

Abstract
Phosphorylated tau (p‐tau) pathology correlates strongly with cognitive decline and is a pathological hallmark of Alzheimer's Disease (AD). In recent years, phosphorylated transactive response DNA‐binding protein (pTDP‐43) has emerged as a common comorbidity, found in up to 70% of all AD cases (Josephs et al., Acta Neuropathol, 131(4), 571–585; Josephs, Whitwell, et al., Acta Neuropathol, 127(6), 811–824). Current staging schemes for pTDP‐43 in AD and primary age‐related tauopathy (PART) track its progression throughout the brain, but the distribution of pTDP‐43 within the entorhinal cortex (EC) at the earliest stages has not been studied. Moreover, the exact nature of p‐tau and pTDP‐43 co‐localization is debated. We investigated the selective vulnerability of the entorhinal subfields to phosphorylated pTDP‐43 pathology in preclinical AD and PART postmortem tissue. Within the EC, posterior‐lateral subfields showed the highest semi‐quantitative pTDP‐43 density scores, while the anterior‐medial subfields had the lowest. On the rostrocaudal axis, pTDP‐43 scores were higher posteriorly than anteriorly (p < 0.010), peaking at the posterior‐most level (p < 0.050). Further, we showed the relationship between pTDP‐43 and p‐tau in these regions at pathology‐positive but clinically silent stages. P‐tau and pTDP‐43 presented a similar pattern of affected subregions (p < 0.0001) but differed in density magnitude (p < 0.0001). P‐tau burden was consistently higher than pTDP‐43 at every anterior–posterior level and in most EC subfields. These findings highlight pTDP‐43 burden heterogeneity within the EC and the posterior‐lateral subfields as the most vulnerable regions within stage II of the current pTDP‐43 staging schemes for AD and PART. The EC is a point of convergence for p‐tau and pTDP‐43 and identifying its most vulnerable neuronal populations will prove key for early diagnosis and disease intervention.
Keywords: aging, entorhinal cortex, immunohistochemistry, neurofibrillary tangles, neuronal cytoplasmic inclusions, pathology concurrence
1. INTRODUCTION
Transactive response DNA binding protein of 43 kDa (TDP‐43) was first identified as the main pathological hallmark of amyotrophic lateral sclerosis and frontotemporal dementia [1, 2]. Numerous studies have since linked TDP‐43 to Alzheimer's disease (AD), which is typically characterized by phosphorylated tau (p‐tau) protein, amyloid‐beta (Aβ) plaques, and neurodegeneration [3, 4, 5, 6, 7]. The prevalence of TDP‐43 in AD depends on factors such as population, stage of disease, antibodies used, comorbidities, among others [8]. For example, multiple reports agree that TDP‐43 can be found in approximately 30%–70% of AD cases [9, 10, 11, 12, 13], and 20%–30% of cognitively normal adults [8, 14, 15, 16]. These data have clinical significance, as AD patients with TDP‐43 pathology present more severe cognitive impairment than patients with no TDP‐43 inclusions [13, 17]. More specifically, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathological change (LATE‐NC) denotes TDP‐43 proteinopathy in older adults (typically >80 years), and is associated with AD‐like amnesic dementia [18]. LATE‐NC is common in brains with coexisting AD and PART pathology, but one is not necessary for the other [18, 19].
Currently the field does not have a reliable imaging or fluid biomarker for TDP‐43, primarily due to poor clinical‐pathological correlations [7, 16]. Histopathological studies have identified five subtypes (A–E) of FTLD TDP‐43 pathology that can be distinguished based on cellular distribution, morphology, and, in some cases, genetic and clinical presentation [13, 20, 21, 22, 23]. TDP‐43 pathological inclusions in AD can be further subdivided into type‐α and type‐β, primarily differentiated by structure, cortical distribution, and association with neurofibrillary tangles (NFTs) [24]. Structurally, TDP‐43 phosphorylated at the serines 409/410 (pTDP‐43) presents similar pathological features as p‐tau, including NFT‐like structures, neuropil threads (NTs), and neuronal cytoplasmic inclusions (NCIs) [13, 25, 26, 27]. P‐tau and pTDP‐43 have been shown to colocalize in hippocampal samples of preclinical and clinical AD and frontotemporal dementia brains [28]. However, the prevalence of pTDP‐43 in most but not all cases with neurodegeneration continues to raise questions regarding the interplay between pTDP‐43 and p‐tau [12, 29].
Josephs and colleagues established a five‐stage rating scheme for pTDP‐43 in AD in which the pathology progresses in a predictable manner, like p‐tau NFTs [11, 30]. Whereas p‐tau progression in the cortex typically begins in the perirhinal region before advancing to the EC [3, 31], the initial stages in Josephs' scheme involve the amygdala first and the EC second (stages I and II) [11, 30]. Zhang et al. created a staging scheme for pTDP‐43 in PART cases, and they identified four stages, beginning in the amygdala (stage I) and progressing into the EC and hippocampal regions (stage II) [27]. Finally, staging for LATE‐NC primarily examines TDP‐43 in amygdala (stage I), hippocampus (stage II), and middle frontal gyrus (stage III), but does not include the EC in its simplified primary criteria [18].
The aim of this report is to demonstrate pTDP‐43 deposition within EC subfields. Out of necessity of the clinical neuropathologic work up, pTDP‐43 staging schemes relegate the EC as a whole entity (if examined at all) and its pathology on a binary basis. In a previous study, we demonstrated the heterogeneity of p‐tau pathology density in the subfields within the EC at the preclinical Braak and Braak stages I and II, and showed that the posterior‐lateral subfields (ECL, ECsLat, and ELc) display a selective vulnerability [32]. Based on those observations we hypothesized that a similar vulnerability exists for pTDP‐43 pathology burden within the EC subfields, peaking at the same subregions as p‐tau. To address this, we applied a semi‐quantitative pathology density protocol and rated pTDP‐43 (serine 409/410) immunostained sections from preclinical AD and PART brains along the rostrocaudal axis; thus, directly comparing p‐tau and pTDP‐43 vulnerabilities in the same case and subfields within the EC. Elucidating EC subfield vulnerabilities to other pathology comorbidities will help decipher early changes in clinically silent neurodegeneration.
2. METHODS
2.1. Brain samples
We used 12 human brain hemispheres for this study (n = 9 left, n = 3 right). All hemispheres were received from the Department of Neuropathology, Autopsy Suite at Massachusetts General Hospital. Sample collection and all experiments were conducted in accordance with the Internal Review Board at Massachusetts General Hospital. Samples comprised seven males and four females, with ages that ranged from 59 to 84 years (mean = 70.73, median = 73, std = 9.54). One sample, case 6, lacked demographic information and was excluded from all analyses concerning age and sex. Brain weight ranged from 1187g to 1414 g (mean = 1288, median = 1319, std = 91.13) and had, with one exception, a postmortem interval under 24 h. Samples were fixed in 10% formalin and stored in 2% periodate‐lysine‐paraformaldehyde solution until sectioning. All demographic information is detailed in Table 1.
TABLE 1.
Demographic information for sample set: Demographic information for the 12 hemispheres studied. Cases are organized according to Braak and Braak staging.
| Case | Hemi | Age | Sex | Dx | BB stage | Aβ | α‐Syn | PMI (hours) | BW (grams) | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | RH | 67 | M | CC | NC | Neg | Neg | 48 | 1380 | Lung cancer |
| 2 | LH | 79 | M | CC | I | Pos | Neg | <24 | 1200 | Surgery Complication |
| 3 | LH | 59 | M | CC | I | Neg | Neg | <24 | 1319 | Surgery complication |
| 4 | RH | 60 | M | CC | I | Neg | Neg | <24 | 1414 | Aortic dissection |
| 5 | LH | 73 | F | CC | II | Neg | Neg | <24 | 1142 | Visceral hemorrhage |
| 6 | LH | n/a | n/a | CC | II | Neg | Neg | <24 | n/a | n/a |
| 7 | LH | 75 | M | CC | II | Pos | Neg | <24 | 1187 | Vascular disease |
| 8 | LH | 84 | F | CC | II | Pos | Neg | <24 | 1221 | Pneumonia |
| 9 | RH | 80 | M | CC | II | Pos | Neg | <24 | 1348 | Aortic dissection |
| 10 | LH | 82 | M | CC | II | Pos | Neg | <24 | 1224 | n/a |
| 11 | LH | 59 | F | CC | II | Pos | Neg | <24 | 1402 | Alveolar damage |
| 12 | LH | 60 | F | CC | II | Neg | Neg | <24 | 1328 | Pancreatic cancer |
Abbreviations: Aβ, amyloid‐beta; α‐Syn, alpha‐synuclein; BB, Braak and Braak tau stage; BW, brain weight; CC, cognitive control; Dx, clinical diagnosis; LH, left hemisphere; NC, normal control; n/a, not available; PMI, post‐mortem interval; RH, right hemisphere.
