

Abstract
Recent larger-scale studies of patients with cancer and longitudinal population cohorts have revealed how age-related expansions of mutant hematopoietic cells (clonal hematopoiesis [CH]) have differential associations with incident and prevalent cancers and their outcomes. Increasing recognition and deeper understanding of genetic subtypes of CH are yielding insights into the tumor-immune interface that may help to explain the heterogeneous impact of CH on tumorigenesis and treatment. Herein, we update the expanding influence of CH in precision oncology and propose important research and clinical questions to address to effectively manage and harness CH in oncology patients.
This article explores the potential consequences of clonal hematopoiesis for patients with solid tumor.
INTRODUCTION
Clonal hematopoiesis (CH) is the clonal expansion of hematopoietic stem cells (HSCs) and their progeny as driven by somatic mutations acquired during aging. CH describes any clonal expansions found in the blood and HSCs driven by somatic mutations and is thus inclusive of hematologic malignancies such as acute myeloid leukemia (AML) but typically refers to patients with premalignancy and no established disease.1-3 A second term, CH of indeterminate potential (CHIP), has been adopted to help differentiate between malignant and nonmalignant CH by specifically referring to the latter and its propensity, but indeterminate potential, for malignancy, and the absence of cytopenia (Fig 1). Between studies, the exact characterization of CH-driving mutations varies, but they are generally identified as cancer-driving single-nucleotide variants or smaller insertions or deletions (indels).1-3 The genes implicated with CH are largely drivers of myeloid hematologic malignancies. Most CH-causing mutations are found on three epigenetic regulator genes—DNMT3A, TET2, and ASXL1. Other frequently mutated gene targets include DNA damage response (DDR) genes such as TP53 and PPM1D, cell growth signalers such as JAK2 and CBL, and RNA splicing factors such as SRSF2, SF3B1, and U2AF1.1-3,5,6 More recent studies have also implicated lymphoid cancer–associated genes and mosaic chromosomal alterations (mCAs)—larger-scale genomic amplification, deletion, and loss of heterozygosity events—in the clonal expansion of HSCs; however, their integration into the literature is ongoing.5,7-9 Another defining characteristic of CH is variant allele frequency (VAF), which describes the fraction or percentage of DNA molecules sequenced that display a given mutation. There are no specific bounds to define CH, while the currently accepted VAF threshold for CHIP is at least 2%, indicating that 4% of total peripheral blood cells are affected, assuming a heterozygous mutation. Most studies have adopted this 2% VAF threshold for identifying CHIP and its corresponding clinical associations; however, this threshold is somewhat arbitrary and was initially determined by the standard limit of detection of next-generation sequencing (NGS) technologies. Newer technologies such as error-corrected (EC)-NGS can detect small CH clones with VAFs as low as 0.01% that normally escape detection, although the clinical implications of these clones should decrease with diminishing clone size.1-3,10,11 Owing to the lack of sufficient differentiation between CH and CHIP in most studies, we refer to both CH and CHIP as CH in this review for its broader definition.
FIG 1.

Schematic overview of clonal expansion of HSCs in CH and the associated clinical consequences of circulating CH-mutant HSC clones. After somatic mutations in a driver gene, the selective advantage created allows the HSC clone to expand in the bone marrow and become overrepresented in the blood, contributing to inflammation and age-related disease while posing a risk of malignant transformation. Created with BioRender.4 CH, clonal hematopoiesis; HSCs, hematopoietic stem cells.
CONTEXT
Key Objective
Clonal hematopoiesis (CH), the common age-associated expansion of somatically mutated blood cells, is associated with immune dysregulation, increased inflammatory disease, and hematologic malignancy risk. This review explores the implications of CH for solid cancers to provide recommendations for future research and practice.
Knowledge Generated
CH is common in patients with solid tumors, attributable to age, cancer treatments, and the possibility that CH is a risk factor for some cancers. Current applications in oncology include the optimization of tumor molecular profiling and management of therapy-related neoplasms. The effects of CH in the tumor microenvironment are diverse, varying between CH driver and cancer type.
Relevance
The frequent copresentation of CH with solid cancer presents a tremendous opportunity to improve clinical outcomes for patients with cancer. Implications of CH for some clinical applications are already clear, although much more research is needed to leverage CH as a tool in precision oncology.
Recent studies have revealed differential associations of CH with incident and prevalent cancers and their outcomes. Increasing recognition and deeper understanding of genetic subtypes of CH are yielding insights at the tumor-immune interface that may help to explain the heterogeneous impact of CH on tumorigenesis and treatment. Herein, we update the expanding influence of CH in precision oncology and propose important research and clinical questions to address in order to effectively manage and harness CH in oncology patients.
