

Abstract
Targeted therapy with a tyrosine kinase inhibitor (TKI) such as imatinib is effective in treating gastrointestinal stromal tumor (GIST), but it is rarely curative. Despite the presence of a robust immune CD8+ T-cell infiltrate, combining a TKI with immune checkpoint blockade (ICB) in advanced GIST has achieved only modest effects. To identify limitations imposed by imatinib on the antitumor immune response, we performed bulk RNA sequencing (RNAseq), single-cell RNAseq, and flow cytometry to phenotype CD8+ T-cell subsets in a genetically engineered mouse model of GIST. Imatinib reduced the frequency of effector CD8+ T cells and increased the frequency of naive CD8+ T cells within mouse GIST, which coincided with altered tumor chemokine production, CD8+ T-cell recruitment, and reduced CD8+ T-cell intracellular PI3K signaling. Imatinib also failed to induce intratumoral T-cell receptor (TCR) clonal expansion. Consistent with these findings, human GISTs sensitive to imatinib harbored fewer effector CD8+ T cells but more naïve CD8+ T cells. Combining an IL15 superagonist (IL15SA) with imatinib restored intratumoral effector CD8+ T-cell function and CD8+ T-cell intracellular PI3K signaling, resulting in greater tumor destruction. Combination therapy with IL15SA and ICB resulted in the greatest tumor killing and maintained an effector CD8+ T-cell population in the presence of imatinib. Our findings highlight the impact of oncogene inhibition on intratumoral CD8+ T cells and support the use of agonistic T-cell therapy during TKI and/or ICB administration.
Keywords: CD8+ T cell, gastrointestinal stromal tumor, imatinib, IL15, single-cell RNA sequencing
Introduction
Gastrointestinal stromal tumor (GIST) is the most common human sarcoma with an annual U.S. incidence of about 6,000 [1,2]. GIST is usually driven by a single mutation in the KIT or PDGFRA tyrosine kinase receptor genes. The introduction into the clinic of imatinib, a tyrosine kinase inhibitor (TKI) of the Kit and Pdgfra oncoproteins, has increased the median survival of patients with advanced GIST from 1 year to over 5 years [3–5]. Unfortunately, imatinib is rarely curative and tumor progression occurs at a median of 18 months, typically due to secondary KIT mutations [6–8]. Second- and third-line TKIs such as sunitinib and regorafenib are effective but the median time to tumor progression is only 5–7 months [9,10]. Conventional cytotoxic chemotherapy is not effective in GIST [11]. Therefore, new approaches are needed for this cancer.
We [12–17], and others [18,19] have reported on the robust immune infiltrate in GIST. CD8+ T cells in GIST restrain tumor growth and have high PD1 expression [12,15]. Further, imatinib potentiates the antitumor T-cell response in part through the inhibition of indoleamine 2,3-dioxygenase (IDO) production [12]. Immunotherapies have been tested in clinical trials involving patients with advanced GIST resistant to more than 2 TKIs. Ipilimumab (anti–CTLA-4) plus dasatinib (TKI) resulted in limited clinical efficacy, although tumor IDO suppression may have correlated with an antitumor response [20]. More recently, nivolumab (anti-PD1) was tested alone and in combination with ipilimumab, but without a TKI. Although the median progression-free survival (PFS) of patients in the nivolumab and nivolumab + ipilimumab arm was limited at 11.7 and 8.3 weeks respectively, there was a subset of patients with a PFS of >2 years (n=3/36) [21]. Thus, immunotherapy has not been effective so far in GIST patients, at least using conventional T-cell checkpoint inhibitors.
Given the efficacy of imatinib in GIST, T-cell immunotherapy would ideally be used in combination with a TKI. However, the interplay between Kit or Pdgfra inhibition and the immune response is complex. First, mutation type (KIT or PDGFRA) influences overall intratumoral immunogenicity, with Pdgfra-mutant GISTs harboring more immune cells with greater cytolytic activity compared to Kit-mutant GISTs [16,18]. Pdgfra- and Kit-mutant GISTs also express distinctive cytokine profiles with different driver-derived neoepitopes, which may impact immune checkpoint blockade (ICB) approach [16]. Second, imatinib therapy in mouse and human GIST reduces the frequency of tumor-associated macrophages (TAMs) and skews them to a M2 phenotype [13]. Finally, imatinib restricts the frequency of intratumoral Batf3+ (CD141+ in humans) dendritic cells (DCs), which limits the antitumor effect of CD8+ T cells [13,14].
In this study, we sought to further examine CD8+ T cells in GIST to better understand and combat the limitation of T-cell immunotherapy in the presence of a TKI. Through traditional and next-generation sequencing methods, we found that intratumoral CD8+ T cells have a clear effector memory profile at baseline that is altered to a naïve subtype following imatinib treatment. Additionally, imatinib therapy reduced intratumoral CD8+ T-cell intracellular PI3K signaling and failed to induce T-cell clonal expansion, likely due to its effects on antigen presenting cells. An IL15 superagonist (IL15SA) supported the antitumor and memory profile of CD8+ T cells in the presence of imatinib, and combination therapy with IL15SA and ICB resulted in the greatest tumor killing and maintained an effector CD8+ T-cell population in the presence of imatinib. Last, we found a similar effect of imatinib therapy on CD8+ T-cell subsets in human GIST surgical specimens. Overall, our results indicate the need for T-cell agonism in the immunotherapy of GIST treated with a targeted molecular agent.
Methods
Mice.
8–12 week old KitV558Δ/+ mice [22] on a C57BL/6 background, C56BL/6 mice, as well as congenic CD45.1 mice (Jackson Laboratory), were maintained in a pathogen–free animal facility and age- and sex-matched for experiments to ensure similar tumor sizes. The animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Treatments.
Imatinib mesylate (Novartis) was dissolved in the drinking water at 600 mg/L and provided ad lib as previously described [14]. For in vivo IL15 stimulation, 1.5 μg IL15 (210-15, Peprotech) was incubated with 7 μg IL15Ra (551-MR, R&D Systems) for 30 minutes at 37°C as previously described [23]. IL15:IL15Ra (referred to as IL15SA) or PBS was administered i.p. on days −2, 2, and 6 relative to initiation of treatment with vehicle/imatinib. For in vivo CD8 depletion during IL15SA or imatinib therapy, 200 μg of anti-mouse CD8α (BE0061, BioXCell) or anti-rat IgG2b (BE0090, BioXCell) was administered i.p. on days −5, −4, −3, 1, and 5 relative to initiation of treatment with vehicle/imatinib. For concurrent IL15SA and anti-PD1 (aPD1) therapy, Il15SA, 250μg aPD1 (RMP1-14, BioXCell), PBS, or anti-rat IgG2a (2A3, BioXCell) was administered i.p. on days −2, 2, 6 relative to initiation of treatment with vehicle/imatinib and every 4 days until day 28.
For adoptive transfer experiments, 200 μg of anti-mouse CD8α (BE0061, BioXCell) was administered to KitV558Δ/+ mice i.p. on day 0, followed by 600 mg/L imatinib or vehicle for 5 days. On day 4, splenic CD8+ T cells were isolated from CD45.1 mice by bead sorting (Miltenyi Biotec, cat. #130-104-075) and labeled with CFSE (Invitrogen, cat. # C34554) according to the manufacturer’s instructions. 2 x 106 CD45.1 CD8+ T cells were adoptively transferred to each KitV558Δ/+ mouse via retroorbital injection. Mice were sacrificed 24 hours later on day 5, and cell number was counted using counting beads (Invitrogen, cat. #C36950).
For in vitro experiments, splenic CD8+ T cells were isolated from C57BL/6 mice by bead sorting (Miltenyi Biotec, cat. #130-104-075). Purity was > 90% by flow cytometry (see Flow cytometry). Using 96-well plates, 8 x 104 CD8+ T cells were cultured in serum-complete RPMI medium (RPMI modified with glutamine, HEPES, and phenol red (Gibco, cat. #22400089), and supplemented with 10% FCS (Lab Force, cat. #FB5002-H), 0.05 mmol/L β-mercaptoethanol (Sigma-Aldrich, cat. #M3148), and 1× penicillin–streptomycin (Gibco, cat. #15140-122)) with CD3/CD28 microbeads (Dynabeads, cat. #11456D), as per the manufacture’s protocol. Cells were treated with vehicle (H2O) or 50nm imatinib for 6 hours prior to analysis.
Preparation of cell suspensions.
Excised tumors, spleens, and lymph nodes were minced and incubated in Liberase TL (Roche, cat. #C790D66), and prepared into single-cell suspensions as previously described [14]. There is one main mesenteric draining lymph node in KitV558Δ/+ mice. Cells were analyzed by flow cytometry or prepared for bulk RNA seq or single-cell RNA sequencing (scRNAseq) (see Flow cytometry and Bulk RNAseq, scRNAseq, and bioinformatics). For isolation of CD8+ T cells from KitV558Δ/+ mice, CD45+CD3+NK1.1−CD4−CD8+ cells were sorted directly into TRIzol LS Reagent (Life Technologies, cat. #10296028) using a FACSAria II flow cytometer (BD Biosciences).
Flow cytometry.
