

Abstract
人类染色体核型分析是诊断遗传疾病的重要手段,染色体图像类型识别是分析过程的关键步骤,准确高效地识别对自动核型分析具有重大意义。本文提出了一种分段重标定的稠密卷积神经网络模型(SR-DenseNet),模型各阶段先利用稠密连接的网络层自动提取染色体不同抽象层次的特征,再用压缩激活(SE)结构对汇集了局部所有特征的层进行特征重标定,对不同特征的重要性显式地构建可学习的结构;提出了一种模型融合方法,构建了染色体识别模型专家组。在国际公开的哥本哈根染色体识别数据集(G 显带)上进行了实验验证,该模型的识别错误率仅为 1.60%;采用模型融合方法后,识别错误率进一步降低到 0.99%。在意大利帕多瓦大学的数据集(Q 显带)上,识别错误率为 6.67%;模型融合后,进一步降低到 5.98%。实验结果表明本文所提方法是有效的,具备实现染色体类型识别自动化的潜力。
Keywords: 染色体识别, 稠密卷积神经网络, 分段重标定, 染色体核型分析
Abstract
Human chromosomes karyotyping is an important means to diagnose genetic diseases. Chromosome image type recognition is a key step in the karyotyping process. Accurate and efficient identification is of great significance for automatic chromosome karyotyping. In this paper, we propose a model named segmentally recalibrated dense convolutional network (SR-DenseNet). In each stage of the model, the dense connected network layers is used to extract the features of different abstract levels of chromosomes automatically, and then the concatenation of all the layers which extract different local features is recalibrated with squeeze-and-excitation (SE) block. SE blocks explicitly construct learnable structures for importance of the features. Then a model fusion method is proposed and an expert group of chromosome recognition models is constructed. On the public available Copenhagen chromosome recognition dataset (G-bands) the proposed model achieves error rate of only 1.60%, and with model fusion the error further drops to 0.99%. On the Padova chromosome dataset (Q-bands) the model gets the corresponding error rate of 6.67%, and with model fusion the error further drops to 5.98%. The experimental results show that the method proposed in this paper is effective and has the potential to realize the automation of chromosome type recognition.
Keywords: chromosome images recognition, dense convolutional network, segmental recalibration, karyotyping
引言
染色体是人类遗传物质的重要载体。正常情况下,体细胞包含 23 对(46 条)无结构改变的染色体。染色体在细胞分裂过程中出现染色体结构或数目异常的疾病,称为染色体病。如果这种情况出现在胚胎发育阶段,严重者会停止发育并流产,少数存活者可能出现机体畸变、智力低下、发育迟缓等情况[1-2]。目前这种疾病无有效的治疗方法,因此对染色体病的遗传分析和产前诊断是重要的预防手段。
染色体核型分析,是临床上诊断染色体是否存在异常的常用方法。该技术以细胞分裂中期的染色体为研究对象,并借助显带技术对染色体进行数字成像。成像后由专业的遗传学医师,人工从图像中分割染色体与其他杂质。再根据图像中各染色体如长短、染色体带型、着丝粒位置、有无随体、长短臂比例特点对染色体半自动分类配对。其中,把分割得到的单条染色体的图像分为 24 类(0~22 号常染色体和 X/Y 性染色体)之一的工作即为染色体图像类型识别(以下简称染色体识别)。得到配对的染色体图像后,按国际人类细胞遗传命名系统(international system for human cytogenetic nomenclature,ISCN)组织排列,再计数、分析以确定是否存在数目和结构异常。
