

Abstract
Hsp70 molecular chaperones play central roles in maintaining a healthy cellular proteome. Hsp70s function by binding to short peptide sequences in incompletely folded client proteins, thus preventing them from misfolding and/or aggregating, and in many cases holding them in a state that is competent for subsequent processes like translocation across membranes. There is considerable interest in predicting the sites where Hsp70s may bind their clients, as the ability to do so sheds light on the cellular functions of the chaperone. In addition, the capacity of the Hsp70 chaperone family to bind to a broad array of clients and to identify accessible sequences that enable discrimination of those that are folded from those that are not fully folded, which is essential to their cellular roles, is a fascinating puzzle in molecular recognition. In this review we discuss efforts to harness computational modeling with input from experimental data to develop predictive understanding of the promiscuous yet selective binding of Hsp70 molecular chaperones to accessible sequences within their client proteins. We trace how an increasing understanding of the complexities of Hsp70-client interaction has led computational modeling to new underlying assumptions and design features. We describe the trend from purely data-driven analysis towards increased reliance on physics-based modeling that deeply integrates structural information and sequence-based functional data with physics-based binding energies. Notably, new experimental insights are adding to our understanding of the molecular origins of “selective promiscuity” in substrate binding by Hsp70 chaperones and challenging the underlying assumptions and design used in earlier predictive models. Taking the new experimental findings together with exciting progress in computational modeling of protein structures leads us to foresee a bright future for predictive understanding of selective-yet-promiscuous binding exploited by Hsp70 molecular chaperones; the resulting new insights will also apply to substrate binding by other chaperones and by signaling proteins.
Graphical Abstract
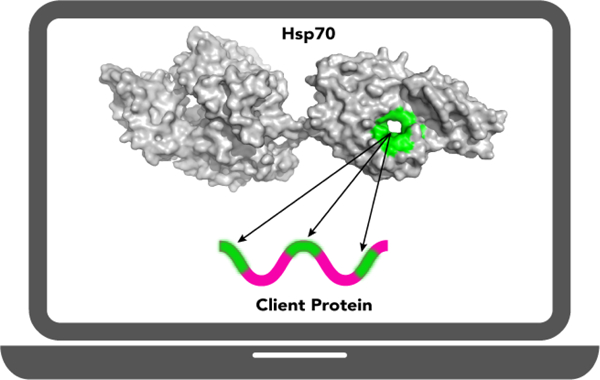
Introduction
Computational modeling of protein folding has played a central role in the evolution of our understanding of this complex and biologically central physicochemical process. From the early days, the research from Harold Scheraga and his coworkers illustrated the power of synergistic and complementary use of experimental data and theoretical approaches to modeling of protein folding1,2. Force fields were parameterized and validated using experimental data, and efforts to describe energy landscapes relied heavily on experimental studies. All the major advances that emerged from this seminal work illustrated that our understanding and predictive ability for protein folding mechanisms would greatly benefit from integration of experiment and theory.
In the decades since the foundational studies of protein folding, we have learned that protein folding in the cell is facilitated by a multicomponent system of molecular chaperones3–5. These players do not alter the sequence-encoded information for protein folding and structure, but they diminish the likelihood of off pathway outcomes such as protein aggregation and misfolded product accumulation. One of the most stunning properties of the protein homeostasis network of chaperones in cells is its ability to favor successful folding of the entire proteome. This ability requires the same chaperone to interact with many different substrates (also called ‘clients’). Our laboratories have focused on the Hsp70 family6–8 of chaperones to better understand what we call “selective promiscuity”9,10, or the ability to interact with many different clients and yet to discriminate proteins that have folded to their native structures and no longer need assistance from those that are incompletely folded.
Hsp70 molecular chaperones play many roles in cells.6–8,10 They greet nascent chains as they emerge from ribosomes and facilitate their folding or hand-off to downstream chaperones. They help to keep proteins from prematurely folding if their eventual destination requires them to traverse a membrane.11–14 They serve as decision points for proteins that may successfully fold,9,15–18 or in some cases, may fail and must be degraded.19–21 They work with other chaperones to disaggregate proteins22–25 and may inhibit aggregation in prior stages, and they help assembly and disassembly of complexes.26–29 These functions rely on the ability of Hsp70 chaperones to bind short extended segments of sequence in their clients, and in an ATP-mediated allosteric step, to release these segments after some dwell time. This allosteric cycle is modulated by co-chaperones: J-proteins30–34, which activate the ATPase activity of Hsp70 to favor its return to a high affinity state, and nucleotide-exchange factors35–37, which catalyze the exchange of ADP for ATP. Binding to the client protein is mediated by the substrate-binding domain (SBD). A cleft is formed by a β-sandwich subdomain in this domain (βSBD), and the binding site is covered by an α-helical lid (Figure 1A). When ATP binds to the adjacent N-terminal nucleotide-binding domain, the helical lid lifts, and the cleft is remodeled to a weaker binding conformation.7,8,38
Figure 1. Structural features of the E. coli Hsp70 (DnaK).
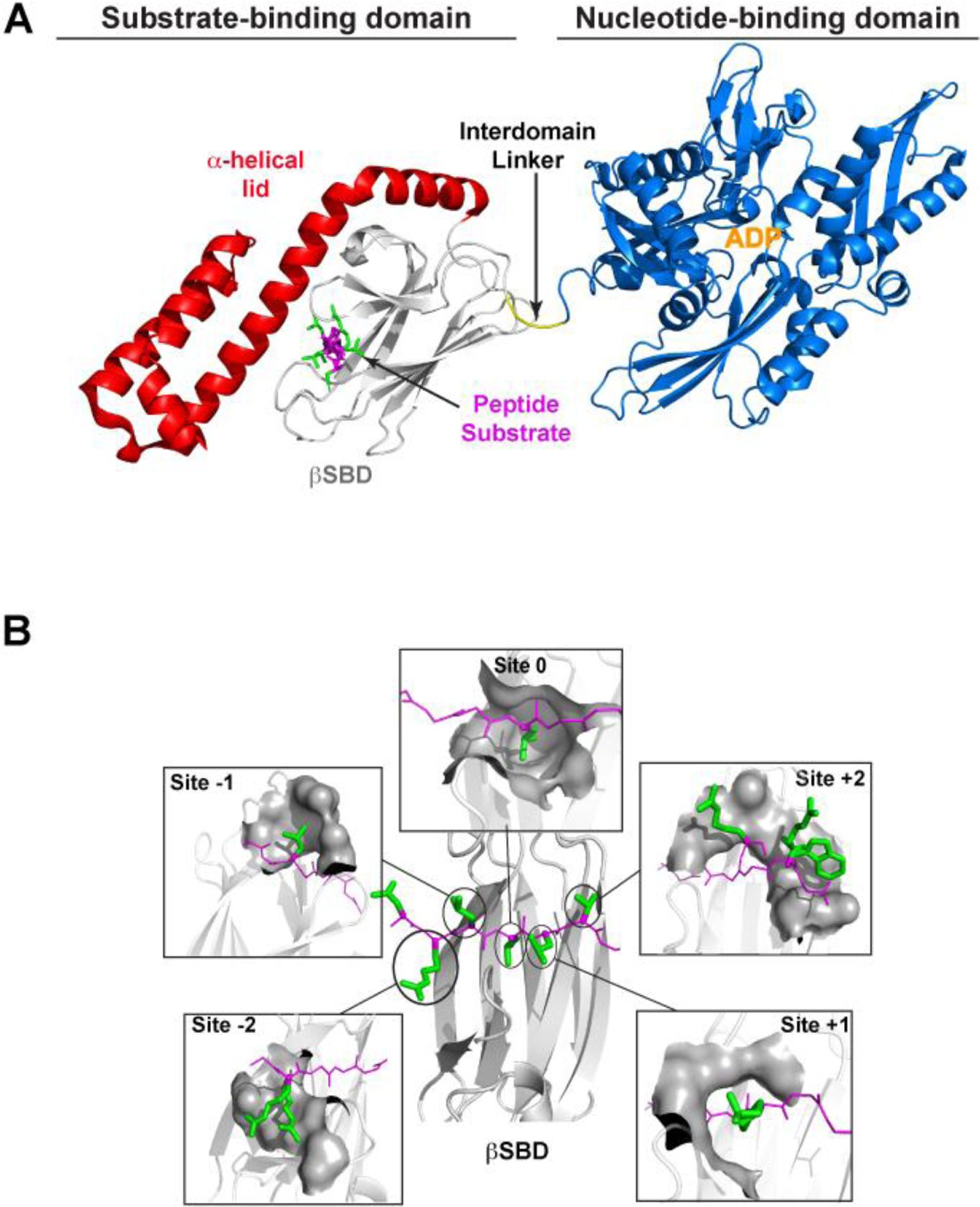
A) Structure of DnaK bound to ADP (PDB ID 2KHO modified to show the peptide substrate using PDB ID 1DKZ). The structure is colored as follows: nucleotide-binding domain (NBD), blue; interdomain linker, yellow; β-sandwich subdomain (βSBD), gray; α-helical lid subdomain, red; substrate peptide NR (NRLLLRTG), magenta (backbone) and green (side chains). B. View of the NR-bound βSBD from the top, where the α-helical lid subdomain was removed for clarity (from PDB ID 1DKZ) (central panel). The peptide backbone (in magenta, with its N-terminus on the left-hand side) lies extended on the binding site projecting its side chains (in green) to discrete “sites” (named “−2”, “−1”, “0”, “+1”, and “+2”) on the βSBD (in gray cartoon). Each surrounding panel shows the sites on the βSBD that act as pockets for each side chain of the bound peptide. The βSBD atoms within 5 Å of the peptide’s side chain are shown as gray surfaces. For sites “−2” and “+2” sidechains from other βSBD-bound peptides are shown to illustrate the different conformations the substrate can adopt (from PDB IDs 4JWD and 4F00 for site “−2”, and PDB IDs 4JWI, 4EZN, 4EZO for site “+2”). This figure is adapted from Nordquist et al.41 Molecular representations are drawn with PyMol.47
The essence of substrate binding in the high affinity state of the Hsp70 SBD is the interaction of five substrate residues through both backbone and side chain moieties with five complementary subsites on the SBD, dubbed “−2”, “−1”, “0”, “+1”, and “+2” (Figure 1B). Throughout the manuscript, we use ‘register’ to refer to which substrate residues interact with the sites in the SBD. Several crystal structures illustrate the binding between the SBD of Hsp70s and representative substrate peptides. Most structures show the peptide bound in the canonical binding orientation (N- to C- on the binding cleft of the BSD, here called “forward”), as shown in the structure of NR-bound SBD.39 Interestingly, a few other structures show a bound peptide in the “reverse” orientation (C- to N-).40 One of the striking findings is that the SBD does very little remodeling to fit different sequences, and different peptides bind in very similar poses to the SBD41. These observations guided the approaches we have used in our work to develop physics-based principles to understand binding energetics.41 Other informative experiments have been based on screening different peptide sequences using peptide arrays to gather sequence binding preferences for Hsp70s.42–46 These data have been exploited to develop predictive algorithms that could identify preferred Hsp70 binding sites in any given protein.