Flow cytometry was performed with an LSR Fortessa (BD) and analyzed using FlowJo v10 (BD). Fc receptor blockade was achieved with anti-CD16/32 (clone 2.4G2, BioXCell). Mouse-specific antibodies conjugated to various fluorochromes were purchased from BioLegend (CD45, clone 30-F11; Tbet, clone 4B10), BD Biosciences (CD44, clone IM7; CD117, clone 2B8; CD62L, clone MEL14; NK1.1, clone PK136; CD4, clone GK1.5; CD3, clone 145-2C11; and CD45.1, clone A20), eBioscience (CD8, clone 53-6.7; CD40, clone 1C10; Ki67, clone SolA15; PD1, clone J43; and CD103, clone 2E7), and Invitrogen (Granzyme B, clone NGZB; Propidium Iodide, cat. #556463; phospho-AKT1, clone SDRNR; phosphor-mTOR, clone MRRBY). Human-specific antibodies conjugated to various fluorochromes were purchased from BioLegend (CD45, clone HI30; CD45RO, clone UCHL1), BD Biosciences (PD1, clone MIH4; CD45RA, clone HI100; CD3, clone SK7; CD56, clone B159; CD19, clone HIB19; CD4, clone RPA-T4; and CD8, clone RPA-T8). Intracellular cytokine staining was performed with the Cytofix/Cytoperm Kit (BD Biosciences). CFSE staining was performed using the Invitrogen CFSE Cell Proliferation Kit (cat. #C34554), as directed. Annexin V staining was performed using the eBioscience Annexin V staining kit (cat. #88-8103-72), as directed. CD8+ effector memory T cells (TEM) were defined as CD45+CD3+NK1.1−CD4−CD8+CD44+CD62L−, central memory (TCM) as CD45+CD3+NK1.1−CD4−CD8+CD44+CD62L+, naïve (TN) as CD45+CD3+NK1.1−CD4−CD8+CD44−CD62L+, and resident as CD103+ [24].
Microscopy.
Tissues were formalin-fixed and embedded in paraffin. 5 μm tissue sections were mounted on glass slides by the Veterinary Medicine Comparative Pathology Core. Routine hematoxylin and eosin staining was performed. Slides were scanned with an Aperio VERSA 200 platform at the University of Pennsylvania School of Veterinary Medicine Comparative Pathology Core.
TCRβ sequencing.
DNA extraction of fresh frozen tumor, spleen, and cecum tissue from KitV558Δ/+ mice treated with either vehicle or imatinib was performed using the DNeasy Blood and Tissue Kit (Qiagen, cat. #69504) according to the manufacturer’s instructions. Samples were analyzed by Adaptive Biotechnologies by high-throughput sequencing of the CDR3 TCRβ region using the ImmunoSEQ assay (Adaptive Biotechnologies). Immunological endpoints including unique productive sequences and clonality were assessed through previously described methods [25].
Bulk RNAseq, scRNAseq, and bioinformatics.
Next-generation RNAseq of approximately 20,000 sorted CD8+ T cells from spleen, tumor-draining lymph node, or tumor from KitV558Δ/+ mice treated with vehicle or imatinib was performed by the institutional Integrated Genomics Operations core facility using an Illumina HiSeq 2500 platform (4/group). Tissue was first processed into single-cell suspensions (see Preparation of cell suspensions) and sorted cells were provided to the Integrated Genomics Operations core for analysis. We obtained a minimum of 40–50 million reads per sample, with a read length of 50 bp with paired ends. Sequencing reads were then aligned to the mouse genome (mus musculus, GRCm38.p6) and gene-level counts were calculated using STAR version 2.6.1b. Raw counts were then normalized, and differential gene expression was performed using the R software package DESeq2. GSEA was performed using the java GSEA software package (version 3.0) and Molecular Signatures Database (Broad Institute).
We processed six tumors from 10-week old KitV558Δ/+ mice into single-cell suspensions (see Preparation of cell suspensions). Mice had been treated with vehicle or imatinib for one week (3/group). FACS on a FACSAria Fusion (BD) was used to collect live 7-AAD−CD45+ cells. Then, purified cell suspensions were submitted to the Center for Applied Genomics at the Children’s Hospital of Philadelphia for scRNAseq on the 10x Genomics platform.
The scRNAseq reads were aligned to the GRCm38/mm10 reference genome and processed using Cellranger (10x Genomics, version 3.1). Downstream analyses were performed with the R package Seurat version 3.0 [26]. Cells with fewer than 200 genes, more than 2,500 genes, or more than 10% mitochondrial RNA content were excluded from further analysis, resulting in 50,263 out of 55,085 cells used for downstream analysis (91.2%).
To cluster all the cell types within the dataset, the package sctransform [27] was used to perform normalization, variance stabilization, and variable feature selection on each sample separately. The function “IntegrateData” was used to combine all cells from the 6 samples into one dataset for downstream clustering and comparison while eliminating batch effects and technical variation, where anchors identified and conserved across all 6 samples are used to map all the cells onto one harmonized dataset. The integrated gene expression data were then used for principal component analysis (PCA). Clusters were identified using shared nearest neighbor (SNN)-based clustering using the first 30 principal components with k = 30 and resolution = 0.6. Unsupervised Uniform Manifold Approximation and Projection (UMAP) were generated from the same principal components with a minimum distance of 0.3 and 30 neighbors. This identified 18 clusters, 3 of which had high expression for Cd3d, Cd3e and Cd3g, constituting a total of 20,447 T cells.
T-cell clustering was performed as described above using PCA and SNN-based clustering using the first 30 components, with k = 30 and resolution = 0.5. For UMAP projection, a minimum distance of 0.1 was used with 20 neighbors. Ten CD8+ T-cell clusters, encompassing 13,369 cells were extracted on the basis of exclusive expression of multiple defining genes, and re-clustered as before to identify heterogeneity within these CD8+ T cells.
Differential expression between clusters was performed in Seurat using “MAST”, a generalized linear model created specifically for scRNAseq data that treats cellular detection rate as a covariate. The R package monocle3 [28] was used to investigate de novo gene modules (genes that correlate together across cells) present within the CD8+ T-cell clusters. The identity of the genes in the modules can be found in Supplemental Table S1. Using Louvain community analysis with a resolution of 0.05, differentially expressed genes (using a q value < 0.05) are clustered and grouped into modules. These modules are then compared across clusters to help inform which clusters are defined by which gene modules.
ProjecTILs T-Cell Classification.
The mouse CD8 scRNA-seq data produced by this study was aligned to a reference tumor-infiltrating lymphocyte dataset using the ProjecTILs R package (version 0.5.1).[29] This dataset and subsequent program was developed using scRNA-seq data from 25 samples of melanoma, colon adenocarcinoma, and tumor draining lymph nodes in mice resulting in 16,803 single cells for this reference atlas. A normalized expression matrix of our GIST CD8+ T cells generated by Seurat version 3.0 was provided as input and additional parameters for the function “make.projection” were not changed from the default settings. Using the function “cellstate.predict”, the cell state of each single cell from our data was predicted using a nearest-neighbor algorithm and then overlaid onto the UMAP of the ProjecTILs T Cell Atlas. This analysis followed the recommended steps as described in the ProjecTILs walkthrough found at https://carmonalab.github.io/ProjecTILs.demo/tutorial.html.
Immunohistochemistry.
IHC was performed on tumors from KitV558Δ/+ mice as described previously [15] using the CD31 (Polyclonal, Abcam, cat. #ab28364) antibody. Slides were scanned with MIRAX scan (Zeiss) and analyzed with Pannoramic Viewer.
Patient samples.
Tumor specimens were obtained from 20 patients with GIST who underwent surgical resection of their tumors after approval by an Institutional Review Board protocol (Supplemental Table S2). Patients provided written informed consent prior to resection, and studies were conducted in accordance with the Declaration of Helsinki. Patient information remained deidentified and encrypted in compliance with Health Insurance Portability and Accountability Act regulations. Tumor tissue was sectioned and digested at the time of collection in 5 mg/ml collagenase IV (Sigma-Aldrich, cat. #C5138-5G) and DNase I (0.5 mg/ml; Roche Diagnostics, cat. #C756V81) in HBSS for 30 min while shaking at 37°C. After procurement, all processed cells were immediately analyzed by flow cytometry.
Statistical analysis.
Comparisons between two groups were performed using unpaired two-tailed Student’s t test. Multiple group comparisons were performed using one-way ANOVA comparisons with a Bonferroni post-test for comparison of individual groups. A difference in proportion analysis was used to compare CD8+ T-cell clusters from scRNAseq. Statistics were performed on datasets using Graph Pad Prism 8.0 (Graph Pad Software). A p value <0.05 was considered significant. Data are shown as mean ± SEM or median.
Data Availability.
Bulk RNAseq and scRNAseq data presented in this work are submitted through the Sequencing Read Archive under accession no. PRJNA824054 and PRJNA859907. The TCRb sequencing data is available on the Immunoseq platform under the project name UPenn_00110053_R-01. The remaining data generated and/or analyzed during the current study are available within the article and its Supplementary Data files or are available from the corresponding author upon reasonable request.
Results
Tumor-infiltrating CD8+ T cells display a distinct phenotype in GIST.
