

Abstract
A 2′-deoxycytidin-N4-yl radical (dC·), a strong oxidant that also abstracts hydrogen atoms from carbon–hydrogen bonds, is produced in a variety of DNA damaging processes. We describe here the independent generation of dC· from oxime esters under UV-irradiation or single electron transfer conditions. Support for this σ-type iminyl radical generation is provided by product studies carried out under aerobic and anaerobic conditions, as well as electron spin resonance (ESR) characterization of dC· in a homogeneous glassy solution at low temperature. Density functional theory (DFT) calculations also support fragmentation of the corresponding radical anions of oxime esters 2d and 2e to dC· and subsequent hydrogen atom abstraction from organic solvents. The corresponding 2′-deoxynucleotide triphosphate (dNTP) of isopropyl oxime ester 2c (5) is incorporated opposite 2′-deoxyadenosine and 2′-deoxyguanosine by a DNA polymerase with approximately equal efficiency. Photolysis experiments of DNA containing 2c support dC· generation and indicate that the radical produces tandem lesions when flanked on the 5′-side by 5′-d(GGT). These experiments suggest that oxime esters are reliable sources of nitrogen radicals in nucleic acids that will be useful mechanistic tools and possibly radiosensitizing agents when incorporated in DNA.
Graphical Abstract
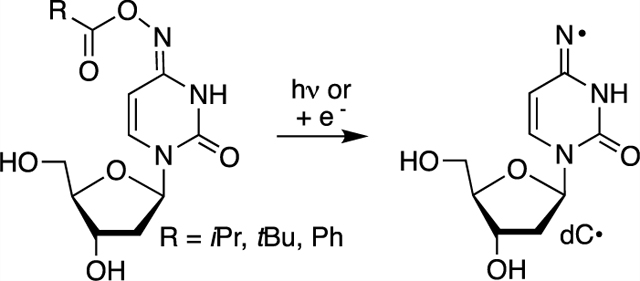
INTRODUCTION
Nucleobase radicals play important roles in nucleic acid damage mediated by ionizing radiation and other oxidizing agents. Ionizing radiation generates nucleobase radicals from native nucleotides by direct oxidation and indirectly through the oxidation of water that yields highly reactive hydroxyl radicals.1 Hydroxyl radicals are also generated by Fe·EDTA, a highly useful reagent for probing nucleic acid structure and noncovalent interactions by producing direct strand breaks and alkali-labile lesions in DNA.2–8 Due to the poor chemoselectivity of ionizing radiation and hydroxyl radicals, the development of photolabile precursors has proven very useful for examining the reactivity of individual radicals in DNA and RNA oxidative damage.9 Experiments using nucleosides or synthetic biopolymers containing radical precursors at specific sites have provided insight into a broad range of questions concerning nucleic acids.9 For instance, independent generation of C4′-nucleotide radicals was foundational in Giese’s seminal studies on charge transfer in DNA.10–13 In another example, this experimental approach revealed pathways to direct strand scission in RNA that are not viable in DNA.14 Ketone photochemistry, specifically the Norrish type I photoreaction, has most frequently been used to generate carbon π-radicals in these studies. This strategy has been expanded to nitrogen nucleobase radical generation via β-fragmentation of a photochemically generated carbon radical.15–17 More often, aminyl and carbon σ-radicals are generated directly via one-electron reduction of alkyl azides and vinyl halides, respectively, as well as UV-photolysis of the latter.18–27 Such precursors are also of interest as radiosensitizing agents for hypoxic cells.27,28 Reduced O2 levels in hypoxic cells enable the precursors to capture solvated electrons produced from ionizing radiation. In contrast to carbon π-type radicals, nitrogen and carbon σ-type radicals directly abstract hydrogen atoms from the 2′-deoxyribose backbone and/or add to nucleobase π-bonds to generate DNA-strand breaks and interstrand cross-links, respectively.1,18,27,29 A 2′-deoxycytidin-N4-yl radical (dC·) is a nitrogen-centered σ-type iminyl radical produced as a result of oxidative stress in DNA.30–33 Herein, we describe the use of oxime esters to generate dC· photochemically and via single electron transfer.
Oxime ethers and esters are useful precursors for σ-type iminyl radicals (Scheme 1). The weak N–O bond (BDE ~ 35 kcal/mol) enables one to generate iminyl radicals via thermolysis although the temperatures required are typically significantly greater than 100 °C.34,35 Direct UV-irradiation (Pyrex filtered light) also yields iminyl radicals.36,37 There are also numerous examples of iminyl radical generation via single electron transfer, particularly for utilization in organic synthesis.38,39 Organometallic catalysts and increasingly photoredox systems have been employed to generate the iminyl radicals from oxime esters via single electron transfer.40,41 We recently generated dC· via direct UV-irradiation from oxime ether 1.42 Although experiments with 1 provided insight regarding the reactivity of dC·, they were complicated in the presence of O2, a necessary component when studying DNA reactivity under biologically relevant conditions. The desire to generate dC· via direct photolysis and/or single electron transfer in water under aerobic conditions led us to investigate the use of oxime esters (2).
Scheme 1.
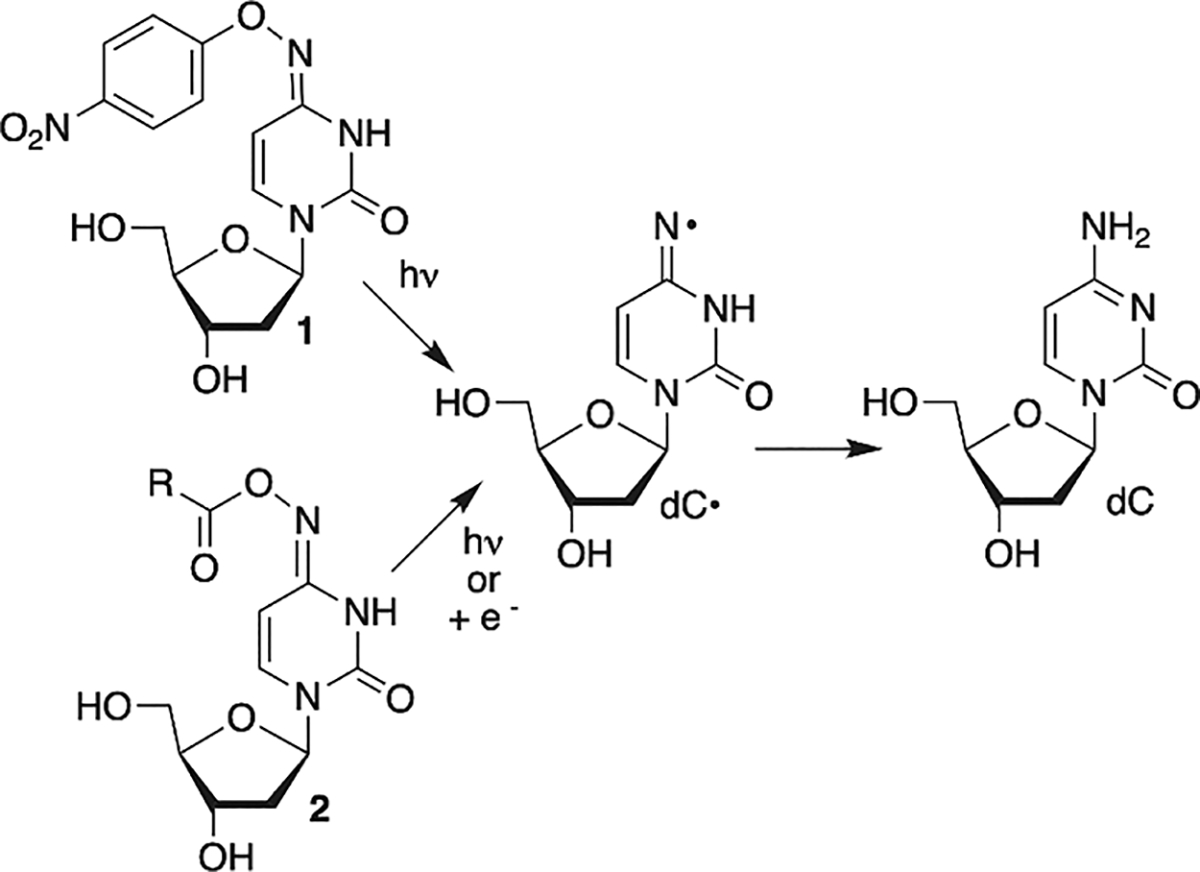
Independent Generation of σ-Type 2′-Deoxycytidin-N4-yl Radical (dC·)
RESULTS AND DISCUSSION
Synthesis of Oxime Esters (2) and Photochemical Generation dC· from Them.
The oxime esters were prepared from 3, as was 2a, which was previously reported (Scheme 2).43 With the exception of 2b (R = Et), acylation was affected using the corresponding acyl chloride. For 2b, acylation was achieved by coupling the carboxylic acid with the water soluble carbodiimide (EDCI). Although the bis-silylated oxime esters (4a–e) could be purified, it was more convenient to deprotect the crude acylated material after aqueous work-up and purify 2a–e. The oxime esters exist as a mixture of aminyl and iminyl (shown) tautomers. The isomers are distinguishable by 1H NMR due to a downfield shift of the vinyl protons of the aminyl tautomer (aromatic) relative to the iminyl form. Consistent with related molecules, the iminyl tautomer is the major isomer in 2a–d (See Supporting Information).16,44 We are unable to determine the tautomeric ratio in 2e, partly due to overlapping signals with the phenyl protons.
Scheme 2.
