

Abstract

Hydrogen-bonded supramolecular assemblies of citric acid, CA, with some heterocyclic compounds (N-donor and N-oxide)—acridine (acr), phenazine (phenz), 1,10-phenanthroline (110phen), 1,7-phenanthroline (17phen), 4,7-phenanthroline (47phen), 1,4-diazabicyclo[2.2.2]octane (dabco), and 4,4′-bipyridyl-N,N′-dioxide (bpydo)—have been reported. Among these, only the N-donors phenz and N-oxide (bpydo) form neutral co-crystals, while the others form salts owing to the deprotonation of −COOH. Thus, depending on the nature of the aggregate (salt/co-crystal), recognition between the co-formers is established through O–H···N/N+–H···O/N+H···O–heteromeric hydrogen bonding. Additionally, CA molecules establish homomeric interactions mediated by O–H···O hydrogen bonds. Moreover, CA forms a cyclic network with the co-formers or on its own, with a noteworthy feature of formation of host–guest networks in the assemblies with acr and phenz (solvated). In the assembly of acr, the CA molecules form a host network and captivate acr molecules as guest species, while in the case of phenz assembly, both the co-formers together encapsulate the solvent in the channels. However, the observed cyclic networks in the other structures form three-dimensional topologies in the form of ladders, a sandwich, lamellar layers, and interpenetrated networks. The structural features of the ensembles are evaluated unequivocally by the single-crystal X-ray diffraction method, while the homogeneity and phase purity are evaluated by using the powder X-ray diffraction method and differential scanning calorimetry. Further, conformational analysis of CA molecules reveals three types of conformations—T-shape (type I), syn–anti (type II), and syn (type III) as also observed in the literature for other CA co-crystals. In addition, the strength of the intermolecular interactions is quantified by performing Hirshfeld analysis.
Introduction
Multi-component solids often referred to as co-crystals1−3 play a pivotal role in the advancement of supramolecular chemistry. These solids are typically prepared by crystallization from a solution or by pulverizing using the mechanochemical approach.4−7 The materials, thus obtained, show improved properties and various applications, mostly in pharmaceutics with the emergence of pharmaceutical co-crystals,8−10 wherein one of the components is an API (active pharmaceutical ingredient), materials science11 (in the domains of mechanical, thermal, electrical, etc.), and so forth. Co-crystals are known not only for their aesthetic and exciting structures12−15 but also for the formation of unique homogeneous products with variable compositions of the co-crystal formers depending on the number of acceptor and donor species present in the native molecules.16
From the literature, it is well established that aromatic organic ligands mediated by the −COOH group show a great significance in the formation of exotic supramolecular architectures like honeycomb, voids, channels, interpenetrating networks, and so forth17−22 through hydrogen bonds. The effectiveness of symmetrically substituted aromatic organic acids like 1,3,5- (trimesic)- and 1,2,4,5-(pyromellitic)-substituted benzenecarboxylic acids23−26 has been well demonstrated in the formation of distinct and targeted supramolecular assemblies.27 Also, analogous cyclic aliphatic acids, for example, 1,2,4,5- or 1,3,5- and some disubstituted (1,2- and 1,3-) cyclohexanecarboxylic acids, were reported in the recent literature to establish the importance of non-aromatic acids in the development of exotic assemblies,28−33 as illustrated in Scheme 1.
Scheme 1. Exotic Channel Architectures Observed in the Supramolecular Assemblies of (a) 1,2,4,5-Benzenetetracarboxylic Acid and (b) cis,cis,cis-1,2,4,5-Cyclohexanetetracarboxylic Acid with a 1,10-Phenanthroline Rigid N-Donor.

In fact, such topological structures have reminiscence to natural zeolites, which possess permanent porosity. However, the arrangements in the organic assemblies are, in general, guest selective, and the hydrogen-bonded networks collapse upon removal of the guest molecules. In the recent literature, however, guest-independent organic ensembles have also been reported, in which porosity remains unaffected by the presence or absence of guests, which, in fact, lead to the exchange of guest moieties with the equivalent ones in dimensions and topology. Such ensembles are referred to as hydrogen-bonded organic framework (HOF) structures.34−38 For example, the hydrogen-bonded organic framework of 1,3,5-tris(4-carboxyphenyl) benzene, with an intricate 8-fold polycatenated assembly, demonstrates exceptional thermal stability, high surface area, and selective separation of C2 and methane gases.
However, aliphatic acyclic compounds which constitute a large portion of the dictionary of organic compounds are not well explored, except for some examples, especially, in the pharmaceutical co-crystals. To name a few, some of the dicarboxylic acids39−42 (fumaric acid, succinic acid, adipic acid, etc.) belong to GRAS (generally recognized as safe), and a few other tri- and tetracarboxylic acid-substituted compounds like aconitic acid (a tricarboxylic entity),43−45 tetracarboxylic acid (butane-1,2,3,4-tetracarboxylic acid),46,47 and so forth are well documented, as retrieved from the Cambridge Structural Database (CSD). A few examples have been projected in Scheme 2.
Scheme 2. Pharmaceutical Co-crystals of (a) Fumaric Acid with Carbamazepine and (b) trans-Aconitic Acid with Pyrazinamide.

It has been well established that the COOH decoration forms complexes with different N-donor compounds through the formation of O–H···N or O–H···N/C–H···O pairwise hydrogen bonds, as a myriad of assemblies are reported in the literature.21,48 In fact, N-donor compounds with binary hydrogen bonding acceptor sites, with variable dimensions, as co-formers, have been demonstrated for their high susceptibility to tune supramolecular architectures.49
Citric acid, CA, a well-known monohydroxy tricarboxylic acid, has gained popularity in recent times in the development of various pharmaceutical co-crystals, including using some N-donor compounds as co-formers, as retrieved from CSD (version, 5.43).50−55 Out of a total of 93 compounds, about 37 are known to form O–H···N hydrogen bonds (Table S3). However, binary binding site-related heterocyclic compounds are not well explored, except for a very few assemblies reported, for example, based on the 4,4′-bipyridine moiety and just reported different crystalline forms with 1,2-bis(4-pyridyl)ethene, which demonstrates the formation of different crystalline forms from the same solution.56−59 Furthermore, fused (aromatic)/bicyclic rings with rigid geometry and possessing binary hydrogen bond acceptors that are known in the gamut of N-donor compounds have also not been well explored.
In fact, the significance of fused N-donors, for example, acridine (acr) and phenazine (phenz), has been demonstrated in the crystal stabilization process through effective calculations of energy parameters also, even with liquid-phase co-formers like APIs.60
Considering these facts, along with the effective role of CAa in biological processes,61−63 the development of several other ensembles of CA with heterocyclic compounds could provide further insights into the preparation and evaluation of tailor-made supramolecular assemblies of CA.
Hence, molecular assemblies of citric acid, CA, with various N-donor ligands, acridine (acr), phenazine (phenz), 1,10-phenanthroline (110phen), 4,7-phenanthroline (47phen), 1,7-phenanthroline (17phen), 1,4-diazabicyclo[2.2.2]octane (dabco), and an N-oxide, 4,4′-bipyridyl N,N′-dioxide (bpydo), as shown in Scheme 3, have been prepared and characterized by the single-crystal X-ray diffraction method to deduce the effective recognition patterns, self-assembly, thermal stability, and so forth.
Scheme 3. Molecular Assemblies of CA with Various Heterocyclic Compounds.

