

Abstract
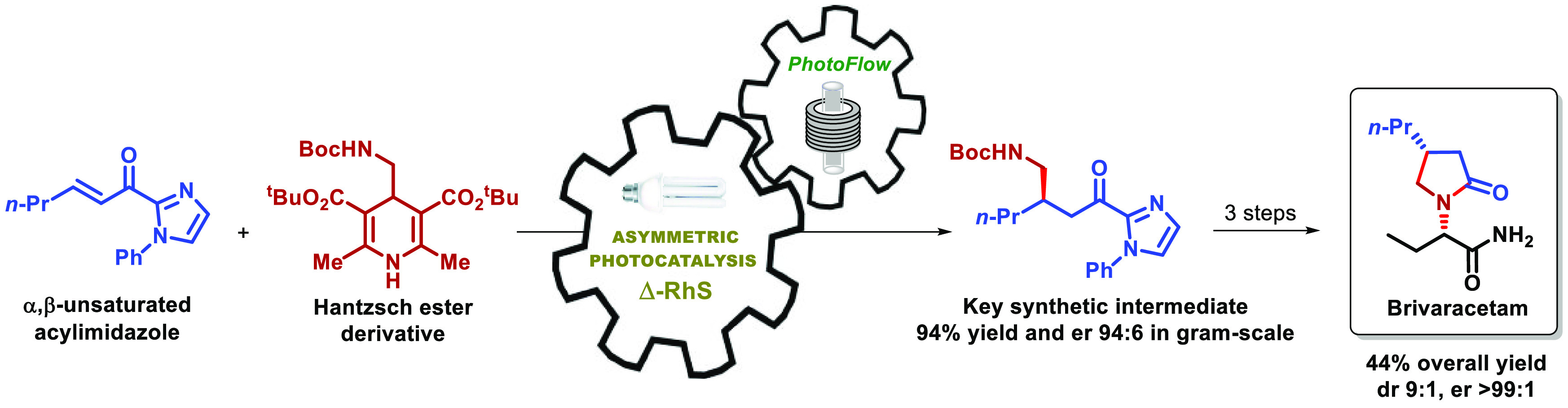
An original total synthesis of the antiepileptic drug brivaracetam (BRV) is reported. The key step in the synthesis consists of an enantioselective photochemical Giese addition, promoted by visible-light and the chiral bifunctional photocatalyst Δ-RhS. Continuous flow conditions were employed to improve the efficiency and allow an easy scale-up of the enantioselective photochemical reaction step. The intermediate obtained from the photochemical step was converted into BRV by two different pathways, followed by one alkylation and amidation, thus giving the desired active pharmaceutical ingredients (API) in 44% overall yield, 9:1 diastereoisomeric ratio (dr) and >99:1 enantiomeric ratio (er).
Introduction
The manufacturing of active pharmaceutical ingredients (API) has been one of the great concerns of nations around the world. The shortage of several medications in many countries during the COVID-19 pandemic demonstrated that this is a matter of sovereignty that requires more of our attention and investment.1 Epilepsy is a condition in which an individual suffers from recurrent unstimulated seizures.2 It is estimated that about 50 million3 people worldwide live with this condition and 40% of all these people do not respond well to the conventional medication used as treatment, a condition known as drug-resistant epilepsy (DRE).4 For this reason, there is a strong search for new antiepileptic drugs (AED) that are capable of acting against DRE. In 2016, the European Union and the United States agencies approved the use of a new AED in cases of DRE called brivaracetam (BRV) (Figure 1).5 BRV was developed from another drug with a very similar structure, levetiracetam (LEV) (Figure 1).
Figure 1.

Structures of levetiracetam (LEV) and brivaracetam (BRV).
The difference between BRV and LEV is the presence of the n-propyl side chain attached to C4, and from the four possible stereoisomers of BRV, the (4R,2’S) shows a much higher affinity for the synaptic vesicle protein 2A (SV2A), which is one of the main therapeutic targets for the search of new AEDs.6 The main challenge associated with the synthesis of BRV is the stereocontrolled installation of the center at C4, and this is why many synthetic routes adopt strategies based on chiral HPLC separation or resolution with chemical or enzymatic agents. In 2008, UCB pharmaceutical demonstrated the preparation of BRV starting from methyl (E)-hex-2-enoate and nitromethane (Scheme 1A).7b Preparation of (R)-4-propylpyrrolidin-2-one is achieved in two steps, followed by chiral HPLC separation. A diastereoisomeric mixture of BRV is obtained after two more steps, which is then subjected again to chiral chromatography separation, affording the final API in 7.2% overall yield. In a different approach, Mankind Pharma demonstrated the preparation of diastereoisomeric BRV in six steps from butyraldehyde e monoethyl malonate (Scheme 1B).7h After a chiral HPLC purification step, BRV was obtained in 12.8% overall yield. A biocatalytic route was reported by Schülé and co-workers, in which proteases B and C from B. subtilis were used in the resolution of the racemic β-carboxyl tert-butyl ester intermediate, affording the corresponding R enantiomer in 42% yield (Scheme 1C).7e The synthesis was completed after a total of seven steps, and BRV was obtained in 9.1% overall yield. Additionally, in 2020, Clininvent Research Pvt. Ltd. reported an alternative route to BRV, based on chemical resolution using S-phenylethylamine (Scheme 1D).7i The diastereoisomeric amide intermediate is purified by recrystallization from di-isopropyl ether, affording the correct stereoisomer that is further converted into BRV. The synthetic route is comprised of nine steps, with 4.3% overall yield. In another example, the synthesis reported by Beijing Abeno Pharmaceutical Co. Ltd. employs a chiral oxazolidine-2-one auxiliary to perform a stereocontrolled alkylation at the α-carbonyl position of the starting material (Scheme 1E).7j−7l The next step in the synthesis involves the cleavage of the auxiliary to deliver the corresponding alcohol, which is further manipulated to obtain the API in 13.7% yield after eight steps. Other examples involving the synthesis of BRV or any relevant intermediate can be found in the specialized literature.6,7
Scheme 1. Reported Approaches for the Total Synthesis of BRV.

In the scenario of modern enantioselective synthesis, asymmetric catalysis has stood out as the most efficient, practical, and sustainable tool that might be used for conducting stereocontrolled transformations. Up to date, only two enantioselective total syntheses of BRV based on asymmetric catalysis have been reported. The first one was reported in 2019 by Jiangxi Qingfeng Pharmaceutical Co. Ltd. and employs the MacMillan asymmetric catalyst to promote the enantioselective reaction between diethyl bromomalonate and pentanal as the key step.8 The reaction uses BiO3 as a photocatalyst, and the product is converted into BRV (58% overall yield) after two more steps (Scheme 2A). In the other approach, reported by two Chinese companies, the synthesis of BRV starts with 3-propylpentanedioic acid and after seven steps, including desymmetrization of the starting acid and chiral resolution, an enantiopure intermediate is obtained, which is converted into BRV after two more steps (Scheme 2B).9 The final API is obtained in 7.2% overall yield.
Scheme 2. Reported Approaches for the Enantioselective Total Synthesis of Brivaracetam Based on Organocatalysis (A) and Desymmetrization/Resolution Strategies (B).