Given that chronic imatinib therapy in mouse and human GIST reduces the frequency of intratumoral TAMs and Batf3+ (CD141+ in humans) DCs [13,14], we sought to determine the consequences on intratumoral CD8+ T cells. We investigated tumors from KitV558Δ/+ mice, which contain a point mutation in Kit exon 11, the most frequently affected site in human GIST. KitV558Δ/+ mice develop a single imatinib-sensitive GIST in the cecum with 100% penetrance [22]. Bulk RNAseq was performed on CD8+ T cells sorted from tumors, tumor-draining lymph nodes (i.e., mesenteric nodes), and spleens of 4 untreated KitV558Δ/+ mice. Intratumoral CD8+ T cells clustered apart from matched spleen and node CD8+ T cells (Fig. 1A–B). Hallmark gene set analysis indicated that compared to CD8+ T cells in the tumor-draining lymph node, those in the tumor had increased inflammatory, IFNγ, and IL2/Stat5 signatures as well as increased glycolysis (Fig. 1C). An analysis of antigen response [30] (Fig. 1D) and chemokine/cytokine genes (Supplemental Fig. S1A) showed higher levels of Tnfrsf9 (i.e., 4-1BB ligand receptor), Ccl3/4/5, Ifng, and Fasl in tumor CD8+ T cells. Additionally, tumor CD8+ T cells had higher expression of downstream markers of antigen response, proliferation, and cytolytic function, including Gzmb, Pdcd1 (i.e., PD1), Tbet, and Eomes (Fig. 1E). In contrast, node and spleen CD8+ T cells expressed more of the naïve T cell genes Ccr7 and Il7r.
Figure 1. Tumor-infiltrating CD8+ T cells display a distinct phenotype in GIST.
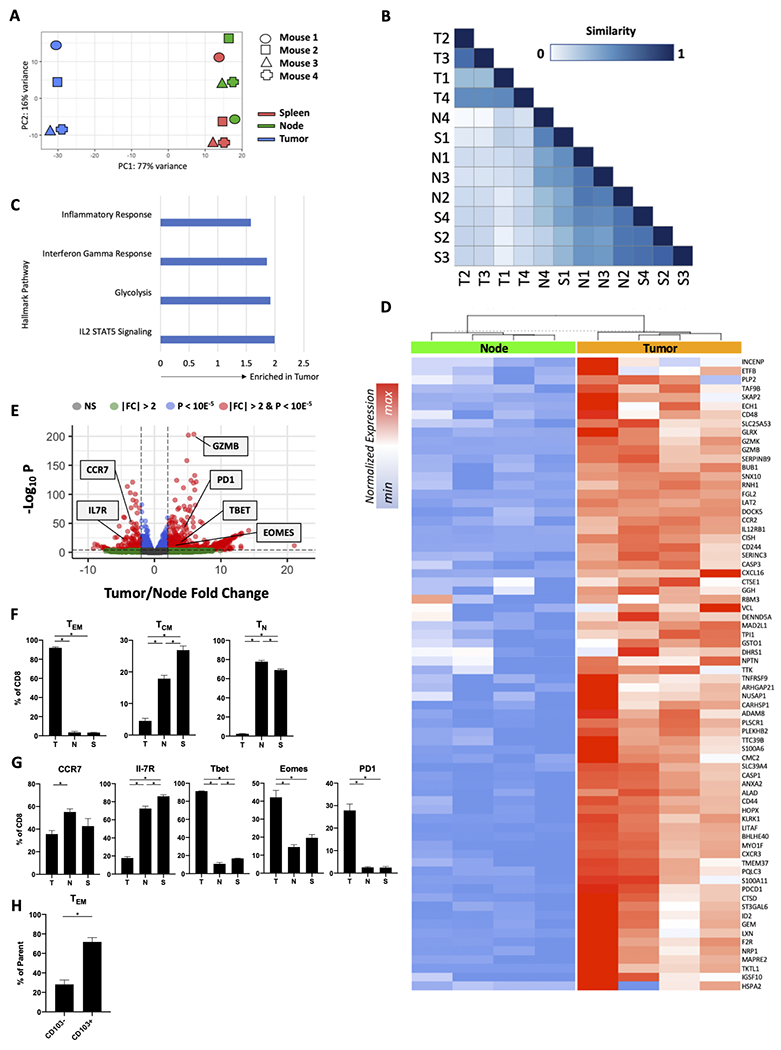
(A-E) Bulk RNAseq was performed on CD8+ T cells sorted from tumors, tumor-draining lymph nodes (i.e., mesenteric nodes), and spleens of 4 untreated KitV558Δ/+ mice. (A) Principal component analysis and (B) similarity of the transcriptomes of the sorted CD8+ T cells from tumor (T), spleen (S), and tumor-draining lymph node (N). (C) Hallmark inflammatory response, interferon gamma response, glycolysis, and IL2/STAT5 signaling pathway gene set enrichment (normalized enrichment score shown; all values p < 0.05), (D) heatmap depicting normalized expression of select antigen response genes, and (E) volcano plot comparing bulk RNAseq of the sorted CD8+ T cells from KitV558Δ/+ tumor or node. (F-H) Flow cytometry of tumors, tumor-draining lymph nodes (i.e., mesenteric nodes), and/or spleens of untreated KitV558Δ/+ mice. (F) Frequency of TEM, TCM, and TN among CD8+ T cells in tumor, spleen, or tumor-draining lymph node (n=4 mice/group). (G) Frequency of CCR7+, IL-7R+, Tbet+, Eomes+, and PD-1+ cells among CD8+ T cells from tumors of KitV558Δ/+ mice (n=4 mice/group). (H) Frequency of CD103+ and CD103− effector CD8+ T cells from tumors of KitV558Δ/+ mice (n=3 mice/group). Each column in (D) represents an individual mouse. NS, non-significant; FC, fold-change. Data represent mean ± SEM; *, p < 0.05.
We confirmed selected findings from the bulk RNAseq analysis with flow cytometry of CD8+ T cells in untreated KitV558Δ/+ tumors. TEM comprised 90% of all CD8+ T cells, while the tumor-draining lymph node and spleen contained mostly TN and TCM subtypes (Fig. 1F, Supplemental Fig. S1B). Consistent with the bulk RNAseq findings, tumor-draining lymph node and spleen CD8+ T cells expressed CCR7 and IL-7R protein, whereas tumor CD8+ T cells expressed the effector/memory markers Tbet, Eomes, and PD1 (Fig. 1G, Supplemental Fig. S1C). We previously found that intratumoral CD8+CD103− cells are likely antitumoral since their presence depended on Batf3+ DCs, which is not the case for CD8+CD103+ cells [14]. CD103+ resident T cells made up about 70% of the TEM population in GIST (Fig. 1H, Supplemental Fig. S1D). Together, these data demonstrate that tumors from KitV558Δ/+ mice contain a distinct population of CD8+ T cells with a predominately effector phenotype that appears more antigen-responsive and proinflammatory than those in the nearby tumor-draining lymph node or spleen.
ScRNAseq reveals intratumoral CD8+ T cell heterogeneity.
The development of high-dimensional profiling techniques such as single-cell transcriptome sequencing has allowed the field of immunology to surpass the traditional and rudimentary definitions of intratumoral CD8+ T cells in both murine and human cancers [29,31]. To investigate the individual gene expression profiles of intratumoral CD8+ T cells, we performed droplet-based scRNAseq of immune cells (CD45+ cells) within KitV558Δ/+ tumors. We obtained transcriptomes from 50,263 tumor-infiltrating immune cells. PCA with dimensional reduction using UMAP and unsupervised clustering identified discrete clusters of T cells, all characterized by high Cd3e, Cd3g, and Cd3d expression (Fig. 2). In untreated tumors, these T-cell clusters comprised 11,641 distinct T cells with expression of Cd4, Cd8a, and/or Foxp3, which we defined as CD4, CD8, and TReg subsets (Fig. 2A–B). Unsupervised clustering of just Cd8a/Cd8b-expressing cells yielded 8,065 cells separated into 10 distinct clusters (Fig. 2C). We then generated gene set modules that contained groups of correlated genes (Supplemental Table S1). This process resulted in 12 modules and we used a heatmap to portray how each module was enriched across the 10 clusters (Fig. 2D). Module 8 included the naïve T-cell markers Ccr7, Sell, Tcf7, and Il7r, which were highly expressed solely in cluster 9 (Fig. 2E, Supplemental Table S1). Clusters 2 and 10 co-expressed Tcf7, Gzmb, Gzmk, low to intermediate Pdcd1, and had a similar association with module 7 genes, suggesting they are TEM [29]. Similarly, clusters 4 and 6 both expressed high levels of granzymes and expressed Pdcd1 and/or Tox, suggesting a terminally differentiated effector subtype [31,32]. Clusters 1 and 5 had low levels of naïve and effector markers and could represent an early activation subset of CD8+ T cells. The resident marker Itgae (CD103) was expressed at an intermediate to high level on many clusters, most notably clusters 3, 4, 6, 7, and 8, consistent with our flow cytometry findings. Overall, these data revealed that CD8+ T cells in GIST exist in diverse stimulated and naïve states.