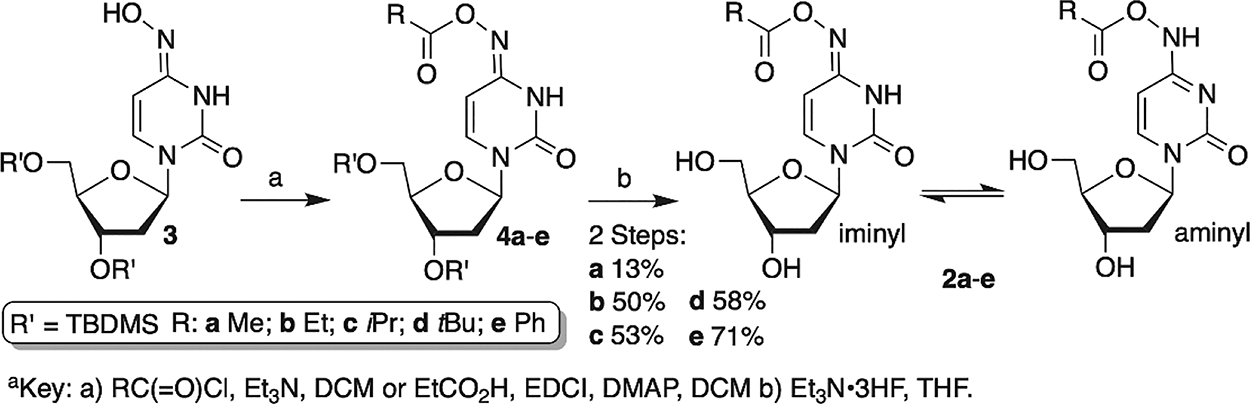
Preparation of Oxime Esters
Oxime ester product studies were carried out in phosphate buffered saline (PBS, 10 mM phosphate (pH 7.2), 100 mM NaCl). Despite its use in DNA experiments, in our hands, the methyl oxime ester (2a) was unstable in PBS.43 Aerobic photolysis (350 nm) of the t-butyl oxime ester (2d) provided dC in 52% on 61% conversion of 2d after 4 h irradiation (Figure 1). dC was the only product identified, and >90% mass balance was maintained throughout the irradiation time. The hydroxylamine (deprotected form of 3) was not detected. No other peaks were evident in the chromatogram. The mass balance was within experimental error when 2d was photolyzed under anaerobic conditions. However, the conversion and dC yield (73% after 4 h irradiation) were consistently higher than when O2 was present during photolysis. Although quenching studies were not carried out, this is consistent with contribution of the triplet excited state to dC· formation, which is quenched by O2. In addition, O2 is not expected to react with the nitrogen-centered radical (dC·), which would affect the yield of dC and the mass balance but not the conversion rate of 2d.16,45 β-Mercaptoethanol (BME, 10 mM) has no effect on dC yield or mass balance under either aerobic or anaerobic conditions (Figure 1). Substituting perdeuterated methanol (MeOH-d4) for methanol resulted in a slight decrease in dC yield due to lower photochemical conversion of 2d and not reduced mass balance. Similarly, using 10% acetonitrile (ACN) as cosolvent also had little effect on the photochemical formation and trapping of dC·. Although isotopic labeling cannot be used, we employed density functional theory (DFT) and high-level Gaussian 4 (G4) calculations to evaluate the ability of hydrogen atom abstraction by dC·.46 Consistent with previous work, the DFT calculations predict that the N–H bond dissociation enthalpy of the parent (1-MeCyt) to π-type aminyl radical is 107 kcal/mol, and the G4 level calculations agree with a value of 106 kcal/mol (Scheme 3 and Figure S8).48,49
Figure 1.
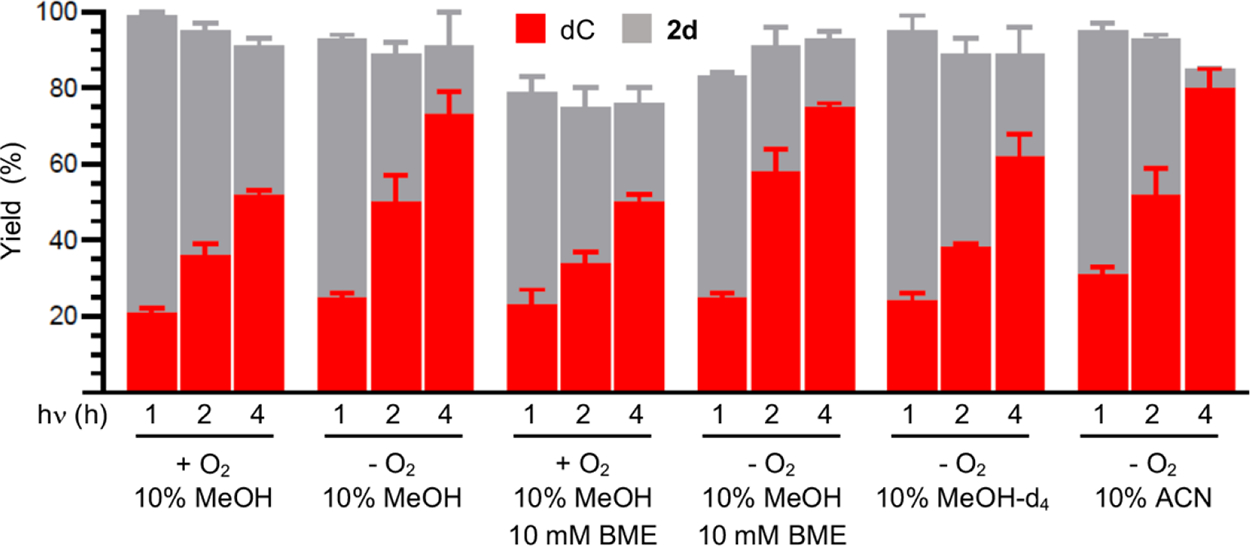
Consumption of t-butyl oxime ester (2d, 0.1 mM) and dC yield as a function of photolysis time and conditions. Yields are the ave. ± std. dev. of three replicates.
Scheme 3.
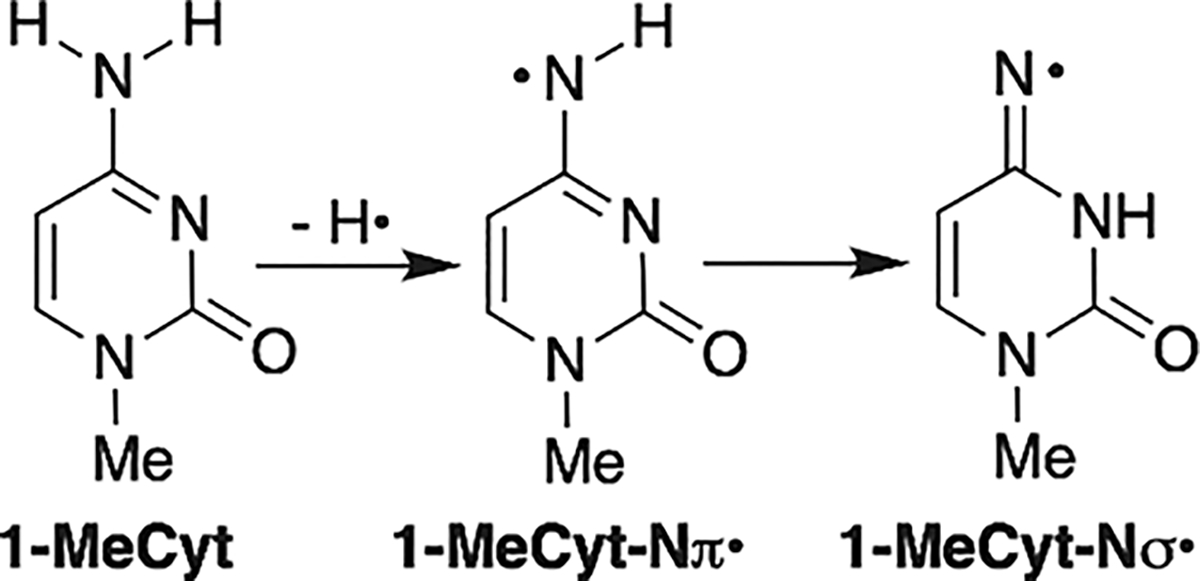
Nitrogen Radical Formation from 1-Methylcytosine
When the exothermic tautomerization of to the iminyl radical () is taken into account, the N–H bond dissociation enthalpy decreases to ~104 kcal/mol (DFT) and 103 kcal/mol (G4) kcal/mol (Figure S8) which are in agreement with previous results.46 Bond strengths of the C–H bonds in ACN and methanol are less than this.48–51 Thus, hydrogen atom abstraction from these organic cosolvents by dC· is exothermic, and these could be the sources of the hydrogen atom required for dC formation. Furthermore, dC· should be able to abstract hydrogen atoms from the 2′-deoxyribose backbone and thymine methyl group in DNA even more readily.47,52
The phenyl oxime ester (2e) photochemistry showed a greater dependence on O2 (Figure 2A). dC was formed in negligible yields under aerobic conditions whether or not BME (10 mM) was present. Although the photochemical conversion of 2e was lower than that of the t-butyl oxime ester (2d) when O2 was present, this was not the sole reason for low dC yields. The mass balance under aerobic conditions was only slightly greater than 50% after 2e was photolyzed for 4 h. The yields of dC increased considerably as did the mass balances when 2e was photolyzed under anaerobic conditions. However, the dC yield (39%) was considerably lower than that obtained from 2d and did not increase when BME (10 mM) was present. The mass balances with and without BME were higher when 2e was photolyzed under anaerobic conditions but were still generally lower than when the t-butyl oxime ester (2d) was used as dC· precursor. Using MeOH-d4 as cosolvent also had no effect on reactivity as expected based upon studies with 2d. We attribute the less productive and efficient photochemistry of the phenyl oxime ester (2e), particularly under aerobic conditions, to differences in excited state chemistry.
Figure 2.
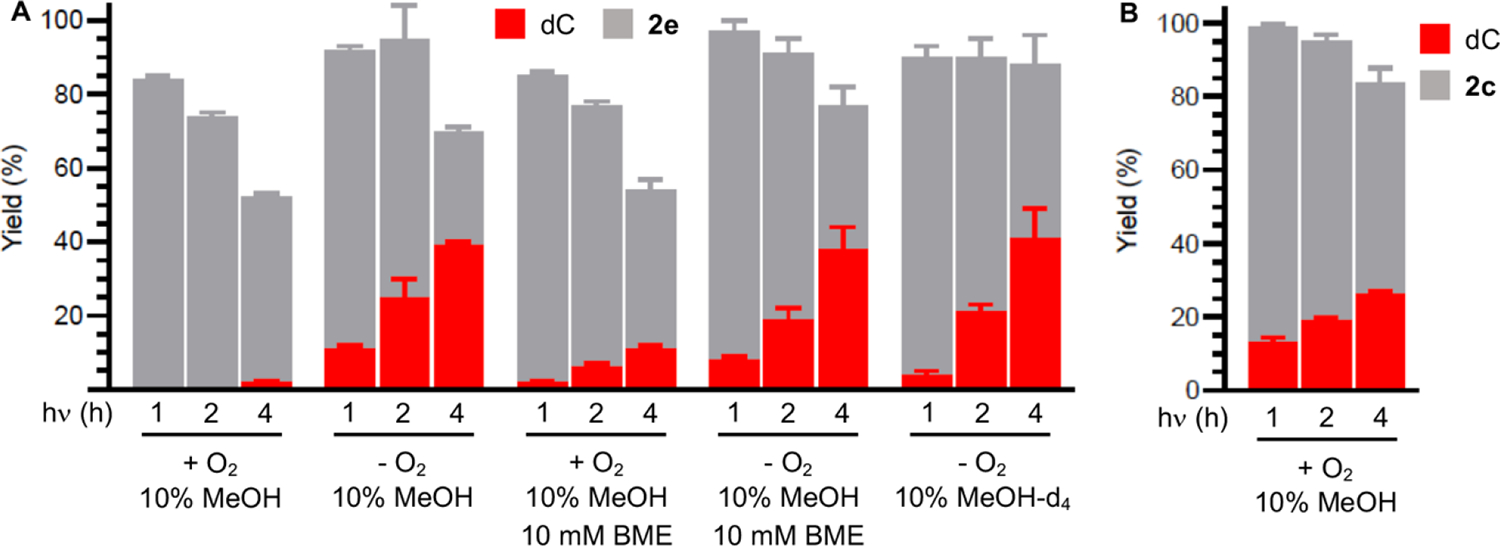
Consumption of oxime esters and dC yield as a function of photolysis time and conditions. (A) Phenyl oxime ester (2e, 0.1 mM). (B) Isopropyl oxime ester (2c, 0.1 mM). Yields are the ave. ± std. dev. of three replicates.