These approaches represent important advances in the synthesis of this API, however, we believe that the development of alternative routes based on asymmetric catalysis is still necessary, not only because the examples reported so far are few but because some improvements are needed, especially to avoid too many steps on the synthesis and a decrease of the catalyst loading.
Aiming at contributing to the expansion of the synthetic literature of BRV, herein we report an original enantioselective synthesis of this API, combining the use of asymmetric photocatalysis and continuous flow. There are several examples where photocatalysis is combined with the continuous flow conditions (photoflow), including the synthesis of many known pharmaceuticals; however, to the best of our knowledge, this is the first time that asymmetric photocatalysis is combined with the continuous flow for the synthesis of an enantiopure API.10
Results and Discussion
Our synthetic design begins with the stereocontrolled installation of the stereogenic center at C4, which can be achieved through an enantioselective conjugate addition of an α-aminoalkyl radical to an α,β-unsaturated carbonyl derivative (Scheme 3A). Cyclization between the deprotected amino group and carbonyl functionality shall assemble the pyrrolidin-2-one core of BRV, which can be coupled to the chiral butanoate fragment by a substitution reaction, followed by functional group interconversion to deliver the target API.
Scheme 3. (A) Synthetic Design for the Synthesis of BRV and (B) Asymmetric Conjugated Addition Approach Previously Reported by Meggers.

Aiming at high levels of stereocontrol for the asymmetric conjugate addition step, the use of the catalyst RhS developed by Meggers and co-workers seems to suit properly as previous studies demonstrate its use in photochemical enantioselective Giese addition reactions with excellent levels of enantioselectivity.11 In a particular example published in 2018,11e the authors describe the β-alkylation of α,β-unsaturated acylimidazoles employing Hantzsch ester derivatives as radical precursors. This reaction was developed in mild conditions with high yields and enantiomeric ratios (ers), using only the chiral catalyst Λ-RhS, without the necessity of an additional photocatalyst (Scheme 3B).
Inspired by this report, we started our synthetic endeavors by investigating the conjugate addition between α,β-unsaturated acylimidazole 1 and Hantzsch ester 2a in the presence of the chiral catalyst Δ-RhS in CH2Cl2 and visible-light irradiation (batch condition).
After the screening of the reaction parameters in the batch regime, we were able to obtain the target product 3 in quantitative yield and an excellent 98:2 enantiomeric ratio (er). This result was achieved by performing the reaction under a 15 W CFL lamp irradiation for 4 h with 1.2 equiv of 2a, 4 mol % of Δ-RhS catalyst, and concentration of 1 at 0.1 M in CH2Cl2; the compound 3 was isolated by column chromatography (Table 1, entry 1). The use of blue LEDs or other CFL lamps had a slightly negative impact in terms of yield and er (Table 1, entries 2–4). Substitution of Hantzsch ester 2a for congeners 2b (R = Me) and 2c (R = Et) showed no influence on the er; however, even after long periods of irradiation, the conversion of substrate 1 was not complete and the yield of 3 decreased drastically (Table 1, entries 5 and 6). Lowering the catalyst loading to 2 mol % made the reaction much slower, requiring 24 h to reach total conversion, and leading to a small decrease in the yield (Table 1, entry 7). The reaction concentration was also proven to be important to ensure an efficient formation of compound 3 as more diluted or more concentrated conditions led both to smaller yields and, for the more concentrated condition, a decreased er (Table 1, entries 8 and 9). Having the optimized conditions in hand, we performed a scale-up experiment in batch using 1.0 mmol of α,β-unsaturated acylimidazole 1 (Table 1, entry 10). We observed an equivalent result (96% yield, 98:2 er) compared to the 0.1 mmol scale (Table 1, entry 1); however, the reaction required much more time to be processed (20 h).
Table 1. Screening of the Reaction Parameters for the Preparation of Compound 3 in Batcha.

| entry | deviation from optimized conditions | yield (%)b | erc |
|---|---|---|---|
| 1 | no changes | quant.d | 98:2 |
| 2 | 25 W CFL instead of 15 W CFL | quant.d | 96:4 |
| 3 | 4 W blue LEDs instead of 15 W CFL | 92 | 96:4 |
| 4 | 30 W blue LEDs instead of 15 W CFL | 96 | 95:5 |
| 5 | 2b (R = Me) instead of 2a | 27 | 98:2 |
| 6 | 2c (R = Et) instead of 2a | 22 | 97:3 |
| 7e | 2 mol % of Δ-RhS instead of 4 mol % | 87 | 96:4 |
| 8 | 0.05 M for 1 instead of 0.1 M | 89 | 98:2 |
| 9 | 0.2 M for 1 instead of 0.1 M | 78 | 91:9 |
| 10 | no changes with scale-up (1 mmol) and 20 h reaction time | 96 | 98:2 |
1 (0.1 mmol), degassed via freeze-pump-thaw three times before irradiation, N2 atmosphere.
Isolated yield.
Determined by chiral HPLC analysis.
Quantitative yield: >99%.
24 h.
Despite the results observed for the scale-up experiment in batch, the elongation of the reaction time might be a problem for the production of greater amounts of 3. Further enlargement of the reaction scale would require the dimensions of the reaction vessel to be enlarged as well, and it is known that such a change in the reaction apparatus usually leads to a significant loss of efficiency for photochemical reactions.12e
With that in mind, we turned our attention to the possibility to use continuous flow reactors12 to improve and scale-up the synthesis of intermediate 3.
Initially, the reaction was transposed from batch to the continuous flow conditions using the racemic rhodium catalyst (rac-RhS). The use of 0.5 and 1.0 mL min–1 provided the desired product in 95 and 96% yield, respectively (Table 2, entries 1 and 2). When the flow rate was increased to 2.0 mL min–1, the yield of compound 3 was lowered to 85% (Table 2, entry 3) and the use of 1.5 mL min–1 afforded 3 in 93% yield (Table 2, entry 4). Aiming at a balance between efficiency and productivity, we selected condition 2 (1.0 mL min–1) as the standard for the incoming reactions using the enantiopure rhodium catalyst.
Table 2. Optimization of the Residence Time in Flow Using rac-RhS.

| entrya | flow rate (mL min–1) | residence time (tR, min) | yield (%)b |
|---|---|---|---|
| 1 | 0.5 | 50 | 95 |
| 2 | 1.0 | 25 | 96 |
| 3 | 2.0 | 12.5 | 85 |
| 4 | 1.5 | 16.7 | 93 |
1 (1.0 equiv, 0.1 mmol, 0.1 M), 2a (1.2 equiv, 0.12 mmol), racemic photocatalyst rac-RhS (4 mol %), CH2Cl2 (1 mL), room temperature (rt), degassed via freeze-pump-thaw three times before irradiation, and light source (blue LED 120 W).
Isolated yield.
