleting CDK2, there was no toxicity specific to any cohort aside from previously reported testicular findings (Berthet et al., 2003; Ortega et al., 2003) (Figure 5B). Another cohort of mice were evaluated 24 weeks after deleting CDK2 to see if more subtle phenotypes would emerge; however, toxicities at 3 weeks after CDK2 KO were identical to those at 24 weeks after CDK2 KO (Figure 5C). Additionally, we saw no changes in the hematology of these mice following CDK2 KO. Clear, potent, multi-organ KO was observed 3 weeks after administering tamoxifen (Figure 5D) and this high level of KO was maintained for 24 weeks (Figure 5E). In contrast to what has been reported for germline CDK2 KO mice (Berthet et al., 2003; Ortega et al., 2003), we did not detect an appreciable phenotype in the ovaries of adult mice after conditional KO of CDK2. This could be due to differences in the timing of KO between these 2 models or due to low penetrance in adult mice. In germline CDK2 KO models, CDK2 is absent throughout development when premature oocytes are being made and CDK2 activity may be necessary. In contrast, in our CDK2 cKO model, all oocytes are already established, but not mature, prior to CDK2 loss.
Figure 5.
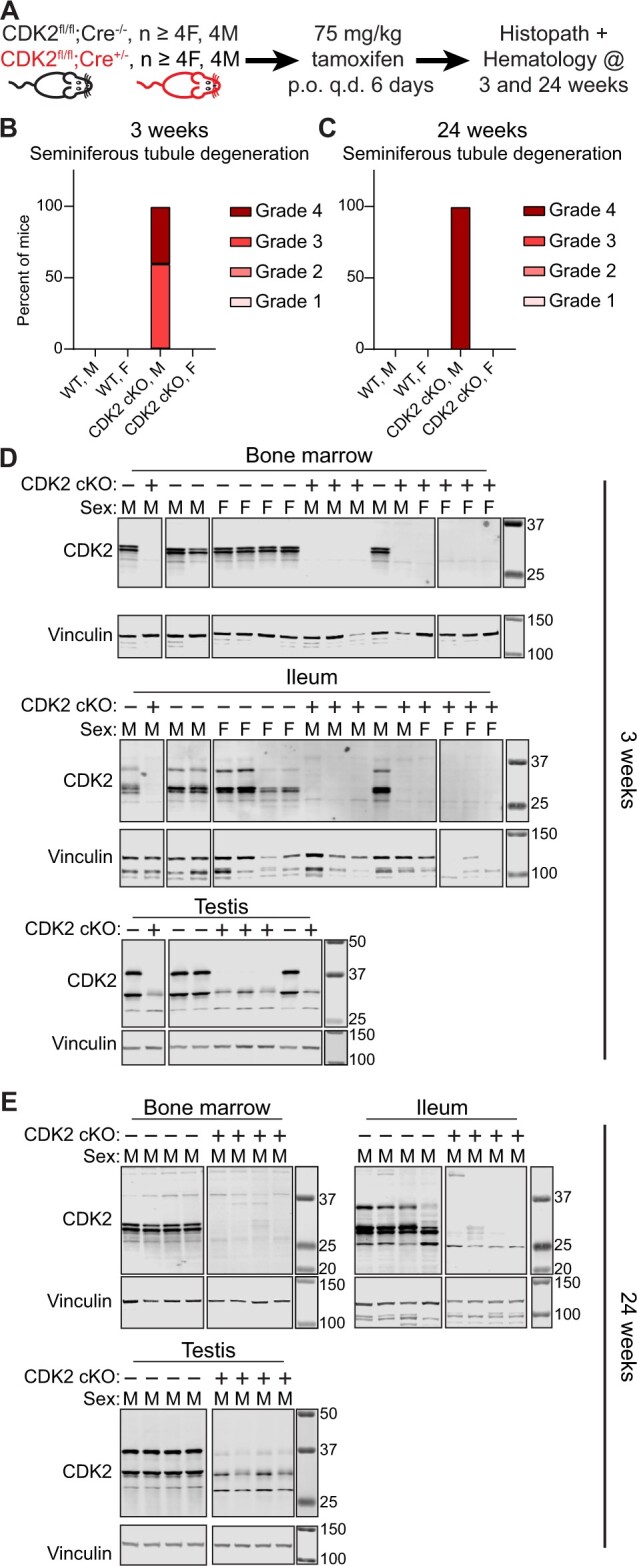
Conditional knockout of CDK2 in adult mice confirms lack of a novel morphological change after CDK2 loss. A–C, Conditional knockout of CDK2 in adult mice using Cre-mediated recombination only causes a morphological change in the testis. A, Study outline. CDK2 cKO (genotype Cdk2fl/fl; Rosa26tm(Cre/ERT2)+/−) or WT (genotype Cdk2fl/fl; Rosa26tm(Cre/ERT2−/−) mice were administered 75 mg/kg tamoxifen by oral gavage once per day for 6 days and mice were analyzed 3 weeks or 24 weeks after tamoxifen administration. B, Summary of histopathology results 3 weeks after deletion of CDK2. H&E slides from at least 4 mice per gender were evaluated by a board-certified veterinary anatomic pathologist. C, Summary of histopathology results 24 weeks after deletion of CDK2. H&E slides from at least 8 mice per gender were evaluated by a board-certified veterinary anatomic pathologist. D and E, Treatment of CDK2 cKO mice with tamoxifen yields potent