

Abstract
Lipid droplets (LDs) form inter‐organelle contacts with the endoplasmic reticulum (ER) that promote their biogenesis, while LD contacts with mitochondria enhance β‐oxidation of contained fatty acids. Viruses have been shown to take advantage of lipid droplets to promote viral production, but it remains unclear whether they also modulate the interactions between LDs and other organelles. Here, we showed that coronavirus ORF6 protein targets LDs and is localized to the mitochondria‐LD and ER‐LD contact sites, where it regulates LD biogenesis and lipolysis. At the molecular level, we find that ORF6 inserts into the LD lipid monolayer via its two amphipathic helices. ORF6 further interacts with ER membrane proteins BAP31 and USE1 to mediate ER‐LDs contact formation. Additionally, ORF6 interacts with the SAM complex in the mitochondrial outer membrane to link mitochondria to LDs. In doing so, ORF6 promotes cellular lipolysis and LD biogenesis to reprogram host cell lipid flux and facilitate viral production.
Keywords: endoplasmic reticulum, lipid droplets, mitochondria, ORF6, organelle interaction
Subject Categories: Metabolism; Microbiology, Virology & Host Pathogen Interaction; Organelles
A lipid droplet‐localized coronaviral protein alters cellular metabolism by promoting their contact formation with the endoplasmic reticulum and mitochondria.
Introduction
Lipid droplets (LDs) are surrounded by a phospholipid monolayer and are responsible for the storage of triacylglycerols (TAGs), cholesteryl esters, and retinyl esters (Ducharme & Bickel, 2008). LD biogenesis starts in the endoplasmic reticulum (ER), where neutral lipid synthesis enzymes, including diglyceride acyltransferase (DGAT), deposit TGs between the leaflets of the ER bilayer and bud out from the ER surface (Olzmann & Carvalho, 2019). LDs can be broken down by two cellular pathways to release free fatty acids, one is lipophagy (LD autophagy; Singh et al, 2009); another is lipolysis, catalyzed by LD‐localized Adipose Triglyceride Lipase (ATGL), the first acting and rate‐limiting intracellular TG hydrolase, following by hormone‐sensitive lipase (HSL) and monoglyceride lipase (MGL; Zechner et al, 2017).
LDs are in contact with many other membrane organelles. Bscl2 (Seipin) helps connect newly formed LDs to the ER and enables lipid transfer to nascent LDs (Szymanski et al, 2007; Fei et al, 2008; Salo et al, 2016; Wang et al, 2016). Spastic paraplegia protein M1 Spastin coordinates fatty acid (FA) trafficking from LDs to peroxisomes for β‐oxidation by forming a tethering complex with peroxisomal ABCD1 and recruiting the membrane‐shaping ESCRT‐III proteins to LDs (Poirier et al, 2006; Chang et al, 2019). LDs can dynamically associate with subpopulations of mitochondria in different physiological contexts and in several tissues (Tarnopolsky et al, 2007; Wang et al, 2011, 2013; Herms et al, 2015; Rambold et al, 2015; Boutant et al, 2017; Benador et al, 2018; Long et al, 2022). In brown adipocytes, FAs are broken down from LDs and move into mitochondria for β‐oxidation to maintain body temperature (Cui et al, 2019). Mitochondria in contact with LD have increased pyruvate oxidation, electron transport, and ATP synthesis capacities, but have reduced β‐oxidation capacity, and support LD expansion (Benador et al, 2018). Starved cells replenish LDs with FAs through autophagic machinery and use LDs as a conduit for supplying mitochondria with FAs for β‐oxidation (Herms et al, 2015; Rambold et al, 2015; Nguyen et al, 2017). However, whether virus infection modulates the contact between LDs and other organelles remains largely unknown.
LDs are associated with multiple cellular anti‐infection processes. Recent studies have highlighted the critical roles of mammalian LDs in the antibacterial response: LDs act as molecular switches in anti‐infection immune response and regulates immune cell metabolism (Anand et al, 2012; Bosch et al, 2020). LDs also play important roles in promoting the early antiviral immune response, particularly by enhancing IFN production after viral infection (Monson et al, 2021). Besides, viral proteins link the replication compartments (RCs) of enteroviruses and LDs to enable the transfer of fatty acids from LDs for RC biogenesis (Laufman et al, 2019). Poliovirus 3A and 3AB bind with HSL and ATGL, recruiting the lipolysis machinery to the LD‐RC contact sites, which is important for virus replication (Laufman et al, 2019). Nucleocapsid core of hepatitis C virus (HCV) recruits viral replication complexes to LDs and also interacts with DGAT1 which is required for its trafficking to LDs and critical for viral replication (Miyanari et al, 2007; Herker et al, 2010). Dengue virus (DENV) infection increases the number of LDs per cell, and LDs sequester the viral capsid protein early during infection (Samsa et al, 2009). In addition, DENV infection induces lipophagy to release free fatty acids (FFAs), resulting in an increase in cellular β‐oxidation and ATP production, which is essential for viral replication (Heaton & Randall, 2010; Zhang et al, 2018). SARS‐CoV‐2 infection increases the expression of key proteins involved in the regulation of lipid metabolism and the number of LDs per cell, as well as viral components associated with LDs in infected cells (Dias et al, 2020). Depletion of fatty acid synthase (FASN) or treatment with fatty acid synthase inhibitors markedly repressed the infection of the SARS‐CoV‐2 (Chu et al, 2021). Several viral proteins such as ORF6 and ORF9C enhance cellular level of TAG, Cer, and polyunsaturated PC, and DGAT1 inhibitors are able to reduce viral production (Farley et al, 2022). These studies indicate possible and essential roles of lipid metabolic reprogramming and LD formation in SARS‐CoV‐2 replication and pathogenesis. However, the mechanism(s) by which LDs modulate SARS‐CoV‐2 production remains poorly understood.
In this study, we showed that the coronavirus ORF6 protein targets LDs and modulates the interactions of ER‐LDs by binding to BAP31 and USE1; the interactions of mitochondria‐LDs by binding to SAM complex. Additionally, ORF6 expression enhances LD biogenesis and lipolysis to reprogram lipid metabolism, which is important for viral production.
Results
Coronavirus ORF6 targets LDs
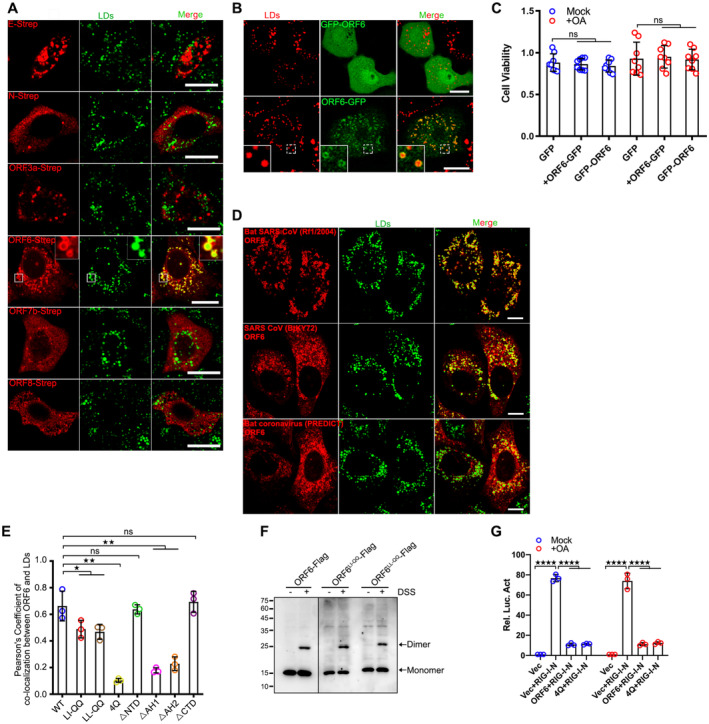
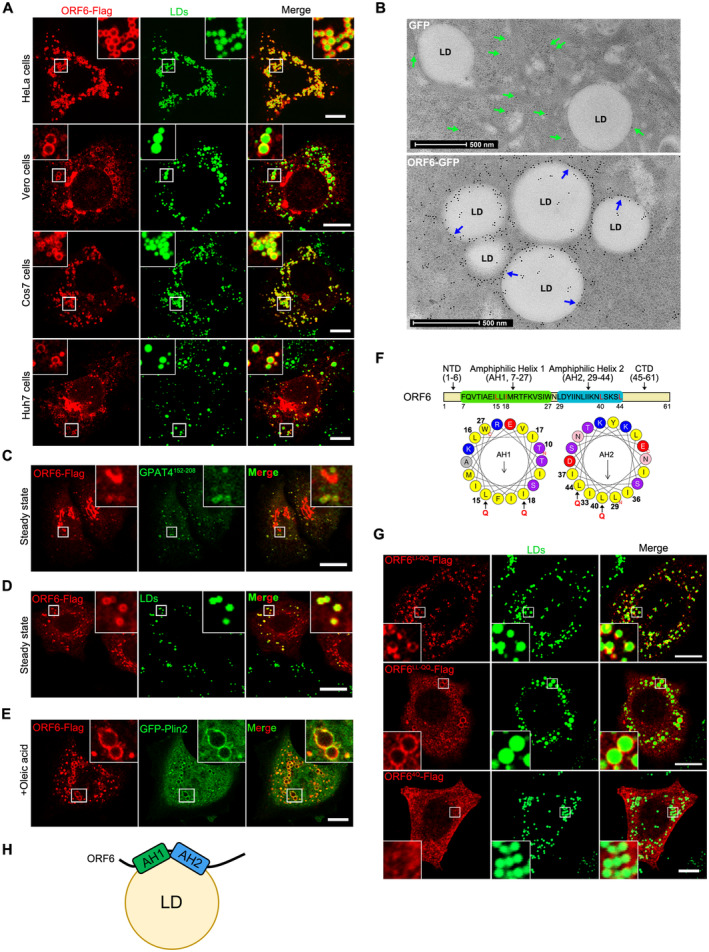
To investigate the viral proteins of SARS‐CoV‐2 targeting to LDs, we first examined the colocalization of boron‐dipyrromethene (BODIPY)‐labeled LDs with Strep‐tagged viral proteins during LD biogenesis induced by oleic acid (OA) treatment via confocal microscopy. Among all SARS‐CoV‐2 viral proteins, E, N, ORF3a, ORF6, ORF7b, and ORF8 showed puncta in cultured HeLa cells. Whereas only ORF6 was remarkably targeted to LDs (Fig EV1A). To confirm that the localization of ORF6 to LDs was not caused by the Strep‐tag, we also expressed ORF6 with a GFP tag at the N‐terminal (GFP‐ORF6) or C‐terminal (ORF6‐GFP), and found that GFP‐ORF6 remained diffused, while ORF6‐GFP showed LD localization (Fig EV1B), suggesting that the N‐terminal GFP‐tag affects the localization of ORF6. It is noted that both ORF6‐GFP and GFP‐ORF6 expression had no effect on the cell viability (Fig EV1C). We further expressed ORF6‐Flag in HeLa, Vero‐E6, Cos7, and Huh7 cells which were treated with OA. ORF6 was targeted to LDs in these cells (Fig 1A). Furthermore, using electron microscopy, we found that GFP‐labeled ORF6 decorated the membrane of LDs, but not GFP control vector (Fig 1B). We also observed the partly colocalization of ORF6 with GPAT4152–208 (Fig 1C), a membrane marker for nascent LDs (Wilfling et al, 2013). ORF6 colocalized with LDs under steady‐state conditions (Fig 1D) and colocalized with perilipin 2 (Plin2)‐labeled LDs under OA treatment (Fig 1E). In addition to SARS‐CoV‐2, we also found that ORF6 proteins of bat SARS‐CoV (Rf1/2006), SARS‐CoV (BtKY72), and Bat coronavirus (PREDICT) are targeted to LDs (Fig EV1D), suggesting that the functions of coronavirus ORF6 proteins are conserved.
Figure EV1. ORF6 targets to LDs.
-
AHeLa cells were transfected with Strep tagged SARS‐CoV‐2 proteins for 12 h and treated with 200 μM OA for another 12 h, then stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
BHeLa cells were transfected with ORF6‐GFP or GFP‐ORF6 for 12 h and treated with 200 μM OA for another 12 h. LDs were labeled with LipidTOX Deep Red (red). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
CHeLa cells were transfected with ORF6‐GFP or GFP‐ORF6 for 12 h and treated or untreated with 200 μM OA for another 12 h. Cell viability was analyzed. Eight biological replicates were performed. Two‐tailed Unpaired Student's t‐test, ns means no significance. Error bars represent the mean ± SD.
-
DHeLa cells were transfected with Flag tagged ORF6 of different coronavirus strains and treated with 200 μM OA for another 12 h, then stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
EColocalization of ORF6 and LDs (Pearson's Coefficient), n = 20 cells, three independent experiments. Two‐tailed Unpaired Student's t‐test, *P < 0.05, **P < 0.01, ns means no significance. Error bars represent the mean ± SD.
-
FHEK293T cells were transfected with ORF6‐Flag or its mutants and treated or untreated with 0.1 mM DSS for 30 min. Cell lysates were analyzed via WB.
-
GLuciferase assays were performed as described in methods. Three biological replicates were performed. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001. Error bars represent the mean ± SD.
Source data are available online for this figure.
Figure 1. SARS‐CoV‐2 ORF6 targets to LDs.
-
ACells were transfected with ORF6‐Flag for 12 h and treated with 200 μM OA for another 12 h, then stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
BImmunogold electron micrograph of GFP or ORF6‐GFP expressed HeLa cells. Blue arrows mark ORF6‐GFP dots are enriched on the surface of LDs. Green arrows mark GFP dots are distributed in the cytoplasm. Scale bar represents 500 nm.
-
C–EHeLa cells stable expressing ORF6‐Flag were treated with or without 200 μM OA for 12 h, then fixed and stained with anti‐Flag (red). Initial LDs were labeled with GFP‐GPAT4152–208 (green), mature LDs were labeled with BODIPY‐493/503 (green) or GFP‐Plin2 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
FSchematic of the domain structure of ORF6 and truncation mutants used in this study. The two predicted amphipathic helical domains of residues 7–27 (helix‐1) and residues 29–44 (helix‐2) of ORF6 were generated via the HeliQuest tool. Point mutations (black arrows) were introduced to the hydrophobic interfaces of these two helices.
-
GHeLa cells were transfected with ORF6‐Flag mutants (L15Q, I18Q in helix‐1 or/and L40Q, L44Q in helix‐2) and were treated with 200 μM OA for 12 h, then fixed and stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
HSchematic of the possible mechanism of ORF6 targeting to LDs.
