

BACKGROUND
Despite favorable clinical outcomes, a subset of patients with thymic epithelial tumors (TETs) develop metastasis. The Cancer Genome Atlas (TCGA) provides genomic data on primary TETs (pTETs). This study assessed the molecular alterations and uncovered targetable pathways in metastatic TETs (mTETs).
METHODS
From 2015 to 2020, 49 patients with stage IV TETs underwent Clinical Laboratory Improvement Amendments–based sequencing using whole-exome sequencing (n = 33), panel-based testing (n = 12), and/or liquid biopsy (n = 24). Specimens were obtained from a metastatic organ (n = 36) or relapsed primary mediastinal mass (n = 10), whereas four patients underwent a liquid biopsy only. Data on pTETs were derived from the TCGA.
RESULTS
Compared with the pTET data set, patients with mTETs were younger (54 years v 60.5 years, P = .009) and had more aggressive histologies, with the most common tumor type being thymic carcinoma (n = 22, 40.7%) and B3 thymoma (n = 15, 27.8%). GTF2I was the most altered gene in primary thymomas (48.80%, n = 60). In metastatic thymoma and thymic carcinoma, TP53 was the most common genetic alteration (31% and 36%, respectively). In mTETs, the genomic alteration occurred in the TP53/CDK, EGFR/RAS, and PI3K/mTOR pathways. Biopsies obtained from distant metastasis were more commonly found to contain targetable mutations. There was an overlap of 61% (22 of 36) between tissue and liquid biopsy genomic alterations.
CONCLUSION
Clinically actionable genomic alterations are frequently observed in mTETs, indicating a value of repeat biopsy (preferably from a metastatic site of TETs for sequencing at the time of recurrence (TCGA data).
INTRODUCTION
Thymomas and thymic carcinoma (TC) are malignant thymic epithelial tumors (TETs) that arise from the anterior mediastinum. The National Cancer Institute's SEER tumor registry estimates the incidence of thymoma in the United States to be approximately 0.15 per 100,000 person-years.1 Although thymoma incidence appears to be decreasing, the SEER data demonstrate that the incidence of TC is rising.2 The WHO classifies TETs into thymoma (on the basis of cell morphology and the ratio of epithelial to lymphoid cells) into various histologic subtypes (A, AB, B1, B2, and B3) and TC.3 Thymomas tend to have an indolent growth pattern, often presenting with early-stage disease, whereas TC typically presents with locally advanced or metastatic disease.4 Anatomic staging by the modified Masaoka classification includes stage I (localized in the thymus), stage II (microscopic invasion), stage III (macroscopic invasion to adjacent organs), and stage IV with metastasis to the pleura (IVa) or other distant sites (IVb).5
CONTEXT
Key Objective
We conducted a retrospective study comparing the genomics of metastatic thymic epithelial tumors (TETs) with that of primary tumors. We assessed the importance of the site of biopsy and the utilization of liquid biopsy in metastatic patients.
Knowledge Generated
Targetable alterations were found frequently in metastatic biopsy compared with the primary tumor biopsy. The DNA repair/TP53, EGFR/RAS, and PIK3/mTOR pathways are altered more in metastatic than in primary TETs. Distant metastasis tissue harbors higher genomic alterations. Patient-matched liquid-tissue biopsy showed an overlap of 60% in targetable genomic alterations.
Relevance
Acquisition of tissue from distant metastasis and genomic sequencing at the time of metastasis is an insightful approach. Liquid biopsy has reasonable concordance with tissue biopsy, but it does not fully reflect the tissue genomic alterations. Time-matched tissue biopsy and liquid biopsy are required to assess the concordance of tissue and liquid genomic findings in metastatic TETs.
Surgery is the mainstay of treatment for localized TETs, and chemotherapy is the standard of care for patients with metastatic tumors.6 Despite the initial high response rate to platinum-based chemotherapy in most patients with stage IVa and IVb disease relapse, treatment options for relapsed/refractory TETs are limited. Small studies have demonstrated the efficacy of various targeted and nontargeted therapies. However, given the rarity of thymic tumors, the discovery of druggable genes has lagged behind that of other malignancies.7
In 2018, The Cancer Genome Atlas (TCGA) provided insights into the genomics of primary TETs (pTETs). One unique mutation was that of GTF2I in WHO type A and AB thymoma, but mutations in HRAS, NRAS, and TP53 were noted. Importantly, the samples from these specimens were obtained from primary sites and not from metastatic sites. Somatic mutations and tumor heterogeneity frequently occur during tumor evolution, contributing to metastasis and death.7 To uncover the potentially actionable genomic pathways and new targets, we explored the genomic alterations in metastatic TETs (stage IVa/b) and compared them with the findings from primary tumors using TCGA.