Figure 2. ScRNAseq reveals intratumoral CD8+ T-cell heterogeneity.
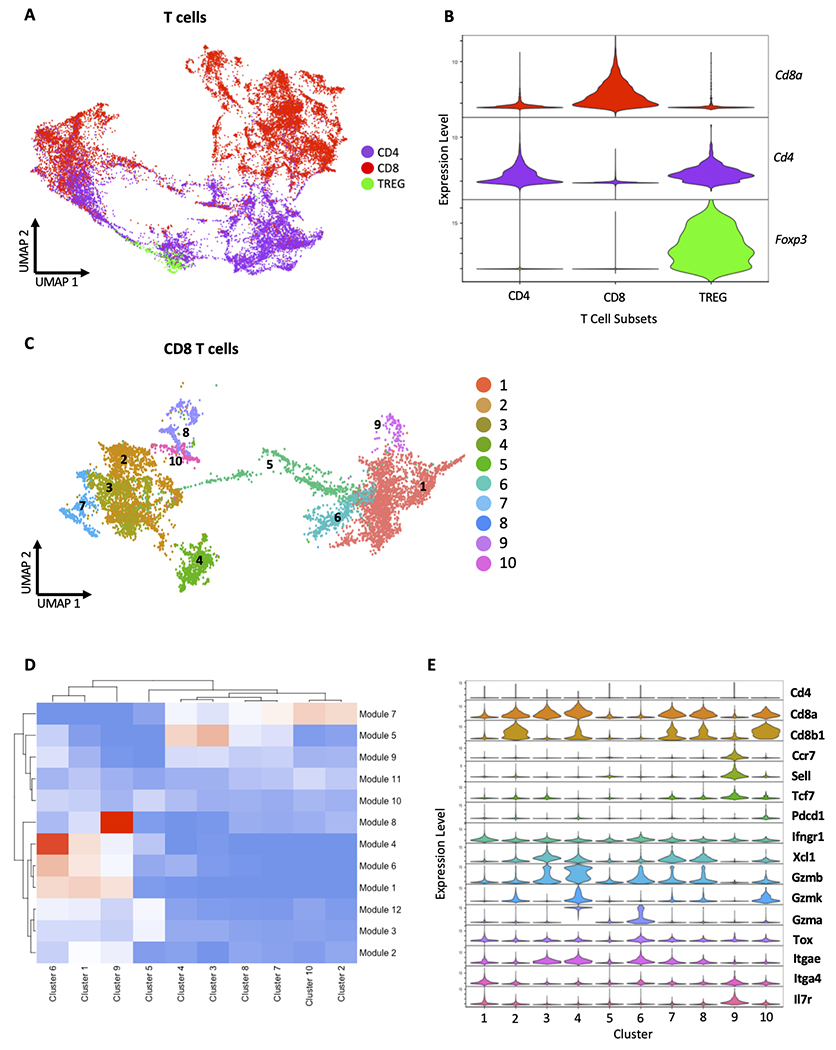
Droplet-based scRNAseq was performed on immune cells (CD45+ cells) sorted from tumors from 3 untreated KitV558Δ/+ mice. (A) Clusters of CD4+ T cells, CD8+ T cells, and Tregs within the CD3+ compartment based on unsupervised clustering by principal component analysis, defined by (B) cd4, cd8a, and foxp3 expression. (C) Numbered clusters within the intratumoral CD8+ T-cell compartment based on unsupervised clustering by principal component analysis. (D) Heatmap of gene expression modules, with stratified module enrichment by CD8+ T-cell clusters. (E) Violin plots of expression of select genes in CD8+ T-cell clusters. (A, C) UMAP plots display scRNAseq profiling of 11,641 cd3d/e/g and 8,065 cd8a/b T cells from 3 mice as detailed in the methods section.
Imatinib expands intratumoral TN and reduces TEM
Once we characterized tumor CD8+ T cells in untreated KitV558Δ/+ mice, we determined the effects of imatinib therapy on intratumoral CD8+ T cells. First, we performed bulk RNAseq on sorted CD8+ populations from KitV558Δ/+ tumors, local tumor-draining lymph nodes, and spleen after vehicle or 1 week of imatinib (n=4 mice/group). PCA demonstrated distinct clustering of CD8+ T-cell populations based on treatment status (Fig. 3A). Although markers of stimulation including Cd44 were upregulated in response to imatinib, a substantial number of effector genes were downregulated, including Gzmb and Tnfrsf9 (Fig. 3B). There was a substantial increase in naïve T cell–signature transcripts in imatinib-treated tumors, including Tcf7 and Sell (Fig. 3B) as well as IL7r (Fig. 3C), which were not altered in tumor-draining lymph node or spleen T cells.
Figure 3. Imatinib expands intratumoral TN and reduces TEM.
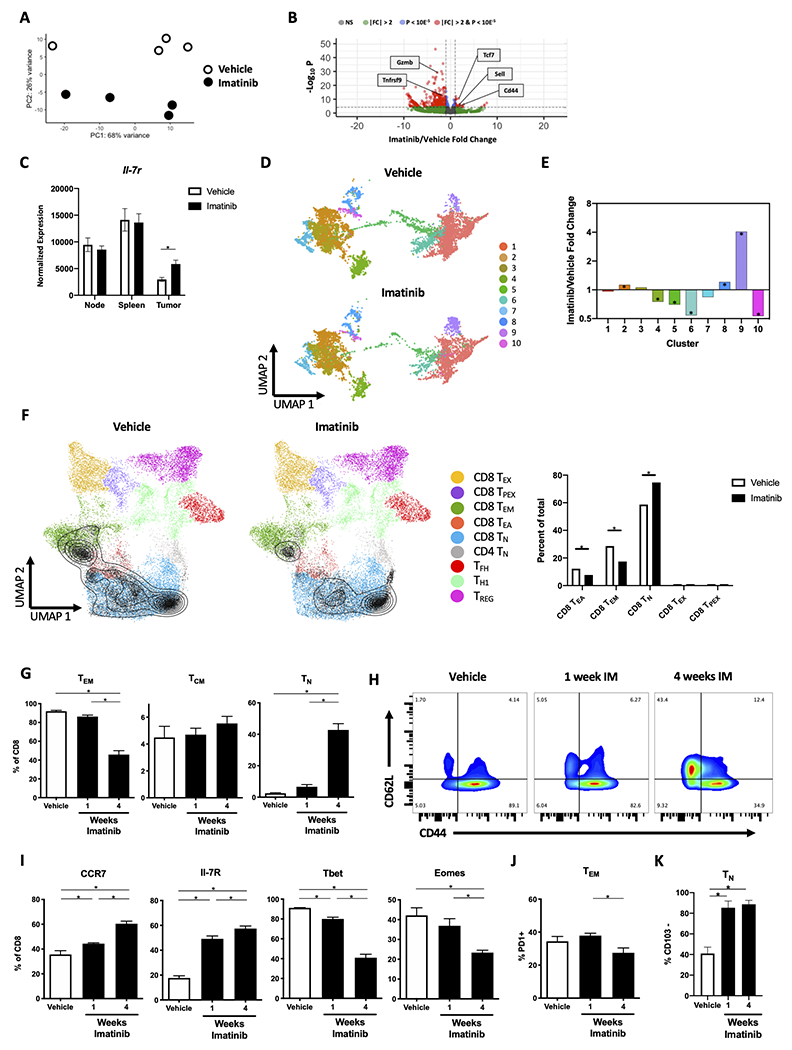
(A-C) KitV558Δ/+ mice were treated with vehicle or imatinib for 1 week (n=4 mice/group). At that time, tumors, tumor-draining lymph nodes (i.e., mesenteric nodes), and spleens were harvested and bulk RNAseq was performed on CD8+ T cells sorted from the tissues. (A) Principal component analysis and (B) volcano plot of sorted CD8+ T cells from tumor. (C) Normalized expression of Il7r in node, spleen, or tumor. Each point in (A) represents a sample from an individual mouse. (D, E) KitV558Δ/+ mice were treated with vehicle or imatinib for 1 week (n=3 mice/group). At that time, scRNAseq was performed on CD45+ cells sorted from the tumors. (D) UMAP plot of CD8+ T-cell clusters and (E) cluster imatinib/vehicle fold change in tumors. (F) CD8+ T-cell clusters from (D), represented as grey triangles/isobars were projected onto a published reference atlas [29] and the difference in proportion of T-cell states including exhausted (TEX), precursor exhausted (TPEX), TEM, early activation (TEA), and TN were compared between vehicle and imatinib groups. (G-J) KitV558Δ/+ mice were divided into 3 groups on Day 0 and all mice were sacrificed on Day 28. Mice were treated with vehicle or imatinib for 1 or 4 weeks and tumors were analyzed by flow cytometry for (G) Frequency of TEM, TCM, and TN among CD8+ T cells; (H) CD44+ and CD62L+ expression in CD8+ T cells; (I) frequency of CCR7+, IL-7R+, Tbet+, Eomes+ cells among CD8+ T cells; (J) frequency of PD1+ cells among TEM CD8+ T cells; and (K) frequency of CD103− TN cells among CD8+ T cells (n=3–4 mice/group). Data represent mean ± SEM; *, p < 0.05.
