

Abstract
Cocaine is one of the most commonly trafficked and abused drugs in the United States, and deployable field tests are important for rapid identification in nonlaboratory settings. At present, colorimetric tests exist for in-field determination, but these fundamentally suffer from interferent effects. Cocaine is an organic salt that is readily water soluble as a cation and almost insoluble in the deprotonated neutral form. Here, we take advantage of the electrochemical window of water to increase the pH at the electrode surface by driving water reduction, effectively electroprecipitating the cocaine base. The precipitate on the electrode surface is then electrochemically oxidized by a voltammetric sweep through sufficiently positive potentials. We demonstrate excellent selectivity to cocaine compared to common adulterants, such as procaine, lidocaine, benzocaine, caffeine, and levamisole. Finally, we detect cocaine on a carbon fiber microelectrode, demonstrating miniaturizability and allowing access to low-resistance media (e.g., tap water).
Graphical Abstract
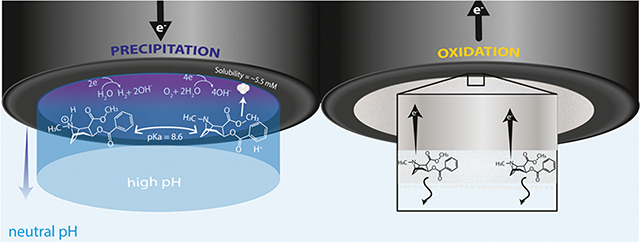
INTRODUCTION
Improving rapid and reliable methods for accurate identifications of illicit drugs in unknown powders is an area of continued interest for analytical chemists.1,2 methods designed for law enforcement officers can inform on the scene for safe practices and streamline judicial processes.3 While there is a thrust to miniaturize and simplify powerful identification techniques so they are suitable for field detection (including handling and interpretation by nonscientists), many of these efforts are still complicated by cost, complex instrumentation, and specificity in mixtures.4–10 The most commonly used rapid field tests for illicit substance identification are colorimetric due to their low cost, simplicity, and rapid readout.2
Cocaine is readily available in the United States and is most often found diluted by other substances. Over the last decade, deaths among Americans involving cocaine abuse have continued to increase each year.11 For detection, suspected cocaine samples are often mixed with cobalt thiocyanate, which interacts to form a blue precipitate. Scott’s test (or a similar version) includes two additional reagent steps to manage interferents: hydrochloric acid and chloroform, where the positive result is indicated by a blue color in the chloroform phase.12 While these reagent steps do increase the specificity,13 carrying chloroform and strong acids to a crime scene presents additional handling and environmental considerations.14–16 addition, there are several steps in Scott’s test that increase the probability of user error: the user must add less than 1 mg of cocaine (this is difficult to judge in the field), 5 drops of cobalt thiocyanate solution, 1 drop of concentrated HCl, and 5 drops of chloroform. Slight deviations from this order, relative ratios of the reagents, or the amount of cocaine samples have been shown to cause false positives and false negatives.13
Electrochemical sensors have been widely used for onsite detections because of their simple electronics, low-cost materials, and easy portability. While electrochemistry also suffers from issues surrounding selectivity, there exist a number of creative solutions to this problem in the literature that allow for cocaine identification in mixtures.17–21 Many of these solutions involve modifying electrodes,22 where modifications include molecularly imprinted polymers,21 aptamers,23 and hydrogels19/films.24 These modifications have the common goal of selective localization of cocaine to the electrode surface. Electrode modifications can be costly, irreproducible, and add a specialization step increasing the activation barrier for introducing and maintaining the procedure.
There are interesting detection modalities that do not require electrode modifications.17 Notably, measurements over an interface between two immiscible electrolyte solutions (ITIESs) can track the charged cocaine molecule moving from one phase to another.25 Other groups simply take advantage of spontaneous adsorption of cocaine to the electrode surface. In 1982, Kalvoda summarized a technique for adsorption accumulation followed by stripping voltammetry detection.26 Most reports on the adsorption accumulation technique for cocaine detection use a mercury electrode and indirectly sense cocaine by cathodic stripping of the spontaneous hydrolysis product, benzoylecgonine.27 The preferential adsorption of benzoylecgonine and electrochemical reduction of the ester group are implicated as the mechanism of detection for such studies. Hanging drop mercury electrodes have a large reduction window useful for passing electrons to benzoylecgonine. In modern day, mercury electrodes have been largely phased out, especially for sensor studies toward deployable applications, and replaced with easier-to-handle, less-toxic electrode options (usually gold, platinum, or carbon). There are a few papers that take this method to more modern electrodes, often employing an empirically determined pH and conditioning voltage to get the best accumulation/signal.28,29 The adsorption process of cocaine hydrochloride (and other drugs) can also be aided by adding surfactant into the test solution to precipitate the molecules onto a screen-printed carbon electrode.30
While there exists a plethora of literature describing the development of electrochemical sensors for cocaine detection,21,23,24,31,32 none are currently employed by the forensic community, as the availability of simple, high-throughput sensors is still lacking.33 Here, we make this technology accessible by demonstrating a cocaine sensor that uses commercial, unmodified electrodes, tap water (which may be available onsite or nearby), and simple techniques available on portable potentiostats.34
By employing a polarization-induced precipitation step followed by a simple voltammetric sweep, we demonstrate that changing the local pH can act as a separation step and allow for the selective detection of low-purity (30–50%) cocaine in complex powder of common adulterants and dilutants. This is demonstrated by accurate detections of cocaine when three pH-altering cutting agents (boric acid, ascorbic acid, and sodium bicarbonate), three local anesthetics with a similar structure (procaine, lidocaine, and benzocaine), and two other drugs (caffeine and levamisole) were mixed with cocaine samples. With this work, we break the status quo in a few ways: (1) using unadjusted water instead of buffer (almost all studies cited up to this point use a buffer around pH 8), (2) forcing the pH well above the pKa to crash the free base out of solution, and (3) changing the pH only locally near the working electrode such that most of the sample is unaffected by the measurement.
