

Abstract

Proteins and their assemblies are fundamental for living cells to function. Their complex three-dimensional architecture and its stability are attributed to the combined effect of various noncovalent interactions. It is critical to scrutinize these noncovalent interactions to understand their role in the energy landscape in folding, catalysis, and molecular recognition. This Review presents a comprehensive summary of unconventional noncovalent interactions, beyond conventional hydrogen bonds and hydrophobic interactions, which have gained prominence over the past decade. The noncovalent interactions discussed include low-barrier hydrogen bonds, C5 hydrogen bonds, C–H···π interactions, sulfur-mediated hydrogen bonds, n → π* interactions, London dispersion interactions, halogen bonds, chalcogen bonds, and tetrel bonds. This Review focuses on their chemical nature, interaction strength, and geometrical parameters obtained from X-ray crystallography, spectroscopy, bioinformatics, and computational chemistry. Also highlighted are their occurrence in proteins or their complexes and recent advances made toward understanding their role in biomolecular structure and function. Probing the chemical diversity of these interactions, we determined that the variable frequency of occurrence in proteins and the ability to synergize with one another are important not only for ab initio structure prediction but also to design proteins with new functionalities. A better understanding of these interactions will promote their utilization in designing and engineering ligands with potential therapeutic value.
1. Noncovalent Interactions
Proteins perform a vast majority of cellular functions that are necessary to sustain life. The three-dimensional (3D) structure adopted by a protein dictates the function that it carries out. Aberration or disruption of the 3D structure can disrupt cellular activities and cause physiological disorders and diseases. Consequently, the adoption and stabilization of functionally relevant protein structures are of utmost importance. While covalent peptide bonds link amino acids linearly to form polypeptide chains, it is the noncovalent interactions that sculpt the 3D structure of proteins by folding the polypeptide chains into a definite structure and providing stability to the structure. Noncovalent interactions also influence aspects related to protein functions, such as (i) the affinities and mode of ligand binding, (ii) the complexation of proteins with other proteins, nucleic acids, lipids, membrane, and carbohydrates, (iii) molecular recognition, and (iv) the enzymatic reaction mechanism and kinetics.
A detailed study of the noncovalent interactions is required to not only understand the protein structure and its function, but also to design proteins with new functions and ligands that modulate protein functions that can potentially serve as therapeutic drugs.
J. D. van der Waals first identified cohesive forces distinct from and weaker than covalent interactions that hold atoms or molecules together, which are referred to as noncovalent interactions.1,2 P. A. Kollman defined noncovalent interactions as “those in which (1) electrons stay paired in reactants and products and (2) there is no net change in chemical bonding.”3 The overall strength of the noncovalent interactions usually ranges from −0.5 to −50 kcal mol–1 and depends on the nature of the interacting atoms or molecules. Unlike covalent interactions, these interactions do not confer much rigidity and are easy to form or break.2,4 This unique feature introduces dynamicity within macromolecules that are integral to their biochemical functions at ambient temperature.2 Additionally, these interactions often work in groups and are critical for determining the structures and functions of macromolecules or their complexes, including catalysis and molecular recognition.4
To understand the nature of the interactions or characterization of the forces involved, the interaction energy between any two interacting atoms/molecules can be broken down into five components: (1) electrostatic, (2) exchange-repulsion, (3) dispersion, (4) polarization, and (5) charge transfer.5−7 Structural, spectroscopic, thermodynamic, database, and ab initio computational analyses are the primary and most valuable tools employed to understand and study noncovalent interactions.2,8,9 For instance, topological features of electron density distribution obtained experimentally from X-ray charge-density analysis are used to understand the nature, strength, and directionality of the noncovalent interactions.10−12 In proteins, the interactions are primarily made by nonpolar groups and polar groups containing hydrogen (H), carbon (C), nitrogen (N), oxygen (O), phosphorus (P), and sulfur (S) atoms.
Hydrogen bonds, ionic bonds, hydrophobic, and van der Waals interactions are noncovalent interactions commonly observed in biomolecules, which have been studied and discussed extensively in various reviews and textbooks. However, over the past decade, the importance of a variety of other noncovalent interactions in biomolecules has been discovered, which have been shown to be important for the structure and function of proteins. This Review focuses on the advances that have taken place, in particular over the past decade, in our understanding of unconventional noncovalent interactions, beyond the conventional H-bonds, ionic bonds, or hydrophobic interactions that are observed in proteins or their complexes with other biomolecules. In particular, chemical basis, experimental evidence, structural features, and the relevance of these interactions in protein structure and function will be discussed. This Review will also discuss recent studies that have contributed to a better understanding of the London dispersion interaction, the attractive component of the van der Waals interaction, and its importance in protein structure and function in light of these findings.
2. Unconventional H-Bonds in Proteins
Hydrogen bonds are one of the most common noncovalent interactions in proteins that play an extremely important role in protein structure and function.13−16 IUPAC definition states that “The hydrogen bond is an attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation.”17 While electrostatic components generally dominate H-bond formation, contributions from charge transfer and polarization interactions are also reported on the basis of the Energy Decomposition Analysis.18,19 D–H···A is a general representation for H-bonds, where D and A are the H-bond donor and acceptor atom, respectively (Figure 1A). Depending on the electronegativity of D and A and their environment, the strength of the H-bonds can be up to −40 kcal mol–1.8 The H-bonds can also be weak, such as the well-studied C–H···O bond.8 On the basis of their strength, H-bonds can be categorized as (i) conventional (−2.4 to −12.0 kcal mol–1), (ii) low-barrier (−12 to 24 kcal mol–1), and (iii) single-well H-bonds (more than −24 kcal mol–1)20 (Figure 1B).
Figure 1.
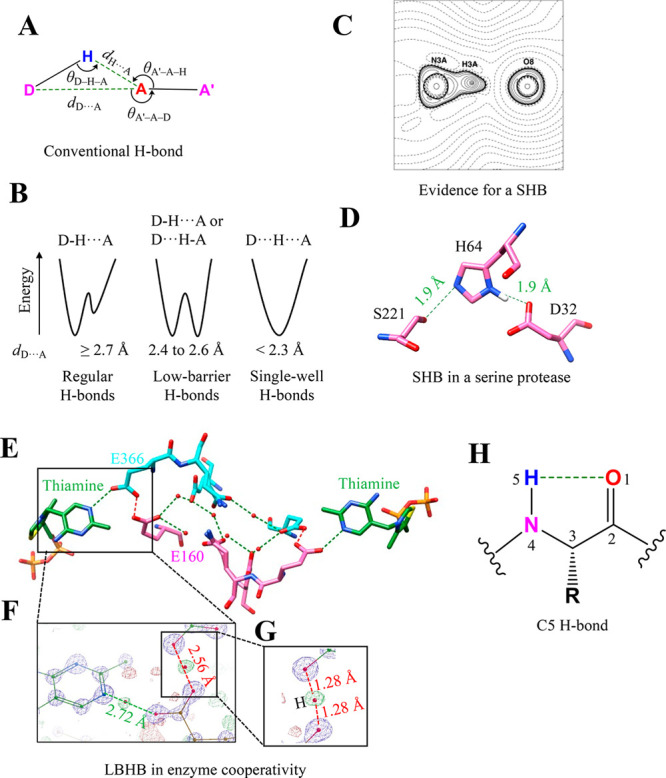
Unconventional H-bonds. (A) Geometrical and stereochemical parameters used to identify a conventional H-bond in biomolecules. (B) Energy profile of a H-bond as a function of the location of the H atom between D and A highlighting the difference between a regular, low-barrier, and single-well H-bond. (C) A charge-density analysis showing a contour plot of the negative of the Laplacian of the electron density in the plane of the N–H···O bond in a model compound mimicking the catalytic triad in serine proteases. Solid lines indicate positive contours, and broken lines indicate a negative contour. Adapted from ref (26). Copyright 2001 Wiley. (D) A 0.78 Å structure showing a short H-bond (dH···O = 1.60 Å) in the Ser-His-Asp catalytic triad of serine protease subtilisin (PDB ID: 1GCI). (E) The 0.97 Å resolution crystal structure of the human transketolase with substrate–thiamine intermediates bound (green) zoomed to show a channel and intervening residues communicating with two active sites of the homodimer (each monomer colored in cyan and pink) (PDB ID: 4KXW). The H-bond network that connects these sites is highlighted in green (regular H-bonds) and red (LBHB). (F) Zoomed section showing residues Glu160, Glu366, and a portion of thiamine with the 2FO – FC electron density map at the 6σ contour level and FO – FC at the 2.7σ contour level. The regular H-bond between thiamine and Glu366 (green) and the LBHB between Glu366 and Glu160 (red) are also highlighted. (G) A magnified section shows the occurrence of electron density almost precisely halfway between Glu366 and Glu160, confirming an LBHB. Mutation of Glu160 to glutamine disrupts the LBHB, resulting in a 5-fold decrease in the enzyme’s catalytic constant (kcat). Parts F and G were adapted from ref (32). Copyright 2019 Springer. (H) Definition and geometry of the C5 H-bond.
2.1. Low-Barrier H-Bond (LBHB)
Energetically, a low-barrier H-bond (LBHB) is formed when a low energy barrier separates two minima of equal depth. Such a situation arises when the pKa values of donor and acceptor atoms are comparable.21−23 In a conventional H-bond, the barrier height is higher than the zero-point energy of the shared hydrogen. Consequently, in a LBHB, the hydrogen atom is located symmetrically between the donor and acceptor atoms and dD–H ≈ dH···A.24 LBHB can be ascertained by locating the position of the hydrogen shared between the donor and the acceptor. Ultra-high-resolution X-ray data, using the electron density for H that can be observed, are required to ascertain LBHB. Neutron crystallography of deuterated proteins is an alternative to locate the H atom positions and identify LBHB. Analyses of ultra-high-resolution protein structures have led to the identification of short H-bonds (SHBs) that have dD···A < 2.7 Å.24,25 When dD···A is within 2.4–2.6 Å, such SHBs could enter the low-barrier H-bond (LBHB) region (Figure 1B).22,24 NMR can also help identify LBHB as the deshielded protons in SHBs result in a characteristic downfield chemical shift for proton spectra. However, the distinction of LBHB from SHB cannot be made solely on the basis of the shorter length obtained from NMR.
In SHBs, sharing of H in the H-bond is because of the decreased potential barrier and has a more significant enthalpy contribution than that seen in conventional H-bonds. A more recent analysis indicates that the strength of such an H-bond could be within −10 to −20 kcal mol–1.23 Not all SHBs are LBHB. The active site of serine protease was proposed to harbor the catalytically important LBHB.20 However, charge-density analysis of small molecules mimicking the active site of this enzyme revealed an intermediate state (strength of −21 kcal mol–1) in comparison to that of the conventional H-bond and LBHB (Figure 1C).26 H in the N–H···O bond was localized in the N shell instead of being shared equally between the N and O atoms, an essential feature of LBHB. This observation agreed with the 0.78 Å crystal structure of the serine protease subtilisin (Figure 1C and D).27 Recently, NMR-based experiments probing chemical shift values supported the existence of LBHB in the active site of the serine protease.28 However, the role of LBHB in the catalytic activity of serine protease is yet to be unambiguously confirmed. Previously, Warshel and Kollman argued that the occurrence of LBHB could destabilize the ionic transition state to become an anticatalyst.29
Cleland and Kreevoy proposed that a strong LBHB formed during the reaction transition state when the pKa values of the donor and acceptor atoms become equal and lower the transition state energy, thus facilitating the proton transfer.21 This hypothesis was tested by measuring the binding affinities of a series of phenolate substrates toward ketosteroid isomerase (KSI) by systematically varying the pKa of phenolates that, in turn, modulated the short H-bond between the O of phenolate and the active site tyrosine.30 This study revealed that the change in the pKa of thiolate could be correlated to a change in enthalpy (ΔH) but not to free energy (ΔG) for substrate binding. This suggested that LBHB may not offer additional stability to the ligand binding as compared to the conventional H-bond but may facilitate the proton transfer by reducing the energy barrier in the reaction catalyzed by KSI.23,30
Highlighting the functional diversity of LBHB, a recent study showed that the interaction can also contribute to ligand binding affinity. For example, a substrate that forms LBHB with aminoglycoside inactivating enzyme has a higher affinity and an increased catalytic turnover as compared to a substrate that forms a conventional H-bond.31 An ultra-high-resolution structure of multimeric human transketolase combined with mutational analysis showed that LBHB contributed to long-range cooperativity between the enzyme’s active sites. The interaction also synchronized the catalysis by the remote active sites (Figure 1E–G).32 It has been demonstrated that LBHB formed by the chromophore of the green fluorescent protein leads to the modulation of the emission wavelength.33,34 A very recent ultra-high-resolution structure of the well-studied light-driven proton pump bacteriorhodopsin revealed a series of conventional H-bonds and LBHBs that was shown to be necessary for signaling and proton storage.35
SHBs or LBHBs, which are enthalpically stronger than the conventional H-bonds, might control the kinetics of the biological processes, such as protein folding, molecular recognition, and ligand binding. It is highly likely that there are many more LBHBs to be discovered that influence protein structure and function. As ultra-high-resolution structures of proteins are a rarity, it is also possible that many functionally important LBHBs remain undiscovered. The identification of LBHB is further compounded by their occurrence in transition states of enzymatic reactions that are not easy to crystallize for structure determination or study by spectroscopic methods. The ongoing revolution in electron cryo-microscopy (cryoEM) has provided the scope to visualize individual atoms in proteins, including a hydrogen atom.36 Consequently, cryoEM is likely to contribute enormously to the identification of LBHB and toward the understanding of its functional role.
