

Abstract
Silanes are important compounds in industrial and synthetic chemistry. Here, we develop a general approach for the synthesis of disilanes as well as linear and cyclic oligosilanes via the reductive activation of readily available chlorosilanes. The efficient and selective generation of silyl anion intermediates, which are arduous to achieve by other means, allows for the synthesis of various novel oligosilanes by heterocoupling. In particular, this work presents a modular synthesis for a variety of functionalized cyclosilanes, which may give rise to materials with distinct properties from linear silanes but remain challenging synthetic targets. In comparison to the traditional Wurtz coupling, our method features milder conditions and improved chemoselectivity, broadening the functional groups that are compatible in oligosilane preparation. Computational studies support a mechanism whereby differential activation of sterically and electronically distinct chlorosilanes are achieved in an electrochemically driven radical-polar crossover mechanism.
Keywords: cross coupling, electrochemistry, disilanes, oligosilanes, electrosynthesis
Graphical Abstract
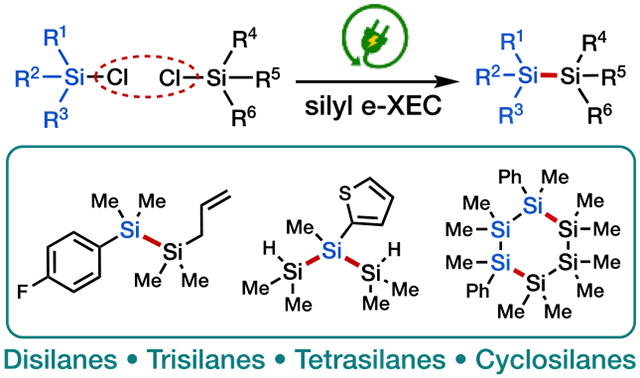
The formation of Si─Si bonds is an important transformation in organic synthesis. Herein, we present a unified electrochemical strategy for the efficient and selective synthesis of disilanes, oligosilanes, and cyclosilanes—which are important precursors for the synthesis of functional organic and polymeric compounds—from readily available chlorosilanes.
Introduction
Linear and cyclic oligosilanes have attracted considerable interest in organic synthesis and material science.[1] Disilanes are versatile reagents for the construction of C─Si bonds via transition-metal catalyzed Si─Si bond activation, which provides convenient access to a range of organosilane building blocks (Scheme 1a).[1],[2] Disilanes are also attractive alternatives to hydrosilane reagents in silylation reactions, owing to their greater stability, ease of handling, and because their second silyl group can sequester the leaving group (e.g., by forming strong Si─O or Si─X bonds) thereby creating a strong thermodynamic driving force for the desired reactivity.[1c] Oligosilanes are precursors to industrially valuable materials such as silicon carbide (Scheme 1b),[3] and their unique electronic[4] and luminescent[5] properties resulting from σ-conjugation of the Si─Si bonds has motivated the development of functional materials incorporating Si chains.[6] The reduced degrees of freedom in cyclic oligosilanes[7] influence conformation-dependent σ-conjugation,[8] and the synthesis of these motifs and related poly(cyclosilane)s has been actively pursued. [6b], [9]
Scheme 1.

(a) Disilanes used as silylation reagents in organic synthesis. (b) Oligosilanes used as material precursors. (c) Previous synthetic strategies to construct Si─Si bonds and this work.
Despite significant and growing interest in the chemistry of disilanes and oligosilanes, strategies for their synthesis remain limited (Scheme 1c). A common approach is the canonical Wurtz coupling of halosilanes using sodium or lithium metal, although this method typically exhibits poor functional group tolerance. [10] A milder approach employing Sm/SmI2 has recently been developed, although this method is currently only used for homodimerization.[11] Transition-metal-catalyzed dehydrogenative coupling has also emerged as a promising approach to constructing Si─Si bonds, [12] but the catalytic systems explored to date often display limited substrate generality (e.g., limited to tri- and dihydrosilanes H3SiR and H2SiR2) as well as limited selectivity in cross-coupling reactions.[13] For cross-coupling reactions, the direct generation of a silyl anion followed by reaction with a chlorosilane is commonly used,[1b] where silyl anions can be prepared by site-selective Si─SiMe3 bond cleavage.[14] Recently, Ito and coworkers disclosed an elegant approach to the synthesis of small oligosilanes (n = 2–4) via activation of silylboron reagents with methyllithium.[15] Although this approach substantially broadens the scope of accessible functionalized oligosilanes, it is currently constrained by the availability of silylboron reagents, which remain difficult to access.