and long-lasting loss of CDK2 protein. D, Western blot for CDK2 in the bone marrow, ileum or testis 3 weeks after WT (“CDK2 cKO –”)or CDK2 cKO (“CDK2 cKO +”) mice have been administered 75 mg/kg tamoxifen for 6 days. E, Western blot for CDK2 in the bone marrow, ileum, or testis 24 weeks after WT or CDK2 cKO mice have been administered 75 mg/kg tamoxifen for 6 days. For each tissue, all samples were ran and transferred on the same gel/membrane but images were cropped to remove failed or unrelated samples. Molecular weight markers are in kDa.
Homozygous CDK2dTAG and CDK5dTAG knock-in mice have reduced fecundity
Unexpectedly during breeding (ie, in the absence of dTAG-13 or any compound treatment), we encountered difficulties crossing HOM CDK2dTAG or CDK5dTAG mice with HOM CDK2dTAG or CDK5dTAG mice, respectively. Breeding HET CDK2dTAG or CDK5dTAG mice yielded a reasonable number of litters and pups; however, breeding HOM CDK2dTAG or CDK5dTAG mice yielded very few litters and pups (Figs. 6A and 6B). Although this may be expected for CDK2dTAG mice as they had reduced total levels of CDK2 protein at baseline and CDK2 KO mice are sterile (Ortega et al., 2003), CDK5 has not been shown to play a role in fertility. For CDK5dTAG mice, crossing male or female HOM CDK5dTAG mice with WT mice largely restored the ability to produce litters; we did not have enough data to perform a similar analysis with CDK2dTAG mice. Reproductive issues can arise via defects in multiple tissues, in a small subset of cells, and at various time points, making it difficult to confidently identify them. As a first pass, we analyzed the sperm from WT, HOM CDK2dTAG or HOM CDK5dTAG mice for gross defects. Compared with sperm from WT mice, sperm from both CDK2dTAG and CDK5dTAG mice were less competent in fertilizing WT oocytes (Figure 6C). Despite this, only sperm from CDK2dTAG mice showed significant obvious, observable differences from sperm from WT mice, such as decreased speed and a slightly abnormal appearance. Given that both CDK2 and CDK5 are still present, albeit at reduced (<60%) levels, complete lack of CDK2 or CDK5 protein would be expected to produce a stronger phenotype.
Figure 6.

