

Introduction
Pediatric opsoclonus-myoclonus-ataxia syndrome (OMAS) is a rare inflammatory neurologic disorder thought to have a primarily immune-mediated etiology. Also known as dancing eyes dancing feet syndrome, OMAS usually presents in developmentally normal children with behavioral changes and irritability as well as motor features of ataxia, often described as imbalance or incoordination, muscle jerking defined as myoclonus, and random, multi-directional darting eyes movements known as opsoclonus.1 Later neuropsychological sequalae have been described as well.1 The most common theorized causes of OMAS include paraneoplastic and idiopathic.2 Opsoclonus-myoclonus-ataxia syndrome is determined to be a paraneoplastic neurological syndrome in about 50% of patients, with neuroblastoma being the most commonly identified oncologic process.3 Of patients with neuroblastoma, between 1.8% and 3% have features of OMAS.4 This report focuses on several instructive features of a child with stage IIB neuroblastoma associated with severe refractory paraneoplastic OMAS.
Case Report and Methods
We present the case of a patient with a history of neuroblastoma, now in remission, associated with severe paraneoplastic OMAS with associated neurobehavioral abnormalities. The patient presented to his primary care office at 15 months with afebrile upper respiratory illness symptoms and 2 days of acute ataxia with shaking and tremors. He had an examination significant for intermittent eye fluttering, ataxic gait, truncal ataxia, dysmetria, generalized tremor, speech regression, and abdominal mass. Past medical history was significant for developmental delay, dysmorphic features including synophrys, epicanthal folds, ptosis, and microcephaly. Patient was also noted to have stagnant growth starting around 9 months of age. The patient had seen a geneticist at 11 months of age with negative serum amino acids, urine organic acids, mitochondrial panel, and karyotype, and microarray interpreted as normal.
He was referred to the emergency department and admitted to the neurology service. Initial broad differential diagnosis included acute post-infectious cerebellar ataxia, toxic ingestion, seizure, OMAS with or without associated malignancy, or an underlying metabolic/genetic disorder such as a channelopathy, mitochondrial disorder, neurotransmitter disorder, or Glucose Transporter 1 deficiency. An electroencephalogram (EEG) was performed which showed no epileptiform activity. Workup including cerebrospinal fluid (CSF) cell counts, glucose, neurotransmitters, and infectious studies, toxicology studies, repeat newborn screen, lactate, pyruvate, Fragile X PCR, and genetic mitochondrial testing were unremarkable. Urine vanillylmandelic acid (VMA) was elevated at 41.6 mg/g creatinine (reference range 7.9–23.0), and urine homovanillic acid (HVA) was elevated at 57.3 mg/g creatinine (reference range 12.2–31.8). Abdominal magnetic resonance imaging revealed a 4.2 × 3.8 × 5.7 cm right infrarenal mass arising from the sympathetic chain, as shown in Figure 1. Findings were consistent with a diagnosis of neuroblastoma with associated opsoclonus-myoclonus syndrome.
Figure 1.