2.2. Comorbidity exclusion
All the patients who provided the samples used in this study were cognitively normal at the time of death, as none had a clinical diagnosis of Alzheimer's disease nor any other neurologic disease. Following guidelines from the NIA Alzheimer's Association guide [33], we controlled for co‐incidental AD mixed pathologies. The Massachusetts General Hospital Autopsy Suite screened all cases and excluded samples with suspected infectious disease (COVID‐19, hepatitis, HIV, and prion diseases) and vascular deficits, such as cerebrovascular strokes, lateral ventricle dilation, white matter discoloration, and lacunar Infarcts (Luxol fast blue/H&E stain). No cases had a clinical diagnosis of Parkinson's disease nor pathological characteristics of the disease, confirmed by negative immunostaining for α‐synuclein (Thermofisher, cat# 32‐8100).
2.3. Histology procedures and Nissl staining
Temporal blocks were excised from the hemispheres and cryoprotected (in a 20% glycerol/2% dimethyl sulfoxide solution) for at least 2 weeks prior to sectioning. Cryoprotected blocks were sectioned coronally at 50 𝜇m on a freezing sliding microtome (Leica Biosystems Richmond, Inc.) with a customized stage, and collected serially. All sections were subsequently frozen at −20°C in cryoprotectant (20% glycerol/2% dimethyl sulfoxide solution). Tissue for Nissl staining was selected every 10 sections (500 𝜇m apart) and mounted onto glass slides coated in a gelatin/chromium potassium sulfate solution. Mounted tissue was dried overnight and stained with thionin for Nissl substance as described in previous work [32, 34].
2.4. Entorhinal subfield parcellation and rostrocaudal level selection
Using a Nikon SMZ100 microscope (Micro Video Instruments, Inc.) we parcellated the subfields within the EC [32, 34, 35]. The parcellated EC subfields comprise olfactory (EO), rostral (ER), lateral‐rostral (ELr), medial‐intermediate (EMI), intermediate (EI), lateral caudal (ELc), caudal (ECs), and caudal‐limiting (ECL) subfields [35] (Figure S1). Based on previous work delineating pathology gradients along the central subfields, we bifurcated the EI and ECs subfields (EIMed/EILat and ECsMed/ECsLat, respectively) [32]. Previously, we identified eight levels along the medial temporal lobe, identified in part by nearby amygdala and hippocampal anatomical landmarks for thorough assessment [32]. Here, we identified a subset of coronal levels to evaluate: level 2 (anterior amygdala), 4 (anterior hippocampal head, genu), 5 (pes region, uncinate gyrus), and 7 (posterior‐most hippocampal head, gyrus intralimbicus). These levels include an ample representation of all EC subfields, and sufficient for a thorough representation of the EC without needing the exhaustiveness of the eight levels.
2.5. Immunohistochemistry
We performed immunohistochemistry on free‐floating sections selected from their respective anterior–posterior levels. The immunostaining series had a neighboring (adjacent or less than 250 𝜇m away) parcellated Nissl‐stained section that confirmed EC subfield cytoarchitecture boundaries. The immunohistochemistry protocol was the same as performed in [32]. Briefly, tissue sections were washed in phosphate buffered saline (PBS) three times to remove cryoprotectant solution. Sections were then incubated with 0.5% TritonX‐100 in 3% hydrogen peroxide for 20 min to quench endogenous peroxide and subsequently blocked in 5% nonfat dry milk in PBS for 2 h at room temperature. Primary antibodies CP13 p‐tau (gift from Dr. Peter Davies) and 409/410 pTDP‐43 (Proteintech, cat# 22309‐1‐AP) were incubated for 2 days in a 1.5% normal goat serum/PBS solution at 4°C. CP13 is ideal for early p‐tau visualization, as it recognizes phosphorylated paired helical filaments at the serine 202 epitope and reveals pretangles, mature NFTs, and NTs [36]. See Figure 1E–H for examples of CP13‐positive p‐tau structures. Sections were incubated in secondary antibodies (Jackson Immunoresearch) at a 1:200 dilution in a 1.5% normal goat serum/PBS solution (goat anti‐mouse for CP13; goat anti‐rabbit for pTDP‐43) for 1 h at room temperature. Tissue was incubated using an ABC kit (Vector laboratories, Burlingame, CA) for 30 min at room temperature and visualized using a 0.003% 3′3‐diaminobenzidine/0.3% hydrogen peroxide/double‐distilled water kit (Vector Laboratories). Stained sections were dehydrated in a 70%, 95%, and 100% ethanol solution progression and cleared with xylene and coverslipped using Permount (Fisher Scientific). Negative (no primary antibody) and positive (tissue with confirmed immunoreactivity) controls were included with every experiment.
FIGURE 1.
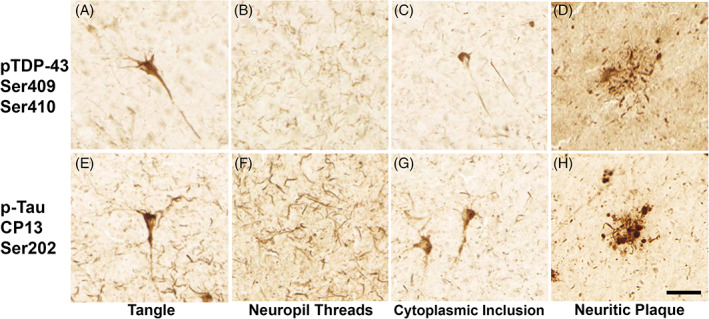
Similar inclusions in p‐tau (CP13) and pTDP‐43 (409/410) pathology: Panels show distinct types of pathologic characteristics observed with antibodies against TDP‐43 phosphorylated at the serines 409/410 (top row) and p‐tau (CP13) at serine 202 (bottom row). Lesions include neurofibrillary tangle‐like structures (A, E), neuropil threads (B, F), neuronal cytoplasmic inclusions (C, G), and immunoreactive material surrounding neuritic plaques (D, H). Magnification bar = 50 𝜇m.
2.6. Neuropathologic diagnosis and staging
CP13 immunostained sections were evaluated for p‐tau severity according to Braak and Braak staging [3, 31]. Perirhinal cortex (Brodmann area 35) was included when evaluating pathology burden for staging. All cases were non‐demented elderly at the time of death and thus presented tau pathology in the preclinical Braak stages BBI and BBII, albeit not dense and isolated in some cases. All cases were examined for CP13‐positive neuropil threads surrounding neuritic plaques. Half of our cases (n = 6) were negative for neuritic plaques, making them primary age‐related tauopathy (PART) cases [37].
pTDP‐43 immunostained tissue was staged according to the revised criteria by Josephs et al. [11]. Josephs defined six stages for AD TDP‐43 pathology that follow a predictable progression through the amygdala (stage I), EC/subiculum (stage II), dentate/occipitotemporal cortex (stage III), insula/ventral striatum/basal forebrain/inferior temporal (stage IV), substantia nigra/inferior olive/midbrain tectum (stage V), and basal ganglia/middle frontal (stage VI) brain regions [11, 30].