Homozygous CDK2dTAG and CDK5dTAG mice have reduced fecundity. A, Breeding of HOM CDK2dTAG and CDK5dTAG mice produces less pups than HET mice. Number of pups born per day from crossing heterozygotes (HET × HET, left) or homozygous (HOM × HOM, right) CDK2dTAG or CDK5dTAG mice. n = number of pups born. B, Breeding of HOM CDK2dTAG or CDK5dTAG mice produces less litters than HET or WT mice. Number of litters born per day from crossing heterozygotes (HET × HET), homozygous (HOM × HOM), or homozygous and wild-type (HOM × WT) CDK2dTAG or CDK5dTAG mice. n is number of litters born. C, Sperm from HOM CDK2dTAG and CDK5dTAG mice are less fertile than WT mice. For fertilization rates, oocytes from WT mice were inseminated with sperm from HOM WT, CDK2dTAG or CDK5dTAG mice and the next day the total number of 2 cell embryos was counted to determine the fertilization rate. For sperm morphological analyses, mice were euthanized and sperm was collected from the caudal epididymis and vas deferens, chilled, and analyzed on an IVOS machine. Liability is the percent of normal sperm in the sample. Data shown are 2 independent runs on the IVOS machine from 4 mice (2 mice per run). *** = p < .01, 2-way ANOVA with Dunnett’s multiple comparison test.
In our studies, the CDK2dTAG and CDK5dTAG mice utilized were 7–10 weeks old and did not show obvious morphologic findings in their testis or ovaries by light microscopy, and we only examined 2–4 mice per sex per genotype. We reasoned that examining a larger number of older mice may reveal more subtle findings that may explain the decreased fecundity of HOM CDK2dTAG and CDK5dTAG mice. We aged male and female HOM WT, CDK2dTAG, and CDK5dTAG mice to approximately 20–22 weeks old and performed histopathology evaluation of pituitary gland, testis, epididymis, prostate, seminal vesicle, ovary, oviduct, uterus, cervix, vagina, and mammary gland. Additionally, we aged a cohort of female CDK5dTAG mice to 32–39 weeks old and examined ovary, uterus, and cervix. In comparison with aged WT mice, aged CDK2dTAG mice showed mild to moderate depletion and degeneration of germ cells in the testes, characterized by depletion of round, elongated and elongating spermatids and spermatocytes, with degeneration and necrosis of round spermatids and pachytene spermatocytes in the seminiferous tubules. This corresponded with decreased sperm content and/or an increase in cellular debris in the epididymis of animals with testicular findings. These findings were very similar to testicular findings in younger, male CDK2dTAG mice treated with dTAG-13, but not vehicle. Other sex organs in aged male or female CDK2dTAG and CDK5dTAG mice were unremarkable. Thus, although reduced fecundity in CDK2dTAG mice may be explained by these testicular findings, the reasons underlying the reduced fecundity of HOM CDK5dTAG mice remains nebulous. It is possible that the reproductive phenotype in CDK5dTAG mice is more subtle than we can detect by light microscopy or that these phenotypes have very low penetrance. The role of CDK5 in fecundity, which has not been explored previously, remains an open question.
Discussion
To ascertain whether toxicity or efficacy from a small molecule inhibitor in animal models is the result of on- or off-target activity, it is essential to have reliable orthogonal approaches that can be utilized in vivo, especially when the small molecule is predicted to have off-target activities. CDK2 is an emerging and attractive target in oncology; however, CDK2 inhibitors (Wells et al., 2020) and degraders (Teng et al., 2020) frequently have off-target activity against CDK5. We used the recently described dTAG system to degrade CDK2 and CDK5 in adult mice and evaluated potential toxicities resulting from the specific loss of CDK2 or CDK5. We specifically chose the dTAG system over traditional methods such as cKO because of its rapid kinetics, reversibility, and specificity.