CH IN INFLAMMATION AND AGE-RELATED DISEASE
CH is notable as a model of somatic mutations and aging in a variety of tissues, but its high prevalence and relationship with various inflammatory processes and age-related diseases makes it of great interest to clinicians and researchers. Initial population screenings for CH surveyed blood-derived genomic sequencing data from more than 30,000 individuals for evidence of clonal expansion. Together, these studies found that CH prevalence increases dramatically with age, detected in 10%-20% of individuals older than 70 years while being almost undetectable in those younger than 40 years. Of great concern, carriers of CH were found to have a 30%-40% increase in all-cause mortality.1-3,6 As noted previously, these estimates rely on older sequencing methods and higher VAF clones. With EC-NGS and the ability to detect lower VAF clones, <2% VAF CH appears almost ubiquitously in younger populations age 50-70 years, reaching rates of 95%, compared with 5% in the aforementioned studies.11
Although CH is a precursor state to hematologic malignancy, not all CH-mutant clones develop into cancer, and the risk of malignant transformation varies between subtypes of CH (reviewed in Bowman et al12). Initial studies revealed an approximately 10-fold increase in relative risk and 1% annual risk of malignant transformation, but it is again important to note the bias toward larger VAF clones because of study methodologies, and increasing CH clone size and the number of mutations were associated with increased risk of hematologic malignancy.1-3 Recent estimates using more sensitive sequencing methods predict a 3 to 5-fold increase in AML risk and when distinguishing between myeloid- and lymphoid-associated CH drivers, a 7-fold and 4.2-fold increase in relative risk of myeloid and lymphoid cancers, respectively.5,13,14 In addition to clone size, the risk of developing a hematologic malignancy is also modified by the affected gene(s). The two most commonly affected genes in CH, DNMT3A and TET2 exhibit some of the lowest rates of AML progression while the less frequently mutated but still prevalent TP53 and U2AF1 demonstrate a much higher risk of malignant transformation and are associated with poorer AML prognosis.1,13-15 Certain mutations within genes also confer varying risks of progression, such as the DNMT3A R882H mutation—the most prevalent CH mutation overall—which is underrepresented in CH versus AML and myelodysplastic syndrome (MDS), indicating a potential for increased progression risk.16
Moving beyond the risk of overt hematologic malignancy, the altered inflammatory milieu imposed by CH promotes systemic inflammation and increased morbidity and mortality.1,17-20 Several studies of CH have characterized a hyperinflammatory environment, including elevated levels of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β through the activation of various inflammatory pathways.1,17-20 The effects of DNMT3A and TET2 mutations have been best characterized in relation to inflammation, although knowledge is still incomplete. These epigenetic regulators have broad roles in restricting inflammation in the immune system, such as in monocyte/macrophages, where loss of function mutations in these genes promote the excessive release of proinflammatory cytokines.1,17,21-23 Importantly, the hyperinflammatory environment created by CH also acts alongside inflammatory stressors such as infection to promote the further development and expansion of CH clones, acting cyclically to exacerbate inflammation (reviewed in Cook et al17). For example, the elevated levels of TNF-α commonly seen in CH have been found to provide a fitness advantage for TET2-mutant HSCs, allowing them to continue to expand and proliferate to occupy a larger proportion of the active HSCs.17,22,24,25
A primary consequence of CH-associated inflammation and one of the main drivers of increased morbidity and mortality in CH carriers is an elevated risk of developing cardiovascular disease (CVD). Initially, the associations between CH and CVD were limited to coronary heart disease and ischemic stroke, where relative risk of incident events were, respectively, 2 and 2.5 in CH carriers, with mutations in JAK2 specifically conferring a 12-fold risk increase for coronary heart disease.1,2,26 Subsequent cohort analyses and mechanistic studies have verified these initial findings while also expanding to show that CH is associated with increased risk of early-onset myocardial infarction, chronic heart failure, and thrombosis.1,23,26-32 CVD is not the only major clinical association of CH aside from hematologic malignancy. CH-associated inflammation has recently been linked to chronic kidney disease, with patients found to have elevated levels of CH versus the general population and had a more than doubled risk of kidney failure during the study follow-up period.33 Other adverse clinical outcomes associated with CH include autoimmune diseases such as antibody-associated vasculitis, chronic obstructive pulmonary disease, osteoporosis, and, albeit controversially, severe COVID-19.34-38 Curiously, the hyperinflammatory milieu conferred by CH may provide a protective effect in some diseases, such as in Alzheimer disease, where CH was found to reduce the risk for dementia and neuropathological features.39
CURRENT INSIGHTS INTO CH AND SOLID CANCER
Presence of CH in Patients With Solid Cancer