2.2. C5 Hydrogen Bond
The intraresidue H-bond between carbonyl O and amino N was recently identified and called a C5 H-bond37,38 (Figure 1H). Here, the main-chain amide group forms a H-bond with the main-chain carbonyl O of the amino acid. The overlap of the p-type lone pair of O with σ* of the N–H bond drives the formation of the C5 H-bond.37 Such a C5 H-bond is prevalent in proteins, particularly in extended β-sheet conformation.37 In a PDB analysis, 5% of all protein residues and 94% of protein structures in a nonredundant set of proteins were found to have a C5 H-bond.37 Almost 62% of these residues were part of β-sheets. The analysis further showed that 14% of residues of antiparallel β-sheets and 9% of residues of parallel β-sheets formed the C5 H-bond. The difference in the C5 H-bond frequency in the two secondary structures is attributed to the difference in the arrangement of the conventional interstrand H-bonds in the two secondary structures.37
The 1H NMR chemical shift and circular dichroism-based thermal denaturation studies of model antiparallel β-hairpins have shown that the perturbation of the C5 H-bond destabilizes peptide structures.37 Using spectroscopy-based experiments along with quantum chemistry calculations, the presence of the C5 H-bond in a model dipeptide molecule in the gas phase was recently confirmed.38 Another study identified that a weak C5 H-bond can have a stabilizing role in cooperation with other conventional H-bonds.39 It was shown that synergy between the two kinds of interactions stabilizes the extended conformation of an α-amino acid peptide in the gas phase and low-polarity solution.39 Although C5 H-bonds are common in proteins, more studies are required to investigate the significance of C5 H-bonds in protein structure and function.
2.3. C–H···π Interaction
The C–H···π interaction involves a C–H group as an H-bond donor and π electrons as an acceptor (Figure 2A). It is geometrically similar to other H-bonds, and their high abundance in proteins makes the C–H···π interactions biologically interesting.40,41 Ab initio calculations suggest that the strength of the interaction ranges from −1.5 to −2.5 kcal mol–1, which is similar to that of a C–H···O interaction.41 Many recent studies showed that dispersion and electrostatic components drive the formation of the interaction.41,42 Surprisingly, contribution due to the hydrophobicity of the CH and π-containing groups in their association to form C–H···π interactions is negligible. Osmometry-based measurements show that the strength of the C–H···π interaction between aliphatic–aromatic motifs is 3-fold higher than that of the hydrophobically driven aliphatic–aliphatic counterparts.43
Figure 2.

C–H···π bond and S-mediated H-bond. (A) Geometry and stereochemical parameters used to identify the C–H···π bond in the biomolecules.44 (B) The observation of J-coupling in an NMR-based experiment between the methyl and π groups in ubiquitin (PDB ID: 1UBQ) because of the C–H···π interaction. The values are adapted from ref (46). (C) NMR structure of a designed miniprotein, PPα-Tyr, and magnified regions showing the C–H···π interactions stabilizing the association of the α-helix and polyproline II helix in PPα-Tyr (PDB ID: 5LO2). The distances for all C–H···π interactions were <2.6 Å. (D) The geometry of aliphatic and aromatic stacking driven by multiple C–H···π interactions. (E) The geometrical and stereochemical parameters used to identify the S-mediated H-bonds in biomolecules.64 The favorable values of these parameters are as follows: dH···S = 2.74 Å, dD···S = 3.52 Å, dH···A = 2.51 Å, dS···A = 3.50 Å, θD–H–S = 141.1°, θH–S–X = 119°, θX–S–Hp = 137°, θA–H–S = 136.5°, and θA′–A–H = 117.4°. (F) A representative example of the S-mediated H-bond (S–H···O interaction) stabilizing the C-terminus of an α-helix by helix capping (PDB ID: 1CPV).
The C–H···π interaction can be identified using three structure-based parameters: (1) the distance between C and the center of mass (X) of the π ring ≤4.5 Å; (2) the angle between C–H···X ≥ 120°; and (3) the distance between Hp and X is within 1.0–1.2 Å, where Hp is H’s vertical projection on the π system (Figure 2A).44 Given the lack of positional information on H atoms in most protein structures, programs, such as CHPI, use the computationally optimized H position to identify and obtain reliable geometry of the C–H···π interaction.45 Observation of J-coupling between the methyl and π group forming the C–H···π interaction has provided direct experimental evidence for the interaction in proteins (Figure 2B).46 Also, an upfield shift for H of Cα of a tripeptide interacting with either tryptophan, phenylalanine, tyrosine, or histidine is additional evidence for a C–H···π interaction in peptides.47 The experimentally measured stability provided to this tripeptide by the C–H···π interaction was measured using NMR to be up to −1.0 kcal mol–1.47
In proteins, three-quarters of tryptophans, one-half of phenylalanines/tyrosines, and one-quarter of histidines form C–H···π interactions, which are often water shielded. The interactions are proposed to contribute to the favorable folding enthalpy.44 Similar to conventional H-bonds, a network of C–H···π interactions, which are frequent in intra- and intersecondary structures, could also stabilize proteins.44,48 For example, the altered thermostability of designed miniproteins by a single C–H···π interaction supports this argument (Figure 2C).49 Apart from this, the interaction also plays a crucial role in biomolecular recognition. In particular, its role in the carbohydrate–protein interaction is well documented.50,51 The stacking of the aliphatic sugar ring of carbohydrates with aromatic protein residues is mainly because of multiple C–H···π interactions. It is considered a driving force behind the carbohydrate–protein association (Figure 2D).50,51 A recent study showed that 39% of the structures of all protein–carbohydrate complexes in PDB have a C–H···π interaction and could contribute up to −8 kcal mol–1 to the overall binding resulting from the sum of all noncovalent interaction energies.52 Similarly, the analysis of 130 structures of protein–DNA complexes from PDB revealed that 40% of all contacts had sugar−π stacking comprising C–H···π and lone pair···π interactions, suggesting its critical role in protein–DNA binding and recognition.53
2.4. S-Mediated H-Bond
Divalent S widely occurs in proteins in the amino acids cysteine and methionine. Historically, the S atom has been acknowledged as a capable H-bond donor or acceptor (Figure 2E).54,55 However, the lower electronegativity of S has led to the assumption, in general, that S-mediated H-bonds are weak, and, consequently, their presence is ignored. Detailed studies, in particular from Wategaonkar’s and Biswal’s laboratories, of model organic molecules using a battery of spectroscopic techniques and quantum chemical calculations have revealed the precise nature and strength of the interaction. The S-mediated H-bonds have strengths in the range of −4.5 to −5.5 kcal mol–1, which is comparable to those of the conventional H-bonds formed by O and N.56−62 The interactions have electrostatic, charge transfer, and dispersion components, with the dispersion component being significantly higher than that observed in conventional H-bonds. S is also involved in forming a weak C–H···S H-bond, which has properties similar to those of the more common and better studied C–H···O H-bond.63
In proteins, the thiol group of cysteine can be a H-bond donor or acceptor, while the S in methionine and disulfide-bonded cystine can be an acceptor. The analysis of a large number of protein structures with computationally optimized hydrogen positions shows that the average dS–H···A is ∼2.8 Å (dS···A ≈ 3.5 Å) and θS–H···A is ∼140° (Figure 2E),64 instead of being close to 180°.13 The longer length of a S-mediated H-bond in comparison to that of a conventional H-bond (dD···A ≈ 2.9–3.0 Å)13 is presumably because of the larger size of the S atom and its diffused electron cloud (Figure 2E).59,65,66 Many protein structures in the PDB have selenomethionine as a replacement for methionine to facilitate structure determination using the anomalous X-ray scattering properties of Se. These structures are a resource to study Se-mediated H-bonds in proteins.62 An analysis of PDB structures reveals a significantly higher number of Se-mediated H-bonds with water than with amino acids.62
Divalent S in proteins has almost a 5-fold higher preference to be a H-bond donor than an acceptor, which makes the sulfhydryl group of reduced cysteine interesting.64 The thiol group of cysteine can also form S–H···π interactions with aromatic amino acids, which can stabilize the protein structures.65−67 The S-mediated H-bond can singularly or together with other interactions play an important role in protein structure and function. For instance, the H-bond between the sulfhydryl group of cysteine (i+4th residue) and carbonyl O (ith residue) stabilizes the C-terminal of α-helices by a mechanism commonly referred to as helix capping (Figure 2F).68,69 Disulfide-bonded and metal-chelated cysteines can participate in N-terminal helix capping.70−73 Removing one such H-bond between the S of iron (Fe) chelated cysteine and the hydroxyl side chain of a threonine/serine in Rieske iron–sulfur protein decreases the midpoint potential of the Fe–S assembly, in turn compromising the activity of cythochrome bc1.74
A charge-density analysis of an iron–sulfur protein using X-ray data at an ultrahigh resolution of 0.48 Å revealed the distorted valency density of S atoms of the iron–sulfur cluster and the coordinating cysteine interacting with H of main-chain amides and also those bonded to C of hydrophobic residues of the protein.75 The analysis indicated modulation in the negative charge of S due to charge transfer from S to the interacting H. Further highlighting the importance of S-mediated H-bonds, charge-density analysis of very-high-resolution X-ray data of the enzyme BacC revealed a network of noncovalent interactions mediated by the thiol group of the three cysteines, which when disrupted by mutagenesis had a significant effect on the enzyme activity.76 Compensatory mutation re-established the network.
3. London Dispersion Interaction
The attractive or negative term in the Lennard-Jones equation (eq 1), which arises from the quantum mechanical electron correlation effect, represents a nondirectional London dispersion interaction.77 This interaction is also referred to as a van der Waals (vdW) dispersion or a van der Waals interaction due to the pioneering work of J. D. van der Waals and F. London.
| 1 |
The original definition provided by London states that it is “an interaction characterized by a short-period mutual perturbation of the inner electron motion of molecules whose magnitude is the major attractive contribution in the simplest nonpolar and also weakly polar molecules.”78,79 The strength of interacting instantaneous fluctuating dipoles between neighboring atoms depends on their polarizability. The distance separating these dipoles is fundamental to the London dispersion interaction, and the interaction energy is proportional to 1/r6 (eq 1).77,80,81
The interaction among nonpolar or weakly polar molecules primarily accounts for the London dispersion interactions.81 The gas-phase calculated strength of the methane dimer (CH4···CH4) is around −0.4 kcal mol–1 and is often considered as a benchmark for the analysis involving London dispersion interactions (Figure 3A).82 The direct correlation between the number of C atoms in n-alkanes and their interaction energy indicates that the London dispersion interaction is additive and nonsaturating in nature. Thus, it is responsible for a linear correlation between the sizes of the hydrocarbons and their increasing melting point.81−83
Figure 3.

London dispersion interaction. (A) The two standard geometries of methane dimer forming the London dispersion interaction. The favorable distances for the C···C interaction are 4.2 (for the geometry in the top panel) and 5.0 Å (for the geometry in the bottom panel), respectively.86 (B) A valence bond model showing the domination of the CH···HC interaction by charge alteration (top panel) for small alkanes and recoupling of bonding electrons to form H···H, C···C, and C···H bonds for large alkanes (bottom panel). Reproduced with pemission from ref (84). Copyright 2013 American Chemical Society. (C) Experimental 3D deformation density map (top panel) and a molecular graph (bottom panel) with bond path and bond critical point for the CH3···CH3 interaction. The red regions in the 3D deformation density map represent charge depletion, and the blue regions represent charge concentration. Adapted from ref (85). Copyright 2019 American Chemical Society. (D) The observation of vdWJCC coupling in the NMR-based experiment between nonpolar residues in protein GB3 (PDB ID: 1IGD) because of the CH···CH London dispersion interaction (the values are from ref (86)). (E) The enthalpy-driven bonding of 2-methoxy-3-isobutylpyrazine (IBMP) to a variant of MUP (PDB ID: 1YP6). IBMP does not form any polar interaction with the surrounding protein residues.
The short C–H···H–C contacts stabilize these n-alkane dimers. The ability of these contacts to stabilize dodecahedrane dimer by ∼ –3 kcal mol–1 is clear evidence for a more substantial effect of the London dispersion interaction in branch chain alkanes and polyhedranes.82 The underlying mechanism for the C–H···H–C contacts depends on the size of the alkanes as per the valence bond model (Figure 3B).84 For smaller alkanes, charge alteration C+–H–···H+–C– dominates the interaction (Figure 3B, top panel), whereas, for large alkanes, the bonding electrons of the CH groups reorganize themselves in a way that recouples these electrons to form H···H, C···C, and C···H bonds (Figure 3B, bottom panel).84 Recently, the electronic nature and features of electron density distribution for such H···H, C···C, and C···H bonds were investigated experimentally in biologically relevant small nonpolar molecules using X-ray diffraction (Figure 3C).85 The CH3···CH3 interaction was found to be stabilized by minimizing the unfavorable electrostatic interaction and enhancing the dispersion interactions (Figure 3C).
A saturated C in the form of aliphatic nonpolar amino acids is common in proteins. An ultra-high-resolution PDB analysis has shown ∼4.5 pairs of contacts per residue with a distance of ∼4.1 and ∼5.4 Å for the two most preferred geometries of C···C interactions (Figure 3A).86 An observation of through-space van der Waals coupling for different C···C interactions provides direct evidence for the London dispersion interaction in proteins (Figure 3D).86 Stabilization of protein structures by −CH2– by up to −1.1 kcal mol–1 could result from the favorable desolvation entropy of nonpolar groups (hydrophobic interaction) followed by an enthalpic contribution from their packing/optimization by the London dispersion interaction at the core of proteins.87
It is challenging to segregate the London dispersion interaction from the hydrophobic interaction.88 However, despite the lack of hydrophobic interaction in membrane proteins, the energetic contribution for packing nonpolar residues in membrane protein is similar to that for soluble proteins. This hints at the possible contribution of a London dispersion interaction in the stability of the protein structures.89 The London dispersion interaction between an electron-deficient carbonyl C and an electron-rich sp3 C of an amino acid side-chain (C···C) is proposed to stabilize the self-assembly of an antiparallel β-sheet in a short peptide noted in its crystal structure.90 This short peptide has a therapeutic application highlighting the role of London dispersion in designing such molecules. The London dispersion interaction can also be crucial for the protein–ligand interaction. The enthalpy-dominated binding of the nonpolar ligands Tween 40 to human serum albumin and of 2-methoxy-3-isobutylpyrazine (IBMP) to the mouse major urinary protein (MUP) suggests a contribution from the London dispersion interaction instead of the expected hydrophobic effect (Figure 3E).91,92 Solvent isotopic substitution isothermal titration calorimetry (ITC) measurements and all-atom molecular dynamics simulations with the inclusion of water confirmed that this enthalpic contribution does not come from ligand or protein pocket desolvation.92
4. Anion···π and Anion–Aromatic Interactions
Aromatic ringed side chains in proteins participate in a variety of interactions, of which π···π stacking and cation···π are the most common and widely discussed.93−103 Another π-mediated interaction that has been studied in detail in the past decade is the anion···π interaction. The interaction forms between an electron-rich ion/molecule (anion) and electron-deficient or π-acidic ring of an aromatic group carrying a positive quadrupole moment (Figure 4A).104 In addition to its electrostatic nature, some contribution from the anion-induced polarization adds to its attractive character.104 The experimentally observed strength of the anion···π interaction between aspartate and phenylalanine is −1.3 kcal mol–1.105 This interaction is widely found in small molecules and extensively used in crystal engineering, supramolecular chemistry, and catalysis.106
Figure 4.