When the reaction was performed using the enantiopure catalyst Δ-RhS, the target product 3 was obtained in 95% yield and 91:9 er (Table 3, entry 1), a slightly lower er compared to the batch results. Aiming at minimizing this deviation on er, we decided to decrease the reaction temperature using a jacketed glass flow reactor.13 However, in both 0 and −20 °C, the yields were similar (91 and 89%, Table 3, entries 2 and 3) and the enantioselectivity was significantly lower (down to 73:27 er). These results could be surprising, but selected examples from the literature revealed that particular reactions can follow a reverse direction on the enantioselectivity as lower the temperature due to the significance of the entropic factor for the stereo-determining step.14 Lower potencies of the blue LEDs were also tested. Using 60 W slightly improved the performance of the reaction (98% yield, 92:8 er, Table 3, entry 4), and no improvements in the enantioselectivity were observed when the temperature was lowered to 0 or −20 °C (Table 3, entries 5 and 6). We also tested the use of 40 W blue LEDs, which afforded product 3 in 91% yield and 93:7 er. (Table 3, entry 7). We then decided to evaluate if a higher temperature could improve the performance of the reaction. Running the reaction at 40 °C provided the target product in 96% yield and er 95:5 (Table 3, entry 8), proving itself as the best condition. Further changes in the temperature did not provide any improvements for the reaction (Table 3, entry 9), and running the reaction at 1.5 mL min–1 flow rate led to a significant decrease in the yield of 3 (Table 3, entry 10).
Table 3. Optimization of the Reaction Conditions in Flow Using Δ-RhSa.

| entry | light source | temp. (°C) | yield (%)b | erc |
|---|---|---|---|---|
| 1 | blue LED 120 W | rt | 95 | 91:9 |
| 2 | blue LED 120 W | 0 °C | 91 | 74:26 |
| 3 | blue LED 120 W | –20 °C | 89 | 73:27 |
| 4 | blue LED 60 W | rt | 98 | 92:8 |
| 5 | blue LED 60 W | 0 °C | 95 | 86:14 |
| 6 | blue LED 60 W | –20 °C | 98 | 77:23 |
| 7 | blue LED 40 W | rt | 91 | 93:7 |
| 8 | blue LED 40 W | 40 °C | 96 | 95:5 |
| 9 | blue LED 40 W | 60 °C | 97 | 95:5 |
| 10d | blue LED 40 W | 60 °C | 77 | 95:5 |
| 11 | blue LED 20 W | 40 °C | 92 | 96:4 |
| 12 | CFL 30 W | 40 °C | 96 | 96:4 |
| 13 | CFL 23 W | 40 °C | 98 | 95:5 |
| 14e | CFL 30 W | 40 °C | 65 | 95:5 |
| 15f | CFL 30 W | 40 °C | 98 | 96:4 |
| 16g | CFL 30 W | 40 °C | 94 | 94:6 |
1 (1.0 equiv, 0.1 mmol, 0.1 M), 2a (1.2 equiv, 0.12 mmol), CH2Cl2 (1 mL), degassed via freeze-pump-thaw three times before irradiation, N2 atmosphere, flow rate (1 mL min–1), residence time (tR = 25 min), and chiral photocatalyst Δ-RhS (4 mol %).
Isolated yield.
Determined by chiral HPLC analysis.
Same as a, but with a total flow of 1.5 mL min–1 (tR = 16.7 min).
Same as a, but with a concentration of 0.05 M to 1 and maintaining the ratio of 2a.
Same as a, but with a concentration of 0.2 M to 1 and maintaining the ratio of 2a.
Same as a, but processing a total 5 mmol of material in total.
Having selected 40 °C as the best temperature for this continuous flow protocol, we kept on trying different light sources. Irradiation with 20 W blue LEDs was tested, affording compound 3 in 92% yield and er 96:4 (Table 3, entry 11). We also tried the use of CFL lamps instead of blue LEDs. Using 30 or 23 W CFL lamps provided equivalent results (Table 3, entries 12 and 13), and we chose to keep the 30 W CFL lamp because of its better stability for constant emission of photons. The last parameter evaluated was the concentration of the reaction concerning the limiting reagent (compound 1). Making the reaction less concentrated resulted in a significantly lower yield of 3 (65%, 95:5 er, Table 3, entry 14); albeit a more concentrated reaction medium did not affect the reaction outcome, the productivity of the process was improved (Table 3, entry 15).
Based on these results, we selected entry 15 as the best condition for running this reaction in flow. The result obtained in the continuous flow setup demonstrated no loss of yield or er compared to the experiment performed in batch but significantly decreased the reaction time from 4 h to only 25 min residence time. Prompted by these results, we performed a gram-scale experiment in flow, processing a total of 5.0 mmol of α,β-unsaturated acylimidazole 1, affording about 1.7 g (94% yield) of product 3 with 94:6 er (Table 3, entry 16). In this experiment, the 5.0 mmol of 1 took only 75 min to be processed under the current residence time, which is much less than the 24 h demanded by the scale-up experiment performed in batch using 1.0 mmol of 1.
Having established a suitable protocol for the gram-scale synthesis of intermediate 3, we moved to the next step in our synthetic approach, the conversion of 3 into lactam 4.
Using the method described by Morimoto and Ohshima,15 acylimidazole 3 was subjected to solvolysis in MeOH at 150 °C in a sealed pressure tube for 48 h. The solvolysis of 3 afforded ester 5 in 71% yield after column chromatography purification. Compound 5 was then treated with TFA in CH2Cl2 at room temperature, providing lactam 4 in 73% yield and 94:6 er after workup, without the necessity of further purification (Scheme 4A).
Scheme 4. (A) Stepwise Conversion of 3 into 4 and (B) Direct Conversion of 3 into 4.

While performing the aforementioned reaction, a careful analysis of the crude mixture containing ester 5 revealed the presence of a small amount of lactam 4, indicating the cleavage of the Boc group and a further cyclization under the solvolysis conditions. We wonder if it would be possible to drive the reaction toward the total conversion of acylimidazole 3 into lactam 4, without isolating ester 5. We found that rising the temperature up to 175 °C and keeping the reaction for 80 h led to a complete conversion of 3 into 4, without any remains of ester 5. Lactam 4 was then isolated by column chromatography in 80% overall yield (Scheme 4B). Our main concern regarding this direct conversion was an eventual racemization of the stereogenic center at β-position of compound 4. Fortunately, we observed no change in the er of 4, compared to 3 (94:6). Although the direct conversion of 3 into 4 was able to provide the target compound with a better yield, we recognize that depending on the circumstances, it might be more suitably performed stepwise, by the isolation of ester 5, mitigating security issues.
Attempts to achieve the direct conversion of 3 into 4 under continuous flow conditions were performed, but unsuccessful. Instead of lactam 4, ester 5 was the main product obtained in the methanolysis of 3, and even after treatment with TFA for a batch cyclization, compound 5 was obtained in only 37% overall yield (1H NMR yield) after 160 min residence time. This result reveals that the long-time demanding batch condition is more suitable for this transformation. With compound 4 in hand, we focus our efforts on the final steps of the total synthesis of BRV, which consists of the incorporation of the chiral butyramide moiety. This transformation is already reported in the literature using a strategy that involves N-alkylation of 4 with a chiral α-carbonyl electrophile, accompanied by manipulation of the carbonyl functionality to access the primary amide.7b,16
We decided to employ the protocol described by UCB S.A.,7b in which lactam 4 is alkylated with racemic bromoester 6, and the methyl ester group is then converted into the desired primary amide by reaction with aqueous ammonia. Unfortunately, in our hands, the reaction of racemic 4 and 6 did not provide the desired ester 7 (Scheme 5).