Experimental Section
All chemicals were purchased from Sigma-Aldrich with >99% purity and have been used without further purification. The solvents employed for crystallization studies were of spectroscopy grade of highest available quality. All co-crystals/salts were prepared by dissolving the respective reactants in an appropriate solvent and allowing the solution to evaporate either at ambient or at low-temperature conditions. In all cases, good-quality single crystals suitable for X-ray diffraction analysis were obtained within 72–96 h.
Crystal Structure Determination
Good-quality single crystals were carefully chosen after being viewed under a Leica microscope supported by a rotatable polarizing stage, glued to glass fiber by using an adhesive, and mounted on the goniometer of a Bruker single-crystal X-ray diffractometer (D8 VENTURE) (Mo Kα radiation, λ = 0.71073 Å) equipped with a PHOTON 100 CMOS detector. All the crystals were stable throughout the data collection period, and data collection was smooth. Data collection was performed using φ and ω scans. The structures were solved by using the intrinsic phasing method followed by full-matrix least-squares refinement against F2 using SHELXTL, embedded within the Bruker suite of programmes.64 All non-hydrogen atoms were refined by the anisotropic method, and hydrogen atoms were either refined or placed in calculated positions (Table S2). All the structural refinements converged to good R factors, as listed in Table 1, and the intermolecular interactions were computed by using PLATON software (see Table 2).65 The packing diagrams were generated by using Diamond (version 4.6.3).66
Table 1. Crystallographic Data for Co-crystals/Salts, 1–7, 2a, and 6a.
| parameters | 1 | 2 | 2a | 3 | 4 | 5 | 6 | 6a | 7 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| formula | C19H17NO7 | C12H12NO7 | C12H12N2O8 | C36H32N4O14 | C18H15N2O7 | C30H24N4O7 | C12H20N2O7 | C12H22N2O8 | C16H16N2O9 | ||||
| Mr | 371.33 | 282.23 | 311.24 | 744.65 | 371.32 | 552.53 | 304.30 | 322.31 | 380.31 | ||||
| crystal color | yellow | yellow | yellow | colorless | brown | brown | colorless | colorless | colorless | ||||
| crystal shape | block | plate | plate | plate | block | plate | plate | plate | rod | ||||
| T/K | 293(2) | 293(2) | 293(2) | 293(2) | 100(2)a | 293(2) | 293(2) | 293(2) | 293(2) | ||||
| λ (Mo Kα)/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | ||||
| crystal system | monoclinic | triclinic | monoclinic | orthorhombic | triclinic | triclinic | monoclinic | orthorhombic | triclinic | ||||
| space group | P21/n | C2/c | Pna21 | P21/n | Pbca | ||||||||
| a/Å | 7.5105(4) | 5.7149(4) | 21.7470(7) | 24.4161(9) | 7.1875(5) | 9.2814(5) | 10.2360(7) | 11.4023(4) | 7.1148(3) | ||||
| b/Å | 12.0169(6) | 10.2355(8) | 5.8877(3) | 7.4403(3) | 10.6542(7) | 10.2674(7) | 7.4616(5) | 13.6103(6) | 10.3487(5) | ||||
| c/Å | 19.3285(9) | 10.8779(8) | 24.4913(10) | 18.4983(6) | 11.8003(8) | 13.4955(9) | 18.8990(9) | 19.3911(9) | 11.2727(7) | ||||
| a/deg | 90 | 73.888(8) | 90 | 90 | 68.142(3) | 82.056(5) | 90 | 90 | 89.991(2) | ||||
| β/deg | 98.282(3) | 84.594(6) | 108.441(4) | 90 | 85.003(3) | 77.442(5) | 97.450(4) | 90 | 78.936(3) | ||||
| γ/deg | 90 | 85.332(5) | 90 | 90 | 81.987(4) | 87.845(7) | 90 | 90 | 83.221(3) | ||||
| V/Å3 | 1726.26(5) | 607.57(8) | 2974.8(2) | 3360.5(2) | 829.87(10) | 1243.21(14) | 1431.26(15) | 3009.3(2) | 808.66(7) | ||||
| Z | 4 | 2 | 8 | 4 | 2 | 2 | 4 | 8 | 2 | ||||
| Dc/g cm–3 | 1.429 | 1.543 | 1.390 | 1.472 | 1.486 | 1.476 | 1.412 | 1.423 | 1.562 | ||||
| μ, mm–1 | 0.110 | 0.129 | 0.118 | 0.115 | 0.116 | 0.107 | 0.117 | 0.120 | 0.130 | ||||
| F(000) | 776 | 294 | 1292 | 1552 | 386 | 576 | 648 | 1376 | 396 | ||||
| θ range [deg] | 2.13–27.16 | 1.95–27.35 | 1.75–27.24 | 1.99–26.35 | 2.22–27.24 | 2.24–26.53 | 2.15–26.41 | 2.55–25.99 | 2.67–28.33 | ||||
| index ranges | –9 ≤ h ≤ 9 | –7 ≤ h ≤ 7 | –27 ≤ h ≤ 28 | –30 ≤ h ≤ 23 | –9 ≤ h ≤ 9 | –11 ≤ h ≤ 11 | –12 ≤ h ≤ 12 | –13 ≤ h ≤ 14 | –9 ≤ h ≤ 9 | ||||
| –15 ≤ k ≤ 15 | –13 ≤ k ≤ 13 | –6 ≤ k ≤ 7 | –9 ≤ k ≤ 8 | –13 ≤ k ≤ 13 | –12 ≤ k ≤ 12 | –9 ≤ k ≤ 9 | –16 ≤ k ≤ 16 | –13 ≤ k ≤ 12 | |||||
| –24 ≤ l ≤ 24 | –13 ≤ l ≤ 13 | –31 ≤ l ≤ 31 | –23 ≤ l ≤ 19 | –13 ≤ l ≤ 15 | –14 ≤ l ≤ 16 | –23 ≤ l ≤ 21 | –19 ≤ l ≤ 23 | –15 ≤ l ≤ 15 | |||||
| total reflections | 16,265 | 10,770 | 25,713 | 19,264 | 15,408 | 18,658 | 24,095 | 17,728 | 14,451 | ||||
| unique reflections | 3258 | 1549 | 3309 | 4943 | 3190 | 3306 | 2584 | 2952 | 3946 | ||||
| no. of parameters | 280 | 199 | 204 | 501 | 310 | 422 | 206 | 210 | 273 | ||||
| goodness-of-fit F2 | 0.986 | 1.002 | 1.010 | 1.001 | 1.003 | 1.002 | 1.008 | 1.002 | 1.004 | ||||
| R1, [I > 2σ(I)] | 0.043 | 0.052 | 0.069 | 0.052 | 0.049 | 0.056 | 0.044 | 0.073 | 0.044 | ||||
| wR2, [I > 2σ(I)] | 0.111 | 0.127 | 0.194 | 0.131 | 0.1450 | 0.144 | 0.116 | 0.2036 | 0.108 | ||||
| Δρmax, Δρmin (e Å–3) | 0.24, −0.23 | 0.41, −0.28 | 0.78, −0.24 | 0.44, −0.26 | 0.33, −0.23 | 0.40, −0.25 | 0.30, −0.19 | 0.90, −0.48 | 0.26, −0.20 | ||||
| CCDC no. | 2242672 | 2242670 | 2245375 | 2242674 | 2242671 | 2242675 | 2242676 | 2245376 | 2242673 |
In order to reduce the disorder of the CA molecule in the structure of 4, data were collected at 100 K.
Table 2. Hydrogen Bond Distances (Å) and Angles (deg) for All the Co-crystals/Salts, 1–7, 2a, and 6aa.
| co-crystal/salt | O–H···N | N+–H···O– | N+–H···O | O–H···O | C–H···O |
|---|---|---|---|---|---|
| 1 | 2.07 2.96(2) 148 | 2.14 2.94(2) 135 | 1.59 2.59(1) 175 | 2.48 3.52(2) 161 | |
| 1.59 2.55(1) 166 | 2.56 3.43(2) 137 | ||||
| 1.75 2.64(2) 151 | 2.74 3.49(2) 154 | ||||
| 2.63 3.52(2) 139 | |||||
| 2 | 1.77 2.74(2) 171 | 1.71 2.68(1) 174 | 2.54 3.61(2) 171 | ||
| 1.71 2.69(2) 172 | 2.56 3.55(2) 153 | ||||
| 2.59 3.39(2) 131 | |||||
| 2.64 3.45(2) 132 | |||||
| 2a | 1.75 2.73(1) 175 | 1.69 2.66(1) 173 | 2.40 3.45(1) 164 | ||
| 1.69 2.66(1) 168 | 2.55 3.46(2) 142 | ||||
| 1.92 2.85(1) 158 | 2.58 3.46(2) 139 | ||||
| 2.59 3.57(1) 150 | |||||
| 3 | 1.94 2.73(1) 133 | 1.48 2.46(1) 173 | 2.51 3.46(1) 147 | ||
| 1.54 2.52(1) 175 | 2.63 3.71(2) 173 | ||||
| 1.55 2.66(2) 167 | 2.63 3.65(2) 158 | ||||
| 1.67 2.61(2) 161 | 2.64 3.61(2) 150 | ||||
| 1.85 2.70(1) 142 | |||||
| 4 | 1.59 2.57(1) 173 | 1.69 2.60(1) 149 | 2.31 3.04(2) 130 | 1.44 2.38(2) 160 | 2.22 3.24(1) 157 |
| 1.94 2.74(2) 137 | 2.26 3.26(2) 153 | ||||
| 2.38 3.41(2) 159 | |||||
| 2.49 3.55(1) 168 | |||||
| 2.52 3.34(2) 139 | |||||
| 2.67 3.68(2) 156 | |||||
| 2.72 3.73(2) 156 | |||||
| 5 | 1.62 2.60(1) 176 | 1.55 2.54(1) 169 | 1.73 2.68(1) 161 | 2.43 3.28(1) 135 | |
| 1.92 2.71(2) 135 | 2.49 3.53(2) 161 | ||||
| 2.54 3.59(2) 163 | |||||
| 2.54 3.58(1) 162 | |||||
| 2.57 3.60(2) 159 | |||||
| 6 | 1.66 2.64(1) 179 | 1.71 2.70(1) 167 | 1.58 2.54(1) 166 | 2.42 3.39(2) 149 | |
| 2.42 3.16(2) 130 | 2.05 2.90(1) 141 | 2.44 3.23(2) 130 | |||
| 2.45 3.43(2) 151 | |||||
| 2.60 3.49(2) 140 | |||||
| 2.67 3.59(2) 143 | |||||
| 2.75 3.69(2) 146 | |||||
| 2.74 3.79(2) 165 | |||||
| 6a | 1.59 2.56(1) 172 | 1.88 2.72(1) 140 | 1.81 2.79(1) 174 | 2.53 3.53(2) 154 | |
| 2.20 2.96(1) 140 | 1.81 2.77(1) 167 | 2.53 3.51(1) 151 | |||
| 1.83 2.80(1) 171 | 2.52 3.37(2) 154 | ||||
| 1.95 2.56(1) 141 | 2.72 3.63(1) 142 | ||||
| 2.85 3.76(2) 143 | |||||
| 2.86 3.79(2) 145 | |||||
| 2.89 3.88(2) 152 | |||||
| 7 | 1.61 2.58(2) 169 | 2.31 3.37(2) 168 | |||
| 1.64 2.60(1) 168 | 2.35 3.43(1) 172 | ||||
| 1.67 2.61(1) 158 | 2.37 3.44(1) 169 | ||||
| 2.46 3.52(2) 169 | |||||
| 2.62 3.50(2) 153 |
For each structure, the three columns represent the distances of H···A, D···A, and angles ∠D–H···A, respectively.
Powder X-ray Diffraction
Powder X-ray diffraction (PXRD) patterns were obtained using a Bruker D8 ADVANCE X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). The voltage and current applied were 40 kV and 30 mA, respectively. Samples were measured in the reflection mode in the 2θ range of 5–40°.
Thermal Analysis
Thermal analysis, differential scanning calorimetry (DSC) and thermogravimetric analyzer (TGA)was carried out on PerkinElmer DSC 8000 and STA 6000 instruments respectively.
In DSC, stainless steel pans were used for the experiment, and the calibration of the instrument was done with the standard sample indium. A sealed empty pan was used as a reference pan, and samples were heated under a nitrogen atmosphere at a scan rate of 5° min–1. In TGA, samples of 2–5 mg were heated in an open alumina crucible under a nitrogen atmosphere at a rate of 5 °C/min from room temperature to a temperature of 350 °C.
Hirshfeld Surface Analysis
The Hirshfeld surface analysis and 2D fingerprint calculations were performed using the Crystal Explorer package version 2167 by importing the atomic coordinates from the CIF files. The Hirshfeld surfaces (separately for each co-former in all structures) are generated, the distance from the nearest nucleus inside and outside the surface was measured and represented by the di and de, respectively, while a normalized contact distance was represented as dnorm. The white, red, and blue colors have been selected for the visualization of dnorm function with very high resolution.
Results and Discussion
Co-crystals of CA with various heterocyclic compounds, as listed in Scheme 3, have been prepared and analyzed by the single-crystal X-ray diffraction method. The pertinent crystallographic parameters are compiled in Table 1. The contents of the asymmetric unit and the corresponding composition are given in the Supporting Information (Table S1) in the form of ORTEP representation with the thermal ellipsoids shown at the 50% probability level for all the structures, 1–7, 2a, and 6a.
In all the ensembles, the primary recognition between CA and the corresponding heterocyclic compounds occurs through the formation of heteromeric hydrogen bonds O–H···N/N+–H···O/N+–H···O– or O–H···O for N-oxide along with homomeric hydrogen bonds between CA molecules. The recognition patterns for structures 1–7, 2a, and 6a are shown in Figure 1. The detailed analysis of each structure is discussed further independently in the following sections.
Figure 1.