To better understand the mechanism responsible for LD‐targeting of ORF6, we identified the LD‐targeting determinants of ORF6. Using the HeliQuest tool (Gautier et al, 2008), two amphipathic helices of SARS‐CoV‐2 ORF6 were identified, with the first helix (AH1) ranging from residues 7 to 27, and the other (AH2) ranging from residues 29 to 44 (Fig 1F). To determine whether these two amphipathic helices are required for ORF6 binding to LDs, point mutations were introduced into the hydrophobic interfaces of each helix: AH1, L15Q and I18Q (ORF6LI‐QQ); AH2, L40Q and L44Q (ORF6LL‐QQ); and L15Q, I18Q, L40Q and L44Q (ORF64Q) to break their structures (Fig 1F). Mutants ORF6LI‐QQ and ORF6LL‐QQ were still localized to LDs, whereas ORF64Q (disruption of both AH1 and AH2) failed to associate with LDs (Figs 1G and EV1E), indicating that the binding of ORF6 to LDs is mediated by the two amphipathic helices. Together, these findings suggest that SARS‐CoV‐2 ORF6 is directly inserted into the LD lipid monolayer via its two amphipathic helices (Fig 1H).
ORF6 homodimerization is important for LD binding
We noticed that ORF6 showed self‐binding in a co‐immunoprecipitation (co‐IP) assay (Fig 2A). In the presence of the cross‐linker disuccinimidyl suberate (DSS), we further confirmed that ORF6 exists in both monomeric and dimeric states (Fig 2B). OA treatment had no effect on the self‐binding of ORF6 (Fig 2C). Next, we determined whether the homodimerization of ORF6 could be important for LD association. We generated several truncations and analyzed their dimerization. Deletion of AH1 or AH2 significantly decreased the self‐interaction (Fig 2D and E). ORF6▵AH1 and ORF6▵AH2 failed to dimerize via the DSS assay (Fig 2F). Intriguingly, ORF6▵AH1 and ORF6▵AH2 remained diffused and failed to associate with LDs (Figs 2G and EV1E), suggesting that ORF6 may be targeted to LDs via amphipathic helices mediated‐homodimerization. Furthermore, using AH1‐GFP, AH2‐GFP and AH1 + AH2‐GFP, we examined the ability of individual AHs to interact with LDs and dimerize. Our data indicate that AH1‐GFP and AH2‐GFP failed to dimerize or localize on LDs while AH1 + AH2‐GFP can form dimerization and target to LDs (Appendix Fig S1A and B). We have shown that ORF64Q (disruption of both AH1 and AH2) failed to associate with LDs (Figs 1G and EV1E). Remarkably, ORF64Q showed less self‐interaction in a co‐IP assay and failed to dimerize (Fig 2H and I), while ORF6LI‐QQ and ORF6LL‐QQ still dimerize (Fig EV1F). Thus, our data indicate that ORF6 homodimerization is important for its association with LD.
Figure 2. ORF6 homodimerization is important for LD targeting.
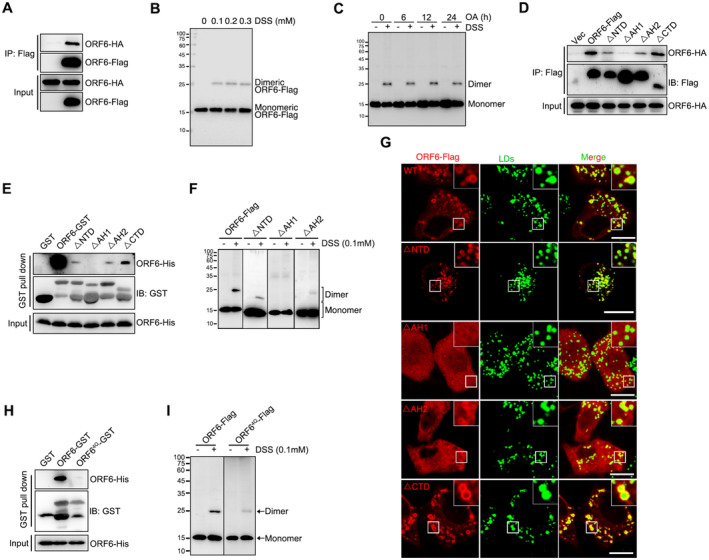
- ORF6‐Flag was co‐expressed in HEK293T cells with ORF6‐HA. Protein interactions were analyzed by immunoprecipitation with anti‐Flag beads and immunoblotting analysis.
- Analysis of the homodimerization of ORF6 by cross‐linking with DSS. HEK293T cells expressing ORF6‐Flag were treated with 0, 0.1, 0.2, and 0.3 mM DSS for 30 min. Cell lysates were analyzed via WB.
- HEK293T cells were transfected with ORF6‐Flag and were incubated with 200 μM OA for indicated times and treated or untreated with 0.1 mM DSS for 30 min. Cell lysates were analyzed via WB.
- The indicated Flag tagged ORF6 or its mutants were co‐expressed in HEK293T cells with HA tagged ORF6. Protein interactions were analyzed by immunoprecipitation with anti‐Flag beads and immunoblotting analysis.
- Purified GST‐ORF6 or indicated mutants were incubated with purified His tagged ORF6, and analysis of the self‐interaction by GST pull‐down.
- HEK293T cells were transfected with ORF6‐Flag or ΔNTD‐Flag or ΔAH1‐Flag or ΔAH2‐Flag and were treated with 0.1 mM DSS for 30 min. Cell lysates were analyzed via WB.
- HeLa cells were transfected with ORF6‐Flag or indicated mutants and were treated with 200 μM OA for 12 h, and then fixed and stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Purified GST‐ORF6 or indicated mutants were incubated with purified His tagged ORF6, and analysis of the self‐interaction by GST pull‐down.
- HEK293T cells were transfected with ORF6‐Flag or ORF64Q‐Flag (L15Q, I18Q, L40Q, L44Q) and were treated with 0.1 mM DSS for 30 min. Cell lysates were analyzed via WB.
Source data are available online for this figure.
ORF6 promotes LD biogenesis
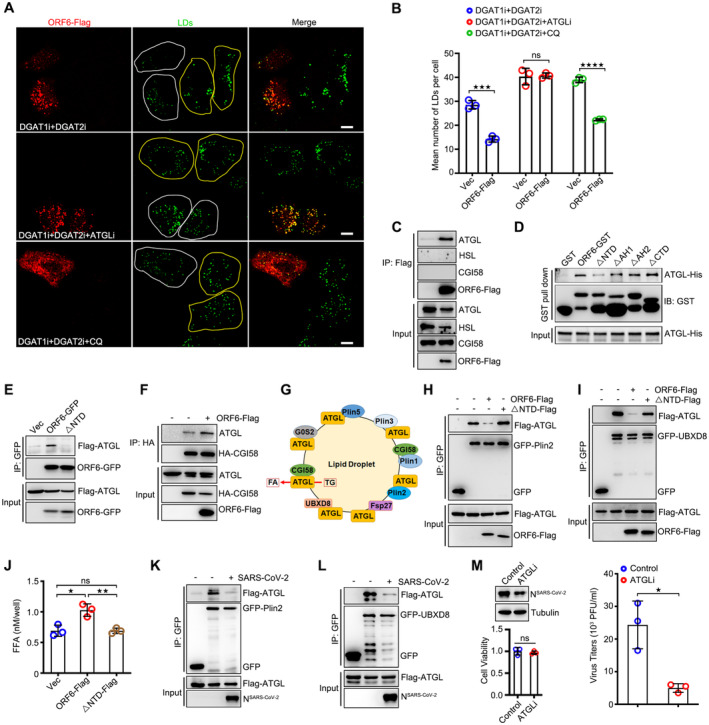
Next, we sought to determine the function of ORF6 in LDs. ORF6 interacts with Rae1 and Nup98 to prevent both nuclear import and export, blocking STAT nuclear import and antagonizing interferon signaling (Miorin et al, 2020; Addetia et al, 2021). Unfortunately, ORF64Q still inhibited IFN signaling (Fig EV1G), indicating that LD association is not required for ORF6 caused IFN inhibition. ORF6 transient expression led to an increase in the number of LDs in both control treated and OA treated wild type cells, whereas ORF64Q expression had no effect on LD abundance (Figs 3A and B, and EV2A). Higher TAG levels were also observed in ORF6 expressed‐cells than in the control cells (Fig 3C). ATGL inhibitor Atglistatin (ATGLi) was used to abolish lipolysis, and ORF6 expression resulted in dramatic LD expansion even in ATGLi‐treated cells (Fig 3D and E). These data indicated that ORF6 expression enhances LD biogenesis.
Figure 3. ORF6 promotes LD biogenesis by binding to DGAT1 and DGAT2.
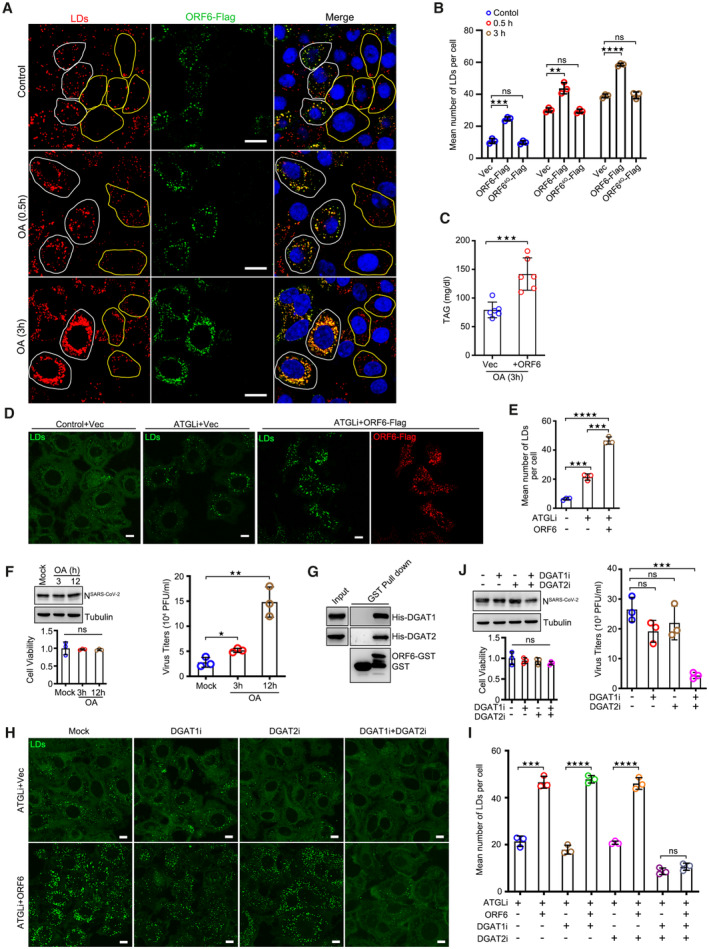
- HeLa cells were transfected with vector or ORF6‐Flag for 24 h, and then treated with 200 μM OA for indicated times. Cells were fixed and stained with anti‐Flag (green). LDs were labeled with LipidTox Deep Red (red). The nuclei were stained with DAPI. Cells were imaged by confocal microscopy. Scale bar represents 10 μm. White ROIs indicate cells expressing ORF6 and yellow ROIs indicate the cells without ORF6 expression.
- Mean number of LDs in each cell in (A) was counted from 25 cells of three independent experiments. Two‐tailed Unpaired Student's t‐test, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns means no significance. Error bars represent the mean ± SD.
- HeLa cells stable expressing the ORF6‐Flag were treated with 200 μM OA for 3 h. The concentration of TAG in cells was analyzed. Error bars, mean ± SD of six independent experiments. Two‐tailed Unpaired Student's t‐test, ***P < 0.001.
- HeLa cells stable expressing vector or ORF6‐Flag were treated or untreated with 50 μM ATGL inhibitor (ATGLi) for 24 h, and then were fixed and stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Mean number of LDs in each cell in (D) was counted from 25 cells of three independent experiments. Two‐tailed Unpaired Student's t‐test, ***P < 0.001, ****P < 0.0001. Error bars represent the mean ± SD.
- SARS‐CoV‐2‐infected Vero‐E6 cells were incubated with 200 μM OA for indicated times. Cell viability was analyzed. Cell lysates were analyzed via WB. Supernatants were determined by plaque assays. Virus titer values represent mean ± SD for three independent experiments. Two‐tailed Unpaired Student's t‐test, *P < 0.05, **P < 0.01, ns means no significance. Error bars represent as the mean ± SD.
- Purified GST‐ORF6 was incubated with His‐DGAT1 or His‐DGAT2, and analysis the interactions by GST pull‐down.
- HeLa cells stable expressing vector or ORF6‐Flag were treated or untreated with 50 μM ATGL inhibitor, along with DMSO or 1 μM DGAT1 inhibitor (DGAT1i) or/and 2 μM DGAT2 inhibitor (DGAT2i) for 24 h. LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Mean number of LDs in each cell in (H) was counted from 35 cells of three independent experiments. Two‐tailed Unpaired Student's t‐test, ***P < 0.001, ****P < 0.0001, ns means no significance. Error bars represent the mean ± SD.
- SARS‐CoV‐2‐infected Vero‐E6 cells were treated with 1 μM DGAT1 inhibitor or/and 2 μM DGAT2 inhibitor for 24 h. Cell viability was analyzed. Cell lysates were analyzed via WB. Supernatants were determined by plaque assays. Virus titer values represent mean ± SD for three independent experiments. Two‐tailed Unpaired Student's t‐test, ***P < 0.001, ns means no significance. Error bars represent the mean ± SD.
Source data are available online for this figure.
Figure EV2. ORF6 promotes LD biogenesis.
-
ACells expressing the ORF64Q‐Flag were treated with 200 μM OA for indicated times, then fixed and stained with anti‐Flag (green). LDs were labeled with LipidTOX Deep Red (red). The nuclei of cells were stained with DAPI. Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
B, CThe interactions of GST tagged ORF6 or the mutants with the His tagged DGAT1 and His tagged DGAT2 were analyzed by GST pull‐down assay.
-
DVero‐E6 cells were infected or non‐infected with SARS‐CoV‐2 for 24 h. Cell lysates were analyzed via WB.
-
EHeLa cells were transfected with gradient of ORF6‐Flag for 24 h. Protein levels of endogenous DGAT1, DGAT2, and ATGL were analyzed via WB.
-
FHeLa cells were transfected with gradient of ORF6‐Flag for 24 h. RNA levels of dgat1 and dgat2 were analyzed via QPCR, three independent experiments. Two‐tailed Unpaired Student's t‐test, ns means no significance. Error bars represent the mean ± SD.
-
GGene Ontology (GO) enrichment analysis was performed on the human interacting proteins of SARS‐CoV‐2 ORF6 protein, P‐values calculated by hypergeometric test and a false discovery rate was used to account for multiple hypothesis testing. The top GO term was shown in the graph.
Source data are available online for this figure.