Several population cohort studies have already begun exploring another significant clinical association of CH—solid cancers. The first project specifically focused on CH in solid cancers was an analysis of 8,810 patients treated at Memorial Sloan Kettering (MSK). CH was common in this cohort, appearing in just over 25% of analyzed patients with solid cancer and associating with age, smoking, and previous exposure to therapy.40 Subsequent analyses of an overlapping but expanded cohort from MSK similarly found the prevalence of CH in patients with cancer to be 30%. Interestingly, the incident risk of CH was not uniform across cancer types—patients with thyroid and ovarian cancer demonstrated an elevated risk of CH, whereas melanoma, prostate cancer, colorectal cancer, and renal cell carcinomas were associated with a lower risk of CH.41 An additional analysis of cancer patient samples identified an increased risk of CH in thymoma patients and a reduced risk in bladder and breast cancers.42 The risk of confounding because of relationships with cancer treatment (see CH as a Predictor of Clinical Outcomes in Patients With Cancer section, below) and shared CH and cancer risk factors such as age and smoking limit the ability of these studies to make causal conclusions about the risk associations between CH and cancer.41
Larger, non–cancer-specific longitudinal studies have the added benefit of monitoring healthy people with CH over an extended period, better modeling incident disease risk. An analysis of 200,453 individuals enrolled in the UK Biobank study found various associations between CH status and the risk of developing a solid tumor. CH, especially with mutation VAF >10%, was linked with incident lung cancer, kidney cancer, lymphoma, and sarcoma. Notably, certain driver genes also carried their own incident risk associations—DNMT3A-mutant CH was additionally associated with incident stomach and bladder cancer while mutations in splicing factors SF3B1 and SRSF2 were uniquely linked with higher rates of colorectal and head/neck cancers.43 A similar analysis using 628,388 individuals from the UK Biobank found relationships between CH status and incident risk of lymphoid cancer, lung cancer in both smokers and nonsmokers, breast cancer, and prostate cancer.44 Strengthening the link between CH and lung cancer, CH conferred a 36% risk increase for disease across several cohorts, even when controlling for other confounders.45 Conversely, a recent evaluation of CH and prostate cancer risk found no association with the risk of prostate cancer.46
CH as a Predictor of Clinical Outcomes in Patients With Cancer
CH can play an important role in predicting various clinical and treatment outcomes after a cancer diagnosis (Fig 2). One of the better-studied implications of CH in this regard is the development of therapy-related myeloid neoplasms (tMNs)—a rare yet severe complication of cancer treatment. tMNs, which include AML, MDS, and MDS/myeloproliferative neoplasms, are gravely dangerous malignancies, with a 5-year survival of just 10%.47 It was previously thought that cancer therapies such as cytotoxic chemotherapy and radiotherapy were directly causing tMNs via mutagenic effects on HSCs; however, recent evidence has demonstrated that the presence of CH in patients with cancer significantly increases the likelihood of tMN development.40,41,47 In fact, the tMN driver mutations in adult patients with cancer are repeatedly found circulating before the receipt of cancer therapy, indicating that cancer therapy is instead providing a selective pressure that favors the growth and ultimately malignant transformation of preexisting CH clones.41 The effects of cancer therapy on CH clones are not uniform, differing on the basis of the role of the mutated driver gene. Studies investigating this phenomenon have recurrently found enrichment of DDR gene-mutant CH in patients after cytotoxic chemotherapy and radiotherapy, with experimental and clinical evidence supporting a fitness advantage for these mutations in the context of therapy.40,41,48-51 This may explain the previously noted enrichment of CH in some groups of patients with solid cancer, with treatments such as radioactive iodine and peptide receptor radionuclides implicated with increased prevalence of CH in thyroid and neuroendocrine tumors, respectively, although the potential roles of CH as a causal driver in tumorigenesis should not be neglected, as seen with lung cancers.45,52,53 Beyond the elevated risk of tMN development, cancer therapy–related CH driven by DDR gene mutations has also been implicated in other treatment complications via excessive inflammation, namely chemotherapy-induced cardiomyopathies and nonischemic heart failure.54-56 Although studies have found that immunotherapies generally have no influence on CH clones in patients with cancer, a case report highlighted a patient with large B-cell lymphoma who experienced the fatal expansion of a TP53-mutant CH clone after anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, demonstrating a need for treatment-specific insights for immunotherapy and clonal dynamics in CH.41,57 In pediatric cancers, chemotherapy and radiation were found to predict CH presence in survivors, albeit these therapy-related clones remained stable in size during longitudinal follow-up.58
FIG 2.
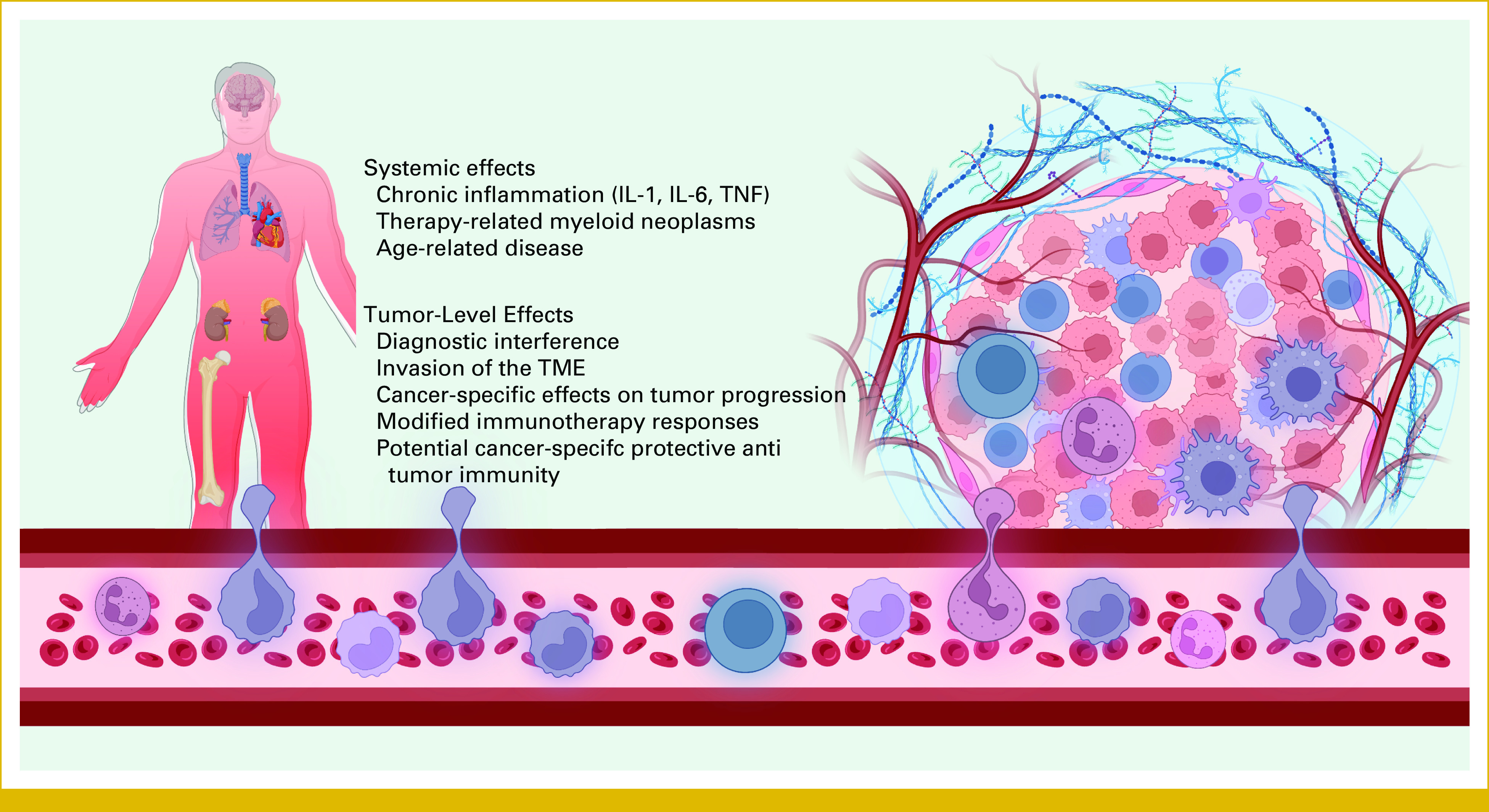
Summary of the systemic and tumor-level effects of CH in patients with solid tumor supported by recent research. Similar to healthy individuals, patients with cancer with CH will experience elevated systemic inflammation, higher risk of age-related conditions, especially cardiovascular disease, and increased hematologic cancer risk, primarily via therapy-related myeloid neoplasm. At the tumor level, CH has a diverse range of effects that vary on the basis of cancer type and CH driver mutation, while also interfering with the diagnosis of tumour mutations. Created with BioRender.4 CH, clonal hematopoiesis; IL, interleukin; TME, tumor microenvironment; TNF, tumor necrosis factor.