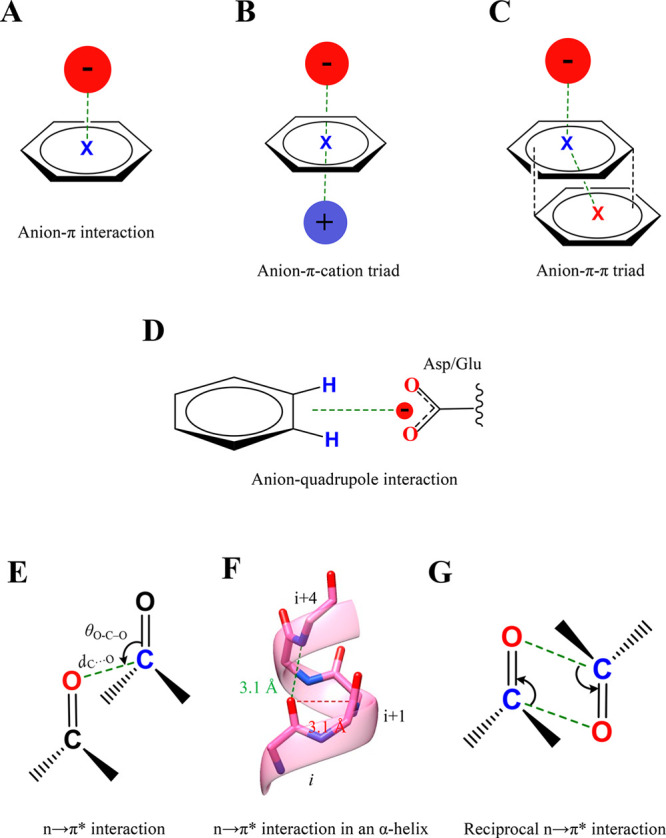
Anion−π, anion–aromatic, and n → π* interactions. (A) A general representation of the anion···π interaction. (B) Anion−π–cation and (C) anion−π–π triads representing the cooperative nature of anion···π interactions. (D) The geometry of an anion–quadrupole interaction. (E) A general representation of the n → π* interaction formed between two carbonyl groups with the geometrical parameters that characterize the interaction. (F) The n → π* interaction formed between the ith and i+1th residues in the α-helix (red). (G) A representation of a reciprocal n → π* interaction.
In PDB, contacts between aromatic residues with chloride and phosphate ions are abundant, mostly within 3.5–4.5 Å.107 Almost 61% of PDB structures are found to have at least one anion−π interaction, primarily involving aspartate or glutamate residues.108 Among the aromatic residues, histidine has a high preference to form anion···π as compared to other aromatic residues. These contacts are presumed to be with protonated histidine, which has an electron-deficient aromatic ring.107 This leads to the question as to how the electron-rich π-ring of other aromatic residues interacts with anions. This appears to be made possible by cooperatively incorporating another stacking interaction forming anion−π–π or anion−π–cation triads (Figure 4B,C).108 The triad formation introduces a positive quadrupole moment for an aromatic ring that interacts with anions. Such an interplay among stacking interactions is well established in small molecules.4 These triads appear essential for folding and specificities in biomolecular structures. The following preference for triad formation is observed: (1) Glu-Tyr-Phe in proteins; (2) Asp-Trp-Phe at protein–protein interfaces; (3) Asp/Glu-Phe-Arg in interchain interactions; (4) Glu-adenine-Arg in the protein–RNA complex; and (5) Asp-His-thymine or Asp-cytosine-adenine in the protein–DNA complex.108
One of the first examples of the functional importance of the anion···π interaction was noted in the hydroxylation of uric acid by molecular O catalyzed by the enzyme urate oxidase. A crystal structure of urate oxidase showed that the bound 8-azaxanthine, an inhibitor and an analogue of uric acid, interacts with molecular O by anion···π interactions.109 The observation of an anion···π interaction between uric acid and cyanide or chloride, which inhibits the enzymatic reaction, in the crystal structure with urate oxidase further substantiated the nature of the interaction.109 A streptavidin variant, obtained using directed evolution, was able to carry out enantioselective addition by stabilizing the anionic transition state via the anion···π interaction.110 The ability of the negatively charged nitrate ion to inhibit the activity confirmed the nature of the interaction. These results indicate the potential use of the anion···π interaction in chemoenzymatic asymmetric synthesis or biocatalysis.110−112
In addition to the anion interacting with the π region of the aromatic rings, its interaction with the edge of the ring is also frequent in proteins and is referred to as the anion–aromatic or anion–quadrupole interaction (Figure 4D).113−115 The distance of ≤4.5 Å between the ring center and anion implies a favorable interaction that is driven by electrostatics.113,114 The calculated strength of this edgewise interaction can be up to −8.0 kcal mol–1.113,114 A PDB survey indicates that aromatic residues prefer an anion–quadrupole more than an anion−π interaction, possibly because of the unfavorable interaction between these electron-rich moieties.115 Of all of the aromatic residues interacting with anions, 62.9% of His, 55.3% of Tyr, 46.4% of Trp, and 27% of Phe residues form the anion–quadrupole interaction.115 On the contrary, only up to ∼8% of these residues interact with anions via the π region to form an anion−π interaction.115 Anion–quadrupole interactions exist at protein–nucleic acids and protein–membrane interfaces, and the significance of the interaction at these interfaces is underappreciated.114 An example of a functionally important anion–quadrupole interaction is the interaction between the catalytic aspartate of ketosteroid isomerase and two neighboring phenylalanine residues that stabilizes the orientation of the aspartate.116 Removing these interactions by mutagenesis compromises the catalytic efficiency of the enzyme.116 The example highlights the potential role of anion–quadrupole interactions in establishing amino acid networks involving negatively charged amino acids and aromatic residues with structural and functional significance. The interaction can also stabilize binding of anion-containing or aromatic ligand binding to proteins with implications to drug design.
5. n → π* Interaction
A carbonyl group (C=O) forms a permanent dipole due to the polarization of electron density toward O as it is more electronegative than C. As a result, the O is partially negative charged and the C is partially positive. Thus, an interaction is established between two carbonyl groups (C=O···C=O), which are abundant in proteins (Figure 4E).117 Earlier, this interaction was considered a dipolar interaction of electrostatic origin.118,119 However, Raines and others confirmed its charge transfer nature that arises from the delocalization of electron density of the lone pair of O to the antibonding orbital (π*) of the carbonyl group.40,120−124 This interaction is now referred to as the n → π* interaction.120,123,125 The estimated interaction energy for a n → π* interaction ranges from −0.3 to −0.7 kcal mol–1.40 One-third of all residues in proteins could participate in the n → π* interaction because of the abundance of the main-chain carbonyl group. Most of them are from α-helices (>70% of all residues in α-helices) and are believed to stabilize the secondary structure in proteins (Figure 4F).117 The favorable distance for the interaction is ∼3.0 Å with θO–C–O ≈ 102.0° (Figure 4F).117 Also, two n → π* interactions formed back and forth within the two carbonyl groups (Figure 4G), referred to as reciprocal n → π* interactions, are often part of polyproline π-helices and can influence protein folding.126
6. σ-Hole Interaction
Besides the noncovalent interactions discussed above, another class of noncovalent interactions formed by P-block elements, called the σ-hole interaction,127−135 also occurs widely in proteins. Politzer and colleagues defined σ-hole as “the electron-deficient outer lobe of a half-filled p (or nearly p) orbital involved in forming a covalent bond. If the electron deficiency is sufficient, there can result a region of positive electrostatic potential which can interact attractively (noncovalently) with negative sites on other molecules (σ-hole bonding).”127,131 Thus, the anisotropic electron density distribution of the covalently bonded atom results in the electron-deficient region (σ-hole) diametrically opposite to the covalent bond (Figure 5A). The interaction of this positive electrostatic potential region with nucleophiles is called the σ-hole interaction.131 The group VII, VI, V, and IV elements from the periodic table can donate the σ-hole (analogous to the H atom in H-bonds) to form σ-hole interactions and are commonly named halogen, chalcogen, pnicogen, and tetrel bonding interactions, respectively134 (Figure 5B). The σ-hole interactions have electrostatic nature with a small contribution from dispersion and charge transfer components.130,136−138 In the following sections, we have presented a summary of this new class of noncovalent interactions and their relevance to protein structure and function.
Figure 5.
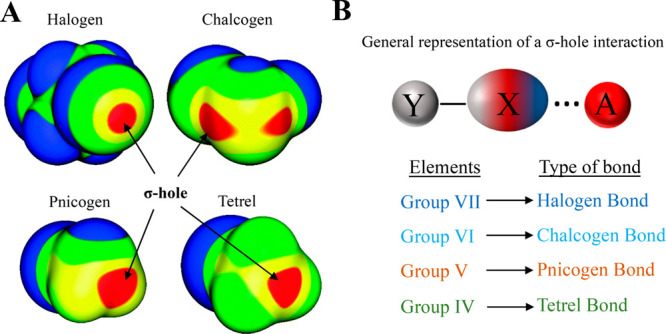
The σ-hole bonding interaction. (A) Molecular electrostatic potential maps for ICF2CF2I, SeFCl, GeH3Br, and PH2Cl adapted from ref (141) (Copyright 2013 Royal Society of Chemistry), showing the σ-hole on a representative example from Type VII (halogen), VI (chalcogen), V (pnicogen), and IV (tetrel) elements, respectively. Maps were computed on the 0.001 au density level. Strength of the σ-holes is highlighted with color coding: red represents >25 kcal mol–1; yellow represents between 15 and 25 kcal mol–1; green represents between 0 and 15 kcal mol–1; and blue represents <0 kcal mol–1. (B) A general representation of the σ-hole bonding interaction formed between a σ-hole donating atom (X) with an electron-rich acceptor atom/molecule (A). Here, Y represents any electron-withdrawing atom/group. The classification of σ-hole bonding interactions on the basis of the type of element from the periodic table that donates the σ-hole.
6.1. Halogen Bonding Interaction
Halogen bonds (X-bonds) are a type of σ-hole interaction formed by chlorine, bromine, and iodine atoms and are common in biomolecules.138−147 According to IUPAC, “a halogen bond occurs when there is evidence of a net attractive interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity.”142 The geometrical parameters to identify the X-bond in biomolecules are provided in Figure 6A.139 Direct imaging of the σ-hole in real space using Kelvin probe force microscopy confirmed the electrostatic nature of the X-bond (Figure 6B).148 Its experimentally measured enthalpic contribution in small molecules is 4–5 kcal mol–1.149
Figure 6.

Halogen bond. (A) The geometric parameters used to investigate the X-bond in biomolecules. The value of dX···O is less than or equal to the sum of the van der Waals radii of X and O. The most favorable values of θ1 and θ2 are ∼170–180° and ∼113°, respectively.139 (B) The top panel shows the molecular electrostatic maps of the tetrakis (4-bromophenyl) methane (4BrPhM) and tetrakis (4-fluorophenyl) methane (4FPhM) molecules, revealing the presence of a σ-hole on Br. The bottom panel shows the 3D representation of the VLCPD maps acquired using Kelvin probe force microscopy for the 4BrPhM and 4FPhM molecules, providing evidence of the presence of a σ-hole on the Br atom. For the VLCPD maps, blue represents low values, whereas red indicates high values. Adapted from ref (148). Copyright 2021 AAAS. (C) A structural snapshot of a derivative of bacterial type II topoisomerase inhibitors (NBTIs) bound to Staphylococcus aureus DNA gyrase. A bifurcated X-bond is shown between Cl (light green) of the derivative of NBTI and the main-chain O of alanine from a symmetry-related protein molecule (PDB ID: 6Z1A). (D) A segment of the 2D static (top panel) and 3D (bottom panel) deformation density maps obtained from the experimentally determined charge-density distribution for 2,5-dichloro-1,4-benzoquinone evidencing C–Cl···O=C halogen bond formation. Adapted from ref (159). Copyright 2011 American Chemical Society. Red regions in the 3D deformation density map represent charge depletion, and blue regions represent charge concentration.
