

Abstract
Low‐efficacy mu‐opioid receptor (MOR) agonists represent promising therapeutics, but existing compounds (e.g., buprenorphine, nalbuphine) span a limited range of low MOR efficacies and have poor MOR selectivity. Accordingly, new and selective low‐efficacy MOR agonists are of interest. A novel set of chiral C9‐substituted phenylmorphans has been reported to display improved MOR selectivity and a range of high‐to‐low MOR efficacies under other conditions; however, a full opioid receptor binding profile for these drugs has not been described. Additionally, studies in mice will be useful for preclinical characterization of these novel compounds, but the pharmacology of these drugs in mice has also not been examined. Accordingly, the present study characterized the binding selectivity and in vitro efficacy of these compounds using assays of opioid receptor binding and ligand‐stimulated [35S]GTPɣS binding. Additionally, locomotor effects were evaluated as a first step for in vivo behavioral assessment in mice. The high‐efficacy MOR agonist and clinically effective antidepressant tianeptine was included as a comparator. In binding studies, all phenylmorphans showed improved MOR selectivity relative to existing lower‐efficacy MOR agonists. In the ligand‐stimulated [35S]GTPɣS binding assay, seven phenylmorphans had graded levels of sub‐buprenorphine MOR efficacy. In locomotor studies, the compounds again showed graded efficacy with a rapid onset and ≥1 h duration of effects, evidence for MOR mediation, and minor sex differences. Tianeptine functioned as a high‐efficacy MOR agonist. Overall, these in vitro and in vivo studies support the characterization of these compounds as MOR‐selective ligands with graded MOR efficacy and utility for further behavioral studies in mice.
Keywords: efficacy, mouse, mu opioid receptor, opioid, locomotor activity, receptor binding
Abbreviations
- CHO
Chinese hamster ovary
- DOR
delta‐opioid receptor
- E max
maximum effect
- EC50
in vitro concentration producing 50% of the E max of the drug
- ED50
in vivo dose producing 50% of the E max of the drug
- [35S]GTPɣS
guanosine 5’‐O‐[gamma‐thio]triphosphate
- DC‐1‐76.1
3‐((1R,5S,9S)‐2‐phenethyl‐9‐vinyl‐2‐azabicyclo[3.3.1]nonan‐5‐yl)phenol
- DC‐1‐76.2
3‐((1R,5S,9R)‐2‐phenethyl‐9‐vinyl‐2‐azabicyclo[3.3.1]nonan‐5‐yl)phenol
- DC‐1‐90.2
3‐((1R,5S,9R)‐2‐phenethyl‐9‐((Z)‐prop‐1‐en‐1‐yl)‐2‐azabicyclo[3.3.1]nonan‐5‐yl)phenol
- DC‐1‐128.1
3‐((1R,5S,9S)‐2‐phenethyl‐9‐((Z)‐prop‐1‐en‐1‐yl)‐2‐azabicyclo [3.3.1]nonan‐5‐yl)phenol
- EG‐1‐203
3‐((1S,5R,9R)‐2‐phenethyl‐9‐propyl‐2‐azabicyclo[3.3.1]nonan‐5‐yl)phenol
- EG‐1‐230
3‐((1S,5R,9R)‐9‐((E)‐3‐hydroxyprop‐1‐en‐1‐yl)‐2‐phenethyl‐2‐azabicyclo[3.3.1]nonan‐5‐yl)phenol
- EWB‐3‐14
(1R,5S,9R)‐(+)‐5‐(3‐hydroxyphenyl)‐9‐methyl‐2‐phenethyl‐2‐azabicyclo[3.3.1]nonane
- JL02‐39
3‐((1S,5R,9R)‐9‐(2‐hydroxyethyl)‐2‐phenethyl‐2‐azabicyclo[3.3.1]nonan‐5‐yl)phenol
- KOR
kappa opioid receptor
- MOR
mu‐opioid receptor
1. INTRODUCTION
Mu‐opioid receptor (MOR) agonists are invaluable as analgesics for the treatment of many different types of pain, but their use is limited by side effects that include lethal respiratory depression, constipation, sedation, and abuse potential. 1 Drugs that bind to the MOR vary in their pharmacodynamic efficacies to activate receptor‐coupled signaling pathways and downstream physiological and behavioral effects. 2 , 3 , 4 The most problematic opioid analgesics (e.g., fentanyl, morphine, oxycodone) have high MOR efficacy sufficient to produce a full spectrum of MOR‐mediated therapeutic effects as well as the most dangerous side effects. By contrast, the lower‐efficacy MOR agonist buprenorphine retains analgesic activity and produces a subset of side effects, but it lacks sufficient efficacy to prouce lethal respiratory depression. 5 Thus, buprenorphine illustrates the general potential to retain analgesic effectiveness and improve safety by developing MOR agonists with relatively low MOR efficacy.
Buprenorphine is one of several lower‐efficacy MOR agonists currently approved by the Food and Drug Administration for clinical use in the United States. Other compounds in this category include nalbuphine, pentazocine, and butorphanol. 1 However, a constellation of factors present barriers to their use. Perhaps most importantly, these compounds all have relatively poor MOR selectivity and also bind to kappa and delta‐opioid receptors (KOR, DOR), and the KOR affinities, in particular, are similar to or only slightly lower than their MOR affinities. 6 , 7 , 8 , 9 Buprenorphine functions largely as an antagonist at these other opioid receptors, but nalbuphine, pentazocine, and butorphanol produce KOR activation that may be associated with undesirable side effects. 10 , 11 , 12 , 13 , 14 Moreover, although these compounds have lower MOR efficacy than their high‐efficacy counterparts, they still have sufficient efficacy to produce a subset of side effects and they represent only a limited range of the full MOR efficacy continuum. As a result, they provide a limited opportunity to explore the degree to which control of MOR efficacy might permit improved control of analgesic effectiveness and safety.
In a series of recent publications, the synthesis and initial pharmacological evaluation was described for a series of chiral C9‐substituted phenylmorphans that include new MOR ligands with graded levels of low, sub‐buprenorphine MOR efficacy and improved MOR selectivity. 15 , 16 , 17 , 18 , 19 However, a full opioid receptor binding profile for these drugs has not been described. Additionally, studies in mice will be useful for preclinical characterization of in vivo effects produced by these novel compounds, but the pharmacology of these drugs in mice has also not been examined. Accordingly, the goal of the present study was to characterize the subset of these compounds shown in Figure 1 in a panel of assays that has been used by us previously to examine other MOR agonists that vary in efficacy. 4 , 20 , 21 Receptor binding was evaluated with competition binding assays in Chinese hamster ovary (CHO) cells expressing MOR, KOR, or DOR. Receptor signaling was evaluated in the same cells using an assay of ligand‐stimulated [35S]GTPɣS binding as the first step in G‐protein‐mediated intracellular signaling. As a first step in characterizing the behavioral pharmacology of these phenylmorphans in male and female mice, the compounds were evaluated using an assay of locomotor activation. Effects of the chiral C9‐substituted phenylmorphans were compared to effects of tianeptine, an antidepressant drug approved for use in Europe and recently discovered to be a high‐efficacy MOR agonist. 22 , 23 , 24 Our results confirm and extend previous work with these compounds and identify a set of selective MOR agonists with graded sub‐buprenorphine efficacies that could be useful both as candidate therapeutics and as tools to examine the role of MOR efficacy as a determinant of therapeutic and undesirable MOR agonist effects.
FIGURE 1.