Consistent with a previous study showing that LDs are necessary for SARS‐CoV‐2 production (Dias et al, 2020), we found that LD biogenesis induced by OA treatment promoted SARS‐CoV‐2 production (Fig 3F). We then sought to determine the mechanism by which ORF6 increases LD abundance. ORF6 showed strong interactions with DGAT1 and DGAT2 in an in vitro GST pull‐down assay (Fig 3G). We then investigated whether ORF6 increases LD biogenesis dependent on DGAT1 and/or DGAT2. ORF6 overexpression has no effect on the association of DGAT1 or DGAT2 with LDs (Appendix Fig S2A and B). ORF6▵AH1 still binds DGAT1, but not DGAT2 (Fig EV2B and C). Cells were treated with ATGLi, further treated with DGAT1 and/or DGAT2 inhibitors and analyzed for LD biogenesis. Interestingly, inhibition of DGAT1 or DGAT2 had minor effects on ORF6‐induced LD abundance, and inhibition of both enzymes was required to strongly revise ORF6‐induced LD biogenesis (Fig 3H and I). Furthermore, inhibition of both DGAT1 and DGAT2 resulted in the reduction of SARS‐CoV‐2 production, whereas inhibition either DGAT1 or DGAT2 had minor effects (Fig 3J). Consistently, compared to non‐infection, SARS‐CoV‐2 infection induced higher protein levels of DGAT1 and DGAT2 (Fig EV2D), while ORF6 expression has no effect on the protein or RNA level of DGAT1 and DGAT2 (Fig EV2E and F), suggest that other viral proteins regulate DGAT1 and DGAT2. Taken together, these data suggest that ORF6 promotes LD biogenesis dependent on DGAT1 and DGAT2 during viral production.
ORF6 promotes LD biogenesis by linking the ER to LDs
LD biogenesis is initiated in the ER bilayer, where fatty acids and cholesterol are converted to TAGs and cholesterol esters. ER‐LD interaction is required for maturation of nascent LDs (Walther et al, 2017). Previous IP/MS data suggested that ORF6 interacts with ER membrane proteins (Fig EV2G; Gordon et al, 2020). Next, we sought to determine whether ORF6 could link the ER to LDs for LD biogenesis. As expected, significant amounts of ORF6‐GFP, but not GFP, were found in the ER fraction, which was marked by calnexin (Fig 4A). Consistently, we found that GFP‐labeled ORF6 decorated the contact between the ER and LD, and enhanced the extent of the ER‐LD interactions in an immune‐electron microscope assay (Fig 4B). Compare to mock cells, more ER‐LD junctions were observed in SARS‐CoV‐2‐infected cells (Fig 4C).
Figure 4. ORF6 links LDs to the ER by binding to BAP31 and USE1.
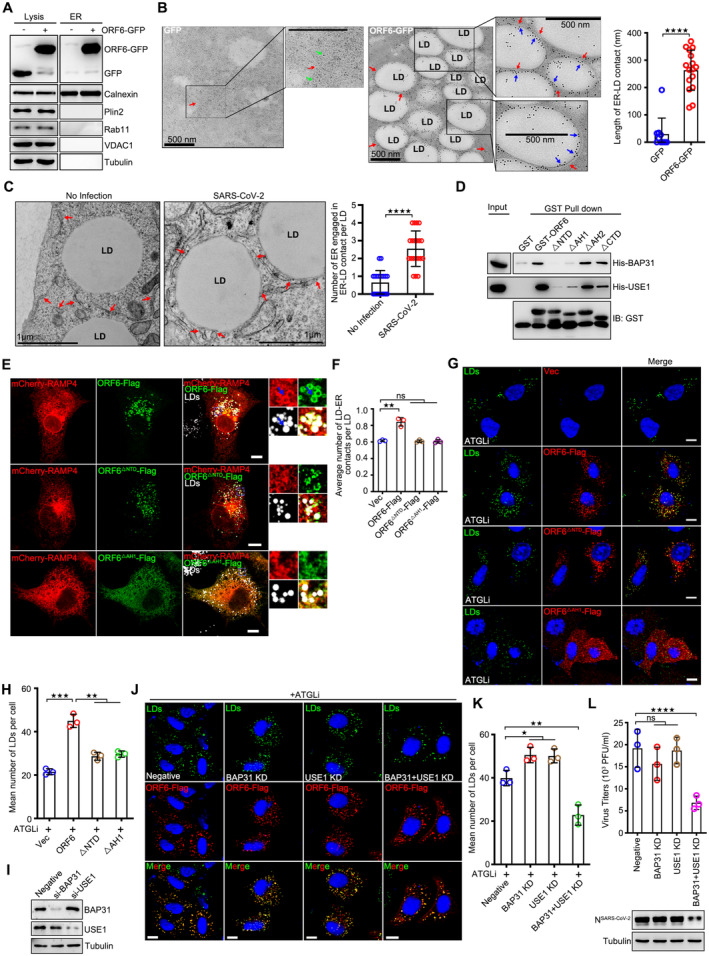
- Subcellular fractions were isolated from GFP or ORF6‐GFP over‐expressed cells. Calnexin represent the ER, Plin2 represent LDs, Rab11 represent endosomes, VDAC represent mitochondria, and Tubulin represents cytoplasm, respectively.
- Immunogold electron micrograph of GFP or ORF6‐GFP expressed HeLa cells. Green arrows mark GFP dots, blue arrows mark ORF6‐GFP dots, red arrows mark ER. Quantification of length of ER‐LD contact via ImageJ. 10 ER‐LD contacts in GFP expressed cells and 16 ER‐LD contacts in ORF6‐GFP expressed cells of two independent experiments were calculated via ImageJ. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001. Error bars represent the mean ± SD.
- Representative transmission electron micrograph of non‐infected or SARS‐CoV‐2 infected Vero‐E6 cells. Red arrows mark the ER. LD, lipid droplets. Scale bar represents 1 μm. Quantification of number of ER engaged in ER‐LD contact per LD. 20 LDs of two independent experiments were counted. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001. Error bars represent as the mean ± SD.
- The interactions of GST tagged ORF6 or the mutants with the His tagged BAP31 and His tagged USE1 were analyzed by GST pull‐down assay.
- ORF6‐Flag or the mutants were co‐expressed with mCheey‐RAMP4. Cells were treated with 200 μM OA for 12 h, and then fixed and stained with anti‐Flag (red). The ER was visualized with mCheey‐RAMP4 (red). LDs were labeled with LipidTOX Deep Red (white). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Quantification of average number of LD‐ER contacts per LD from (E). 25 cells from three independent experiments were calculated. Two‐tailed Unpaired Student's t‐test, **P < 0.01, ns means no significance. Error bars represent the mean ± SD.
- HeLa cells were transfected with ORF6‐Flag or the mutants for 12 h, and were treated with 50 μM ATGL inhibitor for 24 h. Cells were fixed and stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). The nuclei were stained with DAPI. Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Mean number of LDs in each cell in (G) was counted from 25 cells of three independent experiments. Two‐tailed Unpaired Student's t‐test, **P < 0.01, ***P < 0.001. Error bars represent the mean ± SD.
- HeLa cells were transfected with control, or si‐BAP31, or si‐USE1 for 48 h. Cell lysates were analyzed via WB.
- HeLa cells stable expressing ORF6‐Flag were transfected with si‐BAP31 and/or si‐USE1 for 24 h, and were further treated with 50 μM ATGL inhibitor for 24 h. Cells were fixed and stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). The nuclei were stained with DAPI. Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Mean number of LDs in each cell in (J) was counted from 25 cells of three independent experiments. Two‐tailed Unpaired Student's t‐test, *P < 0.05, **P < 0.01. Error bars represent the mean ± SD.
- SARS‐CoV‐2‐infected Vero‐E6 cells were transfected with control, or si‐BAP31 and/or si‐USE1 for 48 h. Cell lysates were analyzed via WB. Supernatants were determined by plaque assays. Virus titer values represent mean ± SD for three independent replicates. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001, ns means no significance. Error bars represent the mean ± SD.
Source data are available online for this figure.
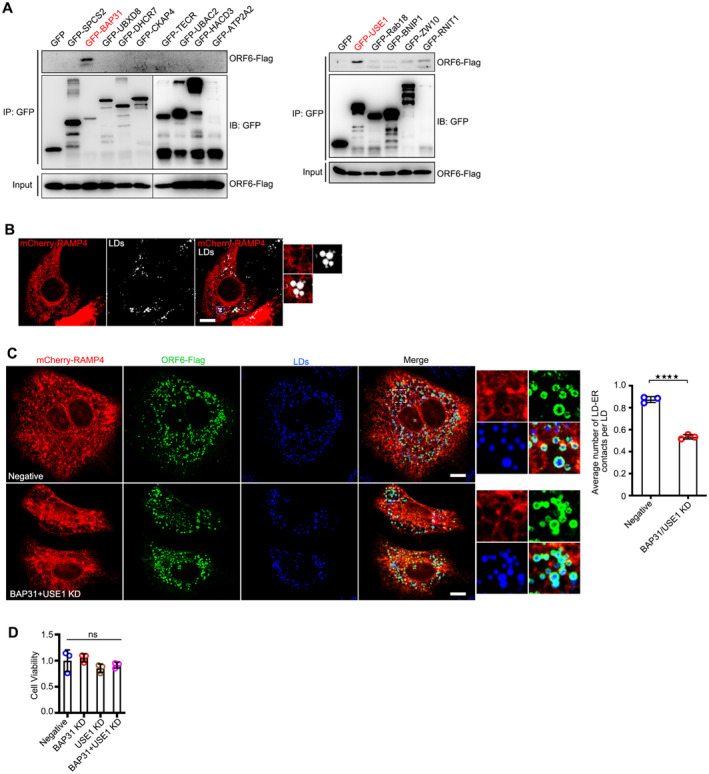
We then examined the interaction between ORF6 and ER membrane proteins using the IP/MS list (Gordon et al, 2020). Two positive candidates were confirmed by in vivo co‐IP and in vitro GST pull‐down: B‐cell receptor‐associated protein 31 (BAP31) and unconventional SNARE in the ER 1 (USE1; Figs EV3A and 4D). The colocalization between ORF6 and BAP31 or USE1 was confirmed by confocal microscopy (Appendix Fig S2C). BAP31 is a transmembrane protein found mainly in the ER, including in mitochondria‐associated membranes (MAMs; Quistgaard, 2021). USE1 forms ER‐associated Q‐SNARE, including Syntaxin 18, USE1, and BNIP1, which bind to LD‐associated Rab18 to mediate ER‐LD contact (Xu et al, 2018). Next, we sought to determine whether ORF6 interacts with BAP31 and/or USE1 to mediate the formation of direct ER‐LD contacts for LD expansion. Depletion of NTD (ORF6▵NTD) significantly abolished the interactions between ORF6‐BAP31 or ORF6‐USE1 (Fig 4D). Using confocal microcopy, we detected obvious colocalization between ER, LDs, and ORF6 (Fig 4E and F). Compare to vector cells, ORF6 expression promoted the formation of direct ER‐LD contacts, but not deleted mutants ORF6▵NTD and ORF6▵AH1 (Figs 4E and F, and EV3B), indicating that the interactions between ORF6‐BAP31 and/or ORF6‐USE1 were important for the formation of direct ER‐LD contact. We further found that in lipolysis‐inhibited cells, only wild‐type ORF6 expression resulted in LD expansion, but not ORF6▵NTD or ORF6▵AH1 (Fig 4G and H). We then investigated whether BAP31 and/or USE1 were required for ORF6‐induced LD biogenesis. siRNAs were used to knockdown BAP31 and USE1 (Fig 4I). Depletion of both BAP31 and USE1 abolished ORF6‐induced ER‐LD junctions (Fig EV3C) and LD biogenesis (Fig 4J and K), whereas knockdown of BAP31 or USE1 had a minor effect (Fig 4J and K). These data indicated that ORF6 directly interacts with BAP31 and USE1 to mediate ER‐LD interactions during LD biogenesis. Moreover, double knockdown of BAP31 and USE1 resulted in a reduction of SARS‐CoV‐2 production while knockdown of BAP31 or USE1 had a minor effect (Figs 4L and EV3D). Together, these data suggest that ORF6 interacts with BAP31 and USE1 to induce LD biogenesis, which is important for viral production.
Figure EV3. ORF6 interacts with BAP31 and USE1.
-
AGFP or GFP tagged ER membrane proteins were co‐expressed in HEK293T cells with ORF6‐Flag. Protein interactions were analyzed by immunoprecipitation with anti‐GFP beads and immunoblotting analysis.
-
BVector were co‐expressed with mCheey‐RAMP4. Cells were treated with 200 μM OA for 12 h, and then fixed. The ER was visualized with mCheey‐RAMP4 (red). LDs were labeled with LipidTOX Deep Red (white). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
CCells co‐expressing the ORF6‐Flag with mCheery tagged RAMP4 were transfected with BAP31 or USE1 siRNA for 36 h and treated with 200 μM OA for 12 h, then fixed and stained with anti‐Flag (green). LDs were labeled with LipidTOX Deep Red (blue). Cells were imaged by confocal microscopy. Scale bar represents 10 μm. Quantification of average number of LD‐ER contacts per LD. 12 cells (Negative) and 25 cells (BAP31/USE1 KD) from three independent experiments were calculated. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001, ns means no significance. Error bars represent the mean ± SD.
-
DCell viability was analyzed in Fig 4L, three independent experiments. Two‐tailed Unpaired Student's t‐test, ns means no significance. Error bars represent the mean ± SD.
Source data are available online for this figure.
ORF6 promotes lipolysis through binding to ATGL
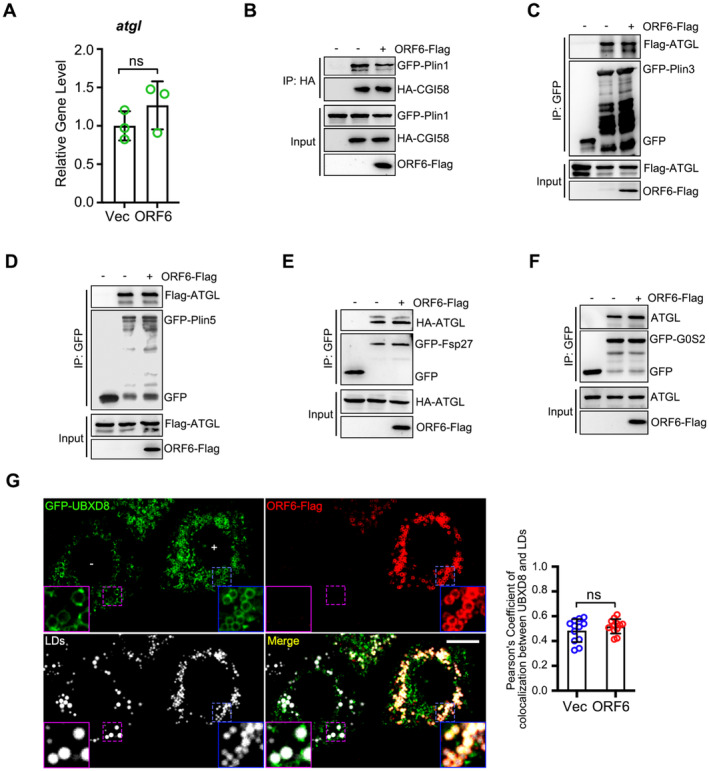
FFAs can be released from LDs through two distinct mechanisms: lipolysis and lipophagy. To determine which of these two mechanisms is responsible for FA delivery to mitochondria under ORF6 expression, cells were pre‐treated with DGAT1 and DGAT2 inhibitors to suppress LD biogenesis, and further treated with an ATGL inhibitor or chloroquine (CQ, which blocks autophagic degradation), examining the LD number in ORF6 expressing cells. We found that with DGAT1 and DGAT2 inhibitor treatment, ORF6 expression resulted in the reduction of LD number, which can be revised by ATGL inhibitor treatment but not CQ (Fig 5A and B), indicating that ORF6 expression positively regulates lipolysis. Next, we determined the mechanism by which ORF6 regulates lipolysis. We first examined the possibility that ORF6 regulates lipolysis by interacting with the lipolysis machinery. To this end, ORF6‐Flag was expressed in HEK293T cells and protein interactions were examined by co‐IP. We found that ORF6 specifically interacted with endogenous ATGL, but not with HSL or CGI58 (Fig 5C). The direct interaction between ORF6 and ATGL was further confirmed using an in vitro GST pull‐down assay (Fig 5D). ORF6▵NTD failed to interact with ATGL in vitro and in vivo (Fig 5D and E), suggesting that ORF6 binds to ATGL via its NTD. Next, we determined whether ORF6 regulates lipolysis by interacting with ATGL. ORF6 expression had no effect on the protein or mRNA levels of ATGL (Figs EV2E and EV4A). Interestingly, ORF6 expression enhanced the interaction between ATGL and its coactivator, CGI58 (Fig 5F). Previous studies have shown that perilipin proteins (Plin2, Plin3, and Plin5), Fsp27, UBXD8, and G0/G1 switch gene 2 (G0S2) bind to ATGL on LDs and negatively regulate ATGL activity by inhibiting the ATGL‐CGI58 interaction (Fig 5G; Yang et al, 2010; Olzmann et al, 2013; Cerk et al, 2018). Remarkably, using co‐IP assays, we observed that ORF6 expression, but not ORF6▵NTD, significantly abrogated the associations of ATGL with Plin2 and UBXD8 (Fig 5H and I). In addition, ORF6 expression had minor effects on the interactions between CGI58‐Plin1, ATGL‐Plin3, ATGL‐Plin5, ATGL‐Fsp27, or ATGL‐G0S2 (Fig EV4B–F). ORF6 expression had a minor effect on LD targeting of UBXD8 (Fig EV4G) and ATGL (Appendix Fig S3A). Consistently, compared to vector cells, wild‐type ORF6 expression resulted in an increase of FFAs, whereas ORF6▵NTD expression had no effect on FFAs production (Fig 5J). Together, these observations demonstrate that ORF6 directly interacts with ATGL to enhance cellular lipolysis by disrupting the interactions of ATGL‐Plin2 and ATGL‐UBXD8, promoting the association of ATGL with CGI58.