METHODS
From 2015 to 2020, 49 patients seen at the Indiana University Simon Comprehensive Cancer Center with advanced or metastatic TETs (stage IVa or IVb) underwent Clinical Laboratory Improvement Amendments–based sequencing (69 samples) using whole-exome sequencing (n = 33), panel-based testing (n = 12), and/or liquid biopsy (n = 24). Liquid biopsy was performed using FoundationOne Liquid CDx (Illumina NovaSeq 6000; San Diego, CA), which assesses the cell-free DNA for more than 324 genes and has been approved by the Food and Drug Administration. This panel has been validated in different cancers and demonstrated high concordance between Liquid CDx and tissue assays for 74 genes. Germline mutations were not removed from this panel. Next-generation sequencing of tissue was performed with FoundationOne or Ashion (Exact Sciences). The hotspot panel was used in FoundationOne where whole-exome sequencing was used in the Ashion tissue panel. Germline mutations were removed from the Ashion panel.
The specimens were taken from a metastatic organ (n = 36, 72%) or relapsed primary mediastinal mass (n = 10, 20%); four (8%) patients had liquid biopsy only. Clinical data were obtained through retrospective chart review. IRB approval was obtained to collect all clinicopathologic and genomic information. Data on pTETs were derived from TCGA. The cBioPortal mutation mapper was used to illustrate the mutations in the lollipop. The TCGA included six patients with pleural or distant metastases (and were excluded from our genomic comparison analysis). We used Polyphen-2228 to assess the pathogenicity of the loss-of-function missense mutations. Ingenuity Pathway Analysis estimated pathway involvement in each subgroup of patients. To identify the correlation between Tp53 mRNA expression and tumor immune markers, the IMmune Estimation Tool (Tumor IMmune Estimation Resource [TIMER])9 was used.
Baseline demographic and disease characteristics are summarized as median (range) for continuous variables and proportions for categorical variables. Comparisons between the pTET and mTET groups were made using chi-square tests for categorical variables or the Wilcoxon test for continuous variables. The chi-square test was performed to compare the TP53 alteration rate between patients with primary and metastatic tumors. The Kaplan-Meier method was used to analyze overall survival (OS) and progression-free survival (PFS) using the log-rank test to compare the groups. Patients who did not die were censored at the last follow-up visit for OS. PFS was measured from the date of diagnosis to the date of first relapse. The median OS and PFS were calculated with 95% confidence intervals along with the 2-, 5-, and 10-year probabilities. The results from a Cox proportional model were used to measure the hazard ratio and 95% confidence interval. Data analysis was conducted using the SAS software version 9.4 (SAS Institute Inc, version 9.4; Cary, NC). A P value of < .05 denoted statistical significance for all tests.
RESULTS
Patient's Clinical Characteristics
TCGA data included 123 patients, of which 117 had stage I-III and six had metastatic (IVa/b) disease (Table 1). In the whole TCGA cohort, the mean age of the patients was 60.5 (17.0, 84.0) years. The population was well-balanced by sex (M/F: 52%/48%). The majority had histology of AB (n = 38, 31.7%) followed by B2 (n = 27, 22.5%) and then A and B3 (each n = 15, 12.5%).
TABLE 1.
Patient's Characteristics: Primary Versus Metastatic Thymic Epithelial Tumors

Patients from our metastatic cohort were younger (54 years v 60.5 years; P = .009) and had a more aggressive histology (P < .0001) than the pTET cohort. The most common tumor type in mTET was TC (n = 20, 40.8%), followed by B3 (n = 13, 26.5%) and B2 (n = 10, 20%). This was compared with the pTET cohort, in which the most common tumor types were AB (n = 28 [57.1%]) followed by B2 (n = 27 [56%]).
Assessing the impact of histology on patients' clinical outcomes, those with metastatic TC had worse OS than patients with metastatic thymomas (hazard ratio, 2.8, P = .04). In our cohort of 49 stage IV mTET, 33 (62%) patients had stage IVb disease, whereas in the TCGA, only 6 (4.3%) had distant metastasis. In our metastatic cohort, 38 patients received chemotherapy before metastasis and tissue rebiopsy and 11 patients had no history of chemotherapy. There was no significant difference in the number of mutations in those who had chemotherapy compared with those with no history of chemotherapy (P = .64). Similarly, a history of prior radiation (yes = 13 v no = 36; P = .53) was not associated with an increase in the tumor mutational rate.