2.7. Semi‐quantitative rating scale: P‐tau and pTDP‐43 density
We applied a semi‐quantitative protocol to determine the density of p‐tau and pTDP‐43 pathology inclusions. Semi‐quantitative protocols have been widely used in neuropathology [3, 28, 31, 38, 39] and allow for reliable replication. Moreover, our group has found strong correlations between quantitative and semi‐quantitative protocols for entorhinal anatomical structures [40] as well as pathological features [32]. The present semi‐quantitative protocol includes NFT‐like structures (Figure 1A,E), neuropil threads (Figure 1B,F), neuronal cytoplasmic inclusions (NCIs; Figure 1C,G), and neuritic plaques (Figure 1D,H). The use of the same semi‐quantitative scale for p‐tau and pTDP‐43 allows for direct comparisons, something not possible in studies that use separate schemes for each [28].
The semi‐quantitative density scheme differentiates between four possible scores: score 0 depicts no visible pathology structures (Figure 2A), score 1 shows isolated NCIs and/or NFT‐like structures and almost no NTs (Figure 2B), score 2 reveals a greater number of more densely packed NCIs and/or NFT‐like structures, NTs in isolated patches (Figure 2C), score 3 indicates a moderate amount of densely packed NCIs and/or NFT‐like structures, usually band‐like in layer II of the EC (Figure 2D), and score 4 shows severe levels of pathology; numerous closely packed NCIs, NFT‐like structures, and NTs along layer II of the EC (Figure 2E).
FIGURE 2.
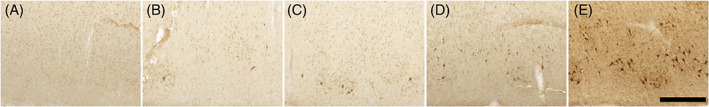
Pathology semi‐quantitative rating protocol: Panels show representative examples of the five pathology density semi‐quantitative scores: (A) score 0 = no visible pathology, (B) score 1 = isolated neuronal cytoplasmic inclusions (NCIs) and/or NFT‐like structures, almost no NTs, (C) score 2 = NCIs and/or NFT‐like structures greater in number and more closely packed than score 1; NTs present in isolated patches, (D) score 3 = moderate amount of densely packed, band‐like NCIs and/or NFT‐like structures, (E) score 4 = severe pathology burden, numerous closely packed NCIs and NFT‐like structures; NTs typically densely packed throughout the layer II region with neuritic plaques. The semi‐quantitative scheme used to assess visual pTDP‐43 pathology density implemented the same density criteria from the protocol for p‐tau applied in [32]. NCI, neuronal cytoplasmic inclusion; NFT, neurofibrillary tangle; NT, neuropil threads. Magnification bar = 500 𝜇m.
2.8. Statistics
Data analysis and presentation was conducted using R‐studio v.1.4 (www.r-studio.com) and Graphpad Prism v.9.1 (Graphpad, www.graphpad.com). First, we assessed differences in pTDP‐43 scores across EC subfields and rostrocaudal levels. Two Kruskal‐Wallis H Tests were computed, utilizing semi‐quantitative pTDP‐43 scores as independent variables, and subregion as well as rostrocaudal level as factors. Post hoc comparisons were conducted and corrected for multiple comparisons using Dunn's tests. Two Wilcoxon Signed‐Rank Tests were used to evaluate differences between medial and lateral portions of the EI and ECs subfields respectively. Then, we investigated if pTDP‐43 burden was associated with age, and four Spearman's correlations were conducted, investigating associations of pTDP‐43 pathology and age on each anterior–posterior level, respectively. Finally, we assessed potential associations and differences of pTDP‐43 and p‐tau semi‐quantitative scores of the same subfields, levels, and cases. A Wilcoxon Signed‐Rank Test compared semi‐quantitative pTDP‐43 and p‐tau scores of each individual subfield. Finally, two Spearman's correlations were computed to reveal associations between pTDP‐43 as well as p‐tau semi‐quantitative scores between each individual subfield, as well as for subfields averaged across cases. All statistical tests were two‐sided and applied an alpha‐value of p < 0.05 as the significance level.
3. RESULTS
3.1. pTDP‐43 pathology in the medial temporal lobe
All our cases presented positive inclusions in the amygdala, entorhinal cortex, and hippocampal regions, giving them at least a stage III on the Josephs' classification [11]. Qualitatively, isolated pTDP‐43 structures were observed in the amygdala, perirhinal, and entorhinal regions among BBI cases, with no major differences in density between them. However, perirhinal and entorhinal regions, respectively, consistently displayed higher pTDP‐43 density than the amygdala among BBII cases.
3.2. pTDP‐43 pathology within the entorhinal cortex
Layer II was the most affected cellular layer in evaluated subfields, primarily exhibiting heterogeneous NFT‐like structures and neuropil threads along the entire extent of the EC. Neuronal cytoplasmic and intranuclear inclusions were present but less common within the EC. Qualitatively, we found a steady stepwise pattern in pTDP‐43 pathology across the four anatomical levels (Figure 3A). This gradient was confirmed with a significant correlation and positive association of the rostrocaudal level and semi‐quantitative pTDP‐43 scores. This correlation was present when investigating subfield individual pTDP‐43 scores (Spearman's correlation; ρ(231) = 0.20, p = 0.002) as well as scores averaged across cases (Spearman's correlation; ρ(46) = 0.36, p = 0.013). We found a significant effect when we analyzed semi‐quantitative scores along anterior–posterior levels (Kruskal‐Wallis H‐test; χ2(3) = 9.62, p = 0.022). Post hoc Dunn's multiple comparison's tests revealed statistical differences between the most posterior level (level 7; posterior‐most hippocampal head) and the most anterior level 2 (p = 0.035; Figure 3B). In the anterior‐most EC anatomical level 2 (near amygdala), pTDP‐43 scores and age of case were significantly correlated (Spearman's correlation; ρ(9) = 0.65, p = 0.033). Figure 3C shows the significant correlation of average level 2 pathology and age of corresponding case. This observation was restricted to the most anterior levels, as correlations between age and average pathology scores for levels 4, 5 and 7 were not significant. Overall, we observed the densest pTDP‐43 pathology at the at the level of the posterior‐most hippocampal head, level 7. Thus, pTDP‐43 pathology occurred in a stepwise relationship with age from anterior to posterior within the entorhinal substructure.
FIGURE 3.
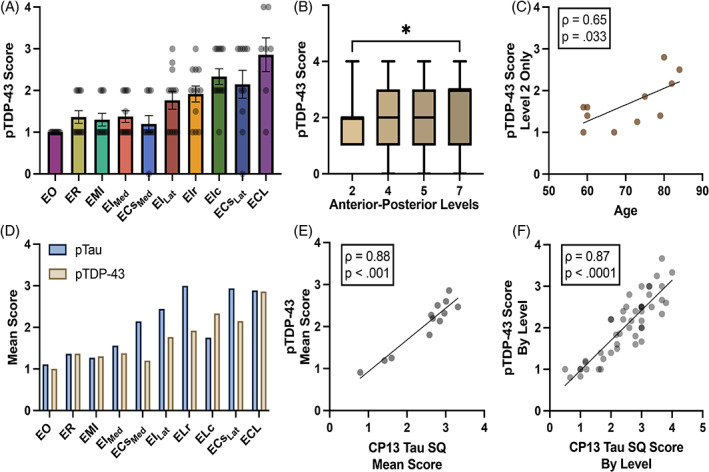
Semi‐quantitative pTDP‐43 and p‐tau scores in the EC: (A) Graph shows mean pTDP‐43 semi‐quantitative scores across all EC subfields and cases (n = 12). Posterior and lateral subfields ECL, ELc, and ECsLat presented the highest pTDP‐43 burden; EO displayed the lowest. Error bars represent standard error mean. (B) pTDP‐43 semi‐quantitative scores are shown across four anterior–posterior levels with horizontal lines that represent the median and whiskers the min/max value. pTDP‐43 pathology burden peaked at the posterior entorhinal (level 7), significantly differing from anterior pathology (level 2) (p = 0.035). (C) Significant Spearman's correlation between anterior pathology at level 2 and age of the individual cases (Spearman's correlation; ρ(9) = 0.65, p = 0.033). (D) Mean pTDP‐43 and p‐tau pathology semi‐quantitative scores compared across the EC subfields. Lateral‐posterior regions (subfields ECL, ECsLat, ELr, and ELc) showed the highest pathology burden for both pTDP‐43 and p‐tau. P‐tau was higher than TDP‐43 in score magnitude in every subfield, except ELc and EMI. (E) Spearman's correlation between mean pTDP‐43 and p‐tau scores per case was strongly significant (Spearman's correlation; ρ(10) = 0.88, p < 0.001). (F) Spearman's correlation comparing mean individual level pTDP‐43 and p‐tau scores per case was also strongly significant (Spearman's correlation; ρ(46) = 0.87, p < 0.0001). *Significance = p < 0.050.