While maintaining low cost (~$60 one-time cost), fast analysis times (less than 2 min), and simplicity, we offer a proof-of-concept electrochemical method that is highly selective for cocaine detection. The proposed method is shown in Scheme 1.
Scheme 1. Proposed Pathway for the Electrochemical Precipitation and Strippinga.
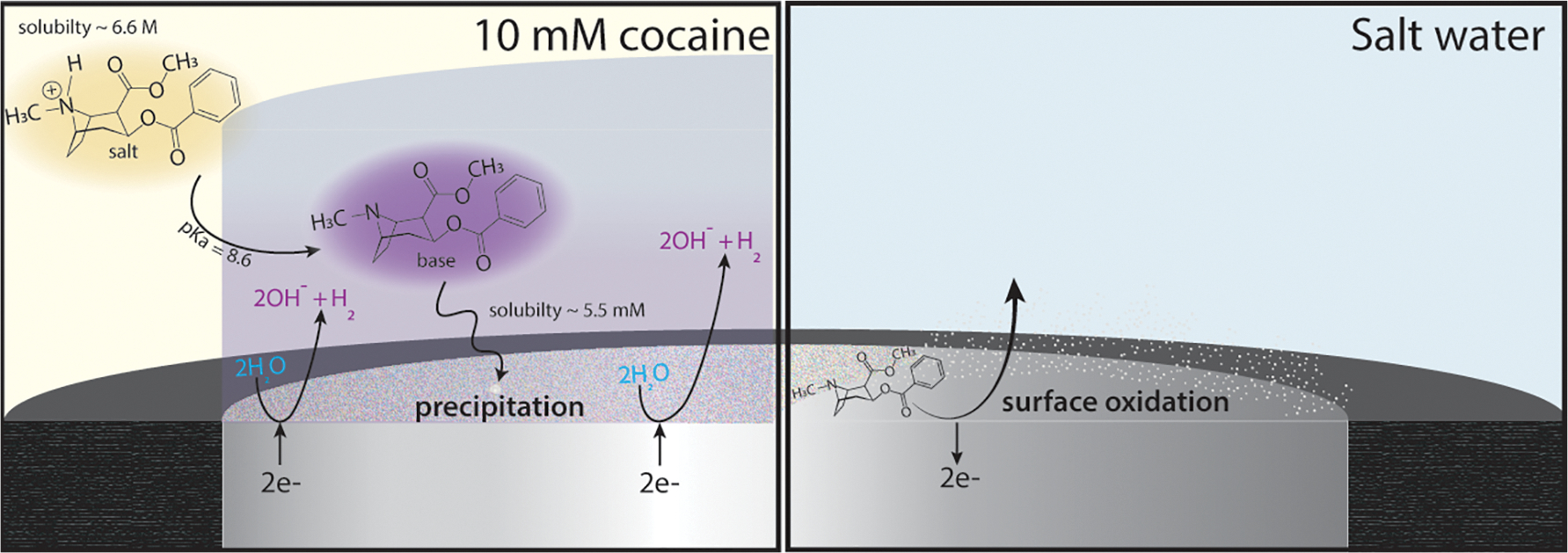
aThe left panel shows the heterogeneous reactions causing a local pH change at the electrode surface in a bulk solution of 10 mM cocaine. The high pH deprotonates the cocaine salt to locally form the cocaine base, which precipitates onto the electrode surface. The right panel shows the oxidation of the cocaine precipitate in a solution of salt water (0.5 M KCl).
MATERIALS AND METHODS
Cocaine hydrochloride C-II, levamisole hydrochloride (United States Pharmacopeia Reference Standard), benzocaine (Supelco, Certified Reference Material), caffeine (Supelco, Certified Reference Material), ascorbic acid, and sodium hydroxide were obtained from Sigma-Aldrich (St. Louis, MO). Procaine hydrochloride (Acros Organics, 99%), lidocaine hydrochloride (99%), sodium bicarbonate, potassium chloride (KCl), and sodium chloride (NaCl) were purchased from Thermo Scientific (Fair Lawn, NJ). Boric acid was obtained from EM Science (Gibbstown, NJ). The cocaine standard solution was obtained from Cerilliant (Round Rock, TX). All solutions were made in water from a Millipore Milli-Q (18.20 MΩ·cm) system unless otherwise stated. Some experiments were performed using tap water (building zip code: 27514). All solutions were prepared using an OHAUS EX125 balance, an Eppendorf research plus pipette set, a benchtop vortex, and standard volumetric glassware. Other typical laboratory equipment (e.g., vortex) was used in the process of experimentation.
All electrochemical experiments were performed by a CHI 601E using a three-electrode system. The glassy carbon electrode (d = 3 mm) and 1 M KCl silver–silver chloride (Ag/AgCl) were purchased from CH Instruments (Austin, TX). The counter electrode was a platinum wire (d = 1 mm) obtained from Goodfellow (Coraopolis, PA) or a glassy carbon rod from Alfa Aesar (Ward Hill, MA). Between experiments, the electrode was polished with 0.05 μm alumina (obtained from CH Instruments) on a wet MicroCloth polishing pad (obtained from Buehler, Lake Bluff, IL) and rinsed with Milli-Q water. The salt bridge was constructed by a glass tube filled with 3% agarose (w/w) in 1 M KCl and stored in 1 M KCl between measurements. A quantitative filter paper (9.0 cm) was obtained from VWR (Radnor, PA). The pH meter was an Orion VeraStar Pro Advanced Electrochemistry Meter from Thermo Scientific (Fair Lawn, NJ). The Pyrex borosilicate capillary (1.5–1.8 mm × 90 mm) was obtained from Corning Inc. (Corning, NY). The Micromanipulator MN-153 was purchased from Narishige International (Amityville, NY). The 3PN 116 Powerstat was obtained from The Superior Electric Company (Bristol, MA). An electrical wire was purchased from Striveday.
The CHI 601E was operated with the associated software provided by CH Instruments. The digital microscopy was performed with a Park Systems microscope and run with the manufacturer-provided CoolingTech or AMCAP software. Adobe Illustrator was used to make all schemes and figures.