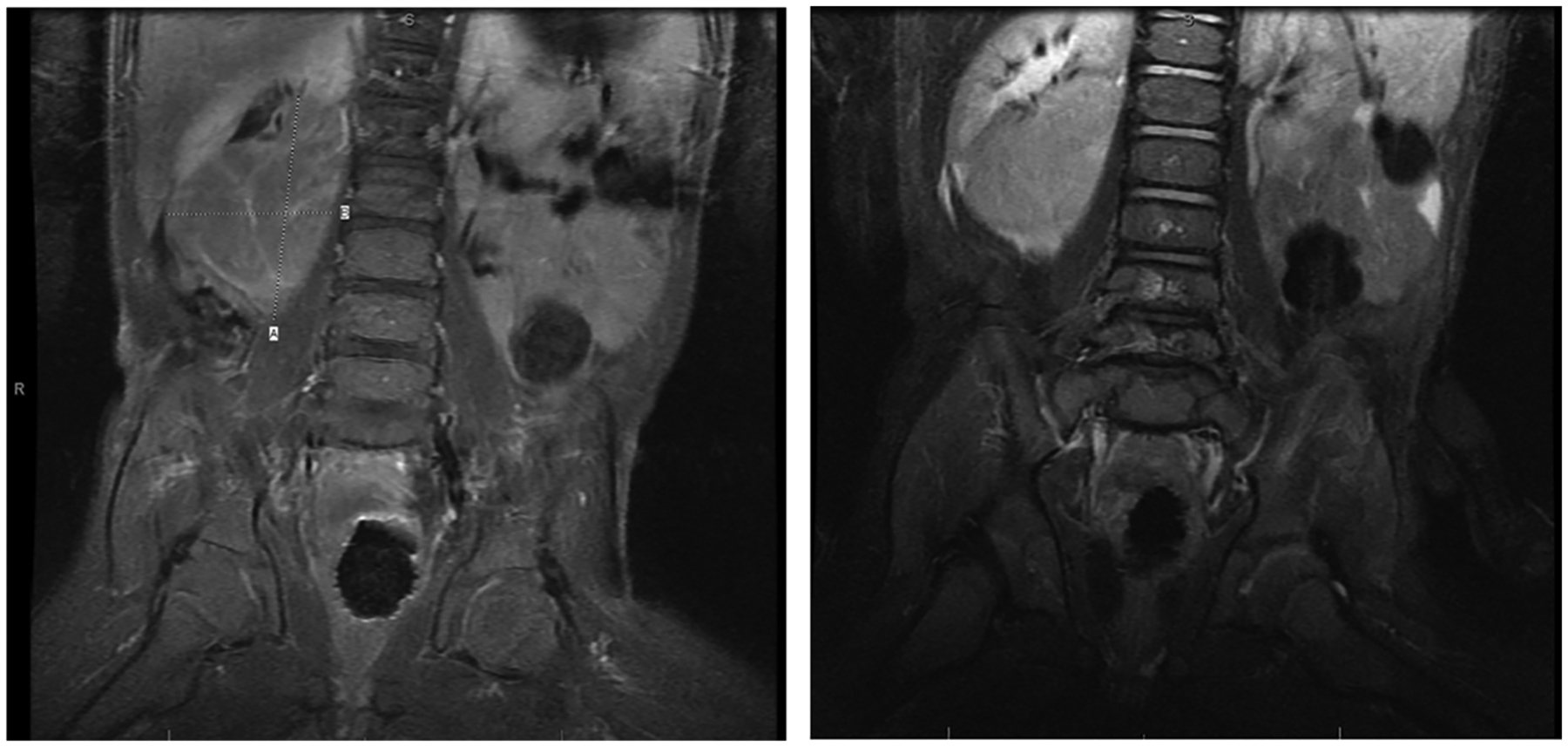
Magnetic resonance imaging abdomen.Lobulated, enhancing right infrarenal mass measuring 4.2 cm in transverse dimension, 3.8 cm in anterior-posterior dimension, and 5.7 cm in craniocaudal dimension displacing the right kidney superiorly and laterally.
Bone marrow aspiration and biopsy as well as CSF were negative for neoplastic process. He underwent complete surgical resection of stage IIB abdominal neuroblastoma. Pathology revealed poorly differentiated neuroblastoma with intermediate mitosis-karyorrhexis index (MKI) and clear margins, as shown in Figure 2. NMYC was negative. Metaiodobenzylguanidine (MIBG) scan performed post-resection did not show evidence of residual or metastatic disease. He did not receive any chemotherapy specifically for the neuroblastoma.
Figure 2.