3.3. pTDP‐43 pathology in posterior‐lateral EC subfields
There was a strong effect of entorhinal subfields on pTDP‐43 semi‐quantitative scores (Figure 3A; Kruskal‐Wallis H‐test; χ2(9) = 38.81, p < 0.0001). Qualitatively, posterior‐lateral entorhinal subfields ECL, ECsLat, and ELc displayed the heaviest pTDP‐43 burden (Figures 3A and 5). Conversely, the anterior‐medial subfields EO and ECsMed had the lowest pTDP‐43 scores, followed by the anterior‐medial subfields EMI, ER, and EIMed (Figures 3A and 5). Medial‐to‐lateral effects in pTDP‐43 were found when testing for differences between bifurcations of the ECs and EI subfields. Lateral portions of the EI and ECs subfields (EILat, ECsLat) presented significantly higher pTDP‐43 density scores when compared to their medial counterparts EIMed (Figure 4C; Wilcoxon; W = 55.00, p = 0.002) and ECsMed (Figure 4D; Wilcoxon; W = 38.00, p = 0.027).
FIGURE 5.
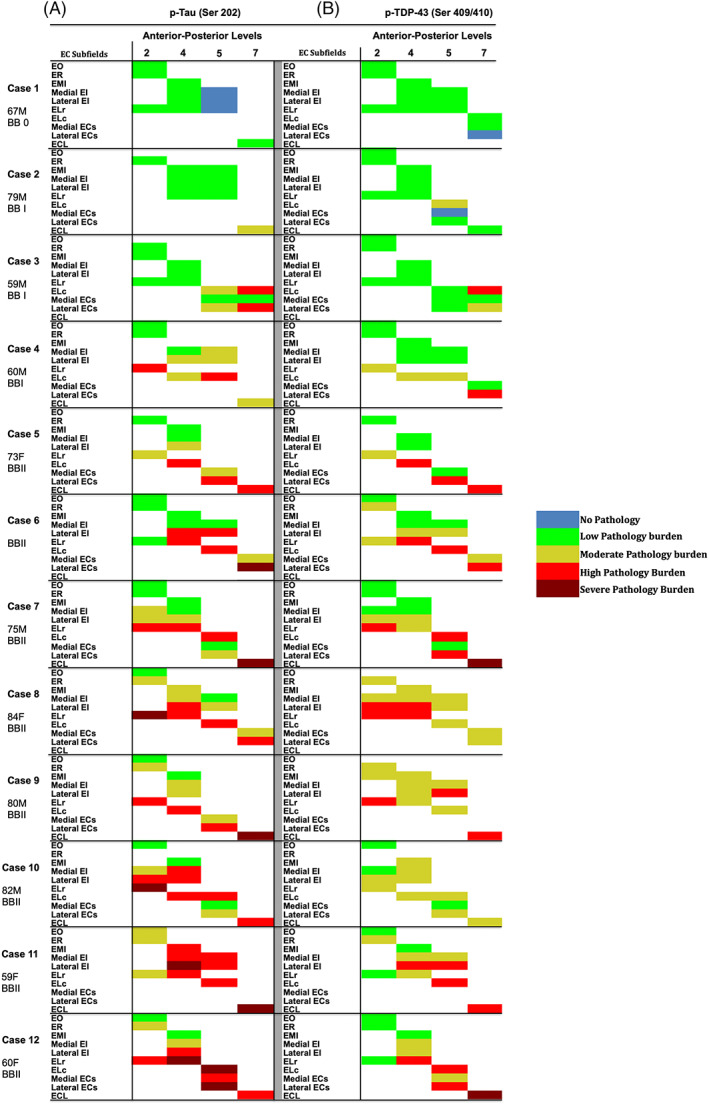
Heat‐map of p‐tau and pTDP‐43 semi‐quantitative scores in entorhinal subfields (n = 12): Figure 5 shows a color‐coded heat‐map of entorhinal subfields for p‐tau and pTDP‐43 pathology semiquantitative scores across cases, levels, and arranged by BB stages (0–II). The left panels show p‐tau SQ scores and the right panels show pTDP‐43 SQ scores. Color code reflects semi‐quantitative scores: blue (score 0, no pathology), green (score 1, isolated pathology inclusions), yellow (score 2, moderate pathology burden), red (score 3, high pathology burden), burgundy (score 4, severe pathology burden). Cases 1 and 2 lacked p‐tau or pTDP‐43 pathology altogether in some entorhinal subfields. Green labels were evenly distributed among the BB0 and BBI cases, and predominantly in medial‐anterior subfields in the BBII cases. Yellow label distribution depended on BB stage, with higher prevalence in anterior regions as overall pathology burden worsened. With few exceptions, red labels were observed predominantly in the lateral posterior EC subfields. Burgundy labels represent severe pathology burden (score 4) and were typically found in the lateral‐posterior subfields among the most affected cases.
FIGURE 4.
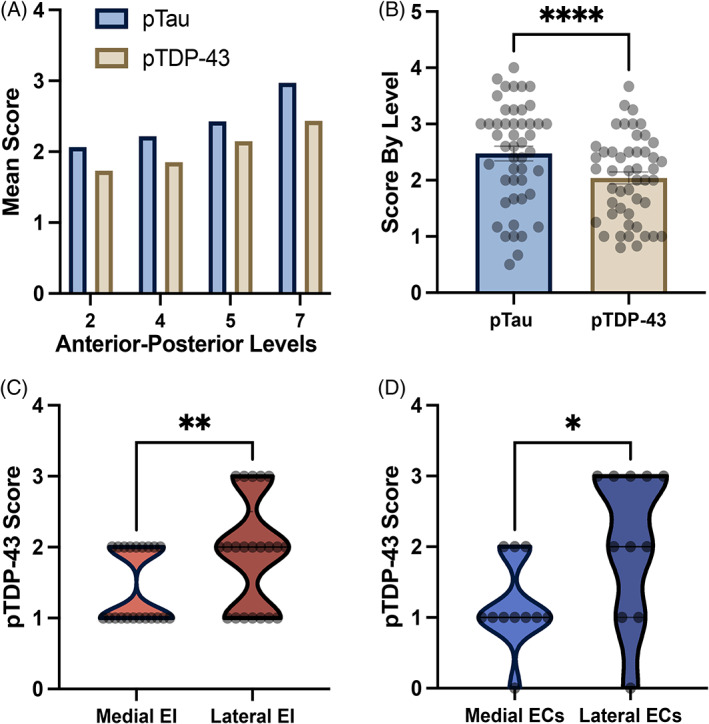
Semi‐quantitative pTDP‐43 and p‐tau scores: (A) Mean semi‐quantitative scores for pTDP‐43 and p‐tau across four anterior–posterior levels. Stepwise rise was demonstrated in both pathologies, peaking posteriorly at level 7. (B) Mean semi‐quantitative scores for pTDP‐43 and p‐tau based on respective immunostains. P‐tau shows significantly higher overall scores (Wilcoxon; W = −842.00, p < 0.0001). Error bars represent standard error mean. (C) We found a significant difference between medial and lateral EI semi‐quantitative scores, with lateral EI presenting a higher pathology burden (Wilcoxon; W = 55.00, p = 0.002). (D) Significant difference between medial and lateral ECs semi‐quantitative scores, with lateral ECs showing a higher pathology burden (Wilcoxon; W = 38.00, p = 0.027). *Significance = p < 0.050.