Computational models, informed and refined by new experimental insights, have played a key role in advancing our understanding of Hsp70-client interaction. The first predictive model of Hsp70’s binding preferences was based on statistical analysis of peptide array data and revealed a preference for branched hydrophobics and positively charged residues, an observation consistent with the structure of the SBD of DnaK (the E. coli Hsp70) in complex with a model peptide (NR, NRLLLTG).39,45 Subsequent predictive models have incorporated empirical48 and statistical energy functions49,50 developed for protein folding and structure prediction, which have consistently improved in the last couple of decades. These have been employed to study Hsp70 binding to substrates51,52 or to inhibitors.53,54 These energy functions have also been successfully combined with binding array data to improve their predictive power and augmented with additional insights obtained directly from the growing number of Hsp70-client structures.55,56
Additionally, as computer processor speeds have increased, molecular dynamics (MD) simulations have provided new insights into substrate binding51,57 and chaperone dynamics.38,58 Harold Scheraga and colleagues conducted the first simulations of complete transitions between the open and closed states of Hsp70s, which identified key internal conformational changes within the NBD likely to facilitate the opening transition and thus important to the allosteric mechanism.59 Within the last few years, physics-based atomistic simulations have started to be directly leveraged to enrich predictive models with insights from protein dynamics inherent to function,41,60–62 particularly to take in account new evidence of heterogeneity in the substrate-binding modes of Hsp70.63,64 Together, these computational tools not only greatly advance our quantitative understanding and prediction of Hsp70-client interaction but can also be adapted to predict the effects of mutations and differences among substrate binding by different Hsp70 homologs.
In this review we recapitulate the development of methods to predict Hsp70 molecular chaperone binding sites within substrates, which are summarized in Table 1. We group the models into three main categories, based primarily on the data they incorporate in the prediction: sequence-based methods, structure-based methods, and physics-based methods. Of course, each successive methodology has built upon the key insights of the previous approaches.
Table 1.
Timeline of methods for Hsp70 binding site prediction and their key insights. The acronym PSSM stands for Position-specific Scoring Matrix. They are 20 by N matrices (20 amino acids, N substrate residues) and are often given in energy units, so that each element in the matrix is the binding affinity per amino acid and substrate position.
| Model or Authors | Year | Chaperone | Key features and insights | Online tool/code | Citation |
|---|---|---|---|---|---|
| Blond-Elguindi et al. | 1993 | BiP | PSSM based on sequence statistics | -- | 42,65 |
| Gragerov et al.; Ivey et al. | 1994 | DnaK | Binding analysis from random peptide phage display. | -- | 43,44 |
| Rüdiger et al. | 1997 | DnaK | Identification of the canonical binding motif | -- | 45 |
| Rüdiger et al.; Srinivasan et al. | 2001; 2012 | DnaJ | DnaJ and DnaK share many targets | -- | 46,66 |
| LIMBO | 2009 | DnaK | FoldX 49 and Structure-based PSSM improves transferability of sequence-based PSSM | https://limbo.switchlab.org/ | 55 |
| BiPPred | 2016 | BiP | MD-based PSSM requires minimal training and no sequence-based PSSM | https://www.bioinformatics.wzw.tum.de/bippred/submit/ | 60 |
| ChaperISM | 2019 | DnaK | Position-Independent Scoring Matrix (PISM) improves agreement with binding array data | https://github.com/BioinfLab/ChaperISM | 56 |
| Paladin | 2021 | DnaK | MD-based PSSM, substrate register and orientation prediction | https://github.com/enordquist/paladin | 41 |
Sequence-based models
Sequence-based models are data-driven without input from any structural or physics-based modeling; most are based on screening sequences either by phage display or peptide arrays. Two early studies used random peptide phage display experiments to elucidate the binding preferences of the endoplasmic reticulum Hsp70, BiP,42 and DnaK.43 The results led to the first sequence-based models of the binding preferences of these two Hsp70 family members.42,44
Probing arrays of overlapping cellulose-bound peptides for binding later emerged as the preferred data source and has been widely applied to study the binding preferences of Hsp70 family members and co-chaperones: DnaK,45 the DnaK co-chaperone DnaJ,66,46 HcsA67 and other chaperones beyond Hsp70s.68–71 A seminal study of DnaK binding preferences was carried out by Rüdiger et al. using a library of cellulose-bound 13-residue peptides derived from a set of 37 proteins.45 Rüdiger et al. analyzed the frequency of residues found in three regions of the peptides: the central five residues as well as the first and last four residues. The frequency was then converted into effective binding affinity by: ΔΔG = -RT log(Fbind/Fnonbind), where Fbind and Fnonbind are the frequencies of binding and nonbinding sequences, respectively. This yields a binding affinity matrix of 20 residues by 3 regions. They incorporated the effective binding affinities in a final predictive algorithm that was fine-tuned to give optimized predictions by giving different weights to each of the eight outer residues. With the weight of the central five-residue region set to 1.0, the weights for the residues as they move away from the central five are: 1.5, 1.0, 0.66, and 0.33. That is, residues just beyond the central five were deemed to contribute significantly to selectivity, likely because they might in fact bind to the central SBD sites. However, the outer residues were hypothesized to contribute minimally to selectivity. Subsequent analysis showed that the model performed very well at reproducing peptide binding array data, including those generated from independent studies.41,55 Notably none of the models that have been developed subsequent to this one has incorporated contributions beyond seven total residues, although new evidence suggests that transient interactions between client residues and unknown binding sites distal to the canonical binding cleft could play a role in modulating the binding register of certain substrate peptides.63
An important insight from Rüdiger et al.’s work45 was the quantification of DnaK’s preference for each amino acid in three key regions. Furthermore, their algorithm defined a clear consensus binding motif in the five central residues, which consisted of branched hydrophobic residues flanked by positively charged residues. Importantly, this motif is consistent with the crystal structure of the peptide-bound SBD of DnaK reported by Zhu et al., which revealed five peptide substrate residues making primarily hydrophobic contacts and β-like hydrogen bonds with the receptor.39 A key prediction from the analysis of Rüdiger et al. is that residues beyond the seven observed directly in the crystal structure also contribute to recognition. A second key prediction is the strong bias against certain residues, namely Glu, Asp, Cys and Trp in the central five residues. The molecular basis for the strong bias against negatively charged residues or Cys in the consensus motif was not entirely clear, but from the structure it was clear that Trp is too bulky for the site 0 binding pocket (Figure 1B). It is possible that these residues have greater uncertainty than more abundantly-occurring residues because the peptide arrays were derived using real protein sequences which represented Trp and Cys at natural abundance. Rüdiger algorithm has been used successfully to screen for candidate Hsp70 binding sites,72–77 to engineer antigenic peptides,78 and to rationally design a protein kinase C mutant with low affinity for Hsp70.79 These successes attest to the importance and potential impacts of this landmark experimentally informed, computationally-aided Hsp70 binding site prediction approach.
Purely sequence-based methods suffer limitations stemming from the quality of data used to derive the model, particularly in dataset diversity and imbalance between binders and nonbinders. Previous studies of major histocompatibility complex binding showed that datasets consisting of more than 200 substrates balanced between binders and nonbinders are critical for transferability beyond the training data.80 Similar conclusions can be drawn in predictors of protein-protein aggregation,81 where more complex, nonlinear models, for example ensemble logistic regression and neural networks, require greater diversity and size of dataset.82,83 Importantly, an imbalance between the number of binding and nonbinding peptides is fundamental to Hsp70, as there are always few binding sites relative to a whole protein’s sequence. Another possible limitation is the discrepancy between the number of interaction sites evident in the original structure with the 13 residues of the peptide binding arrays used in these studies.39,45,84 As a result, sequence-based models are generally agnostic about molecular details including substrate binding registry and orientation, which are also important for understanding the effects of flanking regions in Hsp70 binding.
Structure-based models
The key idea underlying a structure-based (or sequence- and structure-based) method to predict Hsp70 binding to a sequence is to use structure-based modeling together with appropriate effective energy scores to enrich sequence-based data and achieve better predictive power. This approach also offers the opportunity to predict the substrate binding register in addition to binding affinity. Similar approaches have been broadly applied in many contexts such as human mevalonate kinase,85 major histocompatibility complex,86 SH3-ligand complexes,87 G protein-coupled receptor,88 and amyloid formation.89–94 Limbo55 and ChaperISM56 are the two major structure-based models for Hsp70-client interaction prediction.
Limbo was partially built on peptide binding array experiments like those in the original study,45 although a different set of proteins was used, and the peptides in the array were 10-mers. The resulting Position Specific Scoring Matrix (PSSM) was combined with a structure-based score consisting of FoldX-based49 binding affinities calculated based on the X-ray crystal structure of DnaK-NRLLLTG peptide complex.39 FoldX is an empirical energy function designed to score proteins in a particular conformation with a complete free energy by combining empirical and physics-based terms, similar to those in classical force fields, with statistical terms to estimate entropy.49 Specifically, the FoldX energies were calculated by assuming that the binding pose in the available crystal structure was representative for all substrates. As such, only the substrate residues resolved in the structure have relevant interactions, and the interactions of each of the sidechains with DnaK is assumed to occur at conserved sites on DnaK. Furthermore, the extended conformation of the bound substrate suggests that the residue sidechains outside of the central residues do not participate in direct interactions, although this assumption is at odds with evidence that transient or cryptic interactions beyond the core 5–7 sites can contribute to specificity.45,63 From these assumptions and combining the information from the peptide arrays and the FoldX energies derived from the peptide-bound SBD, the authors built a twenty amino acid residue times seven sites PSSM. The elements of the matrix were derived by computing the binding energies of an (Ala)7 peptide and computationally mutating one residue at a time to all the other nineteen, then subtracting the polyalanine energy from a peptide with a mutated residue. This gives the effective contribution of each residue to a total binding energy at each site.