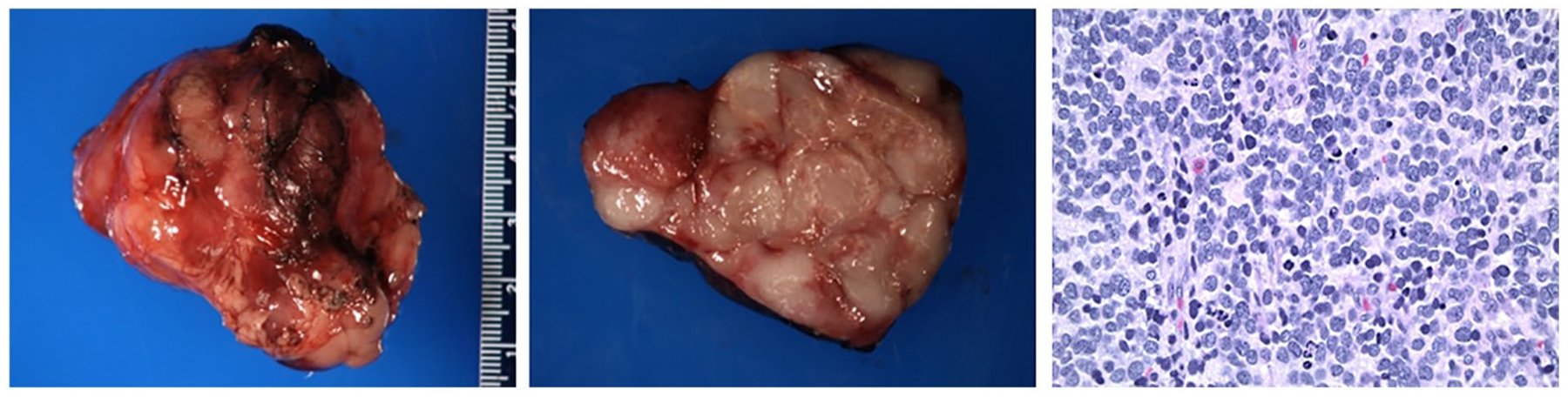
Gross and microscopic pathology. Gross pathology showed an encapsulated mass weighing 40.0 g and measures 5.5 × 4.5 × 3.8 cm. Section of the mass showed a lobulated, tan-white rubbery architecture occupying the vast majority of the specimen with a rim of surrounding capsule. Small foci of yellow discoloration with possible microcalcifications and necrosis were identified. Some nodules showed red discoloration with thin fibrous septae. On microscopic pathology, neuroblastic cells were seen in a background of apoptosis and karyorrhectic debris and scattered lymphoid aggregates. Some degree of nuclear pleomorphism with multinucleated forms and larger cells with hyperchromatic nuclei as well as abundant mitoses were present. The mitotic karyorrhectic index was in the intermediate range.
He started treatment at 16 months of age for his OMAS as per COG protocol ANBL00P3 with cyclophosphamide, prednisone, and IVIG. He appeared to have slight improvement initially with his pretreatment opsoclonus/myoclonus Score decreasing from a total of 7 to 4 three months later.5 However, he would periodically regress with increase in OMS to as high as 9, but then would decrease more toward his baseline of 5. Most of the abnormality of the OMS was attributed to his poor stance and gait. He remained on prednisone, and his symptoms of ataxia and myoclonus slowly improved over time after which his prednisone began to be slowly tapered. At 27 months, the patient continued to be developmentally delayed with significant speech and gross motor delays.
At 28 months of age, his OMAS symptoms worsened in the setting of a viral illness with an OMS of 8. He was started on monthly high-dose dexamethasone (4-day courses of 20 mg/m2/day) for 4 months without any significant improvement. Due to the persistent and refractory nature of his OMAS symptoms with OMS consistently 6–7, at 32 months of age, he received rituximab (375 mg/m2 weekly × 4) with significant improvement in his motor symptoms. Over the next few years, patient was able to walk and run, and had a significant decrease in myoclonus and resolution of opsoclonus.