Patients' Genomic Characteristics
The top five mutations occurring in the four different subgroups (primary thymoma, metastatic, primary TC, and metastatic TC) are shown in Figure 1A.
FIG. 1.

(A) Distinct genomic alterations were observed between primary thymoma, primary TC, metastatic thymoma, and metastatic TC (frequently mutant genes in TETs (%)). IGF2I and CYLD are the most common alterations in primary thymoma and TC, respectively. Tp53 was the frequent mutation in metastatic thymoma/TC. (B) The Ingenuity was used for pathway analysis in metastatic TETs (thymoma, TC). The TP53/CDK, EGFR/RAS, and PI3K/mTOR pathways are the most common altered pathways in metastatic TETs. (C) Pathway analysis in both primary and metastatic TETs. Primary thymoma had more immunologic pathways and primary TC immunologic and metabolic pathway involvement. Metastatic thymoma showed more cell cycles, and mTOR/PI3K and metastatic TC had more defects in the DNA repair pathway, cell cycle checkpoint, and Myc/apoptosis signaling. TC, thymic carcinoma; TETs, thymic epithelial tumors.
Thymoma.
GTF2I was the most altered gene in primary thymoma (TCGA) with a 48.80% (n = 60) mutation rate, whereas only one patient with metastatic thymoma harbored this mutation. The second most common mutation in primary thymoma was HRAS (n = 9, 8.4%). Patients with metastatic thymoma have a much greater incidence of Tp53 mutations than nonmetastatic TCGA patients (n = 10, 31.2% v 0). Notably, there were two Tp53 mutations among the six stage IV patients reported in the TCGA database. Among patients with metastatic thymoma, targetable genes with ≥ 2 mutations included Tp53, mTOR, EGFR, CDK4, ASXL1, and NOTCH1.
TC.
In 11 patients with primary TC, mutations in CYDL were found in three (27%), whereas only two (18%) had Tp53 mutations. By contrast, Tp53 was the most common mutation observed in metastatic TC (Fig 1A). In patients with metastatic TC, we observed frequent alterations in DNA repair pathways (ATM 10%, CHD1 10%, and ARIDA1 10%) and cell cycle checkpoint signaling (Tp53, n = 35%; CDKN2A/B, n = 30%).
Genomic Pathway Analysis
The Ingenuity Pathway Analysis illustrated three pathways that appear to be most involved in metastatic TETs, including the TP53/DNA repair, EGFR/RAS, and PI3K/mTOR pathways (Fig 1B). We have also taken a further step and assessed the involvement of metabolic, immune, and genomic pathways in all four subgroups separately (Fig 1C). The primary thymoma pathway study was more consistent with immune-related tracks (IL2, IL3, IL6, IL4, and JAK2/3), whereas the metastatic thymoma involved the cell cycle, Tp53, telomeres, EGF, PIK3K, and epithelial adhesion signaling.
Defects in the immune pathway (MIF), metabolic pathway (glucocorticoid receptor signaling), AMPK, and P38MAP signaling were more frequently detected in primary TC. By contrast, in mTC, the DNA repair pathway and cell cycle checkpoint signaling were more dominant.
Site-Specific Genomic Alteration
To evaluate the genomic evolution of thymic tumors, we used our metastatic cohort and assessed frequent genomic alterations (two or more alterations) in the primary site and pleural metastatic and nonpleural metastatic specimens (Table 2). There were far fewer genomic mutations (total and targetable mutations) in patients with metastatic tumors who underwent biopsies from the primary site (30% v 10%, respectively). EGFR and ATM alterations were exclusively more common in pleural samples. Although Tp53 was mutated in only 10% of primary and pleural tissue samples, it was dramatically increased in biopsy samples obtained from distant metastasis sites (64.7%; Table 2).
TABLE 2.
Genomic Alterations (≥ 2 Alterations) on the Basis of the Site of Biopsy in Stage IV Patients With Thymic Epithelial Tumors

Comparison of Liquid and Tissue Biopsy Genomic Results
We compared the matched (same patients) liquid- and tissue-targetable mutations (TC = 9, B3 = 6, B2 = 4, B1 = 1, and AB = 2). We included genomic alteration (MAF) with a mean allelic frequency of 20% or more. Our data demonstrated 61% overlap between liquid and blood genomic alterations. The total number of detected targetable mutations was quite similar in liquid biopsies and tissues (17 mut v 19 mut; Fig 2C).