Next, we performed scRNAseq analysis of CD8+ T cells from KitV558Δ/+ tumors to determine the effect of 1 week of imatinib treatment on the 10 clusters. Consistent with our findings with bulk RNAseq, there was a 4-fold increase in the proportion of cells in cluster 9, which contained the highest naïve signature (Fig. 3D–E). We also observed a relative decrease in putative effector clusters with (4 and 6) and without (5 and 10) high Itgae expression. Additionally, we projected our CD8+ T-cell clusters onto a published reference atlas derived from murine tumors and draining lymph node from 6 studies to further identify T-cell states (Fig. 3F) [29]. The major states identified were effector memory, early activation, and naïve subsets, while few cells were characterized as exhausted or progenitor exhausted. The reference atlas identified a larger proportion of naïve T cells at baseline when compared to our flow cytometry results utilizing CD44 and CD62L. Naïve T cells increased in proportion after imatinib therapy, whereas effector memory and early activation T cells decreased.
To confirm our findings, we used flow cytometry to differentiate CD8+ T-cell subsets after 1 and 4 weeks of imatinib treatment (Supplemental Fig. S2A). At 1 week of therapy, tumors were smaller and had a trend toward decreased TEM and increased TN cell populations, and these effects were more pronounced after 4 weeks (Supplemental Fig S2B, Fig. 3G–H). Increases in CCR7 and IL7R and decreases in Tbet and Eomes were also seen (Fig. 3I, Supplemental Fig. S2C). A decreased proportion of CD8+ T cells in the tumor was found at 1 and 4 weeks of imatinib therapy, but this change did not appear to be due to increased apoptosis of CD8+ T cells (Supplemental Fig. S2D–E). Within the TEM subset, there was stable PD1 protein expression at 1 week that decreased by 4 weeks, indicating less differentiation and stimulation over time (Fig. 3J). Consistent with our prior work [14], the TN population at 1 and 4 weeks of imatinib therapy consisted almost entirely of CD103−CD8+ T cells (Fig. 3K) suggesting they were derived peripherally. We considered whether older KitV558Δ/+ mice had altered CD8+ T-cell subsets at baseline compared to younger mice but found no difference (Supplemental Fig. S2F). Additionally, CD31 staining was similar in tumors with and without 4 weeks of imatinib therapy, indicating that the increased TN population was not due to increased tumor vascularity (Supplemental Fig. S2G). Collectively, these data indicated that imatinib therapy causes a phenotypic shift from TEM to TN in the tumor microenvironment, particularly with longer time of treatment.
Imatinib restricts PI3K signaling and clonal expansion in intratumoral CD8+ T cells.
To identify the mechanism of the phenotypic shift from TEM to TN in GIST following imatinib therapy, we first examined our bulk RNAseq data. Hallmark gene analysis established that CD8+ T cells in imatinib-treated tumors at 1 week had many downregulated pathways related to T-cell stimulation, inflammation, and proliferation compared to controls (Fig. 4A). Bulk RNAseq demonstrated that Pdcd1 was upregulated (Fig. 4B), which we suspect was a result of transient stimulation of CD8+ T cells due a decrease in IDO, which we found previously [12]. We found that changes in the CD8+ T-cell subsets coincided with alterations in chemokine production by the tumor. On bulk RNAseq of tumors and sorted CD8+ populations, most chemokines in the tumor were decreased after imatinib treatment, while the majority of their complementary receptors were increased in CD8+ T cells (Supplemental Fig. S3A–B). One chemokine that increased after treatment was Ccl21, which binds Ccr7 and recruits naïve T cells [33]. To test whether imatinib therapy affected the recruitment of CD8+ T cells into the tumor, following CD8+ T-cell depletion we adoptively transferred CSFE-labeled CD8+ T cells from CD45.1 mice into KitV558Δ/+ mice receiving imatinib therapy. We found significantly fewer CD45.1+CSFE+CD8+ T cells per gram of tumor in KitV558Δ/+ mice receiving imatinib compared to controls, while the proportion of CD45.1+CSFE+CD8+ T cells in the spleen was similar between the two groups (Fig. 4C).
Figure 4. Intratumoral CD8+ T cells have restricted PI3K signaling and lack clonal expansion in response to imatinib.
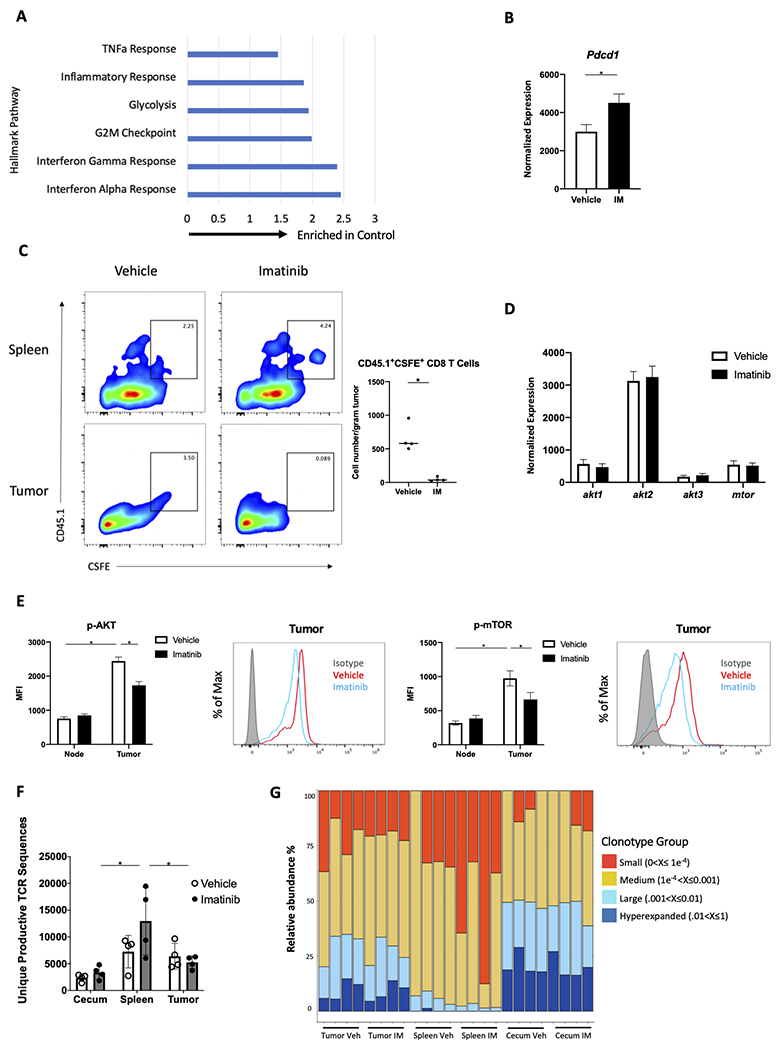
(A-B) KitV558Δ/+ mice were treated with vehicle or imatinib for 1 week (n=4 mice/group) and sorted CD8+ T cells from tumors were analyzed by bulk RNAseq for (A) hallmark TNF alpha response, inflammatory response, glycolysis, G2M checkpoint, interferon gamma response, and interferon alpha response gene set enrichment (normalized enrichment score shown; all values p < 0.05); and (B) Pdcd1 expression. (C) Following CD8+ T-cell depletion with one dose of anti-CD8 on day 0, KitV558Δ/+ mice were treated with vehicle or imatinib for 5 days. On day 4, mice received CSFE-labeled splenic CD8+ T cells from congenic CD45.1 mice by adoptive transfer. On day 5, frequency of CSFE+CD45.1+CD8+ T cells in the spleen and tumor was determined by flow cytometry, as well as the cell count in the tumor (n=4 mice/group, pooled from two independent experiments). (D) Normalized expression of Akt1, Akt2, Akt3, and Mtor in sorted CD8+ T cells in tumors from KitV558Δ/+ mice following bulk RNAseq, as in Fig. 4A,B (n=4 mice/group). (E) Median fluorescence intensity (MFI) and representative histograms of p-AKT+ and p-mTOR+ cells among CD45+CD3+NK1.1−CD4−CD8+ T cells in tumor and tumor-draining lymph node from untreated KitV558Δ/+ mice (5 mice/group, repeated twice). (F, G) KitV558Δ/+ mice were treated with vehicle (Veh) or imatinib (IM) for 1 week (n=4 mice/group). At that time, tumors, spleens and cecum were harvested and analyzed by high-throughput sequencing of the CDR3 TCRβ region. (F) Unique productive TCR sequences and (G) occupied repertoire space of small, medium, large, and hyperexpanded clonotype groups where units represent clonal frequency. Data represent mean ± SEM; *, p < 0.05.