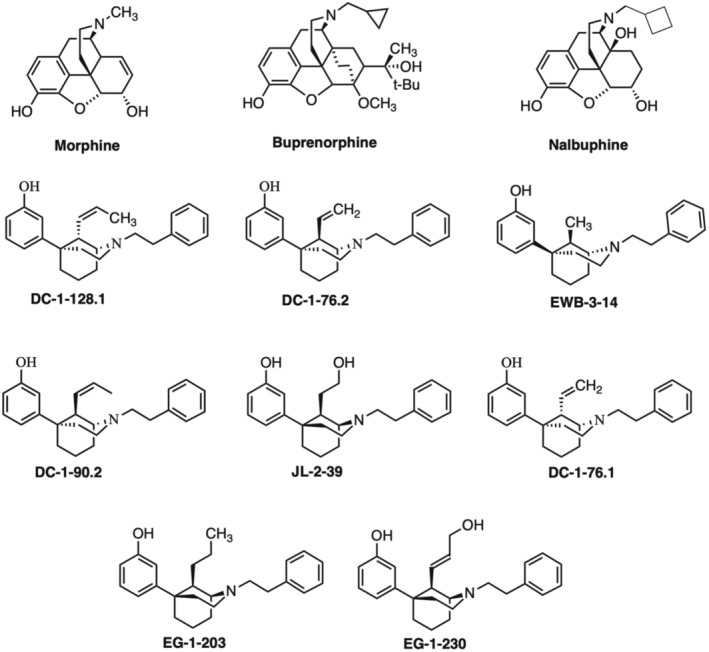
Structures of three reference compounds (morphine, buprenorphine, nalbuphine) and all of the chiral C9‐substituted phenylmorphans investigated in this study.
2. MATERIALS AND METHODS
2.1. In vitro studies of receptor binding and function
2.1.1. Cell culture and membrane preparation
All in vitro assays were performed using CHO cell lines expressing mouse mu‐opioid receptor (mMOR‐CHO), mouse kappa opioid receptor (mKOR‐CHO), or human delta‐opioid receptor (hDOR‐CHO). Cell culture and membrane homogenate preparations were performed as previously described. 21 , 25 All assays were performed in duplicate and repeated at least three times.
2.1.2. Radioligand binding assay
Competition binding assays were performed using mMOR‐, mKOR‐, or hDOR‐CHO membrane homogenates containing 20 μg membrane protein as previously described. 15 , 21 , 25 Homogenates were incubated with approximate KD concentrations of 1.4 nM [3H]naloxone (for mMOR‐CHO), 0.25 nM [3H]diprenorphine (for mKOR‐CHO), or 1 nM [3H]diprenorphine (for hDOR‐CHO) in the presence 0.2nM EGTA (pH 7.4) for 1.5 h at 30°C. Bound radioligand was separated from free radioligand by filtration then radioactivity was determined by liquid scintillation counting. Specific binding was determined as the difference in binding in the absence and presence of 5 μM naltrexone, U50488, or SNC80 for MOR, KOR, or DOR, respectively.
2.1.3. [ 35S]GTPγS binding assay
Membrane homogenates from mMOR‐, mKOR‐, or hDOR‐CHO cells containing 9–15 μg protein, were incubated in Assay Buffer with 100 mM NaCl, 20 μM GDP, and 0.1 nM [35S]GTPγS with and without varying concentrations of test compounds for 1.5 h at 30°C as previously described. 21 , 25 , 26 Additionally, 3 μM DAMGO, 3 μM U50488, or 5 μM SNC80 was included as a reference point for a maximally effective concentration of a full agonist for MOR, KOR, or DOR, respectively. Bound [35S]GTPγS was separated by filtration as described above, and radioactivity was determined by liquid scintillation counting.
2.1.4. Data analysis
Competition binding data were normalized to the binding in the absence of competitor as follows: % Bound = specific dpm bound/specific dpm bound in the absence of competing ligand × 100%. Competition binding was fit by 4‐parameter nonlinear regression analysis with the top and bottom constrained to 100 and 0, respectively, to determine log IC50 values and Hill coefficients. IC50 values were converted to K i values using the Cheng–Prusoff equation. Net stimulation of [35S]GTPγS binding was defined as ligand‐stimulated specific dpm—basal specific dpm. To determine ligand efficacy and potency, [35S]GTPγS binding data were normalized to the maximal net stimulation produced by a full agonist at each receptor type: (net stimulation by ligand/net stimulation by 3 μM DAMGO, 3 μM U50488, or 5 μM SNC80) × 100%. E max, log EC50, and Hill coefficient values were determined by 4‐parameter nonlinear regression analysis with the minimum constrained to 0. Linear regression was used to compare log K i and log EC50 values as in vitro binding and functional measures of drug potency.
2.2. In vivo studies of locomotor activity
2.2.1. Subjects
Subjects were male and female ICR mice (Envigo) that were 6–8 weeks old upon arrival to the laboratory. Males weighed 27–50 g and females weighed 23–38 g throughout the study. Mice were single‐housed in cages with corncob bedding (Envigo), a “nestlet” composed of pressed cotton (Ancare), a cardboard tube for enrichment, and ad libitum access to food (Teklad LM‐485 Mouse/Rat Diet; Envigo). Cages were mounted in racks in a temperature‐controlled room with a 12‐h light/dark cycle (lights on from 6:00 a.m. to 6:00 p.m.) in a facility approved by the American Association for Accreditation of Laboratory Animal Care. All experiments were performed during the light phase of the daily light/dark cycle beginning 1 week after arrival at the laboratory. Ethical animal‐use protocols were approved by the Institutional Animal Care and Use Committee and complied with the National Research Council Guide for the Care and Use of Laboratory Animals.
2.2.2. Apparatus
Horizontal locomotor activity was assessed as described previously 20 during 60‐min sessions in rectangular test boxes (16.8 × 12.7 cm2 floor area × 12.7 cm high) housed in sound‐attenuating chambers (Med Associates) and located in a procedure room separate from the housing room. Each box had black plexiglass walls, a clear plexiglass ceiling equipped with a house light, bar floors, and six photobeams arranged at 3‐cm intervals across the long wall and 1 cm above the floor. Beam breaks were monitored by a microprocessor operating Med Associates software.
2.2.3. Procedure
Procedures were identical to those described previously. 20 Thus, for all drugs except EG‐1‐230, a different group of 12 mice (six females, six males) was used to test each drug. One mouse assigned to the EG‐1‐230 group died before testing began, so this group included 11 mice (six females, five males). Within each group, test sessions were conducted twice a week with at least 48 h between sessions. All mice received a vehicle control and all doses of the designated test drug, and dose order was randomized across mice using a Latin‐square design. The experimenter was not blinded to treatment because data collection was automated by the Med Associates software. There were no exclusion criteria, and all data from all mice were included in final analysis. On test days, mice were brought to the procedure room at least 1 h before session onset. After subcutaneous (SC) test‐drug administration, mice were returned to their home cages for the 5‐min pretreatment interval and then placed into the locomotor activity boxes for a 60‐min test session. Doses for each drug were varied in 0.5 log‐unit increments across a >10‐fold dose range with the intent of progressing from low doses that produced little or no effect to high doses that produced maximal increases in locomotor activation for that drug. The final dose ranges for each drug were as follows: tianeptine (10–100 mg/kg), DC‐1‐128.1 (0.1–3.2 mg/kg), DC‐1‐76.2 (0.1–3.2 mg/kg), EWB‐3‐14 (0.1–32 mg/kg), JL‐2‐39 (1.0–32 mg/kg), DC‐1‐76.1 (0.32–32 mg/kg), EG‐1‐203 (3.2–32 mg/kg), and EG‐1‐230 (3.2–32 mg/kg). For all drugs, antagonism studies were conducted after completion of drug‐alone studies in the same mice using one of two experimental designs. First, to determine effectiveness of the antagonist naltrexone to block effects of higher‐efficacy test compounds, 1.0 mg/kg naltrexone was administered SC 10 min before SC administration of an active dose of the test drug, and test sessions began 5 min after the test drug. Second, to determine effectiveness of lower‐efficacy test compounds to block locomotor‐activating effects of morphine, the test drug was administered SC 10 min before 32 mg/kg SC morphine, and test sessions began 5 min after morphine administration.
2.2.4. Data analysis
The primary dependent variable was the total number of beam breaks, excluding consecutive interruptions of the same beam, during each 60‐min session. To construct and analyze dose‐effect curves for each drug, data were normalized in a two‐step process to account for slight differences in vehicle control data across groups and permit direct comparison to methadone as a high‐efficacy MOR agonist we have examined previously. 20 First, locomotor data in each mouse at each drug dose were expressed as a “Difference Score” relative to vehicle control data in that group using the equation Difference Score = Test − Group Vehicle, where Test equals the number of locomotor counts in a given mouse after a given drug dose, and Group Vehicle equals the mean number of locomotor counts after vehicle treatment in that group. Second, the Difference Score in each mouse at each dose was then expressed as a percentage of the mean maximum Difference Score produced by the reference agonist methadone using the equation % Methadone E max = (Difference Score/Methadone E max) × 100.