At age 6, the patient had several viral illnesses resulting in another relapse of OMAS with ataxia and myoclonus. He was treated acutely with dexamethasone pulses and IVIG for 6 months due to side effects of hypotension and premature ventricular contractions with the infusions and subsequent neutropenia with rituximab and had satisfactory clinical response. Rituximab was again added to treatment regimen at 750 mg/m2 for a total of 2 doses due to persistence of OMAS. He achieved remission. At last follow-up at 10 years of age, balance and gait were much improved, with only minor intermittent hand tremors. He continues to receive services for developmental delay, intellectual disability, and behavioral abnormalities. Repeat surveillance for neuroblastoma recurrence has been negative.
The patient was re-evaluated by a geneticist at 9 years of age. Whole exome sequencing was performed with 3 variants identified. Two variants were autosomal recessive, and 1 variant was autosomal dominant, all reported as variants of unknown significance as of the writing of this article.
Discussion
This case highlights a case of neuroblastoma associated with severe refractory paraneoplastic OMAS first evaluated in the primary care setting.
Opsoclonus-myoclonus-ataxia syndrome most often presents in early childhood, with the median age at disease onset being 18.0 months with a range of 3 months to 8.9 years.6 Opsoclonus-myoclonus-ataxia syndrome is about 10% more common in females than males, and there appears to be no statistical difference in prevalence among racial and ethnic groups.7 In 1 retrospective review, children were noted to be neurodevel-opmentally normal prior to diagnosis.6 A paraneoplastic etiology can be detected in about half of cases, and tumors are most commonly neuroblastic in origin, including neuroblastoma, ganglioneuroblastoma, and ganglioneuroma.7 Of these, neuroblastoma, the most common non-central nervous system solid tumor of childhood, composes the majority of cases.7 Between 1.8% and 3% of neuroblastomas are associated with paraneoplastic OMAS.4
On physical examination, patients exhibit the cardinal motor features of ataxia, myoclonus, and/or opsoclonus. It is important for the general pediatrician to keep OMAS on the differential when these symptoms are noted. Opsoclonus is defined as an involuntary saccadic disturbance with irregular oscillations in all directions, persisting without intersaccadic interval. Myoclonus describes irregular muscle jerks occurring primarily in the trunk. Ataxia may occur in the trunk or limbs, and be described by caregivers as imbalance, incoordination, or refusal to ambulate.3 These features may or may not appear in conjunction with or be preceded by nonmotor features such as behavioral disturbance, irritability, insomnia, or lethargy.7 The unique and varied presentation of OMAS underscores the importance of evaluation by the general pediatrician with a careful physical examination, particularly a thorough abdominal examination which is often difficult in an uncooperative child.
The differential diagnosis for OMAS includes acute postinfectious cerebellar ataxia, as ataxia may precede other features.6 On history, about half of patients may have had a prodromal illness, no significant difference in rate of preceding illness between patients with or without neuroblastoma.6 Purported etiologies of OMAS include oncologic processes, para- or post-infectious states, exposure to toxic metabolites, or may be classified as idiopathic.6 No consistent infectious pathogen has been identified in the pediatric population.1
As noted above, a paraneoplastic etiology to OMAS is detected in about half of cases. Neuroblastoma associated with OMAS can be more difficult to detect, and about 20% of these cases have an atypical presentation. Tumors are typically lower grade and metabolically inactive, with a lower frequency of MIBG scintigraphy uptake and urinary catecholamine secretion.6 Neuroblastoma may also regress spontaneously.1 These factors may lead to delay in diagnosis. Interestingly, the clinical phenotypes of paraneoplastic compared with other etiologies of OMAS were indistinguishable per a large population-based study.1