Scheme 5. Attempt to Prepare Racemic Brivaracetam.

Based on this result, we turned our attention to the possibility of using (R)-2-bromobutyric acid (8) instead of ester 6.16 The racemic lactam 4 (3.0 equiv) was treated with NaH in THF at room temperature, followed by the reaction with the racemic bromoacid 8 (1.0 equiv). The desired acid 9 was obtained and confirmed by 1H NMR analysis of the crude reaction mixture (Scheme 6). The conversion of 9 into racemic BRV was adapted from the original protocol, in which it is achieved by activation of CO2H with ClCO2Et, followed by the reaction with liquid ammonia. Racemic 9 was then subjected to the activation with ClCO2Et in the presence of Et3N in THF at 0 °C, but a commercial solution of ammonia in THF was used in the second step, instead of liquid ammonia (Scheme 6A).
Scheme 6. (A) Conversion of Lactam 4 into Racemic Brivaracetam and (B) Synthesis of BRV.

We were pleased to find that racemic BRV was obtained in 47% overall yield from 4 (Scheme 6A). Before extending this protocol to the preparation enantiopure BRV, we observed problems in the reproducibility of the last steps concerning the conversion of 4 into the final API. We attributed those difficulties to the evaporation of NH3 from the available commercial solution. Seeking alternative and more convenient NH3 sources that might be suitable for the present protocol of activation of the CO2H group, and keeping in mind to avoid the use of liquid ammonia, we found the use of NH4Cl solution as an advantageous option.17 The replacement of the NH3 solution in THF for an aqueous solution of NH4Cl afforded racemic BRV in 58% overall yield from 4 (Scheme 6A).
The use of 4 as the limiting reagent of this transformation was also tested using 3.0 equiv of the bromoacid 8; however, this condition provided BRV in only 30% yield. We also tried to reduce the excess of lactam 4 to 2.0 equiv, and once again, this condition was not beneficial, affording BRV in 32% yield (two last steps).
Next, we moved to the reaction between enantioenriched lactam 4 and 8. The (R)-2-bromobutanoic acid 8 was prepared according to the literature, but all attempts to confirm its enantiomeric purity by chiral HPLC were unsuccessful. However, optical rotation matched the data already reported.18 Using the same conditions established with the racemic compounds, enantioenriched lactam 4 and (R)-2-bromobutanoic acid 8 were reacted, affording BRV in 58% isolated yield (Scheme 6B), with the recovery of the major part (1.8 equiv, 60%) of compound 4 present in excess. The diastereoisomeric ratio (dr) of the final product was determined based on 1H NMR analysis. According to the literature,19 the signals observed at 3.50 and 3.60 ppm correspond to one of the 1H of C5, assigned for the (R,S) and (R,R), respectively (Figure 2A). Integration of these signals provides a 9:1 dr in favor of the desired diastereoisomer. The er was determined by chiral HPLC analysis (Figure 2B).
Figure 2.
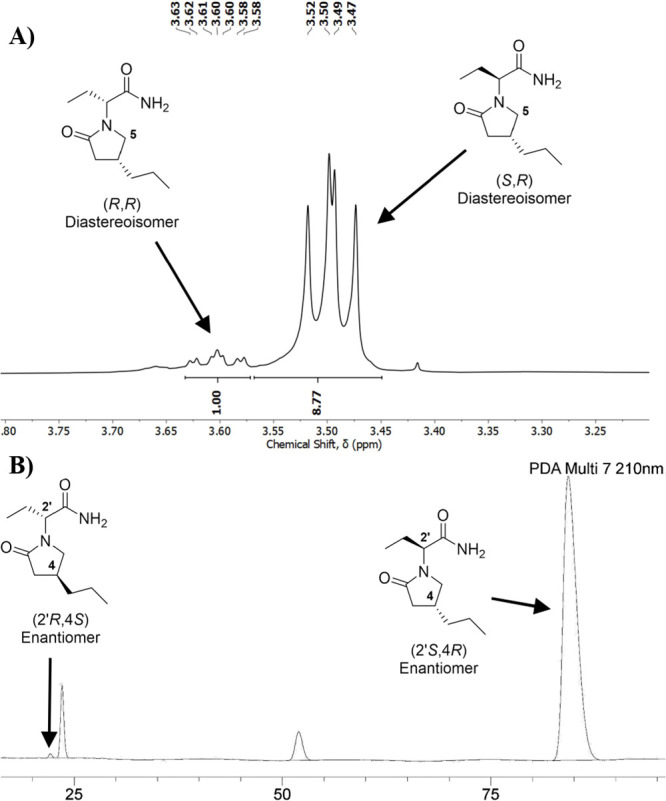
(A) Diastereoisomeric determination by 1H NMR analysis. (B) Peak assignment for BRV HPLC analysis.
It is expected that the combination between the major enantiomer of lactam 4 and acid 8 forms the major stereoisomer observed at 84.3 min in the chromatogram shown in Figure 2B. On the other hand, a combination between the minor enantiomer of 4 and the enantiomer of 8 (which might be present in small amounts) forms the minor stereoisomer observed at 22.1 min. Thus, we know these two peaks correspond to the stereoisomers (4R,2’S) and (4S,2’R). Based on the aforementioned data, a >99:1 er was assigned for the final product of the synthesis. The compound was shown to be levorotatory, which confirms the (4R,2’S) stereoisomer absolute configuration.20
Conclusions
In summary, we were able to synthesize acylimidazole 3 in quantitative yield and 98:2 er in batch conditions, using white visible-light and Δ-RhS as a bifunctional asymmetric catalyst/photocatalyst in only 4 mol %. The synthesis of 3 was successfully adapted to a continuous photoflow platform, which allowed us to scale-up the process to gram scale, without elongating the reaction time nor compromising its efficiency regarding yield and enantioselectivity. The conversion of intermediate 3 into lactam 4 was achieved in a single step with total conservation of the chiral integrity of the compound and 80% yield. Finally, the total synthesis of BRV was accomplished by the alkylation of lactam 4 with bromoacid 8, followed by the reaction with NH4Cl. BRV was obtained in four steps and 44% overall yield from 1, considering that about 60% of lactam 4 was recovered in the preparation of acid 9. The target compound displayed good dr (9:1) and er (>99:1).