The resulting dose‐effect data were then evaluated in a sequence of steps as we have described previously. 20 , 27 First, because sex was not the primary variable of interest, pooled data from both females and males were analyzed by repeated‐measures one‐way ANOVA with dose as the single variable. A significant ANOVA was followed by a Holm‐Sidak post hoc test, and for this and all other parametric statistics, the criterion for significance was p < .05. Second, pooled dose‐effect data were also evaluated to determine E max and ED50 values for each drug. The E max was defined as the mean maximum effect (95% confidence limits [CL]) produced by any drug dose. The ED50 was defined as the dose producing 50% of the E max value for that drug, and ED50 values (95% CL) were determined by linear regression of the linear ascending portion of the dose‐effect curve. E max and ED50 values were considered to be significantly different across drugs if 95% CL did not overlap. Lastly, to provide preliminary information regarding potential sex differences in drug effects, data for each drug were segregated by sex and compared by two‐way ANOVA with sex as a between‐subjects factor and drug dose as a within‐subjects factor. A significant sex × dose interaction was followed by a Holm‐Sidak post hoc test. Additionally, the two‐way ANOVA results were submitted to post hoc power analyses to calculate the Cohen's f effect size, achieved power (1 − β), and the total number of animals predicted as necessary to achieve power ≥0.8.
For antagonism experiments, raw data were analyzed as appropriate by t‐test or by one‐way ANOVA followed by Dunnett's post hoc test. Linear regression was used to compare in vitro and in vivo measures of drug potency (in vitro log EC50 vs. in vivo log ED50 values) and drug efficacy (in vitro and in vivo E max values). Power analysis was conducted using the free statistical analysis program G*Power, 28 and all other analyses were conducted using GraphPad Prism 9.5 (La Jolla, CA).
2.3. Drugs
(±) Methadone HCl and naltrexone HCl were provided by the National Institute on Drug Abuse Drug Supply Program. Tianeptine sodium salt was purchased from Cayman Chemical. The chiral C9‐substituted phenylmorphans were as follows: DC‐1‐128.1, DC‐1‐76.2, DC‐1‐90.2, EWB‐3‐14, JL‐2‐39, DC‐1‐76.1, EG‐1‐203 HBr, and EG‐1‐230 HBr. These compounds were provided by the Drug Design and Synthesis Section, Molecular Targets and Medications Discovery Branch, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism (Bethesda, MD; see Rice et al. 19 for patent information). For in vivo studies of locomotor activity, methadone, naltrexone, tianeptine, and EG‐1‐230 were dissolved in sterile saline. All other compounds were dissolved in a vehicle of 5% ethanol, 5% emulphor, and 90% saline. In vivo doses were calculated using the salt or free‐base form of each drug described above and were administered SC in a volume of 10 mL/kg. Note that DC‐1‐90.2 was included in in vitro studies, but it was not tested in vivo due to its limited solubility.
2.4. Nomenclature of targets and ligands
Key protein targets and ligands in this article ar hyperlinked to corresponding entries in http://guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 29 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 30
3. RESULTS
3.1. In vitro studies of receptor binding and function
In vitro drug effects in competition binding assays and in functional assays of ligand‐stimulated [35S]GTPɣS binding are shown in Figure 2; Tables 1 and 2. In competition binding studies, tianeptine had the lowest MOR affinity of all compounds tested with a K i value of 78 nM, though it had high selectivity for MOR versus KOR, and more than 30‐fold selectivity for MOR versus DOR. The phenylmorphans all showed higher affinity for MOR, with most having subnanomolar affinity similar to buprenorphine, except for DC‐1‐76.1, DC‐1‐90.2, and EG‐1‐230. These three compounds had MOR K i values ranging from ~1.8–4.2 nM, which is similar to or slightly greater than that of morphine. All the phenylmorphans had >10‐fold MOR‐versus‐KOR selectivity and >87‐fold MOR‐versus‐DOR selectivity.
FIGURE 2.
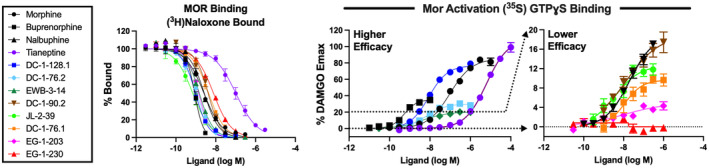
Concentration‐effect curves for competition binding (left panel) and receptor‐mediated G‐protein activation (right panels) in membranes from Chinese hamster ovary (CHO) cells expressing the mouse mu opioid receptor (MOR). In the left panel, [3H]naloxone was used to determine competition binding affinity (K i , nM), and K i values are shown in Table 1 for MOR as well as for kappa opioid receptor (KOR) and delta opioid receptor (DOR). In the right panels, ligand‐modulated [35S]GTPγS binding was used to determine MOR‐mediated G‐protein activation. Data were normalized as a percentage of the stimulation produced by a maximally effective concentration of DAMGO (3 μM; MOR), U50488 (5 μM; KOR) or SNC‐80 (5 μM; DOR). Resulting E max and EC50 values are shown in Table 2 for MOR as well as for KOR and DOR. Note that the middle panel has a Y‐axis range of 0%–120% DAMGO Max and shows effects of higher‐efficacy ligands (tianeptine, morphine, buprenorphine, DC‐1‐128.1, DC‐1‐76.2, EWB‐3‐13), whereas the right panel has a narrower Y‐axis range of 0%–20% DAMGO Max and shows effects of the lower‐efficacy ligands (nalbuphine, DC‐1‐90.2, JL‐2‐39, DC‐1‐76.1, EG‐1‐203, EG‐1‐230). Higher‐efficacy ligands are defined here as having E max >20% DAMGO. All data represent mean values ± SEM (n = 3–9). Specific radioligand binding in MOR‐, KOR‐ and DOR‐CHO cells in the absence of competitor was 1.83 ± 0.12 pmol/mg ([3H]naloxone), 0.602 ± 0.038 pmol/mg ([3H]diprenorphine) and 3.48 ± 0.20 pmol/mg ([3H]diprenorphine), respectively. Basal [35S]GTPγS binding in MOR‐, KOR‐ and DOR‐CHO cells was 44.89 ± 2.16, 51.38 ± 7.23, and 164.1 ± 12.8 fmol/mg, respectively. Full agonist stimulation (% over basal) in MOR‐, KOR‐ and DOR‐CHO cells was 473.5% ± 7.2% (DAMGO), 310.1% ± 16.4% (U50488) and 209.6 ± 20.2% (SNC‐80), respectively.
TABLE 1.
Ligand K i and selectivity values from radioligand competition binding.
| Ligand | MOR K i (nM) | KOR K i (nM) | MOR/KOR | DOR K i (nM) | MOR/DOR |
|---|---|---|---|---|---|
| Morphine a | 1.196 ± 0.163 | 225.5 ± 8.5 | 188.6 | 139.2 ± 4.6 | 116.4 |
| Buprenorphine | 0.391 ± 0.039 | 0.628 ± 0.04 | 1.6 | 10.06 ± 2.05 | 25.7 |
| Nalbuphine | 1.899 ± 0.546 | 16.28 ± 1.761 | 8.6 | 491.0 ± 38.9 | 258.5 |
| Tianeptine | 78.24 ± 9.93 | >100 mM | >50 000 | 2828 ± 416 | 36.2 |
| DC‐1‐128.1 a | 0.562 ± 0.075 | 63.32 ± 7.43 | 112.6 | 317.8 ± 47.0 | 565.2 |
| DC‐1‐76.2 a | 0.497 ± 0.052 | 43.99 ± 2.98 | 88.5 | 347.0 ± 45.6 | 698.5 |
| EWB‐3‐14 | 0.826 ± 0.076 | 12.09 ± 1.41 | 14.6 | 140.5 ± 17.8 | 170.1 |
| DC‐1‐90.2 a | 1.829 ± 0.247 | 34.45 ± 0.77 | 18.8 | 429.8 ± 41.7 | 235.0 |
| JL‐2‐39 | 0.394 ± 0.035 | 17.13 ± 2.22 | 43.4 | 472.9 ± 41.2 | 1199.2 |
| DC‐1‐76.1 a | 1.910 ± 0.137 | 52.89 ± 1.11 | 27.7 | 3387 ± 268 | 1773.3 |
| EG‐1‐203 | 0.761 ± 0.031 | 22.26 ± 4.66 | 29.3 | 66.49 ± 7.70 | 87.4 |
| EG‐1‐230 | 4.216 ± 0.177 | 45.90 ± 7.08 | 10.9 | 1006 ± 147 | 238.5 |
Note: Data are mean K i values ± SEM (n = 3–6) derived from ligand competition curves for [3H]naloxone binding to membranes from MOR‐, or [3H]diprenorphine binding to KOR‐ and DOR‐expressing CHO cells. Fold selectivity for MOR over KOR or DOR was determined by dividing the K i values at KOR or DOR by the K i value at MOR.