Isopropyl oxime ester (2c) behaved more like 2d than the phenyl derivative when photolyzed under aerobic conditions in methanol cosolvent (10%) (Figure 2B). The photochemical conversion of 2c was lower after 4 h photolysis (42%) than the t-butyl oxime ester (61%). However, dC was produced in good yield (26%) and the mass balance was ~84%.
dC· Generation from Oxime Esters by Single Electron Transfer.
The potential use of oxime esters as radiosensitizing agents would be enhanced if they serve as radical precursors under single electron transfer conditions. This would enable them to selectively generate radicals by reacting with solvated electrons that are more available in hypoxic cancer cells (Scheme 4). As proof of principle, we reacted t-butyl (2d) and phenyl (2e) oxime esters with ferrous ammonium sulfate (10 mM). Both esters (0.1 mM; 5% MeOH in PBS) were completely consumed in 1 h. dC was the sole product detected from 2d (75 ± 1%) or 2e (86 ± 2%). We propose that reaction occurs through the radical anion (Scheme 4).
Scheme 4.
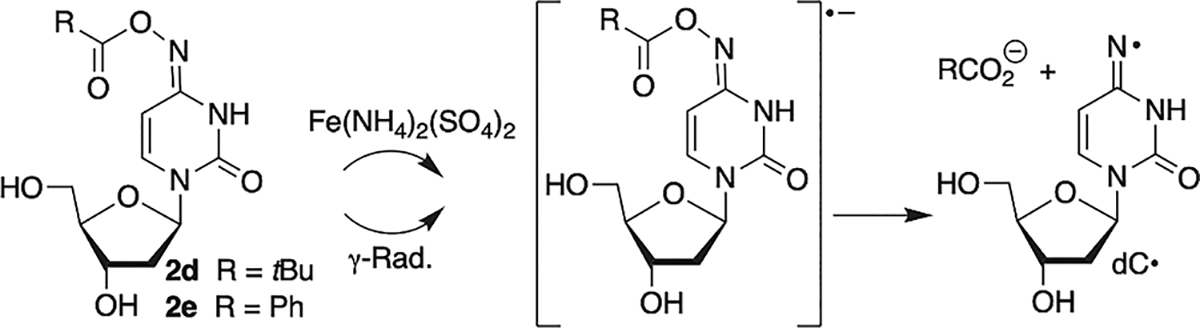
Single Electron Transfer Generation of dC· via Dissociative Electron Attachment
Support for generating dC· via dissociative electron attachment (DEA) was obtained by γ-irradiation (absorbed dose = 600 Gy at 77 K) of a homogeneous supercooled (glassy) solution (7.5 LiCl/D2O) of either 2d or 2e (2 mg/mL), followed by the use of electron spin resonance (ESR) spectroscopy at 77 K (Figure 3).53 Subjecting oxime ether 1 to either the ferrous ion or γ-radiolysis does not yield any evidence for dC· formation (data not shown), highlighting an advantage of the oxime esters as radical precursors via DEA under single electron transfer conditions (Scheme 4).
Figure 3.
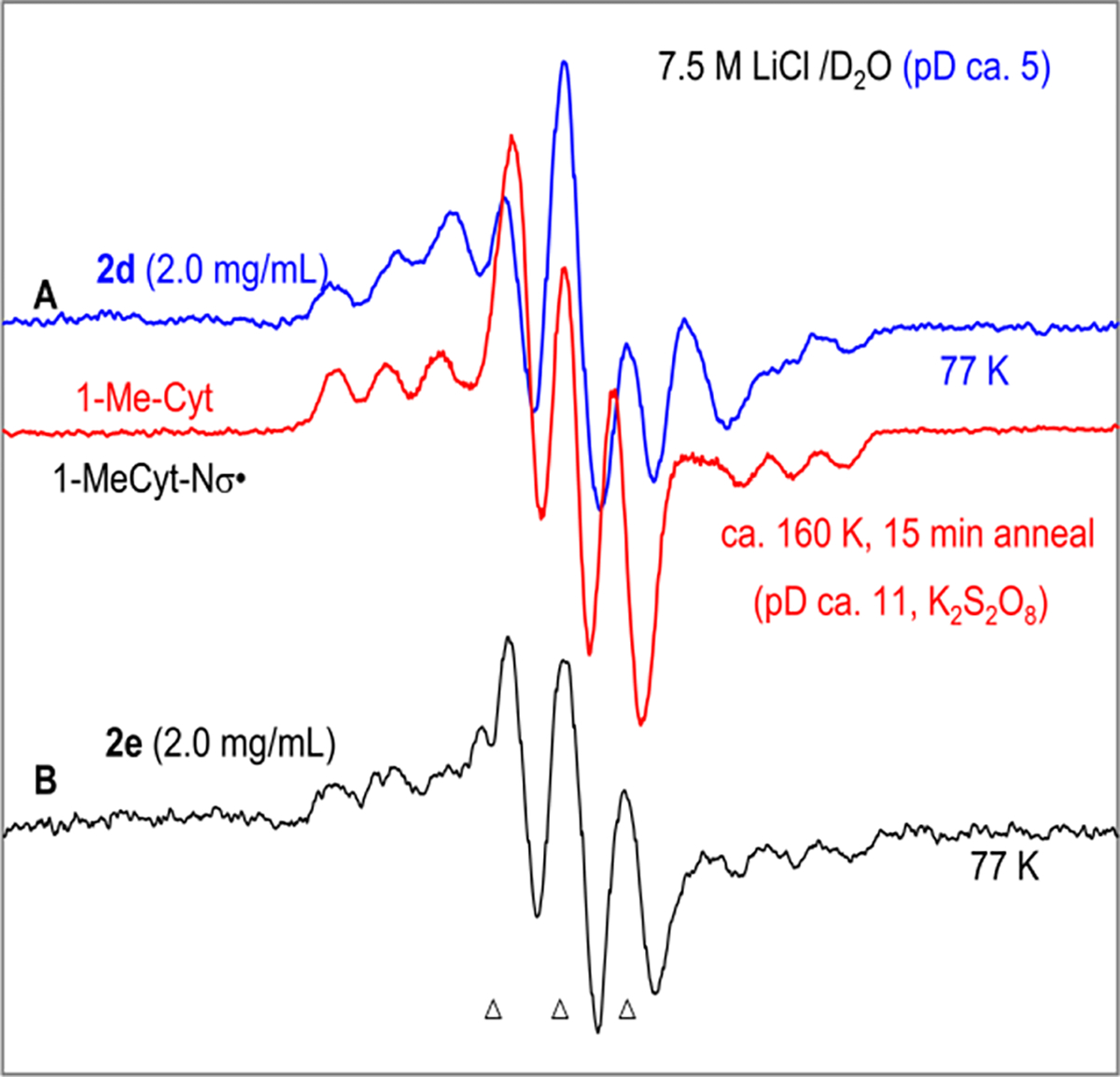
First derivative ESR spectra after one-electron addition (single electron transfer) to (A) 2d (2.0 mg/mL, blue) and to (B) 2e (2.0 mg/mL, black) at 77 K in homogeneous glassy (7.5 M LiCl/D2O, pD ca. 5) solution. Electrons were generated by γ-irradiation (absorbed dose = 600 Gy at 77 K) of the glassy solution. The ESR spectrum (red) (reproduced from ref 53, Copyright [2015] American Chemical Society) shown in panel (A) is that of (Scheme 3) formed by one-electron oxidation of 1-MeCyt by (see ref 53 for details). The three reference markers (indicated by triangles at the bottom of the figure) in this figure are Fremy’s salt resonances. The central marker is at g = 2.0056, and the markers are separated from one another by 13.09 G.18,27,47,53
ESR spectra were recorded of radicals formed by radiation-produced prehydrated electron attachment to 2d (blue spectrum, Figure 3A) and 2e (black spectrum, Figure 3B) in homogeneous glassy (7.5 M LiCl/D2O, pD ca. 5) solutions at 77 K. The total spectral width, hyperfine splittings, line intensities, and g-values at the center of these two spectra establish that each of these spectra is due to two nitrogen hyperfine coupling constant (HFCC) value—one anisotropic N-atom HFCC and one nearly isotropic N-atom HFCC. These spectra match well with the reported ESR spectrum (red spectrum, Figure 3A) of (Scheme 3) that was formed in glassy samples (2 mg/mL) of 1-methylcytosine (1-MeCyt) in 7.5 M LiCl/D2O after one-electron oxidation of the cytosine base by . 53
The 77 K ESR spectra obtained after one-electron reduction of oxime esters 2d and 2e agree well with the benchmark ESR spectrum (red) of obtained via one-electron oxidation of 1-MeCyt. These data confirm that (a) one-electron reduction of 2d and 2e leads to the facile formation of dC· via DEA (Scheme 4) even at 77 K and (b) the electron affinity of the oxime ester group is higher than those of the pyrimidine bases (e.g., cytosine, this work). DFT calculations employing Uωb97xd-IEF-PCM/6–31++G** method and the optimized geometries obtained using the same method show that formation of dC· via DEA (Scheme 4) is the favorable pathway (Figure S9). Moreover, these results explain the formation of dC in high yield (Figure 2) and support the involvement of dC· in the 350 nm photolysis of 2d and 2e in aqueous solution (Scheme 4).
Polymerase Incorporation of Oxime Ester 2c in DNA.