Patients with cancer with >10% VAF CH have been found to experience poorer overall survival than CH-negative patients, even when controlling for factors such as age, sex, and smoking. Strikingly, however, most of this effect was not driven by tMN development, but rather the most common cause of death among patients in the cohort was progression of the primary tumor.40 The reason for this effect on cancer progression is yet to be determined, with different cancer types and CH-driving mutations each experiencing distinct relationships (see CH at the Tumor-Immune Interface section, below). With tumor progression being a potential driver of this relationship between CH and mortality in patients with cancer, the lack of literature examining CH in cancer progression and metastasis is surprising. A small study of patients with metastatic renal cell carcinoma found CH in 43% of patients, negatively affecting overall survival.59 Sequencing of metastatic breast cancer specimens found that CH may be dictating bone metastasis, with enrichment for DNMT3A mutations in the samples found in a pattern resembling CH.60 Although these findings are notable, they are nonetheless incidental, and the conclusions that can be drawn from them are limited. A recent analysis of the results of the FIRE-3 trial for metastatic colorectal cancer was able to focus primarily on CH and outcomes in metastatic cancer, finding that 36% of the patients in the trial had CH and that CH was associated with improved survival outcomes—driven specifically by mutations in DNMT3A.61 In an effort to expand on these findings, the influence of CH on survival was evaluated in metastatic esophagogastric and colorectal cancers. This study found that CH was associated with reduced overall survival in the esophagogastric cancers and had a null effect in colorectal cancers—although DNMT3A mutation status was not independently evaluated as in the FIRE-3 trial analysis.62
CH may also provide value as a predictive biomarker for cancer immunotherapy treatments. A study conducted on the same MSK cohort described above examined the relationship between CH and outcomes for patients undergoing immune checkpoint inhibitor (ICI; anti–PD-1 receptor/ligand) therapy, finding that CH was predictive of poorer survival for most cancer types, with the notable exception of colorectal cancer.63 DNMT3A mutation status also identified a distinct subgroup of patients with metastatic non–small-cell lung cancer that saw improved responses to ICI, although it was unclear whether these mutations were found in tumor cells or CH-mutant tumor-infiltrating leukocytes.64 CH in CAR T-cell therapy has also demonstrated predictive value, although results vary between studies. An initial investigation showed that CH was associated with complete response and cytokine release syndrome but not survival in patients with non-Hodgkin lymphoma and multiple myeloma.65 Another study examining CAR T-cell therapy in non-Hodgkin lymphoma found contradicting results, with CH predicting improved overall survival but no change in response rates.66
Diagnostic Relevance of CH in Solid Cancers
The complete range of prognostic and predictive implications of CH are yet to be fully realized; however, CH has already made an unplanned entry into precision oncology by appearing as an incidental finding in tumor or liquid biopsy genetic testing for patients. As discussed previously, the common mutational drivers of CH fall on common cancer driver genes, and as such, mutations carried by CH clones may be misinterpreted as tumor mutations or even germline events.
Germline interference is generally rare among patients with cancer—CH has been recorded to interfere in just 0.3% and 0.05% of patients in two large studies of germline genetic testing.67,68 Although rare, appropriate measures must be taken to avoid the misdiagnosis of germline conditions such as Li-Fraumeni syndrome (LFS), a condition driven by mutations in the common CH gene TP53. A follow-up study for TP53 variants in germline testing was able to successfully distinguish LFS from CH and other somatic expansions using established LFS diagnostic criteria, tracking of variants through family history, and VAF evaluation.69
CH becomes slightly harder to differentiate when it is discovered alongside other somatic variants sourced from a patient's tumor. If both tumor and matched blood have been sequenced, this distinction can be informed by variations in VAF of the clone between the two samples, with higher blood VAF indicating a clone of hematopoietic origin.40 With tumor-only sequencing, CH mutations may lead to incorrect reporting of tumor variants, potentially leading to recommendations for inappropriate targeted therapies of little to no clinical benefit. In multiple cohorts, CH-associated mutations found in the blood are frequently detected in unpaired tumor sequencing, contributing to the erroneous calling of tumor variants in as many as 5% of patients.70,71 The contamination of liquid biopsies with CH variants is also problematic, with evaluations of cell-free DNA samples showing a significant proportion of somatic mutations that display features consistent with CH.72-75 As an incidental finding, CH with high-risk mutations can act as a point of referral for further hematologic consultation, helping to reveal occult hematological malignancies.76 The possibility of CH contaminating or interfering with diagnostic tests warrants consideration from precision oncologists, although knowledge of CH and the utilization of matched blood and/or tissue normal sequencing alongside tumor sequencing can drastically reduce the risk of any adverse consequences for patients.