Cocaine Electro-Precipitation.
The glassy carbon working electrode (d = 3 mm) is biased at −2.1 V vs Ag/AgCl for 60 s in a solution of 10 mM cocaine hydrochloride in 0.5 M KCl. Reasonable concentrations for this method are discussed in the results section below. The electro-precipitation time and potential were optimized to (1) change the pH and force deposition over the whole electrode surface to maximize the signal, (2) maintain an appreciable rate throughout the duration of the amperometry to ensure that the local pH is sustained, and (3) avoid the creation of interfering bubbles that block the electrode surface.
After the amperometry, the working electrode is disconnected while in solution and precipitation is always visually observed on the electrode surface.
Cocaine Detection by Anodic Sweep Voltammetry.
After precipitation, the working electrode is carefully placed in a solution of 0.5 M KCl. Voltammetry is performed from 0 to 1.4 V vs Ag/AgCl at 0.5 V/s. The scan rate was optimized to give the strongest signal for the peak at ~1.2 V vs Ag/AgCl.
A positive result was attributed to voltammograms that peaked between 1.1 and 1.3 V vs Ag/AgCl with a signal at least 2× the capacitive current.
The precipitation and voltammetric detection were performed on a CHI 601E potentiostat with a glassy carbon working electrode, Ag/AgCl reference electrode, and platinum wire counter electrode.
As this is the initial report of the method, this description by researchers is the proof of concept and future directions will create a device that requires no assembly and automates the peak identification. The important steps are outlined in Scheme 2, where moving the electrodes to a clean solution (step 3) is optional but used throughout this manuscript and very useful for eliminating interferent effects.
Scheme 2. Important Workflow Steps: (1) Dissolve a Sample of Unknown Powder in a Solution of Salt Water, (2) Place the Electrodes in the Solution and Run the Precipitation (60 s), and (3) Place the Electrodes in a New Solution of Salt Water and Run the Voltammetry (3 s)a.

aA positive or negative result is indicated by the presence or absence of a peak between 1.1 and 1.3 V vs Ag/AgCl, respectively.
Interferent Study.
The method for precipitation and detection is the same as written above. For the negative controls, three solutions of each interferent (10 mM) were made in 0.5 M KCl. Each was sonicated until dissolved. Benzocaine was not fully soluble and was filtered with a 1 μM filter before detection. One can visually observe the precipitate on the electrode surface and the solution turning from clear to yellow after amperometry is run in 10 mM procaine solutions. There are reports in the literature of alkaline hydrolysis of procaine.35 The pH of the sodium bicarbonate, boric acid, and ascorbic acid solutions was measured before the experiment. The highest relevant pH for a bulk solution is 8.6 because higher than this pH, the cocaine will be precipitated in bulk and not require local precipitation.
For the positive controls, three solutions of each interferent (10 mM) mixed with 10 mM cocaine hydrochloride were dissolved in 0.5 M KCl. Here, the benzocaine-containing solution was allowed to exceed solubility such that there was visible precipitation during experimentation. The pH of the sodium bicarbonate-, boric acid-, and ascorbic acid-containing solutions was measured before the experiment.
DISCUSSION
Cocaine is electrochemically active, with the commonly proposed oxidation mechanism involving the removal of 2e− and 2H+ to form norcocaine.17 Because the kinetics of these proton-coupled electron transfers are relatively slow at neutral pH on unmodified electrode materials, the current is often indistinguishable from heterogeneous water oxidation. When probed with a glassy carbon electrode in a slightly alkaline solution, a peak for 10 mM cocaine hydrochloride can be easily identified between 0.8 and 1 V vs Ag/AgCl (Figure 1A, red line). Many detection techniques use buffers to keep the pH under the pKa of the amine group (8.6)36 as the uncharged, free base cocaine has low solubility in water (~5.5 mM).37 When the pH of the 10 mM cocaine KCl solution exceeds the pKa of cocaine (8.6),36 there is visible white precipitation (Figure S1) and an additional surface peak arises in the voltammetry. Figure 1A shows the voltammetry of 10 mM cocaine hydrochloride in 0.5 M KCl (pH = 6.2) overlaid with voltammetry of the solution adjusted to just below (pH = 8.4) and just above (pH = 9.0) the pKa.
Figure 1.