FoldX-based binding energies did not have strong predictive power on their own. On the other hand, a sequence-based PSSM derived from the authors’ 10-mer peptide binding scans could reproduce the data it was trained on (cross-validation Mathew’s correlation coefficient (MCC) = 0.708, Table 2) but failed to adequately reproduce separate validation data (validation MCC = 0.106, Table 2). The MCC score ranges from −1 to 1, where 1 is perfect correlation, −1 perfect anticorrelation and 0 for uncorrelated predictions. Note that this sequence-based PSSM itself was in fact biased by structural information and FoldX energies, because it required the knowledge of the DnaK-contacting 7-mer for each 13-mer. The latter was identified as the 7-mer with the most favorable FoldX score. In fact, the validation data must also be fed through FoldX to identify the 7-mer involved in binding, significantly complicating the assessment of Limbo. The authors found that a predictor based on combined sequence- and structure-based PSSM showed the best performance on validation data (validation MCC = 0.375, Table 2), indicating that structure-based information did seem to improve transferability. Optimization of the sequence training data was performed, during which peptides could be removed from the input sequence PSSM if that removal resulted in an improvement in predictor performance across a cycle of cross-validation. The performance on validation data improved for the pure sequence-based PSSM (validation MCC = 0.662) and improved more for the combined sequence- and structure-based PSSM (validation MCC = 0.759). By this optimization method, the authors were able to reduce noise in the peptide array data without significantly biasing the predictor by overly homogenizing the data. Recently, independent tests of the Limbo model showed that it has good performance on data its PSSM was not trained on.41 Specifically, Limbo had a Precision-Recall area-under-the-curve (AUC) of 0.69 on independent peptide array data and predicted the binding orientation of 14 out of 21 peptides derived from crystallographic data.41 This result suggests that indeed the optimized sequence- and structure-based approach improved the model’s transferability across datasets. Limbo has also been used to identify or validate Hsp70 binding sites in many contexts95–99 including for rational design of binding sequences such as inhibitors.100,101
Table 2.
Best Mathews correlation coefficients (MCC) at high specificity (>90%) of the different predictors in the building and final stages of development. This table is adapted from van Durme et al.55
| Benchmark set | Simulated cross-validation | Validation set | |
|---|---|---|---|
| Not optimized PSSMs | |||
| Sequence | 0.792 | 0.708 | 0.106 |
| Structure | 0.588 | - | 0.593 |
| Sequence+Structure | 0.756 | 0.626 | 0.375 |
| Optimized PSSMs | |||
| Sequence | 0.919 | 0.703 | 0.662 |
| Sequence+Structure | 0.819 | 0.703 | 0.759 |
ChaperISM56 is a position-independent scoring matrix (PISM) predictor of Hsp70 binding to a particular sequence derived to reproduce quantitative binding data directly rather than distinguishing how peptides are classified as binders and nonbinders. Based on the same peptide binding data as in the Limbo study55 the source 10-mers were screened using FoldX49 to computationally determine which 7-mers represent maximal affinity to DnaK in the original bound structure.39 The initial PSSM was derived by minimization of the squared error of predicted and experimental binding affinity. Adjustments were made to test the effect of a position-independent matrix and qualitative binding data. PISMs were calculated either by directly minimizing the squared error of a 20×1 site-independent matrix, or by summing the columns of an already-optimized PSSM into a 20×1 PISM. The latter PISM, which includes more parameters during fitting, had the best performance on validation data and did not show signs of overfitting. This is the PISM referred to as ChaperISM. The authors further showed that both the Limbo PSSM and a reduced 10-site Rüdiger PSSM, when converted into PISMs (Figure 2), have minimal loss in performance despite the information loss.56 The key takeaway is that a PISM can capture important features of binding array data, which the authors argue reflects selective promiscuity.
Figure 2. PISMs of Rüdiger et al.,45 Limbo,104 ChaperISM.56.
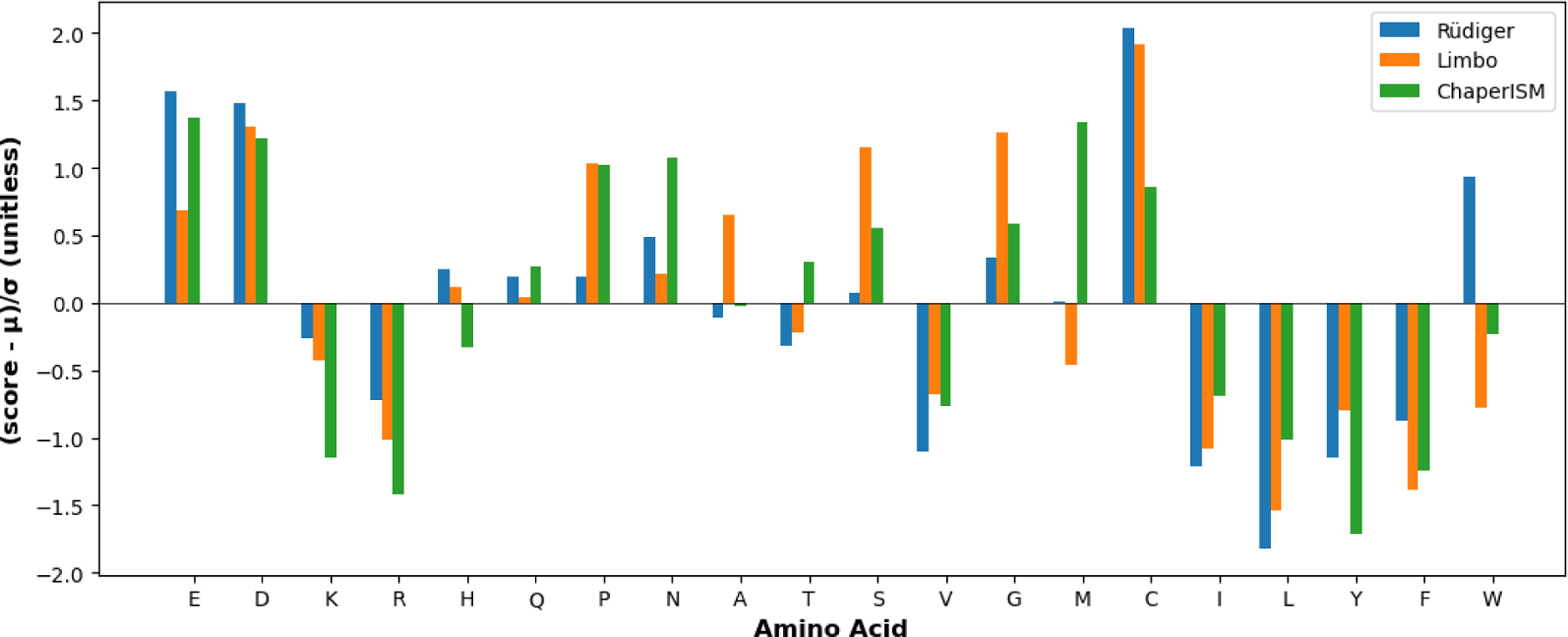
All scores were standardized according to (score - μ)/σ for direct comparison, where μ is the mean score, and σ is the standard deviation. In the case of Limbo and ChaperISM, the default scores have also been changed to negative values to be comparable to those of Rüdiger model, such a lower score corresponds to stronger binding. For Limbo, the PISM is the average score for all seven sites, and for Rüdiger, it is the average score for the inner five residues. This plot is drawn with Matplotlib.105
ChaperISM represents the current state-of-the-art Hsp70 binding hotspot predictor. Evidence in both the ChaperISM paper as well as an independent comparison41 suggests that ChaperISM has better agreement to peptide binding data than Limbo and would therefore be more useful for tasks like hotspot scanning. Like Limbo, ChaperISM has been used to scan for hotspots of amyloid formation102 and more notably was used to validate the glucocorticoid receptor (GR) Hsp70 binding site in the recent cryo-EM structure of Hsp70-Hsp90-Hop-GR complex.103 On the other hand, ChaperISM, or any PISM, does lose some detailed information pertaining to binding register, which Limbo retains.41 The ChaperISM algorithm will predict degenerate binding of NRLLLTG with any of its permutations, including LLTNRGL, which has low affinity.43 Nevertheless, ChaperISM shows state-of-the-art agreement to peptide array scans, which have a similar uncertainty in binding registry.
Physics-based models
Limbo, while an impressively predictive and transferable model, is essentially a weakly-predictive structure-based PSSM of unbiased interaction energies weighted by a sequence-based PSSM with high bias for the training data. In other words, while the sequence-based PSSM suffers from low bias and high variance, and the structure-based PSSM has low variance but high bias, their combination strikes a good balance. Nonetheless, recognizing the potential advantages of the combination suggests an opportunity to greatly improve the transferability of the prediction model by deriving superior structure-based PSSMs, such as using physics-based molecular dynamics simulations. These physics-based models, fine-tuned based on peptide binding array data, may provide an optimal balance in accuracy and transferability as well as in predicting affinities vs molecular details of Hsp70-client interaction.
BiPPred is the first physics-based model of Hsp70-client interaction.60 The central idea is that sampling the interactions between BiP and a client peptide using molecular dynamics would lead to a higher quality PSSM, such that it might require less reweighting from sequence data. Reducing the dependence on sequence-based peptide array data could be important because the peptide array data contain ambiguity with respect to which 7-mers within the 13-mer are involved in binding and the binding orientation is unknown. Additionally, binding affinity measurements in cellulose-bound screening experiments are semi-quantitative at best, and they do not necessarily correlate with measurements in solution.60,65,106,107 A homology model for the SBD of BiP was constructed from the crystal structure of the DnaK/NRLLLTG complex.39 As with Limbo, the goal was to quantify how each residue interacts with BiP by mutating residues on a background (Ala)7 peptide one at a time to all other nineteen and calculate an effective binding affinity. To that end, molecular dynamics followed by Poisson-Boltzmann surface area implicit solvent (MM/PBSA) analysis was used instead of FoldX. Note that MM/PBSA can be highly effective in studies of protein-ligand interactions,51,108,109 even though its accuracy depends sensitively on the choice of the underlying protein force field as well as key parameters of PBSA.110 The classical molecular dynamics force field nonbonded interactions, van der Waals and electrostatics, were also added to the interaction energy matrix. These terms implicitly account for hydrogen bonding interactions that are crucial for Hsp70-substrate binding.
The physics-based PSSM derived from MM/PBSA resulted in minimal predictive power60,108 but could be enhanced by two reweighting methods. Through visual inspection of the MD trajectories, they found a coarse-grained reweighting based on analysis the stability of each sidechain’s interaction with a given binding site (binding site analysis, BA). At site 0, a score of 1.0 is assigned to stable interactions, 0.5 to moderately stable, and 0.0 for no interaction. For other sites, scores are retained for stable interactions, set to 0.5 for moderately stable interactions, and set to 0.0 for no interaction. That gives rise to three types of PSSMs: raw interaction energies (IE), IE reweighted by binding site analysis at site 0 (4th residue in a 7-mer) (IE/4), and IE with binding site analysis at all sites (IE/BA). Each of those three also had two variations, with and without seven site-dependent weights trained by peptide array data. The authors used two types of data, one that was derived from peptide array data, and a smaller one derived from both binding and nonbinding 7-mers in largely solution experiments.65,106,107
Interestingly, an untrained IE PSSM performed the worst on solution data but was able to show improved performance on peptide array data without signs of overtraining. On the other hand, an untrained IE/BA PSSM had an impressive 0.83 AUC on the solution data but had poor agreement with both data types after training the site-dependent weights. This suggests that the peptide array data represent a different picture of binding from the solution experiments. This might also explain the particularly dramatic performance drop of the PSSM IE/BA. The binding site analysis, while not directly biased with respect to the solution dataset, is based on MD sampling of a known-binder peptide that is itself in that dataset.