3.4. pTDP‐43 and p‐tau density scores relate across EC subfields
We compared p‐tau and pTDP‐43, in the same cases using semi‐quantitative protocols with similar criteria. pTDP‐43 semi‐quantitative scores significantly correlated with p‐tau semi‐quantitative scores in the same anterior–posterior levels and sample set. This correlation held true for total average semi‐quantitative scores per case (Figure 3E; Spearman's correlation; ρ(10) = 0.88, p < 0.001) as well as scores for individual anterior–posterior levels (Figure 3F; Spearman's correlation; ρ(46) = 0.87, p < 0.0001). Figure 4A shows this paired stepwise pattern along the rostrocaudal axis. Similarly, a strong correlation was found between p‐tau and pTDP‐43 average semi‐quantitative scores in the EC subfields (Spearman's correlation; ρ(8) = 0.70, p = 0.031), with the lateral subfields ELr and ELc, and the posterior subfields ECL and ECsLat presenting high inclusion density for both pathologies (Figure 3D). Despite p‐tau and pTDP‐43 presenting the same pattern along anterior–posterior levels and a similar pattern among the EC subfields, marked qualitative differences in the absolute magnitude of the two pathologies were observed (Figures 3D, 4A,B and 5). There was a significant difference between average p‐tau and pTDP‐43 level semi‐quantitative scores (Figure 4B; Wilcoxon; W = −842.00, p < 0.0001) and average EC subfield semi‐quantitative pathology ratings (Wilcoxon; W = −39.00, p = 0.049). See Table S1 for descriptive statistics of pTDP‐43 and p‐tau semi‐quantitative pathology scores in the EC subfields.
Figure 5 collectively presents a color‐coded map based on semi‐quantitative scores of p‐tau (Figure 5A) and pTDP‐43 (Figure 5B) densities. Figure 5 illustrates pTDP‐43 pathology density scores for each subfield at every level, and for every case. The color code (blue, green, yellow, red, burgundy = score 0, 1, 2, 3, 4) provides a heat map of pTDP‐43 pathology density and its vulnerability within the preclinical Braak stages.
4. DISCUSSION
Present in as many as 70% of AD cases, transactive response DNA binding protein of 43 kDa (TDP‐43) is a common comorbidity in dementia [9, 10, 11, 12, 13, 30]. In this study, we investigated the selective vulnerability of the entorhinal subfields to phosphorylated TDP‐43 pathology in preclinical AD and PART postmortem tissue. Previous reports suggest that p‐tau and pTDP‐43 aggregates can colocalize in diseased regions [28, 41], but the degree of their neuronal overlap and interaction is still debated. We showed the relationship between pTDP‐43 and p‐tau in the same regions at pathology‐positive but clinically silent stages. P‐tau and pTDP‐43 presented a similar pattern of subregions affected, but different magnitudes in density at this early stage. P‐tau showed a higher or equal magnitude than pTDP‐43 at every anterior–posterior level and in every subfield, except EMI and ELc. These findings suggest that p‐tau and pTDP‐43 aggregates deposit in similar patterns in the EC, and the posterior‐lateral entorhinal neurons show the greatest vulnerability to both.
We found significant differences in pTDP‐43 pathology within the EC. The posterior‐most level (level 7, identified by the nearby posterior‐most hippocampal head and gyrus intralimbicus) presented the highest pathology density (Figure 3B). Within the EC, posterior‐lateral subfields ECL, ELc, and ECsLat, respectively, showed the highest semi‐quantitative pTDP‐43 density scores, while EO had the lowest (Figure 3A). Our finding of a posterior‐lateral EC vulnerability to pTDP‐43 complements molecular characterizations of vulnerable caudal EC neurons [42], PET imaging observations [43], the p‐tau gradient in subfields [32], and the use of lateral EC atrophy as a potential biomarker of preclinical AD [44, 45]. Two reports support a prion‐like seeding mechanism involved in TDP‐43 spread [46, 47], and recent hippocampal data from FTD patients suggest that TDP‐43 pathology spreads along trans‐synaptic connections [48]. However, definitive molecular mechanisms of TDP‐43 spread are still widely unknown. Given that the EC is a pivotal site in the healthy‐aging‐mild cognitive impairment‐Alzheimer's disease continuum, documenting the specific locations of TDP‐43 pathology provides a baseline early in disease progression.
Amygdala, perirhinal, and entorhinal regions displayed isolated pTDP‐43 inclusions among BB0 and BBI cases. However, perirhinal cortex showed the highest overall pathology burden among BBII cases, followed by entorhinal cortex, and finally amygdala in our sample set. This suggests that, while amygdala is the likely origin of pTDP‐43 inclusions, it is surpassed in pathology density by the perirhinal and entorhinal cortices respectively even at early Braak stages.
The present data elucidate the selective vulnerability of pTDP‐43+ neuron populations within stage II of the current AD [11] and PART [27] pTDP‐43 staging schemes. Moreover, the close relationship between TDP‐43 and p‐tau in this region makes the case for the inclusion of the EC in the minimum criteria needed for LATE‐NC staging [18]. Considering the EC as a whole entity in the progression of pTDP‐43 burden may miss early changes. Although EC pTDP‐43 pathology peaked posteriorly, the anterior‐most levels demonstrated pTDP‐43 pathology that significantly associated with increasing age of the case (Figure 3C). Anterograde tracer studies in the monkey have shown that the lateral and basal nuclei of the amygdala (the typical origin of AD pTDP‐43 pathology) project primarily to the subfields EO, ER, and ELr (i.e., anterior EC), with some innervation to ELc and the rostral‐most EI [49]. Since we found a higher pTDP‐43 pathology burden in posterior subregions as opposed to anterior ones, our data suggests an asynchronous initial pathological development, not driven by connectivity. Moreover, the fact that EO (entorhinal olfactory) presented the lowest pathology for both p‐tau and pTDP‐43 should be considered as olfactory assessments for inexpensive tests of cognitive decline continue to be developed [50, 51]. We previously speculated about a first wave of p‐tau pathology affecting posterior EC and a second wave affecting anterior EC [32]. Our data suggests that preclinical pTDP‐43 pathology follows a similar asynchronous pattern in the EC. These findings provide a more detailed analysis of the pathological vulnerabilities within stage II of pTDP‐43 progression in AD, particularly in the context of subregion, density, and age.
Do p‐tau and pTDP‐43 relate to each other in pathologic evaluations? We found strong correlations between pTDP‐43 and p‐tau along the rostrocaudal extent of the EC (Figures 3E,F and 4A) and among the EC subfields themselves (Figure 3D). Mixed findings have surfaced about p‐tau and TDP‐43 confluences in pathology. Tomé and colleagues demonstrated the colocalization of p‐tau and pTDP‐43 at 78.3% in the hippocampus of clinical AD brains [28]. Conversely, Gu and colleagues reported a negative association between pTDP‐43 and p‐tau levels in the human brain, which may be attributed to the potential instability of tau mRNA by TDP‐43 [52]. Double‐label immunofluorescence studies also highlight inconsistencies regarding the co‐localization of pTDP‐43 and p‐tau in the same neurons. While some studies suggest ample concurrence within the same cells [27, 28], while others suggest more independence, with only occasional associations with p‐tau filaments [41, 53, 54]. Higashi and colleagues found that the amygdalae and hippocampi with tau or alpha‐synuclein were also vulnerable to TDP‐43 pathology in AD cases, although the p‐tau and TDP‐43 inclusions hardly overlapped within the same neuron [41]. Nonetheless, given that samples can present p‐tau in the absence of pTDP‐43 and vice versa, it is unlikely that these pathologies necessitate each other. Our findings here suggest that populations of neurons vulnerable to p‐tau within the EC also present pTDP‐43. However, while the deposition patterns correlated, the absolute density of pathology‐positive neurons differed (Figure 4A,B), with a much more severe p‐tau burden present in the same subfields (Figure 3D). This result supports the independence of p‐tau and pTDP‐43 pathologies at the neuronal level, even if their preclinical burden peak at the same EC subfields.