传统的染色体核型分析主要依靠专业医师对采集到的染色体图像进行预处理、识别配对和异常分析[3]。在分割、识别、计数任务中,以识别配对在临床操作中最为耗时;且受染色体图像质量、医师技术经验和操作时精力集中程度等一系列因素的影响,识别任务是染色体核型分析效率较低、容易出错的环节。因而准确高效的染色体识别对自动核型分析具有重大意义。
随着计算机图像识别技术的发展,染色体核型自动分析技术受到了国内外众多研究者的关注[4-8]。2012 年以前的技术大都采用人工设计的特征或浅层人工神经网络提取特征再进行分类识别的方式。郭宏宇等[4]将模糊理论运用到染色体自动识别系统,并与神经网络相结合,提出了一种模糊神经网络模型。蒋欣[5]基于中点法提取染色体中轴,基于几何特征和灰度分布定位着丝粒,采用了平均灰度投影曲线、灰度梯度投影曲线和形状投影曲线提取带纹,再用加权的密度分布(weighted density distribution,WDD)[5]计算和表示带纹特征;然后采用两层分类器进行染色体识别。
深度学习技术的出现,促进了计算机图像研究领域长足的进步,并在大规模自然图像识别竞赛中取得了优异的成绩[9-14]。很多学者把相关技术应用在染色体识别任务中,自动提取图像特征并进行识别,取得了较好的结果[15-20]。Sharma 等[15]提出了结合众包、预处理和深度学习技术的方法,分割并识别染色体。该研究使用众包的方法分割出单条染色体后,对弯曲的染色体进行了预处理,但其预处理方法为填充的部分引入了非真实的像素。Qin 等[16]提出了变焦网络(Varifocal-Net)对染色体进行识别。该网络是一个二阶段的网络结构,包含全局尺度的网络(global-scale network,G-Net)和局部尺度的网络(local-scale network,L-Net)。G-Net 提取全局特征并检测可用于提取精细特征的区域,L-Net 变焦到 G-Net 定位的区域并进一步提取精细的局部特征。谭凯[19]提出了单条染色体图像的伸直处理算法,对弯曲染色体图像进行预处理,以提升后续网络对染色体的识别效果。Lin 等[20]为染色体识别任务设计了改进的开端(Inception)网络,结合提出的数据增强方法,取得了较好的识别效果。
本文充分结合压缩激活网络(squeeze-and-excitation networks,SE-Net)[13]和稠密卷积网络(dense convolutional network,DenseNet)[14]的优点,设计了分段重标定(segmental recalibration,SR)的 DenseNet(SR-DenseNet)模型。该网络利用单条染色体图像和对应的类别标注信息,自动提取染色体特征,并完成染色体图像识别任务。然后,本文选择国际上可公开使用的两个数据集进行实验,分别是:哥本哈根染色体数据集(Copenhagen chromosome dataset,CCD)(网址:ftp://ftp.igmm.ed.ac.uk/pub/CromData/gbands/CPR.data/)和意大利帕多瓦大学染色体分类数据集(Padova chromosome dataset for classification,PCDC)(网址:http://bioimlab.dei.unipd.it/Chromosome%20Data%20Set%204Class.htm)[21]。在这两个数据集上,本文与先进的通用卷积神经网络[9-14]以及 Sharma 等[15]、Qin 等[16]设计的染色体识别网络进行了对比实验。最后,提出了一种模型融合算法,在两个数据集上均进行了实验,进一步降低了染色体识别任务的错误率。实验结果表明,本文所提方法是有效的,具备实现染色体识别自动化的潜力,为进一步实现染色体核型分析的自动化打下基础。
1. SR-DenseNet 网络模型
1.1. SE-Net 的压缩激活结构
传统的卷积神经网络,在局部感受野上同时对不同通道进行卷积运算(convolution,conv),以提取空间和通道之间的信息。SE-Net 的压缩激活结构显式地对通道之间的相关性进行建模,重新标定了各通道之间特征图的重要性,提升了网络的表现能力[13]。该结构首先进行信息压缩(squeeze)操作,通过全局平均池化(global average pooling,GAP)得到各通道特征图的激活平均值,利用统计特性描述了通道的激活程度。然后,作者设计了激活(excitation)操作,以利用通道之间的依赖关系。具体实现时,重标定的网络支路中,GAP 操作之后得到了 1 × 1 × C(其中,C 为通道数)大小的特征图,先使用 1 × 1 conv 对特征图进行降维操作,得到 1 × 1 × C/r(其中,C 为通道数,r 是降维因子)大小的全连接层(fully connected layer,FC);再使用限制线性单元(rectified linear units,ReLU)激活函数得到 1 × 1 × C/r 大小的特征图;接着使用 1 × 1 conv 做升维操作,把降维的特征图还原到 1 × 1 × C 大小;最后使用了 S 状弯曲函数(sigmoid)作激活函数,实现了参数化的门限机制,得到与每个通道对应的 0~1 之间的激活值,该值重标定了特征图的重要性[13]。激活值与对应通道相乘得到重新标定的缩放(scale)层。
1.2. 稠密网络
自 He 等[12]提出残差网络(residual network,ResNet)以来,神经网络层与层之间的“捷径”(shortcut)结构被广泛使用。DenseNet 进一步发展了这种技术,设计了层与层两两之间通过捷径相互连接的局部网络结构,作者称之为稠密块(dense block)[14]。如图 1 下部所示,稠密块中每一层,都汇集其前面各层的特征图作为输入。这些特征图先经批归一化(batch normalization,BN)[22]操作之后,再通过卷积层调整其通道数。第 l 层特征图与前面层的关系如式(1)所示[14]:
图 1.