Figure 5. ORF6 enhances lipolysis by interacting with ATGL.
-
AHeLa cells were transfected with vector or ORF6‐Flag for 12 h and treated with 200 μM OA for another 12 h, the medium was removed and replaced with fresh complete culture containing 1 μM DGAT1 inhibitor and 2 μM DGAT2 inhibitor for 24 h. Meanwhile, 50 μM ATGL inhibitor or 100 μM Chloroquine (CQ) was added and allowed to incubate for 24 or 4 h to block lipolysis or lipophagy, respectively. Cells were fixed and stained with anti‐Flag (red). LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm. White ROIs indicate cells expressing ORF6 and yellow ROIs indicate the cells without ORF6 expression.
-
BMean number of LDs in each cell in (A) was counted from 25 cells of three independent experiments. Two‐tailed Unpaired Student's t‐test, ***P < 0.001, ****P < 0.0001, ns means no significance. Error bars represent the mean ± SD.
-
CHEK293T cells were transfected with ORF6‐Flag. Protein interactions were detected by immunoprecipitation with anti‐Flag beads and immunoblotting analysis with ATGL, HSL, and CGI58 antibodies.
-
DPurified GST‐ORF6 or indicated mutants were incubated with purified His tagged ATGL, and analyzed the interactions by GST pull‐down.
-
EThe interactions of Flag‐ATGL with ORF6‐GFP or the ΔNTD mutant were analyzed by immunoprecipitation.
-
FThe effect of ORF6‐Flag expression on the interaction of HA‐CGI58 with ATGL was analyzed by immunoprecipitation.
-
GModel for the regulation of lipolysis by interacting with ATGL.
-
H, IThe effect of ORF6‐Flag or the ΔNTD mutant on the interactions of Flag‐ATGL with GFP‐Plin2 or GFP‐UBXD8 was analyzed by immunoprecipitation.
-
JHeLa cells were transfected with ORF6‐Flag or the ΔNTD mutant for 6 h and were treated with 200 μM OA for 24 h. The medium was removed and replaced with serum free DMEM for another 4 h. The concentration of FFA in cells was analyzed. Error bars, mean ± SD of three independent experiments. Two‐tailed Unpaired Student's t‐test, *P < 0.05, **P < 0.01, ns means no significance.
-
K, LVero‐E6 cells were transfected with Flag‐ATGL and GFP‐Plin2 or GFP‐UBXD8 for 24 h and infected or non‐infected with SARS‐CoV‐2 for 24 h. Protein interactions were analyzed by immunoprecipitation with anti‐GFP beads and immunoblotting analysis.
-
MSARS‐CoV‐2‐infected Vero‐E6 cells were treated with 50 μM ATGL inhibitor for 24 h. Cell viability was analyzed. Cell lysates were analyzed via WB. Supernatants were determined by plaque assays. Virus titer values represent mean ± SD for three independent replicates. Two‐tailed Unpaired Student's t‐test, *P < 0.05, ns means no significance. Error bars represent as the mean ± SD.
Source data are available online for this figure.
Figure EV4. ORF6 enhances lipolysis.
-
AQPCR analysis of RNA level of atgl in vector and ORF6‐Flag expressed cells, three independent experiments. Two‐tailed Unpaired Student's t‐test, ns means no significance. Error bars represent the mean ± SD.
-
B–FThe effect of ORF6‐Flag on the interactions of GFP‐Plin1 with HA‐CGI58, or GFP‐Plin3 with Flag‐ATGL, or GFP‐Plin5 with Flag‐ATGL, or GFP‐Fsp27 with HA‐ATGL, or G0S2‐GFP with ATGL were analyzed by immunoprecipitation.
-
GHeLa cells were transfected with GFP‐UBXD8 and vector or ORF6‐Flag for 12 h and treated with 200 μM OA for 12 h, then fixed and stained with anti‐Flag (red). LDs were labeled with LipidTOX Deep Red (white). Cells were imaged by confocal microscopy. Scale bar represents 10 μm. Blue ROIs indicate cells expressing ORF6 and purple ROIs indicate the cells without ORF6 expression. Colocalization of UBXD8 and LDs (Pearson's Coefficient), n = 12 cells, two independent experiments. Two‐tailed Unpaired Student's t‐test, ns means no significance. Error bars represent the mean ± SD.
Source data are available online for this figure.
Furthermore, compared to non‐infection, SARS‐CoV‐2 infection subverted the interaction of ATGL with Plin2 and UBXD8 (Fig 5K and L). Higher protein level of ATGL was also observed in SARS‐CoV‐2‐infected cells than non‐infected cells (Fig EV2D). Moreover, ATGL inhibitor treatment resulted in a reduction of SARS‐CoV‐2 production (Fig 5M). These data suggested that SARS‐CoV‐2 up‐regulates ATGL expression and enhances the association of ATGL with CGI58 to promote cellular lipolysis, which is important for viral production.
ORF6 links mitochondria to LDs
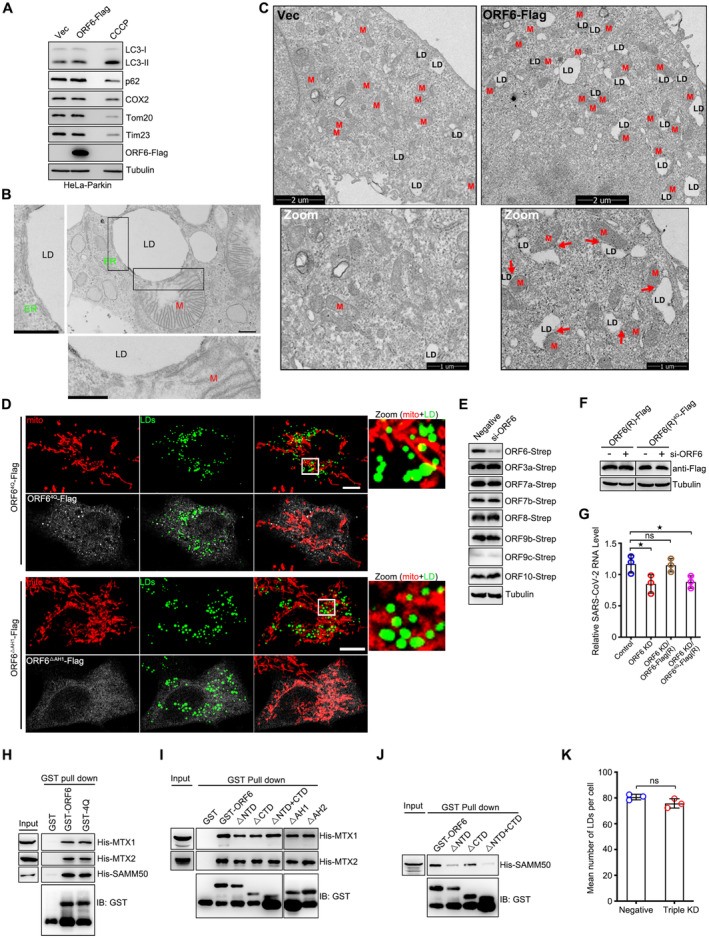
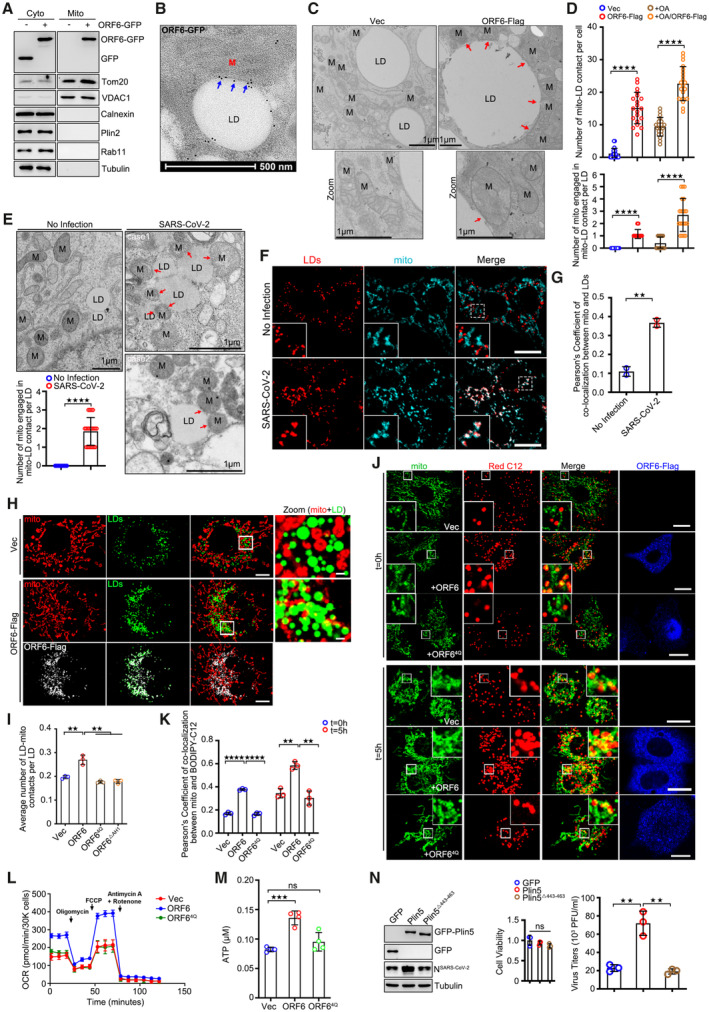
ORF6 was also found to interact with mitochondrial membrane proteins (Fig EV2G; Gordon et al, 2020). ORF6 expression had no effect on mitophagy (Fig EV5A). Intriguingly, we found significant amounts of ORF6‐GFP, but not GFP, floating to the mitochondria fraction, marked by Tom20, VDAC1, and ATP5A1 (Fig 6A). Moreover, using immune electron microscopy, we found that GFP‐labeled ORF6 decorated the contact between mitochondria and LD (Fig 6B). The presence of ORF6 at mitochondria‐LD junctions led us to hypothesize that ORF6 may mediate mitochondria‐LD interactions in SARS‐CoV‐2‐infected cells. Therefore, we expressed ORF6‐Flag in HeLa cells, expanded the LDs with OA treatment, and examined the relationship between mitochondria and LDs using electron microscopy. Consistent with our hypothesis, we found that untransfected cells showed only minimal contact between the two organelles, but ORF6‐Flag expression increased the extent of mitochondria‐LD interactions (Figs 6C and D, and EV5B). Similar results were obtained from ORF6‐Flag‐expressed HeLa cells without OA treatment (Figs 6D and EV5C). In addition, native SARS‐CoV‐2 infection increased the extent of mitochondrial‐LD interactions (Fig 6E–G). We further confirmed the role of ORF6 in the maintenance of mitochondria‐LD junctions using confocal microscopy (Fig 6H and I). ORF64Q or ORF6▵AH1 (failed to associate with LDs)‐expressing cells showed decreased mitochondria‐LD contacts (Figs EV5D and 6I). Together, these findings indicate that SARS‐CoV‐2 ORF6 targets LDs and mediates mitochondrial‐LD interactions.
Figure EV5. ORF6 links LDs to mitochondria.
-
AHeLa‐Parkin cells were transfected with vector or ORF6‐Flag for 24 h, or treated with 10 μM CCCP for 6 h. Cell lysates were analyzed via WB.
-
BRepresentative transmission electron micrograph showing the close contact between LD, ER and mitochondria in ORF6 expressed HeLa cells treated with 200 μM OA for 12 h. LD, lipid droplets. ER, endoplasmic reticulum. M, mitochondria. Scale bars 500 nm.
-
CRepresentative transmission electron micrograph of ORF6‐Flag stable expressing or wild‐type HeLa cells. Red arrows mark the contact sites between LDs and mitochondria. LD, lipid droplets. M, mitochondria.
-
DCos7 cells expressing ORF64Q‐Flag or ORF6▵AH1‐Flag were treated with 200 μM OA for 12 h, and then were fixed and stained with anti‐Flag (white) and anti‐Tom20 (red). Tom20 represents mitochondria marker. LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
ECells were transfected with Negative or si‐ORF6 for 24 h, and then further transfected with ORF6‐Strep, or ORF3a‐Strep, or ORF7a‐Strep, or ORF7b‐Strep, or ORF8‐Strep, or ORF9b‐Strep, or ORF9c‐Strep, or ORF10‐Strep for 24 h. Cell lysates were analyzed via WB.
-
FCells were transfected with Negative or si‐ORF6 for 24 h, and then further transfected with ORF6‐Flag (RNAi resistant), or ORF64Q‐Flag (RNAi resistant) for 24 h. Cell lysates were analyzed via WB.
-
GVero‐E6 cells were transfected with Negative or si‐ORF6 for 24 h, and then further transfected with ORF6‐Flag (RNAi resistant), or ORF64Q‐Flag (RNAi resistant) for 12 h. Cells were further infected with SARS‐CoV‐2 for 24 h. Viral RNA level was determined by RT‐qPCR, three independent replicates. Two‐tailed Unpaired Student's t‐test, *P < 0.05. ns means no significance. Error bars represent the mean ± SD.