CH at the Tumor-Immune Interface
The clinical associations found between CH and cancer are becoming increasingly apparent with each upcoming study, so it is imperative that these are accompanied by basic research to support causal associations and identify potential confounding relationships. By using samples from primary breast tumors, researchers identified that immune cells carrying somatic mutations in CH driver genes were infiltrating the tumor microenvironment (TME).77 Additionally, there is evidence that patients with solid tumors with CH, at least those with TET2 or DNMT3A variants, experience elevated levels of lymphocyte invasion in the TME.78 Given that these mutant immune cells are entering the TME, it is reasonable to predict that they might be disturbing the intricate balance of immunity and eliciting a direct effect on the growth and progression of the tumor.
At the cellular level, changes to immune function induced by CH driver mutations have both protumorigenic and antitumorigenic effects. For example, macrophage-specific mutations in TET2 and PPM1D can drive elevated levels of IL-1β and IL-18 via increased activation of the NLRP3 inflammasome.32,56 The NLRP3 inflammasome has been shown to play a bidirectional role in the antitumor immune response, with activation of the pathway linked to the progression of several types of cancer such as breast cancer, lung cancer, and lymphoma, although of note, NLRP3 activation has been found to be protective in colorectal cancers.79-83 IL-6, another commonly elevated inflammatory marker in CH, is linked with not only numerous detrimental processes in tumorigenesis such as tumor cell proliferation and angiogenesis but also antitumorigenic processes such as T-cell trafficking to the tumor site.84 The disruption of common CH driver genes, namely DNMT3A and TET2, can also directly modulate the function of CAR T cells. With the loss of TET2, CAR T cells demonstrated a central memory phenotype that helped enhance the potency of the cells while DNMT3A regularly plays a role in inducing the epigenetic changes that underlie exhaustion, so deletion facilitated an enhanced antitumor response.85,86 These studies directly reference CH mutations in CAR T cells, although it is yet to be determined how these mutations in normal circulating human T cells can modulate their function.
Animal models have also provided evidence for distinct roles of the CH driver genes in tumorigenesis. TET2 has been most extensively studied in this regard, with one study highlighting a protumorigenic effect of TET2 deletion that operates through increased populations of granulocytic myeloid-derived suppressor cells that deplete CD8+ T cells, driving immunosuppression and tumor growth in models of hepatocellular carcinoma and breast cancer.87 Other models of hematopoietic TET2 depletion have shown varying effects, promoting angiogenesis and tumor progression in lung cancer while reducing tumor burden in melanoma by facilitating a proinflammatory tumor-associated macrophage phenotype that augmented T-cell infiltration.88,89 Less data are available for other common CH drivers, but similar trends appear. DNMT3A loss of function mutations appeared to drive colitis-associated colon cancer growth and progression, although the exact mechanism has not yet been elucidated.90 ASXL1 mutations were found to promote tumorigenesis in a variety of cancer models through disrupted T-cell development and functionality.91 Finally, wild-type p53 in myeloid cells was found to suppress M2 macrophage polarization and tumor invasiveness in an intestinal cancer model, indicating a potential cancer risk with TP53 loss-of-function mutations in CH.92
FUTURE DIRECTIONS AND CHALLENGES
The findings presented here depict an exciting future for research examining the role of CH in solid tumors. Building on this foundation, there are a number of additional steps that need to be taken to translate these findings into precision oncology practice. Primarily, current knowledge of CH in solid cancers depicts a relationship that is as heterogeneous as cancer itself, and more research will be required to better understand this relationship to determine the contexts where CH is helpful, harmful, both, or neutral for patients with solid tumors. Further analyses of longitudinal population and cancer-specific cohorts will provide a better estimate of the risk of specific cancers and clinical outcomes in relation to CH, hopefully while being able to separate confounding effects due to cancer therapy and shared risk factors between both conditions. Although connected by signs of systemic inflammation and immune dysfunction, the variety of CH driver mutations have demonstrated diverse effects on tumorigenesis at different sites, the most notable of which being the distinct protective effect of CH reported in colorectal cancers. These nuances must be studied further if we ever hope to use CH in cancer therapy—both to develop novel strategies and optimize existing ones, whether that be targeting CH directly, incorporating CH as a prognostic and/or predictive biomarker, or leveraging CH in the TME to our benefit.
Targeting CH directly remains elusive, largely driven by a lack of motivation for clinical trials because of the limited risk-benefit profile that has been proposed by existing research. Potential avenues of treatment have been reviewed previously with relevance to precision oncology by Miller and Steensma,93 although some novel approaches have been proposed since then. For example, DNMT3A R882 mutations are noted as the most prevalent of all CH variants across many studies, and the herbal extract, oridonin, has been proposed as a promising candidate to suppress both CH and leukemias that are driven by such mutations.94 One novel approach for TET2-mutant CH is the utilization of eltrombopag, a thrombopoietin receptor agonist that can restrict the growth of malignant TET2-mutant clones while favoring the expansion of healthy cells.95 Targeting of TET2-mutant cells has also shown promise with the mutant IDH1/2 metabolite 2-hydroxyglutarate and similarly engineered small molecule inhibitors, as well as XPO1 inhibitors.96,97 Other advancements include the potential of PARP1 inhibition for antagonizing TET2-mutant CH; however, the risk of hematologic malignancy is elevated in patients undergoing this therapy and thus more research is warranted before this can be applied safely in a clinical setting.44,98 With the implication of inflammasome activity in TET2-mutant and other forms of CH, clinical trials such as IMPACT are now investigating the value of the anti–IL-1β antibody canakinumab in high-risk CH (ie, clonal cytopenia of undetermined significance; ClinicalTrials.gov identifier: NCT05641831).