Josephs et al. recently classified TDP‐43 pathology aggregates into type‐α and type‐β, where the latter is primarily identified by its overlap with NFTs [24]. Some factors, such as prevalent heterogeneous pTDP‐43 NFT‐like structures, suggest that the TDP‐43 pathology observed in our cases most closely resembles the β‐type. This was observed along the entire extent of the EC, independent of subfield (Figures 2 and 6), and was particularly evident in subfields that also presented high levels of p‐tau NFTs (Figures 5 and 6). Moreover, as is the case for type‐β TDP‐43, we noticed significant involvement in limbic regions at early (Josephs stages I–II) TDP‐43 stages [24]. However, we also observed TDP‐43 type‐α features, such as the presence of neuropil threads, albeit less severely than in p‐tau, and occasional neuronal cytoplasmic and intranuclear inclusions in heavily affected regions. Notably, our cases comprise the preclinical Braak stages (I–II), which have been shown to primarily display type‐α TDP aggregates [24]. Though our observed TDP‐43 aggregates categorize more closely to the type‐β classification, future studies will be needed to confirm whether these differences in TDP‐43 correspond to a spectrum or a sharp distinction within AD and non‐demented elderly cases. This division will prove especially valuable in the EC, as it is a point of convergence for p‐tau and TDP‐43 in stage II of their respective pathology scoring [3, 11].
FIGURE 6.
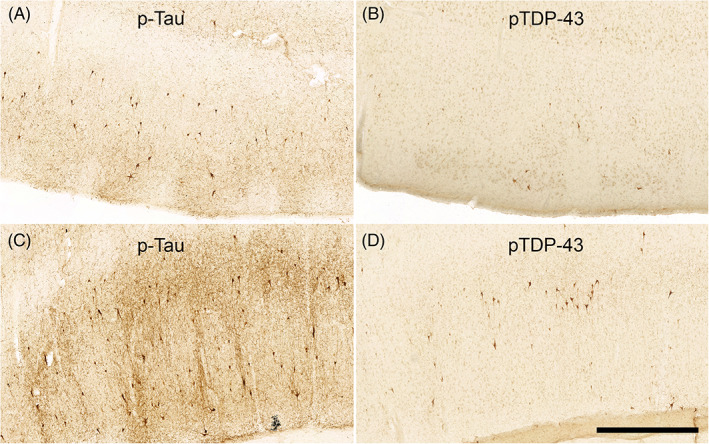
Immunostaining p‐tau and pTDP‐43 shows Qualitative Differences in the Same Subregions: Figure 6 shows representative examples of qualitative differences in the magnitude of p‐tau and pTDP‐43 pathology in the same subregions from the same case (case 12, BBII). Panels A and B show, respectively, p‐tau and pTDP‐43 immunostained tissue from the EC. Panels C and D show, respectively, p‐tau and pTDP‐43 immunostained tissue from the perirhinal cortex. CP13 p‐tau immunostained tissue in the EC and perirhinal cortex displayed a higher pathological burden than the pTDP‐43 burden seen in the same case regions, a widespread phenomenon among the rest of the cases. Magnification bar = 1 mm.
Our work benefitted from several strengths. This annotative study brings together p‐tau and pTDP‐43 not only from a subregional but also from a density perspective. This includes the exact cytoarchitectonic validation of the EC subfields and the correspondence of the p‐tau SQ ratings on the same subfields, anterior–posterior levels, and cases. Assessing the pathology density of similar structures and in the same case subfields allowed for direct comparison of the scores, something not possible in studies with separate scales [28]. This work also examines pTDP‐43 pathology during the preclinical window in cognitively healthy aging, but before Alzheimer's disease, to report the earliest changes in aging. While diagnostic schemes assess pathology on a present‐or‐not basis, we provide a perspective of density.
Limitations include that our cases followed the typical limbic pathology progression, and we did not study AD subtypes [55, 56, 57]. In addition, our sample set contained a mixture of right and left hemispheres. While some reports found that TDP‐43 in some forms of dementia favors the language‐dominant hemisphere [58, 59, 60], the result has not consistently been replicated [48], and it is not yet known if it holds true for typical limbic AD. Finally, while we evaluated pathology on the same subfields on adjacent tissue slides, we did not perform double‐staining to assess the co‐localization of pathology at the individual neuron level.
In summary, we identified a posterior‐lateral vulnerability to pTDP‐43 within the EC. We found an age‐dependent association with anterior EC pTDP‐43 pathology and reported heterogeneity within stage II of the pTDP‐43 staging schemes for AD and PART, which consider the EC as a whole entity [11, 12, 27, 30]. Finally, we demonstrated a close association between p‐tau and pTDP‐43 among the EC subfields, yet p‐tau burden was significantly heavier. This work serves as an observational study that lays groundwork to better understand the deposition of comorbidities in Alzheimer's disease. We provide a reference for future approaches in individualized medicine and highlight early vulnerabilities to two major pathological hallmarks in aging and dementia.
AUTHOR CONTRIBUTIONS
Josué Llamas Rodríguez: conceptualization, formal analysis, investigation, writing – original draft, writing – review & editing, visualization. Jan Oltmer: software, formal analysis. Michael Marshall: resources. Samantha Champion: resources. Matthew P. Frosch: resources. Jean C. Augustinack: conceptualization, investigation, resources, writing – review & editing, supervision, project administration, funding acquisition.
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ETHICS STATEMENT
The studies involving human participants were reviewed and approved by the Internal Review Board at the Massachusetts General Hospital. The patients/participants provided their written informed consent to participate in this study.
Supporting information
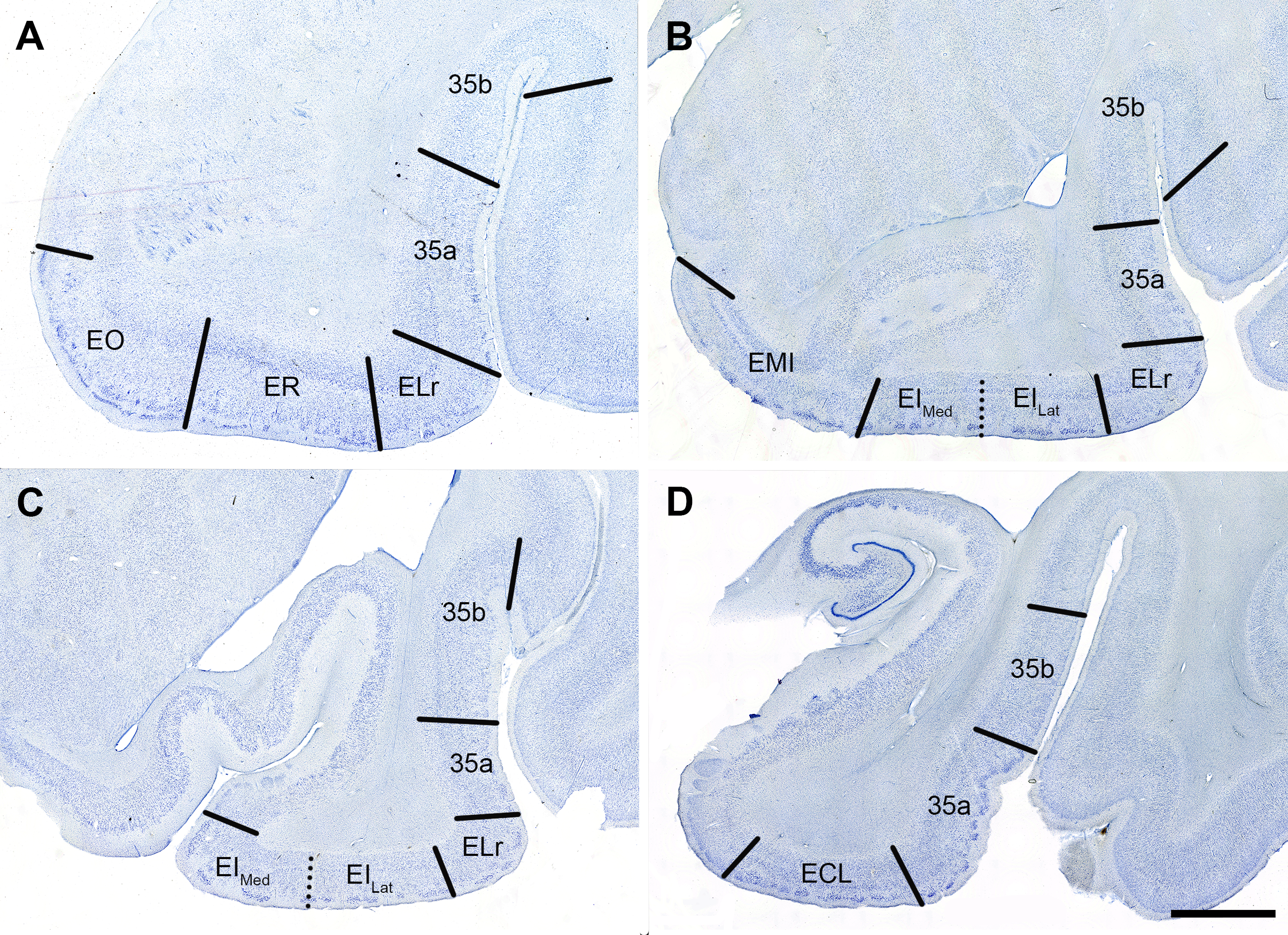
Figure S1. Nissl and pTDP‐43 Stained Tissue from Four Anterior–Posterior levels: Panels (A, C, E, F) show EC subfields in Nissl stained tissue parcellated according to Insausti et al. [35]. All panels show tissue in the coronal plane. Nissl stained tissue served as the cytoarchitectonic basis for the subfield parcellation applied to adjacent pTDP‐43 immunostained tissue. The four anterior–posterior levels shown were adapted from the original eight levels presented in Llamas‐Rodríguez et al. [32] (Table 2): level 2 (panels A, B; anterior amygdala, and typically EC subfields EO, ER, and ELr), level 4 (panels C, D; posterior amygdala and anterior hippocampal head, that is, genu, and typically EC subfields EMI, EI, and ELr), level 5 (panels E, F; hippocampal pes region with gyrus uncinatus, and typically EC subfields ECs, ELc, and occasionally EI), and level 7 (panels G, H; posterior‐most hippocampal head and the gyrus intralimbicus, and typically EC subfield ECL and occasionally ECs). When present, subfields EI and ECs were bifurcated into medial and lateral counterparts. 35a, Brodmann area 35a; 35b, Brodmann area 35b; ECs, entorhinal caudal; ECL, entorhinal caudal limiting; EI, entorhinal intermediate; ELc, entorhinal lateral caudal; ELr, entorhinal lateral rostral; EMI, entorhinal medial intermediate; EO, entorhinal olfactory; ER, entorhinal rostral. Magnification bar = 4 mm.