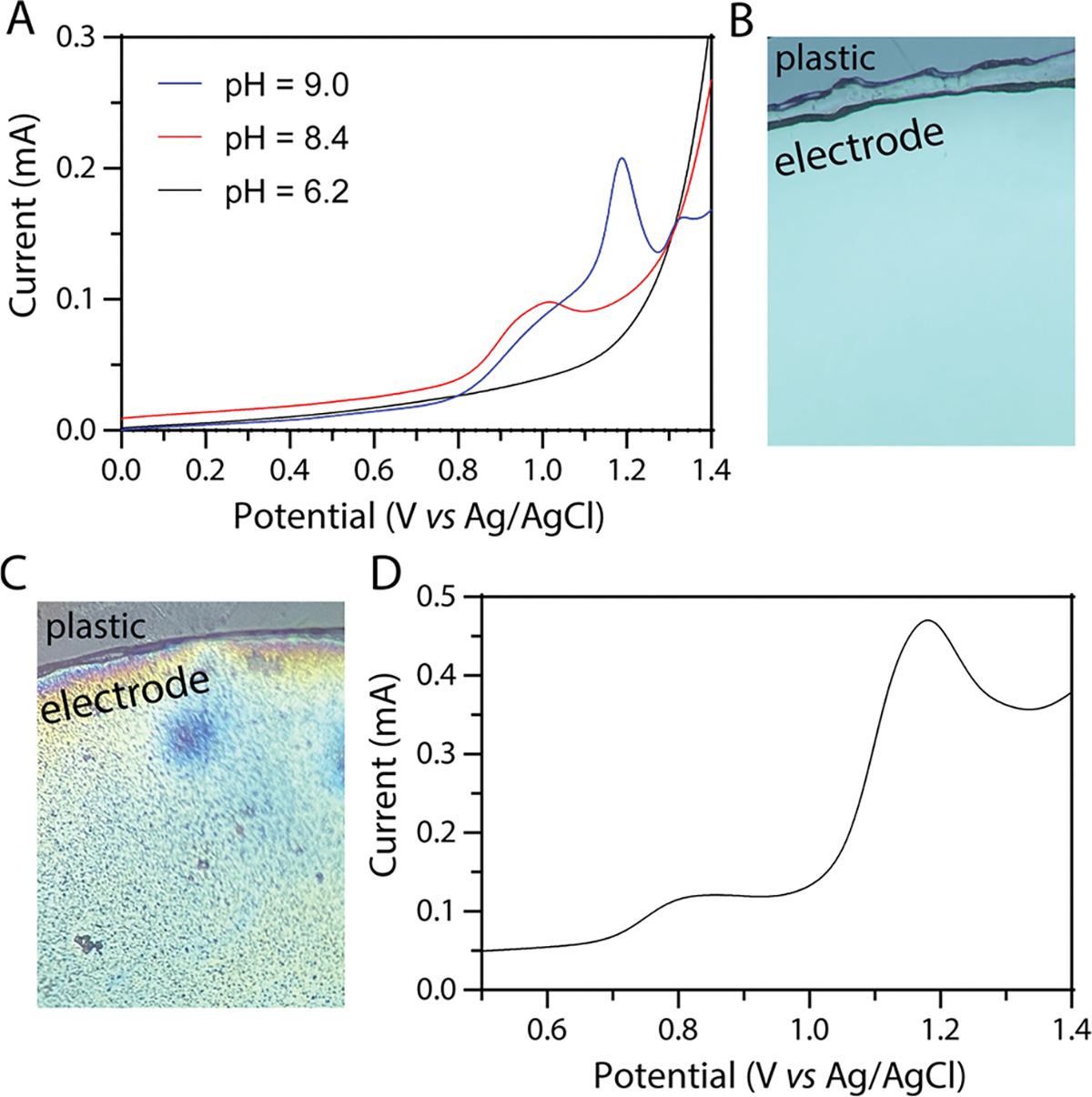
(A) Overlaid voltammograms showing the electrochemical oxidation of 10 mM cocaine in 0.5 M KCl at pH = 7, 8.5, and 9. Voltammetry was collected with a glassy carbon working electrode (d = 3 mm) from 0 to 1.4 V vs Ag/AgCl at 0.5 V/s. (B, C) Photograph of the electrode surface using a 10X objective lens before (B) and after (C) the electro-precipitation where −2.1 V vs Ag/AgCl was applied for 60 s. (D)Representative anodic stripping voltammetry in clean 0.5 M KCl after the the electro-precipitation. Voltammetry was collected with a glassy carbon working electrode (d = 3 mm) from 0.5 to 1.4 V vs Ag/AgCl at 0.5 V/s. In line with the IUPAC convention, for all electrochemical measurements, the anodic current is represented as positive.
In place of adjusting the solution with a strong base, the high pH may be electrogenerated locally by driving water reduction. We expect that oxygen reduction also contributes to the increasing local pH as the solutions were not purged of dissolved oxygen (purging dioxygen in the field may be difficult). However, the electro-precipitation potential was chosen where the rate of reduction is high (current ~ 200 μA) to take advantage of the high molar concentration of water (55.5 M) compared to oxygen (~250 μM).38
Both the reduction of oxygen and water can liberate hydroxide to make the pH at the electrode surface more alkaline.39 Driving water reduction at the working electrode necessitates driving water oxidation at the counter electrode (electroneutrality condition). These reactions rapidly increase and decrease the local pH at the surface of the working and counter electrodes, respectively (Figure S2). For this proof of concept, the electrode is biased at −2.1 V vs Ag/AgCl for 60 s. The precipitation was visualized in real time by video microscopy for optimization (Video S1). Using a highly negative potential ensures that water reduction is occurring at appreciable rates over the experimental time, as shown by the current in Figure S3. Photographs through an inverted microscope lens (Objective 10X) before and after amperometry show visual evidence of precipitate adsorbtion (Figure 1B–C). We show with mass spectrometry that the electrogenerated pH precipitate is free base cocaine (Figure S4) and not the benzoylecgonine (BE) hydrolysis product (MW = 289) that is also minimally soluble in water.
After the electro-precipitation of the free base cocaine, the electrodes are moved into clean 0.5 M KCl for anodic sweep voltammetry. Moving the electrodes to a fresh solution allows for one to see the surface peak while minimizing the signal from diffusion-controlled oxidation. If the electrode is not placed into a fresh solution after precipitation, there is the potential for interferents to influence the determination of cocaine. While many of the substances do not interfere when voltammetry is performed in the same solution, broad peaks are associated with benzocaine, procaine, and ascorbic acid influencing both the positive and negative determination (Table S1 shows voltammetry of each interferent). Figure 1D shows a representative voltammogram for the oxidation of surface adsorbed cocaine in a fresh solution after the precipitation.
The fundamental limit of detection (LOD) for this method is 5.5 mM, requiring the concentration of the cocaine base at the electrode surface to exceed solubility in the test solution. The limit of detection is grounded in an understanding of diffusional flux and concentration profiles. It is possible that clever mass-transfer effects (e.g., convection, electrophoretic migration) could decrease this detection limit by increasing the local concentration compared to the bulk solution. For this initial report, mass-transfer mechanisms are not deeply explored and the cocaine solutions are made at 10 mM. One only needs to sample 3.4–6.8 mg of cocaine (into 2 mL) to make a 5–10 mM solution. This sampling requirement is comparable to the amount used in current presumptive testing, though Scott’s test requires less than 1 mg.40 Especially when employing smaller electrodes (see below), reduced volumes of water could be used to bring the sampling requirement of this technique down to sub-mg amounts. This will be explored in future studies.