Heteromeric and homomeric hydrogen bonds in the supramolecular assemblies: (a) 1, (b) 2, (c) 2a, (d) 3, (e) 4, (f) 5, (g) 6, (h) 6a, and (i) 7.
Channel Structure in the Hydrogen-Bonded Ensembles of 1 (CA and acr)
Co-crystallization of CA and acr in a 1:1 ratio from the CH3CN solution of the co-formers gives good-quality block-shaped yellowish crystals. Due to the proton transfer from one of the −COOH groups of CA to acr, a salt is formed in a 1:1 ratio of the co-formers and crystallizes in a monoclinic space group, P21/n (Table 1), as confirmed by the X-ray diffraction method (Table S1). Analysis of recognition features discloses that the protonated −N atom of acr forms a bifurcated N+–H···O hydrogen bond with both −OH and −COOH groups of CA (H···O 2.07 and 2.14 Å; O···O, 2.96 and 2.94 Å). The complete characteristics of hydrogen bonds are listed in Table 2. Nonetheless, the remaining two −COOH groups of CA further participate in homomeric O–H···O– hydrogen bonds with the adjacent CA molecules with a H···O– distance of 1.59 Å (O···O–, 2.59 Å). The arrangement is shown in Figure 1a.
Such an aggregation further yields a three-dimensional arrangement in the form of a channel structure along the crystallographic axis (a-axis) of the crystal lattice (Figure 2a). In this topology, herein, channels are realized through the stacking of layers of CA molecules, with voids (12 × 11 Å2), formed due to the aggregation of each of six molecules of CA in a cyclic pattern, which are further aligned along the stacking direction. In such channels, acr molecules are captivated as shown in Figure 2b. The cyclic network by CA molecules within the layers is formed through O–H···O and C–H···O (H···O, 2.48 Å, C···O, 3.52 Å) interactions, whereas the acr molecules that reside within the channels are glued to the host network through the N+–H···O hydrogen bonds discussed above for the basic recognition feature between CA and acr. Further, within these channels, acr molecules are stacked through π–π interactions with a separation distance of 3.80 Å. This marks a significant example of an aliphatic compound forming a guest-induced channel structure with voids, mimicking a hydrogen-bonded organic open framework structure as noted in the literature,68 otherwise formed by some symmetrically substituted aromatic acids like trimesic acid.
Figure 2.

(a) Channel structure formed by CA molecules in the crystal lattice 1. (b) Channel being filled by acr molecules.
Layer and Channel Structures of CA and phenz, 2, and 2a
CA and phenz form two different co-crystals in a 2:1 ratio, 2 and 2a (solvated), in the form of good-quality single crystals upon slow evaporation of CH3NO2 solution having co-formers in a 1:1 ratio. The recognition pattern between CA and phenz, evaluated using the structure obtained from the X-ray diffraction method (Table 1), is comparable to the arrangement in 1, with the formation of homomeric and heteromeric hydrogen bonds (see Figure 1b,c). In these assemblies, however, the interaction between CA and phenz is mediated by an O–H···N (H···N, 1.77 Å, O···N, 2.74 Å, 2, H···N, 1.75 Å, O···N 2.73 Å 2a; Table 2) hydrogen bond, which is a typical interaction between −COOH and N-atom in heterocyclic compounds. In addition, the feature emphasizes the formation of a co-crystal rather than a salt as per the nomenclature defined in the literature.69
In these structures of 2 and 2a, CA molecules are bound together by the formation of two cyclic hydrogen bonding patterns between adjacent −COOH groups, as represented by the graph-set notation R22(8). In such centrosymmetric hydrogen bond dimers, the H···O distance is found to be 1.71 Å (O···O, 2.68 Å, 2) and 1.69 Å (O···O, 2.66 Å, 2a). Additionally, a non-centrosymmetric hydrogen bonding pattern is also observed with H···O distances of 1.69 and 1.92 Å (O···O, 2.66 and 2.85 Å) in structure 2a. Interestingly, the potential hydrogen bond donor −OH did not feature in the recognition process in 2 but in 2a as depicted in Figure 1b,c.
The ensembles 2 and 2b are further packed in the crystalline lattices in different forms. In 2, the arrangement is in the form of stacked corrugated sheets, as illustrated in Figure 3a, while in 2a, such an arrangement is observed to be in the form of a host–guest network (Figure 3c). Within a typical sheet, in 2, cyclic networks comprising eight molecules (six of CA and two of phenz) are formed through hydrogen bonds (see Figure 3b) and are further garnered through the interaction between the −COOH groups present in the adjacent networks. In such a cyclic network, voids appear but are being self-filled by part of phenz molecules that also take part in the cyclic network. In the structure of 2a, however, the two co-formers together form a three-dimensional host network along the crystallographic axis (b-axis), with CA molecules alone forming layers that are pillared by phenz molecules, thus creating channels (see Figure 3c), filled by solvent of crystallization molecules. Thus, the structure of 2a may also be regarded as the mimic of a hydrogen-bonded organic framework with a close resemblance to the structure observed in 1.
Figure 3.

(a) Stacked sheets in the crystal lattice 2. (b) Typical interaction between the co-formers within sheets through appropriate hydrogen bonds. (c) Channel structure of CA and phenz in 2a, the channel being filled with the solvent molecule. (d) Interactions of CA molecules in 2a thus to form a cyclohexane chair conformation.
A close examination at Figure 3b,d reveals an intriguing correlation between 2 and 2a: the cyclohexane chair conformation topology observed in 2a, formed by the aggregation of only CA molecules, within the layers, is further expanded in 2 with the insertion of phenz molecules between the CA molecules, thereby masking the channels observed in 2a, leading to the formation of stacked layers in a three-dimensional arrangement in 2.
Sandwich Arrangement in the Supramolecular Assembly of CA with 110phen, 3
The supramolecular assembly of 3 was obtained by slow evaporation from a mixture of acetone and H2O solution, containing the co-formers CA and 110phen in a 1:1 ratio. The resulting crystals were analyzed by the X-ray diffraction method (Table 1) which discloses that the asymmetric unit is in an orthorhombic space group, Pna21 (non-centrosymmetric), with two symmetry-independent molecules (Table S1). As observed in the case of 1, CA with acr, herein also, the co-formers form a salt upon the deprotonation of −COOH to 110phen in both the symmetry-independent molecules. However, the recognition patterns found in this structure show some correlation to the structure of CA and acr (cf. Figure 1d). The primary recognition observed between the co-formers is a single N+–H···O– (H···O–, 1.94 Å, N+···O–, 2.73 Å) formed between N-donor and −COOH of CA, while the CA molecules are stabilized by homomeric single O–H···O– hydrogen bonds, formed by both −OH and −COO– groups, with the respective H···O– being 1.48 Å (O···O–, 2.48 Å) and 1.55 Å (O···O–, 2.66 Å).
Further analysis of the arrangement of co-former aggregates within the crystal lattice perceives that CA molecules form corrugated sheets that are stacked, with 110phen molecules sandwiched between the sheets. The arrangement is shown in Figure 4a, and the interaction among the CA molecules within a typical sheet is projected in Figure 4b. In a representative sheet, each of the two symmetry-independent CA molecules forms chains independently, with the nearby molecules of the same symmetry binding together by a single O–H···O (H···O, 1.48 and 1.54 Å; O···O, 2.46 and 2.52 Å) hydrogen bond. Such juxtaposed chains (symmetry independent), as shown in Figure 4a (space-filled model in different colors), are held together by a non-centrosymmetric R22(10) O–H···O hydrogen bonding pattern with the corresponding H···O distances in the range of 1.67–1.85 Å (O···O, 2.61–2.70 Å), as well as single O–H···O (H···O, 1.55 Å; O···O, 2.66 Å).
Figure 4.