Oxime esters are unstable to the alkaline conditions typically used to deprotect chemically synthesized oligonucleotides. Strategies for nucleobase and phosphate protection exist that avoid such conditions.54,55 However, these require the synthesis of specialized solid-phase synthesis supports and phosphoramidites. This approach also does not provide any insight concerning whether the radical precursors could be incorporated into cellular DNA. For this to be achieved, the corresponding 2′-deoxynucleotide triphosphate (dNTP) must be accepted by DNA polymerase(s). We chose to prepare a duplex containing a dC· oxime ester precursor using the large fragment of DNA polymerase I from E. coli that lacks exonuclease activity (Klenow exo−). Although N4-acylated 2′-deoxycytidine derivatives are incorporated in DNA by polymerases, we sought to identify the oxime ester that provides a compromise between being a good DNA polymerase substrate and sufficient stability in water.56,57 The latter was a concern due to the aforementioned instability of the methyl oxime ester (2a) in our hands, and we assumed that larger alkyl groups would hinder hydrolysis to the hydroxyl amine (the desilylated form of 3). However, we anticipated that oxime esters containing smaller alkyl groups would be superior DNA-polymerase substrates. The phenyl oxime ester (2e) also was not a dNTP candidate because photolysis of this molecule did not produce high yields of dC· as evidenced by the above product studies (Figure 2). Consequently, we examined the stability of the ethyl (2b), isopropyl (2c), and t-butyl (2d) oxime esters in PBS at 25 °C (Table 1). Of these three molecules, the half-lives of the isopropyl and t-butyl oxime esters were between 1 and 4 weeks; only 2b was deemed to hydrolyze to the hydroxylamine too quickly to be viable in this regard.
Table 1.
Hydrolysis Kinetics of Oxime Esters (2b–d) in PBS at 25 °C
| substrate (R) | k (s−1) | t1/2 (h) |
|---|---|---|
| 2b (Et) | 6.8 × 10−6 | 29 |
| 2c (iPr) | 1.1 × 10−6 | 175 |
| 2d (tBu) | 2.8 × 10−7 | 688 |
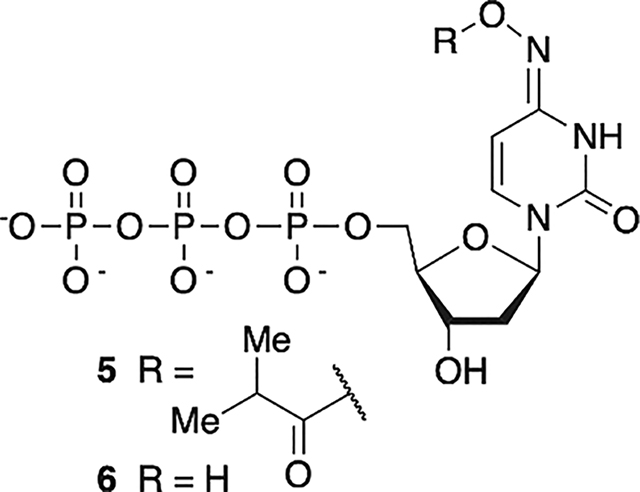
The corresponding nucleotide triphosphate of 2c (5) was chemically synthesized via standard methods.58 Following anion exchange purification, 5 was separated from the hydroxylamine hydrolysis product (6) by C18-reverse phase HPLC. In recognition that 5 readily tautomerizes such that it could present a cytosine (aminyl tautomer)- or thymine (iminyl tautomer)-like Watson-Crick face (Scheme 2), with the latter favored, incorporation of 5 opposite dG and dA by Klenow exo− was quantified under Michaelis–Menten conditions using primer-template complexes 7 and 8 (Table 2).59 The KM for 5 was only ~two-fold higher when the opposing nucleotide is dG (7) than when it is dA (8). The Vmax measurements also indicated that the polymerase modestly preferred 5 as a substrate when dA was the opposing nucleotide. Overall, Klenow exo− accepted 5 as a substrate ~three-fold more efficiently when dA was the opposing nucleotide in the template than when dG was. For comparison, the incorporation kinetics for corresponding cognate native dNTPs were also measured. Although the average Vmax for incorporation of dCTP opposite dG was only slightly greater than for 5 (albeit statistically within experimental error), the KM was almost 1000-times lower. Hence, the specificity constant for dCTP incorporation opposite dG was at least 1000-fold higher than that of 5. In contrast, incorporation kinetics of 5 and TTP opposite dA were much more competitive. TTP was only incorporated opposite dA ~21-fold more efficiently than 5. This was largely due to a smaller difference in KM’s, which was <29-fold higher for 5 incorporation opposite dA than for TTP.
Table 2.
Klenow exo− Incorporation Kinetics
| primer: template | dNTP | N’a | KM (μM)b | Vmax (% min−1)b | Vmax/KM (% min−1 (μM)b |
|---|---|---|---|---|---|
| 7 | 5 | dG | 27.8 ± 1.9 | 19.5 ± 2.1 | 0.7 |
| 7 | dCTP | dG | 0.03 ± 0.01 | 24.9 ± 4.7 | 830 |
| 8 | 5 | dA | 14.3 ± 0.3 | 30.7 ± 2.3 | 2.1 |
| 8 | TTP | dA | 0.5 ± 0.1 | 21.4 ± 6.6 | 42.8 |
N’ is the nucleotide in the DNA template across from which dNTP is added.
Values are the ave. ± std. dev. of three replicates.
Tandem Lesion Formation via dC· Generation from 2c in DNA.
Tandem lesions are defined as two contiguously damaged nucleotides and are a biologically significant hallmark of ionizing radiation.60,61 dC· produces tandem lesions when produced in 5′-d(GGTX) (X = dC·) sequences (Scheme 5), albeit inefficiently.42 Primer template complex 7 was designed such that 5 could be introduced into two such sequences; one resulting from incorporation opposite dG (5′-d(G12G13TX)) and the other opposite dA (5′-d(G16G17YY), Y = T, dC·). A third sequence lacking a thymidine between dG and dC· (5′-d(G20G21AX)) was engineered into the sequence as a negative control for tandem lesion formation. Two contiguous dG’s are present at each site to promote dG oxidation by reducing the ionization potential as a result of π-stacking.62 The primer strand of 7 was 32P-labeled, and 5′−32P-9 was prepared using Klenow exo−, 5, and native dNTPs. Radical precursor 5 (100 μM) is present at saturating concentration to maximize its competition with native dNTPs. Under these conditions, 5′−32P-9 is produced containing indeterminate levels of 2c at up to four positions (please note that for ease of identification, the numbering is the same as the nucleoside) distributed at the positions marked by X (opposite dG) and Y (opposite dA). Enzymatic synthesis of 5′−32P-9 is evident via 20% denaturing PAGE analysis of unphotolyzed, crude product (Figure 4, lane 1) in which full-length and n – 1 mer are detected.
Scheme 5.

Tandem Lesion Formation from dC·
Figure 4.
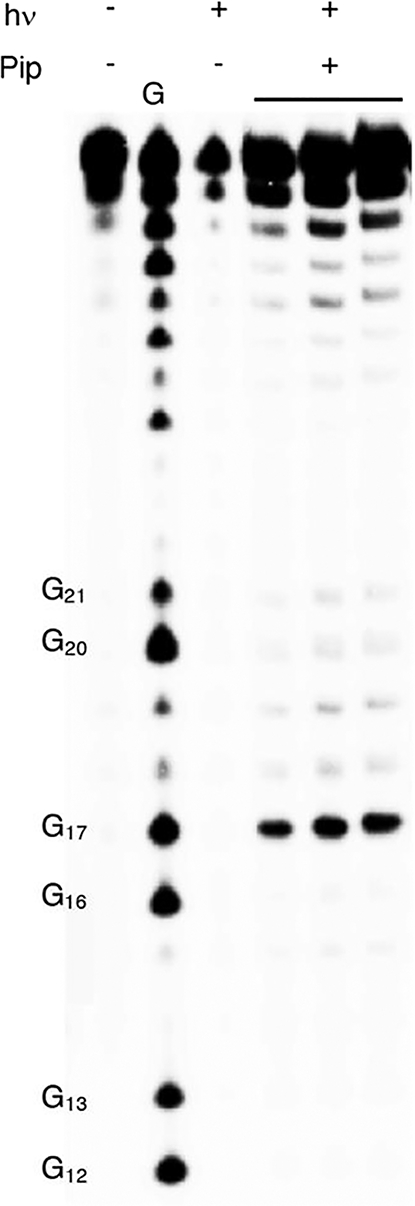
Alkali-labile lesion formation from photolysis of 5′−32P-9. The gel shows three independent replicates of photolyzed 5′−32P-9 treated with piperidine.
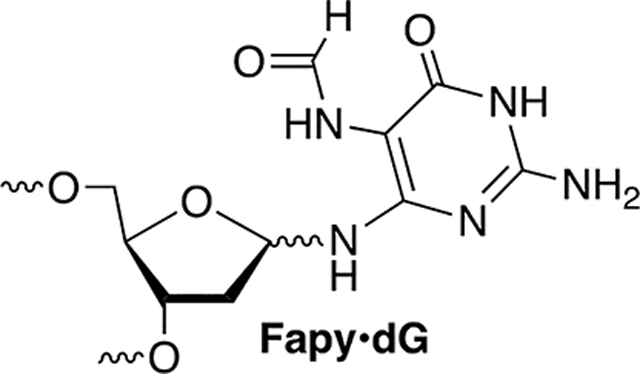
Following aerobic photolysis (350 nm, 3 h), alkali-labile strand damage was not detected at dG21, which is part of the 5′-d(GGAX) control sequence (Figure 4). This is consistent with the expectation that dC· does not react directly with an intrastrand 5′-dG and that damage is not relayed in the absence of a 5′-nucleotide containing a methyl group in the major groove (e.g., thymidine). Piperidine labile cleavage was detected at dG17, consistent with previous studies in which a nucleobase nitrogen radical generated in the major groove abstracts a hydrogen atom from the 5′-thymine (Scheme 5).17,42,63 Based on previous studies, we posit that O2 trapping of T· produces a peroxyl radical that adds to the 5′-adjacent guanine to produce a tandem lesion containing 8-OxodGuo and possibly Fapy·dG.17,64
Interestingly, strand damage was not detected in the region where tandem lesions would emanate from incorporation of 5 opposite dG despite the presence of 5′-adjacent nucleotides in 5′−32P-9 that are conducive to tandem lesion formation. Two explanations, both of which may contribute to the absence of strand damage at dG13, are envisioned. One possibility is that 5 is incorporated in much lower quantities opposite dG than opposite dA due to the greater difference in KM’s for dCTP and 7 (Table 2) opposite dG. In addition, tandem lesion formation when dC· is generated opposite dG could be much less efficient than when dA is the opposing nucleotide due to competing electron transfer to the opposing nucleotide (Scheme 6). Oxidation of an opposing dG is more thermodynamically favorable than dA.65,66 In addition, rapid intra-base pair proton transfer precludes any possibility of back electron transfer and has been shown to decrease the efficiency of DNA damage chemistry associated with dC·.31,42
Scheme 6.