A key takeaway is the importance of selecting the type and quality of data used to construct a model, as well as additional validation that a structure- or physics-based PSSM, similar in nature, can be reweighted to perform well in reproducing those data.55 Importantly BiPPred (IE/BA) achieved its most impressive performance without reweighting based on any sequence data. Since then, BiPPred has been used alongside ChaperISM to validate the binding site on the GR segment bound in Hsp70 in a GR-Hsp90-Hsp70-Hop complex.103 BiPPred is the first model to integrate molecular dynamics simulations for Hsp70 binding prediction and thereby relies less on peptide binding array data, although it is worth noting that similar methods have been used to model binding affinity in other systems.111–114
The development of Paladin, the latest physics-based model, was driven by the need to study the context-dependence of Hsp70 binding. Doing so required knowledge of more molecular details on the binding configuration40,115,116 than were available when Limbo was built. The key experimental insights brought to bear on the problem were three-fold. Firstly, numerous crystal structures reveal a binding pose consisting of five client residues contacting five sites on DnaK.39,40,115,116 Secondly, the reverse binding mode is more prevalent than previously believed in solution and in crystal structures including peptides without Pro.40,63,64 Lastly, peptides binding in the reverse orientation make similar or identical backbone hydrogen bonds as the forward orientation and project their sidechains into the sites in a similar fashion (Figure 3).39–41 Additionally, the BiPPred work made it clear that a properly constructed physics-based PSSM which faithfully accounts for the binding site interactions can be highly predictive without needing to be fit to peptide array data, which can be helpful because peptide array data and other experiments do not necessarily correlate perfectly.
Figure 3. Illustration of peptides bound to the βSBD in opposite orientations.
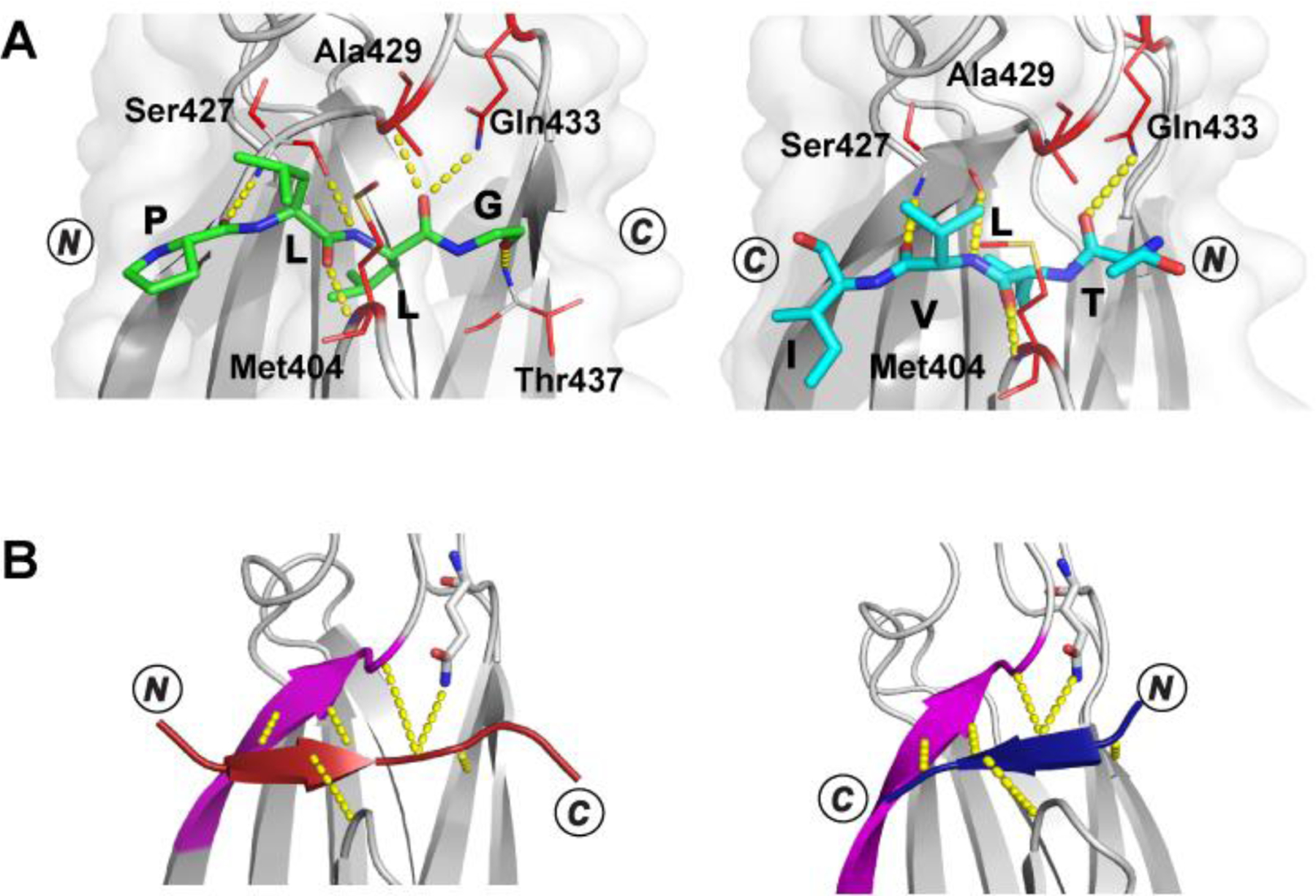
A. Crystal structures (top view, α-helical lid removed for simplicity) of the βSBD (gray) in complex with proPhoA peptide C [left, peptide in green, PDB ID 7N6M], which binds in an N- to C- (forward) orientation, and peptide D [right, peptide in cyan, PDB ID 7JN8], which binds in a C- to N- (reverse) orientation.63 The side chains of the βSBD residues contacting the peptide are shown in red sticks, and hydrogen bonds between the peptide backbone and the βSBD are shown in yellow, dotted lines. B. Schematic showing that the bound peptide interacts with a β-strand of the SBD (magenta) in a parallel or antiparallel strand–strand arrangement for the N- to C- [left; peptide in maroon, PDB ID code 1DKZ] or the C- to N- [right; peptide in blue, PDB ID code 4EZY] binding mode, respectively. The figure is adapted from Nordquist et al. and Clerico et al.41,63 Molecular representations are drawn with PyMol.47
The Paladin model directly incorporates the preceding experimental and computational insights. To account for flexibility in the bound state, both DnaK and the client were harmonically restrained such that the magnitude of structural fluctuations during the MD simulations matches variability present in the available crystallographic structures. The restraint (violation) energy was directly incorporated into the model to account for structural strains required to accommodate a particular sidechain at various sites. Structural analysis revealed that forward and reverse orientations present the side chains for similar interactions with DnaK sites. Indeed, examination of side chain-site interaction energies showed that they were independent of the backbone orientation. As such, the same set of PSSMs could be used regardless of the binding orientation, and a single energy penalty was included to account for the free energy difference of two backbone binding orientations. The latter could be selected to maximize the agreement in predicted and true orientation of all known binding peptides of DnaK from crystallographic data. Finally, a set of weights to rescale energy terms and sites were trained on peptide array data. To deal with the previously discussed ambiguity in peptide arrays, the data were grouped into four classes: strong binders, binders, non-binders, and ambiguous binders. The weights were selected to maximize separations among these four classes. Additionally, the peptides in the peptide array data are 13-mers, but the Paladin model scored each 5-mer and took the lowest energy peptide as the strongest binder rather than scanning through the data set initially and pre-selecting binding peptides based on some energy function.
The Paladin model’s raw energy term PSSM shows modest ability to recapitulate the binding array data without any reweighting. Trained with the peptide array data, Paladin yields true binding predictive power comparable to that of Limbo. Critically, Paladin is able to reproduce high-resolution crystallographic data without any additional training. It reproduces the binding registry of forward orientation peptides well and can resolve the binding orientation with >70% accuracy (Figure 4). However, Paladin’s ability to predict the registry of existing reverse orientation peptides is much poorer, which may be attributed to the presence of Pro at site 0 in many of these peptides. We note that similar challenges also exist for Rüdiger, Limbo and ChaperISM models. The general ability of Paladin to predict binding orientation is yet to be further evaluated as more structural and relevant biophysical data become available.
Figure 4. Paladin can discriminate forward and reverse orientation peptides.