Abbreviations: CHO, Chinese hamster ovary; DOR, delta opioid receptor; KOR, kappa opioid receptor; MOR, mu opioid receptor.
Values at MOR for morphine, DC‐1‐128.1, DC‐1‐76.2, DC‐1‐90.2, and DC‐1‐76.1 were reported in Chambers et al. 15
TABLE 2.
E max and EC50 values from ligand‐modulated [35S]GTPγS binding.
| MOR | KOR | DOR | ||||
|---|---|---|---|---|---|---|
| Ligand | E max (%) | EC50 (nM) | E max (%) | EC50 (nM) | E max (%) | EC50 (nM) |
| Morphine a | 88.30 ± 4.86 | 123.0 ± 23.5 | 54.37 ± 4.24 | 1320 ± 105 | 72.60 ± 11.98 | 502.7 ± 103.9 |
| Buprenorphine | 35.56 ± 1.89 | 0.55 ± 0.04 | 6.60 ± 0.85 | 2.61 ± 1.08 | 11.94 ± 1.89 | 4.69 ± 0.93 |
| Nalbuphine | 18.92 ± 0.64 | 17.35 ± 1.52 | 47.83 ± 2.97 | 86.60 ± 11.45 | 30.13 ± 3.58 | 598.4 ± 184.7 |
| Tianeptine | 109.6 ± 3.6 | 6443 ± 622 | No stim | – | 102.57 ± 3.72 | 19 153 ± 352 |
| DC‐1‐128.1 a | 75.35 ± 3.83 | 8.38 ± 0.77 | No stim | – | 31.40 ± 2.23 | 502.8 ± 117.9 |
| DC‐1‐76.2 a | 29.09 ± 0.78 | 6.73 ± 1.35 | No stim | – | −6.34 ± 0.78 | 160.1 ± 142.4 |
| EWB‐3‐14 | 20.82 ± 1.67 | 7.44 ± 2.67 | No stim | – | −20.32 ± 4.91 | 1309 ± 751 |
| DC‐1‐90.2 a | 17.97 ± 1.45 | 9.64 ± 1.70 | No stim | – | −6.97 ± 0.46 | 252.6 ± 132.7 |
| JL‐2‐39 | 13.04 ± 1.48 | 7.09 ± 3.70 | No stim | – | No stim | – |
| DC‐1‐76.1 a | 10.54 ± 0.82 | 36.97 ± 15.47 | No stim | – | −0.70 ± 2.12 | 1160 ± 214 |
| EG‐1‐203 | 4.79 ± 0.60 | 24.19 ± 13.83 | −3.49 ± 0.67 | 124.9 ± 88.7 | −11.64 ± 3.08 | 37.05 ± 17.74 |
| EG‐1‐230 | 0.67 ± 0.79 | 24.13 ± 12.01 | −3.97 ± 0.65 | 20.71 ± 5.60 | −10.55 ± 3.58 | 517.2 ± 366.2 |
Note: Data are mean E max and EC50 values ± SEM (n = 3–9) derived from concentration‐effect curves as illustrated in Figure 2 for MOR. E max values are expressed as a percent of the stimulation produced by a maximally effective concentration of DAMGO (MOR), U50488 (KOR), or SNC80 (DOR). No stim.: lack of concentration‐dependent stimulation up to at least 3 μM ligand (30 μM for tianeptine).
Abbreviations: CHO, Chinese hamster ovary; DOR, delta opioid receptor; KOR, kappa opioid receptor; MOR, mu opioid receptor.
Values at MOR‐mediated stimulation of GTPɣS binding for morphine, DC‐1‐128.1, DC‐1‐76.2, DC‐1‐90.2, and DC‐1‐76.1 were reported in Chambers et al. 15
The functional [35S]GTPγS assays showed a wide range of potencies and efficacies for the test compounds at MOR. Regarding MOR potencies, tianeptine had the lowest potency, while the phenylmorphans displayed higher potencies ranging from 6.73–36.97 nM. MOR EC50 values for tianeptine and the phenylmorphans were significantly correlated with their MOR K i values in binding studies (R 2 = 0.88, p = .0002; see Figure S1). Regarding efficacy, tianeptine had the highest efficacy at both MOR and DOR with E max values similar to the reference agonists at each receptor and only a 3‐fold higher potency in MOR‐versus‐DOR expressin cells. By contrast, the phenylmorphans had graded lower MOR efficacies and higher levels of MOR selectivity. DC‐1‐128.1 had an MOR E max (75.35%) similar to morphine, but it had a lower E max (31.40%) and more than 60‐fold lower potency at DOR and produced no KOR activation. The remaining phenylmorphans had lower MOR efficacies than buprenorphine and either did not activate or slightly inhibited KOR and DOR signaling. Notably, the lower MOR efficacy phenylmorphans JL‐2‐39 and DC‐1‐76.1 showed detectable but lower MOR efficacy and improved MOR>KOR selectivity relative to the clinically available low‐efficacy opioid nalbuphine.
3.2. In vivo studies of locomotor activity
Table 3 shows the mean ± SEM number of baseline locomotor counts after vehicle administration in each group of mice. There was a significant difference in baseline activity across groups [F (8, 98) = 7.438, p < .0001], and follow‐up analysis by two‐way ANOVA to include sex as a variable confirmed a main effect of group [F(8, 89) = 7.394, p < .0001] but no main effect of sex (p = .195) and no group × sex interaction (p = .560). Raw data to show locomotor‐activating effects of each drug are shown in Figure S2. However, to control for the different levels of baseline activity in each group, raw data for each drug were transformed as to Difference Scores and expressed as a percentage of the mean E max Difference Score produced by the reference drug methadone. Figure 3 uses the data with methadone to illustrate the sequence of data analysis steps that was followed for each drug as described in Methods. Methadone produced a dose‐dependent increase in locomotor activity. Table 3 shows the one‐way ANOVA results, E max and ED50 for methadone pooled across sexes, and Table S1 shows the two‐way ANOVA results and post hoc power analysis for methadone segregated by sex. There was no main effect of sex or sex × dose interaction for methadone.
TABLE 3.
Tabular results for locomotor studies.
| Baseline ± SEM | E max (95%CL) | ED50 (95% CL) | ||
|---|---|---|---|---|
| Drug | # Counts | % Methadone E max | mg/kg | One‐way ANOVA results |
| Methadone | 2350.3 ± 264.7 | 100 (82.8–117.2) | 2.50 (1.61–3.99) | F (2.90, 31.87) = 38.80; p < .0001 |
| Tianeptine | 1248.6 ± 127.6 | 102.8 (84.4–123.2) | 18.28 (16.37–20.46) | F (1.88, 20.73) = 86.74; p < .0001 |
| DC‐1‐128.1 | 2692.3 ± 241.2 | 80.1 (68.9–91.2) | 0.19 (0.10–0.29) | F (2.95, 32.40) = 31.22; p < .0001 |
| DC‐1‐76.2 | 2732.3 ± 706.9 | 87.6 (50.0–125.3) | 0.47 (0.16–0.80) | F (1.94, 21.36) = 19.59; p < .0001 |
| EWB‐3‐14 | 1570.5 ± 51.2 | 101.5 (67.6–135.4) | 1.05 (0.77–1.44) | F (2.82, 31.06) = 22.36; p < .0001 |
| JL‐2‐39 | 3102.8 ± 151.9 | 87.1 (66.6–107.7) | 2.85 (1.31–4.81) | F (2.18, 23.93) = 40.55; p < .0001 |
| DC‐1‐76.1 | 3072.6 ± 156.4 | 34.3 (16.6–52.0) | 0.72 (0.47–1.74) | F (2.28, 25.12) = 7.74; p = .0017 |
| EG‐1‐203 | 989.3 ± 125.2 | 13.0 (2.5–23.5) | 5.42 (not determined) | F (1.75, 19.29) = 5.11; p = .0196 |
| EG‐1‐230 | 1338.8 ± 261.4 | 2.2 (−5.8–10.1) | Inactive | F (2.43, 24.34) = 0.29; p = .7943 |
Note: For each experimental group, data are shown for mean ± SEM number of baseline counts after vehicle administration, E max value (95% CL) for the drug expressed as a % of the methadone E max, and ED50 (95% CL) expressed in mg/kg. The one‐way ANOVA results are also shown for each drug effect. All groups included 12 mice (6 females, 6 males) except EG‐1‐230, which had 11 mice (6 females, 5 males).