According to the Drug Enforcement Administration (DEA) report in 2018, about 40% of wholesale cocaine is adulterated, and levamisole is the most common contaminant in bulk seizures.11,41 From other reports, in addition to levamisole, common adulterants found in cocaine samples include unscheduled additives (e.g., caffeine and paracetamol) and other local anesthetics (often with a similar structure to cocaine), including lidocaine, benzocaine, and procaine.41,42 These adulterants contribute to the effect of the drug. Cocaine is also often diluted, or “cut,” with nonpharmacological substances that are visually indistinguishable from cocaine and intended to stretch the supply. Common cutting agents include boric acid, sugar, laundry detergent, baking soda (sodium bicarbonate), and vitamin C (ascorbic acid).
To test the consequence of mixtures on cocaine identification, eight dilutants with and without cocaine were tested by the sensor. Three cutting agents (boric acid, ascorbic acid, and sodium bicarbonate), three local anesthetics with a similar structure (procaine, lidocaine, and benzocaine), and two other drugs (caffeine and levamisole) were chosen for the interferant study. Because this method relies on the ability to locally change the pH using only a specified voltage, the cutting agents (all weak acids and bases) act as a solution pH control as well. The Supporting Information (Table S1) provides the solvent window and representative amperometric currents (rates) at 2.1 V vs Ag/AgCl across all of the tested samples, highlighting the importance of holding a high overpotential for water reduction. While using a high overpotential makes this generalizable, hydrogen bubbles may also be formed and could contribute to precipitate heterogeneity. At this stage, we did not observe the heterogeneity in this method changing the efficacy of the sensor.
The purity of retail cocaine changes with time but was reported as about 45% in 2010 and 65% in 2018.42 A false positive result is possible if, in the absence of cocaine, an interferent deposits on the electrode surface and is oxidized at a similar potential (1.1–1.3 V). No false positives are observed with this method (see Figure 2A,B for representative voltammetry). This is an improvement when compared to Scott’s test, which has been reported to give false positive results for various adulterants/dilutants, including levamisole, procaine, and lidocaine.32,40 A false negative result can occur when the presence of a dilutant interferes with the signal such that cocaine can no longer be identified in the mixture. To evaluate false negatives, 10 mM of each dilutant is mixed with 10 mM cocaine hydrochloride for cocaine samples of 50% purity. Each mixture was measured three times, and only one measurement (of the 1:1 procaine:cocaine mixtures) gave a false negative result. Figure 2C,D shows representative voltammetry for these studies. The 1:3 false negative rate for the procaine-containing mixtures is caused by wide peaks over the cocaine detection region such that the potential of the peak maxima is difficult to confidently determine (Figure S5). False negatives for 1:1 mixtures of procaine:cocaine have also been reported for Scott’s test.32
Figure 2.
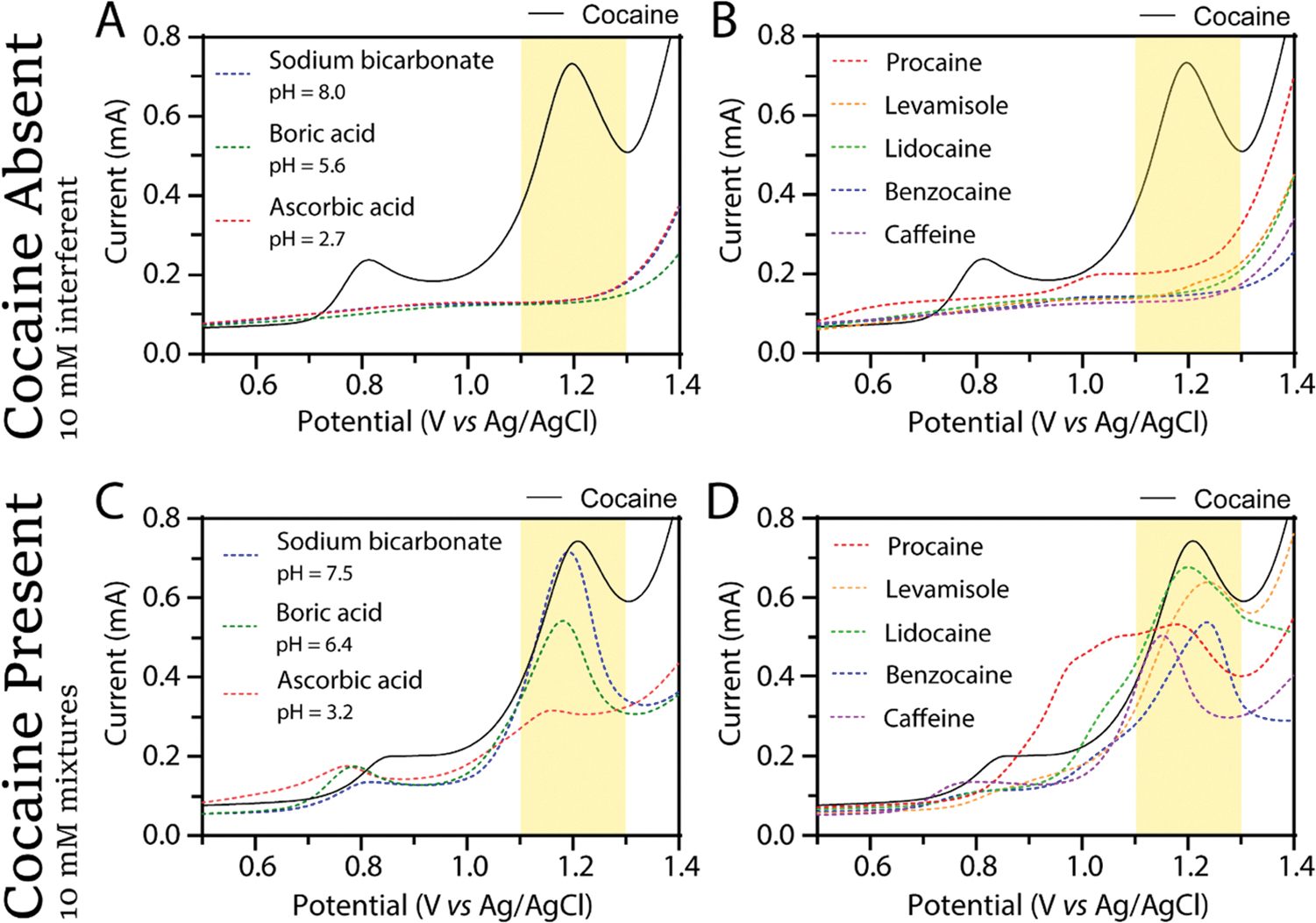
Voltammetry of interferents (A, B) and cocaine mixed with interferents (C, D) overlaid with a reference positive identification of 10 mM cocaine in 0.5 M KCl (solid black line). (A) Representative anodic stripping voltammograms of each weak acid/base cutting agent (10 mM in 0.5 M KCl). (B) Representative anodic stripping voltammograms of each adulterant (10 mM in 0.5 M KCl). (C) Representative anodic stripping voltammograms of 10 mM: 10 mM mixtures of each cutting agent: cocaine in 0.5 M KCl overlaid with a voltammogram of 10 mM cocaine in 0.5 M KCl (solid black line). (D) Representative anodic stripping voltammograms of 10 mM: 10 mM mixtures of adulterant: cocaine in 0.5 M KCl overlaid with a voltammogram of 10 mM cocaine in 0.5 M KCl (solid black line). All voltammetry was collected at a scan rate of 0.5 V/s with a glassy carbon (d = 3 mm) working electrode, a Ag/AgCl reference electrode, and a platinum wire counter electrode. In line with the IUPAC convention, the anodic current is represented as positive in all voltammograms.