{kind=link}
Table S1. Descriptive Statistics of EC Subfield Semi‐Quantitative Pathological Scores: Descriptive information on average EC subfield p‐tau and pTDP‐43 semi‐quantitative scores, confidence intervals, and percentiles. CI, confidence interval; EC, entorhinal cortex; SD, standard deviation; SEM, standard error mean.
ACKNOWLEDGMENTS
We thank the donors for their generous donation. We are grateful to Dr. Peter Davies for the gift of CP13 and his contribution to neuropathology. Grant support for this research was provided by the US National Institute of Health: R01AG057672 and RF1AG072056 for JCA.
Llamas‐Rodríguez J, Oltmer J, Marshall M, Champion S, Frosch MP, Augustinack JC. TDP‐43 and tau concurrence in the entorhinal subfields in primary age‐related tauopathy and preclinical Alzheimer's disease. Brain Pathology. 2023;33(4):e13159. 10.1111/bpa.13159
DATA AVAILABILITY STATEMENT
The original contributions presented in this study are included in the article/supplementary material, further inquiries should be directed to the corresponding author upon reasonable request.
REFERENCES
- 1. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11. 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- 2. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82(4):239–59. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 4. Gómez‐Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41(1):17–24. 10.1002/ana.410410106 [DOI] [PubMed] [Google Scholar]
- 5. Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14(4):535–62. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–800. 10.1212/WNL.58.12.1791 [DOI] [PubMed] [Google Scholar]
- 7. Zetterberg H. Biofluid‐based biomarkers for Alzheimer's disease‐related pathologies: an update and synthesis of the literature. Alzheimers Dement. 2022;18(9):1687–93. 10.1002/alz.12618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nascimento C, Di Lorenzo Alho AT, Amaral, C. BC , Leite REP, Nitrini R, Jacob‐Filho W, et al. Prevalence of transactive response DNA‐binding protein 43 (TDP‐43) proteinopathy in cognitively normal older adults: systematic review and meta‐analysis. Neuropathol Appl Neurobiol. 2018;44(3):286–97. 10.1111/nan.12430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arai T, Mackenzie IRA, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP‐43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117(2):125–36. 10.1007/s00401-008-0480-1 [DOI] [PubMed] [Google Scholar]
- 10. Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L, et al. Rates of hippocampal atrophy and post‐mortem TDP‐43 in Alzheimer's disease: a longitudinal retrospective study. Lancet Neurol. 2017;16(11):917–24. 10.1016/S1474-4422(17)30284-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP‐43 in Alzheimer's disease staging scheme. Acta Neuropathol. 2016;131(4):571–85. 10.1007/s00401-016-1537-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP‐43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127(6):811–24. 10.1007/s00401-014-1269-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meneses A, Koga S, O'Leary J, Dickson DW, Bu G, Zhao N. TDP‐43 pathology in Alzheimer's disease. Mol Neurodegener. 2021;16(1):84. 10.1186/s13024-021-00503-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arnold SJ, Dugger BN, Beach TG. TDP‐43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol. 2013;126(1):51–7. 10.1007/s00401-013-1110-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geser F, Robinson JL, Malunda JA, Xie SX, Clark CM, Kwong LK, et al. Pathological 43‐kDa transactivation response DNA‐binding protein in older adults with and without severe mental illness. Arch Neurol. 2010;67(10):1238–50. 10.1001/archneurol.2010.254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinacker P, Barschke P, Otto M. Biomarkers for diseases with TDP‐43 pathology. Mol Cell Neurosci. 2019;97:43–59. 10.1016/j.mcn.2018.10.003 [DOI] [PubMed] [Google Scholar]
- 17. Spina S, La Joie R, Petersen C, Nolan AL, Cuevas D, Cosme C, et al. Comorbid neuropathological diagnoses in early versus late‐onset Alzheimer's disease. Brain. 2021;144(7):2186–98. 10.1093/brain/awab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27. 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boyle PA, Wang T, Yu L, Wilson RS, Dawe R, Arfanakis K, et al. To what degree is late life cognitive decline driven by age‐related neuropathologies? Brain. 2021;144(7):2166–75. 10.1093/brain/awab092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee EB, Porta S, Baer GM, Xu Y, Suh E, Kwong LK, et al. Expansion of the classification of FTLD‐TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134(1):65–78. 10.1007/s00401-017-1679-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mackenzie IRA, Neumann M, Baborie A, Sampathu DM, Plessis DD, Jaros E, et al. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 2011;122(1):111–3. 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin‐positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169(4):1343–52. 10.2353/ajpath.2006.060438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan RH, Shepherd CE, Kril JJ, McCann H, McGeachie A, McGinley C, et al. Classification of FTLD‐TDP cases into pathological subtypes using antibodies against phosphorylated and non‐phosphorylated TDP43. Acta Neuropathol Commun. 2013;1(1):33. 10.1186/2051-5960-1-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB, et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP‐43 types in non‐FTLD brains. Acta Neuropathol. 2019;137(2):227–38. 10.1007/s00401-018-1951-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64(1):60–70. 10.1002/ana.21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomé SO, Vandenberghe R, Ospitalieri S, Van Schoor E, Tousseyn T, Otto M, et al. Distinct molecular patterns of TDP‐43 pathology in Alzheimer's disease: relationship with clinical phenotypes. Acta Neuropathol Commun. 2020;8(1):61. 10.1186/s40478-020-00934-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang X, Sun B, Wang X, Lu H, Shao F, Rozemuller AJM, et al. Phosphorylated TDP‐43 staging of primary age‐related tauopathy. Neurosci Bull. 2019;35(2):183–92. 10.1007/s12264-018-0300-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tomé SO, Gomes LA, Li X, Vandenberghe R, Tousseyn T, Thal DR. TDP‐43 interacts with pathological τ protein in Alzheimer's disease. Acta Neuropathol. 2021;141(5):795–9. 10.1007/s00401-021-02295-2 [DOI] [PubMed] [Google Scholar]
- 29. Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015;129(4):469–91. 10.1007/s00401-014-1380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP‐43 pathology in Alzheimer's disease. Acta Neuropathol. 2014;127(3):441–50. 10.1007/s00401-013-1211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404. 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Llamas‐Rodríguez J, Oltmer J, Greve DN, Williams E, Slepneva N, Wang R, et al. Entorhinal subfield vulnerability to neurofibrillary tangles in aging and the preclinical stage of Alzheimer's disease. J Alzheimer's Dis. 2022;87(3):1379–99. 10.3233/JAD-215567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1–13. 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oltmer J, Slepneva N, Llamas Rodriguez J, Greve DN, Williams EM, Wang R, et al. Quantitative and histologically validated measures of the entorhinal subfields in ex vivo MRI. Brain Commun. 2022;4(3):fcac074. 10.1093/braincomms/fcac074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Insausti R, Tuñón T, Sobreviela T, Insausti AM, Gonzalo LM. The human entorhinal cortex: a cytoarchitectonic analysis. J Comp Neurol. 1995;355(2):171–98. 10.1002/cne.903550203 [DOI] [PubMed] [Google Scholar]