Experimental Section
General Information
The starting materials and reagents were obtained from commercial suppliers and used without further purification unless stated otherwise. Solvents of technical grade were purified via distillation prior to use (hexane, ethyl acetate, dichloromethane, and methanol), and solvents of PA quality were used without further purification. Reaction monitoring and retention factor (Rf) determination were performed on precoated thin layer chromatography (TLC) sheets (TLC SILICA GEL 60 F254, MERCK) which were visualized either by quenching of ultraviolet light (λmax = 254 nm) or by immersion in potassium permanganate developer solution, followed by heating the plate to stain the spots. Flash column chromatography was performed on silica gel (Silica gel 60, MACHEREY-NAGEL, 230–400 mesh particle size). The ers of the reaction products were determined on Shimadzu UFLC Prominence 20-A HPLC employing a Daicel Chiralpak IG-3 chiral analytical column (150 × 4.6 mm i.d.) using the n-hexane/ethanol isocratic solvent system as mobile phase. The column temperature was 25–30 °C, and UV absorption was measured at 210 or 278 nm. Optical rotations were measured on a Schmidt–Haensch Polartronic E polarimeter equipped with a sodium lamp (589 nm) and reported as follows: [α]DT, (c = g mL–1, solvent). Infrared (IR) spectra were recorded on an ABB FTIR-FTLA2000 spectrometer using KBr pellets. UV–vis spectra were recorded on an Agilent Cary 60 UV–vis spectrophotometer using 1.0 cm quartz cuvettes at 25 °C. The emission spectra of the lamps (LEDs and CFL) were measured using an Ocean Optics (DH2000) USB4000 Fiber Optic spectrometer. Light sources used: blue LED 4 W (Iluctron, 3.8 lux), blue LED 30 W (Cob, 14.5 lux), CFL 15 W (Taschibra, 6.5 lux), and CFL 25 W (Taschibra, 7.5 lux). Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker AVANCE DRX-200 (200 MHz) or Bruker AVANCE DRX-400 (400 MHz) spectrometer. High-resolution mass spectra (HRMS) were recorded on a Bruker microTOF-Q II or Waters Xevo G2-S QTOF mass spectrometer using electrospray ionization (ESI) in positive ion mode (+). The mass error (deviation, in ppm) of the measurements was determined by the difference between the experimentally observed m/z and the theoretical m/z.
Synthesis of Substrates
The substrates (E)-1-(1-phenyl-1H-imidazol-2-yl)hex-2-en-1-one (1),11a Hantzsch ester derivatives (2a–c),11e (R)-2-bromobutanoic acid (8),18 and catalyst Δ-RhS21 were synthesized according to previously reported procedures.
Procedures Involved in the Synthetic Route
Rhodium-Catalyzed Photoredox Reaction in Batch at 0.1 mmol Scale: Synthesis of tert-Butyl(R)-(2-(2-oxo-2-(1-phenyl-1H-imidazol-2-yl)ethyl)pentyl)carbamate (3)
In a 20 mL Schlenk tube, (E)-1-(1-phenyl-1H-imidazol-2-yl)hex-2-en-1-one 1 (24.0 mg, 0.10 mmol, 1 equiv), Hantzsch ester 2a (52.6 mg, 0.12 mmol, 1.2 equiv), the catalyst Δ-RhS (3.4 mg, 4.0 mol %), and HPLC grade CH2Cl2 (1 mL, 0.1 M) were added. The mixture was degassed via freeze-pump-thaw and, after complete degassing of the mixture, the tube was sealed under a nitrogen atmosphere and positioned approximately 5–6 cm from a 15 W CFL lamp. The reaction was stirred at room temperature for 4 h. At the end of this period, the solvent was removed on a rotary evaporator under reduced pressure at 40 °C. The crude product was purified by column chromatography with silica gel using a gradient elution system (hexanes:ethyl acetate, 8:2–7:3). At the end of the isolation step, 3 was obtained as a pale yellow oil (36.9 mg, 0.099 mmol, yield 99%). Enantiomeric ratio established by HPLC analysis using a Chiralcel IG-3 column, er = 98:2 (HPLC: IG-3 column, λ 278 nm, n-hexane/ethanol = 90:10, flow rate 1.0 mL min–1, 27 °C, tr (major) = 7.8 min, tr (minor) = 8.9 min). TLC (Rf): 0.21 (hexane:ethyl acetate = 7:3). [α]D20 = −9.5 (0.02, CH2Cl2). 1H NMR (200 MHz, CDCl3): δ 7.49–7.43 (m, 3H), 7.33–7.26 (m, 3H), 7.18 (s, 1H), 4.98 (s, 1H), 3.24–2.97 (m, 4H), 2.25–2.13 (m, 1H), 1.41 (s, 9H), 1.38–1.30 (m, 4H), 0.88 (t, J = 6.2 Hz, 3H). 13C{1H} NMR (50 MHz, CDCl3): δ 191.1, 156.1, 143.2, 138.4, 129.5, 128.9, 128.7, 127.1, 125.9, 78.8, 43.8, 41.7, 35.2, 34.6, 28.4, 19.9, 14.2. IR (KBr), v̅max (cm–1): 3353, 2972, 2931, 2872, 1691, 1597, 1497, 1448, 1405, 1366, 1248, 1169, 1040, 969, 914, 855, 763, 694. HRMS (ESI+): [M + H]+ = 372.2264 (error = 4.8 ppm), calculated for [C21H30N3O3]+ = 372.2282.
Rhodium-Catalyzed Photoredox Reaction in Batch at 1.0 mmol Scale: Synthesis of tert-Butyl(R)-(2-(2-oxo-2-(1-phenyl-1H-imidazol-2-yl)ethyl)pentyl)carbamate (3)
In a 20 mL Schlenk tube, compound 1 (240.3 mg, 1.0 mmol, 1 equiv), Hantzsch ester 2a (526.0 mg, 1.2 mmol, 1.2 equiv), catalyst Δ-RhS (34.5 mg, 4.0 mol %), and HPLC grade CH2Cl2 (10 mL, 0.1 M) were added. The mixture was degassed via freeze-pump-thaw and, after complete degassing of the mixture, the tube was sealed under a nitrogen atmosphere and positioned approximately 5–6 cm from a 15 W CFL lamp. The reaction was stirred at room temperature for 20 h. Afterward, the solvent was removed on a rotary evaporator under reduced pressure at 40 °C. The crude product was purified by column chromatography over silica gel using a gradient elution system (hexanes:ethyl acetate, 8:2–7:3). At the end of the isolation step, 3 was obtained as a pale yellow oil (356.1 mg, 0.96 mmol, yield 96%). Enantiomeric ratio established by HPLC analysis using a Chiralcel IG-3 column, er = 98:2 (HPLC: IG-3 column, λ 278 nm, n-hexane/ethanol = 90:10, flow rate 1.0 mL min–1, 25 °C, tr (major) = 8.2 min, tr (minor) = 9.4 min).
Rhodium-Catalyzed Photoredox Reaction in Flow at 5.0 mmol Scale: Synthesis of tert-Butyl(R)-(2-(2-oxo-2-(1-phenyl-1H-imidazol-2-yl)ethyl)pentyl)carbamate (3)
In a 50 mL Schlenk tube, compound 1 (1.2 g, 5 mmol, 1 equiv), Hantzsch ester 2a (2.63 g, 6 mmol, 1.2 equiv), catalyst Δ-RhS (172.5 mg, 4.0 mol %), and HPLC grade CH2Cl2 (25 mL, 0.2 M) were added. The mixture was degassed via freeze-pump-thaw and, after complete degassing of the mixture, the mixture was transferred to a PFA coil loop. The mixture was pumped at 1.0 mL min–1 at 40 °C. Then, the solvent was removed on a rotary evaporator under reduced pressure at 40 °C. The crude product was purified by column chromatography over silica gel using a gradient elution system (hexanes:ethyl acetate, 8:2–7:3). At the end of the isolation step, 3 was obtained as a pale yellow oil (1.74 g, 4.7 mmol, yield 94%). Enantiomeric ratio established by HPLC analysis using a Chiralcel IG-3 column, er = 94:6 (HPLC: IG-3 column, λ 278 nm, n-hexane/ethanol = 90:10, flow rate 1.0 mL min–1, 25 °C, tr (major) = 7.5 min, tr (minor) = 8.3 min).