The selectivity of the proposed sensor is very promising, and this methodology has accessible instrumentation and is simple to use. To demonstrate this, we conducted a masked study where a student who was not involved in the sensor development is provided only written instructions and the necessary materials (see the Supporting Information and Figure S6). The unknown powders are comprised of three substances of procaine, benzocaine, lidocaine, levamisole, caffeine, and cocaine in equal parts. Mixtures 1–4 contain 33.3% cocaine and mixtures 5 and 6 contain 0% cocaine (for details on each sample, see the Supporting Information). The amount of pure cocaine in each complex powder was 6.8 grams dissolved in 2 mL. The presence/absence of cocaine for all samples was correctly determined, and the representative voltammograms are shown in Figure S7.
To show the method’s potential for deployability, cocaine detection is performed in tap water instead of Milli-Q water and table salt (NaCl) instead of KCl. As shown in Figure 3A, these measurements are comparable to those in laboratory-grade reagents, suggesting that these identifications could be made without bringing anything but the electrodes, a salt packet, and a portable potentiostat (often battery-powered with Bluetooth connection to smartphones)34 to the field setting. Additional studies to determine the sensor’s efficacy in variable water quality and iodized table salt will be the topic of future investigations.
Figure 3.
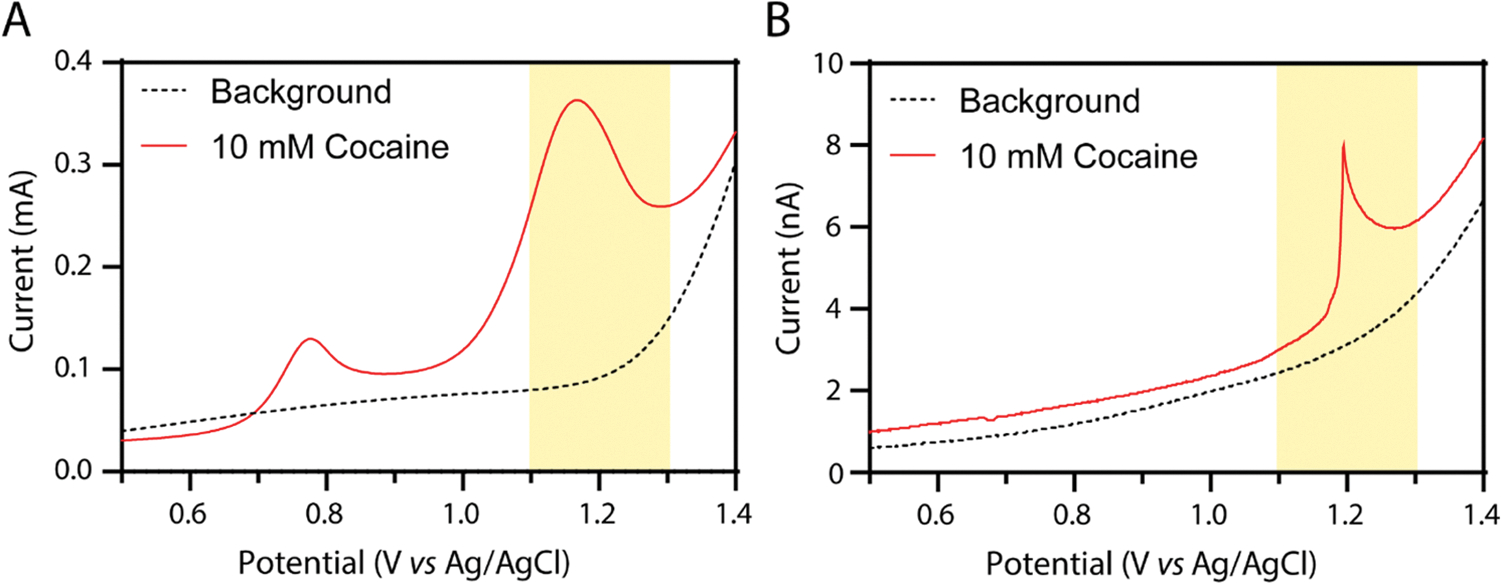
(A) Voltammetry in 0.5 M NaCl tap water after precipitation in 0.5 M NaCl tap water with (red) and without (black dotted) 10 mM cocaine. A glassy carbon (d = 3 mm) working electrode and a scan rate of 0.5 V/s were used. (B) Voltammetry in water containing 10 mM cocaine after 60 s at a potential of −2.2 V vs Ag/AgCl (red) overlayed with a background in the same solution after 60 s at a potential of 0.1 V vs Ag/AgCl (black dotted). A carbon fiber microelectrode (d = 8 μm) working electrode and a scan rate of 0.5 V/s were used. In line with the IUPAC convention, the anodic current is represented as positive in this figure.