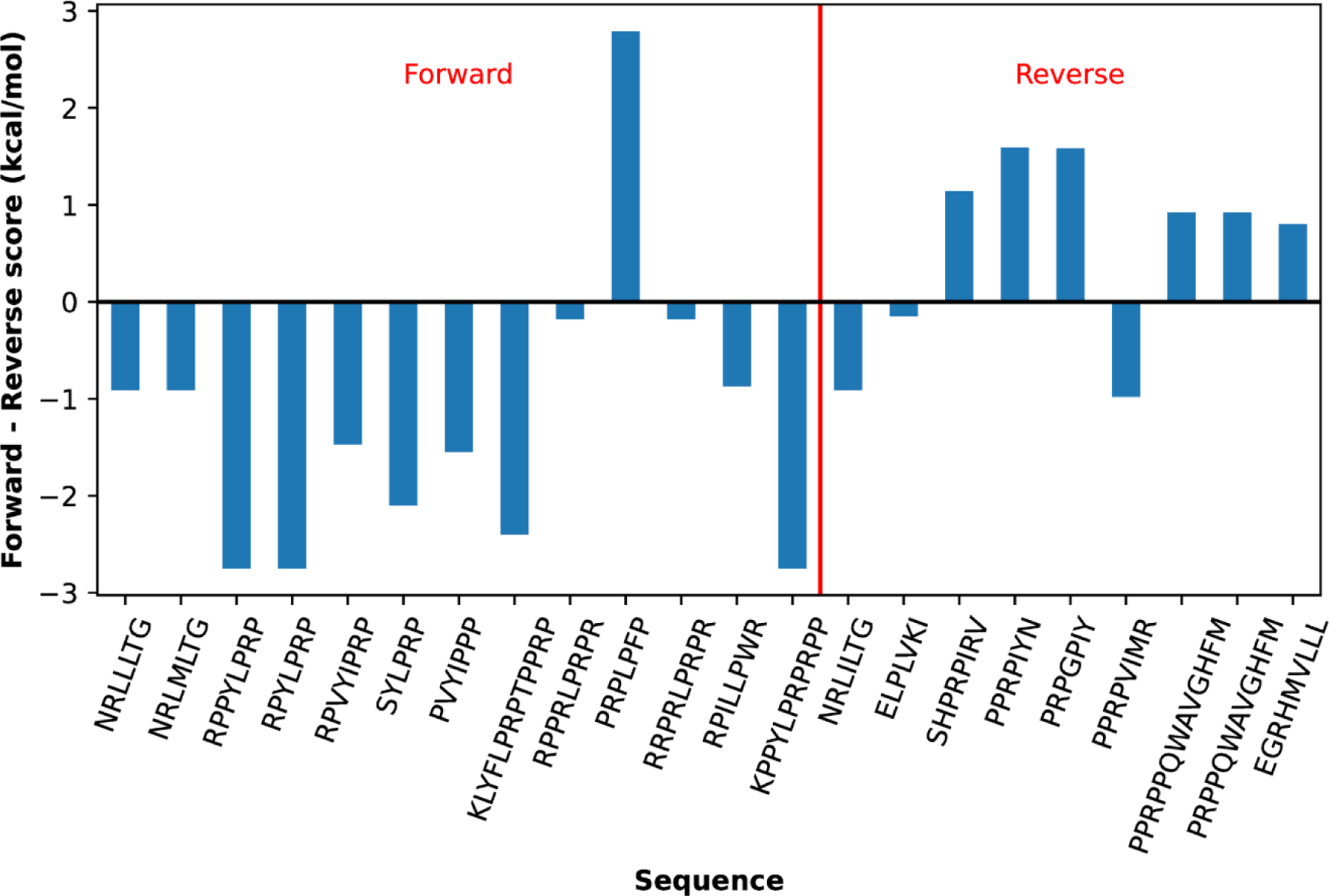
The bars show the difference between forward and reverse scores. A negative difference indicates the forward as the favored orientation, and vice versa. This figure is adapted directly from Nordquist et al.41 This plot is drawn with Matplotlib.105
There still exist several key limitations of the Paladin model. First, methodological refinements could improve the quality of raw PSSMs and alleviate the need for training. Paladin is built on a simple residue-based solvent-accessible surface area (SASA) desolvation term. It could be improved by using atom-based SASA models, which would better resolve the aliphatic and charged group portions of the sidechains of Arg and Lys. In addition, more sophisticated implicit solvent models110 might be needed to better describe the context dependent electrostatic interactions. Both these limitations likely contributed to Paladin’s bias against positively charged residues, a fact that is at odds with the canonical binding motif. A requirement is that the solvation term needs to be decomposable into a residue-specific PSSM. Another remaining limitation of Paladin is the balance of hydrophobic residues like Leu and Ile. Paladin predicts Leu to be preferred over Ile by only ~0.2 kcal/mol, whereas in both the Rüdiger et al. dataset45 and the limited crystal structure dataset Leu is ~1.5 kcal/mol more preferred.41 This discrepancy likely lies at the heart of a key limitation of the Paladin model’s orientation prediction: the confounding, reverse orientation preference for NRLILTG.40 Finally, allowing backbone flexibility to accommodate more favorable interactions with Pro at site 0 could help to reproduce the experimental observations40,116 that currently hamper Paladin’s accuracy, which primarily feature Pro at site 0 in the reverse orientation. Beyond the computational and methodological limitations, additional high-resolution structural data will be the key to validation and further refinement of predictive modeling of Hsp70 binding.
Outlook
Currently, the state-of-the-art for predicting Hsp70 chaperone substrate binding sites and their relative affinity remains less than optimal. ChaperISM can faithfully reproduce the kind of information available in peptide binding array experiments. Methods like Paladin are stretching past this limit towards higher resolution data and more complex problems like client substrate orientation, but there are still several challenges. Consider that the peptide NRLILTG substitutes Leu for Ile and binds in crystal structures in the reverse orientation from NRLLLTG,40 or that several peptides bind with Pro at site 040,116 yet no DnaK-client model predicts that these orientations are favorable.
In fact, the challenge of assigning a single preferred orientation based on structural data may be comparable to the fundamental challenge to assigning single, unique conformations to proteins in general, particularly those that are not globular, and to the bound conformations of clients and their promiscuous chaperones. The energetic differences between different bound states are very small, such that the energy landscape is quite flat for available conformations. It is highly likely that many clients can truly bind in either orientation or both simultaneously in solution.63 This would suggest the need for experiments that can provide detailed information about client orientation and registry with the ability to measure simultaneous occupancies for multiple states in equilibrium.9,63,117,118 Peptide array binding studies are semiquantitative and while they provide insight into the complexities of binding registry, they currently cannot discriminate or control binding orientation making the interpretation of binding registry itself ambiguous.
There are deeper challenges as well, ones which must push the boundaries of experimental techniques before predictive tools can become useful. The first is in the sequence context dependence of binding interactions, which are not well-represented in most crystallographic studies of binding. It has been shown that changing the length of a peptide beyond the five to seven central residues can change its orientation preference.63 There is also evidence that clients, including the canonical NRLLLTG, can sample multiple binding registers and orientations, in some cases simultaneously in a single structure.40,63 One question yet to be addressed is whether known variations in the selectivity of Hsp70 homologs42,65,107 can aide in the rational design of inhibitors targeting the SBD binding site and selective for individual homologs; by definition, the property of selective promiscuity, or a flat binding affinity landscape, makes this seem unlikely. Another question will require a great deal more data: how does the presence of co-chaperones and co-factors modulate specificity?119 Finally, perhaps the greatest challenge is whether we can map out how post-translational modifications (PTM) affect Hsp70 substrate recognition, part of the so-called Chaperone Code.120–122 PTMs are known to specifically affect substrate recognition, binding and unbinding in Hsp70s.123–126 There is abundant PTM data available but a need for better computational tools,127 particularly for Hsp70 chaperones.
Acknowledgement
This work was supported by National Institutes of Health Grants R35 GM144045 (to J.C.) and R35 GM118161 (to L.M.G). E.B.N. received support from the National Research Service Awards T32 GM008515 and T32 GM139789 from the National Institutes of Health.
References
- (1).Momany FA; McGuire RF; Burgess AW; Scheraga HA Energy Parameters in Polypeptides. VII. Geometric Parameters, Partial Atomic Charges, Nonbonded Interactions, Hydrogen Bond Interactions, and Intrinsic Torsional Potentials for the Naturally Occurring Amino Acids. J. Phys. Chem 1975, 79 (22), 2361–2381. 10.1021/j100589a006. [DOI] [Google Scholar]
- (2).Liwo A; Wawak RJ; Scheraga HA; Pincus MR; Rackovsky S Prediction of Protein Conformation on the Basis of a Search for Compact Structures: Test on Avian Pancreatic Polypeptide. Protein Sci 1993, 2 (10), 1715–1731. 10.1002/pro.5560021016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hartl FU; Bracher A; Hayer-Hartl M Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475 (7356), 324–332. 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- (4).Kim YE; Hipp MS; Bracher A; Hayer-Hartl M; Ulrich Hartl F Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem 2013, 82 (1), 323–355. 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- (5).Morimoto RI; Cuervo AM Proteostasis and the Aging Proteome in Health and Disease. J. Gerontol. A. Biol. Sci. Med. Sci 2014, 69 (Suppl 1), S33–S38. 10.1093/gerona/glu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mogk A; Bukau B; Kampinga HH Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69 (2), 214–226. 10.1016/j.molcel.2018.01.004. [DOI] [PubMed] [Google Scholar]
- (7).Mayer MP Intra-Molecular Pathways of Allosteric Control in Hsp70s. Philos. Trans. R. Soc. B Biol. Sci 2018, 373 (1749), 20170183. 10.1098/rstb.2017.0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Clerico EM; Meng W; Pozhidaeva A; Bhasne K; Petridis C; Gierasch LM Hsp70 Molecular Chaperones: Multifunctional Allosteric Holding and Unfolding Machines. Biochem. J 2019, 476 (11), 1653–1677. 10.1042/BCJ20170380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rosenzweig R; Sekhar A; Nagesh J; Kay LE Promiscuous Binding by Hsp70 Results in Conformational Heterogeneity and Fuzzy Chaperone-Substrate Ensembles. eLife 2017, 6, e28030. 10.7554/eLife.28030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rosenzweig R; Nillegoda NB; Mayer MP; Bukau B The Hsp70 Chaperone Network. Nat. Rev. Mol. Cell Biol 2019, 20 (11), 665–680. 10.1038/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- (11).Voos W; Röttgers K Molecular Chaperones as Essential Mediators of Mitochondrial Biogenesis. Biochim. Biophys. Acta BBA - Mol. Cell Res 2002, 1592 (1), 51–62. 10.1016/S0167-4889(02)00264-1. [DOI] [PubMed] [Google Scholar]
- (12).Flores-Pérez Ú; Jarvis P Molecular Chaperone Involvement in Chloroplast Protein Import. Biochim. Biophys. Acta BBA - Mol. Cell Res 2013, 1833 (2), 332–340. 10.1016/j.bbamcr.2012.03.019. [DOI] [PubMed] [Google Scholar]
- (13).Craig EA Hsp70 at the Membrane: Driving Protein Translocation. BMC Biol 2018, 16 (1), 11. 10.1186/s12915-017-0474-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wu X; Cabanos C; Rapoport TA Structure of the Post-Translational Protein Translocation Machinery of the ER Membrane. Nature 2019, 566 (7742), 136–139. 10.1038/s41586-018-0856-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kellner R; Hofmann H; Barducci A; Wunderlich B; Nettels D; Schuler B Single-Molecule Spectroscopy Reveals Chaperone-Mediated Expansion of Substrate Protein. Proc. Natl. Acad. Sci 2014, 111 (37), 13355–13360. 10.1073/pnas.1407086111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lee JH; Zhang D; Hughes C; Okuno Y; Sekhar A; Cavagnero S Heterogeneous Binding of the SH3 Client Protein to the DnaK Molecular Chaperone. Proc. Natl. Acad. Sci 2015, 112 (31), E4206–E4215. 10.1073/pnas.1505173112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sekhar A; Rosenzweig R; Bouvignies G; Kay LE Hsp70 Biases the Folding Pathways of Client Proteins. Proc. Natl. Acad. Sci 2016, 113 (20), E2794–E2801. 10.1073/pnas.1601846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sekhar A; Nagesh J; Rosenzweig R; Kay LE Conformational Heterogeneity in the Hsp70 Chaperone-Substrate Ensemble Identified from Analysis of NMR-Detected Titration Data. Protein Sci 2017, 26 (11), 2207–2220. 10.1002/pro.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gamer J; Bujard H; Bukau B Physical Interaction between Heat Shock Proteins DnaK, DnaJ, and GrpE and the Bacterial Heat Shock Transcription Factor Σ32. Cell 1992, 69 (5), 833–842. 10.1016/0092-8674(92)90294-M. [DOI] [PubMed] [Google Scholar]