-
H–JPurified GST protein or GST‐ORF6 or its mutants were incubated with purified His tagged MTX1, or MTX2, or SAMM50, and analyzed the interactions by GST pull‐down.
-
KQuantification of LD number in (Fig 7F). 25 cells (Negative) and 50 cells (Triple KD) from three independent experiments were calculated. Two‐tailed Unpaired Student's t‐test, ns means no significance. Error bars represent the mean ± SD.
Source data are available online for this figure.
Figure 6. ORF6 links LDs to mitochondria.
-
ASubcellular fractions were isolated from GFP or ORF6‐GFP over‐expressed cells. Tom20 and VDAC1 represent mitochondria markers, Calnexin represents the ER, Plin2 represents LDs, Rab11 represents endosomes, and Tubulin represents cytoplasm, respectively.
-
BRepresentative immunogold electron micrograph of ORF6‐GFP transfected HeLa cell showing ORF6‐GFP dots (blue arrows) are enriched at mitochondria‐LD contact sites. M, mitochondria. LD, lipid droplets. Scale bar represents 500 nm.
-
CRepresentative transmission electron micrograph of ORF6‐Flag stable expressing or vector HeLa cells upon OA treatment. Red arrows mark the contact sites between LDs and mitochondria. M, mitochondria. LD, lipid droplets. Scale bar represents 1 μm.
-
DThe number of mitochondria‐LD contacts per cell in (C and Fig EV5C) was counted from 20 cells. The number of mitochondrial engaged in mitochondrial‐LD contact per LD in (C) was counted from 20 LDs. Two independent experiments. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001. Error bars represent the mean ± SD.
-
ERepresentative transmission electron micrograph of non‐infected or SARS‐CoV‐2 infected Vero‐E6 cells. Red arrows mark the mitochondria‐LD contact. LD, lipid droplets. M, mitochondria. Scale bar represents 1 μm. Quantification of number of mitochondria engaged in mitochondria‐LD contact per LD. 20 LDs of two independent experiments were counted. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001. Error bars represent the mean ± SD.
-
F, GSARS‐CoV‐2‐infected Vero‐E6 cells were fixed and stained with anti‐Tom20 (cyan). Tom20 represents mitochondria marker. LDs were labeled with LipidTOX Deep Red (red). Cells were imaged by confocal microscopy. Scale bar represents 10 μm. Colocalization of LDs and mitochondria (Pearson's Coefficient), n = 20 cells, two independent experiments. Two‐tailed Unpaired Student's t‐test, **P < 0.01. Error bars represent the mean ± SD.
-
HCos7 cells expressing ORF6‐Flag and vector were treated with 200 μM OA for 12 h, and then were fixed and stained with anti‐Flag (white) and anti‐Tom20 (red). Tom20 represents mitochondria marker. LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
IQuantification of average number of LD‐mitochondria contacts per LD from (H and Fig EV5D). 37 cells (Vector), 50 cells (ORF6), 50 cells (ORF64Q), and 50 cells (ORF6▵AH1) from three independent experiments were calculated. Two‐tailed Unpaired Student's t‐test, **P < 0.01. Error bars represent the mean ± SD.
-
JPulse‐chase assays of vector or ORF6‐Flag or ORF64Q‐Flag stable cells with or without starvation treatment. HeLa cells were incubated in complete medium containing 1 μM BODIPY 558/568 C12 (red) for 16 h. Cells were washed with PBS three times and then chased or non‐chased in EBSS for 5 h, and then were fixed and stained with anti‐Flag (blue) and anti‐Tom20 (green). Tom20 represents mitochondria marker. Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
-
KColocalization of BODIPY‐C12 and mitochondria (Pearson's Coefficient), n = 20 cells, three independent experiments. Two‐tailed Unpaired Student's t‐test, **P < 0.01, ****P < 0.0001. Error bars represent the mean ± SD.
-
LSeahorse FAO assays to examine the oxidation of endogenous FAs in vector, or ORF6‐Flag or ORF64Q‐Flag over‐expressed HeLa cells. Three biological replicates were performed with similar results. Error bars represent the mean ± SD.
-
MThe concentration of ATP in cells was analyzed according to manufacturer's instructions. Error bars, mean ± SD of four independent experiments. Two‐tailed Unpaired Student's t‐test, ***P < 0.001, ns means no significance.
-
NVero‐E6 cells stable expressing GFP or GFP‐Plin5 or GFP‐Plin5Δ443–463 were infected with SARS‐CoV‐2 for 24 h. Cell viability was analyzed. Cell lysates were analyzed via WB. Supernatants were determined by plaque assays. Virus titer values represent mean ± SD for three independent replicates. Two‐tailed Unpaired Student's t‐test, **P < 0.01, ns means no significance. Error bars represent the mean ± SD.
Source data are available online for this figure.
FAs transfer from LDs to mitochondria through mitochondria‐LD contacts. We next explored whether mitochondria‐LD interactions mediated by SARS‐CoV‐2 ORF6 play a role in the transfer of FAs. We adopted a pulse‐chase assay using the fluorescent FA analog BODIPY 558/568‐C12 (Red C12; Rambold et al, 2015). Red C12 accumulated in LDs (t = 0 h) and was gradually transferred to mitochondria when cells were further cultured in nutrient‐deleted medium in the absence of Red C12 for 5 h (t = 5 h). Compared to vector‐transfected cells or ORF64Q‐expressed cells, the transfer rate of Red C12 from LDs to mitochondria was significantly higher in ORF6‐Flag expressing cells (Fig 6J and K). FAs traffic from LDs to mitochondria for β‐oxidation and ATP production. Using seahorse FA oxidation assays and ATP kits, we further found that ORF6‐Flag expression, but not ORF64Q‐Flag, resulted in higher β‐oxidation and ATP levels than those in the control (Fig 6L and M). Together, these data suggest that SARS‐CoV‐2 infection and ORF6 expression enhance β‐oxidation level by increasing the mitochondrial‐LD interactions for ATP production.
Plin5 tethers LDs and mitochondria, while truncation lacking the C‐terminal mitochondrial recruiting sequence (Plin5▵443–463) failed to recruit mitochondria to LDs (Benador et al, 2018). Plin5‐overexpressing Vero‐E6 cells exhibited higher SARS‐CoV‐2 production, whereas Plin5▵443–463 overexpression had no effect on viral production (Fig 6N). In addition, we further used siRNA to knockdown ORF6 in SARS‐CoV‐2 infected cells, and rescued wild‐type ORF6 or ORF64Q by using the siRNA resistant constructs (Fig EV5E and F). Only wild‐type could rescue the reduction of viral production in ORF6 KD cells, but not ORF64Q (Fig EV5G). Together, these data indicate that mitochondrial‐LD interactions are important for SARS‐CoV‐2 production.
ORF6 targets mitochondria by interacting with SAM complex
Next, we sought to determine the mechanism(s) by which ORF6 targets mitochondria. ORF6 interacts with mitochondrial membrane proteins (Gordon et al, 2020). Hence, we sought to determine whether ORF6 targets mitochondria by interacting with these membrane proteins. First, we examined the interactions between ORF6 and outer mitochondrial membrane (OMM) proteins from the IP/MS list (Gordon et al, 2020). Metaxin‐2 (MTX2) and SAMM50 interacted with ORF6 in the co‐IP assay (Fig 7A). Metaxin‐1 (MTX1), MTX2, and SAMM50 are core subunits of the sorting and assembly machinery (SAM) complex of the outer mitochondrial membrane (Höhr et al, 2015; Wiedemann & Pfanner, 2017). Using in vitro GST pull‐down assay, we found that ORF6 directly interacted with MTX1, MTX2, and SAMM50 (Fig 7B). The colocalizations between ORF6 and the SAM complex were confirmed by confocal microcopy (Appendix Fig S3B). ORF64Q still interacts with the SAM complex (Fig EV5H), suggesting that LD localization is not required for its interaction with the SAM complex. In addition, deletion of NTD (ORF6▵NTD), AH1 (ORF6▵AH1), AH2 (ORF6▵AH2), CTD (ORF6▵CTD), and NTD plus CTD (ORF6▵NTD+CTD) still interacted with MTX1 and MTX2 (Fig EV5I), indicating that ORF6 binds to MTX1 and MTX2 via its two amphipathic helices. Deletion of NTD (ORF6▵NTD) failed to interact with SAMM50 (Fig EV5J), suggesting that the NTD of ORF6 is required for its interaction with SAMM50.
Figure 7. ORF6 interacts with SAM complex.
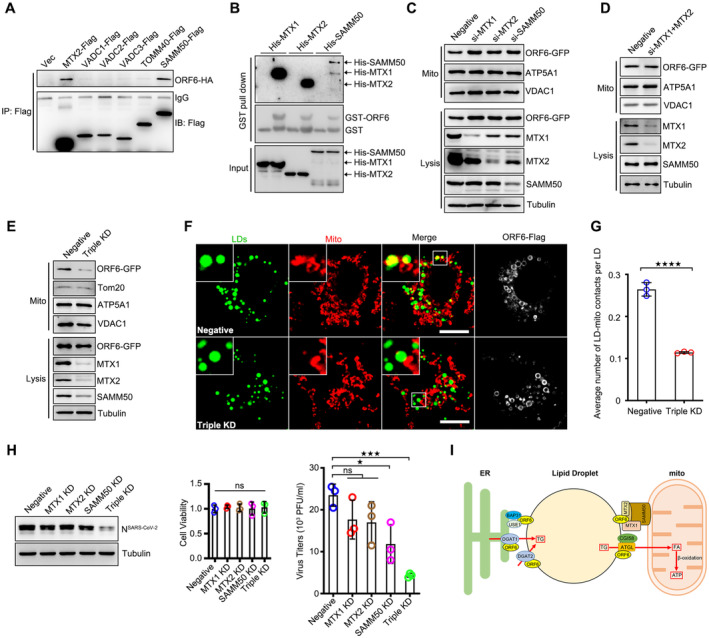
- The Flag tagged mitochondrial outer membrane proteins were co‐expressed in HEK293T cells with ORF6‐HA. Protein interactions were analyzed by immunoprecipitation with anti‐Flag beads and immunoblotting analysis.
- Purified GST protein or GST‐ORF6 were incubated with purified His tagged MTX1, MTX2, or SAMM50, and analyzed the interactions by GST pull‐down.
- HeLa cells were transfected with negative, or si‐MTX1, or si‐MTX2, or si‐SAMM50 for 24 h and further transfected with ORF6‐GFP 24 h. Subcellular fractions were isolated from cells. VDAC1 and ATP5A1 represent mitochondria markers, Tubulin represents cytosolic marker.
- HeLa cells were transfected with negative or si‐MTX1 and si‐MTX2 for 24 h and further transfected with ORF6‐GFP 24 h. Subcellular fractions were isolated from cells. VDAC1 and ATP5A1 represent mitochondria markers, Tubulin represents cytosolic marker.
- HeLa cells were transfected with negative or si‐MTX1/si‐MTX2/si‐SAMM50 (Triple KD) for 24 h and further transfected with ORF6‐GFP 24 h. Subcellular fractions were isolated from cells. VDAC1 and ATP5A1 represent mitochondria markers, Tubulin represents cytosolic marker.
- Cos7 cells expressing ORF6‐Flag were transfected with negative or si‐MTX1/si‐MTX2/si‐SAMM50 (Triple KD) for 36 h and treated with 200 μM OA for another 12 h. Cells were fixed and stained with anti‐Flag (white) and anti‐Tom20 (red). Tom20 represents mitochondria marker. LDs were labeled with BODIPY‐493/503 (green). Cells were imaged by confocal microscopy. Scale bar represents 10 μm.
- Quantification of average number of LD‐mitochondria contacts per LD from (F). 25 cells (Negative), 50 cells (Triple KD) from three independent experiments were calculated. Two‐tailed Unpaired Student's t‐test, ****P < 0.0001. Error bars represent the mean ± SD.
- Vero‐E6 cells were transfected with indicated siRNAs for 24 h and infected with SARS‐CoV‐2 for 24 h. Cell viability was analyzed. Cell lysates were analyzed via WB. Supernatants were determined by plaque assays. Virus titer values represent mean ± SD for three independent replicates. Two‐tailed Unpaired Student's t‐test, *P < 0.05, ***P < 0.001, ns means no significance. Error bars represent the mean ± SD.
- Proposed model for the role of SARS‐CoV‐2 ORF6 links LDs, the ER, and mitochondria, and modulates lipogenesis and lipolysis to facilitate viral production.
Source data are available online for this figure.
We then investigated whether the SAM complex is required for the mitochondrial localization of ORF6. siRNAs were used to knockdown MTX1, MTX2, or SAMM50. Knockdown of MTX1, MTX2, and/or SAMM50 caused minor effect on the number or morphology of LDs and mitochondria (Appendix Fig S3C). ORF6‐GFP was enriched in the mitochondrial fraction, and depletion of all three SAM proteins resulted in a significant reduction of ORF6 in the mitochondrial fraction, whereas suppression of MTX1, MTX2, SAMM50, or MTX1 and MTX2 had a minor effect (Fig 7C–E). Moreover, ORF6 failed to mediate mitochondrial‐LD interactions in SAM complex‐depleted cells (Figs 7F and G, and EV5K). These data indicated that ORF6 directly interacts with the SAM complex to mediate mitochondria‐LD interactions. Given that SAM complex is required for ORF6‐mediated mitochondria‐LDs interaction and that mitochondria‐LD interaction facilitates SARS‐CoV‐2 production, we then examined whether the SAM complex could modulate SARS‐CoV‐2 production. Knockdown of SAM complex resulted in an obvious reduction in SARS‐CoV‐2 production, whereas knockdown of MTX1, MTX2, or SAMM50 failed to suppress viral production (Fig 7H). Together, these data suggest that ORF6 targets mitochondria by interacting with the SAM complex to link LDs to mitochondria for viral production.
Discussion
LDs are associated with SARS‐CoV‐2 production. However, little is known about the underlying mechanisms. In this study, we uncovered novel mechanisms by which SARS‐CoV‐2 ORF6 targets LDs to reprogram cellular metabolism for viral production: (i) ORF6 is directly inserted into the LD lipid monolayer via its two amphipathic helices; (ii) ORF6 enhances LD biogenesis, and linking LD to ER by interacting with USE1 and BAP31; (iii) ORF6 enhances lipolysis by binding to ATGL and enhancing the interaction of ATGL with its coactivator CGI58 by disrupting the interactions of ATGL with two negative regulators UBXD8 and Plin2; and (iv) ORF6 decorates the contact site between mitochondria and LD and interacts with the SAM complex to mediate mitochondria‐LD junctions, facilitating FAs transfer to mitochondria for β‐oxidation and ATP production (Fig 7I).