Beyond traditional chemical synthetic approaches, a select few reports have demonstrated the feasibility of using electrochemistry for the synthesis of di- and polysilanes, although a general strategy enabling rational retrosynthetic disconnection is lacking. Starting in the late 1970s, Hengge and coworkers published two reports on the electroreduction of chlorosilanes to afford symmetric disilanes in good Faradaic yield,[16] although their approach utilized a sacrificial Hg, Pb, or Cd anode, generating highly toxic metal salts as byproducts. In separate advances, Shono,[17] Kunai,[18] and Grogger[19] developed this system further to employ less hazardous electrode materials and expand the reaction scope to heterocoupling.[17b], [18a] However, all these approaches require a large excess of one coupling partner—two or more equivalents—to ensure satisfactory cross-coupling selectivity, as an equal ratio of the two coupling partners usually led to a statistical mixture of the homo- and hetero-coupling products. In addition, the substrate generality remained significantly limited and the functional group compatibility was not demonstrated, especially for functionalities that allow for further functionalization or polymerization such as hydride (Si–H), vinyl, and thienyl groups.[20] To our knowledge, there are very few examples of the use of electrochemistry to synthesize structurally well-defined oligosilanes,[17b], [21] and there is only one report on the electrochemical preparation of cyclosilanes, wherein cyclobutasilanes were synthesized in low product yields (≤ 3%).[16b], [22]
Against this backdrop, we advance herein a unified strategy for the preparation of disilanes by both homo- and cross-coupling, as well as linear and cyclic oligosilanes, via the electroreductive activation and coupling of readily available chlorosilanes, thus providing a new synthetic logic to access a diverse range of known and novel silicon compounds.
Results and Discussion
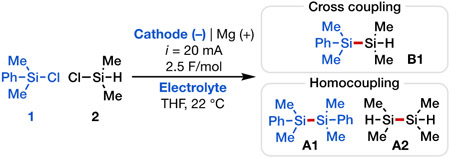
In previous work, our laboratory demonstrated that chlorosilanes can be reduced on a carbon cathode to form silyl radicals in the context of alkene functionalization.[23] We became interested in expanding this electroreductive strategy both mechanistically and synthetically to provide a general and mild approach for the synthesis of disilanes and oligosilanes with high selectivity, with particular application to cyclic oligosilanes that are difficult to prepare by other means. However, cross-coupling of two distinct silyl radicals can be difficult to achieve in a selective manner and is instead likely to lead to a statistical mixture of homo- and hetero-disilanes. To circumvent this challenge, we conceived of a different strategy wherein upon the application of sufficiently reducing potential, a chlorosilane substrate (1) would undergo an overall two-electron reduction via an electrochemical-chemical-electrochemical (ECE) sequence to form a silyl anion (Int2) upon loss of Cl− (Scheme 2a). This ionic intermediate could then react with another chlorosilane partner (2) via nucleophilic substitution to afford the desired cross-coupled product (B1). To achieve high selectivity with this approach, we envisioned exploiting the distinct electronic and steric properties of chlorosilanes with different structures. For example, aryl-substituted chlorosilanes such as chlorodimethylphenylsilane (1) are substantially easier to reduce than alkyl-substituted congeners (computed single-electron reduction potentials Ered = −3.73 V for 1 versus −4.67 V for 2 vs. Fc0/+; these potentials are likely overestimated but the trend is evident;[24] see Supporting Information Section 7). Meanwhile, hydrochlorosilanes such as chlorodimethylsilane (2) would serve as excellent two-electron electrophiles in the desired nucleophilic substitution due to their smaller steric profiles (DFT prediction showed that reaction between Int2 and 2 has a barrier ΔG‡ = 13.7 kcal/mol, vs. ΔG‡ = 17.4 kcal/mol for reaction between Int2 and 1; see Supporting Information Section 7).[25] Importantly, both arylsilyl (Si–Ph) and hydrosilyl (Si–H) groups are useful functionalities that can undergo further derivatization via Si─C or Si─H bond activation under orthogonal conditions,[21], [26] thus rendering the products of the envisioned coupling reaction synthetically valuable. We report here our use of this approach as a generalizable means for the synthesis of a range of diverse silane products, including desirable cyclic oligosilanes (Scheme 2b).