The schema of dense block module
稠密连接模块图示
 |
1 |
其中,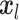 代表第 l 层的特征图,
代表第 l 层的特征图,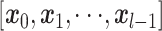 表示对 0 到 l − 1 层的特征进行平铺连接(concatenate,concat),
表示对 0 到 l − 1 层的特征进行平铺连接(concatenate,concat),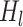 代表非线性变换函数,其结构如图 1 上部
代表非线性变换函数,其结构如图 1 上部 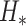 所示。图 1 中,h、w、c 分别特征图的高、宽、通道数,c0 表示稠密块第一层特征图的通道数。
所示。图 1 中,h、w、c 分别特征图的高、宽、通道数,c0 表示稠密块第一层特征图的通道数。
这种方式的连接比简单的通道相加保留了更多的信息,并促使网络选择更有表现力的通道,同时鼓励网络重利用前面层学到的特征,减轻了神经网络中梯度消失的问题,使得网络的训练更加容易。
1.3. SR-DenseNet 网络设计
参考医师既结合不同层次特征又以带型特征为主的分析方法,本文提出了 SR-DenseNet 模型,充分利用了稠密网络的信息融合能力和 SE-Net 的通道重要性的重标定特点,提升了网络的特征表现能力,有利于进一步提高网络对染色体的识别能力。
受到神经网络模块化思想的启发[10, 12-14],本文设计了构建网络的基本模块,称之为重标定的稠密模块。模块结构如图 2 所示,前半部分包含稠密连接的卷积层,后半部分包含重标定部分。其中,稠密连接部分各字母含义与图 1 相同,重标定部分 C 表示通道数,r 表示降维因子。稠密连接部分,前若干层特征图输入该结构后,先经 BN 操作,然后由 1 × 1 大小卷积核调整特征图通道数,接着再进行 BN 操作,并由 3 × 3 大小卷积核提取特征并输出 32 个特征图,形成瓶颈结构;若干 BN-conv1 × 1-BN-conv3 × 3 瓶颈结构再以稠密连接的形式重复堆叠。重标定部分利用压缩激活结构自适应地对各通道特征的重要性进行评价,充分利用对识别有利的特征。
图 2.

The schema of recalibrated dense module
重标定的稠密模块
基于上述模块,本文设计了 125 层的 SR-DenseNet 网络模型。网络第一层是包含 64 个 7 × 7 大小卷积核的卷积层。随后是 4 个阶段重标定的稠密模块,各阶段稠密模块包含的瓶颈结构数量分别为 6、12、24、16。每个模块的低层特征图与高层特征图在稠密部分的顶层平铺连接后,再自适应地重标定。需要注意的是,与 SE-Net 不同,稠密部分顶层包含了该模块不同层次的特征,本文网络不对稠密部分中间层的特征图实施重标定。最后的分类层包含 24 个神经元,对应染色体的 24 个类别。
1.4. 多模型融合
本文希望得到一个模型,它对各类染色体的识别精度都达到最高。但在实验过程中,本文发现不同的模型对不同类别染色体的识别错误率有差异。由于训练过程的随机性,重复实验时这些相对差异不能稳定不变,为避免读者误解为某个模型能一直保持相同的识别偏好,后文以 A、B、C、D、E 指代本次实验中的各模型,并对其进行分析。如表 1 所示,模型 A 对 0 号染色体识别较差,但对 1 号染色体较好;模型 D 对 0 号染色体和 1 号染色体识别错误率都达到最低,但模型 D 对 5 号染色体识别错误率却很高。出现该问题的原因是由于在神经网络训练过程中的随机性造成了不同模型学习到的特征偏好于部分类别。由以上可知,单一模型同时面对不同类别难识别样本时,很难完全平衡。当医师面临难以识别的染色体图像时,会由几名医师商议后共同决策。受此启发,本文推测,在染色体图像识别时,如果能综合利用各模型各自的优势,可以进一步降低模型的识别错误率。以表 1 所示,对 0 号染色体识别,模型 B、C、D 更有优势。对新的样本,如果识别结果为 0 号染色体,B、C、D 这三个模型对该类识别结果的置信度更高。模型融合时,以识别结果的置信度形成综合意见,如果多数模型都识别正确(类似投票策略)或少数模型识别的置信度相对更高,都有可能纠正其中某个模型对个别染色体图像的识别错误,从而可降低识别错误率。当然,也存在染色体图像被所有模型都识别错误,此时模型融合就会失效。
表 1. Example of recognition error rates by different models (%).