Finally, we demonstrate the ability to determine cocaine in tap water with no added salt by the use of a microelectrode (Figure 3B). Microelectrodes are convenient for miniaturizing systems for deployed applications (see the Supporting Information and Figure S8 for microelectrode characterization).43,44 Additionally, microelectrodes have been used to make electrochemical measurements in the absence of salt.43,45–47 Owing to the smaller electrode radii (radii < 10 μm compared to macroelectrodes with radii from ~1 mm), smaller currents are passed, and the measurement is less dependent on solution conductivity (iR drop). Fundamentally, the radial diffusion profile at microelectrodes quickly cycles reactants and products to and from the electrode without building a time-dependent depletion layer (in contrast with macroelectrodes). The experiments in Figure 3 demonstrate that it is possible to electrogenerate a sufficient local concentration of hydroxide to crash out the cocaine base. This detection was performed in the absence of additional salt.
Carbon microelectrodes are available in many cost-effective, commercially available forms (e.g., screen-printed, graphite, and carbon fiber) and offer the appealing prospect of detection without the demand of bringing any reagents to the scene. While this method demonstrates the use of carbon fiber microelectrodes, disposable screen-printed carbon microelectrodes have been shown in the literature to be easy to fabricate and useful for electrochemical stripping analyses.48
CONCLUSIONS
We present a robust detection strategy for in-field cocaine identification using pH electro-precipitation and voltammetric detection. While most electrochemistry is done in spite of the solvent window, we use water’s faradaic exchange to add a separation step for the electrochemical detection of cocaine. Because of this, we can detect cocaine at neutral pH, avoiding the usual requirement of changing the solution pH with the addition of acid/base chemicals. These are the first steps to developing a specific electrochemical sensor that uses an unmodified electrode in an essentially reagentless solution. Finally, we demonstrate that a sensor based on this methodology is highly specific and may be useful for deployable sensing in complex and highly impure powder mixtures.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge support from the National Institutes of Health under Grant 1-R35- GM138133-01. K.J.V. acknowledges the National Institute of Justice for fellowship support under Grant 2020-R2-CX-0036. The authors acknowledge the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory, especially Erin Tracy, for their assistance with mass spectrometry analysis. They thank Sondrica Goines for helpful discussions surrounding the microelectrode fabrication.
Funding
J.E.D. and K.J.V. are coinventors on a provisional patent application.
Footnotes
The authors declare the following competing financial interest(s): The authors, Jeffrey Dick and Kathryn Vannoy, are co-inventors on a provisional patent application.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.2c01630.
Photographs of cocaine solutions at various bulk pH, photographs of the litmus paper, mass spectrum of the precipitate, cyclic voltammetry of interferents and information on the structure and pKas, materials used and voltammetry for the masked study, more voltammetry examples of the cocaine and procaine mixture, voltammetric characterization of the carbon fiber microelectrode, and precipitation video (PDF)
Precipitation was visualized in real time by video microscopy for optimization (MP4)
Contributor Information
Kathryn J. Vannoy, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Lynn E. Krushinski, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Edgar F. Kong, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Jeffrey E. Dick, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Lineberger Comprehensive Cancer Center, School of Medicine, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
REFERENCES
- (1).Krauss ST; Remcho TP; Lipes SM; Aranda R; Maynard HP; Shukla N; Li J; Tontarski RE Jr.; Landers HP Anal. Chem. 2016, 88, 8689–8697. [DOI] [PubMed] [Google Scholar]
- (2).Philp M; Fu S Drug Test. Anal. 2018, 10, 95–108. [DOI] [PubMed] [Google Scholar]
- (3).National Drug Threat Assessment Drug Enforcement Adminstration; U.S. Department of Justice, 2018. [Google Scholar]
- (4).Yu B; Ge M; Li P; Xie Q; Yang L Talanta 2019, 191, 1–10. [DOI] [PubMed] [Google Scholar]
- (5).Postigo C; Lopez de Alda MJ; Viana M; Querol X; Alastuey A; Artinaño B; Barceló D Anal. Chem. 2009, 81, 4382–4388. [DOI] [PubMed] [Google Scholar]
- (6).Chen X; Wu X; Luan T; Jiang R; Ouyang GJ Chromatogr. A 2021, 1640, No. 461961. [DOI] [PubMed] [Google Scholar]
- (7).de Araujo WR; Cardoso TMG; da Rocha RG; Santana MHP; Muṅoz AA; Richter EM; Paixāo TRLC; Coltro KT Anal. Chim. Acta 2018, 1034, 1–21. [DOI] [PubMed] [Google Scholar]