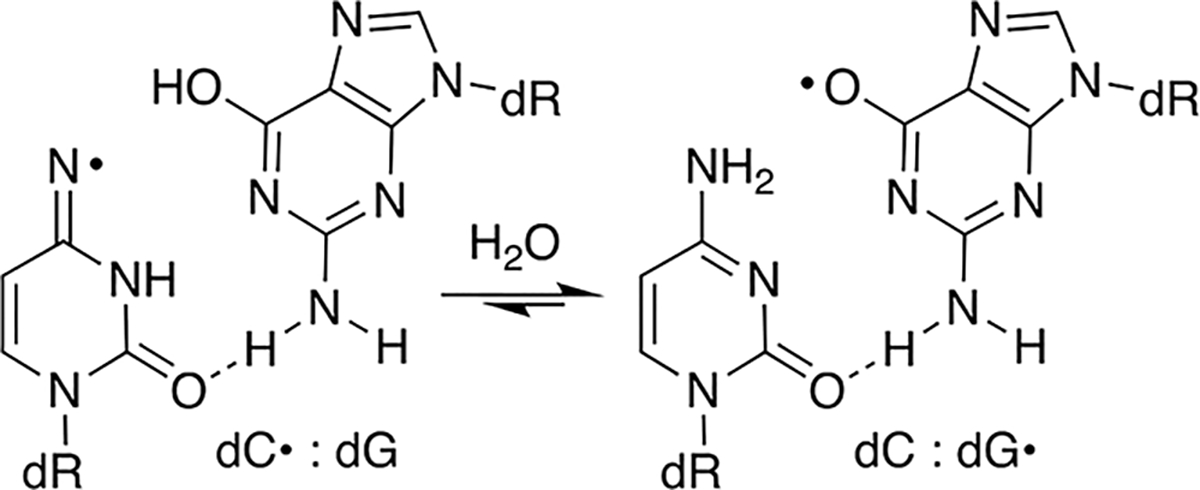
Intra-Base Pair Electron Transfer
SUMMARY
Oxime esters, particularly alkyl derivatives, provide high chemical yields of dC· independent of O2 under photolysis conditions (350 nm, <4 h) that do not damage DNA. dC· is also cleanly generated from oxime esters under single electron transfer conditions via DEA. The value of at least one of the alkyl oxime esters is increased further by the demonstration that the corresponding nucleotide triphosphate is accepted as a substrate by a DNA polymerase. The oxime is incorporated in DNA and produces tandem lesions when part of an appropriate sequence. Tandem lesions are a form of DNA damage associated with ionizing radiation that are often more difficult to repair than the isolated lesions they are comprised.60,61 The chemical and biochemical properties of the oxime esters indicate that they will be useful mechanistic tools for studying nucleic acid oxidation and potentially as radiosensitizing agents.
EXPERIMENTAL SECTION
General Methods.
THF was distilled over Na/benzophenone. MeCN, DCM, Et3N, and pyridine were distilled over CaH2. The other reagents were obtained from commercial sources and were directly used without further purification unless noted otherwise. All reactions were performed under a positive pressure of argon atmosphere. Klenow exo− and T4 polynucleotide kinase were purchased from New England Biolabs (NEB). γ−32P-ATP was purchased from Perkin Elmer. C18-Sep-Pak cartridges were obtained from Waters, and Poly-Prep columns from Bio-Rad. Nuclear magnetic resonance spectra were acquired on Bruker 400 MHz for 1H, 101 MHz for 13C, and 162 MHz for 31P. High-resolution ESI mass spectra for HRMS of synthesized molecules were recorded on a Waters Acquity/Xevo-G2 UPLC-MS system in positive mode. Photolyses were carried out in Pyrex tubes using a Rayonet photochemical reactor (Southern New England Ultraviolet) equipped with a merrygo-round apparatus and 16 lamps having a maximum output at 350 nm at 25 °C. The flux within the photoreactor was measured using a UVX radiometer and determined to be 1.91 mW/cm2 at 360 nm and 60 μW/cm2 at 310 nm. Temperature was maintained using the fan at the bottom of the unit provided by the manufacturer and a Dayton 239 CFM AC axial fan at the top facing such that air flowing through the unit is drawn out through the top.
Preparation of 3′,5′-O-Di(tert-butyldimethylsilyl)-2′-deoxyuridine 4-Oxime (3).
1,2,4-Triazole (1.52 g, 22.0 mmol) was suspended in MeCN at 0 °C for 5 min.43 Phosphoryl chloride (0.73 g, 4.75 mmol) was added dropwise into the reaction flask via syringe. The mixture was stirred at 0 °C for 10 min. Triethylamine (2.23 g, 22.0 mmol) was slowly added to the reaction flask at 0 °C (it is important to add the triethylamine slowly). The mixture was stirred for another 20 min at 0 °C. 3′,5′-O-Di(tert-butyldimethylsilyl)-2′-deoxyuridine (0.55 g, 1.21 mmol) was dissolved in MeCN (1 mL) and added to the reaction flask. After stirring for 3 h at room temperature, the reaction mixture was diluted with TEAA (pH 7, 50 mL). The resulting solution was partitioned in EtOAc (40 mL). The organic layer was washed with NaHCO3, NaCl, and dried over Na2SO4. After filtering, the solution was evaporated to dryness under vacuum. The crude residue was dissolved in pyridine (3 mL). NH2OH·HCl (0.13 g, 1.92 mmol) was added. The reaction was stirred at 25 °C overnight. The resulting solution was partitioned in EtOAc. The organic layer was washed with NaHCO3, NaCl, and dried over Na2SO4. The solvent was removed to yield 3 (0.36 g, 56%, 2 steps) as a white foam. TLC (EtOAc/hex: 1/2) Rf = 0.2. 1H NMR (DMSO-d6, 400 MHz) δ 10.00 (s, 1H), 9.53 (s, 1H), 6.92 (d, J = 8.3 Hz, 1H), 6.16–6.09 (m, 1H), 5.55–5.47 (m, 1H), 4.34 (dd, J = 5.9, 3.0 Hz, 1H), 3.16 (d, J = 5.3 Hz, 3H), 2.16–2.08 (m, 1H), 0.87 (d, J = 4.6 Hz, 18H), 0.13–0.01 (m, 12H).
Preparation of Methyl Oxime Ester (2a).
Hydroxylamine 3 (70.7 mg, 0.15 mmol), Et3N (20.3 mg, 0.2 mmol), and acetyl chloride (16.5 mg, 0.2 mmol) were added to DCM (1 mL).43 The reaction mixture was stirred at room temperature overnight. After the starting material disappeared, the reaction was passed through a short silica gel column (EtOAc in hexane, 1:2; 30 mL). The solvents were removed under reduced pressure. The residue was dissolved in THF (0.5 mL). Et3N· 3HF (75 mg, 0.45 mmol) was slowly added. The reaction mixture was stirred at room temperature overnight. The solvent was removed under vacuum. The residue was purified by flash chromatography on a silica column (MeOH in DCM, 3–10%) to yield 2a as a colorless oil (5.5 mg, 13%). 1H NMR (MeOH-d4, 400 MHz) δ 7.51(d, J = 8.2 Hz, 1H), 6.29 (t, J = 6.9 Hz, 1H), 5.72 (d, J = 8.2 Hz, 1H), 4.39 (m, 1 H), 3.91 (m, 1H), 3.74 (m, 2H), 2.23–2.20 (m, 2H), 2.20 (s, 3H).
Preparation of Ethyl Oxime Ester (2b).
Hydroxylamine 3 (472 mg, 1 mmol), propanoic acid (109 mg, 1.47 mmol), EDCI (223.8 mg, 1.5 mmol), and DMAP (12.2 mg, 0.1 mmol) were added to DCM (3 mL). The reaction mixture was stirred at room temperature overnight. After the starting material disappeared, the reaction mixture was washed with water (3 × 5 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was dissolved in THF (3 mL). Et3N·3HF (484 mg, 3 mmol) was slowly added. The reaction mixture was stirred at room temperature overnight. The solvent was removed under vacuum. The residue was purified by flash chromatography on a silica column (MeOH in DCM, 3–10%) to yield 2b as a white solid (140 mg, 50% yield two steps). 1H NMR (DMSO-d6, 400 MHz). Major tautomer: δ 10.83 (bd s, 1H), 7.40 (d, J = 8.2 Hz, 1H), 6.15 (t, J = 7 Hz, 1H), 5.72 (d, J = 8.2 Hz, 1H), 5.22 (d, J = 4.2 Hz, 1H), 4.97 (s, 1H), 4.21 (m, 1 H), 3.74 (m, 1H), 3.53 (m, 2H), 2.43 (m, 2H), 2.04 (m, 2H), 1.05 (t, J = 7.6 Hz, 3H). Minor tautomer: δ 11.13 (bd s, 1H), 7.47 (d, J = 8.2 Hz, 1H), 6.15 (t, J = 7 Hz, 1H), 6.02 (d, J = 8.2 Hz), 5.22 (d, J = 4.2 Hz, 1H), 4.97 (s, 1H), 4.21 (m, 1 H), 3.74 (m, 1H), 3.53 (m, 2H), 2.43 (m, 2H), 2.04 (m, 2H), 1.05 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (DMSO-d6, 101 MHz) δ 171.6, 155.6, 150.1, 149.3, 136.8, 134.5, 99.8, 92.1, 87.7, 87.6, 84.2, 84.1, 71.1, 71.0, 61.91, 61.86, 39.5, 26.0, 25.8, 9.5. HRMS (ESI TOF) m/z Calcd for (C12H17N3O6Na)+ (M + Na)+ = 322.1014, found m/z = 322.1010.
Preparation of Isopropyl Oxime Ester (2c).