X-bonds are prevalent in protein–ligand complexes.146,150−154 The appearance of halogen atoms in ∼50% of high-throughput drug screens that could potentially form X-bonds makes them pharmacologically relevant.147 Thus, incorporating X-bonds in force field methods becomes essential to model them accurately.155−157 For instance, a symmetrically bifurcated X-bond by −Cl of an inhibitor of the DNA gyrase with the protein’s main-chain O reveals the mechanism of action of the inhibitor (Figure 6C).158 Topological features such as bond critical points, deformation density, and electrostatic potential obtained from the X-ray charge-density analysis provide experimental evidence for the C–Cl···O=C bond in small molecules (Figure 6D).159 An example of the utility of X-bond in drug design is the incorporation of the interaction in designing better derivatives of selective serotonin reuptake inhibitor toward their target serotonin transporters.160
A high abundance of intra- or intermolecular X-bonds made by halogenated bases in nucleic acid in PDB suggests its role in controlling conformation or participating in protein–nucleic acid recognition.161−164 H- and X-bonds can compete for a common acceptor,165 and it has been found that polar solvents favor the X-bond over the H-bond, while an opposite trend has been noted for nonpolar solvents.166 The synergistic relation between the H- and X-bond167 opens the possibility of modulating the stability and function of biomolecules in an aqueous environment. For instance, re-engineering of the X-bond could alter the local fold of the proteins168 or the thermal stability, activity, and substrate selectivity of the enzymes.169−171
6.2. Chalcogen Bonding Interaction
Group VI elements, mainly divalent S, Se, or Te, can interact with various nucleophiles to form another set of σ-hole interactions called chalcogen bonds (Ch-bond) (Figure 5B).130,134,136−138 IUPAC defines the Ch-bond as a “net attractive interaction between an electrophilic region associated with a chalcogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity.”172 Although σ-hole interactions are considered to be of an electrostatic origin, an NMR-based experiment suggested that charge transfer is the primary stabilizing force for the Ch-bond.173 This charge transfer arises from electron donation from nucleophiles to the antibonding orbital (σ*) of the chalcogen atom (n → σ*).173 Although this observation and a few theoretical analyses resulted in a debate on the origin of the Ch-bond, its electrostatic nature is still widely acknowledged.174−178
The divalent S is abundantly present in proteins in the form of methionine and cysteine. CSD and PDB data analyses have provided many instances of a Ch-bond in small and protein molecules68,178−180 (Figure 7A). Statistical analysis of proteins revealed that 22% of S from methionines had a short contact with carbonyl O where dS···O was <5 Å180 (Figure 7B). Most of these contacts had θ as being greater than 40° and Φ between 30° and 60°. A more comprehensive analysis revealed that the divalent S preferred to approach the π region over the lone pair region of carbonyl O, indicating the importance of directionality for the formation of the interaction in proteins.181 A recent study showed that this directional selectivity originates from the electrostatic and cooperative nature of the Ch-bond.182 Also, 1.3% and 8.9% of methionine and cysteine in proteins could participate in Ch-bond formation, respectively, suggesting their role in stabilizing protein structures.181 Furthermore, S could interact with −OH or – NH groups in proteins and could participate in either H- or Ch-bond formation.69 The lack of positional information on H in most of the structures determined using X-ray crystallography or electron cryo-microscopy makes the identification of the interaction nontrivial. To overcome this, a directional-based strategy, independent of the positional information on H, has been devised to identify the nature of the S-mediated interactions69 (Figure 7C).
Figure 7.

Chalcogen and tetrel bonding interaction. (A) A representation showing the distinct approach of electrophiles and nucleophiles toward divalent S. (B) The geometric parameters used to investigate the Ch-bond in proteins. The most common value of dS···O is 3.6 Å, where θ > 50° and φ ranges from 30°–60°.180 (C) The definition of angular parameters used to delineate the S-mediated Ch- from the H-bond. The most favorable values of θ are within 95–145°, whereas values of δ range from −90° to −50° or from 50° to 90° for H-bonds in proteins.69 In the case of the Ch-bond, the most common values of θ are from 115° to 155°, and δ ranges from −50° ≤ δ ≤ 50°.69 (D) Static 3D deformation density map for acetazolamide (left) and a molecular graph showing the bond path and critical bond point in the dimer of acetazolamide (right), evidencing intramolecular Ch-bond formation. The BCP for the S···O interaction is highlighted with black arrows. Adapted from ref (187). Copyright 2015 Royal Society of Chemistry. Red regions in the 3D deformation density map represent charge depletion, and blue regions represent charge concentration. (E) An electrostatic S+···Oδ− interaction at the active site of histone-lysine N-methyltransferase SET7/9 (PDB ID: 4J83). (F) A representative example showing the conserved C···O tetrel bond in AdoMet-dependent methyltransferase (PDB ID: 5VSC). (G) A segment of the 3D deformation density map and the experimentally obtained bond path with BCP C···O C-bonding in dimethylammonium 4-hydroxybenzoate. Red regions in the 3D deformation density map represent charge depletion, and blue regions represent charge concentration. Adapted from ref (200). Copyright 2014 Royal Society of Chemistry. (H) The geometric parameters used to investigate the C-bond in proteins. The following distance and angular criteria are used to find the C-bond in proteins: dC···O = 2.5–3.6 Å, θ1 that is within 160–180°, and θ2 that is within 160–180°. Adapted from ref (198). Copyright 2018 Wiley.
Divalent selenium (Se) from selenomethionine is also abundant in PDB. The Ch-bond formed by Se from selenomethionine is found to have geometrical parameters similar to but of higher strength than the Ch-bond formed by S and is proposed to stabilize the proteins containing them.183,184 Similarly, a PDB survey found that complexes of Se-containing carbohydrates with proteins could stabilize their complexes via Ch-bond,185 thus participating in protein–carbohydrate recognition. In addition, the analysis of PDB data suggests that Ch-bonds are common in protein–ligand complexes that involve aromatic S.183,186 The Ch-bond between S-containing aromatic ligands, many of which are drug molecules, with protein amino acids has been confirmed experimentally (Figure 7D), thus indicating the importance of the interaction for target-based drug design.187,188
Akin to H-bonds, a recent PDB and computational analysis carried out by us showed that the Ch-bond can stabilize protein secondary structures.69,189 The Ch-bonds are found to be conserved in phospholipase A2 domains, ribonuclease A, lysozyme, and proteins of the insulin family.190,191 These Ch-bonds are proposed to contribute to their enzymatic activities, conformational rearrangement at the active site, thermodynamic stability, or folding.190,191 The Ch-bonds have a favorable interaction energy (up to −3 kcal mol–1), have highly directional properties, and are independent of solvent polarity. These features make the interaction useful for establishing molecular specificity and recognition.173,181 Interactions different from the Ch-bond can also exist between S and O. Fick et al. showed that the sulfonium ion of S-adenosylmethionine (AdoMet) forms a strong S···O bond with a residue of the SET7/9 protein because of the positively charged trivalent S. This allows SET7/9 to discriminate between product and reactant192 (Figure 7E). However, this interaction differs in origin and features from a σ-hole-mediated Ch-bond.
6.3. Tetrel Bonding Interaction
Group IV elements (C, Si, Ge, Sn, or P) can also donate a σ-hole to form an interaction analogous to those of the X- and Ch-bonds, which is referred to as a tetrel bonding interaction.134,141,193−195 Among the various group IV elements, tetrel bonds formed by the C-sp3 atom, particularly by the methyl group, with O or N or S (X–Csp3···O/N/S interaction) are frequent in proteins. Hence, the interaction is sometimes also called a carbon bond (C-bond).196−198 The calculated strength of the X–C···O/N/S interaction, which can be up to −4 kcal mol–1, depends on the electron-withdrawing capacity of X.196 The strong electron-withdrawing ability of sulfonium cation in AdoMet was found to strengthen the C-bond formed between the methyl group of AdoMet and O of ligand or water at the active site of methyltransferases (MTases) (Figure 7F).197,199 As a result, the tetrel-bond was found to be conserved in the structures of AdoMet-dependent MTases in PDB and proposed to be necessary for stabilizing the SN2 transition state for methyl group transfer.199 Charge-density analysis of small molecules of biological relevance has provided direct experimental evidence for the tetrel-bonding interaction200 (Figure 7G).
A recent statistical analysis revealed 8358 contacts for C-bond that satisfied the distance and angular criteria (refer to Figure 7H) among the protein structures in the PDB.198 The analysis revealed alanine, leucine, aspartate, and glutamate to have a high preference (up to ∼40% together) to act as a tetrel-bond acceptor, whereas lysine was the dominant tetrel-bond donor.198 Although a tetrel-bond was found in all types of secondary structures, residues in coils and turns had a higher preference to be donors, while those in α-helices were acceptors for tetrel-bond formation.198 Another PDB survey found that the O atom of water dominated the formation of the tetrel-bond in proteins (∼57%), followed by main-chain O (∼24%), side-chain O (∼15%), and S (∼4%). The distribution could be correlated with the frequency of these nucleophiles in protein structures.197,201 The tetrel-bond was also found in protein–ligand complexes, although the exact contribution of the interaction toward ligand affinity is yet to be experimentally quantified.202 Interestingly, an enthalpy-driven tetrel-bond and an entropy-driven hydrophobic interaction, both of which involve C atoms, are found to coexist abundantly in proteins. Understanding the effect of the interplay between the tetrel-bond and hydrophobic interactions on the protein structure and stability would be of broad interest in protein engineering and ligand/inhibitor design.
7. Conclusions
Accurately predicting protein structures or their biochemical or biophysical properties, including enzymatic reaction mechanisms, kinetics of protein folding, ligand binding, and enzymatic reactions, from sequence information has been a long challenge.203−209 Progress in ab initio protein structure prediction based on physicochemical principles has been slow, hindered by an incomplete description of the force field and high computational cost.40 A detailed understanding of noncovalent interactions that pervade protein structure and function is essential for an improved description of the force field. The force fields are important for quantum mechanical calculations and molecular dynamics simulations that are the computational tools for understanding protein structure and dynamics, predicting ligand binding, and deciphering the reaction mechanism and kinetics.
The recently developed template-guided or neural network/machine learning-based approaches, such as the artificial intelligence program AlphaFold and RoseTTAFold, have had a revolutionary impact on the prediction of three-dimensional structures of protein with high accuracy.203,210−213 AlphaFold, for example, integrates machine learning strategies and an experimental database for sequence and structural information.214,215 The success of AlphaFold in determining the 3D structures of proteins with reasonably high accuracy has empowered researchers to obtain atomic structures of their protein of interest effortlessly and with great confidence. A recent version of the program has, to a limited extent, been successful in identifying partner proteins and predicting structures of their complexes.
The main limitation of the program so far has been its poor performance in predicting the structures of intrinsically disordered proteins and of multimeric proteins or complexes of proteins with ligands, cofactors, or nucleic acids. In addition, the cofactor/ligand-dependent folding, protein dynamics, and accurate side-chain conformation prediction are also integral to protein structure and function, which current AI-based programs, such as AlphaFold, do with a varying degree of accuracy and requires to be improved.214 Combining structural information from prediction software such as AlphaFold with molecular dynamics simulations and quantum mechanical calculations that take into consideration the various noncovalent interactions and their synergy can address these challenges.
Scrutinizing the forces that stabilize or destabilize the proteins or their complexes and their inclusion into the force field is an important step toward achieving this goal.37,40,60,115,118 So far, the favorable contribution of H-bond and hydrophobic interactions to compensate for the unfavorable conformational entropy toward folding the peptide chain and making the folding process favorable is well considered.216 However, their insufficiency in explaining the discrepancies in computational and experimental results highlights the necessity to include other noncovalent interactions, including those summarized in this Review. Although many of these individual interactions appear diminutive in strength, their cumulative effect could strongly alter the stability and biochemical properties of proteins because of their frequent occurrence. Raines in a recent review on secondary forces in proteins concluded that the cumulative enthalpic contribution of these interactions is around 25% as compared to conventional H-bonds or hydrophobic interactions.40 Furthermore, recognizing the significance of these unconventional noncovalent interactions is important to better understand ligand binding, reaction mechanisms, and kinetics.
Precise knowledge of these forces is essential not only to accurately determine protein structures ab initio but also to predict their dynamics or function, which most AI-based programs perform poorly in their current versions. Over the past decade, extensive research efforts employing detailed and multipronged experimental and computational approaches have led to the discovery of new noncovalent interactions and their importance in chemistry and biology. Estimating the critical properties of these interactions precisely, such as the experimental energy of stabilization, environment dependence, and synergy between the interactions, is a topic that requires further attention. We believe that the revolutionary improvement in the resolution of 3D structural data of proteins determined using cryoEM will aid in the identification of unconventional noncovalent interactions in proteins and reveal new roles of these interactions in protein function. With many more protein structures being determined at very- and ultra-high resolution, use of charge-density analysis combined with detailed quantum chemical calculations, such as Atoms In Molecule analysis, will provide unprecedented insights into the nature and function of noncovalent interactions in proteins.75,76,182 We believe that with newer and improved experimental and computational techniques being used for probing noncovalent interactions in small molecules and proteins, our understanding of the nature of these interactions, their role, and significance is only on the rise.
Acknowledgments
The area of noncovalent interactions is an extensively researched topic with scientists working around the world. We apologize to our colleagues from across the globe whose studies on noncovalent interactions could not be cited here due to the focus of this Review and restrictions on the manuscript length. V.A.A. would like to thank the Council of Scientific and Industrial Research (CSIR) for a fellowship. K.S. would like to thank the Department of Biotechnology, Government of India, for a grant to the Indian Institute of Science Education and Research Pune, Department of Biotechnology (DBT), India, under BICB centre grant (BT/PR40262/BTIS/137/38/2022).
The authors declare no competing financial interest.