FIGURE 3.
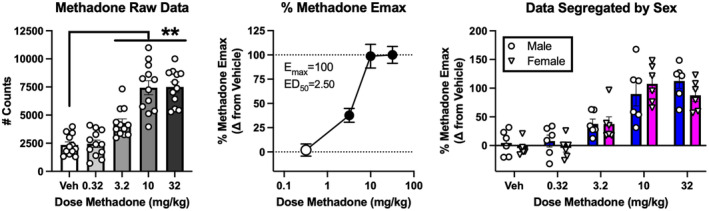
Experimental design and analysis illustrated with methadone. Drugs were tested in separate groups of 12 mice (6 per sex) using a within‐subjects repeated‐measures design. The left panel shows that initial analysis pooled raw data from both sexes. Abscissa: dose methadone in mg/kg administered SC. Veh = vehicle. Ordinate: Total locomotor activity counts during a 60‐min test session. Bars show mean ± SEM, and points show individual data. **indicate different from vehicle as indicated by one‐way ANOVA followed by a Holm‐Sidak post hoc test, p < .01. The middle panel shows calculation of dose‐effect parameters (E max, ED50). Data for each mouse at each dose were transformed to % Methadone E max using the equation [(Drug − Veh)/(5149.8)] × 100, where Drug = total locomotor counts in a given mouse after a drug dose, Veh = mean locomotor counts after vehicle in that group, and 5149.8 = the mean maximum increase in locomotor counts produced by methadone in the methadone group (7500.1 counts at 32 mg/kg vs. 2350.3 counts after saline vehicle). Filled symbols indicate different from vehicle as indicated by one‐way ANOVA followed by a Holm‐Sidak post hoc test, p < .05. The right panel shows the same data as the middle panel segregated by sex and analyzed by two‐way ANOVA. In this case, there was a main effect of dose [F (2.82, 28.2) = 38.74, p < .0001], but no main effect of sex [F (1, 10) = 0.42, p = .53] and no sex × dose interaction [F (4, 40) = 0.98, p = .43].
Figure 4 shows dose‐effect curves for data pooled across sexes for each test drug. E max values, ED50 values, and one‐way ANOVA results are shown in Table 3. The potency rank order of all compounds as determined by ED50 values was DC‐1‐128.1 > DC‐1‐76.2 ≥ DC‐1‐76.1 ≥ EWB‐3‐14 > methadone ≥ JL‐2‐39 > EG‐1‐203 > tianeptine. An ED50 value for EG‐1‐230 could not be determined because it was inactive when administered alone. Regarding efficacy, all drugs except EG‐1‐230 produced dose‐dependent and significant increases in locomotor activity. E max values for the reference agonist methadone and the test compounds tianeptine, DC‐1‐128.1, DC‐1‐76.2, EWB‐3‐14, and JL‐2‐39 were statistically similar as indicated by overlapping 95% confidence limits. Conversely, the test compounds DC‐1‐76.1, EG‐1‐203, and EG‐1‐230 had lower E max values than methadone and the other test compounds (except for an overlap in E max 95% CL for DC‐1‐76.2 ≥ DC‐1‐76.1). Lastly, the E max for EG‐1‐230 was also lower than that for DC‐1‐76.1.
FIGURE 4.
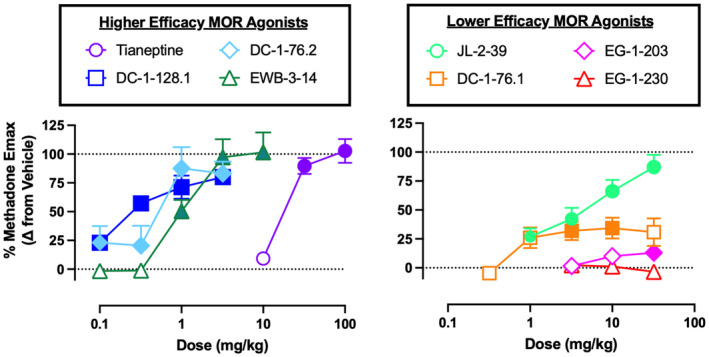
Locomotor‐activating effects of the novel opioids in female and male ICR mice. Abscissae: dose in mg/kg administered SC (log scale). Ordinates: Locomotor‐activating effects expressed as a percent of the methadone E max. Points show mean ± SEM and filled points indicate doses that produced effects significantly greater than vehicle (p < .05). Data were further segregated by sex and analyzed by two‐way ANOVA, and these results are shown in Table S1.
Two‐way ANOVA results for data segregated by sex for each drug are shown in Table S1. For most groups, there was not a significant main effect of sex or sex × dose interaction. As the only exception, there was a significant sex × dose interaction for EG‐1‐203 [F (3, 30) = 3.77; p = .0208]; however, even here, post hoc analysis did not indicate a significant effect of sex at any dose. Table S1 also shows post hoc power analysis of results.
Figure 5 compares the time courses of vehicle and the E max drug dose in each group during the 60‐min session. Vehicle‐treated animals had high initial locomotor activity followed by a decline to lower levels later in the session. Drug‐induced increases in locomotor activity were generally observed within the first 10–15 min of the session and were sustained for the duration of the session.
FIGURE 5.
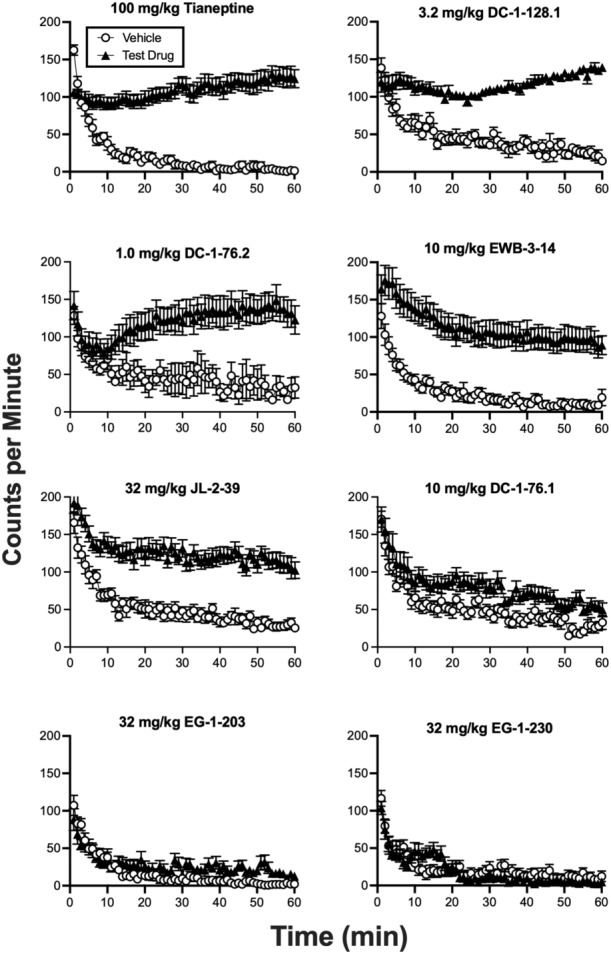
Time course of effects produced by vehicle and the peak locomotor‐activating dose of each drug. The identity and dose of each test drug is shown in the header to each panel. Abscissae: Time in min of the 60‐min session, which began 5 min after drug administration. Ordinates: Number of locomotor counts per minute. All points show mean ± SEM.