There has been limited characterization of the dTAG system in vivo. Unexpectedly, our CDK2dTAG and CDK5dTAG knock-in mice had reduced levels of the dTAG fusion protein. The recently described NELFBdTAG knock-in mice also had reduced expression of the NELFBdTAG fusion protein (Abuhashem et al., 2022). We believe that the tendency of the dTAG to reduce the protein level of the fusion partner is a common phenomenon. In our hands, knock-in of the dTAG onto CDK2, CDK5, or other targets in mammalian cell lines tends to decrease expression of the fusion protein, potentially by decreasing protein stability (data not shown). Improving the stability of dTAG fusion proteins while maintaining ligandability would greatly enhance the utility of this system. An additional challenge in using the dTAG system in vivo is safe and efficient delivery of the dTAG degrader dTAG-13, which has a high molecular weight (1049 g/mol) and poorly solubility in aqueous solutions at concentrations needed to overcome its rapid metabolism and excretion in vivo. We employed 2 formulations, one that facilitated potent protein degradation but caused injection site toxicity after repeat dosing (Formulation no. 4), and another that facilitated more modest protein degradation but had less toxicity (Formulation no. 1). Both formulations could be used in different applications for early-stage drug discovery where the caveats of either formulation are not an issue. The formulation with increased exposure and toxicity (Formulation no. 4) could be used in short-term studies looking at modulation of an early/fast maker or phenotype (toxicology- or efficacy-based) that is not associated with any toxicities identified in this study. The formulation with decreased exposure and toxicity (Formulation no. 1) may be better suited for longer duration studies looking at gross phenotypic changes related to efficacy or toxicity, especially when strong target modulation is not desired or necessary. Future work to improve the stability of dTAG fusion proteins and better molecules and/or formulations to promote degradation in vivo would improve the utility of the dTAG system in vivo. We believe that neither challenge is insurmountable and this is exemplified by the identification of dTAGv-1, a dTAG-targeted PROTAC that has slightly better in vivo properties (Nabet et al., 2020).
Our findings suggest that CDK2 and CDK5 are not required in vivo for most adult tissues. We found that CDK2 is only required in the testis of adult mice, as was previously reported using conditional KO mice (Berthet et al., 2003; Ortega et al., 2003). Degradation of CDK5 caused no appreciable phenotype in any tissue evaluated. A caveat of our study is that we were unable to assess the role of CDK2 and CDK5 in the adult brain as treatment of CDK2dTAG or CDK5dTAG mice with dTAG-13 did not cause protein degradation in the brain, likely because dTAG-13 was unable to cross the blood brain barrier. The lack of phenotypic changes after degrading or deleting CDK2 is not surprising because CDK2 germline KO mice only have appreciable defects in their testis or ovaries (Berthet et al., 2003; Ortega et al., 2003). However, this is surprising for CDK5, which has purported roles outside of the brain based on various tissue-specific knockouts (Liebl et al., 2015; Mangold et al., 2021; Merk et al., 2016). A key difference between these prior studies and our study is that these prior studies knocked out CDK5 during development, whereas we degraded it in an adult mouse. Additionally, some of these purported phenotypes may be too subtle for us to detect via standard light microscopy, hematology, and serum metabolite measurements. Regardless, our results suggest that loss of CDK2 or CDK5 in all organs other than the brain in adult mice causes very little novel phenotypic changes. Future work to corroborate these findings, determine the role of CDK2 and CDK5 in the brains of adult mice, and the role of CDK5 in reproductive health, should be undertaken to fully understand the biology of CDK2 and CDK5 in adult mice.
Supplementary Material
Contributor Information
Paul Yenerall, Pfizer, San Diego, California 92121, USA.
Tae Sung, Pfizer, San Diego, California 92121, USA.
Kiran Palyada, Pfizer, San Diego, California 92121, USA.
Jessie Qian, Pfizer, Groton, Connecticut 06340, USA.
Seda Arat, Pfizer, Groton, Connecticut 06340, USA.
Steven W Kumpf, Pfizer, Groton, Connecticut 06340, USA.
Shih-Wen Wang, Pfizer, Cambridge, Massachusetts 02139, USA.
Kathleen Biddle, Pfizer, Groton, Connecticut 06340, USA.
Carlos Esparza, Pfizer, San Diego, California 92121, USA.
Stephanie Chang, Pfizer, San Diego, California 92121, USA.
Wesley Scott, Pfizer, San Diego, California 92121, USA.
Walter Collette, Pfizer, San Diego, California 92121, USA.
Taylor-Symon Winrow, Pfizer, San Diego, California 92121, USA.
Tim Affolter, Pfizer, San Diego, California 92121, USA.
Norimitsu Shirai, Pfizer, Groton, Connecticut 06340, USA.
Stephane Thibault, Pfizer, San Diego, California 92121, USA.
Julia Wang, Pfizer, Cambridge, Massachusetts 02139, USA.
Ling Liu, Pfizer, San Diego, California 92121, USA.
Mary Bauchmann, Pfizer, Groton, Connecticut 06340, USA.