Beyond cancer, developments in our understanding of the biology of CH will also prove beneficial in a variety of clinical settings. For example, the implications of more novel CH drivers such as lymphoid cancer–associated genes and mCAs are not yet fully elucidated in a more general sense, let alone in the context of cancer. Looking toward the future, the influence of CH in the era of precision medicine is expanding rapidly, and patients with solid cancer await the research and development of effective strategies for managing CH in a clinical oncology setting.
SUPPORT
M.M.B. is supported by a Canada Graduate Scholarship-Master's (CGS-M) from the Canadian Institutes of Health Research (CIHR) and M.J.R. by a CIHR Project Grant (Application No. 451137).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
No other potential conflicts of interest were reported.
REFERENCES
- 1. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.BioRender. http://biorender.com
- 5. Niroula A, Sekar A, Murakami MA, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med. 2021;27:1921–1927. doi: 10.1038/s41591-021-01521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saiki R, Momozawa Y, Nannya Y, et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat Med. 2021;27:1239–1249. doi: 10.1038/s41591-021-01411-9. [DOI] [PubMed] [Google Scholar]
- 8. Loh P-R, Genovese G, McCarroll SA. Monogenic and polygenic inheritance become instruments for clonal selection. Nature. 2020;584:136–141. doi: 10.1038/s41586-020-2430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gao T, Ptashkin R, Bolton KL, et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat Commun. 2021;12:338. doi: 10.1038/s41467-020-20565-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Young AL, Challen GA, Birmann BM, et al. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell. 2018;22:157–170. doi: 10.1016/j.stem.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559:400–404. doi: 10.1038/s41586-018-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Desai P, Mencia-Trinchant N, Savenkov O, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24:1015–1023. doi: 10.1038/s41591-018-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buscarlet M, Provost S, Zada YF, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130:753–762. doi: 10.1182/blood-2017-04-777029. [DOI] [PubMed] [Google Scholar]
- 17. Cook EK, Luo M, Rauh MJ. Clonal hematopoiesis and inflammation: Partners in leukemogenesis and comorbidity. Exp Hematol. 2020;83:85–94. doi: 10.1016/j.exphem.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 18. Cook EK, Izukawa T, Young S, et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019;3:2482–2486. doi: 10.1182/bloodadvances.2018024729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med. 2021;218:e20201544. doi: 10.1084/jem.20201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yura Y, Sano S, Walsh K. Clonal hematopoiesis: A new step linking inflammation to heart failure. JACC Basic Transl Sci. 2020;5:196–207. doi: 10.1016/j.jacbts.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cobo I, Tanaka T, Glass CK, et al. Clonal hematopoiesis driven by DNMT3A and TET2 mutations: Role in monocyte and macrophage biology and atherosclerotic cardiovascular disease. Curr Opin Hematol. 2022;29:1–7. doi: 10.1097/MOH.0000000000000688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cull AH, Snetsinger B, Buckstein R, et al. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70.e13. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 23. Sano S, Oshima K, Wang Y, et al. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res. 2018;123:335–341. doi: 10.1161/CIRCRESAHA.118.313225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hormaechea-Agulla D, Matatall KA, Le DT, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell. 2021;28:1428–1442.e6. doi: 10.1016/j.stem.2021.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abegunde SO, Buckstein R, Wells RA, et al. An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp Hematol. 2018;59:60–65. doi: 10.1016/j.exphem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 26. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dorsheimer L, Assmus B, Rasper T, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:25. doi: 10.1001/jamacardio.2018.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10:eaan8292. doi: 10.1126/scitranslmed.aan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marnell CS, Bick A, Natarajan P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J Mol Cell Cardiol. 2021;161:98–105. doi: 10.1016/j.yjmcc.2021.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abplanalp WT, Mas-Peiro S, Cremer S, et al. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol. 2020;5:1170–1175. doi: 10.1001/jamacardio.2020.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sano S, Oshima K, Wang Y, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71:875–886. doi: 10.1016/j.jacc.2017.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vlasschaert C, McNaughton AJM, Chong M, et al. Association of clonal hematopoiesis of indeterminate potential with worse kidney function and anemia in two cohorts of patients with advanced chronic kidney disease. J Am Soc Nephrol. 2022;33:985–995. doi: 10.1681/ASN.2021060774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arends CM, Weiss M, Christen F, et al. Clonal hematopoiesis in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Haematologica. 2020;105:e264–e267. doi: 10.3324/haematol.2019.223305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller PG, Qiao D, Rojas-Quintero J, et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood. 2022;139:357–368. doi: 10.1182/blood.2021013531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim PG, Niroula A, Shkolnik V, et al. Dnmt3a-mutated clonal hematopoiesis promotes osteoporosis. J Exp Med. 2021;218:e20211872. doi: 10.1084/jem.20211872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bolton KL, Koh Y, Foote MB, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. Nat Commun. 2021;12:5975. doi: 10.1038/s41467-021-26138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou Y, Shalhoub RN, Rogers SN, et al. Clonal hematopoiesis is not significantly associated with Covid-19 disease severity. Blood. 2022;140:14. doi: 10.1182/blood.2022015721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouzid H, Belk JA, Jan M, et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. medRxiv. [DOI] [PMC free article] [PubMed]