The mechanisms by which proteins target LDs are not fully understood, however, several studies are emerging (Walther & Farese Jr., 2012). Viral proteins have been identified to target to LDs through the amphipathic helices, and depletion of the amphipathic helices abolished the LD association (Barba et al, 1997; Laufman et al, 2019). In this study, we also found that ORF6 of SARS‐CoV‐2 directly inserts into the LD lipid monolayer through its two amphipathic helices. ORF6 was also found to directly interact with ATGL and DGAT2, two LD‐bound proteins. Under steady state, ORF6 showed partial non‐LD localization, and it is possible that ORF6 could be recruited to LD from other organelles by interacting with ATGL and DGAT2 upon OA stimulation. ORF6 exists in both monomeric and dimeric states, and we found that the first helix‐mediated homodimerization is essential for LD association. Poliovirus 2C proteins showed self‐interaction, which mediated LD clustering (Laufman et al, 2019). We also observed LD clustering in several ORF6 expressing cells, but not all ORF6 targeted LDs showed clustering. It will be of interest to determine whether LD clustering play a role in SARS‐CoV‐2 production. In addition to SARS‐CoV‐2, we also found that ORF6 proteins of bat SARS‐CoV (Rf1/2006), SARS‐CoV (BtKY72), and bat coronavirus (PREDICT) are targeted to LDs. It would be interesting to find out whether these viruses may also use the same mechanism to reprogram lipid flux. Additionally, we noted that the N‐terminal tagged ORF6 remained diffused (Fig EV1B), suggesting that the N‐terminal tag can affect the localization of ORF6. To explain this observation, we propose that two amphipathic helices are localized in the N‐terminal region of ORF6, and that an N‐terminal tag may break the amphipathic characteristic of helices. Furthermore, we only showed that dimerization and LD binding are correlated but were short of direct evidences to conclude that homodimerization is required for LD binding. Further in vitro simplified system using synthetic peptides (AH1 or AH2) and artificial LDs could help understand the relationship between homodimerization and LD localization.
SARS‐CoV‐2 infection increases the expression of key proteins involved in the regulation of lipid metabolism and the number of LDs per cell (Dias et al, 2020). Treatment with fatty acid synthase inhibitors also markedly suppresses SARS‐CoV‐2 production (Chu et al, 2021). Here, we confirmed that the pharmacological inhibition of both DGAT1 and DGAT2 blocks SARS‐CoV‐2 production and SARS‐CoV‐2 production enhances the expression of DGAT1 and DGAT2. These findings indicated that LDs positively regulate SARS‐CoV‐2 production. A previous study showed that DGAT1 interacts with the nucleocapsid of HCV and is required for trafficking of core to LDs, which is critical for viral production (Herker et al, 2010). We found that ORF6 binds to both DGAT1 and DGAT2, and pharmacological inhibition of DGAT1 and DGAT2 abolishes LD biogenesis induced by ORF6 expression, suggesting that ORF6 enhances LD biogenesis by targeting DGAT1 and DGAT2. It should be noted that in contrast to the ER‐resident DGAT1, DGAT2 adopts a hairpin structure and is able to target to the LD surface upon OA treatment (Wilfling et al, 2013), indicating that ORF6 targets both DGAT1 mediated ER‐resident synthesis and DGAT2 mediated local synthesis. Furthermore, although our results demonstrated that ORF6 binds to DGAT1 and DGAT2, and both are required for SARS‐CoV‐2 production, more direct evidence is required to further confirm that SARS‐CoV‐2 and ORF6 expression directly modulate DGAT activity to promote LD biogenesis.
LDs are formed from the ER, and several cellular proteins have been identified to generate close contact between the ER and LD to facilitate lipid incorporation and LD biogenesis. However, whether viral proteins can tether ER‐LD and establish ER‐LD contact is poorly understood. USE1, a component of ER‐associated Q‐SNAREs (USE1, Syntaxin18, and BNIP1), plays a potential role in Golgi‐ER retrograde vesicle trafficking and ER‐LD interactions. BAP31 is the most abundant ER membrane protein, including in mitochondria‐associated membrane, where it contributes to mitochondrial tethering by interacting with mitochondrial proteins Fis1 and TOM40 (Iwasawa et al, 2011; Namba, 2019). E5 of human papillomaviruses (HPVs) were found to interact and colocalize with Bap31 in perinuclear patterns consistent with localization to the ER, and this interaction is required for the proliferative capacity of HPV‐positive keratinocytes following differentiation (Regan & Laimins, 2008). Simian virus 40 (SV40) VP2 also binds to BAP31, which is essential for the dislocation of SV40 from the ER to the cytosol (Geiger et al, 2011). In this study, we found that ORF6 localizes at ER‐LD junctions, and ER membrane proteins USE1 and BAP31 interacts with ORF6 and act as receptors on the ER to mediate ER‐LD contact in SARS‐CoV‐2 infected cells. Furthermore, ORF6 failed to bind and colocalize with Seipin (Appendix Fig S4A and B). Thus, these findings suggest that ORF6 can target DGAT1 mediated ER‐resident LD biogenesis by interacting with USE1 and BAP31 to enhance the connection between ER and LDs.
Viral proteins were found to physically interact with the lipolysis machinery, and these interactions mediate the LD‐viral production compartment (RC) contacts, facilitating FAs transfer from LDs to RCs, thereby providing lipids essential for RC biogenesis (Laufman et al, 2019). However, whether viral proteins modulate and exploit host lipolysis pathway for viral production, and the underlying mechanisms remain unclear. Similar to Poliovirus, we confirmed that lipolysis is essential for SARS‐CoV‐2 production. A previous study has shown that SARS‐CoV‐2 components are associated with LDs in infected cells (Dias et al, 2020). It would be interesting to determine whether the interaction between ORF6 and ATGL could be responsible for this contact. Alternatively, we propose that ORF6 binds to ATGL and enhances the interaction of ATGL with its coactivator CGI58 by disrupting the interactions of ATGL with two negative regulators, UBXD8 and Plin2, thus increasing lipolysis. We further found that lipolysis‐released FAs are sent to mitochondria and peroxisomes for ATP production via β‐oxidation. In addition, LD can also be degraded by autophagy, DENV infection induce lipophagy for viral production (Heaton & Randall, 2010). We showed that CQ treatment had a minor effect on LD reduction caused by ORF6 expression when lipogenesis was inhibited, suggesting that ORF6 only induces LD degradation via lipolysis. However, it is unclear whether other viral proteins can induce lipophagy. ORF3a of SARS‐CoV‐2 inhibits autophagic degradation by blocking the fusion of autophagosomes with lysosomes (Miao et al, 2021), indicating that even if lipophagy was induced by SARS‐CoV‐2, LD can only be engulfed into autophagosomes but failed to fusion with lysosomes for degradation.
FAs transfer to mitochondria through LD‐mitochondria junctions. Several proteins have been identified that mediate such conjunctions (Wang et al, 2011, 2021; Freyre et al, 2019). In virus‐infected cells, the molecular machinery that promotes FA transfer at mitochondria‐LD contact has not yet been identified. We found that SARS‐CoV‐2 infection increased the interaction between mitochondria and LDs. ORF6 interacts with many OMM proteins (Gordon et al, 2020). SAM complex (SAMM50, MTX1, and MTX2) associates with the mitochondrial contact site, and are thought to mediate insertion of β‐barrel proteins into the OMM (van der Laan et al, 2016). In addition, the SAM complex also plays a critical role in formation of the mitochondrial membrane contact sites to the inner membrane and the ER membrane (van der Laan et al, 2012). Pathogens, such as Toxoplasma gondii, target SAMM50 and TOM70 to remodel the outer mitochondrial membrane and impede the biogenesis of mitochondria that function as nutrient competitors or immune signaling hubs during infection (Li et al, 2022). However, it is unclear whether the SAM complex is required for viral production. The interactions of ORF6 with the SAM complex are required for the maintenance of mitochondria‐LD junctions, indicating that ORF6 expression hijacks the SAM complex for mitochondria‐LD interactions. The conclusion that ORF6 promotes LD‐mitochondrial contact may be due to increased LD number or increased TAG content, but not necessarily due to an interaction with the SAM complex. We found that overexpression of DGAT1 or DGAT2 increased the number of LD‐mitochondrial contacts per cell, but have minor effect on the average number of LD‐mitochondrial contacts per LD (Appendix Fig S5A and B). Additionally, it should be noted that although our results demonstrate that ORF6 links mitochondria to LDs and enhances lipolysis to release FAs for mitochondria oxidation, but do not indicate the role of ORF6 in FAs trafficking. More evidence is required to investigate the mechanism by which FAs transfer into mitochondria.
This study had several limitations. We failed to prove that during infection indeed endogenous ORF6 localizes clearly on LD. The viral titer decreased in KD of BAP31/USE1 and in SAMM50/MTX1/MTX2 may be due to effects on ER or mitochondrial function, respectively, rather than loss of binding to ORF6. Further work on constructing interfering peptides based on ORF6 NTD and AH domains to inhibit the binding of BAP31/USE1 and SAM complex, and evaluating the anti‐viral effect of these peptides should be performed. Although we have shown that SARS‐CoV‐2 infection promotes ER‐LD and mitochondria‐LD interactions, lipogenesis, and lipolysis, which are required for SARS‐CoV‐2 production, for obtaining most conclusions we relied on an overexpression system rather than wild‐type viruses. Further experiments, such as those involving the use of the real recombinant virus (ORF64Q), should be performed to confirm that SARS‐CoV‐2 targets LDs to reprogram cellular metabolism.
Overall, our study proposes an interesting and novel model in which viral protein modulates LD biogenesis and lipolysis pathways, linking LDs to the ER and mitochondria to reprogram cellular metabolism, and facilitates ATP production for viral production. These studies indicate the possible and essential roles of lipid metabolic reprogramming in SARS‐CoV‐2 production and pathogenesis, suggesting potential strategies for blocking viral production by targeting LD metabolism.
Materials and Methods
Cell cultures
HEK293T, HeLa, Vero‐E6 and COS7 cells were cultured in Dulbecco's modified Eagle's medium (11995065, Gibco) supplemented with 10% fetal bovine serum (12303C, Sigma‐Aldrich) and 1% penicillin–streptomycin (15140163, Gibco) at 37°C with 5% CO2. All cells were tested for mycoplasma negative.
Antibodies and reagents
Antibodies against HA (mouse, AE008), Flag (mouse, AE005), Flag (rabbit, AE004), GFP (mouse, AE012), GFP (rabbit, AE011), GST (mouse, AE001), VDAC1 (rabbit, A19707), MTX1 (rabbit, A7912), MTX2 (rabbit, A7958), SAMM50 (rabbit, A3401), ATGL (rabbit, A51265), HSL (rabbit, A15686), DGAT2 (rabbit, A13891), and CGI58 (rabbit, A8673) were purchased from Abclonal. Antibodies against His (mouse, sc‐53073), Tom20 (mouse, sc‐17764), Tim23 (mouse, sc‐514463), LAMP2 (mouse, sc‐18822), Plin2 (mouse, sc‐377429) and DGAT1 (mouse, sc‐271934) were obtained from Santa Cruz Biotechnology. Antibodies against Calnexin (rabbit, #2433), Myc (mouse, #2276), ATGL (rabbit, #2138), BSCL2 (Rabbit, #23846) and Rab11 (rabbit, #5589) were purchased from Cell Signaling Technology. Antibody against SARS‐CoV‐2 Nucleocapsid (rabbit, 40143‐R001) was purchased from Sino Biological. Antibody against Tubulin (mouse, E7) was purchased from Developmental Studies Hybridoma Bank. Antibody against ATP5A1 (mouse, 43‐9800) was obtained from Invitrogen. Antibodies against Strep (mouse, ab184224), cox2(mouse, ab110258), PMP70 (rabbit, AB3421), GFP (rabbit, ab6556) were purchased from Abcam. Antibody against LC3 (rabbit, L7543) was purchased from Sigma‐Aldrich. Antibody against p62 (mouse, H00008878‐M01) was obtained from ABnOVA. HRP‐conjugated goat anti‐mouse or rabbit IgG were obtained from Abclonal. Alexa Fluor 488/568/647‐conjugated goat anti‐mouse or rabbit IgG were purchased from Life Technologies. Anti‐Flag (B26102) and anti‐HA magnetic beads (B26202) were obtained from Bimake. BODIPY 493/503 (D3922), BODIPY 558/568 C12 (D3835), LipidTOX Deep Red neutral lipid stain (H34477), disuccinimidyl suberate (DSS, 21555) were purchased from ThermoFisher Scientific. Oleic acid (O1008), DGAT2 inhibitor (PF‐06424439), Chloroquine (CQ, C6628) and carbonyl cyanide m‐chlorophenyl hydrazone (CCCP, C2759) were purchased from Sigma‐Aldrich. Glutathione resin (L00206) was obtained from GeneScript. Anti‐GFP affinity beads 4FF (SA070001) was obtained from Smart‐Lifesciences Biotechnology. DAPI stain solution (G1012) was obtained from Servicebio. ATGL inhibitor (Atglistatin) and DGAT1 inhibitor (PF‐04620110) were purchased from Selleck Chemicals.
Virus infection
Vero‐E6 Cells were infected with SARS‐CoV‐2 (WBP‐1) at a multiplicity of infection (MOI) of 0.5 for 1 h. Cells were cultured with fresh medium supplemented with 2% FBS. All experiments with the SARS‐CoV‐2 virus were conducted in the BSL‐3 laboratory of Hubei Provincial Center for Disease Control and Prevention.
Plaque assays
Vero‐E6 cells were infected with virus preparations that had been serially diluted. Approximately 2 h post‐infection, the infection medium was removed and replaced with methylcellulose, cells were cultured at 37°C with 5% CO2 for another 3–4 days until visible viral plaques were detected. Plates were stained with 0.5% crystal violet for 4 h at room temperature. Stained cells were rinsed with water, plaques were counted and titers of infectious virus were calculated.
DNA construction
The plasmids encoding for the C‐terminus Strep tagged SARS‐CoV‐2 proteins were generous gifts from Nevan J. Krogan (UCSF). ORF6‐Strep was used as a template to generate wild‐type the C‐terminus tagged ORF6 mammalian expression plasmids pCDNA4‐ORF6‐Flag, pCDNA4‐ORF6‐HA, pCDNA4‐ORF6‐EGFP; and the N‐terminus tagged expression plasmid pCDNA4‐GFP‐ORF6. Point and truncation mutations of ORF6 were generated by PCR‐based mutagenesis from ORF6‐Flag and ORF6‐EGFP. The initial LD marker GFP‐GPAT4152–208 was generated by cloning sequences encoding the hairpin domain of GPAT4 from pEGFP‐N1‐GPAT4 (a gift from Peng Li, Tsinghua University) into pCDNA4‐EGFP vector. HA tagged ATGL and CGI58 were provided from Peng Li (Tsinghua University). Flag tagged ATGL was generated by cloning sequences encoding the ATGL from HA‐ATGL into pCDNA4 vector. GFP tagged Plin2, Plin5, Fsp27, SNAREs, and NRZ complex were gifts from Peng Li (Tsinghua University). GFP‐DGAT2, Flag‐DGAT1 and Flag‐DGAT2 were gifts from Qing Zhong (Shanghai Jiaotong University School of Medicine). Full‐length cDNAs encoding various human proteins were amplified by PCR from cDNAs of 293T cells. cDNAs encoding Plin1, Plin3, SPCS2, BAP31, UBXD8, DHCR7, CKAP4, TECR, UBAC2, HACD3 and ATP2A2 were cloned into pCDNA4 vector with a GFP tagged at its N‐terminus. cDNAs encoding BAP31 was cloned into pCDNA4 vector with a GFP tagged at its C‐terminus. cDNA encoding G0S2 was cloned into pCDNA4 vector with a GFP tagged at its C‐terminus. cDNA encoding RAMP4 was cloned into pCDNA4 vector with a mCherry tagged at its N‐terminus. cDNAs encoding MTX1, MTX2, VDAC1, VDAC2, VDAC3, Seipin and SAMM50 were cloned into pCDNA4‐Flag vector. Synonymous mutation of RNAi resistant ORF6(R)‐Flag or ORF6(R)4Q‐Flag were generated by PCR‐based mutagenesis from pCDNA4‐ORF6‐Flag. Primers used for the generation of referenced constructs are listed in Table EV1.