FIG. 2.

(A) Distribution of targetable mutations and their frequency in metastatic TETs (thymoma plus TC; targetable mutations rate). It showed that 60% of patients harbor actable mutations at the time of metastasis. (B) The rate and type of targeted therapies used in the cohort of metastatic TETs. More than 60% of patients received at least one targeted therapy. STAT3 inhibitor was used in the context of a clinical trial. Despite the considerable risk of autoimmune flare, all patients with high TMB received immunotherapy in different trials. (C) Assessment of paired patients (same patients, n = 22) tissue and blood genomic tests. The liquid and tissue biopsies are from patients with metastatic TET. To remove the nonpathologic mutations, we included alterations with a mean allelic frequency of 20% or higher. Notably, there was a 61% overlap between liquid and blood genomic targetable alterations. Total numbers of detected targetable mutations were quite the same in liquid biopsy and tissue biopsy (17 mut v 19 mut). Tp53 mutations were reported slightly more frequently in liquid biopsy (liquid n = 8 v tissue n = 6). TC, thymic carcinoma; TETs, thymic epithelial tumors; TMB, tumor mutational burden.
Targetable Mutations/Targeted Therapies in the Indiana University Metastatic Cohort
Importantly, in the cohort of metastatic TETs, 60% of the patients had molecular alterations with known or potential therapeutic implications (Fig 2A). Although not necessarily using these specific findings, 54% of our cohort received one or more than one targeted therapy during the subsequent course of treatment (Fig 2B). Eighteen percentage of patients (n = 10) had high tumor mutational burden (TMB) > 10 mut/Mb, and all received immunotherapy in a clinical trial during their treatment course. The mean relapse-free interval (retrospective analysis) after starting immunotherapy was 18 months (Appendix Table A2).
Other patients in the metastatic cohort received targeted therapy in clinical trials with napabucasin (STAT3 inhibitor) plus paclitaxel and buparlisib (an oral pan-PI3K inhibitor). By contrast, additional patients received sunitinib (multiple receptor tyrosine kinase inhibitor) and everolimus (mTOR inhibitor) on the basis of published data supporting their efficacy in recurrent disease. Of 25 patients who received targeted therapies, treatment was stopped in 12 patients because of toxicity and 12 patients because of disease progression and one patient is still on rucaparib. The patient has been on this therapy for 32 months. The mean time to disease progression in those whose treatment was stopped because of disease progression (not toxicity) was 9.9 months (Appendix Table A3).
Tp53 was the most altered gene in metastatic thymoma (31%) and TC (36%). To investigate the different types and locations of gene mutations, we mapped Tp53 alteration. As expected for a tumor suppressor, it spread through the gene (Fig 3A). Most Tp53 mutations (20 of 22) were detected to be missense, followed by one fusion and one deletion. V173M and R273H/Z mutations were detected in two patients. To interrogate the impact of missense mutations on Tp53 protein function, we used Polyphen-2 algorithm analysis, which showed that 83% (18 of 20) of mutations were likely to have a highly damaging effect on Tp53 protein function or structure (Appendix Table A1). To assess the potential effect of Tp53 mutations on patients' clinical outcomes, we separately compared the Tp53-mutant group with the wild types in the univariate analysis in thymoma and TC. Despite the early curve separation in each group, there was no significant difference in OS (Fig 3C1-C2). To further assess the impact of Tp53 on immune cells in primary thymic tumors, we used TIMER for primary tumors (TCGA data). TIMER analysis showed that Tp53 transcriptomic expression has significant positive correlation with macrophage (r = 0.42, P = 2.75E-06), dendritic cell (r = 0.70, P = 1.29E-18), CD4+ T cell (r = 0.61, P = 7.26E-13), and CD8+ T cell (r = 0.42, P = 1.85E-06) infiltration (Fig 3B). Lower expression of Tp53 was associated with lower T-cell infiltrative lymphocytes in primary thymoma.
FIG 3.