Hydroxylamine 3 (473 mg, 1 mmol), Et3N (174 mg, 1.7 mmol), and isobutyryl chloride (163 mg, 1.5 mmol) were added to DCM (1.5 mL) and reacted as described for the preparation of 2a. The residue was dissolved in THF (3 mL). Et3N·3HF (484 mg, 3 mmol) was slowly added. The reaction was carried out as described for the preparation of 2a. The residue was purified by flash chromatography on a silica column (MeOH in DCM, 3–10%) to yield 2c as a white solid (163 mg, 53% yield two steps). UV–VIS (H2O) λmax = 274 nm, ε = 1.03 × 104 M−1 cm−1. 1H NMR (MeOH-d4, 400 MHz). Major tautomer: δ 7.50 (d, J = 8.3 Hz, 1H), 6.29 (t, J = 6.9 Hz, 1H), 5.72 (d, J = 8.3 Hz, 1H), 4.38 (m, 1 H), 3.90 (m, 1 H), 3.74 (m, 2H), 2.84 (hept, J = 6.9 Hz, 1H), 2.20 (m, 2H), 1.23 (d, J = 6.9 Hz, 6H). Minor tautomer: δ 7.57 (d, J = 8.3 Hz, 1H), 7.24 (d, J = 8.3 Hz, 1H), 6.29 (t, J = 6.9 Hz, 1H), 4.38 (m, 1 H), 3.90 (m, 1 H), 3.74 (m, 2H), 2.84 (hept, J = 6.9 Hz, 1H), 2.20 (m, 2H), 1.23 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (MeOH-d4, 101 MHz) δ 174.8, 149.8, 149.1, 134.4, 117.1, 96.1, 87.2, 84.6, 71.0, 61.7, 39.3, 32.3, 18.2, 18.0. UV (H2O:MeOH, 9:1) λmax = 274 nm, ε = 1.03 × 104 M−1 cm−1. HRMS (ESI TOF) m/z Calcd for (C14H21N3O6Na)+ (M + Na)+ = 350.1328, found m/z = 350.1314.
Preparation of t-Butyl Oxime Ester (2d).
Hydroxylamine 3 (70 mg, 0.15 mmol), Et3N (20.3 mg, 0.2 mmol), and trimethyl acetyl chloride (24.5 mg, 0.2 mmol) were added to DCM (1 mL). The reaction was carried out as described for the preparation of 2a. The crude bis-silylated oxime ester was dissolved in THF (0.5 mL), and Et3N·3HF (75 mg, 0.45 mmol) was slowly added. The reaction was carried out as described for the preparation of 2a. The residue was purified by flash chromatography on a silica column (MeOH in DCM, 3–10%) to yield 2d as a white solid (28.3 mg, 58% yield two steps). 1H NMR (MeOH-d4, 400 MHz). Major tautomer: δ 7.52 (d, J = 8.3 Hz, 1H), 6.32–6.23 (m, 1H), 5.75 (d, J = 8.3 Hz, 1H), 4.39 (m, 1H), 3.91 (m, 1H), 3.75 (m, 2H), 2.28–2.17 (m, 2H), 1.34 (s, 9H). Minor tautomer: δ 7.59 (d, J = 8.3 Hz, 1H), 6.32–6.23 (m, 1H), 6.09 (d, J = 8.3 Hz, 1H), 4.39 (m, 1H), 3.91 (m, 1H), 3.75 (m, 2H), 2.28–2.17 (m, 2H), 1.30 (s, 9H). 13C{1H} NMR (acetone-d6, 101 MHz) δ 174.8, 149.9, 149.4, 134.9, 97.4, 88.6, 85.5, 72.4, 63.0, 40.7, 39.4, 27.6. UV (H2O:MeOH, 9:1) λmax = 274 nm, ε = 1.17 × 104 M−1 cm−1. HRMS (ESI TOF) m/z Calcd for (C14H21N3O6Na)+ (M + Na)+ = 350.1328, found m/z = 350.1314.
Preparation of Phenyl Oxime Ester (2e).
Hydroxylamine 3 (141 mg, 0.3 mmol), Et3N (44 mg, 0.43 mmol), benzoyl chloride (60.5 mg, 0.43 mmol), and DMAP (5 mg, 0.03 mmol) were added to DCM (1 mL). The reaction was carried out as described for the preparation of 2a. The residue was dissolved in THF (0.7 mL), and Et3N·3HF (215 mg, 1.35 mmol) was slowly added. The reaction was carried out as described for the preparation of 2a. The residue was purified by flash chromatography on a silica column (MeOH in DCM, 3–10%) to yield 2e as a colorless solid (72.9 mg, 71% two steps). 1H NMR (MeOH-d4, 400 MHz). Major tautomer: δ 8.23–8.20 (m, 2H), 7.70–7.63 (m, 1H), 7.58–7.53 (m, 3H), 6.31 (t, J = 7.0 Hz, 1H), 5.82 (d, J = 8.2 Hz, 1H), 4.41 (m, 1H), 3.92 (m, 1H), 3.80–3.71 (m, 2H), 2.26–2.22 (m, 2H). Minor tautomer: δ 8.10–8.08 (m, 2H), 7.70–7.63 (m, 1H), 7.58–7.53 (m, 3H), 6.31 (t, J = 7.0 Hz, 1H), 5.82 (d, J = 8.2 Hz, 1H), 4.41 (m, 1H), 3.92 (m, 1H), 3.80–3.71 (m, 2H), 2.26–2.22 (m, 2H). 13C{1H} NMR (acetone-d6, 101 MHz) δ 164.3, 151.1, 149.5, 135.3, 134.0, 130.6, 130.3, 129.3, 97.2, 88.53, 85.51, 72.3, 63.0, 40.7. UV (H2O:MeOH, 9:1) λmax = 282 nm, ε = 1.72 × 104 M−1 cm−1. HRMS (ESI-TOF) m/z Calcd for (C16H17N3O6Na)+ (M + Na)+ = 370.1015, found m/z = 370.1013.
Preparation of Oxime Ester Nucleotide Triphosphate (5).
Trimethyl phosphate (1 mL) was added to a mixture of 2c (37.6 mg, 0.12 mmol) and proton sponge (41 mg, 0.2 mmol). The resulting solution was stirred on ice for 10 min. Phosphoryl chloride (27 mg, 0.2 mmol) was added to the reaction flask, and stirring was continued at 0 °C for 1 h. A prechilled cocktail containing tributylammonium pyrophosphate 0.2 M in DMF (3.6 mL, 0.72 mmol) and tributylamine (132 mg, 0.72 mmol) was added to the reaction mixture, and stirring was continued for 30 min at 0 °C. The reaction was quenched by slow addition of TEAB (pH 8, 12 drops) and water (5 mL). The aqueous layer was extracted with EtOAc (6 × 5 mL) and concentrated under reduced pressure. The resulting crude product was redissolved in water (3 mL) and purified by Mono Q 5/50 GC column (5 × 50 mm) at 5 °C using a gradient of 0.1 to 1.0 M TEAB (pH 8; flow rate, 1 mL/min). The desired fractions (based on retention time of dCTP, tR = 15 min) were collected and lyophilized overnight. The residue was further purified by reversed-phase HPLC while being monitored at 260 nm and 284 nm. HPLC was performed on a Phenomenex Luna C-18 column (A, 10 mM TEAA; B, ACN; 3% B from t = 0 to t = 2 min; 3–40% B linearly over 38 min; 40–3% B linearly over 2 min; 3% B from t = 42 to t = 45 min; flow rate, 1 mL/min). After the solvent was removed under vacuum, 5 was obtained as a white foam (1.6 mg, 3%). UV–VIS (H2O) λmax = 274 nm. Due to the low mass of 5, the extinction coefficient was assumed to be the same as 2c. 31P NMR (162 MHz, D2O) δ −6.55 (d, J = 15.7 Hz), −11.27 (d, J = 20.6 Hz), −22.56 (t, J = 20.6 Hz). ESI-MS m/z Calcd for (C13H21N3O15P3)− (M-H)− = 552.02, found m/z = 552.09.
Nucleoside Photolysis Experiments.
Reaction mixtures (50 μL) containing precursor (100 μM), internal standard (thymidine, 100 μM), and additives (e.g., reducing agents and organic solvent) in buffer (10 mM phosphate, 100 mM NaCl, pH 7.2) were photolyzed at room temperature under aerobic or anaerobic conditions. Samples for anaerobic reactions were degassed by three freeze-pump-thaw cycles at 2 mTorr and flame sealed under vacuum. The reaction mixtures (including unphotolyzed controls) were analyzed by C18-reverse phase HPLC while being monitored at 260 and 284 nm. HPLC was performed on a Phenomenex Luna C-18 column (4.6 × 250 mm) (A, 10 mM ammonium formate; B, ACN; 3% B from t = 0 to t = 1 min; 3–28% B linearly over 9 min; 28–97% B linearly over 5 min; 97% B from t = 25 to t = 20 min; 97–3% B linearly over 2 min; 3% B from t = 22 to t = 40 min; flow rate, 1 mL/min). Response factors (Rf) for compound 2c–e and dC (“X”) versus thymidine was calculated using the following formula: ([X]/[dT]) = Rf(A(X)/A(dT)), where [X] is the concentration of compound X and [dT] is the concentration of dT. A(X) and A(dT) are the areas under the peaks corresponding to X versus dT. The peaks were integrated and quantified against the internal standard (thymidine). The measured response factors are: dC: 1.56, 2c: 1.24, 2d: 1.67, and 2e: 1.08.
Oxime Ester Hydrolysis in PBS.
Reaction mixtures (50 μL) containing precursor 2b–d (100 μM) and internal standard (thymidine, 100 μM) in buffer (10 mM phosphate, 100 mM NaCl, pH 7.2) were incubated at room temperature. Aliquots were removed at various times and analyzed by C18-reverse phase HPLC as described above. The disappearance of 2b–d was fit to first-order kinetics.
ESR Spectroscopy.
Glassy Sample Preparation, γ-Irradiation, and Storage. Preparation of Homogeneous Solutions:
Homogeneous solution was prepared by dissolving ca. 2–2.5 mg/mL of either 2d or 2e in 7.5 M LiCl in D2O. The native pH of 7.5 M LiCl in D2O is ca. 5, and pH of these solutions was not adjusted.18,27,47,53
Preparation of Glassy Samples:
Following previous reports,18,27,47,53 the oxime ester solutions were thoroughly sparged with nitrogen. The sparged solutions were immediately drawn into Suprasil quartz tubes (4 mm diameter, catalog no. 734-PQ-8, WILMAD Glass Co., Inc., Buena, NJ, USA) and immersed in liquid nitrogen (77 K). Owing to rapid cooling at 77 K, the homogeneous liquid solutions formed transparent homogeneous glassy solutions.
γ-Irradiation and Storage of These Samples:
Following previous reports,18,27,47,53 the transparent glassy solutions were γ-irradiated (absorbed dose = 600 Gy) at 77 K to generate samples for ESR spectral studies. All glassy samples were stored in the dark at 77 K in Teflon containers prior to and after γ-irradiation.
ESR Spectroscopic Studies.
Following previous studies,18,27,47,53 a Varian Century Series X-band (9.3 GHz) ESR spectrometer with an E-4531 dual cavity, 9-inch magnet, and a 200 mW Klystron was used to carry out the ESR spectroscopic studies. For field calibration, Fremy’s salt (gcenter = 2.0056, A(N) = 13.09 G) was employed. All ESR spectra were recorded at 77 K and at 45 dB (6.3 μW). We note here that recording of ESR spectra at 77 K maximizes the signal height and allows for comparison of signal intensities.8,27,47,53
Theoretical Calculations.