不同模型对染色体识别错误率示例(%)
| 模型 | 染色体编号 | ||
| 0 | 1 | 5 | |
| 模型 A | 5.88 | 2.94 | 0 |
| 模型 B | 2.94 | 5.88 | 0 |
| 模型 C | 2.94 | 5.88 | 0 |
| 模型 D | 2.94 | 2.94 | 2.86 |
| 模型 E | 5.88 | 2.94 | 2.86 |
由以上分析,本文提出了以识别置信度为基础的多模型融合方法,形成染色体图像识别模型专家组。组中各模型识别染色体图像后,各自得到一个 24 维的识别置信度向量,该向量的 0~24 维分别描述了图像归属 0~24 类染色体的置信度。各向量对应维度相加,得到模型融合的综合置信度向量,如式(2)所示:
 |
2 |
其中,I 表示送入模型的单条染色体图像, 是模型专家组中单个模型对图像 I 的识别置信度向量
是模型专家组中单个模型对图像 I 的识别置信度向量 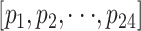 (
(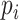 表示图像属于第 i 类的置信度,
表示图像属于第 i 类的置信度,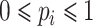 ),M 表示专家组中模型的数量,在本文中实验中 M = 5。
),M 表示专家组中模型的数量,在本文中实验中 M = 5。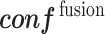 体现了模型专家组对图像 I 识别置信度的综合意见。
体现了模型专家组对图像 I 识别置信度的综合意见。
得到综合的识别置信度后,如式(3)所示,计算模型融合后的识别结果。
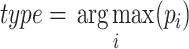 |
3 |
其中,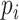 是模型融合后图像 I 被模型识别为类别 i 的置信度,type 是图像 I 最终被识别的类别。
是模型融合后图像 I 被模型识别为类别 i 的置信度,type 是图像 I 最终被识别的类别。
2. 实验数据集和结果
2.1. 数据集
本文选择在国际上可公开使用的人类染色体识别数据集 CCD 和 PCDC 上进行对比实验。实验中采用的图像数据均以编号命名,核型图像中也不存在任何与送检人员直接相关的信息,且数据仅供图像识别研究使用,保证了样本来源者的个人隐私。
2.1.1. CCD 染色体图像数据集
CCD 数据集,由 Lundsteen 等[23]在哥本哈根的瑞斯医院(Rigshospitalet)收集样本并标注类别[24]。该数据集包含 180 个细胞的染色体核型图像,经分割得到 8 106 条染色体的图像[25]。在本文的实验中,将该数据集按 9∶1 的比例分为训练集和验证集两部分,分别包含 7 295 张和 811 张已标注的染色体图像样本;由于样本数量有限,验证集同时也作为测试集。本文将原图像样本置于黑色背景中,统一制作成分辨率为 200 × 200 的图像样本,以满足神经网络需要固定大小输入图像的条件。部分调整后的数据集样本如图 3 所示。
图 3.

Partial samples of CCD
CCD 数据集部分样本
2.1.2. PCDC 染色体分类数据集
PCDC 染色体数据集的显带方式是 Q 显带[21]。如图 4 所示,Q 显带的染色体条带特征不如 G 显带的条带特征明显。该数据集来源于 119 张符合 ISCN 标准的染色体核型图像,经分割标注,得到总计 5 474 张染色体图像样本。与 CCD 数据集类似,按 9∶1 比例将 PCDC 数据集分为训练集和验证集(测试集),分别包含 4 922 张和 552 张图像样本。本文按识别需要重新组织为 24 类,且对方向不正的样本进行了调整。最终得到了类别组织、染色体方向、图像大小与 CCD 保持一致的数据集。
图 4.