(A) cBioPortal mutation mapper showing distribution and types of Tp53 mutations throughout the gene in primary and metastatic thymic tumors. Missense mutations are labeled in green. Truncating mutations: nonsense, frameshift deletion, frameshift insertion, and splice site given in black. All other types of mutations are given in purple. Patients with metastatic TC had more frequent Tp53 mutations. The majority of mutations are missense and distributed throughout the genes, indicating the tumor suppressor pattern. (B) The TCGA data (pTETs) TIMER result on the correlation of the Tp53 expression with the tumor purity, B cell, CD8+ T cell, CD4+ T cell, macrophage, neutrophil, and dendritic cell in particular. In pTETs, there was a very strong positive correlation between the dendritic cell infiltration and the TP53 mRNA expression level. Neutrophil infiltration showed a significant inverse correlation with the Tp53 expression level. (C) The OS of patients with Tp53 mutations compared with wild-type (C1) thymoma and (C2) TC. Despite the early separation in the curve, there was no significant survival difference in our cohort. aKaplan-Meier method; bCox model; cLogrank test; dWald chi-square test. OS, overall survival; pTETs, primary TETs; TC, thymic carcinoma; TCGA, The Cancer Genomic Atlas; TETs, thymic epithelial tumors; TIMER, Tumor Immune Estimation Resource.
DISCUSSION
In this study, we investigated the genomic landscape of patients with metastatic TETs. In contrast to TCGA analysis, which predominantly provides data on primary tumors and treatment-naive patients, our study highlights specific genomic alterations after tumor evolution, including those with chemotherapy and radiation exposure. We observed that metastatic tumors have molecular characteristics very distinct from those of pTETs. The Tp53 gene was the most altered gene in metastatic thymoma and TC, whereas GTF2I was the most common mutation in primary thymoma and CYLD in primary TC. Patients with mTETs and a history of chemotherapy or radiation therapy before tissue biopsy did not have higher rate of genomic mutations.
Notably, we found that 60% of mTETs harbor targetable driver mutations or have high TMB. We aligned these gene alterations to three significant pathways: DNA repair/CDK, EGFR/RAS, and mTOR/PI3K. Furthermore, we discovered that immunologic and metabolic pathways are more striking in pTET, whereas DNA damage repair pathways and cell cycle checkpoint signaling are more striking in mTETs. In addition, the paired liquid-tissue comparison showed more than a 60% overlap between tissue and liquid biopsy genomic alterations.
Thymomas have been reported to have fewer recurrent genomic alterations than TC.10,11 In TCGA data (n = 117), the majority of primary tumors had thymomas with more favorable histology (ie, WHO types A and A/B), in which GTF2I was the most mutated gene (39%), followed by HRAS (8.1) and TP53 (3%).12 Notably, GTF2I was not reported in our cohort of patients with metastatic thymic tumors. The GTF2I mutation is uniquely seen in thymic tumors and has not been reported in any other solid tumors within the entirety of the TCGA.12 Similarly, Petrini et al10 showed that GTF2I is the only gene with frequent mutations in thymoma (82% in thymoma A and 74% in thymoma A/B). Collectively, these observations highlight the benign behavior of GTF2I mutations, despite their clonality.
In the metastatic TETs panel, Tp53 was the most commonly mutated gene, indicating the role of Tp53 in tumor progression rather than tumor initiation. We also found that Tp53 mutations have a higher rate of alteration in mTC compared with metastatic thymomas. This is in line with a previous study showing Tp53 mutation rates of 26% in TCs and 3% in thymomas.13 Tp53 is a guardian of the genome, and the activation of wild-type Tp53 induces cell cycle arrest in response to expressing or intrinsic stress.14 Tumor progression and metastasis are associated with Tp53 loss-of-function mutations.15 Indeed, Tp53 mutations cause stem-cell gene signatures in epithelial tumors and play a role in the late stages of tumorigenesis.16 Interestingly, we had one patient with a Tp53 V273H missense mutation, a known mutation for tumor cell migration, which plays a role in late tumorigenesis.17 There is evidence supporting Tp53 prognostication exclusively in TC.13 Therefore, we evaluated the OS on the basis of Tp53 status in patients with TC and B3 separately, and there was no adverse survival impact in Tp53-mutant patients with either thymoma or TC histology subtype. Our current survival finding is consistent with the report by Sakane et al,18 which showed that Tp53 plays an essential role in tumor progression with no predictive value in TC.