- (8).Fedick PW; Pu F; Morato NM; Cooks RG J. Am. Soc. Mass Spectrom. 2020, 31, 735–741. [DOI] [PubMed] [Google Scholar]
- (9).Ifa DR; Jackson AU; Paglia G; Cooks RG Anal. Bioanal. Chem. 2009, 394, 1995–2008. [DOI] [PubMed] [Google Scholar]
- (10).Harper L; Powell J; Pijl EM Harm Reduct. J. 2017, 14, No. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).National Drug Threat Assessment Drug Enforcement Adminstration; U.S. Department of Justice, 2021. [Google Scholar]
- (12).Tettey J; Collins M; Salouras H; Swan H; Wang Y Recommended Methods for the Identification and Analysis of Cocaine in Seized Materials. In Laboratory and Scientific Section United Nations Office on Drugs and Crime; United Nations, Vienna, 2012. [Google Scholar]
- (13).Marcelo MCA; Mariotti KC; Ortiz RS; Ferrão MF; Anzanello MJ Microchem. J. 2016, 127, 87–93. [Google Scholar]
- (14).McCulloch A Chemosphere 2003, 50, 1291–1308. [DOI] [PubMed] [Google Scholar]
- (15).Evans CD; Monteith DT; Fowler D; Cape JN; Brayshaw S Environ. Sci. Technol. 2011, 45, 1887–1894. [DOI] [PubMed] [Google Scholar]
- (16).Kodavanti PRS; Loganathan BG Organohalogen Pollutants and Human Health. In Encyclopedia of Public Health; Academic Press: Oxford, 2017; pp 359–366. [Google Scholar]
- (17).Poltorak L; Sudhölter EJR; de Puit M TrAC, Trends Anal. Chem. 2019, 114, 48–55. [Google Scholar]
- (18).Ahmed SR; Chand R; Kumar S; Mittal N; Srinivasan S; Rajabzadeh AR TrAC, Trends Anal. Chem. 2020, 131, No. 116006. [Google Scholar]
- (19).de Jong M; Sleegers N; Kim J; Durme FV; Samyn N; Wang J; De Wael K Chem. Sci. 2016, 7, 2364–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).De Rycke E; Stove C; Dubruel P; De Saeger S; Beloglazova N Biosens. Bioelectron. 2020, 169, No. 112579. [DOI] [PubMed] [Google Scholar]
- (21).Florea A; Cowen T; Piletsky S; De Wael K Analyst 2019, 144, 4639–4646. [DOI] [PubMed] [Google Scholar]
- (22).Ren S; Zeng J; Zheng Z; Shi H Sens. Actuators, A 2021, 329, No. 112821. [Google Scholar]
- (23).Swensen JS; Xiao Y; Ferguson BS; Lubin AA; Lai RY; Heeger AJ; Plaxco KW; Soh HT J. Am. Chem. Soc. 2009, 131, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).de Oliveira LS; Balbino MA; de Menezes MMT; Dockal ER; de Oliveira MF Microchem. J. 2013, 110, 374–378. [Google Scholar]
- (25).Samec Z; Langmaier J; Trojánek A; Samcová E; Málek J Anal. Sci. 1998, 14, 35–41. [Google Scholar]
- (26).Kalvoda R Anal. Chim. Acta 1982, 138, 11–18. [Google Scholar]
- (27).Pavlova V; Mirčeski V; Komorsky-Lovrić Š; Petrovska-Jovanović S; Mitrevski B Anal. Chim. Acta 2004, 512, 49–56. [Google Scholar]
- (28).Ahmar H; Tabani H; Hossein Koruni M; Davarani SSH; Fakhari AR Biosens. Bioelectron. 2014, 54, 189–194. [DOI] [PubMed] [Google Scholar]
- (29).Ott CE; Cunha-Silva H; Kuberski SL; Cox JA; Arcos-Martinez MJ; Arroyo-Mora LEJ Electroanal. Chem. 2020, 873, No. 114425. [Google Scholar]
- (30).Parrilla M; Joosten F; De Wael K Sens. Actuators, B 2021, 348, No. 130659. [Google Scholar]
- (31).Asturias-Arribas L; Alonso-Lomillo MA; Domínguez-Renedo O; Arcos-Martínez MJ Anal. Chim. Acta 2014, 834, 30–36. [DOI] [PubMed] [Google Scholar]
- (32).de Jong M; Florea A; Eliaerts J; Van Durme F; Samyn N; De Wael K Anal. Chem. 2018, 90, 6811–6819. [DOI] [PubMed] [Google Scholar]
- (33).Brown K; Dennany L Forensic Analytical Method; The Royal Society of Chemistry, 2019; pp 115–139. [Google Scholar]
- (34).Glasscott MW; Verber MD; Hall JR; Pendergast AD; McKinney CJ; Dick JE J. Chem. Educ. 2020, 97, 265–270. [Google Scholar]
- (35).Aven M; Foldes FF Science 1951, 114, 206–208. [DOI] [PubMed] [Google Scholar]
- (36).Karaffa LS The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; RSC Publishing, 2013. [Google Scholar]
- (37).Gillams RJ; Lorenz CD; McLain SE Chem. Phys. Lett. 2017, 676, 58–64. [Google Scholar]
- (38).Xing W; Yin G; Zhang J Rotating Electrode Methods and Oxygen Reduction Electrocatalysts; Elsevier Science, 2014; pp 1–31. [Google Scholar]
- (39).Bard AJ; Faulkner LR Russ. J. Electrochem. 2002, 38, 1364–1365. [Google Scholar]
- (40).Tsumura Y; Mitome T; Kimoto S Forensic Sci. Int. 2005, 155, 158–164. [DOI] [PubMed] [Google Scholar]
- (41).United Nations publication. World Drug Report, Sales No. E.21.XI.8, 2021.
- (42).UNODC and EUROPOL, The illicit trade of cocaine from Latin America to Europe–from oligopolies to free-for-all? Cocaine Insights 1; UNODC: Vienna, September 2021. [Google Scholar]
- (43).Glasscott MW; Vannoy KJ; Kazemi R; Verber MD; Dick JE Environ. Sci. Technol. Lett. 2020, 7, 489–495. [Google Scholar]
- (44).Lin Z; Takahashi Y; Kitagawa Y; Umemura T; Shiku H; Matsue T Anal. Chem. 2008, 80, 6830–6833. [DOI] [PubMed] [Google Scholar]
- (45).Bond AM; Fleischmann M; Robinson JJ Electroanal. Chem. Interfacial Electrochem. 1984, 168, 299–312. [Google Scholar]
- (46).Ciszkowska M; Stojek ZJ Electroanal. Chem. 1999, 466, 129–143. [Google Scholar]
- (47).Howell JO; Wightman RM Anal. Chem. 1984, 56, 524–529. [Google Scholar]
- (48).Kadara RO; Jenkinson N; Banks CE Electrochem. Commun. 2009, 11, 1377–1380. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.