Jessica Frey, Pfizer, San Diego, California 92121, USA.
Stefanus Steyn, Pfizer, Cambridge, Massachusetts 02139, USA.
Aida Sacaan, Pfizer, San Diego, California 92121, USA.
Allison Vitsky, Pfizer, San Diego, California 92121, USA.
Youngwook Ahn, Pfizer, Cambridge, Massachusetts 02139, USA.
Tom Paul, Pfizer, San Diego, California 92121, USA.
Lawrence Lum, Pfizer, San Diego, California 92121, USA.
Jon Oyer, Pfizer, San Diego, California 92121, USA.
Amy Yang, Pfizer, San Diego, California 92121, USA.
Wenyue Hu, Pfizer, San Diego, California 92121, USA.
Supplementary data
Supplementary data are available at Toxicological Sciences online.
Declaration of conflicting interests
All authors are employees of Pfizer and have no other conflicts of interest.
References
- Abuhashem A., Lee A. S., Joyner A. L., Hadjantonakis A. K. (2022). Rapid and efficient degradation of endogenous proteins in vivo identifies stage-specific roles of RNA pol II pausing in mammalian development. Dev. Cell. 57, 1068–1080.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulbach A., Vitsky A., Arndt T., Ramaiah L., Logan M., Siska W., Cregar L., Tripathi N., Adedeji A., Provencher A., et al. (2019). Interpretative considerations for clinical pathology findings in nonclinical toxicology studies. Vet. Clin. Pathol. 48, 383–388. [DOI] [PubMed] [Google Scholar]
- Banaszynski L. A., Sellmyer M. A., Contag C. H., Wandless T. J., Thorne S. H. (2008). Chemical control of protein stability and function in living mice. Nat. Med. 14, 1123–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C., Aleem E., Coppola V., Tessarollo L., Kaldis P. (2003). CDK2 knockout mice are viable. Curr. Biol. 13, 1775–1785. [DOI] [PubMed] [Google Scholar]
- Bondeson D. P., Mullin-Bernstein Z., Oliver S., Skipper T. A., Atack T. C., Bick N., Ching M., Guirguis A. A., Kwon J., Langan C., et al. (2022). Systematic profiling of conditional degron tag technologies for target validation studies. Nat. Commun. 13, 5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L., Raina K., Darricarrere N., Hines J., Gustafson J. L., Smith I. E., Miah A. H., Harling J. D., Crews C. M. (2015). Haloprotacs: use of small molecule protacs to induce degradation of halotag fusion proteins. ACS Chem. Biol. 10, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H. K., Jacobs C. L., Huo Y., Yang J., Krumm S. A., Plemper R. K., Tsien R. Y., Lin M. Z. (2015). Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 11, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felker A., Nieuwenhuize S., Dolbois A., Blazkova K., Hess C., Low L. W., Burger S., Samson N., Carney T. J., Bartunek P., et al. (2016). In vivo performance and properties of tamoxifen metabolites for creert2 control. PLoS One. 11, e0152989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Marth J. D., Orban P. C., Mossmann H., Rajewsky K. (1994). Deletion of a DNA polymerase beta gene segment in t cells using cell type-specific gene targeting. Science 265, 103–106. [DOI] [PubMed] [Google Scholar]
- Lau P. M., Stewart K., Dooley M. (2004). The ten most common adverse drug reactions (ADRs) in oncology patients: do they matter to you? Support. Care Cancer 12, 626–633. [DOI] [PubMed] [Google Scholar]
- Liebl J., Zhang S., Moser M., Agalarov Y., Demir C. S., Hager B., Bibb J. A., Adams R. H., Kiefer F., Miura N., et al. (2015). CDK5 controls lymphatic vessel development and function by phosphorylation of Foxc2. Nat. Commun. 6, 7274. [DOI] [PubMed] [Google Scholar]