- 40. Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374–382.e4. doi: 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bolton KL, Ptashkin RN, Gao T, et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet. 2020;52:1219–1226. doi: 10.1038/s41588-020-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pich O, Reyes-Salazar I, Gonzalez-Perez A, et al. Discovering the drivers of clonal hematopoiesis. Nat Commun. 2022;13:4267. doi: 10.1038/s41467-022-31878-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kar SP, Quiros PM, Gu M, et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat Genet. 2022;54:1155–1166. doi: 10.1038/s41588-022-01121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kessler MD, Damask A, O’Keeffe S, et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature. 2022;612:301–309. doi: 10.1038/s41586-022-05448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tian R, Wiley B, Liu J, et al. Clonal hematopoiesis and risk of incident lung cancer. J Clin Oncol. 2023;41:1423–1433. doi: 10.1200/JCO.22.00857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang A, Xu Y, Yu Y, et al. Clonal hematopoiesis and risk of prostate cancer in large samples of European ancestry men. Hum Mol Genet. 2022;32:489–495. doi: 10.1093/hmg/ddac214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat Rev Cancer. 2017;17:513–527. doi: 10.1038/nrc.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sperling AS, Guerra VA, Kennedy JA, et al. Lenalidomide promotes the development of TP53-mutated therapy-related myeloid neoplasms. Blood. 2022;140:1753–1763. doi: 10.1182/blood.2021014956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hsu JI, Dayaram T, Tovy A, et al. PPM1D mutations drive clonal hematopoiesis in response to cytotoxic chemotherapy. Cell Stem Cell. 2018;23:700–713.e6. doi: 10.1016/j.stem.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kahn JD, Miller PG, Silver AJ, et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood. 2018;132:1095–1105. doi: 10.1182/blood-2018-05-850339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boucai L, Falcone J, Ukena J, et al. Radioactive iodine–related clonal hematopoiesis in thyroid cancer is common and associated with decreased survival. J Clin Endocrinol Metab. 2018;103:4216–4223. doi: 10.1210/jc.2018-00803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh A, Mencia-Trinchant N, Griffiths EA, et al. Mutant PPM1D- and TP53-driven hematopoiesis populates the hematopoietic compartment in response to peptide receptor radionuclide therapy. JCO Precis Oncol. 2022 doi: 10.1200/PO.21.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evans MA, Sano S, Walsh K. Cardiovascular disease, aging, and clonal hematopoiesis. Annu Rev Pathol Mech Dis. 2020;15:419–438. doi: 10.1146/annurev-pathmechdis-012419-032544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sano S, Wang Y, Ogawa H, et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight. 2021;6:e146076. doi: 10.1172/jci.insight.146076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yura Y, Miura-Yura E, Katanasaka Y, et al. The cancer therapy-related clonal hematopoiesis driver gene Ppm1d promotes inflammation and non-ischemic heart failure in mice. Circ Res. 2021;129:684–698. doi: 10.1161/CIRCRESAHA.121.319314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eder LN, Martinovic D, Mazzeo P, et al. Fatal progression of mutated TP53-associated clonal hematopoiesis following anti-CD19 CAR-T cell therapy. Curr Oncol. 2023;30:1146–1150. doi: 10.3390/curroncol30010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hagiwara K, Natarajan S, Wang Z, et al. Dynamics of age- versus therapy-related clonal hematopoiesis in long-term survivors of pediatric cancer. Cancer Discov. 2023;13:844–857. doi: 10.1158/2159-8290.CD-22-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bacon JVW, Annala M, Soleimani M, et al. Plasma circulating tumor DNA and clonal hematopoiesis in metastatic renal cell carcinoma. Clin Genitourin Cancer. 2020;18:322–331.e2. doi: 10.1016/j.clgc.2019.12.018. [DOI] [PubMed] [Google Scholar]
- 60. Rinaldi J, Sokol ES, Hartmaier RJ, et al. The genomic landscape of metastatic breast cancer: Insights from 11,000 tumors. PLoS One. 2020;15:e0231999. doi: 10.1371/journal.pone.0231999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Arends CM, Dimitriou S, Stahler A, et al. Clonal hematopoiesis is associated with improved survival in patients with metastatic colorectal cancer from the FIRE-3 trial. Blood. 2022;139:1593–1597. doi: 10.1182/blood.2021014108. [DOI] [PubMed] [Google Scholar]
- 62. Diplas BH, Ptashkin R, Chou JF, et al. Clinical importance of clonal hematopoiesis in metastatic gastrointestinal tract cancers. JAMA Netw Open. 2023;6:e2254221. doi: 10.1001/jamanetworkopen.2022.54221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hsiehchen D, Sfreddo HJ, Zhao K, et al. Clonal hematopoiesis and differential outcomes after immune checkpoint blockade. Cancer Cell. 2022;40:1071–1072. doi: 10.1016/j.ccell.2022.08.024. [DOI] [PubMed] [Google Scholar]
- 64. Ricciuti B, Alessi JVM, Li YY, et al. DNMT3A mutation to identify a subset of non-small cell lung cancers with increased sensitivity to PD-(L)1 blockade. J Clin Oncol. 2021;39 suppl 15; abstr 9113. [Google Scholar]
- 65. Miller PG, Sperling AS, Brea EJ, et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 2021;5:2982–2986. doi: 10.1182/bloodadvances.2021004554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Teipel R, Kroschinsky F, Kramer M, et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Adv. 2022;6:1941–1946. doi: 10.1182/bloodadvances.2021005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maani N, Panabaker K, McCuaig JM, et al. Incidental findings from cancer next generation sequencing panels. npj Genomic Med. 2021;6:63. doi: 10.1038/s41525-021-00224-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weitzel JN, Chao EC, Nehoray B, et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med. 2018;20:809–816. doi: 10.1038/gim.2017.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schwartz AN, Hyman SR, Stokes SM, et al. Evaluation of TP53 variants detected on peripheral blood or saliva testing: Discerning germline from somatic TP53 variants. JCO Precis Oncol. 2021 doi: 10.1200/PO.21.00278. [DOI] [PubMed] [Google Scholar]