DNA transfection and RNA interference
Plasmid DNAs were transfected into 293T and Cos7 cells using Polyethylenimine Linear (40815ES03, YEASEN) or HeLa cells using Lipofectamine 2000 (11668019, Invitrogen) according to the manufacturer's instructions.
siRNAs were introduced into cells using riboFECT CP Transfection Kit (C10511‐05, RiboBio). Cells were then cultured for 48 h and harvested. The knockdown efficiency for each protein was evaluated by Western blot analysis. siRNAs targeting for BAP31 (5′‐GGUGAACCUCCAGAACAAU‐3′), USE1 (5′‐GGGAAAGUCUGCUUGAGUA‐3′), MTX1 (5′‐AAGUGGUAUGCAGAGGCUAUG‐3′), MTX2 (5′‐GGGAAGUCAAACGUAAGAUGA‐3′), SAMM50 (5′‐GGACAUUCACUGAAAUCAUCU‐3′) and ORF6 (5′‐CCGAGAATAAGTATTCTCAGTTGGA‐3′) were synthesized by GenePharma.
Establishment of stable cell‐line
For generation of stable ORF6‐Flag overexpressed HeLa cell line, pLenti‐ORF6‐Flag was generated by cloning sequences encoding the ORF6 into pLenti‐Flag vector. Lentivirus were produced by cotransfecting pLenti‐ORF6‐Flag with packaging plasmids pMD2.G and psPAX2 (gifts from Weike Ji, HUST) into 293T cells. The supernatant from transfected 293T cells were collected 48 h post initial transfection, followed by filtering through a syringe with 0.45 μm filters. Adding filtered supernatant to HeLa cells, after HeLa cells were infected with Lentivirus for 24 h, puromycin (5 μg/ml) was added to select positive cells for 2–3 days. The positive cells were verified by imaging and Western blot. For generation of stable GFP‐Plin5 or GFP‐Plin5Δ443–463 overexpressed Vero‐E6 cell lines, GFP‐Plin5 or GFP‐Plin5Δ443–463 were subcloned into pLenti‐EGFP vector. Lentivirus were produced by transfecting pLenti‐GFP‐Plin5 or pLenti‐GFP‐Plin5Δ443–463 with packaging plasmids pMD2.G and psPAX2 into 293T cells. Following procedure of generation of stable Vero‐E6 cell lines was similar to that of stable ORF6‐Flag overexpressed HeLa cell line except that virus‐infected Vero‐E6 cells were grown in medium containing 8 μg/ml puromycin.
Immunofluorescence microscopy
For immunostaining, cells cultured on coverslips were washed twice with PBS (135 mM NaCl, 4.7 mM KCl, 10 mM Na2HPO4, 2 mM NaH2PO4), then fixed with 4% paraformaldehyde for 15 min, and permeabilized with 0.1% Triton X‐100 in PBS for 10 min. Cells were blocked with 1% BSA in PBS for 30 min and incubated with primary antibodies in 1% BSA overnight at 4°C and then with secondary antibodies for 1 h at room temperature. LDs were stained with BODIPY 493/503, BODIPY 558/568 C12, LipidTOX Deep Red in PBS for another 30 min at room temperature. After washing with PBS three times, samples were mounted on slides with antifade mounting medium (BL701A, Biosharp). Cells were imaged with a laser scanning confocal microscope (LSM780, Zeiss, Germany) equipped with multiple excitation lasers (405, 458, 488, 514, 561, and 633 nm).
Image analysis
All image analysis and processing were performed using ImageJ. Colocalization‐based analysis of the contact sites of mitochondria‐LDs and ER‐LDs were performed using a plugin named colocalization in ImageJ with the following settings: Ratio (0–100%): 50; Threshold channel 1(0–255): 50; Threshold channel 2 (0–255): 50; Display value (0–255): 255. Contacts automatically identified by colocalization plugin with white pixels representing potential contact sites. Average number of LD‐ER contacts or LD‐mitochondria contacts per LD were calculated (contact number divided by LD number). Pearson's Coefficient of co‐localization analysis were performed using a plugin named JACoP in ImageJ. For the measurement of the length of ER‐LD contacts in TEM images, a line encompassing the ER‐LD contacts was manually selected, and was measured via ImageJ.
Pulse‐chase assay
HeLa cells were incubated in complete medium (DMEM with 10% fetal bovine serum) containing 1 μM BODIPY 558/568 C12 for 16 h. Cells were washed with PBS three times and then chased in EBSS for the time indicated (0 and 5 h).
Transmission electron microscopy
Cells were fixed by a fixative liquid (3% paraformaldehyde, 1.5% glutaraldehyde, 2.5% [wt/vol] sucrose in 0.1 M sodium phosphate buffer, pH 7.4) for 2 h at room temperature. Then the cells were collected and centrifuged at 4°C. Post fixed with 1% osmium tetroxide for 1 h on ice under dark conditions, cells were incubated with 2% uranyl acetate overnight, dehydrated in increasing concentrations of ethanol (50, 75, 95, and 100%), and processed for embedding in epoxy resin. Ultrathin (70‐nm) sections were collected on uncoated 200‐mesh copper grids, stained with uranyl acetate and lead citrate, and observed by transmission electron microscopy.
Immunoelectron microscopy
High pressure freezing
The HeLa cells were cultured on 3 mm sapphire discs. Sapphire disc was placed with cells facing up on flat aluminum planchette and another aluminum planchette with 25‐μm depth inner space was used as a cover. The spaces between the two aluminum planchettes were filled with 1‐hexadecane. Then the samples were frozen immediately using the EM ICE high pressure freezing machine (Leica) and rapidly transferred into liquid nitrogen for storage.
Freeze substitution and ultra‐thin section
After all of the samples were frozen, the samples were transferred into the EM ASF2 (Leica) for substitution. Samples were incubated for 48 h in acetone contained 0.2%UA at −90°C. Then the temperature was raised to −50°C in 4 h. After incubated in acetone contained 0.2%UA for another 12 h, the temperature was raised to −30°C in 4 h. After 2 h incubation at −30°C, the samples were rinsed three times with pure acetone (15 min each). Then the samples were gradually infiltrated in HM20 resin with grades of 25, 50,75% and pure resin (2 h each) at −30°C. After infiltrated in pure resin overnight, the samples were embedded in gelatin capsules. The samples were polymerized under UV light for 48 h at −30°C and 12 h at 25°C. After polymerization the samples were trimmed and ultra‐thin sectioned with a microtome (Leica UC7). Serial thin sections (100 nm thick) were collected onto formvar‐coated nickel grids.
Immunogold labeling
The formvar‐coated nickel grids with sections were incubated in 0.01 M PBS contain 1%BSA, 0.05%Triton X‐100 and 0.05% Tween20 for 5 min. Then the sections were incubated in the GFP antibody (Abcam, ab6556) diluted in 0.01 M PBS contain 1%BSA and 0.05% Tween20 at 4°C overnight. After washed six times (2 min each) with 0.01 M PBS, the sections were incubated in the secondary antibody (goat anti‐rabbit conjugated with 10 nm gold) diluted in 0.01 M PBS containing 1% BSA and 0.05% Tween20 (1:50) for 2 h at RT. After washed six times (2 min each) with 0.01 M PBS and four times (2 min each) with distill water, the sectioned were dried in the RT and examined in a transmission electron microscopy (Thermo Fisher/FEI Talos L 120C).
Western blot
The samples were denatured by heating at 100°C for 10 min in the SDS‐PAGE sample loading buffer (P0015L, Beyotime Biotechnology). Protein samples were electrophoresed on 10 or 13% gradient SDS‐PAGE gels depending on the molecular weight of the relevant proteins and transferred to nitrocellulose membranes (HATF00010, Millipore). Membranes were blocked with 5% skim milk in PBST (135 mM NaCl, 4.7 mM KCl, 10 mM Na2HPO4, 2 mM NaH2PO4, 0.1% Tween‐20) for 30 min at room temperature and then incubated with the relevant primary antibodies in the primary antibody dilution buffer (ABS954‐1L, Absin) overnight at 4°C. Membranes were washed three times with PBST for 5 min each and incubated with HRP‐conjugated secondary antibodies in blocking solution for 1 h at room temperature. Membranes were washed and signals were developed using ECL Plus Western blotting substrate (BL520B, Biosharp).
Immunoprecipitation
293T cells were transfected with the indicated constructs for 36 h and collected in cold PBS and centrifuged at 16,060 g for 5 min. The supernatant was removed and cells were re‐suspended in TAP lysis buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% NP‐40, 1 mM EDTA) supplemented with protease inhibitor cocktail and incubated for 30 min on ice. Then samples were centrifuged at 16,060 g for 20 min at 4°C, 10% of the supernatant was saved for analysis by immunoblotting of the input fraction, 5‐μl beads were washed with lysis buffer and added to the remaining protein samples and incubated overnight at 4°C. The beads were washed with lysis buffer three times before adding 1× loading buffer. Proteins co‐IP were analyzed by Western blot.
GST pull‐down
GST or His tagged proteins were constructed into pGEX‐4T‐1 vector or pCOLD‐6 × His vector. Proteins were purified from Escherichia coli strain Rosetta (DE3). Bacteria were grown at 37°C to an OD600 of 0.6–0.8, then protein expression was induced with 0.4 mM IPTG for 20 h at 16°C. Bacteria were harvested, resuspended, and then lysed in lysis buffer (20 mM Tris, pH 7.4, 300 mM NaCl, 0.1% NP‐40, 1 mM PMSF) by sonication. Lysates were centrifuged at 10,000 g for 10 min and collected the supernatant, mixed the indicated protein samples and incubated with Glutathione resins overnight at 4°C, and then the resins were washed with lysis buffer three times before adding 1× loading buffer. Samples were analyzed by Western blot.
Subcellular fractionation
Mitochondrial Isolation and Protein Extraction Kit (PK10016, Proteintech) was used to extract mitochondria according to manufacturer's instructions. Briefly, cells were collected in cold PBS and centrifuged at 500 g for 5 min at 4°C, removed the supernatant and resuspended the cells in 1 ml buffer A (containing 1 mM PMSF) and homogenized on ice by ultrasonic. The homogenates were laid on the same volume of buffer B slowly and centrifuged at 600 g for 10 min at 4°C, then collected the supernatant as cytosol. Two‐thirds of the cytosol fraction was further centrifuged at 10,000 g for 10 min at 4°C, the pellet was mitochondria.
For isolation of the ER, cells were washed with cold PBS, harvested the cells and centrifuged at 600 g for 5 min at 4°C, resuspended the cells in 1 ml buffer E (20 mM HEPES, 250 mM sucrose, 1 mM EDTA, pH 7.4, 1 mM PMSF) and homogenized on ice by ultrasonic. The homogenates were centrifuged at 1,000 g for 10 min at 4°C, the supernatant was cytosol fraction. Two‐thirds of the cytosol fraction was further centrifuged at 8,000 g for 20 min at 4°C, then the supernatant was transferred to ultracentrifuge tube and ultracentrifuged at 100,000 g for 1 h at 4°C. The obtained pellet was collected as the ER fraction.
For isolation of the ER and mitochondria, cells were washed with cold PBS, harvested the cells and centrifuged at 500 g for 10 min at 4°C, resuspended the cells in 1 ml buffer (10 mM Tris–HCl, 250 mM sucrose, 2 mM MgCl2, 10 mM KCl, pH 7.4, 1 mM PMSF) and homogenized on ice by ultrasonic. The homogenates were centrifuged at 1,000 g for 10 min at 4°C, the supernatant was cytosol fraction. Two‐thirds of the cytosol fraction was further centrifuged at 5,000 g for 10 min at 4°C, the supernatant was collected to a new tube, the pellet was resuspended in 1 ml buffer and centrifuged at 10,000 g for 2 min at 4°C, and the obtained pellet was collected as the mitochondria fraction. Then the supernatant in the new tube was centrifuged at 8,000 g for 10 min at 4°C, the supernatant was transferred to ultracentrifuge tube and ultracentrifuged at 100,000 g for 1 h at 4°C, the obtained pellet was collected as the ER fraction.
Quantitative RT‐PCR
HeLa cells were transfected with the indicated constructs for 24 h, RNA was isolated with Total RNA Isolation Reagent (BS259A, Biosharp) according to the instructions from the manufacturer. cDNA was reverse‐transcribed using Reverse Transcription Kit (BL699A, Biosharp) according to the directions of the manufacturer. The cDNA was analyzed using quantitative PCR with qPCR Mix (1725120, Bio‐Rad). The relative quantification values were normalized by those of an endogenous control, GAPDH. Sequences of the primers were as follows: for gapdh 5′‐GACAAGCTTCCCGTTCTCAG‐3′(forward), 5′‐GAGTCAACGGATTTGGTCGT‐3′ (reverse); for dgat1 5′‐CCTACCGCGATCTCTACTACTT‐3′ (forward), 5′‐GGGTGAAGAACAGCATCTCAA‐3′ (reverse); for dgat2 5′‐GCTGACCACCAGGAACTATATC‐3′(forward), 5′‐GGGAACTTCTTGCTCACTTCT‐3′ (reverse); for atgl 5′‐GTGTCAGACGGCGAGAATG‐3′ (forward), 5′‐TGGAGGGAGGGAGGGATG‐3′ (reverse).
Cellular triglyceride assay
Cells were treated with 200 μM OA for 3 h before collecting. Cells were washed with PBS for three times, collected cells and measured triglyceride content according to manufacturer's instructions (Triglyceride Colorimetric Assay Kit, 10010303, Cayman). Briefly, cells were collected by centrifugation at 1,000 × g for 10 min at 4°C, then the cells were resuspended in 1 ml diluted Standard Diluent and homogenized on ice by ultrasonic, the cell suspension was centrifuged at 10,000 × g for 10 min at 4°C to obtain the supernatant. For triglyceride assay, 10 μl standard or sample was added to the 96 well plate, the reaction was initiated by adding 150 μl diluted Enzyme Mixture solution to each well. The plate was incubated for 30 min at 37°C, the absorbance at 530–550 nm was measured.
ATP assay
Cells were treated with 200 μM OA overnight to enlarge LDs, then washed the cells and cultured the cells in complete medium for another 24 h, intracellular ATP content was measured using the Luminescent ATP Detection Assay Kit (ab113849, Abcam). Briefly, 50 μl detergent was added into each well, the 96 well plate was shaken for 5 min in an orbital shaker at 600–700 rpm to lyse the cells and stabilize the ATP. 100 μl ATP standard was added into the plate, 50 μl Substrate Solution was added to each of the wells, the plate was shaken for 5 min in an orbital shaker at 600–700 rpm and covered for 10 min to adapt dark, the luminescence was measured.