(a) Sandwich arrangement of 110phen molecules between the corrugated layers of CA molecules in 3. (b) Typical arrangement of CA molecules within the corrugated sheets.
Self-Aligned Ladders’ Three-Dimensional Assembly in the Structure of CA and 47phen, 4
Upon slow evaporation of CH3OH solution containing CA and 47phen, in a 1:1 ratio, the co-formers
crystallize in a triclinic space group,  . Complete structure determination parameters
are listed in Table 1. Among the structures studied so far, herein only, CA molecules present in a disordered form, around one of the −COOH
groups, reflecting the facile conformation flexibility of CA, and the same is shown in ORTEP drawing (Table S1). For the brevity purpose, the disordered moiety is disregarded
in all subsequent discussions. Similar to the other structures, like 1 and 3, herein also, deprotonation of one of
the −COOH groups leads to the formation of a salt between the
co-formers. The recognition features, which are depicted in Figure 1e, reveal that two
−COOH groups of CA molecules indeed establish heteromeric interaction with 47phen through
O–H···N (H···N, 1.59 Å,
N···O, 2.57 Å) and N+–H···O– (H···O–, 1.69 Å,
N+···O–, 2.60 Å).
Furthermore, similar to 1, here also, the protonated
−N atom of 47phen involved
in bifurcated N+–H···O hydrogen bonds
with both −COOH (N+–H···O–) and −OH groups of CA by the N+–H···O hydrogen bond (H···O
2.31 Å; O···O, 3.04 Å), apart from the third
−COOH group forming homomeric hydrogen bonds
(O–H···O–, H···O–, 1.44 Å, O···O, 2.38 Å) with
the adjacent CA molecules. The ensembles of co-formers
further self-assemble in the crystal lattice in the form of infinite
ladders connecting the adjacent ladders together through common slide
rails. A typical representation is shown in Figure 5a. In such an arrangement, while the rails
are being formed by the chains of CA molecules, the rungs
are being mimicked by 47phen molecules through the
hydrogen bonds discussed above through the representation in Figure 1e. In a three-dimensional
arrangement, such ladders are stacked, as presented in Figure 5b, by self-filling the voids
between the rungs with the molecules from the adjacent ladders above
and below, thus establishing close packing effectively. The stacked
layers are further stabilized by C–H···O hydrogen
bonds with H···O distances in the range of 2.22–2.38
Å (C···O, 3.24–3.68 Å).
. Complete structure determination parameters
are listed in Table 1. Among the structures studied so far, herein only, CA molecules present in a disordered form, around one of the −COOH
groups, reflecting the facile conformation flexibility of CA, and the same is shown in ORTEP drawing (Table S1). For the brevity purpose, the disordered moiety is disregarded
in all subsequent discussions. Similar to the other structures, like 1 and 3, herein also, deprotonation of one of
the −COOH groups leads to the formation of a salt between the
co-formers. The recognition features, which are depicted in Figure 1e, reveal that two
−COOH groups of CA molecules indeed establish heteromeric interaction with 47phen through
O–H···N (H···N, 1.59 Å,
N···O, 2.57 Å) and N+–H···O– (H···O–, 1.69 Å,
N+···O–, 2.60 Å).
Furthermore, similar to 1, here also, the protonated
−N atom of 47phen involved
in bifurcated N+–H···O hydrogen bonds
with both −COOH (N+–H···O–) and −OH groups of CA by the N+–H···O hydrogen bond (H···O
2.31 Å; O···O, 3.04 Å), apart from the third
−COOH group forming homomeric hydrogen bonds
(O–H···O–, H···O–, 1.44 Å, O···O, 2.38 Å) with
the adjacent CA molecules. The ensembles of co-formers
further self-assemble in the crystal lattice in the form of infinite
ladders connecting the adjacent ladders together through common slide
rails. A typical representation is shown in Figure 5a. In such an arrangement, while the rails
are being formed by the chains of CA molecules, the rungs
are being mimicked by 47phen molecules through the
hydrogen bonds discussed above through the representation in Figure 1e. In a three-dimensional
arrangement, such ladders are stacked, as presented in Figure 5b, by self-filling the voids
between the rungs with the molecules from the adjacent ladders above
and below, thus establishing close packing effectively. The stacked
layers are further stabilized by C–H···O hydrogen
bonds with H···O distances in the range of 2.22–2.38
Å (C···O, 3.24–3.68 Å).
Figure 5.

(a) Ladder-type arrangement formed in the crystal structure 4. (b) Stacking of the molecules in three-dimensional arrangement masking the voids from the adjacent ladders.
Ladderane Topology of CA and 17phen in the Crystal Structure of 5
Co-crystallization
of CA and 17phen, in a 1:1 ratio, from
an acetone solution of the co-formers gives good-quality plate-shaped
single crystals, consisting of a salt of the co-formers in a 1:2 ratio.
The molecules in the asymmetric unit, crystallized in a triclinic
space group,  , are portrayed in Table S1, which illustrates that out of the two 17phen molecules, only one of it is protonated. The pertinent crystallographic
details are listed in Table 1. The contents establish recognition with each of the CA molecule connected to two 17phen molecules
via O–H···N and N+–H···O– (heteromeric hydrogen bonds), with
the corresponding distances H···N, 1.62 Å (N···O,
2.60 Å) and H···O– 1.55 Å
(N+···O–, 2.54 Å).
In addition, CA molecules form homomeric hydrogen bonds through a centrosymmetric cyclic pattern of O–H···O– with an H···O– distance
of 1.73 Å (O···O–, 2.68 Å).
Also, a noteworthy feature to be noted is the potential hydrogen bond
donor −OH that does not participate in the recognition process.
The recognition pattern is represented in Figure 1f.
, are portrayed in Table S1, which illustrates that out of the two 17phen molecules, only one of it is protonated. The pertinent crystallographic
details are listed in Table 1. The contents establish recognition with each of the CA molecule connected to two 17phen molecules
via O–H···N and N+–H···O– (heteromeric hydrogen bonds), with
the corresponding distances H···N, 1.62 Å (N···O,
2.60 Å) and H···O– 1.55 Å
(N+···O–, 2.54 Å).
In addition, CA molecules form homomeric hydrogen bonds through a centrosymmetric cyclic pattern of O–H···O– with an H···O– distance
of 1.73 Å (O···O–, 2.68 Å).
Also, a noteworthy feature to be noted is the potential hydrogen bond
donor −OH that does not participate in the recognition process.
The recognition pattern is represented in Figure 1f.
Furthermore, in the crystal lattice, the ensembles self-assembled into a ladder-like structure, as shown in Figure 6b, with CA molecules being rails, while the N-donor as rungs. The interaction between CA molecules along the rails is shown in Figure 6c, wherein two cyclic hydrogen bonding patterns could be visualized, exclusively formed by O–H···O– and C–H···O hydrogen bonds in the graphical notation forms R22(14) and R2(8), respectively, with the corresponding distances of H···O– 1.73 Å (O···O–, 2.68 Å) and H···O, 2.54 Å (C···O, 3.59 Å), respectively. In addition to interacting with CA molecules in rails, the 17phen molecules establish homomeric interactions through C–H···N hydrogen bonds, with the H···N distance being 2.67 Å (C···N, 3.68 Å), in such aggregations.
Figure 6.

(a) 3D sheet arrangement of molecules present in the crystal lattice 5. (b) Interaction of CA rails in ladderane topology. (c) Ladderane arrangement of molecules, CA as rails, and 17phen as rungs.
Interwoven and Sheet Arrangements in Supramolecular Assemblies, 6 and 6a, of CA and dabco
The heterocyclic compounds employed in the structures discussed above, 1–5 and 2a, indeed, are aromatic in character. However, several non-aromatic equivalents, for example, dabco (a bicyclic rigid N-donor) employed thoroughly in numerous molecular recognition studies, are also well known.70 Thus, co-crystallization of CA with dabco, in fact, shows distinct features in both the crystallization process and structural aspects (formation of stacked layer structures). CA and dabco form two different crystal types depending on the solvent of crystallization. From the CH3OH solution of 1:1 co-former, co-crystals are also realized in a 1:1 ratio (see ORTEP in Table S1). However, similar co-crystals, but as a hydrate, are observed from a solution of CH3OH and water mixture of the same co-formers in a 1:1 ratio, as depicted in ORTEP (Table S1). The anhydrous and hydrate forms are labeled 6 and 6a, respectively, for the ease of discussion. The crystallographic parameters are enumerated in Table 1, while the molecular recognition features and packing of molecules in the lattices are shown in Figures 1g,h and 7, respectively.
Figure 7.