References
- van der Waals D. J.On the Continuity of the Gaseous and Liquid States; Leiden University: Leiden, The Netherlands, 1873. [Google Scholar]
- Müller-Dethlefs K.; Hobza P.; Mu K.; Hobza P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100 (1), 143–168. 10.1021/cr9900331. [DOI] [PubMed] [Google Scholar]
- Kollman P. A. Noncovalent Interactions. Acc. Chem. Res. 1977, 10 (10), 365–371. 10.1021/ar50118a003. [DOI] [Google Scholar]
- Mahadevi A. S.; Sastry G. N. Cooperativity in Noncovalent Interactions. Chem. Rev. 2016, 116 (5), 2775–2825. 10.1021/cr500344e. [DOI] [PubMed] [Google Scholar]
- Kollman P. Chapter 2 Non-Covalent Forces of Importance in Biochemistry. New Compr.Biochem. 1984, 6, 55–71. 10.1016/S0167-7306(08)60373-7. [DOI] [Google Scholar]
- Morokuma K. Molecular Orbital Studies of Hydrogen Bonds. III. C=O ···H-O HydrogenBond in H2CO···H2O and H2CO ···2H2O. J. Chem. Phys. 1971, 55 (3), 1236. 10.1063/1.1676210. [DOI] [Google Scholar]
- Ziegler T.; Rauk A. On the Calculation of Bonding Energies by the Hartree Fock Slater Method. Theor. Chim. Acta 1977, 46, 1–10. 10.1007/BF02401406. [DOI] [Google Scholar]
- Desiraju G. R.; Steiner T.. The Weak Hydrogen Bond: In Structural Chemistry and Biology; International Union of Crystallography, Monographs on Crystallography, 1999. [Google Scholar]
- Mati I. K.; Cockroft S. L. Molecular Balances for Quantifying Non-Covalent Interactions. Chem. Soc. Rev. 2010, 39, 4195–4205. 10.1039/b822665m. [DOI] [PubMed] [Google Scholar]
- Koritsanszky T. S.; Coppens P. Chemical Applications of X-Ray Charge-Density Analysis. Chem. Rev. 2001, 101 (6), 1583–1628. 10.1021/cr990112c. [DOI] [PubMed] [Google Scholar]
- Dittrich B.; Matta C. F. Contributions of Charge-Density Research to Medicinal Chemistry. IUCrJ. 2014, 1 (6), 457–469. 10.1107/S2052252514018867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. P.; Dikundwar A. G.; Sarkar S.; Pavan M. S.; Pal R.; Hathwar V. R.; Row T. N. G. The Relevance of Experimental Charge Density Analysis in Unraveling Noncovalent Interactions in Molecular Crystals. Molecules 2022, 27, 3690. 10.3390/molecules27123690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey G. A.; Saenger W.. Hydrogen Bonding in Biological Structures; Springer-Verlag: New York, 1994. [Google Scholar]
- van der Lubbe S. C. C.; Fonseca Guerra C. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem.-Asian J. 2019, 16, 2760–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. M.; Graf J. F.; Painter P. C. The Nature of the Hydrogen Bond. Specif. Interact. Miscibility Polym. Blends 2017, 157–170. 10.1201/9780203738986-3. [DOI] [Google Scholar]
- Herschlag D.; Pinney M. M. Hydrogen Bonds: Simple after All?. Biochemistry. 2018, 57 (24), 3338–3352. 10.1021/acs.biochem.8b00217. [DOI] [PubMed] [Google Scholar]
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; Hobza P.; Kjaergaard H. G.; Legon A. C.; Mennucci B.; Nesbitt D. J. Definition of the Hydrogen Bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83 (8), 1637–1641. 10.1351/PAC-REC-10-01-02. [DOI] [Google Scholar]
- Umeyama H.; Morokuma K. The Origin of Hydrogen Bonding. An Energy Decomposition Study. J. Am. Chem. Soc. 1977, 99 (5), 1316–1332. 10.1021/ja00447a007. [DOI] [Google Scholar]
- Kollman P. A.; Allen L. C. The Theory of the Hydrogen Bond. Chem. Rev. 1972, 72 (3), 283–303. 10.1021/cr60277a004. [DOI] [Google Scholar]
- Frey P. A.; Whitt S. A.; Tobin J. B. A Low-Barrier Hydrogen Bond in the Catalytic Triad of Serine Proteases. Science. 1994, 264 (5167), 1927–1930. 10.1126/science.7661899. [DOI] [PubMed] [Google Scholar]
- Cleland W. W.; Kreevoy M. M. Low-Barrier Hydrogen Bonds and Enzymic Catalysis. Science. 1994, l264 (5167), 1887–1890. 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- Hosur M. V.; Chitra R.; Hegde S.; Choudhury R. R.; Das A.; Hosur R. V. Low-Barrier Hydrogen Bonds in Proteins. Crystallogr. Rev. 2013, 19 (1), 3–50. 10.1080/0889311X.2013.771633. [DOI] [Google Scholar]
- Kemp M. T.; Lewandowski E. M.; Chen Y. Low Barrier Hydrogen Bonds in Protein Structure and Function. Biochimica et Biophysica Acta - Proteins and Proteomics 2021, 1869 (1), 140557. 10.1016/j.bbapap.2020.140557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.; Wang L. Unraveling the Structural and Chemical Features of Biological Short Hydrogen Bonds. Chem. Sci. 2019, 10, 7734–7745. 10.1039/C9SC01496A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal S.; Vishveshwara S. Short Hydrogen Bonds in Proteins. FEBS J. 2005, 272 (8), 1819–1832. 10.1111/j.1742-4658.2005.04604.x. [DOI] [PubMed] [Google Scholar]
- Overgaard J.; Schiøtt B.; Larsen F. K.; Iversen B. B. The Charge Density Distribution in a Model Compound of the Catalytic Triad in Serine Proteases. Chem. - A Eur. J. 2001, 7 (17), 3756–3767. . [DOI] [PubMed] [Google Scholar]
- Kuhn P.; Knapp M.; Soltis S. M.; Ganshaw G.; Thoene M.; Bott R. The 0.78 Å Structure of a Serine Protease: Bacillus Lentus Subtilisin. Biochemistry 1998, 37 (39), 13446–13452. 10.1021/bi9813983. [DOI] [PubMed] [Google Scholar]
- Agback P.; Agback T. Direct Evidence of a Low Barrier Hydrogen Bond in the Catalytic Triad of a Serine Protease. Sci. Rep. 2018, 8, 10078. 10.1038/s41598-018-28441-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Papazyan A.; Kollman P. A.; Cleland W. W.; Kreevoy M. M.; Frey P. A. On Low-Barrier Hydrogen Bonds and Enzyme Catalysis. Science 1995, 269 (5220), 102–106. 10.1126/science.7661987. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Sigala P. A.; Pybus B.; Liu C. W.; Ringe D.; Petsko G. A.; Herschlag D. Testing Electrostatic Complementarity in Enzyme Catalysis: Hydrogen Bonding in the Ketosteroid Isomerase Oxyanion Hole. PLoS Biol. 2006, 4 (4), e99. 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Agarwal P. K.; Waddell M. B.; Mittag T.; Serpersu E. H.; Cuneo M. J. Low-Barrier and Canonical Hydrogen Bonds Modulate Activity and Specificity of a Catalytic Triad. Angew. Chemie - Int. Ed. 2019, 58 (45), 16260–16266. 10.1002/anie.201908535. [DOI] [PubMed] [Google Scholar]
- Dai S.; Funk L. M.; von Pappenheim F. R.; Sautner V.; Paulikat M.; Schröder B.; Uranga J.; Mata R. A.; Tittmann K. Low-Barrier Hydrogen Bonds in Enzyme Cooperativity. Nature. 2019, 573, 609–613. 10.1038/s41586-019-1581-9. [DOI] [PubMed] [Google Scholar]
- Di Donato M.; Van Wilderen L. J. G. W.; Van Stokkum I. H. M.; Stuart T. C.; Kennis J. T. M.; Hellingwerf K. J.; Van Grondelle R.; Groot M. L. Proton Transfer Events in GFP. Phys. Chem. Chem. Phys. 2011, 13, 16295–16305. 10.1039/c1cp20387h. [DOI] [PubMed] [Google Scholar]
- Oltrogge L. M.; Boxer S. G. Short Hydrogen Bonds and Proton Delocalization in Green Fluorescent Protein (GFP). ACS Cent. Sci. 2015, 1 (3), 148–156. 10.1021/acscentsci.5b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borshchevskiy V.; Kovalev K.; Round E.; Efremov R.; Astashkin R.; Bourenkov G.; Bratanov D.; Balandin T.; Chizhov I.; Baeken C.; Gushchin I.; Kuzmin A.; Alekseev A.; Rogachev A.; Willbold D.; Engelhard M.; Bamberg E.; Büldt G.; Gordeliy V. True-Atomic-Resolution Insights into the Structure and Functional Role of Linear Chains and Low-Barrier Hydrogen Bonds in Proteins. Nat. Struct. Mol. Biol. 2022, 29, 440–450. 10.1038/s41594-022-00762-2. [DOI] [PubMed] [Google Scholar]
- Yip K. M.; Fischer N.; Paknia E.; Chari A.; Stark H. Atomic-Resolution Protein Structure Determination by Cryo-EM. Nature 2020, 587, 157–161. 10.1038/s41586-020-2833-4. [DOI] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. A Prevalent Intraresidue Hydrogen Bond Stabilizes Proteins. Nat. Chem. Biol. 2016, 12, 1084–1088. 10.1038/nchembio.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Mishra K. K.; Singh S. K.; Borish K.; Dey S.; Sarkar B.; Das A. Observation of a Weak Intra-Residue C5 Hydrogen-Bond in a Dipeptide Containing Gly-Pro Sequence. J. Chem. Phys. 2019, 151, 104309. 10.1063/1.5115040. [DOI] [PubMed] [Google Scholar]
- Mundlapati V. R.; Imani Z.; D’Mello V. C.; Brenner V.; Gloaguen E.; Baltaze J. P.; Robin S.; Mons M.; Aitken D. J. N-H···X Interactions Stabilize Intra-Residue C5 Hydrogen Bonded Conformations in Heterocyclic α-Amino Acid Derivatives. Chem. Sci. 2021, 12, 14826–14832. 10.1039/D1SC05014A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. Secondary Forces in Protein Folding. ACS Chem. Biol. 2019, 14 (8), 1677–1686. 10.1021/acschembio.9b00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M.; Umezawa Y.; Fantini J.; Weiss M. S.; Chakrabarti P. CH-π Hydrogen Bonds in Biological Macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. 10.1039/C4CP00099D. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S. CH/π Interactions. Annu. Reports Prog. Chem. - Sect. C 2012, 108, 69–95. 10.1039/c2pc90003c. [DOI] [Google Scholar]
- Cheng X.; Shkel I. A.; O’Connor K.; Thomas Record M. Experimentally Determined Strengths of Favorable and Unfavorable Interactions of Amide Atoms Involved in Protein Self-Assembly in Water. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (44), 27339–27345. 10.1073/pnas.2012481117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl M.; Weiss M. S.; Jabs A.; Sühnel J.; Hilgenfeld R. C-H···π-Interactions in Proteins. J. Mol. Biol. 2001, 307 (1), 357–377. 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- Umezawa Y.; Nishio M. CH/π Interactions in the Crystal Structure of TATA-Box Binding Protein/DNA Complexes. Bioorg. Med. Chem. 2000, 8 (11), 2643–2650. 10.1016/S0968-0896(00)00197-8. [DOI] [PubMed] [Google Scholar]
- Plevin M. J.; Bryce D. L.; Boisbouvier J. Direct Detection of CH/π Interactions in Proteins. Nat. Chem. 2010, 2 (6), 466–471. 10.1038/nchem.650. [DOI] [PubMed] [Google Scholar]
- Ganguly H. K.; Majumder B.; Chattopadhyay S.; Chakrabarti P.; Basu G. Direct Evidence for CH···π Interaction Mediated Stabilization of Pro-CisPro Bond in Peptides with Pro-Pro-Aromatic Motifs. J. Am. Chem. Soc. 2012, 134 (10), 4661–4669. 10.1021/ja209334v. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Balaji P. V. C–H···π Interactions in Proteins: Prevalence, Pattern of Occurrence, Residue Propensities, Location, and Contribution to Protein Stability. J. Mol. Model. 2014, 20, 2136. [DOI] [PubMed] [Google Scholar]
- Baker E. G.; Williams C.; Hudson K. L.; Bartlett G. J.; Heal J. W.; Goff K. L. P.; Sessions R. B.; Crump M. P.; Woolfson D. N. Engineering Protein Stability with Atomic Precision in a Monomeric Miniprotein. Nat. Chem. Biol. 2017, 13 (7), 764–770. 10.1038/nchembio.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio J. L.; Ardá A.; Cañada F. J.; Jiménez-Barbero J. Carbohydrate-Aromatic Interactions. Acc. Chem. Res. 2013, 46 (4), 946–954. 10.1021/ar300024d. [DOI] [PubMed] [Google Scholar]
- Hudson K. L.; Bartlett G. J.; Diehl R. C.; Agirre J.; Gallagher T.; Kiessling L. L.; Woolfson D. N. Carbohydrate-Aromatic Interactions in Proteins. J. Am. Chem. Soc. 2015, 137 (48), 15152–15160. 10.1021/jacs.5b08424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser J.; Kozmon S.; Mishra D.; Hammerová Z.; Wimmerová M.; Koča J. The CH−π Interaction in Protein–Carbohydrate Binding: Bioinformatics and In Vitro Quantification. Chem. - A Eur. J. 2020, 26 (47), 10769–10780. 10.1002/chem.202000593. [DOI] [PubMed] [Google Scholar]
- Wilson K. A.; Kellie J. L.; Wetmore S. D. DNA-Protein π-Interactions in Nature: Abundance, Structure, Composition and Strength of Contacts between Aromatic Amino Acids and DNA Nucleobases or Deoxyribose Sugar. Nucleic Acids Res. 2014, 42 (10), 6726–6741. 10.1093/nar/gku269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley M. J.; Marvel C. S.; Ginsberg E. Hydrogen Bonding by S-h. VII. Aryl Mercaptans. J. Am. Chem. Soc. 1939, 61 (11), 3161–3162. 10.1021/ja01266a052. [DOI] [Google Scholar]
- Heafield T. G.; Hopkins G.; Hunter L. Hydrogen Bonds Involving the Sulphur Atom [1]. Nature. 1942, 149, 218. 10.1038/149218a0. [DOI] [Google Scholar]