- Mangold N., Pippin J., Unnersjoe-Jess D., Koehler S., Shankland S., Brähler S., Schermer B., Benzing T., Brinkkoetter P. T., Hagmann H. (2021). The atypical cyclin-dependent kinase 5 (CDK5) guards podocytes from apoptosis in glomerular disease while being dispensable for podocyte development. Cells 10, 2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk H., Zhang S., Lehr T., Müller C., Ulrich M., Bibb J. A., Adams R. H., Bracher F., Zahler S., Vollmar A. M., et al. (2016). Inhibition of endothelial CDK5 reduces tumor growth by promoting non-productive angiogenesis. Oncotarget 7, 6088–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Ferguson F. M., Seong B. K. A., Kuljanin M., Leggett A. L., Mohardt M. L., Robichaud A., Conway A. S., Buckley D. L., Mancias J. D., et al. (2020). Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 11, 4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Roberts J. M., Buckley D. L., Paulk J., Dastjerdi S., Yang A., Leggett A. L., Erb M. A., Lawlor M. A., Souza A., et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natrajan R., Mackay A., Wilkerson P. M., Lambros M. B., Wetterskog D., Arnedos M., Shiu K. K., Geyer F. C., Langerød A., Kreike B., et al. (2012). Functional characterization of the 19Q12 amplicon in grade III breast cancers. Breast Cancer Res. 14, R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T., Ward J. M., Huh C. G., Longenecker G., Pant H. C., Brady R. O., Martin L. J., Kulkarni A. B., Veeranna . (1996). Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. U.S.A. 93, 11173–11178., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S., Prieto I., Odajima J., Martín A., Dubus P., Sotillo R., Barbero J. L., Malumbres M., Barbacid M. (2003). Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25–31. [DOI] [PubMed] [Google Scholar]
- Pandey K., Park N., Park K. S., Hur J., Cho Y. B., Kang M., An H. J., Kim S., Hwang S., Moon Y. W. (2020). Combined CDK2 and CDK4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence. Cancers 12, 3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi K., Cawley S., Yates P. D., Chang C., Funk C., Niosi M., Lin J., Di L. (2015). Plasma protein binding of challenging compounds. J. Pharm. Sci. 104, 2627–2636. [DOI] [PubMed] [Google Scholar]
- Rubinson D. A., Dillon C. P., Kwiatkowski A. V., Sievers C., Yang L., Kopinja J., Rooney D. L., Zhang M., Ihrig M. M., McManus M. T., et al. (2003). A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat. Genet. 33, 401–406. [DOI] [PubMed] [Google Scholar]
- Santamaría D., Barrière C., Cerqueira A., Hunt S., Tardy C., Newton K., Cáceres J. F., Dubus P., Malumbres M., Barbacid M. (2007). CDK1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815. [DOI] [PubMed] [Google Scholar]
- Sharma S., Sicinski P. (2020). A kinase of many talents: non-neuronal functions of CDK5 in development and disease. Open Biol. 10, 190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. C., Tsai L. H. (2011). Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 27, 465–491. [DOI] [PubMed] [Google Scholar]
- Teng M., Jiang J., He Z., Kwiatkowski N. P., Donovan K. A., Mills C. E., Victor C., Hatcher J. M., Fischer E. S., Sorger P. K., et al. (2020). Development of CDK2 and CDK5 dual degrader TMX-2172. Angew. Chem. Int. Ed. 59, 13865–13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N. C., Liu Y., Zhu Z., Loi S., Colleoni M., Loibl S., DeMichele A., Harbeck N., André F., Bayar M. A., et al. (2019). Cyclin e1 expression and palbociclib efficacy in previously treated hormone receptor-positive metastatic breast cancer. J. Clin. Oncol. 37, 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., Jaenisch R. (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C. I., Vasta J. D., Corona C. R., Wilkinson J., Zimprich C. A., Ingold M. R., Pickett J. E., Drewry D. H., Pugh K. M., Schwinn M. K., et al. (2020). Quantifying CDK inhibitor selectivity in live cells. Nat. Commun. 11, 2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Wang H., Shivalila C. S., Cheng A. W., Shi L., Jaenisch R. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Fang D., Chen H., Lu Y., Dong Z., Ding H. F., Jing Q., Su S. B., Huang S. (2015). Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 6, 20801–20812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesbolatova A., Saito Y., Kitamoto N., Makino-Itou H., Ajima R., Nakano R., Nakaoka H., Fukui K., Gamo K., Tominari Y., et al. (2020). The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 11, 5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.