Calculations were performed using DFT and employing Uωb97xd functional, 6–31++G** basis set in the aqueous phase employing the integral equation formalism of the polarized continuum model (IEF-PCM)67 Gaussian 16 suite of programs68 and using the optimized geometries of the compound and the radicals applying the same method.
32P-Radiolabeling and Single Nucleotide Incorporation Kinetics.
The primer strand (50 pmol) was labeled at the 5′-terminus with γ−32P-ATP using T4 PNK (30 units) in T4 PNK buffer (70 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 5 mM DTT, 2 h, 37 °C). The labeled primer was hybridized to the template (1.5 equiv) in PBS by heating at 90 °C for 5 min and slowly cooling to room temperature in a thermal cycler to provide 5′−32P-7 or 5′−32P-8. The primer-template was diluted into a 2 X cocktail containing Klenow exo-, Klenow exoreaction buffer (20 mM Tris–HCl, pH 7.9, 20 mM MgCl2, 2 mM DTT, 100 mM NaCl), and bovine serum albumin (BSA, 0.2 mg/mL). A reaction mixture (10 μL) was prepared by mixing 2X cocktail (5 μL) and 2X substrate (dNTP) solutions at different concentrations (5 μL) for various reaction times. After incubation at 37 °C, the reaction mixture was quenched with 5 mM EDTA in 95% formamide (10 μL), heated at 90 °C for 1 min, incubated on ice for 1 min, and loaded on a 20% denaturing PAGE gel. Velocities were determined as previously described and fit using the Michaelis–Menten equation.59 Note: enzymes were diluted and stored as aliquots (1 μM, 2 μL) in 30% glycerol and reaction buffer (1×) to ensure consistent concentration in various experiments.
Preparation and Photolysis of 5′−32P-9.
A solution (20 μL) containing DNA primer-template 5′−32P-7 (200 nM), Klenow exo− (20 nM), Klenow exo-reaction buffer (10 mM Tris–HCl, pH 7.9, 10 mM MgCl2, 1 mM DTT, 100 mM NaCl), BSA (0.1 mg/mL), 5 (100 μM), and d(A,T,G)TP (25 μM each) was incubated at 37 °C for 2 h. The solution was diluted to 50 nM 5′−32P-9 in PBS buffer (100 mM NaCl, 10 mM sodium phosphate, pH 7.2) before photolysis. All photolyses were carried out in Pyrex tubes for 3 h. Aliquots (10 μL) from photolyzed solutions or unphotolyzed controls were treated with piperidine (2 M, 10 μL) for 30 min at 90 °C. Piperidine treated samples were evaporated to dryness under vacuum and washed with water (2 × 10 μL), which was also removed under vacuum. Samples were analyzed by dissolving/resuspending in formamide loading buffer prior to analyzing by 20% denaturing PAGE.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for support from the National Institute of General Medical Sciences (GM-131736) to M.M.G., S.W., A.K., M.D.S., and A.A. thank the National Cancer Institute of the National Institutes of Health (Grant No. RO1CA045424) for support. A.A. thanks the National Science Foundation under Grant No. CHE-1920110.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.3c00646.
NMR spectra of 2a–e, 3, and 5, DFT calculations, and complete citation for ref 68 (PDF)
Contributor Information
Haihui Peng, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States; Present Address: State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China (H.P.).
Son Vu, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Parker Retes, Department of Chemistry, Oakland University, Rochester, Michigan 48309, United States.
Samuel Ward, Department of Chemistry, Oakland University, Rochester, Michigan 48309, United States.
Anil Kumar, Department of Chemistry, Oakland University, Rochester, Michigan 48309, United States.
Michael D. Sevilla, Department of Chemistry, Oakland University, Rochester, Michigan 48309, United States
Amitava Adhikary, Department of Chemistry, Oakland University, Rochester, Michigan 48309, United States.
Marc M. Greenberg, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- (1).von Sonntag C Free-Radical-Induced DNA Damage and Its Repair; Springer-Verlag: Berlin, 2006. [Google Scholar]
- (2).Pogozelski WK; McNeese TJ; Tullius TD What Species Is Responsible for Strand Scission in the Reaction of [Fe(2)EDTA]2− and H2O2 with DNA? J. Am. Chem. Soc. 1995, 117, 6428–6433. [Google Scholar]
- (3).Ingle SS; Azad RN; Jain SS; Tullius TD Chemical Probing of RNA with the Hydroxyl Radical at Single-Atom Resolution. Nucleic Acids Res. 2014, 42, 12758–12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Price MA; Tullius TD Using Hydroxyl Radical to Probe DNA Structure. Methods Enzymol. 1992, 212, 194–219. [DOI] [PubMed] [Google Scholar]
- (5).Hayes JJ; Tullius TD; Wolffe AP The Structure of DNA in a Nucleosome. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 7405–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dervan PB Design of Sequence-Specific DNA-Binding Molecules. Science 1986, 232, 464–471. [DOI] [PubMed] [Google Scholar]
- (7).Hao Y; Bohon J; Hulscher R; Rappe MC; Gupta S; Adilakshmi T; Woodson SA Time-Resolved Hydroxyl Radical Footprinting of Rna with X-Rays. Curr. Protoc. Nucleic Acid Chem. 2018, 73, No. e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hulscher RM; Bohon J; Rappe MC; Gupta S; D’Mello R; Sullivan M; Ralston CY; Chance MR; Woodson SA Probing the Structure of Ribosome Assembly Intermediates in Vivo Using Dms and Hydroxyl Radical Footprinting. Methods 2016, 103, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Greenberg MM Reactivity of Nucleic Acid Radicals. Adv. Phys. Org. Chem. 2016, 50, 119–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Giese B Long-Distance Electron Transfer through DNA. Annu. Rev. Biochem. 2002, 71, 51–70. [DOI] [PubMed] [Google Scholar]
- (11).Giese B Long-Distance Charge Transport in DNA: The Hopping Mechanism. Acc. Chem. Res. 2000, 33, 631–636. [DOI] [PubMed] [Google Scholar]
- (12).Meggers E; Michel-Beyerle ME; Giese B Sequence Dependent Long Range Hole Transport in DNA. J. Am. Chem. Soc. 1998, 120, 12950–12955. [Google Scholar]
- (13).Meggers E; Kusch D; Spichty M; Wille U; Giese B Electron Transfer through DNA in the Course of Radical-Induced Strand Cleavage. Angew. Chem., Int. Ed. 1998, 37, 460–462. [DOI] [PubMed] [Google Scholar]
- (14).Paul R; Greenberg MM Rapid Rna Strand Scission Following C2′-Hydrogen Atom Abstraction. J. Am. Chem. Soc. 2015, 137, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zheng L; Dai X; Su H; Greenberg MM Independent Generation and Time-Resolved Detection of 2′-Deoxyguanosin-N2-Yl Radicals. Angew. Chem., Int. Ed. 2020, 59, 13406–13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zheng L; Griesser M; Pratt DA; Greenberg MM Aminyl Radical Generation Via Tandem Norrish Type I Photocleavage, Β-Fragmentation: Independent Generation and Reactivity of the 2′-Deoxyadenosin- N6-Yl Radical. J. Org. Chem. 2017, 82, 3571–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zheng L; Greenberg MM Traceless Tandem Lesion Formation in DNA from a Nitrogen-Centered Purine Radical. J. Am. Chem. Soc. 2018, 140, 6400–6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mudgal M; Rishi S; Lumpuy DA; Curran KA; Verley KL; Sobczak AJ; Dang TP; Sulimoff N; Kumar A; Sevilla MD; Wnuk SF; Adhikary A Prehydrated One-Electron Attachment to Azido-Modified Pentofuranoses: Aminyl Radical Formation, Rapid H-Atom Transfer, and Subsequent Ring Opening. J. Phys. Chem. B 2017, 121, 4968–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dextraze M-E; Gantchev T; Girouard S; Hunting D DNA Interstrand Cross-Links Induced by Ionizing Radiation: An Unsung Lesion. Mutat. Res., Rev. Mutat. Res. 2010, 704, 101–107. [DOI] [PubMed] [Google Scholar]
- (20).Ito T; Rokita SE Excess Electron Transfer from an Internally Conjugated Aromatic Amine to 5-Bromo-2′-Deoxyuridine in DNA. J. Am. Chem. Soc. 2003, 125, 11480–11481. [DOI] [PubMed] [Google Scholar]
- (21).Hong H; Wang Y Formation of Intrastrand Cross-Link Products between Cytosine and Adenine from UV Irradiation of D(Brca) and Duplex DNA Containing a 5-Bromocytosine. J. Am. Chem. Soc. 2005, 127, 13969–13977. [DOI] [PubMed] [Google Scholar]
- (22).Zeng Y; Wang Y Facile Formation of an Intrastrand Cross-Link Lesion between Cytosine and Guanine Upon Pyrex-Filtered UV Light Irradiation of D(Brcg) and Duplex DNA Containing 5-Bromocytosine. J. Am. Chem. Soc. 2004, 126, 6552–6553. [DOI] [PubMed] [Google Scholar]
- (23).Sannohe Y; Kizaki S; Kanesato S; Fujiwara A; Li Y; Morinaga H; Tashiro R; Sugiyama H Controlling Electron Rebound within Four-Base π-Stacks in Z-DNA by Changing the Sugar Moiety from Deoxy- to Ribonucleotide. Chem. – Eur. J. 2014, 20, 1223–1225. [DOI] [PubMed] [Google Scholar]
- (24).Morinaga H; Kizaki S; Takenaka T; Sannohe Y; Kanesato S; Sugiyama H Photoreaction of 5-Bromouracil in RNA. Photomed. Photobiol. 2012, 34, 13–14. [Google Scholar]
- (25).Watanabe T; Tashiro R; Sugiyama H Photoreaction at 5′-(G/C)Aabr Ut-3′ Sequence in Duplex DNA: Efficient Generation of Uracil-5-Yl Radical by Charge Transfer. J. Am. Chem. Soc. 2007, 129, 8163–8168. [DOI] [PubMed] [Google Scholar]
- (26).Chen T; Cook GP; Koppisch AT; Greenberg MM Investigation of the Origin of the Sequence Selectivity for the 5-Halo-2′-Deoxyuridine Sensitization of DNA to Damage by UV-Irradiation. J. Am. Chem. Soc. 2000, 122, 3861–3866. [Google Scholar]