In line with our discovery of frequent alterations in EGFR/RAS and PI3K pathways in metastatic TETs, Wang et al13 recently showed recurrent genomic alterations in PI3K, RAS, DNA damage, DNA methylation pathways, and epigenetic dysregulation such as ASXL1, BAP1, BRCA2, CDKN2A, CYLD, HRAS, KIT, SDHA, TET2, and SMARCA4 in patients with TC. In addition, most thymic tumors develop pleural involvement before distant metastasis. However, our preliminary data from a large cohort of metastatic TETs specified a subset of patients (approximately 20%) who developed distant metastasis without preceding pleural involvement (data not shown). Here, we demonstrated a distinct and higher rate of mutations in distant metastasis than in primary and pleural tumor tissues. Therefore, the biopsy site is one of the main determinants for detecting genomic alterations, and it is valuable to consider biopsies from distant metastatic sites.
Our data demonstrate a high correlation between tissue-liquid biopsy genomic alterations (> 60% in alterations with MAF more than 20%). However, the proof of concept of using liquid biopsy as a surrogate for tissue biopsy is yet to be evaluated in a larger cohort. Of note, 50% of tissue and liquid biopsies were performed at the same time. Thus, the difference between genomic findings (approximately 40% of alterations) might be due to the dynamic landscape of the cancer genome over time and further assessment of the concordance between liquid and tissue biopsies is required in the future.
One limitation of this study is using hotspot panels, which prioritize targetable mutations. In addition, considering the retrospective nature of the present study, we did not have a uniform genomic analysis platform or tissue sampling criteria that could impose a heterogeneous result. In addition, there is a lack of matched time-series tumor sequencing in the same patient. Applying matched primary and metastatic samples will decrease interpersonal variability and map the cancer progression trajectory.
In conclusion, clinically actionable genomic alterations are frequently seen in patients with mTETs, indicating the value of the routine sequencing at the time of metastasis. Despite the reasonable concordance in tissue-liquid biopsy genomic findings, liquid biopsy dose not fully reflect the tissue genomic alterations and it is worth considering both studies to fully picture the tumor heterogeneity and discover the potential druggable alterations. Immune and metabolic-related pathways are more involved in pTETs, whereas DNA repair, EGFR/RAS, and PIK3/mTOR are frequently mutated in metastatic TETs. Further studies to characterize the serial genomic evolution in individual patients may be helpful to better understand the molecular underpinnings for the development of metastatic tumors in this rare malignancy.
APPENDIX
TABLE A1.
Missense Mutation Functionality Assessment With Polyphen-2
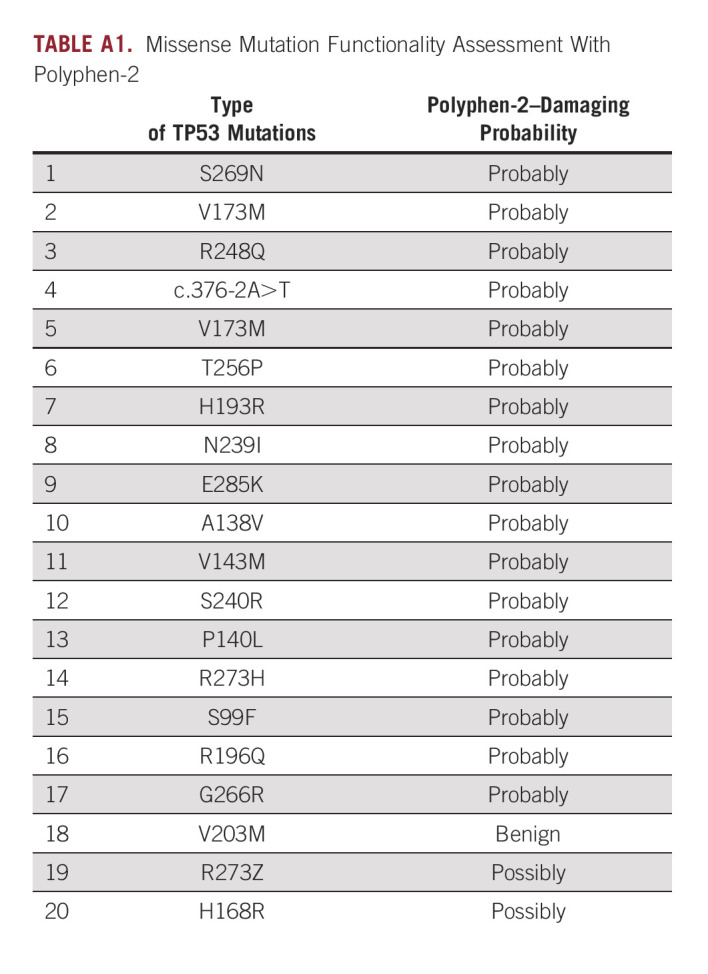
TABLE A2.