- Biswal H. S.; Gloaguen E.; Loquais Y.; Tardivel B.; Mons M. Strength of NH···S Hydrogen Bonds in Methionine Residues Revealed by Gas-Phase IR/UV Spectroscopy. J. Phys. Chem. Lett. 2012, 3 (6), 755–759. 10.1021/jz300207k. [DOI] [PubMed] [Google Scholar]
- Biswas H. S.; Wategaonkar S. Nature of the N-H···S Hydrogen Bond. J. Phys. Chem. A 2009, 113 (46), 12763–12773. 10.1021/jp907658w. [DOI] [PubMed] [Google Scholar]
- Howard D. L.; Kjaergaard H. G. Hydrogen Bonding to Divalent Sulfur. Phys. Chem. Chem. Phys. 2008, 10, 4113–4118. 10.1039/b806165c. [DOI] [PubMed] [Google Scholar]
- Rao Mundlapati V.; Ghosh S.; Bhattacherjee A.; Tiwari P.; Biswal H. S. Critical Assessment of the Strength of Hydrogen Bonds between the Sulfur Atom of Methionine/Cysteine and Backbone Amides in Proteins. J. Phys. Chem. Lett. 2015, 6 (8), 1385–1389. 10.1021/acs.jpclett.5b00491. [DOI] [PubMed] [Google Scholar]
- Chand A.; Sahoo D. K.; Rana A.; Jena S.; Biswal H. S. The Prodigious Hydrogen Bonds with Sulfur and Selenium in Molecular Assemblies, Structural Biology, and Functional Materials. Acc. Chem. Res. 2020, 53 (8), 1580–1592. 10.1021/acs.accounts.0c00289. [DOI] [PubMed] [Google Scholar]
- Mishra K. K.; Borish K.; Singh G.; Panwaria P.; Metya S.; Madhusudhan M. S.; Das A. Observation of an Unusually Large IR Red-Shift in an Unconventional S-H···S Hydrogen-Bond. J. Phys. Chem. Lett. 2021, 12 (4), 1228–1235. 10.1021/acs.jpclett.0c03183. [DOI] [PubMed] [Google Scholar]
- Metya S.; Das A. S-H···O Hydrogen Bond Can Win over O-H···S Hydrogen Bond: Gas-Phase Spectroscopy of 2-Fluorothiophenol···H2O Complex. J. Phys. Chem. A 2022, 126 (49), 9178–9189. 10.1021/acs.jpca.2c06083. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Chopra P.; Wategaonkar S. C-H···S Interaction Exhibits All the Characteristics of Conventional Hydrogen Bonds. Phys. Chem. Chem. Phys. 2020, 22, 17482–17493. 10.1039/D0CP01508C. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Tian F.; Lv F.; Shang Z. Geometric Characteristics of Hydrogen Bonds Involving Sulfur Atoms in Proteins. Proteins Struct. Funct. Bioinforma. 2009, 76 (1), 151–163. 10.1002/prot.22327. [DOI] [PubMed] [Google Scholar]
- Forbes C. R.; Sinha S. K.; Ganguly H. K.; Bai S.; Yap G. P. A.; Patel S.; Zondlo N. J. Insights into Thiol-Aromatic Interactions: A Stereoelectronic Basis for S-H/π Interactions. J. Am. Chem. Soc. 2017, 139 (5), 1842–1855. 10.1021/jacs.6b08415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobecq H.; Boll E.; Sénéchal M.; Desmet R.; Saliou J. M.; Lacapère J. J.; Mougel A.; Vicogne J.; Melnyk O. A Central Cysteine Residue Is Essential for the Thermal Stability and Function of SUMO-1 Protein and SUMO-1 Peptide-Protein Conjugates. Bioconjugate Chem. 2016, 27 (6), 1540–1546. 10.1021/acs.bioconjchem.6b00211. [DOI] [PubMed] [Google Scholar]
- Gómez-Tamayo J. C.; Cordomí A.; Olivella M.; Mayol E.; Fourmy D.; Pardo L. Analysis of the Interactions of Sulfur-Containing Amino Acids in Membrane Proteins. Protein Sci. 2016, 5 (8), 1517–1524. 10.1002/pro.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoret L. M.; Rader S. D.; Fletterick R. J.; Cohen F. E.; Robert R.; Gregoret L. M.; Rader S. D.; Fletterick R. J.; Cohen F. E. Hydrogen Bonds Involving Sulfur Atoms in Proteins. Proteins: Struct., Funct., Bioinf. 1991, 107, 99–107. [DOI] [PubMed] [Google Scholar]
- Sticke D. F.; Presta L. G.; Dill K. A.; Rose G. D. Hydrogen Bonding in Globular Proteins. J. Mol. Biol. 1992, 226 (4), 1143–1159. 10.1016/0022-2836(92)91058-W. [DOI] [PubMed] [Google Scholar]
- Aurora R.; Rose G. D. Helix Capping. Protein Sci. 1998, 7 (1), 21–38. 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisenant J.; Burgess K. Synthetic Helical Peptide Capping Strategies. Chemical Society Reviews. 2022, 51, 5795–5804. 10.1039/D1CS01175H. [DOI] [PubMed] [Google Scholar]
- Adhav V. A.; Shelke S. S.; Balanarayan P.; Saikrishnan K. Sulfur-Mediated Chalcogen versus Hydrogen Bonds in Proteins: A Seesaw Effect in the Conformational Space. QRB Discovery 2023, 1–30. 10.1017/qrd.2023.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbalsyah T. M.; Moutevelis E.; Warwicker J.; Errington N.; Doig A. J. The CXXC Motif at the N Terminus of an α-Helical Peptide. Protein Sci. 2006, 15 (8), 1945–1950. 10.1110/ps.062271506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denke E.; Merbitz-Zahradnik T.; Hatzfeld O. M.; Snyder C. H.; Link T. A.; Trumpower B. L. Alteration of the Midpoint Potential and Catalytic Activity of the Rieske Iron-Sulfur Protein by Changes of Amino Acids Forming Hydrogen Bonds to the Iron-Sulfur Cluster. J. Biol. Chem. 1998, 273 (15), 9085–9093. 10.1074/jbc.273.15.9085. [DOI] [PubMed] [Google Scholar]
- Hirano Y.; Takeda K.; Miki K. Charge-Density Analysis of an Iron-Sulfur Protein at an Ultra-High Resolution of 0.48 Å. Nature 2016, 534, 281–284. 10.1038/nature18001. [DOI] [PubMed] [Google Scholar]
- Perinbam K.; Balaram H.; Guru Row T. N.; Gopal B. Probing the Influence of Non-Covalent Contact Networks Identified by Charge Density Analysis on the Oxidoreductase BacC. Protein Eng. Des. Sel. 2017, 30 (3), 265–272. 10.1093/protein/gzx006. [DOI] [PubMed] [Google Scholar]
- Wagner J. P.; Schreiner P. R. London Dispersion in Molecular Chemistry - Reconsidering Steric Effects. Angew. Chemie - Int. Ed. 2015, 54 (42), 12274–12296. 10.1002/anie.201503476. [DOI] [PubMed] [Google Scholar]
- London F. Zur Theorie Und Systematik Der Molekularkräfte. Zeitschrift für Phys. 1930, 63, 245. 10.1007/BF01421741. [DOI] [Google Scholar]
- Wagner J. P.; Schreiner P. R. London Dispersion Decisively Contributes to the Thermodynamic Stability of Bulky NHC-Coordinated Main Group Compounds. J. Chem. Theory Comput. 2016, 12 (1), 231–237. 10.1021/acs.jctc.5b01100. [DOI] [PubMed] [Google Scholar]
- Hwang J.; Li P.; Smith M. D.; Shimizu K. D. Distance-Dependent Attractive and Repulsive Interactions of Bulky Alkyl Groups. Angew. Chemie - Int. Ed. 2016, 55 (28), 8086–8089. 10.1002/anie.201602752. [DOI] [PubMed] [Google Scholar]
- Reilly A. M.; Tkatchenko A. Van Der Waals Dispersion Interactions in Molecular Materials: Beyond Pairwise Additivity. Chemical Science. 2015, 6, 3289–3301. 10.1039/C5SC00410A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverría J.; Aullón G.; Danovich D.; Shaik S.; Alvarez S. Dihydrogen Contacts in Alkanes Are Subtle but Not Faint. Nat. Chem. 2011, 3 (4), 323–330. 10.1038/nchem.1004. [DOI] [PubMed] [Google Scholar]
- Parks G. S. Selected Values of Physical and Thermodynamic Properties of Hydrocarbons and Related Compounds. J. Am. Chem. Soc. 1954, 76 (7), 2032–2032. 10.1021/ja01636a111. [DOI] [Google Scholar]
- Danovich D.; Shaik S.; Neese F.; Echeverría J.; Aullón G.; Alvarez S. Understanding the Nature of the CH···HC Interactions in Alkanes. J. Chem. Theory Comput. 2013, 9 (4), 1977–1991. 10.1021/ct400070j. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Thomas S. P.; Potnuru L. R.; Edwards A. J.; Grosjean A.; Ramanathan K. V.; Guru Row T. N. Experimental Insights into the Electronic Nature, Spectral Features, and Role of Entropy in Short CH3···CH3 Hydrophobic Interactions. J. Phys. Chem. Lett. 2019, 10 (22), 7224–7229. 10.1021/acs.jpclett.9b02734. [DOI] [PubMed] [Google Scholar]
- Li J.; Wang Y.; An L.; Chen J.; Yao L. Direct Observation of CH/CH van Der Waals Interactions in Proteins by NMR. J. Am. Chem. Soc. 2018, 140 (9), 3194–3197. 10.1021/jacs.7b13345. [DOI] [PubMed] [Google Scholar]
- Nick Pace C.; Martin Scholtz J.; Grimsley G. R. Forces Stabilizing Proteins. FEBS Lett. 2014, 588, 2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Adam C.; Nichol G. S.; Cockroft S. L. How Much Do van Der Waals Dispersion Forces Contribute to Molecular Recognition in Solution?. Nat. Chem. 2013, 5 (12), 1006–1010. 10.1038/nchem.1779. [DOI] [PubMed] [Google Scholar]
- Joh N. H.; Oberai A.; Yang D.; Whitelegge J. P.; Bowie J. U. Similar Energetic Contributions of Packing in the Core of Membrane and Water-Soluble Proteins. J. Am. Chem. Soc. 2009, 131 (31), 10846–10847. 10.1021/ja904711k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra S.; Singh P.; Mahata R. N.; Brandaõ P.; Roy S.; Mahapatra A. K.; Nanda J. Supramolecular Antiparallel β-Sheet Formation by Tetrapeptides Based on Amyloid Sequence. J. Phys. Chem. B 2021, 125 (17), 4274–4285. 10.1021/acs.jpcb.0c10920. [DOI] [PubMed] [Google Scholar]
- Ghosh N.; Mondal R.; Deshmukh A.; Dutta S.; Mukherjee S. Weak Interactive Forces Govern the Interaction between a Non-Ionic Surfactant with Human Serum Albumin. Chem. Phys. Lett. 2015, 634, 77–82. 10.1016/j.cplett.2015.05.046. [DOI] [Google Scholar]
- Barratt E.; Bingham R. J.; Warner D. J.; Laughton C. A.; Phillips S. E. V.; Homans S. W. Van Der Waals Interactions Dominate Ligand-Protein Association in a Protein Binding Site Occluded from Solvent Water. J. Am. Chem. Soc. 2005, 127 (33), 11827–11834. 10.1021/ja0527525. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R.; Samanta U. Aromatic-Aromatic Interactions in and around α-Helices. Protein Eng. 2002, 15, 91–100. [DOI] [PubMed] [Google Scholar]
- Burley S. K.; Petsko G. A. Amino-Aromatic Interactions in Proteins. FEBS Lett. 1986, 203 (2), 139–143. 10.1016/0014-5793(86)80730-X. [DOI] [PubMed] [Google Scholar]
- Zacharias N.; Dougherty D. A. Cation-π Interactions in Ligand Recognition and Catalysis. Trends Pharmacol. Sci. 2002, 23 (6), 281–287. 10.1016/S0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]
- Carter-Fenk K.; Herbert J. M. Reinterpreting π-Stacking. Phys. Chem. Chem. Phys. 2020, 22, 24870–24886. 10.1039/D0CP05039C. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Golovin A. Cation-π Interactions in Protein-Protein Interfaces. Proteins Struct. Funct. Genet. 2005, 59 (2), 231–239. 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. The Cation-π Interaction. Acc. Chem. Res. 2013, 46 (4), 885–893. 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty D. A. Cation-π Interactions in Chemistry and Biology: A New View of Benzene, Phe, Tyr, and Trp. Science. 1996, 271 (5246), 163–168. 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- Gallivan J. P.; Dougherty D. A. Cation-Pi Interactions in Structural Biology. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (17), 9459–9464. 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M.; Muhseen Z.; Junaid M.; Zhang H. The Aromatic Stacking Interactions Between Proteins and Their Macromolecular Ligands. Curr. Protein Pept. Sci. 2015, 16 (6), 502–512. 10.2174/138920371606150702131516. [DOI] [PubMed] [Google Scholar]
- Salonen L. M.; Ellermann M.; Diederich F. Aromatic Rings Aromatic Rings in Chemical and Biological Recognition : Energetics and Structures Angewandte 2011, 50 (21), 4808–4842. 10.1002/anie.201007560. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S.; Honda K.; Uchimaru T.; Mikami M.; Tanabe K. Origin of Attraction and Directionality of the π/π Interaction: Model Chemistry Calculations of Benzene Dimer Interaction. J. Am. Chem. Soc. 2002, 124 (1), 104–112. 10.1021/ja0105212. [DOI] [PubMed] [Google Scholar]
- Schottel B. L.; Chifotides H. T.; Dunbar K. R. Anion-π Interactions. Chemical Society Reviews. 2008, 37, 68–83. 10.1039/B614208G. [DOI] [PubMed] [Google Scholar]
- Smith M. S.; Lawrence E. E. K.; Billings W. M.; Larsen K. S.; Bécar N. A.; Price J. L. An Anion-π Interaction Strongly Stabilizes the β-Sheet Protein WW. ACS Chem. Biol. 2017, 12 (10), 2535–2537. 10.1021/acschembio.7b00768. [DOI] [PubMed] [Google Scholar]
- Ahmad Rather I.; Ali R.. Anion-π Catalysis: A Novel Supramolecular Approach for Chemical and Biological Transformations. Current Topics in Chirality - From Chemistry to Biology; IntechOpen, 2021. [Google Scholar]
- Robertazzi A.; Krull F.; Knapp E. W.; Gamez P. Recent Advances in Anion-π Interactions. CrystEngComm. 2011, 13, 3293–3300. 10.1039/c0ce00819b. [DOI] [Google Scholar]