Figure 6 shows the results of antagonism studies to determine receptor mechanisms of drug action. Naltrexone significantly attenuated the effects of locomotor‐activating doses of tianeptine, DC‐1‐128.1, DC‐1‐76.2, EWB‐3‐14, JL‐2‐39, and DC‐1‐76.1. Reciprocally, the lower‐efficacy compounds EG‐1‐203 and EG‐1‐230 both significantly attenuated the locomotor‐activating effects of morphine.
FIGURE 6.
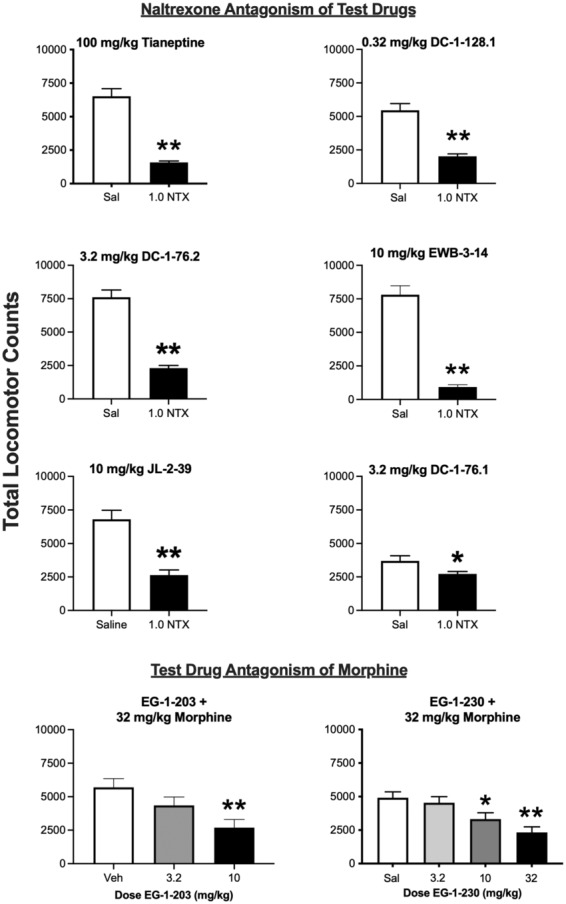
Antagonism studies with test drugs. For the six higher‐efficacy test drugs (top six panels), a locomotor‐activating dose of the drug was tested after pretreatment with either saline (Sal) or 1.0 mg/kg naltrexone (1.0 NTX). For the two lowest efficacy drugs (bottom two panels), a locomotor‐activating dose of morphine (32 mg/kg) was administered after pretreatment with either vehicle (Veh) or increasing test‐drug doses. All points show mean ± SEM. *p < .05, **p < .01 compared to saline (top panels) or vehicle (bottom panels) after t‐test or one‐way ANOVA as appropriate.
3.3. Comparison of in vitro and in vivo results
Figure 7 shows the relationship between in vitro drug potency and effectiveness to stimulate in vitro [35S]GTPɣS binding in mMOR‐CHO cells and in vivo potency and effectiveness to stimulate locomotor activity in ICR mice. Regarding potencies, the correlation between in vitro log EC50 and in vivo log ED50 values approached but did not meet the criterion for significance (R 2 = 0.54, p = .0612). Tianeptine displayed the lowest potency both in vitro and in vivo, but there was less consistency for in vitro versus in vivo relative potencies for the phenylmorphans. For example, JL‐2‐39 was ~5‐fold more potent than DC‐1‐76.1 in vitro but ~4‐fold less potent than DC‐1‐76.1 in vivo. Regarding efficacies, increases in [35S]GTPɣS binding up to approximately 21% of the DAMGO E max were associated with increases in locomotor activity (region denoted by the gray box in Figure 6 right panel). Linear regression analysis for the data in this range indicated a significant correlation of in vitro and in vivo E max values (R 2 = 0.89, p = .0165), and in vitro activity (95% confidence limits) of 10.4 (4.7–16.7) % DAMGO E max was sufficient to produce in vivo locomotor activation of 50% methadone E max. However, compounds that produced progressively higher maximum levels of [35S]GTPɣS binding above 21% of the DAMGO E max did not produce further increases in locomotor activation.
FIGURE 7.
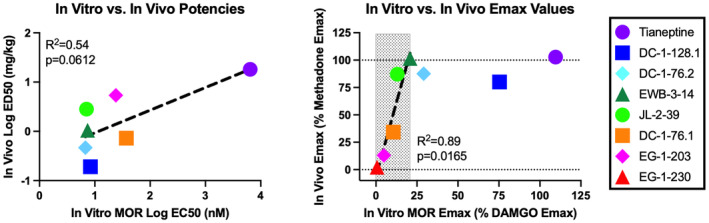
Relationship between in vitro and in vivo potency and E max values. The left panel shows the potency relationship between in vitro log EC50 values on the abscissa and in vivo log ED50 values on the ordinate. The dotted line shows the linear regression and the correlation approached but did not achieve the criterion for significance (R 2 = 0.54, p = .0612). The right panel shows the efficacy relationship between in vitro E max values on the abscissa and in vivo E max values on the ordinate. The hatched box shows the range of in vitro E max values associated with increasing in vivo E max values. The dotted line shows the linear regression for these data, which did yield a significant correlation (R 2 = 0.89, p = .0165). Higher in vitro E max values were not associated with further increases in the in vivo E max.
4. DISCUSSION
This study characterized the MOR selectivity and efficacy of a series of chiral C9‐substituted phenylmorphans. The opioid antidepressant tianeptine was included as a comparator. There were four main findings. First, all the phenylmorphans bound with high affinity to MOR, and all had >10‐fold selectivity for MOR versus KOR and DOR. Second, the phenylmorphans had graded levels of high‐to‐low MOR efficacies evident in both the in vitro and in vivo assays. Third, the in vivo locomotor studies in mice provided evidence for rapid onset, modest duration, and MOR mediation of effects with no apparent sex differences. Lastly, tianeptine functioned as a low‐potency but high‐efficacy MOR agonist. Overall, these findings support further research with these chiral C9‐substituted phenymorphans as candidate low‐efficacy opioid therapeutics and as tools to investigate MOR efficacy as a determinant of opioid effects.
4.1. MOR selectivity of the phenylmorphans
The present results confirm and extend previous findings to suggest that the novel phenylmorphans investigated here have relatively high MOR selectivity. Binding studies at MOR, KOR, and DOR had been conducted previously for only EWB‐3‐14, 17 and results here with EWB‐3‐14 are consistent in showing subnanomolar MOR affinity and selectivity for MOR>KOR>DOR. Although binding across all three opioid receptor subtypes has not been reported previously for the other phenylmorphans, these compounds were shown to have functional MOR selectivity in an in vitro assay of forskolin‐stimulated cAMP accumulation. Thus, all of these compounds displayed higher potency to inhibit cAMP accumulation in cells expressing MOR than to function as either an agonist or antagonist for inhibition of cAMP accumulation in cells expressing KOR or DOR. 15 , 16 , 18 Of particular relevance for the development of these compounds as candidate therapeutics, the lower‐efficacy phenylmorphans all showed improved MOR selectivity relative to nalbuphine as an existing, clinically available, low MOR efficacy opioid. Thus, nalbuphine displayed only 8.6‐fold MOR‐versus‐KOR binding selectivity in the present study, whereas all the phenylmorphans displayed >10‐fold selectivity. Moreover, results from the in vitro assay of ligand‐stimulated GTPɣS binding indicated that nalbuphine had partial KOR agonist activity, whereas none of the phenylmorphans had KOR agonist activity and some functioned as weak inverse agonists. Taken together, these findings suggest that the lower‐efficacy phenymorphans may be devoid of the KOR agonist effects that appear to be a limitation for existing, clinically approved, low‐efficacy opioids including nalbuphine, butorphanol, and pentazocine. 10 , 11 , 12 , 13 , 14
4.2. MOR efficacy of the phenylmorphans
In addition to this evidence for improved MOR selectivity, the present study also provides new evidence for the graded MOR efficacies of these compounds. As described previously using in vitro assays of ligand‐induced inhibition of cAMP accumulation or stimulation of GTPɣS binding, 15 , 16 , 17 , 18 the phenylmorphans studied here displayed varying degrees of MOR efficacy, and most had lower MOR efficacy than either buprenorphine or nalbuphine as examples of existing, clinically available low‐efficacy opioids. The locomotor studies in mice illustrated the functional importance of this graded MOR efficacy for expression of in vivo behavioral effects. Thus, the five compounds tested in vivo with the lowest MOR efficacies (from highest to lowest: EWB‐3‐14, JL‐2‐39, DC‐1‐76.1, EG‐1‐203, and EG‐1‐230) showed a graded and correlated decline in both in vitro stimulation of GTPɣS binding and in vivo locomotor activation. These results agree with our earlier findings using other opioids and opioid agonist/antagonist mixtures to show that locomotor activation in mice is dependent on the MOR efficacy of the opioid. 20 Moreover, these data agree with other in vivo data that have been reported for a subset of these compounds. For example, the antinociceptive and respiratory depressant effects of DC‐1‐76.2, JL‐2‐39, and DC‐1‐76.1 have been examined in squirrel monkeys. 15 , 18 Although results across compounds were not compared statistically, they showed a general trend of declining antinociceptive and respiratory depressant effects in monkeys similar to their declining locomotor stimulant effects in mice.