- 36. Moloney CM, Lowe VJ, Murray ME. Visualization of neurofibrillary tangle maturity in Alzheimer's disease: a clinicopathologic perspective for biomarker research. Alzheimers Dement. 2021;17(9):1554–74. 10.1002/alz.12321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755–66. 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alosco ML, Cherry JD, Huber BR, Tripodis Y, Baucom Z, Kowall NW, et al. Characterizing tau deposition in chronic traumatic encephalopathy (CTE): utility of the McKee CTE staging scheme. Acta Neuropathol. 2020;140(4):495–512. 10.1007/s00401-020-02197-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravikumar S, Wisse LEM, Lim S, Ittyerah R, Xie L, Bedard ML, et al. Ex vivo MRI atlas of the human medial temporal lobe: characterizing neurodegeneration due to tau pathology. Acta Neuropathol Commun. 2021;9(1):173. 10.1186/s40478-021-01275-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Augustinack JC, Huber KE, Postelnicu GM, Kakunoori S, Wang R, van der Kouwe AJW, et al. Entorhinal verrucae geometry is coincident and correlates with Alzheimer's lesions: a combined neuropathology and high resolution ex vivo MRI analysis. Acta Neuropathol. 2012;123(1):85–96. 10.1007/s00401-011-0929-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, et al. Concurrence of TDP‐43, tau and alpha‐synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–94. 10.1016/j.brainres.2007.09.048 [DOI] [PubMed] [Google Scholar]
- 42. Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer's disease. Nat Neurosci. 2021;24(2):276–87. 10.1038/s41593-020-00764-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berron D, Vogel JW, Insel PS, Pereira JB, Xie L, Wisse LEM, et al. Early stages of tau pathology and its associations with functional connectivity, atrophy and memory. Brain. 2021;144(9):2771–83. 10.1093/brain/awab114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holbrook AJ, Tustison NJ, Marquez F, Roberts J, Yassa MA, Gillen DL, et al. Anterolateral entorhinal cortex thickness as a new biomarker for early detection of Alzheimer's disease. Alzheimer's Dement. 2020;12(1):e12068. 10.1002/dad2.12068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, et al. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer's disease. Nat Neurosci. 2014;17(2):304–11. 10.1038/nn.3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA‐binding proteins with prion‐like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smethurst P, Sidle KCL, Hardy J. Review: prion‐like mechanisms of transactive response DNA binding protein of 43 kDa (TDP‐43) in amyotrophic lateral sclerosis (ALS). Neuropathol Appl Neurobiol. 2015;41(5):578–97. 10.1111/nan.12206 [DOI] [PubMed] [Google Scholar]
- 48. Jamshidi P, Kim G, Shahidehpour RK, Bolbolan K, Gefen T, Bigio EH, et al. Distribution of TDP‐43 pathology in hippocampal synaptic relays suggests Transsynaptic propagation in frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2020;79(6):585–91. 10.1093/jnen/nlaa029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pitkänen A, Kelly JL, Amaral DG. Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the entorhinal cortex in the macaque monkey. Hippocampus. 2002;12(2):186–205. 10.1002/hipo.1099 [DOI] [PubMed] [Google Scholar]
- 50. Kim SM, Kim HR, Min HJ, Kim KS, Jin J‐C, Han DH. A novel olfactory threshold test for screening cognitive decline among elderly people. PLoS One. 2021;16(7):e0254357. 10.1371/journal.pone.0254357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stamps JJ, Bartoshuk LM, Heilman KM. A brief olfactory test for Alzheimer's disease. J Neurol Sci. 2013;333:19–24. 10.1016/j.jns.2013.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gu J, Wu F, Xu W, Shi J, Hu W, Jin N, et al. TDP‐43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017;45(10):6177–93. 10.1093/nar/gkx175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Amador‐Ortiz C, Lin W‐L, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP‐43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61(5):435–45. 10.1002/ana.21154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin W‐L, Dickson DW. Ultrastructural localization of TDP‐43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116(2):205–13. 10.1007/s00401-008-0408-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferreira D, Nordberg A, Westman E. Biological subtypes of Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2020;94(10):436–48. 10.1212/WNL.0000000000009058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murray ME, Graff‐Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–96. 10.1016/S1474-4422(11)70156-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27(5):871–81. 10.1038/s41591-021-01309-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim G, Ahmadian SS, Peterson M, Parton Z, Memon R, Weintraub S, et al. Asymmetric pathology in primary progressive aphasia with progranulin mutations and TDP inclusions. Neurology. 2016;86(7):627–36. 10.1212/WNL.0000000000002375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim G, Bolbolan K, Shahidehpour R, Jamshidi P, Gefen T, Ayala IA, et al. Morphology and distribution of TDP‐43 pre‐inclusions in primary progressive aphasia. J Neuropathol Exp Neurol. 2019;78(3):229–37. 10.1093/jnen/nlz005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim G, Vahedi S, Gefen T, Weintraub S, Bigio EH, Mesulam M‐M, et al. Asymmetric TDP pathology in primary progressive aphasia with right hemisphere language dominance. Neurology. 2018;90(5):e396–403. 10.1212/WNL.0000000000004891 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Nissl and pTDP‐43 Stained Tissue from Four Anterior–Posterior levels: Panels (A, C, E, F) show EC subfields in Nissl stained tissue parcellated according to Insausti et al. [35]. All panels show tissue in the coronal plane. Nissl stained tissue served as the cytoarchitectonic basis for the subfield parcellation applied to adjacent pTDP‐43 immunostained tissue. The four anterior–posterior levels shown were adapted from the original eight levels presented in Llamas‐Rodríguez et al. [32] (Table 2): level 2 (panels A, B; anterior amygdala, and typically EC subfields EO, ER, and ELr), level 4 (panels C, D; posterior amygdala and anterior hippocampal head, that is, genu, and typically EC subfields EMI, EI, and ELr), level 5 (panels E, F; hippocampal pes region with gyrus uncinatus, and typically EC subfields ECs, ELc, and occasionally EI), and level 7 (panels G, H; posterior‐most hippocampal head and the gyrus intralimbicus, and typically EC subfield ECL and occasionally ECs). When present, subfields EI and ECs were bifurcated into medial and lateral counterparts. 35a, Brodmann area 35a; 35b, Brodmann area 35b; ECs, entorhinal caudal; ECL, entorhinal caudal limiting; EI, entorhinal intermediate; ELc, entorhinal lateral caudal; ELr, entorhinal lateral rostral; EMI, entorhinal medial intermediate; EO, entorhinal olfactory; ER, entorhinal rostral. Magnification bar = 4 mm.
Table S1. Descriptive Statistics of EC Subfield Semi‐Quantitative Pathological Scores: Descriptive information on average EC subfield p‐tau and pTDP‐43 semi‐quantitative scores, confidence intervals, and percentiles. CI, confidence interval; EC, entorhinal cortex; SD, standard deviation; SEM, standard error mean.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries should be directed to the corresponding author upon reasonable request.