Our previous work demonstrated that imatinib therapy caused both an M1 to M2 transition of intratumoral macrophages as well as depletion of Batf3+ DCs [13,14], so we suspected decreased antigen presentation and/or co-stimulation contributed to intratumoral CD8+ naivety following imatinib treatment. To explore this, we assessed TCR and/or CD28 costimulation by measuring downstream PI3K signaling, as others have done [34]. First, we used our bulk RNAseq data to evaluate Akt and mTor transcription in imatinib-treated tumors, which showed no difference compared to controls (Fig. 4D). By flow cytometry, tumor CD8+ T cells had higher baseline levels of p-Akt and p-mTor compared to CD8+ T cells from tumor-draining lymph node, and imatinib decreased phosphorylation, signifying a reduction in PI3K signaling (Fig. 4E). To determine whether imatinib had a direct effect on CD8+ T cells, isolated splenic CD8+ T cells were cultured in the presence of CD3/CD28 beads with or without imatinib, which demonstrated no difference in p-AKT and p-mTOR MFI (Supplemental Fig. S3C). There was, however decreased transcription of the scaffold adaptor for PI3K (Pik3ap1) (Supplemental Fig. S3D), which is critical for propagating CD8+ T-cell PI3K signaling and clonal expansion [35].
Next, we assessed TCR cell clonal expansion in matched KitV558Δ/+ tumors, spleen, and cecum by ImmunoSEQ assay (Adaptive Biotechnologies). There was a trend towards more unique productive TCR sequences in spleen CD8+ T cells than tumor or cecum CD8+ T cells at baseline, and this difference was significant following imatinib treatment (Fig. 4F). The occupied repertoire space of large and hyperexpanded clonotypes was unchanged after treatment, as was clonality, as indicated by Simpson clonality (Fig. 4G, Supplemental Fig. S3E), indicating a lack of TCR clonal expansion. TCRs were very similar in tumor and cecum from the same mouse, and TCRs in untreated tumors were similar at baseline and after imatinib treatment (Supplemental Fig. S3F). Thus, intratumoral CD8+ T cells have diminished chemokine and PI3K signaling and lack clonal expansion following imatinib therapy, supporting the hypothesis that both recruitment and deficiency of antigen presentation/costimulation contribute to the changes in the CD8+ T-cell subsets following imatinib therapy.
Human GIST display differential CD8+ T-cell subsets.
To determine the clinical relevance of our findings from KitV558Δ/+ mice, we performed flow cytometry on 20 freshly obtained GIST specimens from 20 patients (Supplemental Table S2). Tumors were classified as untreated, sensitive, or resistant to imatinib at the time of surgery based on serial radiological assessment, as described previously [12]. Since we found that the TEM and TN populations in murine GIST were significantly altered in imatinib-treated tumors, we first evaluated expression of the human naïve/memory markers CD45RA and CD45RO. We found that the proportion of CD45RA+CD45RO−CD8+ T cells was significantly increased in sensitive tumors after a median of 12 months of imatinib treatment (Fig. 5A, Supplemental Fig. S4). Further analysis of the CD8+ TN (CD45RA+CCR7+), TCM (CD45RA−CCR7+), TEM (CD45RA−CCR7−), and effector memory cells re-expressing CD45RA (TEMRA, CD45RA+CCR7 −) showed that the abundance of TN and TEM cells was higher and lower respectively between the untreated and sensitive groups (Fig. 5B). These changes were not present in the resistant group. There was an increase in terminally differentiated TEMRA cells in the sensitive group compared to the untreated group, indicating that the sensitive group had a terminally differentiated CD8+ T-cell subset of uncertain proliferative potential [36,37]. Overall, sensitive human GISTs had a lower and higher proportion of TEM and TN CD8+ T cells compared to untreated GIST.
Figure 5. Human GIST display differential CD8+ T-cell subsets.
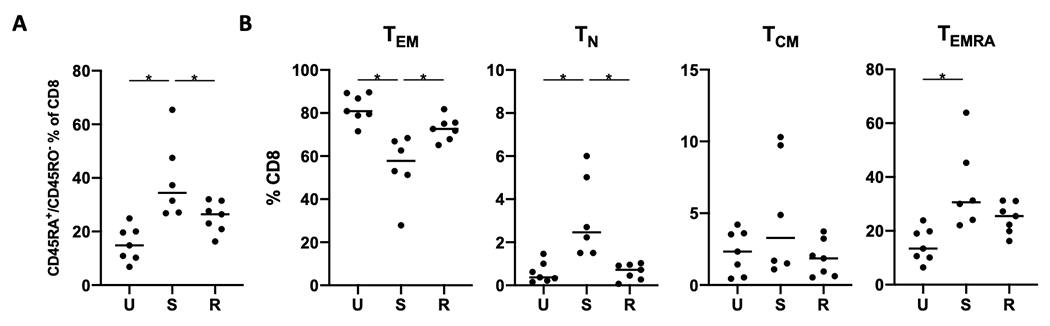
CD8+ T cells from human GIST specimens (20 tumor specimens from 20 patients) were analyzed by flow cytometry for (A) frequency of CD45RA and CD45RO expression among CD8+ T cells and (B) frequency of memory subsets among the CD8+ T-cell population. Line indicates median; *, p < 0.05
IL15-mediated stimulation with ICB improves the response to imatinib.
Given the seemingly negative effects of imatinib on intratumoral CD8+ T cells, particularly after longer treatment times, we investigated methods to improve the antitumoral immune response. We considered stimulation with IL15 given its known influence on the PI3K pathway in T cells [38] and because it was most highly expressed by intratumoral Batf3+ DCs by scRNAseq, and these cells are reduced with imatinib (Supplemental Fig. S5A) [14]. We investigated which of our scRNAseq CD8+ clusters expressed Il2rb and Il2rg, which comprise the heterodimer required for IL15 signaling. Although Il2rg expression was similar among all clusters, Il2rb was highly expressed in the effector cluster 6 and expressed at the lowest level in the naïve cluster 9 (Supplemental Fig. S5B). Hence, we speculated that IL15 may preferentially propagate an effector T-cell response. The IL15/IL15Rα superagonist (IL15SA) has been demonstrated to increase the half-life of IL15 and support naïve and memory CD8+ T-cell proliferation, and new formulations have been used with limited side effects in early clinical trials [39,40]. Therefore, we treated KitV558Δ/+ mice with imatinib and IL15SA for 1 week (Fig. 6A). At 1 week, there were additive effects of IL15SA with imatinib therapy, as tumors weighed 30% less than after treatment with imatinib alone (Fig. 6B, Supplemental Fig. S5C). IL15SA therapy alone did not reduce tumor size (Supplemental Fig. S5D). There was a similar histological appearance of the tumors among treatment groups (Supplemental Fig. S5E). We also considered the effect of IL15SA on natural killer (NK) cells, as they express Il2rb in GIST (Supplemental Fig. S5F). However, NK cells are infrequent in GIST, did not expand after treatment with imatinib and IL15SA compared to imatinib alone (Supplemental Fig. S5G), and as we showed previously, NK-cell depletion in KitV558Δ/+ mice does not affect tumor size [12]. Combination therapy increased TEM compared to imatinib alone and blunted the increase in TN induced by imatinib (Fig. 6C, Supplemental Fig. S5H).
Figure 6. IL15-mediated stimulation with immune checkpoint blockade improves the response to imatinib.
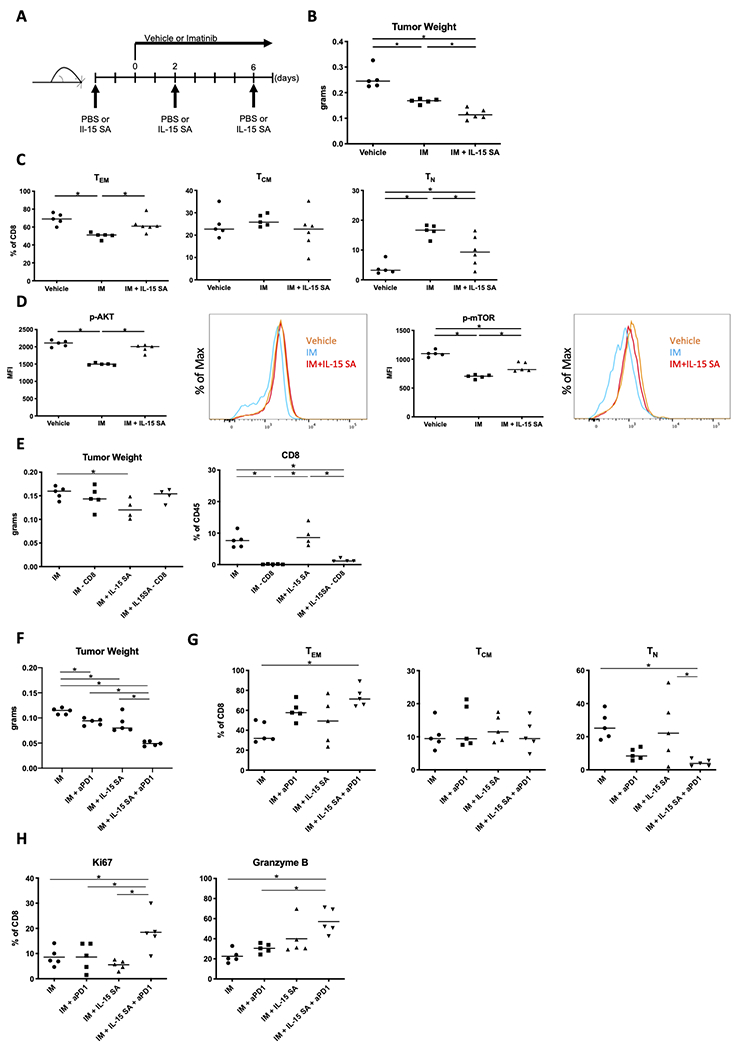
(A) KitV558Δ/+ mice were treated with IL15SA three times over the course of 9 days and given imatinib daily for 7 days. Controls received PBS for 9 days and vehicle or imatinib from days 0 to 7. (B) Tumor weights of KitV558Δ/+ GIST treated as described in (A) (n=5–6 mice/group, repeated twice). Flow cytometry of tumors was performed to determine: (C) Frequency of TEM, TCM, and TN among intratumoral CD8+ T cells; and (D) Median fluorescence intensity (MFI) and representative histogram of p-AKT+ or p-mTOR+ CD8+ T cells in tumors from KitV558Δ/+ mice (n=5 mice/group, repeated twice). (E) KitV558Δ/+ mice were treated with IM (imatinib), IM + depleting anti-CD8 (−CD8), IM + IL15SA, or IM + IL15SA + −CD8 and assessed for tumor weight and frequency of CD8+ T cells among CD45+ cells, as assessed by flow cytometry (4–5 mice/group). (F) KitV558Δ/+ mice were treated with IM, IM + aPD1 (anti-PD1), IM + IL15SA, or IM + IL15SA + aPD1 for 4 weeks and assessed for tumor weight (5 mice/group, pooled from two independent experiments). Flow cytometry was used to assess these mice for (G) CD8+ T-cell subsets and (H) frequency of Ki67+ and Granzyme B+ cells among CD8+ T. Line indicates median; *, p < 0.05.