Free fatty acid assay
Cells were treated with 200 μM OA for 24 h, then the medium was removed and incubated cells with serum free DMEM for another 4 h, cellular free fatty acid was measured using the Free Fatty Acid Assay Kit (ab65341, Abcam). Briefly, the cells were harvested in the cold PBS and centrifuged at 600 × g for 10 min at 4°C, then, the cells were resuspended in 200 μl chloroform/Triton X‐100 (1% Triton X‐100 in pure chloroform), homogenized by pipetting up and down and incubated on ice for 20 min. The extract was centrifuged for 10 min at top speed in a microcentrifuge, the lower phase was collected and dried at 50°C in a fume hood and vacuum dryer. The dried lipids were dissolved in 200 μl Fatty Acid Assay Buffer by vortexing extensively for 5 min. 50 μl standard or sample was added into 96 well plate, 2 μl ACS Reagent was added into all standard and sample wells, the plate was incubated for 30 min at 37°C. Then, 50 μl Reaction Mix was added to each well, the plate was protected from light and incubated at 37°C for 30 min, the absorbance at 570 nm was measured.
Measurement of cellular energetics by Agilent seahorse instrument
The experiment was carried out according to Agilent Seahorse protocol. In detail, HeLa cells were transfected with indicated constructs (1 μg/one 12‐well plate well using PolyJet DNA Transfection Reagent (SignaGen, MD)), and seeded to an Agilent Seahorse XF24 cell culture microplate at 30,000 cells per well (Wells A1, B4, C3, and D6 are used for the background without cells) the second day. In parallel, an Agilent Seahorse XFe24 extracellular flux assay plate with 1 ml Seahorse XF Calibrant Solution was equilibrated overnight at 37°C. The next day, Mito Stress Test was performed as described in In vitro Seahorse XF Cell Mito Stress Test Kit with sequential injection of the following compounds: Port A: Oligomycin, a final concentration of 2 μM; Port B: FCCP, a final concentration of 1 μM; Port C: Rotenone, a final concentration of 10 μM and Antimycin A, a final concentration of 40 μM.
Luciferase assay
For luciferase assay, Dual Luciferase Reporter Gene Assay Kit (11402ES60, YEASEN) was used. Briefly, HEK293T cells in 24 well plates were transfected with IFN‐β reporter and TK reporter with RIG‐I‐N expressing plasmid, together with vector or plasmids expressing wild‐type ORF6 or ORF64Q. Cells transfected with only IFN‐β reporter and TK reporter as negative control. For OA treatment groups, 200 μM OA was added for 12 h. At 24 h after transient transfection, cells were washed with cold PBS and incubated with 200 μl cell lysates on ice for 5 min to fully lyse the cells. 20 μl of lysate was added to the opaque white 96 well plate and mixed with 100 μl 1× Firefly luciferase reaction solution, the activity of Firefly luciferase was detected. Then, 100 μl of 1× Renilla luciferase reaction solution was added and mixed well by shaking, the activity of Renilla luciferase was detected. Firefly luciferase values were normalized to Renilla, and the relative luciferase activity was calculated as the ratio of experimental group versus negative control.
Cell viability assay
For cell viability assay, Cell Counting Kit‐8 (40203ES80, YEASEN) was used. Briefly, 2,000 cells were seeded into 96 well plate and grown in medium overnight. Cells were transfected with indicated plasmids or siRNA or treated with indicated drug. The medium was removed and changed with fresh complete medium before 10 μl CCK‐8 assay reagent was added into the wells, after the cells were incubated for another 4 h, the absorbance at 450 nm was measured.
Quantification and statistical analysis
Data are expressed as mean ± standard deviation (SD). The significance of the variability between different groups was determined by two‐way analyses of variance using GraphPad Prism software. Error bars, mean ± SD of two or three independent experiments. Two‐tailed Unpaired Student's t‐test, a P‐value of < 0.05 was considered statistically significant and a P‐value of > 0.05 was considered statistically non‐significant (ns).
Author contributions
Mengzhen Yue: Data curation; formal analysis; investigation; methodology. Bing Hu: Resources; investigation. Jiajia Li: Investigation; writing – original draft; writing – review and editing. Ruifeng Chen: Validation; investigation. Zhen Yuan: Validation; investigation. Hurong Xiao: Validation; investigation. Haishuang Chang: Investigation; methodology. Yaming Jiu: Resources; writing – review and editing. Kun Cai: Resources; investigation. Binbin Ding: Conceptualization; data curation; formal analysis; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are deeply grateful to Qing Zhong (Shanghai Jiaotong University School of Medicine) and Mingzhou Chen (Wuhan University) for critical suggestions on the current work. We thank Yi Zhu (Baylor College of Medicine) for Seahorse analysis; Weike Ji (HUST) for helpful suggestions on quantifications; Nevan J. Krogan (UCSF) for providing SARS‐CoV‐2 viral proteins expression plasmids; Pei Zhang (Wuhan Institute of Virology) for TEM assistance; the Electron Microscopy center of Shanghai Institute of Precision Medicine, Shanghai Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, for their technical support and assistance in the electron microscopy. We would like to thank Editage (www.editage.cn) for English language editing. This work was mainly supported by the Major Research Plan of the National Natural Science Foundation of China (92054107), the National Natural Science Foundation of China (U22A20337), partly supported by the National Natural Science Foundation of China (92054104, 32222022) and Open Research Fund Program of the State Key Laboratory of Virology of China (2022KF006).
The EMBO Journal (2023) 42: e112542
Data availability
This study includes no data deposited in external repositories.
References
- Addetia A, Lieberman NAP, Phung Q, Hsiang TY, Xie H, Roychoudhury P, Shrestha L, Loprieno MA, Huang ML, Gale M Jr et al (2021) SARS‐CoV‐2 ORF6 disrupts bidirectional nucleocytoplasmic transport through interactions with Rae1 and Nup98. mBio 12: e00065‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Cermelli S, Li Z, Kassan A, Bosch M, Sigua R, Huang L, Ouellette AJ, Pol A, Welte MA et al (2012) A novel role for lipid droplets in the organismal antibacterial response. Elife 1: e00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, Eder G, Schaff Z, Chapman MJ, Miyamura T et al (1997) Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci USA 94: 1200–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, Acín‐Pérez R, Shum M, Oliveira MF, Cinti S et al (2018) Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab 27: 869–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Sánchez‐Álvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, Moreira L, López JA, Campo R, Marí M et al (2020) Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science (New York, NY) 370: eaay8085 [DOI] [PubMed] [Google Scholar]
- Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera‐Alberni M, Combe R, Zorzano A, Cantó C (2017) Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J 36: 1543–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerk IK, Wechselberger L, Oberer M (2018) Adipose triglyceride lipase regulation: an overview. Curr Protein Pept Sci 19: 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Weigel AV, Ioannou MS, Pasolli HA, Xu CS, Peale DR, Shtengel G, Freeman M, Hess HF, Blackstone C et al (2019) Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT‐III. J Cell Biol 218: 2583–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Xing C, Du Y, Duan T, Liu S, Zhang P, Cheng C, Henley J, Liu X, Qian C et al (2021) Pharmacological inhibition of fatty acid synthesis blocks SARS‐CoV‐2 replication. Nat Metab 3: 1466–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Mirza AH, Zhang S, Liang B, Liu P (2019) Lipid droplets and mitochondria are anchored during brown adipocyte differentiation. Protein Cell 10: 921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias SSG, Soares VC, Ferreira AC, Sacramento CQ, Fintelman‐Rodrigues N, Temerozo JR, Teixeira L, Nunes da Silva MA, Barreto E, Mattos M et al (2020) Lipid droplets fuel SARS‐CoV‐2 replication and production of inflammatory mediators. PLoS Pathog 16: e1009127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme NA, Bickel PE (2008) Lipid droplets in lipogenesis and lipolysis. Endocrinology 149: 942–949 [DOI] [PubMed] [Google Scholar]
- Farley SE, Kyle JE, Leier HC, Bramer LM, Weinstein JB, Bates TA, Lee JY, Metz TO, Schultz C, Tafesse FG (2022) A global lipid map reveals host dependency factors conserved across SARS‐CoV‐2 variants. Nat Commun 13: 3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W, Shui G, Gaeta B, Du X, Kuerschner L, Li P, Brown AJ, Wenk MR, Parton RG, Yang H (2008) Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J Cell Biol 180: 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyre CAC, Rauher PC, Ejsing CS, Klemm RW (2019) MIGA2 links mitochondria, the ER, and lipid droplets and promotes de novo lipogenesis in adipocytes. Mol Cell 76: 811–825 [DOI] [PubMed] [Google Scholar]
- Gautier R, Douguet D, Antonny B, Drin G (2008) HELIQUEST: a web server to screen sequences with specific alpha‐helical properties. Bioinformatics 24: 2101–2102 [DOI] [PubMed] [Google Scholar]
- Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A (2011) BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol 13: 1305–1314 [DOI] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O'Meara MJ, Rezelj VV, Guo JZ, Swaney DL et al (2020) A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton NS, Randall G (2010) Dengue virus‐induced autophagy regulates lipid metabolism. Cell Host Microbe 8: 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV Jr, Ott M (2010) Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase‐1. Nat Med 16: 1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, Rupérez C, Fernández‐Vidal A, Ferguson C, Rentero C, Tebar F et al (2015) AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 6: 7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höhr AI, Straub SP, Warscheid B, Becker T, Wiedemann N (2015) Assembly of β‐barrel proteins in the mitochondrial outer membrane. Biochim Biophys Acta 1853: 74–88 [DOI] [PubMed] [Google Scholar]
- Iwasawa R, Mahul‐Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria‐ER interface to establish a platform for apoptosis induction. EMBO J 30: 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufman O, Perrino J, Andino R (2019) Viral generated inter‐organelle contacts redirect lipid flux for genome replication. Cell 178: 275–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Straub J, Medeiros TC, Mehra C, den Brave F, Peker E, Atanassov I, Stillger K, Michaelis JB, Burbridge E et al (2022) Mitochondria shed their outer membrane in response to infection‐induced stress. Science 375: eabi4343 [DOI] [PubMed] [Google Scholar]
- Long M, Sanchez‐Martinez A, Longo M, Suomi F, Stenlund H, Johansson AI, Ehsan H, Salo VT, Montava‐Garriga L, Naddafi S et al (2022) DGAT1 activity synchronises with mitophagy to protect cells from metabolic rewiring by iron depletion. EMBO J 41: e109390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao G, Zhao H, Li Y, Ji M, Chen Y, Shi Y, Bi Y, Wang P, Zhang H (2021) ORF3a of the COVID‐19 virus SARS‐CoV‐2 blocks HOPS complex‐mediated assembly of the SNARE complex required for autolysosome formation. Dev Cell 56: 427–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miorin L, Kehrer T, Sanchez‐Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E et al (2020) SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA 117: 28344–28354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K (2007) The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9: 1089–1097 [DOI] [PubMed] [Google Scholar]
- Monson EA, Crosse KM, Duan M, Chen W, O'Shea RD, Wakim LM, Carr JM, Whelan DR, Helbig KJ (2021) Intracellular lipid droplet accumulation occurs early following viral infection and is required for an efficient interferon response. Nat Commun 12: 4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T (2019) BAP31 regulates mitochondrial function via interaction with Tom40 within ER‐mitochondria contact sites. Sci Adv 5: eaaw1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, Olzmann JA (2017) DGAT1‐dependent lipid droplet biogenesis protects mitochondrial function during starvation‐induced autophagy. Dev Cell 42: 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Carvalho P (2019) Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 20: 137–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Richter CM, Kopito RR (2013) Spatial regulation of UBXD8 and p97/VCP controls ATGL‐mediated lipid droplet turnover. Proc Natl Acad Sci USA 110: 1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier Y, Antonenkov VD, Glumoff T, Hiltunen JK (2006) Peroxisomal β‐oxidation—a metabolic pathway with multiple functions. Biochim Biophys Acta 1763: 1413–1426 [DOI] [PubMed] [Google Scholar]
- Quistgaard EM (2021) BAP31: physiological functions and roles in disease. Biochimie 186: 105–129 [DOI] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, Lippincott‐Schwartz J (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32: 678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan JA, Laimins LA (2008) Bap31 is a novel target of the human papillomavirus E5 protein. J Virol 82: 10042–10051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo VT, Belevich I, Li S, Karhinen L, Vihinen H, Vigouroux C, Magré J, Thiele C, Hölttä‐Vuori M, Jokitalo E et al (2016) Seipin regulates ER‐lipid droplet contacts and cargo delivery. EMBO J 35: 2699–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa MM, Mondotte JA, Iglesias NG, Assunção‐Miranda I, Barbosa‐Lima G, Da Poian AT, Bozza PT, Gamarnik AV (2009) Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5: e1000632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, Garg A, Anderson RG, Goodman JM (2007) The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci USA 104: 20890–20895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak‐Tarnopolsky SN, Devries MC, Hamadeh MJ (2007) Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol 292: R1271–R1278 [DOI] [PubMed] [Google Scholar]
- van der Laan M, Bohnert M, Wiedemann N, Pfanner N (2012) Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol 22: 185–192 [DOI] [PubMed] [Google Scholar]
- van der Laan M, Horvath SE, Pfanner N (2016) Mitochondrial contact site and cristae organizing system. Curr Opin Cell Biol 41: 33–42 [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV Jr (2012) Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 81: 687–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Chung J, Farese RV Jr (2017) Lipid droplet biogenesis. Annu Rev Cell Dev Biol 33: 491–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet‐associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52: 2159–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sreenivasan U, Gong DW, O'Connell KA, Dabkowski ER, Hecker PA, Ionica N, Konig M, Mahurkar A, Sun Y et al (2013) Cardiomyocyte‐specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J Lipid Res 54: 953–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK et al (2016) Seipin is required for converting nascent to mature lipid droplets. eLife 5: e16582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Fang N, Xiong J, Du Y, Cao Y, Ji WK (2021) An ESCRT‐dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D‐TSG101 interactions. Nat Commun 12: 1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Pfanner N (2017) Mitochondrial machineries for protein import and assembly. Annu Rev Biochem 86: 685–714 [DOI] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng JX, Graham M, Christiano R, Fröhlich F et al (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24: 384–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Li Y, Wu L, Li Y, Zhao D, Yu J, Huang T, Ferguson C, Parton RG, Yang H et al (2018) Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J Cell Biol 217: 975–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lu X, Lombès M, Rha GB, Chi YI, Guerin TM, Smart EJ, Liu J (2010) The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab 11: 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Madeo F, Kratky D (2017) Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol 18: 671–684 [DOI] [PubMed] [Google Scholar]
- Zhang J, Lan Y, Li MY, Lamers MM, Fusade‐Boyer M, Klemm E, Thiele C, Ashour J, Sanyal S (2018) Flaviviruses exploit the lipid droplet protein AUP1 to trigger Lipophagy and drive virus production. Cell Host Microbe 23: 819–831 [DOI] [PubMed] [Google Scholar]