- Lucas X.; Bauzá A.; Frontera A.; Quiñonero D. A Thorough Anion-π Interaction Study in Biomolecules: On the Importance of Cooperativity Effects. Chem. Sci. 2016, 7, 1038–1050. 10.1039/C5SC01386K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estarellas C.; Frontera A.; Quiñonero D.; Deyà P. M. Relevant Anion-π Interactions in Biological Systems: The Case of Urate Oxidase. Angew. Chem. 2011, 123 (2), 435–438. 10.1002/ange.201005635. [DOI] [PubMed] [Google Scholar]
- Cotelle Y.; Lebrun V.; Sakai N.; Ward T. R.; Matile S. Anion-π Enzymes. ACS Cent. Sci. 2016, 2 (6), 388–393. 10.1021/acscentsci.6b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estarellas C.; Frontera A.; Quiñonero D.; Deyà P. M. Anion-π Interactions in Flavoproteins. Chem. - An Asian J. 2011, 6 (9), 2316–2318. 10.1002/asia.201100285. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Cotelle Y.; Liu L.; López-Andarias J.; Bornhof A. B.; Akamatsu M.; Sakai N.; Matile S. The Emergence of Anion-π Catalysis. Acc. Chem. Res. 2018, 51 (9), 2255–2263. 10.1021/acs.accounts.8b00223. [DOI] [PubMed] [Google Scholar]
- Philip V.; Harris J.; Adams R.; Nguyen D.; Spiers J.; Baudry J.; Howell E. E.; Hinde R. J. A Survey of Aspartate-Phenylalanine and Glutamate-Phenylalanine Interactions in the Protein Data Bank: Searching for Anion-π Pairs. Biochemistry. 2011, 50 (14), 2939–2950. 10.1021/bi200066k. [DOI] [PubMed] [Google Scholar]
- Chakravarty S.; Ung A. R.; Moore B.; Shore J.; Alshamrani M. A Comprehensive Analysis of Anion-Quadrupole Interactions in Protein Structures. Biochemistry 2018, 57 (12), 1852–1867. 10.1021/acs.biochem.7b01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzniak-Glanowska E.; Glanowski M.; Kurczab R.; Bojarski A. J.; Podgajny R. Mining Anion-Aromatic Interactions in the Protein Data Bank. Chem. Sci. 2022, 13 (14), 3984–3998. 10.1039/D2SC00763K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Lassila J. K.; Gonzalez A.; Tsai Y.; Herschlag D. Use of Anion-Aromatic Interactions to Position the General Base in the Ketosteroid Isomerase Active Site. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (28), 11308–11313. 10.1073/pnas.1206710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. J.; Choudhary A.; Raines R. T.; Woolfson D. N. N→π* Interactions in Proteins. Nat. Chem. Biol. 2010, 6 (8), 615–620. 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F. R.; Wood P. A.; Allen F. H.; Diederich F. Orthogonal Dipolar Interactions between Amide Carbonyl Groups. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (45), 17290–17294. 10.1073/pnas.0806129105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley B.; Richard G.; Harbison G. S.; Powers R. 13C NMR Reveals No Evidence of N-Π* Interactions in Proteins. PLoS One 2012, 7 (8), e42075. 10.1371/journal.pone.0042075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Gandla D.; Krow G. R.; Raines R. T. Nature of Amide Carbonyl-Carbonyl Interactions in Proteins. J. Am. Chem. Soc. 2009, 131 (21), 7244–7246. 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Vanveller B.; Guzei I. A.; Raines R. T. N →π* Interactions of Amides and Thioamides: Implications for Protein Stability. J. Am. Chem. Soc. 2013, 135 (21), 7843–7846. 10.1021/ja4033583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. K.; Mishra K. K.; Sharma N.; Das A. Direct Spectroscopic Evidence for an N→π* Interaction. Angew. Chemie - Int. Ed. 2016, 55 (27), 7801–7805. 10.1002/anie.201511925. [DOI] [PubMed] [Google Scholar]
- Sahariah B.; Sarma B. K. Relative Orientation of the Carbonyl Groups Determines the Nature of Orbital Interactions in Carbonyl-Carbonyl Short Contacts. Chem. Sci. 2019, 10, 909–917. 10.1039/C8SC04221G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahariah B.; Sarma B. K. Spectroscopic Evidence of n → Π* Interactions Involving Carbonyl Groups. Phys. Chem. Chem. Phys. 2020, 22, 26669–26681. 10.1039/D0CP03557B. [DOI] [PubMed] [Google Scholar]
- Kamer K. J.; Choudhary A.; Raines R. T. Intimate Interactions with Carbonyl Groups: Dipole-Dipole or n →π*?. J. Org. Chem. 2013, 78 (5), 2099–2103. 10.1021/jo302265k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim A.; Saha P.; Jha K. K.; Sukumar N.; Sarma B. K. Reciprocal Carbonyl–Carbonyl Interactions in Small Molecules and Proteins. Nat. Commun. 2017, 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13 (2), 291–296. 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Concha M. C. σ-Hole Bonding between like Atoms; a Fallacy of Atomic Charges. J. Mol. Model. 2008, 14, 659–665. 10.1007/s00894-008-0280-5. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. σ-Hole Bonding: A Physical Interpretation. Top. Curr. Chem. 2014, 358, 19–42. 10.1007/128_2014_568. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T.; Resnati G. The σ-Hole Revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. 10.1039/C7CP06793C. [DOI] [PubMed] [Google Scholar]
- Murray J. S.; Lane P.; Politzer P. Expansion of the σ-Hole Concept. In. J. Mol. Model. 2009, 15, 723–729. 10.1007/s00894-008-0386-9. [DOI] [PubMed] [Google Scholar]
- Murray J. S.; Lane P.; Clark T.; Riley K. E.; Politzer P. σ-Holes, π-Holes and Electrostatically-Driven Interactions. J. Mol. Model. 2012, 18, 541–548. 10.1007/s00894-011-1089-1. [DOI] [PubMed] [Google Scholar]
- Hennemann M.; Murray J. S.; Politzer P.; Riley K. E.; Clark T. Polarization-Induced σ-Holes and Hydrogen Bonding. J. Mol. Model. 2012, 18 (6), 2461–2469. 10.1007/s00894-011-1263-5. [DOI] [PubMed] [Google Scholar]
- Bauzá A.; Mooibroek T. J.; Frontera A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16 (12), 2496–2517. 10.1002/cphc.201500314. [DOI] [PubMed] [Google Scholar]
- Lim J. Y. C.; Beer P. D. Sigma-Hole Interactions in Anion Recognition. Chem. 2018, 4 (4), 731–783. 10.1016/j.chempr.2018.02.022. [DOI] [Google Scholar]
- Murray J. S.; Lane P.; Clark T.; Politzer P. σ-Hole Bonding: Molecules Containing Group VI Atoms. J. Mol. Model. 2007, 13 (10), 1033–1038. 10.1007/s00894-007-0225-4. [DOI] [PubMed] [Google Scholar]
- Murray J. S.; Lane P.; Politzer P. A Predicted New Type of Directional Noncovalent Interaction. Int. J. Quantum Chem. 2007, 107 (12), 2286–2292. 10.1002/qua.21352. [DOI] [Google Scholar]
- Politzer P.; Murray J. S. Halogen Bonding: An Interim Discussion. ChemPhysChem. 2013, 14 (2), 278–294. 10.1002/cphc.201200799. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. Halogen Bonds in Biological Molecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (48), 16789–16794. 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metrangolo P.; Resnati G. Chemistry: Halogen versus Hydrogen. Science. 2008, 321 (5891), 918–919. 10.1126/science.1162215. [DOI] [PubMed] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. Halogen Bonding and Other σ-Hole Interactions: A Perspective. Phys. Chem. Chem. Phys. 2013, 15 (27), 11178–11189. 10.1039/c3cp00054k. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R.; Shing Ho. P.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar]
- Costa P. J. The Halogen Bond: Nature and Applications. Phys. Sci. Rev. 2019, 2, 20170136. [Google Scholar]
- Czarny R. S.; Ho A. N.; Shing Ho P. A Biological Take on Halogen Bonding and Other Non-Classical Non-Covalent Interactions. Chemical Record. 2021, 21 (5), 1240–1251. 10.1002/tcr.202100076. [DOI] [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116 (4), 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56 (4), 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Hernandes M.; Cavalcanti S. M.; Moreira D. R.; de Azevedo Junior W.; Leite A. C. Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11 (3), 303–314. 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- Mallada B.; Gallardo A.; Lamanec M.; de la Torre B.; Špirko V.; Hobza P.; Jelinek P. Real-Space Imaging of Anisotropic Charge of σ-Hole by Means of Kelvin Probe Force Microscopy. Science. 2021, 374 (6569), 863–867. 10.1126/science.abk1479. [DOI] [PubMed] [Google Scholar]
- Pike S. J.; Hunter C. A.; Brammer L.; Perutz R. N. Benchmarking of Halogen Bond Strength in Solution with Nickel Fluorides: Bromine versus Iodine and Perfluoroaryl versus Perfluoroalkyl Donors. Chem. - A Eur. J. 2019, 25 (39), 9237–9241. 10.1002/chem.201900924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.; Tian F.; Zou J.; Shang Z. Rediscovery of Halogen Bonds in Protein-Ligand Complexes. Mini-Reviews Med. Chem. 2010, 10 (4), 309–314. 10.2174/138955710791331016. [DOI] [PubMed] [Google Scholar]
- Riley K. E.; Hobza P. Strength and Character of Halogen Bonds in Protein-Ligand Complexes. Cryst. Growth Des. 2011, 11 (10), 4272–4278. 10.1021/cg200882f. [DOI] [Google Scholar]
- Shinada N. K.; De Brevern A. G.; Schmidtke P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62 (21), 9341–9356. 10.1021/acs.jmedchem.8b01453. [DOI] [PubMed] [Google Scholar]
- Margiotta E.; Van Der Lubbe S. C. C.; De Azevedo Santos L.; Paragi G.; Moro S.; Bickelhaupt F. M.; Fonseca Guerra C. Halogen Bonds in Ligand-Protein Systems: Molecular Orbital Theory for Drug Design. J. Chem. Inf. Model. 2020, 60 (3), 1317–1328. 10.1021/acs.jcim.9b00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontera A.; Bauzá A. Biological Halogen Bonds in Protein-Ligand Complexes: A Combined QTAIM and NCIPlot Study in Four Representative Cases. Org. Biomol. Chem. 2021, 19, 6858–6864. 10.1039/D1OB01212F. [DOI] [PubMed] [Google Scholar]
- Kolár M.; Hobzaa P.; Bronowska A. K. Plugging the Explicit σ-Holes in Molecular Docking. Chem. Commun. 2013, 49, 981–983. 10.1039/C2CC37584B. [DOI] [PubMed] [Google Scholar]
- Nunes R.; Vila-Viçosa D.; Machuqueiro M.; Costa P. J. Biomolecular Simulations of Halogen Bonds with a GROMOS Force Field. J. Chem. Theory Comput. 2018, 14 (10), 5383–5392. 10.1021/acs.jctc.8b00278. [DOI] [PubMed] [Google Scholar]
- Ford M. C.; Rappé A. K.; Ho P. S. A Reduced Generalized Force Field for Biological Halogen Bonds. J. Chem. Theory Comput. 2021, 17 (8), 5369–5378. 10.1021/acs.jctc.1c00362. [DOI] [PubMed] [Google Scholar]
- Kolarič A.; Germe T.; Hrast M.; Stevenson C. E. M.; Lawson D. M.; Burton N. P.; Vörös J.; Maxwell A.; Minovski N.; Anderluh M. Potent DNA Gyrase Inhibitors Bind Asymmetrically to Their Target Using Symmetrical Bifurcated Halogen Bonds. Nat. Commun. 2021, 12, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathwar V. R.; Gonnade R. G.; Munshi P.; Bhadbhade M. M.; Row T. N. G. Halogen Bonding in 2,5-Dichloro-1,4-Benzoquinone: Insights from Experimental and Theoretical Charge Density Analysis. Cryst. Growth Des. 2011, 11, 1855–1862. [Google Scholar]
- Staroń J.; Pietruś W.; Bugno R.; Kurczab R.; Satała G.; Warszycki D.; Lenda T.; Wantuch A.; Hogendorf A. S.; Hogendorf A.; Duszyńska B.; Bojarski A. J. Tuning the Activity of Known Drugs via the Introduction of Halogen Atoms, a Case Study of SERT Ligands – Fluoxetine and Fluvoxamine. Eur. J. Med. Chem. 2021, 220, 113533. 10.1016/j.ejmech.2021.113533. [DOI] [PubMed] [Google Scholar]
- Kolář M. H.; Tabarrini O. Halogen Bonding in Nucleic Acid Complexes. J. Med. Chem. 2017, 60 (21), 8681–8690. 10.1021/acs.jmedchem.7b00329. [DOI] [PubMed] [Google Scholar]
- Frontera A.; Bauzá A. Halogen Bonds in Protein Nucleic Acid Recognition. J. Chem. Theory Comput. 2020, 16 (7), 4744–4752. 10.1021/acs.jctc.0c00431. [DOI] [PubMed] [Google Scholar]
- Mu K.; Zhu Z.; Abula A.; Peng C.; Zhu W.; Xu Z. Halogen Bonds Exist between Noncovalent Ligands and Natural Nucleic Acids. J. Med. Chem. 2022, 65 (6), 4424–4435. 10.1021/acs.jmedchem.1c01854. [DOI] [PubMed] [Google Scholar]
- Piña M. D. L. N.; Frontera A.; Bauzá A. Quantifying Intramolecular Halogen Bonds in Nucleic Acids: A Combined Protein Data Bank and Theoretical Study. ACS Chem. Biol. 2020, 15 (7), 1942–1948. 10.1021/acschembio.0c00292. [DOI] [PubMed] [Google Scholar]
- Aakeröy C. B.; Fasulo M.; Schultheiss N.; Desper J.; Moore C. Structural Competition between Hydrogen Bonds and Halogen Bonds. J. Am. Chem. Soc. 2007, 129 (45), 13772–13773. 10.1021/ja073201c. [DOI] [PubMed] [Google Scholar]