The comparison of type 1 chromosome from two datasets
两种数据集 1 号染色体样本对比
2.2. 实验结果对比与分析
2.2.1. 评价指标
为了定量地评价各模型和模型融合方法,本文采用了在测试集上的总体识别错误率和各类别染色体识别错误率来评价模型的识别能力。模型总体识别错误率如式(4)所示:
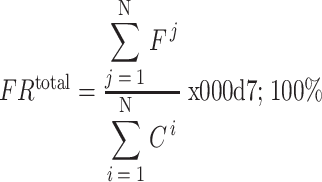 |
4 |
其中,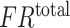 表示总体识别错误率,
表示总体识别错误率,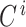 表示测试集第 i 类染色体的数量,
表示测试集第 i 类染色体的数量,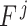 表示第 j 类染色体被错分的数量,N 为染色体类别总数(N = 24,为常数)。该指标以数据集染色体的标注类别为准,按类别 j 比较识别结果与标注类别是否一致。若二者不同,
表示第 j 类染色体被错分的数量,N 为染色体类别总数(N = 24,为常数)。该指标以数据集染色体的标注类别为准,按类别 j 比较识别结果与标注类别是否一致。若二者不同,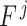 计数加一。然后,累加各类别错分计数
计数加一。然后,累加各类别错分计数 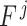 ,以累加值比测试集样本总数得到总体识别错误率。
,以累加值比测试集样本总数得到总体识别错误率。
模型各类识别错误率如式(5)所示:
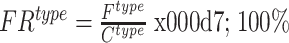 |
5 |
其中,type 表示染色体类别,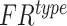 表示第 type 类的错误识别率,
表示第 type 类的错误识别率,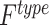 为第 type 类被模型错分的样本数量,
为第 type 类被模型错分的样本数量, 为第 type 类的样本数量。该指标以数据集染色体的标注类别为准,按类别 type 比较识别结果与标注类别是否一致,若二者不同,
为第 type 类的样本数量。该指标以数据集染色体的标注类别为准,按类别 type 比较识别结果与标注类别是否一致,若二者不同,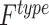 计数加一。然后,各类别分别以错分样本数比该类样本数量得到 type 类的识别错误率。
计数加一。然后,各类别分别以错分样本数比该类样本数量得到 type 类的识别错误率。
2.2.2. CCD 数据集识别效果对比
2012 年以来,图像识别主要是基于深度神经网络模型,期间在自然图像识别数据集(ImageNet)上出现了大量优秀的模型,如亚历克斯网络模型(AlexNet)[9]、谷歌网络模型(GoogLenet)[10]和视觉几何组网络模型(visual geometry group,VGG)[11]等,这些模型代表了通用分类模型的最高水平。Sharma 等[15]设计的模型(Sharma 模型)发表在计算机视觉与模式识别会议(conference on computer vision and pattern recognition,CVPR),该会议是计算机视觉顶级会议,代表了染色体识别的较高水平。Varifocal-Net 是一个二阶段的模型,为了公平地比较,本文在单模型对比时仅采用 Varifocal-Net 中的 G-Net 部分进行比较。
模型训练阶段,本文分别按初始学习率为 0.1、0.01、0.001 和 0.000 1 从头开始训练,每 25 个周期学习率减小 10 倍,共训练 100 个周期。训练完成后,选择各模型表现最好的结果记入表 2。各模型在测试集上的总体识别错误率如表 2 所示。
表 2. Recognition error rate of different models on CCD.
CCD 数据集上各模型识别错误率
| 识别模型 | 总体识别错误率(%) | 网络层数/层 |
| Sharma | 2.47 | 11 |
| G-Net | 1.36 | 28 |
| AlexNet | 3.45 | 8 |
| GoogLenet | 2.59 | 22 |
| VGG16 | 2.47 | 16 |
| ResNet18 | 1.97 | 18 |
| DenseNet | 1.97 | 121 |
| SE-Net | 2.34 | 101 |
| SR-DenseNet | 1.60 | 125 |
| 融合 | 0.99 | — |
如表 2 所示,本文提出的模型在染色体识别数据集 CCD 上识别错误率仅为 1.60%,与 Sharma 模型相比,在其基础上降低了 35.2%,略逊于 G-net;与 DenseNet 相比,总体识别错误率降低了 0.37%;与 SE-Net 相比,总体识别错误率降低了 0.74%。从实验结果来看,随着模型层数的加深,模型的识别错误率呈下降趋势。这说明染色体图像的形态变化十分复杂,需要较深的神经网络模型来逐层完成从低级局部形态特征到高层类别语义特征的提取。与 DenseNet 相比,本文提出的 SR 稠密模块具有更强的特征表现能力。该模块既汇集了不同层次的局部特征,又自适应地标定了各通道特征的重要性,使网络具备了专业遗传学医师的分析能力。在识别染色体图像时,该模块更多地关注最具辨识度的特征,并按权重同时兼顾其他特征。各个模型在 CCD 数据集上的单类别识别错误率比较结果如表 3 所示。
表 3. Recognition error rate about each type of different models on CCD.