(a) Cyclic network arrangement observed in crystal lattice 6. (b) Highlighting of a two-fold interpenetrated network and (c) corresponding 3D arrangement of 6. (d) Two-dimensional crinkle ribbon in 6a. (e) Three-dimensional sheet arrangement in 6a.
In both the forms, having similar recognition patterns as in 4 and 5, a −COOH group of CA forms heteromeric hydrogen bonds with the co-former dabco, as shown in Figure 1g,h, through the formation O–H···N/N+–H···O hydrogen bonds (1.66 Å (O···N, 2.64 Å) and 1.71 Å (N+···O–, 2.70 Å) 6; 1.59 Å (O···N, 2.56 Å) and 1.88 Å (N+···O–, 2.72 Å) 6a), while the other −COOH group of CA molecules is involved in homomeric hydrogen bonds (H···O–, 1.58 Å; O···O–, 2.54 Å 6 and 1.95 Å, O···O–, 2.56 Å 6a).
Such recognition patterns ultimately yield distinctly distinguishable structures as represented in Figure 7. In the anhydrous form, 6, the CA and dabco molecules form 10-membered cyclic networks consisting of 8 CA and 2 dabco molecules which are further connected by non-centrosymmetric R22(10) hydrogen bonding patterns (see Figure 7a). These networks are arranged in three dimensions in the form of an interpenetrated sheet structure (see Figure 7b,c). However, in 6a, the packing of the molecules is in the form of regular stacked sheets (Figure 7d), with the crinkled ribbons within each layer being held together by H2O molecules (Figure 7e).
Cyclic Network in a Two-dimensional Arrangement in the Crystals of 7 (CA and bpydo)
Upon co-crystallization of bpydo, an N-oxide, with CA in a 1:1 ratio from CH3OH
solution, good-quality crystals were obtained through slow evaporation.
Analysis by the X-ray diffraction method corroborates the formation
of co-crystals in a triclinic space group, 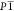 , with a 1:1 ratio of co-formers in the
asymmetric (Table S1). The requisite crystallographic
details are given in Table 1. The molecules in the crystal lattice are bound to each other
through the recognition patterns formed by different types of hydrogen
bonds (heteromeric and homomeric), as illustrated in Figure 1i, which shows two O–H···O (H···O,
1.61 and 1.67 Å; O···O, 2.58 and 2.61 Å)
hydrogen bonds formed by two bpydo with CA molecules as well as a centrosymmetric R22(8) O–H···O
hydrogen bonding pattern with a H···O distance of 1.64
Å (O···O, 2.60 Å).
, with a 1:1 ratio of co-formers in the
asymmetric (Table S1). The requisite crystallographic
details are given in Table 1. The molecules in the crystal lattice are bound to each other
through the recognition patterns formed by different types of hydrogen
bonds (heteromeric and homomeric), as illustrated in Figure 1i, which shows two O–H···O (H···O,
1.61 and 1.67 Å; O···O, 2.58 and 2.61 Å)
hydrogen bonds formed by two bpydo with CA molecules as well as a centrosymmetric R22(8) O–H···O
hydrogen bonding pattern with a H···O distance of 1.64
Å (O···O, 2.60 Å).
The recognition patterns, thus observed, self-assemble in the crystal lattice to yield a cyclic network composed of four CA and two bpydo molecules. The feature is portrayed in Figure 8a. Within each cyclic network, the dimers of CA molecules that are held together by R22(8) hydrogen bonding patterns are further connected to bpydo molecules through pairwise O–H···O and C–H···O (H···O, 2.43 Å; C···O, 3.20 Å) hydrogen bonds (Table 2).
Figure 8.

(a) Basic six-membered cyclic recognition pattern between the co-formers in the crystal lattice 7. (b) Three-dimensional packing observed in the co-crystal.
Furthermore, the cyclic networks are glued together and extended to an infinite arrangement. In 3D, those cyclic networks are further stabilized by stacking such that the void space within each sheet is being filled by the molecules from the adjacent layers by C–H···O (2.30–2.61 Å; C···O, 3.37–3.50 Å) hydrogen bonds by utilizing the −OH group of CA. Such an arrangement is shown in Figure 8b.
Phase Purity and Homogeneity Analysis
The structural information discussed above is based on the X-ray diffraction data obtained from a single crystal of the samples, which provides valuable insights into the structural properties. In the case of compounds with multiple recognition moieties, such as CA (with a −OH group and three −COOH groups) and N-hetero compounds, there would be a high possibility to have other crystals with different compositions or polymorphic forms that may appear even in the same external morphology. In addition, phase transformations, both at ambient and non-ambient conditions, may also possibly account for some deviations than the final co-crystals observed, especially as noted in various pharmaceutical ensembles,15 and in fact recently reported transformations involving citric acid in its co-crystals with 1,2-bis(4-pyridyl)ethene.59
PXRD and DSC can be used to refute such complexity and establish the homogeneity of the product formation. Thus, the PXRD patterns have been recorded on the ground samples of crystals obtained from solution growth and also by neat grinding the co-formers in a ball-mill grinder. Both the types of patterns are found to be similar and corroborate with the simulated PXRD patterns from the three-dimensional structures, thus confirming the formation of sole products as shown in Section S2 in the Supporting Information, except in 2 and 2a, as well as 6 and 6a. For the former, the patterns of the neat ground and even solvent drop ground samples did not match with the simulated patterns (Figure S1b), and for the latter, the neat ground mixture of CA and dabco shows a combination of patterns from both 6 and 6a (Figure S1f).
In addition, thermal analysis by DSC, as shown in Figure 9, gives only melting endotherms, thereby confirming the stability of all the co-crystals/salts, at non-ambient conditions (high temperature), without undergoing any phase transformations. From the analysis, it is further noted that in majority of the structures, the observed melting points are between the melting points of their respective co-formers, as shown in Table 3, which are in consistent with most of the co-crystals reported in the literature. However, co-crystals 1, 4, and 6 melt at high melting points compared to their respective co-formers. The observed variations may be due to their robust packing arrangements and strong heteromeric interactions, as compared to their respective homomeric interactions in pure form. The robust packing arrangements, such as the channel formation in 1 and the two-folded interpenetrated network observed in 6, may be accounted for the anomalous high melting point, whereas the disordered structure in 4 may be responsible for its high melting point.
Figure 9.

DSC thermograms for all the co-crystals/salts, 1–7.
Table 3. Melting Point of the Co-crystals and Salts 1–7.
| co-former melting point (°C) |
|||
|---|---|---|---|
| co-crystal/salt | CA | co-former | melting point (°C) |
| 1 | 152 | 110 | 178 |
| 2 | 152 | 177 | 155 |
| 3 | 152 | 117 | 139 |
| 4 | 152 | 172 | 194 |
| 5 | 152 | 79 | 133 |
| 6 | 152 | 161 | 180 |
| 7 | 152 | 220 | 199 |
Furthermore, as it is a well-established fact that salts melt at higher temperatures, the observed high melting in structures 1, 4, and 6 is self-explanatory as these are salts over the assemblies of 2 and 7 which are co-crystals. However, despite being salts, the structures 3 and 5 exhibit lower melting points than their corresponding co-formers, which may be accounted for the recognition patterns and structural packing. For example, in the structures of 3 and 5, though they have two hydrogen bond N-donor sites, only one of this actively involved in the recognition patterns; in contrast, such binary sites are fully involved in hydrogen bonding in structures 1, 4, and 6. In addition, acr with a mono N-donor atom melts at higher temperature as the −N atom herein is involved in bifurcating hydrogen bonding, which is also seen in the case of 47phen. Such observations are already formalized with the fact that as there are more number of strong interactions between the co-formers, the co-crystals/salts will melt at higher temperature than their individual co-formers.
The contributions of the hydrogen bonds and variations observed between the two categories of structures (1, 4, and 6 vs 2, 3, 5, and 7) are well reflected with the Hirshfeld analysis carried out, considering the N-donor as the reference molecule, as described in the following sections.
Moreover, the solvated crystals 2a and 6a were analyzed by thermogravimetric analysis (TGA) to evaluate the percentage of contribution of solvent molecules to stabilize the observed crystal system. In all the cases, the calculated and experimental weight losses were found to be well in agreement. The TGA curves are illustrated in Figure S2a (2a) and S2b (6a). In 2a, the observed weight loss of 8.1% (calcd 9.2%), in the temperature range of 70–120 °C, possibly may be accounted to a disordered CH3NO2 molecule. Similarly, the weight loss of 4.5% (calcd 5.9%) in 6a corresponds to a H2O molecule in the temperature range of 50–100 °C.
Conformational Analysis of CA in Its Native Structure and Supramolecular Assemblies, 1–7, 2a, and 6a
Conformation flexibility is generally considered as the propensity of organic molecules, which is well studied in all states of matter. Polymorphism, the well-studied and current highly populated research area in the domain of organic crystals, indeed, is due to such conformational flexibility of the molecules. The specific conformation of a molecule in the solid state (crystals) is the geometry with the lowest energy, which is influenced by the interactions established with the neighboring molecules. Thus, in co-crystals having co-formers of different nature, the potential for conformational variations would be at high stake. In the current study, the common co-former CA is a monohydroxy tricarboxylic acid with flexible C–C bonds, which could yield different conformations in the vicinity of different rigid N-donor and N-oxide compounds.
A collective conformational analysis of all the co-crystals reported herein, along with the conformation of CA in its native form, discloses that CA adopts three distinct conformations in these assemblages, taking into account the position of three −COOH groups by aligning along the −OH group. An overlay drawing of the conformations of CA is shown in Figure 10.
Figure 10.

(a) Overlay diagram of different conformations of CA in the crystal structure of co-crystals along with the native structure. Color codes: red, native CA; blue 1; orange, 2; light blue, 2a; magenta 3; brown, 4; green, 5; gray, 6; light pink, 6a; and black, 7. (b) Three different conformations of the CA molecule.
The geometry of native CA resembles “T” shape (see type I in Figure 10), with the −COOH groups at all terminal positions showing a small degree of torsion around the C–C bonds and also having a notable twist around the −COOH group. The calculated torsion angles and the schematic diagram of CA are shown in Table 4. It is apparent from Figure 10 that CA molecules in the co-crystals, 2 (orange), 2a (light blue), and 7 (black), are found to be aligned with the conformation of the native (red) CA, albeit with a modest variation of the torsion angles (see Table 4). However, in the remaining co-crystals, 5 (green) forms a standalone conformation in the syn–anti form (type II) with respect to the −OH group with the two −COOH groups at linear ends of T are twisted into opposite directions, while 1 (blue), 3 (magenta), 4 (brown), 6 (gray), and 6a (light pink) adopt syn conformation (type III), with respect to the −OH group as only one −COOH group along the linear direction of T is twisted. The distinctions in the recognition patterns of the co-formers, which are compiled in Table S1, may account for these different conformations. Thus, neutral co-crystals (2, 2a, and 7) represent type I, while all the structures with deprotonated CA represent type III or type II.
Table 4. Torsion Angles of CA in Its Native Structure and in the Co-crystals/Salts, Around Different C–C Bonds.