- Robertson C. C.; Wright J. S.; Carrington E. J.; Perutz R. N.; Hunter C. A.; Brammer L. Hydrogen Bonding: Vs. Halogen Bonding: The Solvent Decides. Chem. Sci. 2017, 8, 5392–5398. 10.1039/C7SC01801K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riel A. M. S.; Rowe R. K.; Ho E. N.; Carlsson A. C. C.; Rappé A. K.; Berryman O. B.; Ho P. S. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52 (10), 2870–2880. 10.1021/acs.accounts.9b00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danelius E.; Andersson H.; Jarvoll P.; Lood K.; Gräfenstein J.; Erdélyi M. Halogen Bonding: A Powerful Tool for Modulation of Peptide Conformation. Biochemistry 2017, 56 (25), 3265–3272. 10.1021/acs.biochem.7b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A. C. C.; Scholfield M. R.; Rowe R. K.; Ford M. C.; Alexander A. T.; Mehl R. A.; Ho P. S. Increasing Enzyme Stability and Activity through Hydrogen Bond-Enhanced Halogen Bonds. Biochemistry 2018, 57 (28), 4135–4147. 10.1021/acs.biochem.8b00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholfield M. R.; Ford M. C.; Carlsson A. C. C.; Butta H.; Mehl R. A.; Ho P. S. Structure-Energy Relationships of Halogen Bonds in Proteins. Biochemistry 2017, 56 (22), 2794–2802. 10.1021/acs.biochem.7b00022. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Zhang L.; Cui D.; Yao Z.; Gao B.; Lin J.; Wei D. The Important Role of Halogen Bond in Substrate Selectivity of Enzymatic Catalysis. Sci. Rep. 2016, 6, 34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aakeroy C. B.; Bryce D. L.; Desiraju G. R.; Frontera A.; Legon A. C.; Nicotra F.; Rissanen K.; Scheiner S.; Terraneo G.; Metrangolo P.; Resnati G. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91 (11), 1889–1892. 10.1515/pac-2018-0713. [DOI] [Google Scholar]
- Pascoe D. J.; Ling K. B.; Cockroft S. L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139 (42), 15160–15167. 10.1021/jacs.7b08511. [DOI] [PubMed] [Google Scholar]
- Tarannam N.; Shukla R.; Kozuch S. Yet Another Perspective on Hole Interactions. Phys. Chem. Chem. Phys. 2021, 23, 19948–19963. 10.1039/D1CP03533A. [DOI] [PubMed] [Google Scholar]
- Bunchuay T.; Docker A.; Eiamprasert U.; Surawatanawong P.; Brown A.; Beer P. D. Chalcogen Bond Mediated Enhancement of Cooperative Ion-Pair Recognition. Angew. Chemie - Int. Ed. 2020, 59 (29), 12007–12012. 10.1002/anie.202001125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov A. V.; Katlenok E. A.; Zhmykhova M. V.; Ivanov A. Y.; Kuznetsov M. L.; Bokach N. A.; Kukushkin V. Y. Metal-Involving Chalcogen Bond. The Case of Platinum(II) Interaction with Se/Te-Based σ-Hole Donors. J. Am. Chem. Soc. 2021, 143 (38), 15701–15710. 10.1021/jacs.1c06498. [DOI] [PubMed] [Google Scholar]
- Scilabra P.; Terraneo G.; Resnati G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52 (5), 1313–1324. 10.1021/acs.accounts.9b00037. [DOI] [PubMed] [Google Scholar]
- Docker A.; Guthrie C. H.; Kuhn H.; Beer P. D. Modulating Chalcogen Bonding and Halogen Bonding Sigma-Hole Donor Atom Potency and Selectivity for Halide Anion Recognition. Angew. Chemie - Int. Ed. 2021, 60 (40), 21973–21978. 10.1002/anie.202108591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfield R. E.; Parthasarathy R.; Dunitz J. D. Directional Preferences of Nonbonded Atomic Contacts with Divalent Sulfur. 1. Electrophiles and Nucleophiles. J. Am. Chem. Soc. 1977, 99 (14), 4860–4862. 10.1021/ja00456a072. [DOI] [Google Scholar]
- Pal D.; Chakrabarti P. Non-Hydrogen Bond Interactions Involving the Methionine Sulfur Atom. J. Biomol. Struct. Dyn. 2001, 19 (1), 115–128. 10.1080/07391102.2001.10506725. [DOI] [PubMed] [Google Scholar]
- Iwaoka M.; Takemoto S.; Tomoda S. Statistical and Theoretical Investigations on the Directionality of Nonbonded S···O Interactions. Implications for Molecular Design and Protein Engineering. J. Am. Chem. Soc. 2002, 124 (35), 10613–10620. 10.1021/ja026472q. [DOI] [PubMed] [Google Scholar]
- Adhav V. A.; Pananghat B.; Saikrishnan K. Probing the Directionality of S···O/N Chalcogen Bond and Its Interplay with Weak C-H···O/N/S Hydrogen Bond Using Molecular Electrostatic Potential. J. Phys. Chem. B 2022, 126 (40), 7818–7832. 10.1021/acs.jpcb.2c03745. [DOI] [PubMed] [Google Scholar]
- Biswal H. S.; Sahu A. K.; Galmés B.; Frontera A.; Chopra D. Se···O/S and S···O Chalcogen Bonds in Small Molecules and Proteins: A Combined CSD and PDB Study. ChemBioChem 2022, 23, e202100498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carugo O.; Resnati G.; Metrangolo P. Chalcogen Bonds Involving Selenium in Protein Structures. ACS Chem. Biol. 2021, 16 (9), 1622–1627. 10.1021/acschembio.1c00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández Riveras J. A.; Frontera A.; Bauzá A. Selenium Chalcogen Bonds Are Involved in Protein-Carbohydrate Recognition: A Combined PDB and Theoretical Study. Phys. Chem. Chem. Phys. 2021, 23, 17656–17662. 10.1039/D1CP01929E. [DOI] [PubMed] [Google Scholar]
- Kristian K.; Fanfrlík J.; Lepšík M. Chalcogen Bonding in Protein–Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. 10.1002/cphc.201800409. [DOI] [PubMed] [Google Scholar]
- Thomas S. P.; Jayatilaka D.; Guru Row T. N.; Row T. N. G.; Guru Row T. N. S···O Chalcogen Bonding in Sulfa Drugs: Insights from Multipole Charge Density and X-Ray Wavefunction of Acetazolamide. Phys. Chem. Chem. Phys. 2015, 17, 25411–25420. 10.1039/C5CP04412J. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Gong Z.; Li J.; Lu T. Intermolecular Sulfur···Oxygen Interactions: Theoretical and Statistical Investigations. J. Chem. Inf. Model. 2015, 55 (10), 2138–2153. 10.1021/acs.jcim.5b00177. [DOI] [PubMed] [Google Scholar]
- Adhav V. A.; Shelke S. S.; Balanarayan P.; Saikrishnan K. Sulfur-Mediated Chalcogen versus Hydrogen Bonds in Proteins: A Seesaw Effect in the Conformational Space. bioRxiv 2023, 1. 10.1017/qrd.2023.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaoka M.; Isozumi N. Hypervalent Nonbonded Interactions of a Divalent Sulfur Atom. Implications in Protein Architecture and the Functions. Molecules. 2012, 17 (6), 7266–7283. 10.3390/molecules17067266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaoka M.; Isozumi N. Possible Roles of S···O and S···N Interactions in the Functions and Evolution of Phospholipase A2. Biophysics (Oxf) 2006, 2, 23–34. 10.2142/biophysics.2.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fick R. J.; Kroner G. M.; Nepal B.; Magnani R.; Horowitz S.; Houtz R. L.; Scheiner S.; Trievel R. C. Sulfur-Oxygen Chalcogen Bonding Mediates AdoMet Recognition in the Lysine Methyltransferase SET7/9. ACS Chem. Biol. 2016, 11 (3), 748–754. 10.1021/acschembio.5b00852. [DOI] [PubMed] [Google Scholar]
- Esrafili M. D.; Mousavian P. Strong Tetrel Bonds: Theoretical Aspects and Experimental Evidence. Molecules 2018, 23 (10), 2642. 10.3390/molecules23102642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner S. Origins and Properties of the Tetrel Bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. 10.1039/D1CP00242B. [DOI] [PubMed] [Google Scholar]
- Scheiner S. Tetrel Bonding as a Vehicle for Strong and Selective Anion Binding. Molecules 2018, 23 (5), 1147. 10.3390/molecules23051147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani D.; Arunan E. The X-C···Y (X = O/F, y = O/S/F/Cl/Br/N/P) “carbon Bond” and Hydrophobic Interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. 10.1039/c3cp51658j. [DOI] [PubMed] [Google Scholar]
- Walker M. G.; Mendez C. G.; Ho P. S. Non-Classical Non-Covalent σ-Hole Interactions in Protein Structure and Function: Concepts for Potential Protein Engineering Applications. Chem.-Asian J. 2023, 18, e202300026. [DOI] [PubMed] [Google Scholar]
- Mundlapati V. R.; Sahoo D. K.; Bhaumik S.; Jena S.; Chandrakar A.; Biswal H. S. Noncovalent Carbon-Bonding Interactions in Proteins. Angew. Chemie - Int. Ed. 2018, 57 (50), 16496–16500. 10.1002/anie.201811171. [DOI] [PubMed] [Google Scholar]
- Trievel R. C.; Scheiner S. Crystallographic and Computational Characterization of Methyl Tetrel Bonding in S-Adenosylmethionine-Dependent Methyltransferases. Molecules 2018, 23 (11), 2965. 10.3390/molecules23112965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S. P.; Pavan M. S.; Guru Row T. N. Experimental Evidence for ‘Carbon Bonding’ in the Solid State from Charge Density Analysis. Chem. Commun. 2014, 50, 49–51. 10.1039/C3CC47226D. [DOI] [PubMed] [Google Scholar]
- Mooibroek T. J. Intermolecular Non-Covalent Carbon-Bonding Interactions with Methyl Groups: A CSD, PDB and DFT Study. Molecules 2019, 24 (18), 3370. 10.3390/molecules24183370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauzá A.; Frontera A. RCH3· · ·O Interactions in Biological Systems: Are They Trifurcated H-Bonds or Noncovalent Carbon Bonds?. Crystals 2016, 6 (3), 26. 10.3390/cryst6030026. [DOI] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Freddolino P. L.; Zhang Y.. Ab Initio Protein Structure Prediction. From Protein Structure to Function with Bioinformatics, 2nd ed.; Springer: Dordrecht, 2017. [Google Scholar]
- Marks D. S.; Hopf T. A.; Sander C. Protein Structure Prediction from Sequence Variation. Nat. Biotechnol. 2012, 30, 1072–1080. 10.1038/nbt.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov S.; Park H.; Varghese N.; Huang P. S.; Pavlopoulos G. A.; Kim D. E.; Kamisetty H.; Kyrpides N. C.; Baker D. Protein Structure Determination Using Metagenome Sequence Data. Science. 2017, 355 (6322), 294–298. 10.1126/science.aah4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijević V.; Renfrew P. D.; Kosciolek T.; Leman J. K.; Berenberg D.; Vatanen T.; Chandler C.; Taylor B. C.; Fisk I. M.; Vlamakis H.; Xavier R. J.; Knight R.; Cho K.; Bonneau R. Structure-Based Protein Function Prediction Using Graph Convolutional Networks. Nat. Commun. 2021, 12, 3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulmanov M.; Hoehndorf R. DeepGOPlus: Improved Protein Function Prediction from Sequence. Bioinformatics 2020, 36 (2), 422–429. 10.1093/bioinformatics/btz595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisstock J. C.; Lesk A. M. Prediction of Protein Function from Protein Sequence and Structure. Q. Rev. Biophys. 2003, 36 (3), 307–340. 10.1017/S0033583503003901. [DOI] [PubMed] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W. R.; Bridgland A.; Penedones H.; Petersen S.; Simonyan K.; Crossan S.; Kohli P.; Jones D. T.; Silver D.; Kavukcuoglu K.; Hassabis D. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Pearce R.; Zhang Y. Deep Learning Techniques Have Significantly Impacted Protein Structure Prediction and Protein Design. Curr. Opin. Struct. Biol. 2021, 68, 194–207. 10.1016/j.sbi.2021.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl C. A.; Strauss C. E. M.; Misura K. M. S.; Baker D. Protein Structure Prediction Using Rosetta. Methods Enzymol. 2004, 383, 66–93. 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Baek M.; DiMaio F.; Anishchenko I.; Dauparas J.; Ovchinnikov S.; Lee G. R.; Wang J.; Cong Q.; Kinch L. N.; Dustin Schaeffer R.; Millán C.; Park H.; Adams C.; Glassman C. R.; DeGiovanni A.; Pereira J. H.; Rodrigues A. V.; Van Dijk A. A.; Ebrecht A. C.; Opperman D. J.; Sagmeister T.; Buhlheller C.; Pavkov-Keller T.; Rathinaswamy M. K.; Dalwadi U.; Yip C. K.; Burke J. E.; Christopher Garcia K.; Grishin N. V.; Adams P. D.; Read R. J.; Baker D. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373 (6557), 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A.; Sixma T. K. AI Revolutions in Biology: The Joys and Perils of AlphaFold. EMBO Rep. 2021, 22, e54046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouatta N.; Sorger P.; AlQuraishi M. Protein Structure Prediction by AlphaFold2: Are Attention and Symmetries All You Need?. Acta Crystallogr. Sect. D Struct. Biol. 2021, D77, 982–991. 10.1107/S2059798321007531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. N. Energetics of Protein Hydrogen Bonds. Nature Structural and Molecular Biology 2009, 16, 681–682. 10.1038/nsmb0709-681. [DOI] [PubMed] [Google Scholar]