Demographic and Duration of Response to Immunotherapy in the Indiana University Metastatic Patient Cohort

TABLE A3.
Demographic and Duration of Response to Targeted Therapy in the Indiana University Metastatic Patient Cohort

Bryan P. Schneider
This author is an associate editor of JCO Precision Oncology. Journal policy recused the author from having any role in the peer review of this manuscript.
Honoraria: Lilly, Research to Practice
Research Funding: Genentech/Roche, Pfizer, Foundation Medicine, Epic Sciences
Fabiana Perna
Honoraria: AstraZeneca
Consulting or Advisory Role: AstraZeneca
Research Funding: Lonza, NGM Biopharmaceuticals
Patents, Royalties, Other Intellectual Property: Royalties through work at MSKCC
Milan Radovich
Employment: Caris Life Sciences
Leadership: Caris Life Sciences
Stock and Other Ownership Interests: LifeOmic
Research Funding: Lilly (Inst)
Patents, Royalties, Other Intellectual Property: Combination therapy for the treatment of TNBC with a PI3K pathway inhibitor that targets PI3KDelta and PI3KGamma (Inst)
Travel, Accommodations, Expenses: Caris Life Sciences
Ashiq Masood
Honoraria: Bristol Myers Squibb, Boehringer Ingelheim
Speakers' Bureau: Bristol Myers Squibb, Boehringer Ingelheim
Research Funding: Boston Biomedical (Inst), Ipsen (Inst), Seattle Genetics (Inst), Novocure (Inst), MacroGenics (Inst), Merck (Inst), SIGNATERA (Inst), Genentech/Roche (Inst), Exelixis (Inst), Astellas Pharma (Inst), Debiopharm Group (Inst), PRA Health (Inst), CytomX Therapeutics (Inst), Calithera Biosciences (Inst), Proteus Digital Health (Inst), Tempus, Gritstone Bio (Inst)
Patrick J. Loehrer
Research Funding: Novartis (Inst), Lilly Foundation (Inst), Taiho Pharmaceutical (Inst)
Patents, Royalties, Other Intellectual Property: US PPA/61/499,988 Gene Expression Analysis of Thymic Neoplasms Inventors: Sunil Badve, Yesim Gokmen-Polar, Patrick Loehrer (Inst)
Other Relationship: National Cancer Institute
No other potential conflicts of interest were reported.
SUPPORT
Supported in part by the Hochberg Family Foundation and NCI P30 CA082709.
AUTHOR CONTRIBUTIONS
Conception and design: Fatemeh Ardeshir-Larijani, Milan Radovich, Ashiq Masood, Huda Salman
Provision of study materials or patients: Fatemeh Ardeshir-Larijani
Collection and assembly of data: Fatemeh Ardeshir-Larijani, Milan Radovich
Data analysis and interpretation: Fatemeh Ardeshir-Larijani, Bryan P. Schneider, Sandra K. Althouse, Fabiana Perna, Huda Salman
Manuscript writing: Fatemeh Ardeshir-Larijani, Bryan P. Schneider, Sandra K. Althouse, Milan Radovich, Ashiq Masood, Fabiana Perna, Huda Salman
Final approval of manuscript: Fatemeh Ardeshir-Larijani, Bryan P. Schneider, Sandra K. Althouse, Milan Radovich, Ashiq Masood, Fabiana Perna, Huda Salman
Accountable for all aspects of the work: Fatemeh Ardeshir-Larijani, Bryan P. Schneider, Sandra K. Althouse, Milan Radovich, Ashiq Masood, Fabiana Perna, Huda Salman
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Bryan P. Schneider
This author is an associate editor of JCO Precision Oncology. Journal policy recused the author from having any role in the peer review of this manuscript.