| compound | conformation | (τ1) | (τ2) | (τ3) | (τ4) |
|---|---|---|---|---|---|
| native CA | I | 8.25 | 50.44 | 5.31 | 171.12 |
| 1 | III | 121.01 | 25.44 | 115.89 | 179.07 |
| 2 | I | 24.14 | 161.00 | 178.98 | 179.53 |
| 2a | I | 49.62 | 41.13 | 177.27 | 175.33 |
| 3 | III | 91.22 | 30.99 | 121.82 | 6.23 |
| 4 | III | 47.35 | 161.64 | 107.13 | 171.55 |
| 5 | II | 160.12 | 79.78 | 115.41 | 121.58 |
| 6 | III | 90.87 | 3.58 | 117.68 | 11.47 |
| 6a | III | 68.17 | 55.34 | 103.59 | 3.92 |
| 7 | I | 57.16 | 129.15 | 4.66 | 176.58 |
Furthermore, to assess the population of the observed conformations in a large data set, a search performed on the Cambridge Structural Database (CSD), version, 5.43,71 retrieves 93 co-crystals of CA. Conformational analysis of CA molecules in all these structures reveals type I conformation as predominant with 52 structures expressing it, while type II is least preferred with only five structures displaying it and with type III found in 27 structures, thus closely mapping with the observations in this study as type II is reflected only in one structure among either structure.
Hirshfeld Surface Analysis
All the co-crystals reported in this study show abundant interactions including both neutral (co-crystals) and ionic (salts) hydrogen bonds, along with π–π interactions and van der Waals forces. Furthermore, as described in the previous sections, since some structures of salts have unusual higher melting points than the corresponding co-formers, which may be attributed to the strength of the intermolecular interactions (herein, hydrogen bonds), and CA was noted to be in three distinct conformations influenced by these interactions. Hence, an exercise is carried out to further evaluate the contributions of all types of intermolecular interactions in each structure of the co-crystals using Crystal Explorer21.
The Hirshfeld surfaces are generated around each co-former separately in all the structures; this process produces the dnorm surfaces, which provides the normalized distance between a point on the surface and the nearest nucleus inside (di contacts) and outside (de contacts) the surface. In addition, corresponding 2D fingerprint plots are also developed, which clearly exhibit the quantitative weightage as well as visualization of all the interactions present in the structures. In the dnorm surface, the strength of intermolecular interactions is represented by different color codes: strong interactions (or short contacts) in deep bright red on the surface (representing donor and acceptor atoms), while white and blue regions on the surface expressing the moderate to very weak interactions (longer contacts), respectively. In the following sections, the details of the dnorm surface and the corresponding 2D fingerprint plots of H···O/O···H for each of the co-crystals/salts (1–7, 2a, and 6a) are shown Table 5, while the remaining 2D finger plots are shown in Section S7.
Table 5. 3D dnorm Surfaces for CA and N-Donor or N-Oxide, along with 2D Fingerprint Plots for Both Considering Only O···H/H···O Interactions for 1–7, 2a, and 6a.

Further analysis reveals that in all the structures, the O···H/H···O interactions are the main contributors to the total Hirshfeld area, 55–63% (Figure 11). These interactions arise mainly from O–H···O, N+–H···O–, N+–H···O, and C–H···O hydrogen bonds, which appear as bright-red spots for the respective donor and acceptor atoms on the Hirshfeld surface. In the 2D fingerprint plots, the prominent donor and acceptor regions appear as long spikes at (de + di ∼ 1.7 Å) in the upper left and lower right, respectively. The second highest contribution to the total Hirshfeld area (22–34%) corresponds to H···H contacts, and these appear as diffused at the middle portion (de = di ∼ 1.2 Å) of the fingerprint plots (Table S5). Thus, major structural stabilization (∼90%) in all structures may be accounted for strong O···H/H···O, compared to dispersive H···H, as shown in Table S6.
Figure 11.

Relative contribution of different intermolecular interactions in crystal lattices taking CA as a dominant surface of 1–7, 2a, and 6a.
From this analysis, a noteworthy observation is that herein the crystal structures are stabilized by strong O···H/H···O interactions as these contribute the highest percentage to the surface compared to the dispersive H···H contacts, which are generally dominant interactions, in majority of organic crystal structures.
In fact, the significance of effective contribution of O···H/H···O interactions further reflects clearly in the 2D fingerprint plots of the co-formers (heterocyclic), as shown in Table 6, which explains the high melting point of 1, 4, and 6 than the corresponding co-formers. Because in these structures, the O···H/H···O contribution (29, 32, and 43%, respectively, highlighted in red) is higher compared to other structures. This is further clearly visible in the dnorm plots also, as presented in Table 5, with appropriate intense red spots and more in number.
Table 6. Contribution of Different Intermolecular Interactions on the Hirshfeld Surface on N-Donor and N-Oxide Co-formers.