Synthesis of (R)-4-Propylpyrrolidin-2-one (4)
In a 10 mL Schlenk tube, compound 3 (356.1 mg, 0.96 mmol) and methanol (4.8 mL, 0.2 M) were added. Then, the flask was sealed and, in an oil bath preheated to 175 °C, the reaction mixture was kept under stirring for 80 h. At the end of the mentioned period, the reaction mixture was cooled to room temperature, diluted with CH2Cl2, adsorbed onto silica gel by solvent evaporation, and then purified by column chromatography over silica gel using a gradient elution system (hexanes:ethyl acetate, 1:1–1:9). At the end of the isolation step, (R)-4-propylpyrrolidin-2-one (4) was obtained as a yellow oil (99.2 mg, 0.78 mmol, yield 80%). Enantiomeric ratio established by HPLC analysis using a Chiralcel IG-3 column, er = 97:3 (HPLC: IG-3 column, λ 210 nm, n-hexane/ethanol = 90:10, flow rate 1.0 mL min–1, 25 °C, tr (minor) = 8.4 min, tr (major) = 9.4 min). TLC (Rf): 0.26 (hexanes:ethyl acetate = 2:8). 1H NMR (400 MHz, CDCl3): δ 6.15 (s, 1H), 3.52–3.46 (m, 1H), 3.02 (dd, J = 9.4, 6.9 Hz, 1H), 2.50–2.40 (m, 2H), 2.06–1.98 (m, 1H), 1.48–1.42 (m, 2H), 1.38–1.30 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 178.6, 48.2, 36.9, 36.8, 34.9, 20.8, 14.2. IR (KBr), v̅max (cm–1): 3231, 2958, 2929, 2872, 1701, 1489, 1456, 1379, 1285, 1065, 742, 696, 504. HRMS (ESI+): [M + Na]+ = 150.0890 (error = 0.7 ppm), calculated for [C7H13NONa]+ = 150.0889.
Synthesis of Methyl 3-(((tert-Butoxycarbonyl)amino)methyl)hexanoate (5)
In a 10 mL Schlenk tube, compound 3 (37.2 mg, 0.10 mmol) and methanol (0.2 M, 0.5 mL) were added. Then, the flask was sealed and the reaction mixture was stirred for 48 h at 150 °C. At the end of the mentioned period, the reaction mixture was cooled to room temperature, diluted with CH2Cl2, adsorbed on silica gel by solvent evaporation, and then purified by column chromatography with silica gel using a gradient elution system (hexanes:ethyl acetate, 8:2–7:3). Finally, ester 5 was obtained as a colorless oil (18.4 mg, 0.07 mmol, 71% yield). TLC (Rf): 0.55 (hexane:ethyl acetate = 7:3). 1H NMR (200 MHz, CDCl3): δ 4.62 (s, 1H), 3.61 (s, 3H), 3.19–2.88 (m, 2H), 2.22 (d, J = 6.6 Hz, 2H), 2.05–1.89 (m, 1H), 1.37 (s, 10H), 1.29–1.16 (m, 4H), 0.83 (t, J = 6.6 Hz, 3H). 13C{1H} NMR (50 MHz, CDCl3): δ 173.8, 156.2, 79.3, 51.7, 44.2, 37.1, 35.9, 34.5, 28.5, 20.0, 14.3. HRMS (ESI+): [M + Na]+ = 282.1672 (error = 1.4 ppm), calculated for [C13H25NO4Na]+ = 282.1676.
Synthesis of (S)-2-((R)-2-Oxo-4-propylpyrrolidin-1-yl)butanamide (BRV)
In a 20 mL dry Schlenk tube sealed with a rubber septum under N2 atmosphere, containing a NaH suspension (60% oily dispersion, 2.8 mmol, 7.0 equiv) in THF (4 mL), a solution of (R)-4-propylpyrrolidin-2-one (4) (153.0 mg, 1.2 mmol, 3.0 equiv) in tetrahydrofuran (2 mL) was added via syringe. The mixture was stirred at room temperature and, after the evolution of gas had ceased, a solution of (R)-2-bromobutanoic acid (8) (66.8 mg, 0.4 mmol, 1.0 equiv) in THF (2 mL) was added. The reaction mixture was stirred for 24 h. Subsequently, the mixture was extracted with aqueous NaOH solution (1 M) and CH2Cl2 (3 × 5 mL). The organic phase was dried over anhydrous Na2SO4 and filtered through filter paper, and the organic solvent was removed on a rotary evaporator under reduced pressure at 40 °C to recover 4. The aqueous phase was acidified with HCl solution (1 M, to pH = 2) and extracted with CH2Cl2 (3 × 5 mL). The organic phase was dried over Na2SO4, filtered, and concentrated on a rotary evaporator under reduced pressure at 40 °C providing the substitution product, which was used without purification in the next step. Subsequently, the crude product obtained in the previous step was transferred to a 10 mL Schlenk tube and dissolved with THF (2.3 mL), and the resulting solution was cooled to 0 °C. Then, ethyl chloroformate (35.6 mg, 0.33 mmol, 1.4 equiv) was added followed by Et3N (0.7 mmol, 3.0 equiv). After 0.5 h of stirring at 0 °C, an aqueous solution of NH4Cl 1 M (0.35 mL, 0.35 mmol, 1.5 equiv) was added dropwise at 0 °C to the suspension. The mixture was stirred for 3 h at 0 °C. Then, the resulting solution was extracted with brine and CH2Cl2 (3 × 10 mL), and the organic phase was dried over anhydrous Na2SO4, filtered, concentrated on a rotary evaporator at 40 °C, and purified by column chromatography with silica gel using a gradient elution system (CH2Cl2:MeOH, 9.9:0.1–9:1). After the isolation step, the BRV was obtained as a white solid (49.4 mg, 0.23 mmol, 58% yield relative to 8). TLC (Rf): 0.17 (hexane:ethyl acetate = 1:9). Diastereoisomeric ratio established by 1H NMR analysis, dr = 9:1 (δ (peak 1) = 3.50 ppm, δ (peak 2) = 3.60 ppm). Enantiomeric ratio established by HPLC analysis using a Chiralcel IG-3 column, er = >99:1 (HPLC: IG-3 column, λ 278 nm, n-hexane/ethanol = 80:20, flow rate 0.5 mL min–1, 25 °C, tr (major diastereoisomer) (peak 1) = 22.1 min, (peak 2) = 84.3 min, tr (minor diastereoisomer) (peak 1) = 23.6 min, tr (peak 2) = 52 min). 1H NMR (400 MHz, CDCl3): δ 6.65 (s, 1H), 6.06 (s, 1H), 4.49 (dd, J = 8.9, 6.8 Hz, 1H), 3.50 (dd, J = 9.8, 7.9 Hz, 1H), 3.08 (dd, J = 9.8, 7.2 Hz, 1H), 2.57 (dd, J = 16.7, 8.6 Hz, 1H), 2.40–2.28 (m, 1H), 2.15–2.05 (m, 1H), 1.99–1.88 (m, 1H), 1.74–1.63 (m, 1H), 1.47–1.30 (m, 4H), 0.95–0.88 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 175.7, 172.5, 55.9, 49.6, 37.9, 36.6, 32.0, 21.1, 20.6, 14.1, 10.5. IR (KBr), v̅max (cm–1): 3349, 3204, 2958, 2921, 2874, 1683, 1432, 1393, 1352, 1281, 1242, 706, 667, 583, 563. HRMS (ESI+): [M + Na]+ = 235.1418 (error = 0.4 ppm), calculated for [C11H20N2O2Na]+ = 235.1417.