- (20).Liberek K; Galitski TP; Zylicz M; Georgopoulos C The DnaK Chaperone Modulates the Heat Shock Response of Escherichia Coli by Binding to the Sigma 32 Transcription Factor. Proc. Natl. Acad. Sci 1992, 89 (8), 3516–3520. 10.1073/pnas.89.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liberek K; Georgopoulos C Autoregulation of the Escherichia Coli Heat Shock Response by the DnaK and DnaJ Heat Shock Proteins. Proc. Natl. Acad. Sci 1993, 90 (23), 11019–11023. 10.1073/pnas.90.23.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schumacher RJ; Hansen WJ; Freeman BC; Alnemri E; Litwack G; Toft DO Cooperative Action of Hsp70, Hsp90, and DnaJ Proteins in Protein Renaturation. Biochemistry 1996, 35 (47), 14889–14898. 10.1021/bi961825h. [DOI] [PubMed] [Google Scholar]
- (23).Deuerling E; Patzelt H; Vorderwülbecke S; Rauch T; Kramer G; Schaffitzel E; Mogk A; Schulze-Specking A; Langen H; Bukau B Trigger Factor and DnaK Possess Overlapping Substrate Pools and Binding Specificities. Mol. Microbiol 2003, 47 (5), 1317–1328. 10.1046/j.1365-2958.2003.03370.x. [DOI] [PubMed] [Google Scholar]
- (24).Tyedmers J; Mogk A; Bukau B Cellular Strategies for Controlling Protein Aggregation. Nat. Rev. Mol. Cell Biol 2010, 11 (11), 777–788. 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- (25).Aprile FA; Arosio P; Fusco G; Chen SW; Kumita JR; Dhulesia A; Tortora P; Knowles TPJ; Vendruscolo M; Dobson CM; Cremades N Inhibition of α-Synuclein Fibril Elongation by Hsp70 Is Governed by a Kinetic Binding Competition between α-Synuclein Species. Biochemistry 2017, 56 (9), 1177–1180. 10.1021/acs.biochem.6b01178. [DOI] [PubMed] [Google Scholar]
- (26).Schlossman DM; Schmid SL; Braell WA; Rothman JE An Enzyme That Removes Clathrin Coats: Purification of an Uncoating ATPase. J. Cell Biol 1984, 99 (2), 723–733. 10.1083/jcb.99.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xing Y; Böcking T; Wolf M; Grigorieff N; Kirchhausen T; Harrison SC Structure of Clathrin Coat with Bound Hsc70 and Auxilin: Mechanism of Hsc70-Facilitated Disassembly. EMBO J 2010, 29 (3), 655–665. 10.1038/emboj.2009.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Böcking T; Aguet F; Harrison SC; Kirchhausen T Single-Molecule Analysis of a Molecular Disassemblase Reveals the Mechanism of Hsc70-Driven Clathrin Uncoating. Nat. Struct. Mol. Biol 2011, 18 (3), 295–301. 10.1038/nsmb.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sousa R; Liao H-S; Cuéllar J; Jin S; Valpuesta JM; Jin AJ; Lafer EM Clathrin-Coat Disassembly Illuminates the Mechanisms of Hsp70 Force Generation. Nat. Struct. Mol. Biol 2016, 23 (9), 821–829. 10.1038/nsmb.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kityk R; Kopp J; Mayer MP Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69 (2), 227–237.e4. 10.1016/j.molcel.2017.12.003. [DOI] [PubMed] [Google Scholar]
- (31).Qian YQ; Patel D; Hartl F-U; McColl DJ Nuclear Magnetic Resonance Solution Structure of the Human Hsp40 (HDJ-1) J-Domain. J. Mol. Biol 1996, 260 (2), 224–235. 10.1006/jmbi.1996.0394. [DOI] [PubMed] [Google Scholar]
- (32).Suh W-C; Burkholder WF; Lu CZ; Zhao X; Gottesman ME; Gross CA Interaction of the Hsp70 Molecular Chaperone, DnaK, with Its Cochaperone DnaJ. Proc. Natl. Acad. Sci 1998, 95 (26), 15223–15228. 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pellecchia M; Szyperski T; Wall D; Georgopoulos C; Wüthrich K NMR Structure of the J-Domain and the Gly/Phe-Rich Region of TheEscherichia ColiDnaJ Chaperone. J. Mol. Biol 1996, 260 (2), 236–250. 10.1006/jmbi.1996.0395. [DOI] [PubMed] [Google Scholar]
- (34).Laufen T; Mayer MP; Beisel C; Klostermeier D; Mogk A; Reinstein J; Bukau B Mechanism of Regulation of Hsp70 Chaperones by DnaJ Cochaperones. Proc. Natl. Acad. Sci 1999, 96 (10), 5452–5457. 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bracher A; Verghese J The Nucleotide Exchange Factors of Hsp70 Molecular Chaperones. Front. Mol. Biosci 2015, 2. [DOI] [PMC free article] [PubMed]
- (36).Stürner E; Behl C The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci 2017, 10. [DOI] [PMC free article] [PubMed]
- (37).Yakubu UM; Morano KA Roles of the Nucleotide Exchange Factor and Chaperone Hsp110 in Cellular Proteostasis and Diseases of Protein Misfolding. Biol. Chem 2018, 399 (10), 1215–1221. 10.1515/hsz-2018-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).English CA; Sherman W; Meng W; Gierasch LM The Hsp70 Interdomain Linker Is a Dynamic Switch That Enables Allosteric Communication between Two Structured Domains. J. Biol. Chem 2017, 292 (36), 14765–14774. 10.1074/jbc.M117.789313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhu X; Zhao X; Burkholder WF; Gragerov A; Ogata CM; Gottesman ME; Hendrickson WA Structural Analysis of Substrate Binding by the Molecular Chaperone DnaK. Science 1996, 272 (5268), 1606–1614. 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zahn M; Berthold N; Kieslich B; Knappe D; Hoffmann R; Sträter N Structural Studies on the Forward and Reverse Binding Modes of Peptides to the Chaperone DnaK. J. Mol. Biol 2013, 425 (14), 2463–2479. 10.1016/j.jmb.2013.03.041. [DOI] [PubMed] [Google Scholar]
- (41).Nordquist EB; English CA; Clerico EM; Sherman W; Gierasch LM; Chen J Physics-Based Modeling Provides Predictive Understanding of Selectively Promiscuous Substrate Binding by Hsp70 Chaperones. PLoS Comput. Biol 2021, 17 (11), e1009567. 10.1371/journal.pcbi.1009567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Blond-Elguindi S; Cwirla SE; Dower WJ; Lipshutz RJ; Sprang SR; Sambrook JF; Gething M-JH Affinity Panning of a Library of Peptides Displayed on Bacteriophages Reveals the Binding Specificity of BiP. Cell 1993, 75 (4), 717–728. 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- (43).Gragerov A; Zeng L; Zhao X; Burkholder W; Gottesman ME Specificity of DnaK-Peptide Binding. J. Mol. Biol 1994, 235 (3), 848–854. 10.1006/jmbi.1994.1043. [DOI] [PubMed] [Google Scholar]
- (44).Ivey RA; Subramanian C; Bruce BD Identification of a Hsp70 Recognition Domain within the Rubisco Small Subunit Transit Peptide. Plant Physiol 2000, 122 (4), 1289–1299. 10.1104/pp.122.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rüdiger S; Germeroth L; Schneider-Mergener J; Bukau B Substrate Specificity of the DnaK Chaperone Determined by Screening Cellulose-Bound Peptide Libraries. EMBO J 1997, 16 (7), 1501–1507. 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Srinivasan SR; Gillies AT; Chang L; Thompson AD; Gestwicki JE Molecular Chaperones DnaK and DnaJ Share Predicted Binding Sites on Most Proteins in the E. Coli Proteome. Mol. Biosyst 2012, 8 (9), 2323. 10.1039/c2mb25145k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.8, 2015. [Google Scholar]
- (48).Lazaridis T; Karplus M Effective Energy Function for Proteins in Solution. Proteins Struct. Funct. Bioinforma 1999, 35 (2), 133–152. . [DOI] [PubMed] [Google Scholar]
- (49).Schymkowitz J; Borg J; Stricher F; Nys R; Rousseau F; Serrano L The FoldX Web Server: An Online Force Field. Nucleic Acids Res 2005, 33 (Web Server), W382–W388. 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Alford RF; Leaver-Fay A; Jeliazkov JR; O’Meara MJ; DiMaio FP; Park H; Shapovalov MV; Renfrew PD; Mulligan VK; Kappel K; Labonte JW; Pacella MS; Bonneau R; Bradley P; Dunbrack RL; Das R; Baker D; Kuhlman B; Kortemme T; Gray JJ The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput 2017, 13 (6), 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Bhattacharjee R; Devi A; Mishra S Molecular Docking and Molecular Dynamics Studies Reveal Structural Basis of Inhibition and Selectivity of Inhibitors EGCG and OSU-03012 toward Glucose Regulated Protein-78 (GRP78) Overexpressed in Glioblastoma. J. Mol. Model 2015, 21 (10), 272. 10.1007/s00894-015-2801-3. [DOI] [PubMed] [Google Scholar]
- (52).de Oliveira AA; Faustino J; de Lima ME; Menezes R; Nunes KP Unveiling the Interplay between the TLR4/MD2 Complex and HSP70 in the Human Cardiovascular System: A Computational Approach. Int. J. Mol. Sci 2019, 20 (13), 3121. 10.3390/ijms20133121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Elengoe A; Naser MA; Hamdan S Modeling and Docking Studies on Novel Mutants (K71L and T204V) of the ATPase Domain of Human Heat Shock 70 KDa Protein 1. Int. J. Mol. Sci 2014, 15 (4), 6797–6814. 10.3390/ijms15046797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sangeetha K; Sasikala RP; Meena KS Pharmacophore Modeling, Virtual Screening and Molecular Docking of ATPase Inhibitors of HSP70. Comput. Biol. Chem 2017, 70, 164–174. 10.1016/j.compbiolchem.2017.05.011. [DOI] [PubMed] [Google Scholar]