CCD 数据集上各模型的单类别识别错误率
| 类别 | 染色体单类别识别错误率(%) | |||||||||
| Sharma | G-Net | AlexNet | GoogLenet | VGG16 | ResNet18 | DenseNet | SE-Net | SR-DenseNet | 融合 | |
| 1 | 2.94 | 2.94 | 5.88 | 2.94 | 5.88 | 2.94 | 5.88 | 5.88 | 5.88 | 2.94 |
| 2 | 2.94 | 2.94 | 5.88 | 5.88 | 5.88 | 5.88 | 5.88 | 5.88 | 2.94 | 2.94 |
| 3 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 4 | 8.57 | 2.86 | 11.43 | 5.71 | 5.71 | 5.71 | 5.71 | 8.57 | 2.86 | 2.86 |
| 5 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 6 | 5.71 | 2.86 | 2.86 | 0.00 | 2.86 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 7 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 8 | 5.71 | 2.86 | 5.71 | 8.57 | 2.86 | 2.86 | 2.86 | 2.86 | 2.86 | 2.86 |
| 9 | 0.00 | 0.00 | 2.86 | 5.71 | 2.86 | 2.86 | 0.00 | 0.00 | 2.86 | 0.00 |
| 10 | 2.78 | 2.78 | 5.56 | 5.56 | 2.78 | 5.56 | 5.56 | 8.33 | 5.56 | 2.78 |
| 11 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 12 | 2.86 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 2.86 | 0.00 | 0.00 | 0.00 |
| 13 | 2.86 | 5.71 | 5.71 | 5.71 | 2.86 | 2.86 | 2.86 | 5.71 | 2.86 | 2.86 |
| 14 | 2.78 | 0.00 | 2.78 | 2.78 | 2.78 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 15 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 16 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 17 | 0.00 | 0.00 | 0.00 | 0.00 | 2.78 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 18 | 5.56 | 5.56 | 8.33 | 2.78 | 5.56 | 5.56 | 2.78 | 5.56 | 2.78 | 2.78 |
| 19 | 2.78 | 0.00 | 2.78 | 2.78 | 0.00 | 2.78 | 2.78 | 5.56 | 5.56 | 0.00 |
| 20 | 0.00 | 0.00 | 2.78 | 2.78 | 2.78 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 21 | 2.78 | 0.00 | 0.00 | 0.00 | 2.78 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 22 | 2.78 | 0.00 | 5.56 | 2.78 | 5.56 | 2.78 | 0.00 | 2.78 | 0.00 | 0.00 |
| X | 7.69 | 3.85 | 15.38 | 7.69 | 3.85 | 7.69 | 3.85 | 3.85 | 3.85 | 3.85 |
| Y | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 22.22 | 0.00 | 0.00 | 0.00 |
2.2.3. PCDC 数据集实验结果
在 PCDC 数据集上,本文采用了与 CCD 数据集上相同的实验方法。在该测试集上,各模型的总体识别错误率如表 4 所示。
表 4. Recognition error rate of different models on PCDC.
PCDC 数据集各模型识别错误率
| 识别模型 | 总体识别错误率(%) | 网络层数/层 |
| Sharma | 8.15 | 11 |
| G-Net | 6.78 | 28 |
| AlexNet | 11.81 | 8 |
| GoogLenet | 7.94 | 22 |
| VGG16 | 7.86 | 16 |
| ResNet-18 | 6.88 | 18 |
| DenseNet | 6.88 | 121 |
| SE-Net | 6.96 | 101 |
| SR-DenseNet | 6.67 | 125 |
| 融合 | 5.98 | — |
如表 4 所示,本文提出的模型取得了最低的识别错误率,仅为 6.67%,比 DenseNet 提高了 0.21%;与 Sharma 模型相比,提升了 1.48%,与 G-Net 相比提升了 0.11%。实验说明本文设计的模块,既汇集了不同抽象层次特征又关注其中的重要特征,在面临复杂的染色体样本图像时,模型特征表现能力更强,更有利于识别任务。与 CCD 数据集相比,各模型的表现都要差一点,这是因为 PCDC 数据集样本图像条带特征不明显所致,与前文对比两数据集样本特点的结论一致。
2.3. 多模型融合方法的实验结果
模型融合后,CCD 数据集上的整体识别错误率仅为 0.99%(如表 2 所示),在本文模型的总体识别错误率基础上进一步降低了 38.12%。如表 3 所示,模型融合后,各类别的识别错误率均能达到最低。通过模型融合,PCDC 数据集上的整体识别错误率仅为 5.98%(如表 4 所示),在本文模型基础上降低了 10.3%。以图 5 为例,模型融合前,本文设计的模型把弯曲的 1 号染色体错误地识别为 3 号染色体;模型融合后,正确识别了弯曲的 1 号染色体,减小了错误率。从实验结果看,本文提出的模型融合方法,充分挖掘了不同模型对识别任务有利的建模能力,构建了识别模型专家组,降低了总体识别错误率和单类别识别错误率。
图 5.