Acknowledgments
The authors thank FAPESC (2020TR1451), FAPESP (2019/27176-8, 2020/06874-6, 2020/04114-4, and 2018/00879-6), and INCT-CATÁLISE/FAPESC/CNPq for the financial support, as well as CNPq and CAPES. M.S.F. thanks CAPES (633/2020) for the fellowship. The authors also acknowledge UFSC and UFSCar for the infrastructure, LAMRMN for the NMR analyses, LabCroMS for the HPLC analyses, and LABIME for the HRMS analyses.
Data Availability Statement
The data underlying this study are available in the published article and its online Supplementary Material.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02134.
Experimental procedures; analytical data; copies of NMR, IR, chromatograms, and HRMS of compounds; and FAIR data, including the primary NMR FID files, for compounds 3, 4, and 5, and BRV (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Piatek O. I.; Ning J. C.-M.; Touchette D. R. National drug shortages worsen during COVID-19 crisis: Proposal for a comprehensive model to monitor and address critical drug shortages. Am. J. Health Syst. Pharm. 2020, 77, 1778–1785. 10.1093/ajhp/zxaa228. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Choo E. K.; Rajkumar S. V. Medication shortages during the COVID-19 crisis: What we must do. Mayo Clin. Proc. 2020, 95, 1112–1115. 10.1016/j.mayocp.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abram M.; Jakubiec M.; Kamiński K. Chirality as an important factor for the development of new antiepileptic drugs. Chem. Med. Chem. 2019, 14, 1744–1761. 10.1002/cmdc.201900367. [DOI] [PubMed] [Google Scholar]
- https://www.who.int/health-topics/epilepsy#tab=tab_1, Accessed in February of 2023.
- a Kwan P.; Schachter S. C.; Brodie M. J. Drug-resistant epilepsy. N. Engl. J. Med. 2011, 365, 919–926. 10.1056/NEJMra1004418. [DOI] [PubMed] [Google Scholar]; b Engel J. What can we do for people with drug-resistant epilepsy? The 2016 Wartenberg Lecture. Neurology 2016, 87, 2483–2489. 10.1212/WNL.0000000000003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Markham A. Brivaracetam: First global approval. Drugs 2016, 76, 517–522. 10.1007/s40265-016-0555-6. [DOI] [PubMed] [Google Scholar]; b Stephen L. J.; Brodie M. J. Brivaracetam: A novel antiepileptic drug for focal-onset seizures. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617742081 10.1177/1756285617742081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenda B. M.; Matagne A. C.; Talaga P. E.; Pasau P. M.; Differding E.; Lallemand B. I.; Frycia A. M.; Moureau F. G.; Klitgaard H. V.; Gillard M. R.; Fuks B.; Michel P. Discovery of 4-Substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J. Med. Chem. 2004, 47, 530–549. 10.1021/jm030913e. [DOI] [PubMed] [Google Scholar]
- a Surtees J.; Lurquin F.; Diouf O.. Process for preparing 2-oxo-1-pyrrolidine derivatives. WO 2005028435A1, 2005.; b Ates C.; Lurquin F.; Quesnel Y.; Schule A.. 4-Substituted pyrrolidin-2-ones and their use. UCB Pharma S.A. WO 2007031263A1, 2007.; c Ates C.; Schule A.. Preparation of 1-carbamoylalkyl-3-carboxy-2-oxopyrrolidine. WO 2007065634A1, 2007.; d Defrance T.; Septavaux J.; Nuel D.. Procédé de préparation de Brivaracétam. WO 2017076738A1, 2017.; e Schülé A.; Merschaert A.; Szczepaniak C.; Maréchal C.; Carly N.; O’Rourke J.; Ates C. A Biocatalytic route to the novel antiepileptic drug Brivaracetam. Org. Process Res. Dev. 2016, 20, 1566–1575. 10.1021/acs.oprd.6b00094. [DOI] [Google Scholar]; f Wang P.; Li P.; Wei Q.; Liu Y.. Procédés de production du Brivaracetam. WO 2016191435A1, 2016.; g Gayke M.; Narode H.; Eppa G.; Bhosale R. S.; Yadav J. S. Synthetic approaches toward the synthesis of Brivaracetam: An antiepileptic drug. ACS Omega 2022, 7, 2486–2503. 10.1021/acsomega.1c05378. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Kumar S.; Ankit J.; Bhaskar B.; Kumar A.. A novel compound used for the preparation of brivaracetam and its preparation thereof. IN 201711038420, 2019.; i Sarabindu R. A. G.; Ramesh D. K.; Mutyala V. P.; Gopal P.; Abhishek G.; Sourav C.; Animesh H.; Ajay K. Y.; Subho R.. Enantioselective synthesis of brivaracetam and intermediates thereof. WO 2020148787 A1, 2020.; j Liang M.Novel preparation method of brivaracetam. CN 108689903 B, 2019.; k Liang M.Method for preparing optically pure (R)-4-n-propyl-dihydrofuran-2(3H)-one. WO 2018152949 A1, 2018.; l Liang M.New method for preparing brivaracetam. WO 2018152950 A1, 2018.
- Difa L.; Ting O.; Zhibin H.; Xunfa G.; Zhengmao L.; Panxing Y.; Min F.. A kind of preparation method of Brivaracetam and its intermediate. CN 109574778A, 2019.
- Peng G.; Fengwei L.; Jianxiao R.; Wenfeng Z.. Preparation method for Brivaracetam and intermediate thereof. WO 2020051796A1, 2020.