Sequalae including attention-deficit disorder, learning and memory disorders, motor deficits, and cognitive impairment may emerge as later term effects, and often, these sequalae are managed in the primary care setting.7 Those diagnosed at a young age and with more severe initial symptoms are at greater risk of developing these neuropsychological effects.6 Furthermore, relapses of motor symptoms may be triggered by intercurrent illnesses, weaning of steroid treatment, or general anesthesia.6
Treatment for OMAS varies, but is comprised of immunomodulatory treatments such as intravenous immune globulin (IVIg), steroids (adrenocorticotropic hormone [ACTH], prednisone, or dexamethasone) along with rituximab or cyclophosphamide.2 With appropriate treatment, almost all cases of OMAS had improvement of neurological symptoms. Previously, many patients with OMAS had persistence of neuropsychological disturbances, with neurodevelopmental recovery requiring long-term supportive intervention in several cases.2 The risk of a chronic relapsing rather than monophasic disease course was associated with motor and speech abnormalities, learning disability, and behavioral disturbance.6 However, in recent years, more aggressive immunosuppression has led to improved functional outcomes.8 The importance of IVIG was recently demonstrated in a Children’s Oncology Group randomized therapeutic trial showing patients who received IVIG in addition to prednisone and risk-adapted chemotherapy had a higher rate of recovery from their OMAS symptoms.5 Weaning from initial treatment with corticosteroids or ACTH often led to relapse of OMAS symptoms.8 Recently, early introduction of rituximab/cyclophosphamide has been shown to not only decrease symptom burden of OMAS but also decrease the rate of relapse, leading to improved developmental outcomes.8
Conclusion
Opsoclonus-myoclonus-ataxia syndrome highlights the complex interplay between oncology, the central nervous system, and our immune response. Our case demonstrates a presentation of severe relapsing paraneoplastic OMAS and the importance of recognition by the general pediatrician.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Pranzatelli MR, Tate ED, McGee NR. Multifactorial analysis of opsoclonus-myoclonus syndrome etiology (“Tumor” vs. Pediatr Blood Cancer. 2018;65(8):e29721. doi: 10.1002/pbc.27097. [DOI] [PubMed] [Google Scholar]
- 2.Patel A, Fischer C, Lin YC, et al. Treatment and revacci-nation of children with paraneoplastic opsoclonus-myoclonus-ataxia syndrome and neuroblastoma: the Memorial Sloan Kettering experience. Pediatr Blood Cancer. 2020;67(8):e28319. doi: 10.1002/pbc.28319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaes F, Dharmalingam B. Childhood opsoclonus-myoclonus syndrome: diagnosis and treatment. Expert Rev Neurother. 2016;16(6):641–648. doi: 10.1080/14737175.2016.1176914. [DOI] [PubMed] [Google Scholar]
- 4.Gorman MP. Update on diagnosis, treatment, and prognosis in opsoclonus-myoclonus-ataxia syndrome. Curr Opin Pediatr. 2010;22(6):745–750. doi: 10.1097/MOP.0b013e32833fde3f. [DOI] [PubMed] [Google Scholar]
- 5.de Alarcon PA, Matthay KK, London WB, et al. Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Health. 2018;2(1):25–34. doi: 10.1016/S2352-4642(17)30130-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunklaus A, Pohl K, Zuberi SM, de Sousa C. Outcome and prognostic features in opsoclonus-myoclonus syndrome from infancy to adult life. Pediatrics. 2011;128(2):e388–e394. doi: 10.1542/peds.2010-3114. [DOI] [PubMed] [Google Scholar]
- 7.Pranzatelli MR, Tate ED, McGee NR. Demographic, clinical, and immunologic features of 389 children with opsoclonus-myoclonus syndrome: a cross-sectional study. Front Neurol. 2017;8:468. doi: 10.3389/fneur.2017.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell WG, Wooten AA, O’Neil SH, Rodriguez JG, Cruz RE, Wittern R. Effect of increased immunosuppression on developmental outcome of opsoclonus myoclonus syndrome (OMS). J Child Neurol. 2015;30(8):976–982. doi: 10.1177/0883073814549581. [DOI] [PubMed] [Google Scholar]