- 70. Ptashkin RN, Mandelker DL, Coombs CC, et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 2018;4:1589–1593. doi: 10.1001/jamaoncol.2018.2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Coombs CC, Gillis NK, Tan X, et al. Identification of clonal hematopoiesis mutations in solid tumor patients undergoing unpaired next-generation sequencing assays. Clin Cancer Res. 2018;24:5918–5924. doi: 10.1158/1078-0432.CCR-18-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Razavi P, Li BT, Brown DN, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hu Y, Ulrich BC, Supplee J, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24:4437–4443. doi: 10.1158/1078-0432.CCR-18-0143. [DOI] [PubMed] [Google Scholar]
- 74. Jensen K, Konnick EQ, Schweizer MT, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol. 2021;7:107. doi: 10.1001/jamaoncol.2020.5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kotecha RR, Gedvilaite E, Ptashkin R, et al. Matched molecular profiling of cell-free DNA and tumor tissue in patients with advanced clear cell renal cell carcinoma. JCO Precis Oncol. 2022 doi: 10.1200/PO.22.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aldea M, Tagliamento M, Bayle A, et al. Liquid biopsies for circulating tumor DNA detection may reveal occult hematologic malignancies in patients with solid tumors. JCO Precis Oncol. 2023 doi: 10.1200/PO.22.00583. [DOI] [PubMed] [Google Scholar]
- 77. Kleppe M, Comen E, Wen HY, et al. Somatic mutations in leukocytes infiltrating primary breast cancers. npj Breast Cancer. 2015;1:15005. doi: 10.1038/npjbcancer.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Severson EA, Riedlinger GM, Connelly CF, et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood. 2018;131:2501–2505. doi: 10.1182/blood-2018-03-840629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hamarsheh S, Zeiser R. NLRP3 inflammasome activation in cancer: A double-edged sword. Front Immunol. 2020;11:1444. doi: 10.3389/fimmu.2020.01444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Guo B, Fu S, Zhang J, et al. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. 2016;6:36107. doi: 10.1038/srep36107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang Y, Kong H, Zeng X, et al. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol Rep. 2016;35:2053–2064. doi: 10.3892/or.2016.4569. [DOI] [PubMed] [Google Scholar]
- 82. Zhao X, Zhang C, Hua M, et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget. 2017;8:108571–108583. doi: 10.18632/oncotarget.21010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity. 2015;43:751–763. doi: 10.1016/j.immuni.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 84. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. 2014;26:38–47. doi: 10.1016/j.smim.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Prinzing B, Zebley CC, Petersen CT, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. 2021;13:eabh0272. doi: 10.1126/scitranslmed.abh0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li S, Feng J, Wu F, et al. TET2 promotes anti-tumor immunity by governing G-MDSCs and CD8+ T-cell numbers. EMBO Rep. 2020;21:e49425. doi: 10.15252/embr.201949425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nguyen YTM, Fujisawa M, Nguyen TB, et al. Tet2 deficiency in immune cells exacerbates tumor progression by increasing angiogenesis in a lung cancer model. Cancer Sci. 2021;112:4931–4943. doi: 10.1111/cas.15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pan W, Zhu S, Qu K, et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity. 2017;47:284–297.e5. doi: 10.1016/j.immuni.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Feng Y, Newsome R, Robinson T, et al. Dnmt3a mutations in the hematopoietic system promote colitis-associated colon cancer: A model of clonal hematopoiesis in solid tumors. Blood. 2021;138 suppl 1; abstr 2161. [Google Scholar]
- 91. Liu X, Sato N, Shimosato Y, et al. CHIP-associated mutant ASXL1 in blood cells promotes solid tumor progression. Cancer Sci. 2022;113:1182–1194. doi: 10.1111/cas.15294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. He X-Y, Xiang C, Zhang C-X, et al. p53 in the myeloid lineage modulates an inflammatory microenvironment limiting initiation and invasion of intestinal tumors. Cell Rep. 2015;13:888–897. doi: 10.1016/j.celrep.2015.09.045. [DOI] [PubMed] [Google Scholar]
- 93. Miller PG, Steensma DP. Implications of clonal hematopoiesis for precision oncology. JCO Precis Oncol. 2020 doi: 10.1200/PO.20.00144. [DOI] [PubMed] [Google Scholar]
- 94. Liao M, Dong Q, Chen R, et al. Oridonin inhibits DNMT3A R882 mutation-driven clonal hematopoiesis and leukemia by inducing apoptosis and necroptosis. Cell Death Discov. 2021;7:297. doi: 10.1038/s41420-021-00697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guan Y, Hasipek M, Jiang D, et al. Eltrombopag inhibits TET dioxygenase to contribute to hematopoietic stem cell expansion in aplastic anemia. J Clin Invest. 2022;132:e149856. doi: 10.1172/JCI149856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Guan Y, Tiwari AD, Phillips JG, et al. A therapeutic strategy for preferential targeting of TET2-mutant and TET dioxygenase–deficient cells in myeloid neoplasms. Blood Cancer Discov. 2021;2:146–161. doi: 10.1158/2643-3230.BCD-20-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jing C-B, Fu C, Prutsch N, et al. Synthetic lethal targeting of TET2-mutant hematopoietic stem and progenitor cells (HSPCs) with TOP1-targeted drugs and PARP1 inhibitors. Leukemia. 2020;34:2992–3006. doi: 10.1038/s41375-020-0927-5. [DOI] [PubMed] [Google Scholar]
- 98. Moore KN, Mirza MR, Matulonis UA. The poly (ADP ribose) polymerase inhibitor niraparib: Management of toxicities. Gynecol Oncol. 2018;149:214–220. doi: 10.1016/j.ygyno.2018.01.011. [DOI] [PubMed] [Google Scholar]