Example of model fusion
模型融合效果示例
3. 讨论
从实验结果来看,本文提出的方法在基线方法(DenseNet 和 SE-Net)上取得了一定的提升,但仍存在需继续研究的问题。存在部分样本,本文的方法重复实验多次都不能正确识别(对比方法也存在相同的问题),如图 6 所示。这些染色体样本主要有两个特点,分别是呈弯曲形态和成像质量欠佳。
图 6.

Misrecognition samples from CCD
CCD 数据集中错误识别样本
染色体是属于非刚性物体,成像后的形态变化多样(如图 3 所示),其中尤其以弯曲形态最难识别。造成识别困难的本质原因有两个:第一,数据集中弯曲的样本的数量不足,造成卷积神经网络不能充分学习这些弯曲样本的特征;第二,卷积神经网络的卷积核以滑窗形式在图像中提取特征,且卷积核有内在的方向性,其从竖直形态染色体学习到的特征不能适用于弯曲形态染色体。如图 6 所示,成像质量欠佳的染色体条带特征难以辨认,网络也难以识别。综上,本文认为未来研究可从以下两方面着手:① 从数据集的角度看,未来的研究应该构建规模更大的公开数据集,并尽可能地包含各种弯曲形态的染色体图像样本,以适应当前深度学习技术的数据需求并促进该领域的发展。② 从识别技术角度看,未来的研究或需要先识别出弯曲形态的染色体,转换为竖直形态后再进行染色体识别;对成像质量欠佳的染色体识别,未来的研究可综合核型图中其他染色体的识别结果,推断出可能的类别。
4. 结论
本文提出了一种 SR-DenseNet 模型,该网络包含 4 个阶段重标定的稠密模块。每个模块前面部分包含若干稠密连接的卷积层,以提取不同层次的特征,后面部分以压缩激活结构重标定提取到特征。该模块既能充分提取不同层次的特征,又重点利用了更具表现力的特征。实验结果表明,该网络结构有更好的特征提取能力,识别错误率更低。以本文设计的 SR-DenseNet 为主,构建了识别模型专家组,进一步降低了识别错误率。实验发现,识别错误的样本大多为形态各异的弯曲样本,未来的工作需要更加关注这些样本。
利益冲突声明:本文全体作者均声明不存在利益冲突。
致谢:诚挚地感谢爱丁堡西部综合医院(Western General Hospital,Edinburgh)的 Jim Piper 和奥尔堡大学(Aalborg University)的 Erik Granum 名誉教授,两位学者在本文获取 CCD 数据集时提供了热情帮助和详细的解答。
Funding Statement
国家重点研发计划项目(2016YFB1102304);2017年广东省产学研合作项目(2017B090901040);2019年广东省人工智能重大科技专项(2018B010108001);2017年广东省重大科技专项(2017B030306017)
Ministry of Science and Technology of the People’s Republic of China; Science and Technology planning Project of Guangdong Province; Science and Technology planning Project of Guangdong Province; Science and Technology planning Project of Guangdong Province
References
- 1.曾秋伊, 朱素优 绒毛染色体核型分析在产前诊断及自然流产中的临床应用. 中国优生与遗传杂志. 2019;27(6):679–681. [Google Scholar]
- 2.闫梅, 李辉, 陈佛兰, 等 广东惠州地区 2117 例孕妇羊水染色体结果分析. 检验医学与临床. 2019;16(15):2171–2174. doi: 10.3969/j.issn.1672-9455.2019.15.018. [DOI] [Google Scholar]
- 3.Britto A P, Ravindran G A review of cytogenetics and its automation. J Med Sci. 2007;7:1–18. [Google Scholar]
- 4.郭宏宇, 鲍旭东, 蒋春涛 基于模糊人工神经网络的染色体识别. 中国生物医学工程学报. 2004;23(2):116–120, 126. doi: 10.3969/j.issn.0258-8021.2004.02.004. [DOI] [Google Scholar]
- 5.蒋欣. 人类染色体图像自动分析技术研究. 武汉: 华中科技大学, 2007.