- Ötvös S. B.; Kappe C. O. Continuous flow asymmetric synthesis of chiral active pharmaceutical ingredients and their advanced intermediates. Green Chem. 2021, 23, 6117–6138. 10.1039/D1GC01615F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Huo H.; Harms K.; Meggers E. Catalytic, enantioselective addition of alkyl radicals to alkenes via visible-light-activated photoredox catalysis with a chiral Rhodium complex. J. Am. Chem. Soc. 2016, 138, 6936–6939. 10.1021/jacs.6b03399. [DOI] [PubMed] [Google Scholar]; b Wang C.; Harms K.; Meggers E. Catalytic asymmetric Csp3–H functionalization under photoredox conditions by radical translocation and stereocontrolled alkene addition. Angew. Chem., Int. Ed. 2016, 55, 13495–13498. 10.1002/anie.201607305. [DOI] [PubMed] [Google Scholar]; c Ma J.; Xie X.; Meggers E. Catalytic asymmetric synthesis of fluoroalkyl–containing compounds by three-component photoredox chemistry. Chem. – Eur. J. 2018, 24, 259–265. 10.1002/chem.201704619. [DOI] [PubMed] [Google Scholar]; d Ma J.; Lin J.; Zhao L.; Harms K.; Marsch M.; Xie X.; Meggers E. Synthesis of β-substituted γ-aminobutyric acid derivatives through enantioselective photoredox catalysis. Angew. Chem., Int. Ed. 2018, 57, 11193–11197. 10.1002/anie.201804040. [DOI] [PubMed] [Google Scholar]; e de Assis F. F.; Huang X.; Akiyama M.; Pilli R. A.; Meggers E. Visible-light-activated catalytic enantioselective β-alkylation of α,β-unsaturated 2-acyl imidazoles using Hantzsch esters as radical reservoirs. J. Org. Chem. 2018, 83, 10922–10932. 10.1021/acs.joc.8b01588. [DOI] [PubMed] [Google Scholar]
- a Buglioni L.; Raymenants F.; Slattery A.; Zondag S. D. A.; Noël T. Technological innovations in photochemistry for organic synthesis: Flow chemistry, high-throughput experimentation, scale-up, and photoelectrochemistry. Chem. Rev. 2022, 122, 2752–2906. 10.1021/acs.chemrev.1c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Neyt N. C.; Riley D. L. Application of reactor engineering concepts in continuous flow chemistry: A review. React. Chem. Eng. 2021, 6, 1295–1326. 10.1039/D1RE00004G. [DOI] [Google Scholar]; c Donnelly K.; Baumann M. Scalability of photochemical reactions in continuous flow mode. J. Flow Chem. 2021, 11, 223–241. 10.1007/s41981-021-00168-z. [DOI] [Google Scholar]; d Braga F. C.; Ramos T. O.; Brocksom T. J.; de Oliveira K. T. Synthesis of Fentanyl under continuous photoflow conditions. Org. Lett. 2022, 24, 8331–8336. 10.1021/acs.orglett.2c03338. [DOI] [PubMed] [Google Scholar]; e Su Y.; Straathof N. J. W.; Hessel V.; Noël T. Photochemical transformations accelerated in continuous flow reactors: Basic concepts and applications. Chem. – Eur. J. 2014, 20, 10562–10589. 10.1002/chem.201400283. [DOI] [PubMed] [Google Scholar]
- de Souza A. A. N.; Silva N. S.; Müller A. V.; Polo A. S.; Brocksom T. J.; de Oliveira K. T. Porphyrins as Photoredox Catalysts in Csp2–H Arylations: Batch and Continuous Flow Approaches. J. Org. Chem. 2018, 83, 15077–15086. 10.1021/acs.joc.8b02355. [DOI] [PubMed] [Google Scholar]
- a Chan V. S.; Chiu M.; Bergman R. G.; Toste F. D. Development of Ruthenium Catalysts for the Enantioselective Synthesis of P-Stereogenic Phosphines via Nucleophilic Phosphido Intermediates. J. Am. Chem. Soc. 2009, 131, 6021–6032. 10.1021/ja9014887. [DOI] [PubMed] [Google Scholar]; b Matusmoto A.; Fujiwara S.; Hiyoshi Y.; Zawatzky K.; Makarov A. A.; Welchc C. J.; Soai K. Unusual reversal of enantioselectivity in the asymmetric autocatalysis of pyrimidyl alkanol triggered by chiral aromatic alkanols and amines. Org. Biomol. Chem. 2017, 15, 555–558. 10.1039/C6OB02415G. [DOI] [PubMed] [Google Scholar]; c Yang H.; Jönsson A.; Wehtje E.; Adlercreutz P.; Mattiasson B. The enantiomeric purity of alcohols formed by enzymatic reduction of ketones can be improved by optimisation of the temperature and by using a high co-substrate concentration. Biochim. Biophys. Acta Gen. Subj. 1997, 1336, 51–58. 10.1016/S0304-4165(97)00010-X. [DOI] [PubMed] [Google Scholar]; d Otera J.; Sakamoto K.; Tsukamoto T.; Orita A. Temperature-effected tuning of enantioselectivity in asymmetric catalysis. Tetrahedron Lett. 1998, 39, 3201–3204. 10.1016/S0040-4039(98)00459-6. [DOI] [Google Scholar]; e Sibi M. P.; Gorikunti U.; Liu M. Temperature dependent reversal of stereochemistry in enantioselective conjugate amine additions. Tetrahedron 2002, 58, 8357–8363. 10.1016/S0040-4020(02)00988-2. [DOI] [Google Scholar]; f Buschmann H.; Scharf H.-D.; Hoffmann N.; Esser P. The Isoinversion Principle—a General Model of Chemical Selectivity. Angew. Chem., Int. Ed. 1991, 30, 477–515. 10.1002/anie.199104771. [DOI] [Google Scholar]
- Xin H.-L.; Pang B.; Choi J.; Akkad W.; Morimoto H.; Ohshima T. C–C bond cleavage of unactivated 2-acylimidazoles. J. Org. Chem. 2020, 85, 11592–11606. 10.1021/acs.joc.0c01458. [DOI] [PubMed] [Google Scholar]
- Surtees J.; Bouvy D.; Thomas A.; Combert Y.; Frank M.; Schmidt G.; Duchene G.. Process for the preparation of 2-oxo-1-pyrrolidine derivatives. US US20080009638 A1, 2006.
- Noguchi T.; Sekine M.; Yokoo Y.; Jung S.; Imai N. Convenient preparation of primary amides via activation of carboxylic acids with ethyl chloroformate and triethylamine under mild conditions. Chem. Lett. 2013, 42, 580–582. 10.1246/cl.130096. [DOI] [Google Scholar]
- Chen X.; Klöckner J.; Holze J.; Zimmermann C.; Seemann W. K.; Schrage R.; Bock A.; Mohr K.; Tränkle C.; Holzgrabe U.; Decker M. Rational design of partial agonists for the muscarinic M1 Acetylcholine receptor. J. Med. Chem. 2015, 58, 560–576. 10.1021/jm500860w. [DOI] [PubMed] [Google Scholar]
- Liao S.; Chen H.; Wang G.; Wu S.; Yang Z.; Luo W.; Liu Z.; Gao X.; Qin J.; Li C.-H.; Wang Z. Identification, characterization, synthesis and strategy for minimization of potential impurities observed in the synthesis of Brivaracetam. Tetrahedron 2020, 76, 131273 10.1016/j.tet.2020.131273. [DOI] [Google Scholar]
- Qiu S.; Tehrani K. A.; Sergeyev S.; Bultinck P.; Herrebout W.; Mathieu B. Stereochemistry of the Brivaracetam diastereoisomers. Chirality 2016, 28, 215–225. 10.1002/chir.22558. [DOI] [PubMed] [Google Scholar]
- Ma J.; Zhang X.; Huang X.; Luo S.; Meggers E. Preparation of chiral-at-metal catalysts and their use in asymmetric photoredox chemistry. Nat. Protoc. 2018, 13, 605–632. 10.1038/nprot.2017.138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online Supplementary Material.