Conclusions
Supramolecular assemblies of citric acid, CA, with various N-donor and N-oxide compounds are prepared and characterized by X-ray diffraction techniques and DSC thermal analysis. These structural analysis demonstrates the ability of CA to form diverse structural topologies such as channel structures which mimic the HOF structures, ladders, a sandwich, lamellar layers, and interwoven networks. Also, a significant feature realized from the analysis is that in most of the structures, despite the −OH group being a strong hydrogen bond donor functionality, its role is noted to be subtle, limiting itself to mere crystal lattice stabilization rather directly playing a pivotal role in the molecular recognition between the co-formers, except in the case of 1 and 4. Further, conformational analysis reveals the formation of three distinct conformations of CA (type I, type II, and type III), as observed in other co-crystals of CA retrieved from CSD. In addition, the thermal analysis suggests that the obtained co-crystals/salts are thermodynamically stable with only a single endothermic peak being observed, in all the thermograms, which may be accounted for their stability without enduring any phase transformations. In addition, the Hirshfeld surface area analysis confirms that all the structures are stabilized by the strong O···H/H···O interactions rather than dispersive H···H contacts, which is not very commonly observed in the crystal structures. A noteworthy feature that may be deduced (not very commonly observed) further is that, in the assemblies 1, 4, and 6, such O···H/H···O interactions’ contribution is much higher than in other structures, leading to an unusual meting behavior with higher melting temperature than the corresponding co-formers.
Acknowledgments
We acknowledge IIT Bhubaneswar for infrastructure and DST-SERB, New Delhi, for the financial support.
Glossary
Abbreviations
- CA
citric acid
- acr
acridine
- phenz
phenazine
- 110phen
1,10-phenanthroline
- 47phen
4,7-phenanthroline
- 17phen
1,7-phenanthroline
- dabco
1,4-diazabicyclo[2.2.2]octane
- bpydo
4,4′-bipyridyl N,N′-dioxide
- DSC
differential scanning calorimetry
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c03446.
ORTEP plots, powder X-ray diffraction patterns, TGA thermograms, CSD analysis, details of interactions of CA with different heterocyclic compounds, and Hirshfeld surface analysis (PDF)
Crystallographic data for CA with 17phen, 47phen, acridine, bpydo, dabco, dabco hydrate, phenazine, phenazine solvate, and 110phen (ZIP)
The authors declare no competing financial interest.
Footnotes
In the metabolism of most organisms such as being a natural intermediate in Krebs cycle and regulating the size of apatite crystals in bones through citrate, as well as multiple functions as an environmentally benign cleaning agent, antioxidant capabilities, and FDA (Food and Drug Administration)-approved GRAS substance.
Supplementary Material
References
- Bond A. D. What is a co-crystal?. CrystEngComm 2007, 9, 833–834. 10.1039/b708112j. [DOI] [Google Scholar]
- Zukerman-Schpector J.; Tiekink E. R. What is a co-crystal?. Z. Kristallogr.—Cryst. Mater. 2008, 223, 233–234. 10.1524/zkri.2008.0021. [DOI] [Google Scholar]
- Friščić T.; Fabian L.; Burley J. C.; Reid D. G.; Duer M. J.; Jones W. Exploring the relationship between cocrystal stability and symmetry: is Wallach’s rule applicable to multi-component solids?. Chem. Commun. 2008, 1644–1646. 10.1039/b717532a. [DOI] [PubMed] [Google Scholar]
- Braga D.; Maini L.; Grepioni F. Mechanochemical preparation of co-crystals. Chem. Soc. Rev. 2013, 42, 7638–7648. 10.1039/c3cs60014a. [DOI] [PubMed] [Google Scholar]
- Bond A. D.Fundamental aspects of salts and co-crystals. Pharmaceutical Salts and Co-crystals; The Royal Society of Chemistry: Cambridge, 2011; pp 9–28. [Google Scholar]
- Friščić T.; Jones W. Recent Advances in Understanding the Mechanism of Cocrystal Formation via Grinding. Cryst. Growth Des. 2009, 9, 1621–1637. 10.1021/cg800764n. [DOI] [Google Scholar]
- Friščić T.; Fábián L.; Burley J. C.; Jones W.; Motherwell W. D. S. Exploring cocrystal–cocrystal reactivity via liquid-assisted grinding: the assembling of racemic and dismantling of enantiomeric cocrystals. Chem. Commun. 2006, 5009–5011. 10.1039/b613073a. [DOI] [PubMed] [Google Scholar]
- Park S. K.; Varghese S.; Kim J. H.; Yoon S.-J.; Kwon O. K.; An B.-K.; Gierschner J.; Park S. Y. Tailor-Made Highly Luminescent and Ambipolar Transporting Organic Mixed Stacked Charge-Transfer Crystals: An Isometric Donor–Acceptor Approach. J. Am. Chem. Soc. 2013, 135, 4757–4764. 10.1021/ja312197b. [DOI] [PubMed] [Google Scholar]
- McNeil S. K.; Kelley S. P.; Beg C.; Cook H.; Rogers R. D.; Nikles D. E. Cocrystals of 10-Methylphenthiazine and 1,3-Dinitrobenzene: Implications for the Optical Sensing of TNT-Based Explosives. ACS Appl. Mater. Interfaces 2013, 5, 7647–7653. 10.1021/am401961s. [DOI] [PubMed] [Google Scholar]
- Wang F.; Du G.; Liu X.; Shao M.; Zhang C.; Chen L. Molecular dynamics application of cocrystal energetic materials: A review. Nanotechnol. Rev. 2022, 11, 2141–2153. 10.1515/ntrev-2022-0124. [DOI] [Google Scholar]
- Yan D.; Evans D. G. Molecular crystalline materials with tunable luminescent properties: from polymorphs to multi-component solids. Mater. Horiz. 2014, 1, 46–57. 10.1039/c3mh00023k. [DOI] [Google Scholar]
- Vishweshwar P.; McMahon J. A.; Bis J. A.; Zaworotko M. J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. 10.1002/jps.20578. [DOI] [PubMed] [Google Scholar]
- Karimi-Jafari M.; Padrela L.; Walker G. M.; Croker D. M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. 10.1021/acs.cgd.8b00933. [DOI] [Google Scholar]
- Shan N.; Zaworotko M. J. The role of cocrystals in pharmaceutical science. Drug Discov. Today 2008, 13, 440–446. 10.1016/j.drudis.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Bolla G.; Sarma B.; Nangia A. K. Crystal engineering of pharmaceutical cocrystals in the discovery and development of improved drugs. Chem. Rev. 2022, 122, 11514–11603. 10.1021/acs.chemrev.1c00987. [DOI] [PubMed] [Google Scholar]
- Aitipamula S.; Antonijevic I.; Baruah J. B.; Berryman O. B.; Boldyreva E. V.; Decato D. A.; Fourmigue M.; Groeneman R. H.; Gryl M.; Hutchins K. M.. Multi-Component Crystals: Synthesis, Concepts, Function; Walter de Gruyter GmbH & Co KG, 2017. [Google Scholar]
- Delori A.; Jones W. A hydrogen bonded cocrystal with an unusual interweaving between the adjacent triple-helices. CrystEngComm 2011, 13, 6315–6318. 10.1039/c1ce05534h. [DOI] [Google Scholar]
- Marivel S.; Suresh E.; Pedireddi V. Molecules to supermolecules and self assembly: a study of some cocrystals of cyanuric acid. Tetrahedron Lett. 2008, 49, 3666–3671. 10.1016/j.tetlet.2008.03.143. [DOI] [Google Scholar]
- Adachi T.; Ward M. D. Versatile and resilient hydrogen-bonded host frameworks. Acc. Chem. Res. 2016, 49, 2669–2679. 10.1021/acs.accounts.6b00360. [DOI] [PubMed] [Google Scholar]
- Sharma C. V. K.; Zaworotko M. J. X-Ray crystal structure of C6H3(CO2H)3-1, 3, 5· 1.5 (4, 4′-bipy): a ‘super trimesic acid’chicken-wire grid. Chem. Commun. 1996, 2655–2656. 10.1039/cc9960002655. [DOI] [Google Scholar]
- Bhogala B. R.; Nangia A. Cocrystals of 1, 3, 5-Cyclohexanetricarboxylic Acid with 4, 4 ‘-Bipyridine Homologues: Acid Pyridine Hydrogen Bonding in Neutral and Ionic Complexes. Cryst. Growth Des. 2003, 3, 547–554. 10.1021/cg034047i. [DOI] [Google Scholar]
- Shattock T. R.; Vishweshwar P.; Wang Z.; Zaworotko M. J. 18-Fold interpenetration and concomitant polymorphism in the 2: 3 Co-crystal of trimesic acid and 1, 2-bis (4-pyridyl) ethane. Cryst. Growth Des. 2005, 5, 2046–2049. 10.1021/cg0501985. [DOI] [Google Scholar]
- Almeida Paz F. A.; Klinowski J. Supramolecular architecture of a novel salt of trimesic acid and 1, 2-bis (4-pyridyl) ethane. CrystEngComm 2003, 5, 238–244. 10.1039/b302949b. [DOI] [Google Scholar]
- Griessl S.; Lackinger M.; Edelwirth M.; Hietschold M.; Heckl W. M. Self-assembled two-dimensional Molecular host-guest architectures from trimesic acid. Single Mol. 2002, 3, 25–31. . [DOI] [Google Scholar]
- Arora K. K.; Pedireddi V. R. A Rational Study of Crystal Engineering of Supramolecular Assemblies of 1,2,4,5-Benzenetetracarboxylic Acid. J. Org. Chem. 2003, 68, 9177–9185. 10.1021/jo034434z. [DOI] [PubMed] [Google Scholar]
- Ravat P.; SeethaLekshmi S.; Biswas S. N.; Nandy P.; Varughese S. Equivalence of ethylene and azo-bridges in the modular design of molecular complexes: role of weak interactions. Cryst. Growth Des. 2015, 15, 2389–2401. 10.1021/acs.cgd.5b00183. [DOI] [Google Scholar]
- Du M.; Zhang Z.-H.; Zhao X.-J. Cocrystallization of trimesic acid and pyromellitic acid with bent dipyridines. Cryst. Growth Des. 2005, 5, 1247–1254. 10.1021/cg0495680. [DOI] [Google Scholar]
- Shan N.; Bond A. D.; Jones W. Supramolecular architectures of cyclohexane-1, 3cis, 5cis-tricarboxylic acid in acid–base complexes. New J. Chem. 2003, 27, 365–371. 10.1039/b208686g. [DOI] [Google Scholar]
- Bhogala B. R.; Nangia A. Ternary and quaternary co-crystals of 1, 3-cis, 5-cis-cyclohexanetricarboxylic acid and 4, 4′-bipyridines. New J. Chem. 2008, 32, 800–807. 10.1039/b800293b. [DOI] [Google Scholar]
- Giri L.; Pedireddi V. Exploration of supramolecular assemblies of rac-1, 3-cyclohexanedicarboxylic acid. J. Mol. Struct. 2015, 1100, 455–463. 10.1016/j.molstruc.2015.07.064. [DOI] [Google Scholar]
- Zheng L. L.; Zhou A. J.; Hu S. Co-crystallization of a Versatile Building Block Benzoguanamine with Two Flexible Cyclohexanedicarboxylic Acids. J. Chem. Crystallogr. 2018, 48, 117–124. 10.1007/s10870-018-0718-2. [DOI] [Google Scholar]
- Bhogala B. R.; Basavoju S.; Nangia A. Tape and layer structures in cocrystals of some di-and tricarboxylic acids with 4, 4′-bipyridines and isonicotinamide. From binary to ternary cocrystals. CrystEngComm 2005, 7, 551–562. 10.1039/b509162d. [DOI] [Google Scholar]
- Raut M. D.; Giri L.; Pedireddi V. R. Supramolecular Assemblies of cis, cis, cis-1, 2, 4, 5-Cyclohexanetetracarboxylic Acid with Various Aza-Donors. ChemistrySelect 2018, 3, 3194–3202. 10.1002/slct.201702990. [DOI] [Google Scholar]