The present studies in mice also provide additional insights regarding the in vivo pharmacology of these phenylmorphans. First, all compounds with significant agonist activity produced a relatively rapid onset of effects with a duration of at least 60 min. Given other evidence to suggest that MOR agonist‐induced locomotor stimulation in mice is mediated by receptors in the central nervous system (e.g., Ref. [31]), these results suggest that all of these compounds distribute to the brain after systemic administration and have a modest duration of action similar to clinically available opioids like morphine. 20 The lack of a significant correlation between in vitro and in vivo potencies suggests that there may be modest differences in pharmacokinetics. For example, JL‐2‐39 had ~5‐fold higher MOR affinity and was ~5‐fold more potent to stimulate GTPɣS binding in MOR CHO cells than DC‐1‐76.1, but JL‐2‐39 was ~4‐fold less potent than DC‐1‐76.1 to stimulate locomotor activity. This suggests that JL‐2‐39 may not distribute across the blood–brain barrier as effectively or may be metabolized more rapidly than DC‐1‐76.1. Future pharmacokinetic studies would be required to clarify the role of these factors.
Second, antagonism studies suggest that effects of these compounds were mediated by MORs and not by non‐opioid off‐target receptors. Thus, effects of the six higher‐efficacy phenylmorphans were blocked by naltrexone similarly to naltrexone blockade of morphine‐induced locomotor activation. 20 Reciprocally, the two lower‐efficacy phenylmorphans blocked the effects of morphine similarly to antagonist effects of other low‐efficacy MOR ligands. 20
Third, the mouse locomotor studies also provide additional support for the in vivo functional relevance of MOR selectivity. In particular, we reported previously that nalbuphine produced significant but weak effects in this same behavioral assay of locomotor activation in mice. 20 The other clinically available low‐efficacy MOR agonists butorphanol and pentazocine also produce weak locomotor activation in mice. 32 , 33 These low levels of locomotor activation could be influenced by their low MOR selectivity. In particular, the KOR‐mediated effects of these compounds might oppose and limit MOR‐mediated hyperactivity. 31 In the present study, JL‐2‐39 displayed lower MOR efficacy but higher MOR selectivity than nalbuphine, and it produced a higher locomotor E max than nalbuphine and similar to much higher MOR efficacy opioids. DC‐1‐76.1 had even lower MOR efficacy, but it also has higher MOR selectivity than nalbuphine and produced a higher locomotor E max than nalbuphine. Overall, these results suggest that low MOR‐versus‐KOR selectivity may limit some MOR‐mediated effects of existing low‐efficacy MOR agonists like nalbuphine, and the more MOR‐selective phenylmorphans studied here can produce greater MOR‐mediated effects despite their lower MOR efficacy.
Lastly, the present study was not intended or powered to detect sex differences in drug effects, but both females and males were included, and results provide preliminary evidence on the extent of sex differences in drug effects. 27 There was not a main effect of sex for any of the phenylmorphans, and in the only instance of a significant sex × dose interaction (for EG‐1‐203), the post hoc test did not reveal an effect of sex at any dose. These results should be interpreted with caution given that achieved power was often less than 0.8 as a common criterion to protect against a Type II error (i.e., concluding that an effect is absent when it is in fact present). Nonetheless, these results add to our previous finding that sex differences in MOR ligand effects on mouse locomotor activity are rare. 20
4.3. Tianeptine
Tianeptine was included in this study as a putative high MOR efficacy comparator, and results are consistent with other recent studies to indicate that tianeptine functions as a low‐potency, high‐efficacy MOR/DOR agonist. 22 , 23 , 24 Insofar as DOR activation has been linked to antidepressant effects, 34 , 35 these findings support the proposition that clinical antidepressant effects of tianeptine may involve DOR as well as MOR effects. The present study extended these previous results in finding no sex difference in tianeptine effects. Thus, as with the phenylmorphans in this study and with other opioids tested previously, 20 tianeptine also appears to produce similar locomotor activation in both sexes.
5. CONCLUSION
In conclusion, this study further characterized the pharmacology of a series of chiral C9‐substituted phenylmorphans. Relative to existing low‐efficacy opioid analgesics, these phenylmorphans display relatively high MOR selectivity (which can reduce off‐target and particularly KOR‐mediated side effects) and graded MOR efficacies (which provides greater opportunity to control efficacy in therapeutic or experimental applications). Compounds like JL‐2‐39, DC‐1‐76.1, and EG‐1‐203, which have lower MOR efficacy than buprenorphine or nalbuphine but retain in vivo MOR‐mediated effects, may be of particular interest as novel candidate opioid therapeutics.
AUTHOR CONTRIBUTIONS
Participated in research design: Edna J. Santos, Nima Nassehi, Dana E. Selley, S. Stevens Negus. Conducted experiments: Edna J. Santos, Nima Nassehi, Samuel A. Marsh. Contributed new reagents or analytic tools: Eric W. Bow, Dana R. Chambers, Eugene S. Gutman, Arthur E. Jacobson, Joshua A. Lutz, Kenner C. Rice, Agnieszka Sulima. Performed data analysis: Edna J. Santos, Nima Nassehi, Dana E. Selley, S. Stevens Negus. Wrote or contributed to writing of the manuscript: Edna J. Santos, Nima Nassehi, Eric W. Bow, Dana R. Chambers, Eugene S. Gutman, Arthur E. Jacobson, Joshua A. Lutz, Kenner C. Rice, Agnieszka Sulima, Dana E. Selley, S. Stevens Negus.
FUNDING INFORMATION
This work was supported by the National Institutes of Health via National Institute on Drug Abuse Grants P30DA033934 and T32DA007027 (PI: Dewey WL), National Institute on Neurological Disorders and Stroke Grant F31NS122525 (PI: Santos EJ), and National Institute of General Medical Sciences Grant R25GM090084 (PI: Akbarali HI). Additionally, a portion of this work was supported by the NIH Intramural Research Programs of the National Institute on Drug Abuse and the National Institute of Alcohol Abuse and Alcoholism.
DISCLOSURE
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
ETHICS STATEMENT
Patents have been obtained for some of the compounds (see Rice et al. 19 ).
Supporting information
Data S1.
ACKNOWLEDGMENTS
The authors thank Rolando E. Mendez for technical assistance with binding assays.
Santos EJ, Nassehi N, Bow EW, et al. Role of efficacy as a determinant of locomotor activation by mu‐opioid receptor (MOR) ligands in female and male mice. II. Effects of novel MOR‐selective phenylmorphans with high‐to‐low MOR efficacy. Pharmacol Res Perspect. 2023;11:e01111. doi: 10.1002/prp2.1111
Edna J. Santos and Nima Nassehi are co‐first authors.
DATA AVAILABILITY STATEMENT
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Data.