Since we found that imatinib therapy decreased Akt and mTor signaling in intratumoral CD8+ T cells, we investigated the effect of IL15SA combination therapy on the PI3K pathway. Adding IL15SA to imatinib therapy fully restored CD8+ T-cell p-Akt and partially restored p-mTor expression by flow cytometry (Fig. 6D). To determine whether the antitumor effects of imatinib and IL15SA combination therapy depended on CD8+ T cells, we used an antibody to deplete the CD8+ cells. This abrogated the benefit of adding IL15SA during imatinib therapy as there was no incremental change in tumor weight and CD8+ T cells remained significantly depleted despite IL15SA therapy (Fig. 6E, Supplemental Fig. S5I). Further, we sought to test the effect on imatinib therapy of Il15SA, ICB, or both IL15SA and ICB, particularly at longer time points. Although tumors treated with IL15SA and imatinib for 4 weeks were smaller than those treated with imatinib alone (Fig. 6F), the proportion of intratumoral TEM was not increased as was seen at 1 week of therapy, possibly due to tachyphylaxis. Combined therapy using IL15SA and anti-PD1 with imatinib increased the proportion of intratumoral TEM and decreased TN at 4 weeks (Fig. 6G, Supplemental Fig. S5J), as well as had the greatest tumor killing. Consistent with these findings, triple therapy resulted in the highest proportion of CD8+ T cells expressing Ki67 and granzyme B at 4 weeks of treatment (Fig. 6G, Supplemental Fig. S5K). Therefore, treatment of GIST with imatinib and IL15SA improved tumor response and rescued PI3K signaling in CD8+ T cells at an early time point. Combination therapy with IL15SA and ICB was needed to maximize tumor killing and effector CD8+ T cell subsets for longer durations of therapy.
Discussion
We investigated the molecular signatures of intratumoral CD8+ T cells in KitV558Δ/+ mice at the single-cell level. We found that CD8+ T cells in GIST have a distinctive molecular profile consistent with an effector phenotype and cytotoxic ability compared to matched CD8+ T cells from secondary lymphoid organs. CD8+ T cells from the local lymph node were more similar to CD8+ T cells from the spleen than the tumor. These findings suggest that there may not be communication between the tumor and draining lymph node, which is consistent with the fact that GISTs, like most sarcomas, do not metastasize to lymph nodes. Our TCR sequencing results support the existence of tumor-specific antigens, as the relative abundance of hyperexpanded TCR clones was higher in untreated tumors, where there are more mature CD8+ T cells compared to spleen. Immunogenic antigens have not been identified in GIST, although mutant KIT or PDGFRA peptides are attractive possibilities. We previously estimated that the majority of patients with an oncogenic mutation in PDGFRA or KIT produced at least one high-affinity neoepitope [16]. That TCR sequences were similar between cecum and tumor suggests that T cells from the surrounding intestine infiltrate GIST. Despite the immunogenicity of intratumoral CD8+ T cells and their activation following IDO inhibition with imatinib [12], they did not appear to undergo clonal expansion. Overall, our findings highlight the limitation of using imatinib as a sole immune modulator.
The present data build on our previous work [12–14,41] demonstrating the widespread effects of imatinib on the tumor immune microenvironment. Here, we have further investigated the effects of this TKI on intratumoral CD8+ T cells with respect to naïve and effector subsets, gene expression at the bulk and single-cell levels, and TCR phenotype. Our previous work investigated the effect of IDO inhibition through imatinib treatment on CD8+ T-cell functionality at a short time point (1 week), whereas here we have discovered that chronic (4 weeks) imatinib therapy increases the proportion of naïve CD8+ T cells within the tumor in our mouse model of GIST, which may have major implications for the combined use of TKIs and immunotherapy. To attempt to maximize the antitumor effect of CD8+ T cells during imatinib therapy, we treated GIST with imatinib and IL15SA therapy, with or without concomitant anti-PD1 therapy.
The immune response and its modulation by TKI treatment in GIST is complex. Utilizing scRNAseq in our mouse model of GIST allowed a unique view into the early effects of imatinib on molecularly defined CD8+ T-cell subsets. scRNAseq in melanoma responsive to PD1 blockade has demonstrated clonal replacement of CD8+ T cells by newly infiltrating T cells [41]. In keeping with our integration method of scRNAseq, we did not find a discrete CD8+ T-cell cluster solely defined by either vehicle or imatinib treated cells in our model. Rather, we observed a shift in the relative frequencies of our defined clusters, which mimicked our flow cytometry data. We also saw a similar shift in T-cell subsets when analyzing a previously published reference atlas [29]. scRNAseq is revealing the intricacies of the tumor immune response and challenging decades of research using traditional techniques. One study used scRNAseq to associate T-cell states in primary human melanoma and metastatic lymph nodes to checkpoint blockade response. Through this analysis [42] and others [29], these authors concluded that tumor-infiltrating lymphocytes (TIL) expressing TCF7, and not conventional markers of tumor response including PD1, were enriched in responders vs. non-responders both pre- and post-treatment. Further, when TILs from primary melanoma and metastatic lymph node were projected onto a T-cell reference atlas of 25 scRNAseq samples from six studies, responders had a relative 2–4 fold increase in naïve CD4+ and CD8+ T cells compared to non-responders [29]. In our study, we suspected that the naïve CD8+ T-cell infiltrate following imatinib limited an effective immune response. However, it is evident that predictors of response to immunotherapy are both tumor- and agent-specific. To further investigate the potential interaction of our CD8+ T-cell clusters within the tumor immune environment, we plan to apply the novel technique of spatially resolved transcriptomics, which is beyond the scope of this report.
There have been few studies evaluating TILs in human GIST, but the presence of TILs has been positively correlated with progression-free survival [43]. We previously reported that effector CD8+CD45RO+CCR7− T cells are enriched in GIST compared to matched blood, and PD-L1 is expressed in GIST but reduced with imatinib [14,15]. Others have compared GIST to various sarcomas and found that GISTs have a high number of infiltrating CD8+ T cells comprised mostly of TEM and TEMRA subtypes, consistent with our findings [44]. GIST also has lower expression of co-stimulatory ligands and a higher polyclonal T-cell repertoire compared to other sarcomas, with 5 of 9 GISTs in this study having received imatinib [44]. Tumor mutational burden (TMB) has recently been used to predict response to immunotherapy. However, TMB may not be a useful marker in GIST, which is typically driven by a single mutation and has a low TMB of 1.8 mutations/megabase [45]. In fact, GIST, and other tumors driven by activating mutations, may prove to be an exception to the normal relationship between TMB and immune response [46]. There have been no studies using TMB to evaluate immunotherapy response in GIST. In addition, TMB will need to be validated as a predictor of response to immunotherapy when combined with targeted molecular agents such as TKIs [47].
Imatinib has changed the standard of care for patients with GIST in both the adjuvant and neoadjuvant setting, and it has prolonged median survival in advanced GIST from a historical 16–19 months to 5 years [3,48]. The interpretation of our human data is limited because patients with imatinib-sensitive GISTs in this study had already been treated with the TKI for a median of 12 months. Furthermore, we cannot determine whether the observed changes in CD8+ T-cell subsets were directly related to imatinib therapy. Nonetheless, there were correlations in the changes of TN and TEM in tumors of KitV558Δ/+ mice and sensitive human GIST. CD8+ T cells in patients with resistant GIST (progressing at the time of surgery despite being treated with a TKI) reverted to levels seen in untreated GISTs, emphasizing the central role of oncogenic signaling in GIST. Given imatinib’s broad impact, we venture whether the future of GIST treatment and other cancers treated with TKIs lies in alternating immunotherapy with TKIs in order to maximize the benefit of both therapies.