Honoraria: Lilly, Research to Practice
Research Funding: Genentech/Roche, Pfizer, Foundation Medicine, Epic Sciences
Fabiana Perna
Honoraria: AstraZeneca
Consulting or Advisory Role: AstraZeneca
Research Funding: Lonza, NGM Biopharmaceuticals
Patents, Royalties, Other Intellectual Property: Royalties through work at MSKCC
Milan Radovich
Employment: Caris Life Sciences
Leadership: Caris Life Sciences
Stock and Other Ownership Interests: LifeOmic
Research Funding: Lilly (Inst)
Patents, Royalties, Other Intellectual Property: Combination therapy for the treatment of TNBC with a PI3K pathway inhibitor that targets PI3KDelta and PI3KGamma (Inst)
Travel, Accommodations, Expenses: Caris Life Sciences
Ashiq Masood
Honoraria: Bristol Myers Squibb, Boehringer Ingelheim
Speakers' Bureau: Bristol Myers Squibb, Boehringer Ingelheim
Research Funding: Boston Biomedical (Inst), Ipsen (Inst), Seattle Genetics (Inst), Novocure (Inst), MacroGenics (Inst), Merck (Inst), SIGNATERA (Inst), Genentech/Roche (Inst), Exelixis (Inst), Astellas Pharma (Inst), Debiopharm Group (Inst), PRA Health (Inst), CytomX Therapeutics (Inst), Calithera Biosciences (Inst), Proteus Digital Health (Inst), Tempus, Gritstone Bio (Inst)
Patrick J. Loehrer
Research Funding: Novartis (Inst), Lilly Foundation (Inst), Taiho Pharmaceutical (Inst)
Patents, Royalties, Other Intellectual Property: US PPA/61/499,988 Gene Expression Analysis of Thymic Neoplasms Inventors: Sunil Badve, Yesim Gokmen-Polar, Patrick Loehrer (Inst)
Other Relationship: National Cancer Institute
No other potential conflicts of interest were reported.
REFERENCES
- 1. Engels EA, Pfeiffer RM. Malignant thymoma in the United States: Demographic patterns in incidence and associations with subsequent malignancies. Int J Cancer. 2003;105:546–551. doi: 10.1002/ijc.11099. [DOI] [PubMed] [Google Scholar]
- 2. Hsu C-H, Chan JK, Yin C-H, et al. Trends in the incidence of thymoma, thymic carcinoma, and thymic neuroendocrine tumor in the United States. PLoS One. 2019;14:e0227197. doi: 10.1371/journal.pone.0227197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quintanilla-Martinez L, Harris NL, Wilkins EW, Jr, et al. Thymoma: Histologic subclassification is an independent prognostic factor. Cancer. 1994;74:606–617. doi: 10.1002/1097-0142(19940715)74:2<606::aid-cncr2820740212>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 4. Scorsetti M, Leo F, Trama A, et al. Thymoma and thymic carcinomas. Crit Rev Oncol/Hematol. 2016;99:332–350. doi: 10.1016/j.critrevonc.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 5. Masaoka A, Monden Y, Nakahara K, Tanioka T. Follow-up study of thymomas with special reference to their clinical stages. Cancer. 1981;48:2485–2492. doi: 10.1002/1097-0142(19811201)48:11<2485::aid-cncr2820481123>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 6. Kalhor N, Moran CA. Thymoma and thymic carcinoma: A perspective on the NCCN clinical practice guidelines in oncology. Mediastinum. 2018;2:49. [Google Scholar]
- 7. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li T, Fan J, Wang B, et al. TIMER: A web server for comprehensive analysis of tumor‐infiltrating immune cell. Cancer Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Petrini I, Meltzer PS, Kim I-K, et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat Genet. 2014;46:844–849. doi: 10.1038/ng.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rajan A, Zhao C. Deciphering the biology of thymic epithelial tumors. Mediastinum. 2019;3:36. doi: 10.21037/med.2019.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radovich M, Pickering CR, Felau I, et al. The integrated genomic landscape of thymic epithelial tumors. Cancer Cell. 2018;33:244–258.e10. doi: 10.1016/j.ccell.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Thomas A, Lau C, et al. Mutations of epigenetic regulatory genes are common in thymic carcinomas. Scientific Rep. 2014;4:7336. doi: 10.1038/srep07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 15. Nakayama M, Hong CP, Oshima H, et al. Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties. Nat Commun. 2020;11:2333. doi: 10.1038/s41467-020-16245-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Solomon H, Dinowitz N, Pateras IS, et al. Mutant p53 gain of function underlies high expression levels of colorectal cancer stem cells markers. Oncogene. 2018;37:1669–1684. doi: 10.1038/s41388-017-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lv T, Lv H, Fei J, et al. p53-R273H promotes cancer cell migration via upregulation of neuraminidase-1. J Cancer. 2020;11:6874–6882. doi: 10.7150/jca.44718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakane T, Sakamoto Y, Masaki A, et al. Mutation profile of thymic carcinoma and thymic neuroendocrine tumor by targeted next-generation sequencing. Clin Lung Cancer. 2021;22:92–99.e4. doi: 10.1016/j.cllc.2020.11.010. [DOI] [PubMed] [Google Scholar]