- (55).Van Durme J; Maurer-Stroh S; Gallardo R; Wilkinson H; Rousseau F; Schymkowitz J Accurate Prediction of DnaK-Peptide Binding via Homology Modelling and Experimental Data. PLoS Comput. Biol 2009, 5 (8), e1000475. 10.1371/journal.pcbi.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Gutierres MBB; Bonorino CBC; Rigo MM ChaperISM: Improved Chaperone Binding Prediction Using Position-Independent Scoring Matrices. Bioinformatics 2019, btz670. 10.1093/bioinformatics/btz670. [DOI] [PubMed]
- (57).Azoulay I; Kucherenko N; Nachliel E; Gutman M; Azem A; Tsfadia Y Tracking the Interplay between Bound Peptide and the Lid Domain of DnaK, Using Molecular Dynamics. Int. J. Mol. Sci 2013, 14 (6), 12675–12695. 10.3390/ijms140612675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Assenza S; Sassi AS; Kellner R; Schuler B; De Los Rios P; Barducci A Efficient Conversion of Chemical Energy into Mechanical Work by Hsp70 Chaperones. eLife 2019, 8, e48491. 10.7554/eLife.48491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gołaś E; Maisuradze GG; Senet P; Ołdziej S; Czaplewski C; Scheraga HA; Liwo A Simulation of the Opening and Closing of Hsp70 Chaperones by Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput 2012, 8 (5), 1750–1764. 10.1021/ct200680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Schneider M; Rosam M; Glaser M; Patronov A; Shah H; Back KC; Daake MA; Buchner J; Antes I BiPPred: Combined Sequence- and Structure-Based Prediction of Peptide Binding to the Hsp70 Chaperone BiP. Proteins Struct. Funct. Bioinforma 2016, 84 (10), 1390–1407. 10.1002/prot.25084. [DOI] [PubMed] [Google Scholar]
- (61).Antes I; Marion A; Zheng C; Melse O Accurate Prediction of Protein-Ligand Binding by Combined Molecular Dynamics-Based Docking and QM/MM Methods. Biophys. J 2018, 114 (3), 42A–42A. [Google Scholar]
- (62).Zalewski M; Kmiecik S; Koliński M Molecular Dynamics Scoring of Protein–Peptide Models Derived from Coarse-Grained Docking. Molecules 2021, 26 (11), 3293. 10.3390/molecules26113293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Clerico EM; Pozhidaeva AK; Jansen RM; Özden C; Tilitsky JM; Gierasch LM Selective Promiscuity in the Binding of E. Coli Hsp70 to an Unfolded Protein. Proc. Natl. Acad. Sci. U. S. A 2021, 118 (41), e2016962118. 10.1073/pnas.2016962118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Tapley TL; Cupp-Vickery JR; Vickery LE Sequence-Dependent Peptide Binding Orientation by the Molecular Chaperone DnaK †. Biochemistry 2005, 44 (37), 12307–12315. 10.1021/bi051145r. [DOI] [PubMed] [Google Scholar]
- (65).Knarr G; Modrow S; Todd A; Gething MJ; Buchner J BiP-Binding Sequences in HIV Gp160. Implications for the Binding Specificity of Bip. J. Biol. Chem 1999, 274 (42), 29850–29857. 10.1074/jbc.274.42.29850. [DOI] [PubMed] [Google Scholar]
- (66).Rüdiger S; Schneider-Mergener J; Bukau B Its Substrate Specificity Characterizes the DnaJ Co-Chaperone as a Scanning Factor for the DnaK Chaperone. EMBO J 2001, 20 (5), 1042–1050. 10.1093/emboj/20.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Tapley TL; Cupp-Vickery JR; Vickery LE Structural Determinants of HscA Peptide-Binding Specificity †. Biochemistry 2006, 45 (26), 8058–8066. 10.1021/bi0606187. [DOI] [PubMed] [Google Scholar]
- (68).Hoff KG; Ta DT; Tapley TL; Silberg JJ; Vickery LE Hsc66 Substrate Specificity Is Directed toward a Discrete Region of the Iron-Sulfur Cluster Template Protein IscU *. J. Biol. Chem 2002, 277 (30), 27353–27359. 10.1074/jbc.M202814200. [DOI] [PubMed] [Google Scholar]
- (69).Hennecke G; Nolte J; Volkmer-Engert R; Schneider-Mergener J; Behrens S The Periplasmic Chaperone SurA Exploits Two Features Characteristic of Integral Outer Membrane Proteins for Selective Substrate Recognition *. J. Biol. Chem 2005, 280 (25), 23540–23548. 10.1074/jbc.M413742200. [DOI] [PubMed] [Google Scholar]
- (70).Chatellier J; Buckle AM; Fersht AR GroEL Recognises Sequential and Non-Sequential Linear Structural Motifs Compatible with Extended β-Strands and α-Helices11Edited by J. Karn. J. Mol. Biol 1999, 292 (1), 163–172. 10.1006/jmbi.1999.3040. [DOI] [PubMed] [Google Scholar]
- (71).Lentze N; Narberhaus F Detection of Oligomerisation and Substrate Recognition Sites of Small Heat Shock Proteins by Peptide Arrays. Biochem. Biophys. Res. Commun 2004, 325 (2), 401–407. 10.1016/j.bbrc.2004.10.043. [DOI] [PubMed] [Google Scholar]
- (72).Mogk A; Tomoyasu T; Goloubinoff P; Rüdiger S; Röder D; Langen H; Bukau B Identification of Thermolabile Escherichia Coli Proteins: Prevention and Reversion of Aggregation by DnaK and ClpB. EMBO J 1999, 18 (24), 6934–6949. 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Vega CA; Kurt N; Chen Z; Rüdiger S; Cavagnero S Binding Specificity of an Alpha-Helical Protein Sequence to a Full-Length Hsp70 Chaperone and Its Minimal Substrate-Binding Domain. Biochemistry 2006, 45 (46), 13835–13846. 10.1021/bi061432a. [DOI] [PubMed] [Google Scholar]
- (74).Vandenbroeck K; Alloza I; Brehmer D; Billiau A; Proost P; McFerran N; Rüdiger S; Walker B The Conserved Helix C Region in the Superfamily of Interferon-γ/Interleukin-10-Related Cytokines Corresponds to a High-Affinity Binding Site for the HSP70 Chaperone DnaK*. J. Biol. Chem 2002, 277 (28), 25668–25676. 10.1074/jbc.M202984200. [DOI] [PubMed] [Google Scholar]
- (75).Foth BJ; Ralph SA; Tonkin CJ; Struck NS; Fraunholz M; Roos DS; Cowman AF; McFadden GI Dissecting Apicoplast Targeting in the Malaria Parasite Plasmodium Falciparum. Science 2003, 299 (5607), 705–708. 10.1126/science.1078599. [DOI] [PubMed] [Google Scholar]
- (76).Anwar A; Siegel D; Kepa JK; Ross D Interaction of the Molecular Chaperone Hsp70 with Human NAD(P)H:Quinone Oxidoreductase 1*. J. Biol. Chem 2002, 277 (16), 14060–14067. 10.1074/jbc.M111576200. [DOI] [PubMed] [Google Scholar]
- (77).Kmiecik S; Breton Laura Le; Mayer Matthias P. Feedback Regulation of Heat Shock Factor 1 (Hsf1) Activity by Hsp70-Mediated Trimer Unzipping and Dissociation from DNA. EMBO J 2020, 39 (14), e104096. 10.15252/embj.2019104096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Waku T; Watanabe Y; Haida H; Kunugi S; Tanaka N Designing Antigenic Peptides with Dual Binding Affinities for HSP70 and MHC. Chem. Lett 2015, 44 (10), 1377–1379. 10.1246/cl.150526. [DOI] [Google Scholar]
- (79).Gao T; Newton AC Invariant Leu Preceding Turn Motif Phosphorylation Site Controls the Interaction of Protein Kinase C with Hsp70*. J. Biol. Chem 2006, 281 (43), 32461–32468. 10.1074/jbc.M604076200. [DOI] [PubMed] [Google Scholar]
- (80).Roomp K; Antes I; Lengauer T Predicting MHC Class I Epitopes in Large Datasets. BMC Bioinformatics 2010, 11 (1), 90. 10.1186/1471-2105-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Prabakaran R; Rawat P; Kumar S; Gromiha MM Evaluation of in Silico Tools for the Prediction of Protein and Peptide Aggregation on Diverse Datasets. Brief. Bioinform 2021, 22 (6), bbab240. 10.1093/bib/bbab240. [DOI] [PubMed] [Google Scholar]
- (82).Família C; Dennison SR; Quintas A; Phoenix DA Prediction of Peptide and Protein Propensity for Amyloid Formation. PLOS ONE 2015, 10 (8), e0134679. 10.1371/journal.pone.0134679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Prabakaran R; Rawat P; Kumar S; Michael Gromiha M ANuPP: A Versatile Tool to Predict Aggregation Nucleating Regions in Peptides and Proteins. J. Mol. Biol 2021, 433 (11), 166707. 10.1016/j.jmb.2020.11.006. [DOI] [PubMed] [Google Scholar]
- (84).Rüdiger S; Buchberger A; Bukau B Interaction of Hsp70 Chaperones with Substrates. Nat. Struct. Mol. Biol 1997, 4 (5), 342–349. 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- (85).Browne C; Timson DJ In Silico Prediction of the Effects of Mutations in the Human Mevalonate Kinase Gene: Towards a Predictive Framework for Mevalonate Kinase Deficiency. Ann. Hum. Genet 2015, 79 (6), 451–459. 10.1111/ahg.12126. [DOI] [PubMed] [Google Scholar]
- (86).Antes I; Siu SWI; Lengauer T DynaPred: A Structure and Sequence Based Method for the Prediction of MHC Class I Binding Peptide Sequences and Conformations. Bioinformatics 2006, 22 (14), E16–E24. 10.1093/bioinformatics/btl216. [DOI] [PubMed] [Google Scholar]
- (87).Brannetti B; Via A; Cestra G; Cesareni G; Citterich MH SH3-SPOT: An Algorithm to Predict Preferred Ligands to Different Members of the SH3 Gene Family. J. Mol. Biol 2000, 298 (2), 313–328. 10.1006/jmbi.2000.3670. [DOI] [PubMed] [Google Scholar]
- (88).Nemoto W; Yamanishi Y; Limviphuvadh V; Saito A; Toh H GGIP: Structure and Sequence-Based GPCR–GPCR Interaction Pair Predictor. Proteins Struct. Funct. Bioinforma 2016, 84 (9), 1224–1233. 10.1002/prot.25071. [DOI] [PubMed] [Google Scholar]
- (89).Maurer-Stroh S; Debulpaep M; Kuemmerer N; de la Paz ML; Martins IC; Reumers J; Morris KL; Copland A; Serpell L; Serrano L; Schymkowitz JWH; Rousseau F Exploring the Sequence Determinants of Amyloid Structure Using Position-Specific Scoring Matrices. Nat. Methods 2010, 7 (3), 237–242. 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- (90).Thompson MJ; Sievers SA; Karanicolas J; Ivanova MI; Baker D; Eisenberg D The 3D Profile Method for Identifying Fibril-Forming Segments of Proteins. Proc. Natl. Acad. Sci 2006, 103 (11), 4074–4078. 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Zhang Z; Chen H; Lai L Identification of Amyloid Fibril-Forming Segments Based on Structure and Residue-Based Statistical Potential. Bioinformatics 2007, 23 (17), 2218–2225. 10.1093/bioinformatics/btm325. [DOI] [PubMed] [Google Scholar]