Historically, T-cell agonism in cancer therapy was achieved with IL2, which induced durable responses in patients with metastatic melanoma and renal cell carcinoma, albeit with some toxicity [49]. Recombinant IL15 has also been trialed, but due to the variable expression of IL15R, which acts to stabilize and increase the biological activity of IL15, high doses were required to achieve responses [50]. The IL15SA ALT-803 has been tested in patients as a monotherapy and in combination with PD1 and CTLA-4 checkpoint blockade [40,51,52]. ALT-803 alone increased circulating NK and CD8+ T cells, as well as Ki-67 expression on CD8+ T cells [40]. In a study of non-small cell lung cancer [52], ALT-803 induced objective responses after treatment relapse or failure following anti-PD1 immunotherapy. The same group also found that ALT-803 decreased the clonality of peripheral TCRs, which could imply an increase in neoantigen-reactive T cells. We found a decrease in clonality in our spleen samples following imatinib treatment, suggesting imatinib could have a similar effect. In addition, we found that IL15SA therapy in GIST had a pan-CD8+ T-cell effect despite our hypothesis that it would preferentially augment effector cells. This finding may be beneficial in GIST, since imatinib alters the relative frequencies of CD8+ T cells including decreasing the TEM population. Collectively, our results provide the rationale for including an IL15SA in addition to checkpoint blockade in future trials of GIST.
Supplementary Material
SYNOPSIS.
The tyrosine kinase inhibitor imatinib is shown to alter intratumoral CD8+ T-cell subtypes and antitumor activity in gastrointestinal stromal tumor. Combining T-cell agonistic therapy with imatinib improves antitumor responses in a mouse model of the disease.
Acknowledgments
The investigators were supported by NIH grants R01 CA102613 and T32 CA251063, the David Foundation, Betsy Levine-Brown and Marc Brown, and the GIST Cancer Research Fund (RPD).
The authors are grateful to the assistance of the Office of Laboratory Animal Welfare, University Laboratory Animal Resources, and flow cytometry staff at the University of Pennsylvania.
Disclosures:
Research grant from BluePrint Medicines (RPD). The authors declare no potential conflicts of interest.
References
- 1.Mastrangelo G, Coindre JM, Ducimetière F, Dei Tos AP, Fadda E, Blay JY, et al. Incidence of soft tissue sarcoma and beyond: a population-based prospective study in 3 European regions. Cancer. 2012;118(21):5339–48. [DOI] [PubMed] [Google Scholar]
- 2.Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–5. [DOI] [PubMed] [Google Scholar]
- 4.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. [DOI] [PubMed] [Google Scholar]
- 5.Gold JS, van der Zwan SM, Gonen M, Maki RG, Singer S, Brennan MF, et al. Outcome of metastatic GIST in the era before tyrosine kinase inhibitors. Ann Surg Oncol. 2007;14(1):134–42. [DOI] [PubMed] [Google Scholar]
- 6.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–34. [DOI] [PubMed] [Google Scholar]
- 7.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–90. [DOI] [PubMed] [Google Scholar]
- 9.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. [DOI] [PubMed] [Google Scholar]
- 10.Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2002;33(5):466–77. [DOI] [PubMed] [Google Scholar]
- 12.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. 2013;210(13):2873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina BD, Liu M, Vitiello GA, Seifert AM, Zeng S, Bowler T, et al. Oncogenic kinase inhibition limits Batf3-dependent dendritic cell development and antitumor immunity. J Exp Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ, et al. PD-1/PD-L1 Blockade Enhances T-cell Activity and Antitumor Efficacy of Imatinib in Gastrointestinal Stromal Tumors. Clin Cancer Res. 2017;23(2):454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vitiello GA, Bowler TG, Liu M, Medina BD, Zhang JQ, Param NJ, et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J Clin Invest. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang JQ, Zeng S, Vitiello GA, Seifert AM, Medina BD, Beckman MJ, et al. Macrophages and CD8(+) T Cells Mediate the Antitumor Efficacy of Combined CD40 Ligation and Imatinib Therapy in Gastrointestinal Stromal Tumors. Cancer Immunol Res. 2018;6(4):434–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gasparotto D, Sbaraglia M, Rossi S, Baldazzi D, Brenca M, Mondello A, et al. Tumor genotype, location, and malignant potential shape the immunogenicity of primary untreated gastrointestinal stromal tumors. JCI Insight. 2020;5(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mao X, Yang X, Chen X, Yu S, Yu S, Zhang B, et al. Single-cell transcriptome analysis revealed the heterogeneity and microenvironment of gastrointestinal stromal tumors. Cancer Sci. 2021;112(3):1262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Angelo SP, Shoushtari AN, Keohan ML, Dickson MA, Gounder MM, Chi P, et al. Combined KIT and CTLA-4 Blockade in Patients with Refractory GIST and Other Advanced Sarcomas: A Phase Ib Study of Dasatinib plus Ipilimumab. Clin Cancer Res. 2017;23(12):2972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh AS, Hecht JR, Rosen L, Wainberg ZA, Wang X, Douek M, et al. A Randomized Phase 2 Study Of Nivolumab Monotherapy Or Nivolumab Combined with Ipilimumab In Patients with Advanced Gastrointestinal Stromal Tumors. Clin Cancer Res. 2021. [DOI] [PubMed] [Google Scholar]
- 22.Sommer G, Agosti V, Ehlers I, Rossi F, Corbacioglu S, Farkas J, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci U S A. 2003;100(11):6706–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epardaud M, Elpek KG, Rubinstein MP, Yonekura AR, Bellemare-Pelletier A, Bronson R, et al. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. 2008;68(8):2972–83. [DOI] [PubMed] [Google Scholar]
- 24.Jameson SC, Masopust D. Understanding Subset Diversity in T Cell Memory. Immunity. 2018;48(2):214–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114(19):4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32(4):381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreatta M, Corria-Osorio J, Muller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun. 2021;12(1):2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldrath AW, Luckey CJ, Park R, Benoist C, Mathis D. The molecular program induced in T cells undergoing homeostatic proliferation. Proc Natl Acad Sci U S A. 2004;101(48):16885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carmona SJ, Siddiqui I, Bilous M, Held W, Gfeller D. Deciphering the transcriptomic landscape of tumor-infiltrating CD8 lymphocytes in B16 melanoma tumors with single-cell RNA-Seq. Oncoimmunology. 2020;9(1):1737369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571(7764):270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9(9):618–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity. 2016;44(3):609–21. [DOI] [PubMed] [Google Scholar]
- 35.Singh MD, Ni M, Sullivan JM, Hamerman JA, Campbell DJ. B cell adaptor for PI3-kinase (BCAP) modulates CD8(+) effector and memory T cell differentiation. J Exp Med. 2018;215(9):2429–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101(11):4260–6. [DOI] [PubMed] [Google Scholar]
- 37.Verma K, Ogonek J, Varanasi PR, Luther S, Bunting I, Thomay K, et al. Human CD8+ CD57- TEMRA cells: Too young to be called “old”. PLoS One. 2017;12(5):e0177405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci U S A. 2010;107(38):16601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoklasek TA, Schluns KS, Lefrancois L. Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo. J Immunol. 2006;177(9):6072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margolin K, Morishima C, Velcheti V, Miller JS, Lee SM, Silk AW, et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2018;24(22):5552–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu M, Etherington MS, Hanna A, Medina BD, Vitiello GA, Bowler TG, et al. Oncogenic KIT Modulates Type I IFN-Mediated Antitumor Immunity in GIST. Cancer Immunol Res. 2021;9(5):542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175(4):998–1013 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013;73(12):3499–510. [DOI] [PubMed] [Google Scholar]
- 44.Klaver Y, Rijnders M, Oostvogels A, Wijers R, Smid M, Grunhagen D, et al. Differential quantities of immune checkpoint-expressing CD8 T cells in soft tissue sarcoma subtypes. J Immunother Cancer. 2020;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, et al. Tumor Mutation Burden and Efficacy of EGFR-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res. 2019;25(3):1063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jardim DL, Goodman A, de Melo Gagliato D, Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39(2):154–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231(1):51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayashi H, Carrasquillo JA, Paik CH, Waldmann TA, Tagaya Y. Differences of biodistribution, pharmacokinetics, and tumor targeting between interleukins 2 and 15. Cancer Res. 2000;60(13):3577–83. [PubMed] [Google Scholar]
- 51.Pinette A, McMichael E, Courtney NB, Duggan M, Benner BN, Choueiry F, et al. An IL-15-based superagonist ALT-803 enhances the NK cell response to cetuximab-treated squamous cell carcinoma of the head and neck. Cancer Immunol Immunother. 2019;68(8):1379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19(5):694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNAseq and scRNAseq data presented in this work are submitted through the Sequencing Read Archive under accession no. PRJNA824054 and PRJNA859907. The TCRb sequencing data is available on the Immunoseq platform under the project name UPenn_00110053_R-01. The remaining data generated and/or analyzed during the current study are available within the article and its Supplementary Data files or are available from the corresponding author upon reasonable request.