REFERENCES
- 1. Yaksh T, Wallace W. Opioids, analgesia, and pain management. In: Brunton LL , Hilal‐Dandan R, Knollman BC, eds. Goodman and Gilman's: the Pharmacological Basis of Therapeutics, 13e. McGraw‐Hill; 2018:355‐386. [Google Scholar]
- 2. Gillis A, Sreenivasan V, Christie MJ. Intrinsic efficacy of opioid ligands and its importance for apparent bias, operational analysis, and therapeutic window. Mol Pharmacol. 2020;98:410‐424. [DOI] [PubMed] [Google Scholar]
- 3. Kelly E, Conibear A, Henderson G. Biased agonism: lessons from studies of opioid receptor agonists. Annu Rev Pharmacol Toxicol. 2023;63:491‐515. [DOI] [PubMed] [Google Scholar]
- 4. Selley DE, Liu Q, Childers SR. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor‐stimulated [35S]GTP gamma S binding in mMOR‐CHO cells and rat thalamus. J Pharmacol Exp Ther. 1998;285:496‐505. [PubMed] [Google Scholar]
- 5. Dahan A, Yassen A, Romberg R, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth. 2006;96:627‐632. [DOI] [PubMed] [Google Scholar]
- 6. Raynor K, Kong H, Chen Y, et al. Pharmacological characterization of the cloned kappa‐, delta‐, and mu‐opioid receptors. Mol Pharmacol. 1994;45:330‐334. [PubMed] [Google Scholar]
- 7. Wentland MP, Lou R, Lu Q, et al. Syntheses of novel high affinity ligands for opioid receptors. Bioorg Med Chem Lett. 2009a;19:2289‐2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wentland MP, Lou R, Lu Q, et al. Syntheses and opioid receptor binding properties of carboxamido‐substituted opioids. Bioorg Med Chem Lett. 2009b;19:203‐208. [DOI] [PubMed] [Google Scholar]
- 9. Wood PL, Charleson SE, Lane D, Hudgin RL. Multiple opiate receptors: differential binding of mu, kappa and delta agonists. Neuropharmacology. 1981;20:1215‐1220. [DOI] [PubMed] [Google Scholar]
- 10. Pick CG, Paul D, Pasternak GW. Nalbuphine, a mixed kappa 1 and kappa 3 analgesic in mice. J Pharmacol Exp Ther. 1992;262:1044‐1050. [PubMed] [Google Scholar]
- 11. Preston KL, Bigelow GE, Bickel WK, Liebson IA. Drug discrimination in human postaddicts: agonist‐antagonist opioids. J Pharmacol Exp Ther. 1989;250:184‐196. [PubMed] [Google Scholar]
- 12. Remmers AE, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Opioid efficacy in a C6 glioma cell line stably expressing the human kappa opioid receptor. J Pharmacol Exp Ther. 1999;288:827‐833. [PubMed] [Google Scholar]
- 13. Schmidt WK, Tam SW, Shotzberger GS, Smith DH Jr, Clark R, Vernier VG. Nalbuphine. Drug Alcohol Depend. 1985;14:339‐362. [DOI] [PubMed] [Google Scholar]
- 14. Vivian JA, DeYoung MB, Sumpter TL, Traynor JR, Lewis JW, Woods JH. Kappa‐opioid receptor effects of butorphanol in rhesus monkeys. J Pharmacol Exp Ther. 1999;290:259‐265. [PubMed] [Google Scholar]
- 15. Chambers DR, Sulima A, Luo D, et al. A journey through diastereomeric space: the design, synthesis, in vitro and in vivo pharmacological activity, and molecular modeling of novel potent diastereomeric MOR agonists and antagonists. Molecules. 2022;27:6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gutman ES, Bow E, Li F, et al. G‐protein biased opioid agonists: 3‐hydroxy‐N‐phenethyl‐5‐phenylmorphans with three‐carbon chain substituents at C9. RSC Med Chem. 2020;11:896‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hiebel AC, Lee YS, Bilsky E, et al. Probes for narcotic receptor mediated phenomena. 34. Synthesis and structure‐activity relationships of a potent mu‐agonist delta‐antagonist and an exceedingly potent antinociceptive in the enantiomeric C9‐substituted 5‐(3‐hydroxyphenyl)‐N‐phenylethylmorphan series. J Med Chem. 2007;50:3765‐3776. [DOI] [PubMed] [Google Scholar]
- 18. Lutz JA, Sulima A, Gutman ES, et al. Discovery of a potent highly biased MOR partial agonist among diastereomeric C9‐hydroxyalkyl‐5‐phenylmorphans. Molecules. 2023, 28(12), 4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rice KC, Jacobson AE, Li F, et al. Biased potent opioid‐like agonists as improved medications to treat chronic and acute pain and methods of using the same. 2019. US Application Serial No.: 62/644,791 filed on March 19, 2018. US Patent 11,352,365 issued 6‐7‐2022. International Publication WO 2019/182950 A1 September 26, 2019, USA, pp. 91.
- 20. Santos EJ, Banks ML, Negus SS. Role of efficacy as a determinant of locomotor activation by mu opioid receptor ligands in female and male mice. J Pharmacol Exp Ther. 2022;382:44‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Selley DE, Banks ML, Diester CM, et al. Manipulating pharmacodynamic efficacy with agonist + antagonist mixtures: In vitro and In vivo studies with opioids and cannabinoids. J Pharmacol Exp Ther. 2021;376:374‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baird TR, Akbarali HI, Dewey WL, et al. Opioid‐like adverse effects of tianeptine in male rats and mice. Psychopharmacology (Berl). 2022;239:2187‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gassaway MM, Rives ML, Kruegel AC, Javitch JA, Sames D. The atypical antidepressant and neurorestorative agent tianeptine is a mu‐opioid receptor agonist. Transl Psychiatry. 2014;4:e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Samuels BA, Nautiyal KM, Kruegel AC, et al. The behavioral effects of the antidepressant tianeptine require the mu‐opioid receptor. Neuropsychopharmacology. 2017;42:2052‐2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li G, Aschenbach LC, Chen J, et al. Design, synthesis, and biological evaluation of 6alpha‐ and 6beta‐N‐heterocyclic substituted naltrexamine derivatives as mu opioid receptor selective antagonists. J Med Chem. 2009;52:1416‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuan Y, Li G, He H, et al. Characterization of 6alpha‐ and 6beta‐N‐heterocyclic substituted naltrexamine derivatives as novel leads to development of mu opioid receptor selective antagonists. ACS Chem Nerosci. 2011;2:346‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Diester CM, Banks ML, Neigh GN, Negus SS. Experimental design and analysis for consideration of sex as a biological variable. Neuropsychopharmacology. 2019;44:2159‐2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faul F, Erdfelder E, Lang AG, Buchner A. G*power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175‐191. [DOI] [PubMed] [Google Scholar]
- 29. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander SP, Christopoulos A, Davenport AP, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein‐coupled receptors. Br J Pharmacol. 2021;178(Suppl 1):S27‐S156. [DOI] [PubMed] [Google Scholar]
- 31. Narita M, Takahashi Y, Takamori K, et al. Effects of kappa‐agonist on the antinociception and locomotor enhancing action induced by morphine in mice. Jpn J Pharmacol. 1993;62:15‐24. [DOI] [PubMed] [Google Scholar]
- 32. Filibeck U, Castellano C, Oliverio A. Differential effects of opiate agonists‐antagonists on morphine‐induced hyperexcitability and analgesia in mice. Psychopharmacology (Berl). 1981;73:134‐136. [DOI] [PubMed] [Google Scholar]
- 33. Sansone M, Castellano C, Libri V. Tripelennamine enhances buprenorphine‐, but not pentazocine‐induced hyperactivity in mice. Psychopharmacology (Berl). 1988;95:176‐179. [DOI] [PubMed] [Google Scholar]
- 34. Jutkiewicz EM. The antidepressant ‐like effects of delta‐opioid receptor agonists. Mol Interv. 2006;6:162‐169. [DOI] [PubMed] [Google Scholar]
- 35. Peppin JF, Raffa RB. Delta opioid agonists: a concise update on potential therapeutic applications. J Clin Pharm Ther. 2015;40:155‐166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Data.