- (92).Trovato A; Seno F; Tosatto SCE The PASTA Server for Protein Aggregation Prediction. Protein Eng. Des. Sel 2007, 20 (10), 521–523. 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- (93).Bui JM; Cavalli A; Gsponer J Identification of Aggregation-Prone Elements by Using Interaction-Energy Matrices. Angew. Chem. Int. Ed 2008, 47 (38), 7267–7269. 10.1002/anie.200802345. [DOI] [PubMed] [Google Scholar]
- (94).Jr AWB; Menke M; Cowen LJ; Lindquist SL; Berger B BETASCAN: Probable β-Amyloids Identified by Pairwise Probabilistic Analysis. PLOS Comput. Biol 2009, 5 (3), e1000333. 10.1371/journal.pcbi.1000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Gersing SK; Wang Y; Grønbæk-Thygesen M; Kampmeyer C; Clausen L; Willemoës M; Andréasson C; Stein A; Lindorff-Larsen K; Hartmann-Petersen R Mapping the Degradation Pathway of a Disease-Linked Aspartoacylase Variant. PLOS Genet 2021, 17 (4), e1009539. 10.1371/journal.pgen.1009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Baaklini I; Gonçalves C. de C.; Lukacs GL; Young JC Selective Binding of HSC70 and Its Co-Chaperones to Structural Hotspots on CFTR. Sci. Rep 2020, 10 (1), 4176. 10.1038/s41598-020-61107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Imamoglu R; Balchin D; Hayer-Hartl M; Hartl FU Bacterial Hsp70 Resolves Misfolded States and Accelerates Productive Folding of a Multi-Domain Protein. Nat. Commun 2020, 11 (1), 365. 10.1038/s41467-019-14245-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Guin D; Gelman H; Wang Y; Gruebele M Heat Shock-Induced Chaperoning by Hsp70 Is Enabled in-Cell. PLOS ONE 2019, 14 (9), e0222990. 10.1371/journal.pone.0222990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Peffer S; Gonçalves D; Morano KA Regulation of the Hsf1-Dependent Transcriptome via Conserved Bipartite Contacts with Hsp70 Promotes Survival in Yeast. J. Biol. Chem 2019, 294 (32), 12191–12202. 10.1074/jbc.RA119.008822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Jong WSP; ten Hagen-Jongman CM; Vikström D; Dontje W; Abdallah AM; de Gier J-W; Bitter W; Luirink J Mutagenesis-Based Characterization and Improvement of a Novel Inclusion Body Tag. Front. Bioeng. Biotechnol 2020, 7. [DOI] [PMC free article] [PubMed]
- (101).Dalphin MD; Stangl AJ; Liu Y; Cavagnero S KLR-70: A Novel Cationic Inhibitor of the Bacterial Hsp70 Chaperone. Biochemistry 2020, 59 (20), 1946–1960. 10.1021/acs.biochem.0c00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Cereghetti G; Wilson-Zbinden C; Kissling VM; Diether M; Arm A; Yoo H; Piazza I; Saad S; Picotti P; Drummond DA; Sauer U; Dechant R; Peter M Reversible Amyloids of Pyruvate Kinase Couple Cell Metabolism and Stress Granule Disassembly. Nat. Cell Biol 2021, 23 (10), 1085–1094. 10.1038/s41556-021-00760-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Wang RY-R; Noddings CM; Kirschke E; Myasnikov AG; Johnson JL; Agard DA Structure of Hsp90–Hsp70–Hop–GR Reveals the Hsp90 Client-Loading Mechanism. Nature 2022, 601 (7893), 460–464. 10.1038/s41586-021-04252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).van der Kant R; van Durme J; Rousseau F; Schymkowitz J SolubiS: Optimizing Protein Solubility by Minimal Point Mutations. In Protein Misfolding Diseases: Methods and Protocols; Gomes CM, Ed.; Methods in Molecular Biology; Springer: New York, NY, 2019; pp 317–333. 10.1007/978-1-4939-8820-4_21. [DOI] [PubMed] [Google Scholar]
- (105).Hunter JD Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 2007, 9 (3), 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- (106).Knarr G; Gething MJ; Modrow S; Buchner J BiP Binding Sequences in Antibodies. J. Biol. Chem 1995, 270 (46), 27589–27594. 10.1074/jbc.270.46.27589. [DOI] [PubMed] [Google Scholar]
- (107).Marcinowski M; Rosam M; Seitz C; Elferich J; Behnke J; Bello C; Feige MJ; Becker CFW; Antes I; Buchner J Conformational Selection in Substrate Recognition by Hsp70 Chaperones. J. Mol. Biol 2013, 425 (3), 466–474. 10.1016/j.jmb.2012.11.030. [DOI] [PubMed] [Google Scholar]
- (108).Wang C; Greene D; Xiao L; Qi R; Luo R Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci 2018, 4, 87. 10.3389/fmolb.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Liu X; Liu J; Zhu T; Zhang L; He X; Zhang JZH PBSA_E: A PBSA-Based Free Energy Estimator for Protein-Ligand Binding Affinity. J. Chem. Inf. Model 2016, 56 (5), 854–861. 10.1021/acs.jcim.6b00001. [DOI] [PubMed] [Google Scholar]
- (110).Chen J; Brooks CL; Khandogin J Recent Advances in Implicit Solvent-Based Methods for Biomolecular Simulations. Curr. Opin. Struct. Biol 2008, 18 (2), 140–148. 10.1016/j.sbi.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Bordner AJ Towards Universal Structure-Based Prediction of Class II MHC Epitopes for Diverse Allotypes. PLOS ONE 2010, 5 (12), e14383. 10.1371/journal.pone.0014383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Karakulak T; Rifaioglu AS; Rodrigues JPGLM; Karaca E Predicting the Specificity-Determining Positions of Paralogous Complexes. bioRxiv January 27, 2021, p 2021.01.26.428202. 10.1101/2021.01.26.428202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Zhang H; Wang P; Papangelopoulos N; Xu Y; Sette A; Bourne PE; Lund O; Ponomarenko J; Nielsen M; Peters B Limitations of Ab Initio Predictions of Peptide Binding to MHC Class II Molecules. PLOS ONE 2010, 5 (2), e9272. 10.1371/journal.pone.0009272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Kar P; Ruiz-Perez L; Arooj M; Mancera RL Current Methods for the Prediction of T-Cell Epitopes. Pept. Sci 2018, 110 (2), e24046. 10.1002/pep2.24046. [DOI] [Google Scholar]
- (115).Liebscher M; Roujeinikova A Allosteric Coupling between the Lid and Interdomain Linker in DnaK Revealed by Inhibitor Binding Studies. J. Bacteriol 2009, 191 (5), 1456–1462. 10.1128/JB.01131-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Zahn M; Kieslich B; Bethold N; Daniel Knappe; Ralf Hoffman; Norbert Strater. Structural Identification of DnaK Binding Sites within Bovine and Sheep Bactenecin Bac7. Protein Pept Lett 2014, 21 (4), 407–412. 10.2174/09298665113206660111. [DOI] [PubMed] [Google Scholar]
- (117).Sekhar A; Rosenzweig R; Bouvignies G; Kay LE Mapping the Conformation of a Client Protein through the Hsp70 Functional Cycle. Proc. Natl. Acad. Sci 2015, 112 (33), 10395–10400. 10.1073/pnas.1508504112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Sekhar A; Velyvis A; Zoltsman G; Rosenzweig R; Bouvignies G; Kay LE Conserved Conformational Selection Mechanism of Hsp70 Chaperone-Substrate Interactions. eLife 2018, 7, e32764. 10.7554/eLife.32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Calloni G; Chen T; Schermann SM; Chang H; Genevaux P; Agostini F; Tartaglia GG; Hayer-Hartl M; Hartl FU DnaK Functions as a Central Hub in the E. Coli Chaperone Network. Cell Rep 2012, 1 (3), 251–264. 10.1016/j.celrep.2011.12.007. [DOI] [PubMed] [Google Scholar]
- (120).Cloutier P; Coulombe B Regulation of Molecular Chaperones through Post-Translational Modifications: Decrypting the Chaperone Code. Biochim. Biophys. Acta 2013, 1829 (5), 443–454. 10.1016/j.bbagrm.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Griffith AA; Holmes W Fine Tuning: Effects of Post-Translational Modification on Hsp70 Chaperones. Int. J. Mol. Sci 2019, 20 (17), 4207. 10.3390/ijms20174207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Nitika; Porter CM; Truman AW; Truttmann MC Post-Translational Modifications of Hsp70 Family Proteins: Expanding the Chaperone Code. J. Biol. Chem 2020, 295 (31), 10689–10708. 10.1074/jbc.REV120.011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Gao T; Newton AC The Turn Motif Is a Phosphorylation Switch That Regulates the Binding of Hsp70 to Protein Kinase C. J. Biol. Chem 2002, 277 (35), 31585–31592. 10.1074/jbc.M204335200. [DOI] [PubMed] [Google Scholar]
- (124).Preissler S; Rato C; Chen R; Antrobus R; Ding S; Fearnley IM; Ron D AMPylation Matches BiP Activity to Client Protein Load in the Endoplasmic Reticulum. eLife 2015, 4, e12621. 10.7554/eLife.12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Jakobsson ME; Moen A; Bousset L; Egge-Jacobsen W; Kernstock S; Melki R; Falnes PØ Identification and Characterization of a Novel Human Methyltransferase Modulating Hsp70 Protein Function through Lysine Methylation*. J. Biol. Chem 2013, 288 (39), 27752–27763. 10.1074/jbc.M113.483248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Lim S; Kim DG; Kim S ERK-Dependent Phosphorylation of the Linker and Substrate-Binding Domain of HSP70 Increases Folding Activity and Cell Proliferation. Exp. Mol. Med 2019, 51 (9), 1–14. 10.1038/s12276-019-0317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Ramazi S; Zahiri J Post-Translational Modifications in Proteins: Resources, Tools and Prediction Methods. Database 2021, 2021, baab012. 10.1093/database/baab012. [DOI] [PMC free article] [PubMed]