- (27).Wen Z; Peng J; Tuttle PR; Ren Y; Garcia C; Debnath D; Rishi S; Hanson C; Ward S; Kumar A; Liu Y; Zhao W; Glazer PM; Liu Y; Sevilla MD; Adhikary A; Wnuk SF Electron-Mediated Aminyl and Iminyl Radicals from C5 Azido-Modified Pyrimidine Nucleosides Augment Radiation Damage to Cancer Cells. Org. Lett. 2018, 20, 7400–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rudra A; Hou D; Zhang Y; Coulter J; Zhou H; DeWeese TL; Greenberg MM Bromopyridone Nucleotide Analogues, Anoxic Selective Radiosensitizing Agents That Are Incorporated in DNA by Polymerases. J. Org. Chem. 2015, 80, 10675–10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ma J; Bahry T; Denisov SA; Adhikary A; Mostafavi M Quasi-Free Electron-Mediated Radiation Sensitization by C5-Halopyrimidines. J. Phys. Chem. A 2021, 125, 7967–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang Y; Zhao H; Yang C; Jie J; Dai X; Zhou Q; Liu K; Song D; Su H Degradation of Cytosine Radical Cations in 2′-Deoxycytidine and in I-Motif DNA: Hydrogen-Bonding Guided Pathways. J. Am. Chem. Soc. 2019, 141, 1970–1979. [DOI] [PubMed] [Google Scholar]
- (31).Anderson RF; Shinde SS; Maroz A Cytosine-Gated Hole Creation and Transfer in DNA in Aqueous Solution. J. Am. Chem. Soc. 2006, 128, 15966–15967. [DOI] [PubMed] [Google Scholar]
- (32).Shao F; O’Neill MA; Barton JK Long-Range Oxidative Damage to Cytosines in Duplex DNA. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17914–17919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hawkins CL; Davies MJ Hypochlorite-Induced Damage to DNA, RNA, and Polynucleotides: Formation of Chloramines and Nitrogen-Centered Radicals. Chem. Res. Toxicol. 2002, 15, 83–92. [DOI] [PubMed] [Google Scholar]
- (34).Singh J; Nelson TJ; Mansfield SA; Nickel GA; Cai Y; Jones DD; Small JE; Ess DH; Castle SL Microwave- and Thermally Promoted Iminyl Radical Cyclizations: A Versatile Method for the Synthesis of Functionalized Pyrrolines. J. Org. Chem. 2022, 87, 16250–16262. [DOI] [PubMed] [Google Scholar]
- (35).Blake JA; Pratt DA; Lin S; Walton JC; Mulder P; Ingold KU Thermolyses of O-Phenyl Oxime Ethers. A New Source of Iminyl Radicals and a New Source of Aryloxyl Radicals. J. Org. Chem. 2004, 69, 3112–3120. [DOI] [PubMed] [Google Scholar]
- (36).Walton JC The Oxime Portmanteau Motif: Released Heteroradicals Undergo Incisive EPR Interrogation and Deliver Diverse Heterocycles. Acc. Chem. Res. 2014, 47, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).McCarroll AJ; Walton JC Exploitation of Aldoxime Esters as Radical Precursors in Preparative and EPR Spectroscopic Roles. J. Chem. Soc., Perkin Trans. 2 2000, 2399–2409. [Google Scholar]
- (38).Yu X-Y; Zhao Q-Q; Chen J; Xiao W-J; Chen J-R When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [DOI] [PubMed] [Google Scholar]
- (39).Morcillo SP Radical-Promoted C–C Bond Cleavage: A Deconstructive Approach for Selective Functionalization. Angew. Chem., Int. Ed. 2019, 58, 14044–14054. [DOI] [PubMed] [Google Scholar]
- (40).Shen X; Zhao J-J; Yu S Photoredox-Catalyzed Intermolecular Remote C–H and C–C Vinylation Via Iminyl Radicals. Org. Lett. 2018, 20, 5523–5527. [DOI] [PubMed] [Google Scholar]
- (41).Zhao J-F; Duan X-H; Gu Y-R; Gao P; Guo L-N Iron-Catalyzed Decarboxylative Olefination of Cycloketone Oxime Esters with Α,Β-Unsaturated Carboxylic Acids Via C–C Bond Cleavage. Org. Lett. 2018, 20, 4614–4617. [DOI] [PubMed] [Google Scholar]
- (42).Peng H; Jie J; Mortimer IP; Ma Z; Su H; Greenberg MM Reactivity and DNA Damage by Independently Generated 2′-Deoxycytidin-N4-yl Radical. J. Am. Chem. Soc. 2021, 143, 14738–14747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Münzel M; Szeibert C; Glas AF; Globisch D; Carell T Discovery and Synthesis of New UV-Induced Intrastrand C(4–8)G and G(8–4)C Photolesions. J. Am. Chem. Soc. 2011, 133, 5186–5189. [DOI] [PubMed] [Google Scholar]
- (44).Stolarski; Kierdaszuk B; Hagberg CE; Shugar D Hydroxylamine and Methoxyamine Mutagenesis: Displacement of the Tautomeric Equilibrium of the Promutagen N6-Methoxyadenosine by Complementary Base Pairing. Biochemistry 1984, 23, 2906–2913. [DOI] [PubMed] [Google Scholar]
- (45).Al-Sheikhly M The Reactivity of Adenyl and Guanyl Radicals Towards Oxygen. Radiat. Phys. Chem. 1994, 44, 297–301. [Google Scholar]
- (46).Huang Y; Kenttämaa H Theoretical and Experimental Investigations on the Chemical Reactions of Positively Charged Phenyl Radicals with Cytosine and 1-Methylcytosine. J. Am. Chem. Soc. 2003, 125, 9878–9889. [DOI] [PubMed] [Google Scholar]
- (47).Adhikary A; Becker D; Collins S; Koppen J; Sevilla MD C5′- and C3′-Sugar Radicals Produced Via Photo-Excitation of One-Electron Oxidized Adenine in 2′-Deoxyadenosine and Its Derivatives. Nucleic Acids Res. 2006, 34, 1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Evans ME; Li T; Vetter AJ; Rieth RD; Jones WD Thermodynamic Trends in Carbon—Hydrogen Bond Activation in Nitriles and Chloroalkanes at Rhodium. J. Org. Chem. 2009, 74, 6907–6914. [DOI] [PubMed] [Google Scholar]
- (49).Tóth P; Szűcs T; Czakó G. Benchmark Ab Initio Characterization of the Abstraction and Substitution Pathways of the Cl + CH3CN Reaction. J. Phys. Chem. A 2022, 126, 2802–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cruickshank FR; Benson SW Carbon-Hydrogen Bond Dissociation Energy in Methanol. J. Phys. Chem. 1969, 73, 733. [Google Scholar]
- (51).Ruscic B Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J. Phys. Chem. A 2015, 119, 7810–7837. [DOI] [PubMed] [Google Scholar]
- (52).Li M-J; Liu L; Wei K; Fu Y; Guo Q-X Significant Effects of Phosphorylation on Relative Stabilities of DNA and RNA Sugar Radicals: Remarkably High Susceptibility of H-2′ Abstraction in RNA. J. Phys. Chem. B 2006, 110, 13582–13589. [DOI] [PubMed] [Google Scholar]
- (53).Adhikary A; Kumar A; Bishop CT; Wiegand TJ; Hindi RM; Adhikary A; Sevilla MD π-Radical to σ-Radical Tautomerization in One-Electron-Oxidized 1-Methylcytosine and Its Analogs. J. Phys. Chem. B 2015, 119, 11496–11505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shahsavari S; Eriyagama DNAM; Chen J; Halami B; Yin Y; Chillar K; Fang S Sensitive Oligodeoxynucleotide Synthesis Using Dim and Dmoc as Protecting Groups. J. Org. Chem. 2019, 84, 13374–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Venkatesan H; Greenberg MM Improved Utility of Photolabile Solid Phase Synthesis Supports for the Synthesis of Oligonucleotides Containing 3′-Hydroxyl Termini. J. Org. Chem. 1996, 61, 525–529. [DOI] [PubMed] [Google Scholar]
- (56).Jakubovska J; Tauraitė D; Birštonas L; Meškys R N4-Acyl-2′-Deoxycytidine-5′-Triphosphates for the Enzymatic Synthesis of Modified DNA. Nucleic Acids Res. 2018, 46, 5911–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Jakubovska J; Tauraitė D; Meškys R Transient N4-Acyl-DNA Protection against Cleavage by Restriction Endonucleases. ChemBioChem 2019, 20, 2504–2512. [DOI] [PubMed] [Google Scholar]
- (58).Kore AR; Shanmugasundaram M; Senthilvelan A; Srinivasan B An Improved Protection-Free One-Pot Chemical Synthesis of 2′-Deoxynucleoside-5′-Triphosphates. Nucleosides, Nucleotides Nucleic Acids 2012, 31, 423–431. [DOI] [PubMed] [Google Scholar]
- (59).Petruska J; Goodman MF; Boosalis MS; Sowers LC; Cheong C; Tinoco I Jr. Comparison between DNA Melting Thermodynamics and DNA Polymerase Fidelity. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 6252–6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Patrzyc HB; Dawidzik JB; Budzinski EE; Freund HG; Wilton JH; Box HC Covalently Linked Tandem Lesions in DNA. Radiat. Res. 2012, 178, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sage E; Harrison L Clustered DNA Lesion Repair in Eukaryotes: Relevance to Mutagenesis and Cell Survival. Mutat. Res. 2011, 711, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Sugiyama H; Saito I Theoretical Studies of GG-Specific Photocleavage of DNA Via Electron Transfer: Significant Lowering of Ionization Potential and 5′-Localization of Homo of Stacked Gg Bases in B-Form DNA. J. Am. Chem. Soc. 1996, 118, 7063–7068. [Google Scholar]
- (63).Zheng L; Greenberg MM DNA Damage Emanating from a Neutral Purine Radical Reveals the Sequence Dependent Convergence of the Direct and Indirect Effects of Γ-Radiolysis. J. Am. Chem. Soc. 2017, 139, 17751–17754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Robert G; Wagner JR Tandem Lesions Arising from 5-(Uracilyl)Methyl Peroxyl Radical Addition to Guanine: Product Analysis and Mechanistic Studies. Chem. Res. Toxicol. 2020, 33, 565–575. [DOI] [PubMed] [Google Scholar]
- (65).Steenken S; Jovanovic SV How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar]
- (66).Psciuk BT; Lord RL; Munk BH; Schlegel HB Theoretical Determination of One-Electron Oxidation Potentials for Nucleic Acid Bases. J. Chem. Theory Comput. 2012, 8, 5107–5123. [DOI] [PubMed] [Google Scholar]
- (67).Tomasi J; Mennucci B; Cammi R Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [DOI] [PubMed] [Google Scholar]
- (68).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; et al. Gaussian 16, Revision A.03; Gaussian.Com; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.