- Ranganathan A.; Pedireddi R.; Chatterjee S.; Rao C. N. R.; Rao N. R. C. An organic channel structure formed by the supramolecular assembly of trithiocyanuric acid and 4,4′-bipyridyl. J. Mater. Chem. 1999, 9, 2407–2411. 10.1039/a905344a. [DOI] [Google Scholar]
- Song X.; Wang Y.; Wang C.; Wang D.; Zhuang G.; Kirlikovali K. O.; Li P.; Farha O. K. Design Rules of Hydrogen-Bonded Organic Frameworks with High Chemical and Thermal Stabilities. J. Am. Chem. Soc. 2022, 144, 10663–10687. 10.1021/jacs.2c02598. [DOI] [PubMed] [Google Scholar]
- Giri L.; Mohanty B.; Thapa R.; Jena B. K.; Pedireddi V. R. Hydrogen-Bonded Organic Framework Structure: A Metal-Free Electrocatalyst for the Evolution of Hydrogen. ACS Omega 2022, 7, 22440–22446. 10.1021/acsomega.2c01585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner C. A.; Lai H. W. H.; Greenfield J. T.; Wiscons R. A.; Zeller M.; Campana C. F.; Talu O.; FitzGerald S. A.; Rowsell J. L. C. High surface area and Z′ in a thermally stable 8-fold polycatenated hydrogen-bonded framework. Chem. Commun. 2015, 51, 11642–11645. 10.1039/c5cc04219d. [DOI] [PubMed] [Google Scholar]
- Yoon T.-U.; Baek S. B.; Kim D.; Kim E.-J.; Lee W.-G.; Singh B. K.; Lah M. S.; Bae Y.-S.; Kim K. S. Efficient separation of C2 hydrocarbons in a permanently porous hydrogen-bonded organic framework. Chem. Commun. 2018, 54, 9360–9363. 10.1039/c8cc04139c. [DOI] [PubMed] [Google Scholar]
- Devarapalli R.; Indukuri A.; Bollineni M.; Mondal A.; Reddy C. M.; Chennuru R. Investigation of poor solubility of a salt-cocrystal hydrate: A case study of the common-ion effect in Betrixaban, an anticoagulant drug. Mol. Pharm. 2021, 18, 1138–1149. 10.1021/acs.molpharmaceut.0c01045. [DOI] [PubMed] [Google Scholar]
- Kansız S.; Azam M.; Dege N.; Ermiş N.; Al-Resayes S. I.; Alam M. Supramolecular assembly in designing co-crystals of fumaric acid and pyrimidine/picolinate derivatives. Green Chem. Lett. Rev. 2022, 15, 825–836. 10.1080/17518253.2022.2130016. [DOI] [Google Scholar]
- Rahim S. A.; Hammond R. B.; Sheikh A. Y.; Roberts K. J. A comparative assessment of the influence of different crystallization screening methodologies on the solid forms of carbamazepine co-crystals. CrystEngComm 2013, 15, 3862–3873. 10.1039/c3ce26878k. [DOI] [Google Scholar]
- Remenar J. F.; Morissette S. L.; Peterson M. L.; Moulton B.; MacPhee J. M.; Guzmán H. R.; Almarsson Ö. Crystal Engineering of Novel Cocrystals of a Triazole Drug with 1,4-Dicarboxylic Acids. J. Am. Chem. Soc. 2003, 125, 8456–8457. 10.1021/ja035776p. [DOI] [PubMed] [Google Scholar]
- Wang J.-R.; Ye C.; Zhu B.; Zhou C.; Mei X. Pharmaceutical cocrystals of the anti-tuberculosis drug pyrazinamide with dicarboxylic and tricarboxylic acids. CrystEngComm 2015, 17, 747–752. 10.1039/c4ce02044h. [DOI] [Google Scholar]
- Sugden I. J.; Braun D. E.; Bowskill D. H.; Adjiman C. S.; Pantelides C. C. Efficient Screening of Coformers for Active Pharmaceutical Ingredient Cocrystallization. Cryst. Growth Des. 2022, 22, 4513–4527. 10.1021/acs.cgd.2c00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Yang Y.; Xia M.; Dai W.; Zhu B.; Mei X. Improving the dissolution behaviors and bioavailability of abiraterone acetate via multicomponent crystal forms. Int. J. Pharm. 2022, 614, 121460. 10.1016/j.ijpharm.2022.121460. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gao X.; Fang W.; Liu B.; Jin S.; Wang D.; Xu W. Crystal structure of five solid forms from isonicotinamide and carboxylic acids assembled by classical hydrogen bonds and other noncovalent interactions. J. Mol. Struct. 2021, 1233, 130048. 10.1016/j.molstruc.2021.130048. [DOI] [Google Scholar]
- Stevens J. S.; Newton L. K.; Jaye C.; Muryn C. A.; Fischer D. A.; Schroeder S. L. Proton transfer, hydrogen bonding, and disorder: Nitrogen near-edge X-ray absorption fine structure and X-ray photoelectron spectroscopy of bipyridine–acid salts and co-crystals. Cryst. Growth Des. 2015, 15, 1776–1783. 10.1021/cg5018278. [DOI] [Google Scholar]
- Shan N.; Bond A. D.; Jones W. Crystal engineering using 4,4′-bipyridyl with di- and tricarboxylic acids. Cryst. Eng. 2002, 5, 9–24. 10.1016/s1463-0184(02)00002-3. [DOI] [Google Scholar]
- Li X.; Zhang S.; Li J.; Qian Y.; Duan W.; Zeng Q. Advances in the regulation of bipyridine derivatives on two-dimensional (2D) supramolecular nanostructures. New J. Chem. 2019, 43, 13315–13325. 10.1039/c9nj02027f. [DOI] [Google Scholar]
- Golovnev N. N.; Vasiliev A. D.; Kirik S. D. Enrofloxacinium citrate monohydrate: Preparation, crystal structure, thermal stability and IR-characterization. J. Mol. Struct. 2012, 1021, 112–117. 10.1016/j.molstruc.2012.04.059. [DOI] [Google Scholar]
- Stainton P.; Grecu T.; McCabe J.; Munshi T.; Nauha E.; Scowen I. J.; Blagden N. First Comparative Study of the Three Polymorphs of Bis(isonicotinamide) Citric Acid Cocrystals and the Concomitant Salt 4-Carbamoylpyridinium Citrate Isonicotinamide. Cryst. Growth Des. 2018, 18, 4150–4159. 10.1021/acs.cgd.8b00597. [DOI] [Google Scholar]
- Djaló M.; Cunha A. E. S.; Luís J. P.; Quaresma S.; Fernandes A.; André V.; Duarte M. T. Sparfloxacin Multicomponent Crystals: Targeting the Solubility of Problematic Antibiotics. Cryst. Growth Des. 2021, 21, 995–1005. 10.1021/acs.cgd.0c01346. [DOI] [Google Scholar]
- Fang Z.-Y.; Zhang B.-X.; Xing W.-H.; Jia H.-L.; Wang X.; Gong N.-B.; Lu Y.; Du G.-H. A series of stable, metastable and unstable salts of Imatinib with improved solubility. Chin. Chem. Lett. 2022, 33, 2159–2164. 10.1016/j.cclet.2021.10.056. [DOI] [Google Scholar]
- Barbas R.; Prohens R.; Bauzá A.; Franconetti A.; Frontera A. H-Bonded anion–anion complexes in fentanyl citrate polymorphs and solvates. Chem. Commun. 2019, 55, 115–118. 10.1039/c8cc09028a. [DOI] [PubMed] [Google Scholar]
- Elbagerma M. A.; Edwards H. G. M.; Munshi T.; Scowen I. J. Identification of a new cocrystal of citric acid and paracetamol of pharmaceutical relevance. CrystEngComm 2011, 13, 1877–1884. 10.1039/c0ce00461h. [DOI] [Google Scholar]
- Soleimannejad J.; Aghabozorg H.; Najafi S.; Nasibipour M.; Attar Gharamaleki J. 4, 4′-Bipyridine–2-hydroxypropane-1, 2, 3-tricarboxylic acid (3/2). Acta Crystallogr., Sect. E: Struct. Rep. Online 2009, 65, o532–o533. 10.1107/s1600536809004607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.-L. Piperazine-1,4-diium 2-(carboxymethyl)-2-hydroxybutanedioate monohydrate. Acta Crystallogr., Sect. E: Struct. Rep. Online 2010, 66, o2191. 10.1107/s1600536810030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M.; Zhang Z.-H.; Zhao X.-J. A Search for Predictable Hydrogen-Bonding Synthons in Cocrystallization of Unusual Organic Acids with a Bent Dipyridine. Cryst. Growth Des. 2006, 6, 390–396. 10.1021/cg0503292. [DOI] [Google Scholar]
- Easmin S.; Pedireddi V. R. Unusual In Situ Transformation of Metastable Co-Crystal Forms into a Stable Form, in Solution and Solid State, and the [2 + 2] Photochemical Reactivity in the Crystalline Forms. Cryst. Growth Des. 2023, 23, 2802–2811. 10.1021/acs.cgd.3c00004. [DOI] [Google Scholar]
- Mazzeo P. P.; Canossa S.; Carraro C.; Pelagatti P.; Bacchi A. Systematic coformer contribution to cocrystal stabilization: energy and packing trends. CrystEngComm 2020, 22, 7341–7349. 10.1039/d0ce00291g. [DOI] [Google Scholar]
- Ciriminna R.; Meneguzzo F.; Delisi R.; Pagliaro M. Citric acid: emerging applications of key biotechnology industrial product. Chem. Cent. J. 2017, 11, 22. 10.1186/s13065-017-0251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa K.; Amino Y.; Kohmura M.; Ueda Y.; Kuroda M.. 4.16 - Human–Environment Interactions – Taste. In Comprehensive Natural Products II; Liu H.-W., Mander L., Eds.; Elsevier: Oxford, 2010; pp 631–671. [Google Scholar]
- Lambros M.; Tran T.; Fei Q.; Nicolaou M. Citric acid: A multifunctional pharmaceutical excipient. Pharmaceutics 2022, 14, 972. 10.3390/pharmaceutics14050972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. SHELXT: Integrating space group determination and structure solution. Acta Crystallogr., Sect. A: Found. Crystallogr. 2014, 70, C1437. 10.1107/s2053273314085623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/s0021889802022112. [DOI] [Google Scholar]
- Bergerhoff G.; Berndt M.; Brandenburg K. Evaluation of crystallographic data with the program DIAMOND. J. Res. Natl. Inst. Stand. Technol. 1996, 101, 221. 10.6028/jres.101.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spackman P. R.; Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Jayatilaka D.; Spackman M. A. CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. 10.1107/s1600576721002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Lin R.-B.; Zhang Z.; Xiang S.; Chen B. Hydrogen-Bonded Organic Frameworks as a Tunable Platform for Functional Materials. J. Am. Chem. Soc. 2020, 142, 14399–14416. 10.1021/jacs.0c06473. [DOI] [PubMed] [Google Scholar]
- Aitipamula S.; Banerjee R.; Bansal A. K.; Biradha K.; Cheney M. L.; Choudhury A. R.; Desiraju G. R.; Dikundwar A. G.; Dubey R.; Duggirala N.; Ghogale P. P.; Ghosh S.; Goswami P. K.; Goud N. R.; Jetti R. R. K. R.; Karpinski P.; Kaushik P.; Kumar D.; Kumar V.; Moulton B.; Mukherjee A.; Mukherjee G.; Myerson A. S.; Puri V.; Ramanan A.; Rajamannar T.; Reddy C. M.; Rodriguez-Hornedo N.; Rogers R. D.; Row T. N. G.; Sanphui P.; Shan N.; Shete G.; Singh A.; Sun C. C.; Swift J. A.; Thaimattam R.; Thakur T. S.; Kumar Thaper R.; Thomas S. P.; Tothadi S.; Vangala V. R.; Variankaval N.; Vishweshwar P.; Weyna D. R.; Zaworotko M. J. Polymorphs, Salts, and Cocrystals: What’s in a Name?. Cryst. Growth Des. 2012, 12, 2147–2152. 10.1021/cg3002948. [DOI] [Google Scholar]
- Ye H.-Y.; Ge J.-Z.; Chen F.; Xiong R.-G. Reversible phase transition of the 1 : 1 co-crystal of 1,4-diazabicyclo-[2.2.2]octane (DABCO) and hydroquinone. CrystEngComm 2010, 12, 1705–1708. 10.1039/b923314h. [DOI] [Google Scholar]
- Ferrence G. M.; Tovee C. A.; Holgate S. J. W.; Johnson N. T.; Lightfoot M. P.; Nowakowska-Orzechowska K. L.; Ward S. C. CSD Communications of the Cambridge Structural Database. IUCrJ 2023, 10, 6–15. 10.1107/s2052252522010545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.