- 6.Hu R L, Karnowski J, Fadely R, et al. Image segmentation to distinguish between overlapping human chromosomes. arXiv preprint, 2017. arXiv: 1712.07639.
- 7.Munot M V Development of computerized systems for automated chromosome analysis: current status and future prospects. International Journal of Advanced Research in Computer Science. 2018;9(1):782–791. doi: 10.26483/ijarcs.v9i1.5436. [DOI] [Google Scholar]
- 8.Qiu Y, Lu X, Yan S, et al. Applying deep learning technology to automatically identify metaphase chromosomes using scanning microscopic images: an initial investigation//Biophotonics and Immune Responses XI. International Society for Optics and Photonics, San Francisco: SPIE, 2016, 9709: 97090K.
- 9.Krizhevsky A, Sutskever I, Hinton G E. ImageNet classification with deep convolutional neural networks//Advances in Neural Information Processing Systems. Stateline: NIPS, 2012: 1097-1105.
- 10.Szegedy C, Liu W, Jia Y, et al. Going deeper with convolutions//2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston: IEEE, 2015: 1-9.
- 11.Simonyan K, Zisserman A. Very deep convolutional networks for large-scale image recognition//International Conference on Learning Representations (ICLR), 2015. arXiv: 1409.1556
- 12.He Kaiming, Zhang Xiangyu, Ren Shaoqing, et al. Deep residual learning for image recognition//2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas: IEEE, 2016: 770-778.
- 13.Hu Jie, Shen Li, Sun Gang. Squeeze-and-Excitation networks//2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, Salt Lake City: IEEE, 2018: 7132-7141.
- 14.Huang G, Liu Z, van der Maaten L, et al. Densely connected convolutional networks//Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu: IEEE, 2017: 4700-4708.
- 15.Sharma M, Saha O, Sriraman A, et al. Crowdsourcing for chromosome segmentation and deep classification//2017 IEEE Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), Honolulu: IEEE, 2017: 786-793.
- 16.Qin Y, Wen J, Zheng H, et al Varifocal-Net: A chromosome classification approach using deep convolutional networks. IEEE Trans Med Imaging. 2019;38(11):2569–2581. doi: 10.1109/TMI.2019.2905841. [DOI] [PubMed] [Google Scholar]
- 17.Jindal S, Gupta G, Yadav M, et al. Siamese networks for chromosome classification//2017 IEEE International Conference on Computer Vision Workshops (ICCVW), Honolulu: IEEE, 2017: 72-81.
- 18.Gagula-Palalic S, Can M. Human chromosome classification using competitive neural network teams (CNNT) and nearest neighbor//IEEE-EMBS International Conference on Biomedical and Health Informatics (BHI), Valencia: IEEE, 2014: 626-629.
- 19.谭凯. 染色体图像智能分析的综合方法研究及应用. 成都: 电子科技大学, 2020.
- 20.Lin C, Zhao G, Yang Z, et al. CIR-net: automatic classification of human chromosome based on Inception-ResNet architecture. IEEE/ACM Trans Comput Biol Bioinform, 2020, PP. DOI: 10.1109/TCBB.2020.3003445.
- 21.Poletti E, Grisan E, Ruggeri A. Automatic classification of chromosomes in Q-band images//2008 30th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Vancouver: IEEE, 2008: 1911-1914.
- 22.Ioffe S, Szegedy C. Batch normalization: accelerating deep network training by reducing internal covariate shift//International Conference on Machine Learning. Lille: IMLS, 2015: 448-456.
- 23.Lundsteen C, Lind A M, Granum E Visual classification of banded human chromosomes. I. Karyotyping compared with classification of isolated chromosomes. Ann Hum Genet. 1976;40(1):87–97. doi: 10.1111/j.1469-1809.1976.tb00167.x. [DOI] [PubMed] [Google Scholar]
- 24.Sweeney Jr W P. Musavi M T, Guidi J N Classification of chromosomes using a probabilistic neural network. Cytometry: the Journal of the International Society for Analytical Cytology. 1994;16(1):17–24. doi: 10.1002/cyto.990160104. [DOI] [PubMed] [Google Scholar]
- 25.Piper J, Granum E On fully automatic feature measurement for banded chromosome classification. Cytometry. 1989;10(3):242–255. doi: 10.1002/cyto.990100303. [DOI] [PubMed] [Google Scholar]