Scheme 2.

(a) Design principle of the silyl cross electrophile coupling. (b) This work.
To establish the feasibility of the proposed approach, we initially studied the reductive coupling of chlorodimethylphenylsilane (1) and chlorodimethylsilane (2), and we discovered that in an undivided cell using a sacrificial Mg anode and graphite cathode in a THF solution of tetrabutylammonium perchlorate (TBAClO4) electrolyte, the desired heterodisilane B1 was generated in 65% yield (Table 1, entry 1). Additionally, good cross-coupling selectivity was attained using a relatively small excess of 2 (1.5 equiv) with respect to 1, with homodisilane A1 as the main side product (13:1 ratio), in addition to a negligible amount of homodisilane A2. We further surveyed Ni, Mo, Pt, and Ag as alternative working electrodes and found that nickel and molybdenum promote the formation of B1 in excellent yields (68–70%) with excellent selectivity (>20:1) over homocoupling (Table 1, entries 4–5).[27] Of note, the electrolyte concentration could be reduced by half without decreasing the reaction yield when tetrabutylammonium tetraphenylborate (TBABPh4) was used instead of TBAClO4 (Table 1, entries 9–10), likely a result of greater dissociation of TBABPh4 versus TBAClO4 in the THF solvent.[28] Further, comparable yield and selectivity magnitudes for B1 were achieved with as little as 1.25 equiv of 2 with TBABPh4 as electrolyte (Table 1, entry 10). Control experiments run in the absence of an electric current or in the presence of magnesium powder as the chemical reductant in lieu of electrolysis showed no product formation (see Supporting Information Section 4). Of note, reaction using N,N-diisopropylethylamine as a sacrificial reductant instead of a Mg sacrificial anode produced B1 in comparable yield and synthetically useful selectivity (Table 1, entry 11), providing a homogenous system for disilane synthesis that is potentially amenable to scaling up using flow electrochemistry. Ultimately, we identified two sets of optimal reaction conditions using magnesium sacrificial anode, either with molybdenum cathode with TBABPh4 as electrolyte or graphite cathode with TBAClO4 as electrolyte, to achieve efficient Si–Si coupling (see Supporting Information Section 3 for full details of reaction optimization).
Table 1.
Optimization of conditions for cross-electrophile coupling of chlorosilanes
| ||||
|---|---|---|---|---|
| Entry | Cathode | Electrolyte | Yield (B1) | Selectivity (B1:A1) |
| 1 | Graphite | TBACIO4 | 65% | 13:1 |
| 2 | Pt | TBACIO4 | 51% | 5:1 |
| 3 | Ag | TBACIO4 | 62% | 8:1 |
| 4 | Ni | TBACIO4 | 68% | >20:1 |
| 5 | Mo | TBACIO4 | 70% (67%) | >20:1 |
| 6 | Mo | TBAPF6 | 48% | 6:1 |
| 7 | Mo | TBABF4 | 23% | 15:1 |
| 8 | Mo | TBABPh4 | 66% | >20:1 |
| 9[a] | Mo | TBABPh4 | 67% | >20:1 |
| 10 [b] | Mo | TBABPh4 | 65% | >20:1 |
| 11[c] | Graphite | TBACIO4 | 54% | 7:1 |
Reactions conditions: 1 (1 mmol, 1 equiv), 2 (1.5 mmol, 1.5 equiv) and electrolytes (2 mmol, 2 equiv, 0.2 M) in THF (8 mL) in an undivided cell; constant current of 20 mA, passing 2.5 F/mol of charge (3.5h). Yields were determined by 1H NMR using dibromomethane as internal standard. Isolated yields are shown in parentheses. Selectivities were determined by 1H NMR spectroscopy for B1 versus A1; A2 was detected in trace amounts by GC (see SI Section 4).
0.1 M of TBABPh4.
0.1 M TBABPh4 and 1.25 equiv of 2.
Reaction using sacrificial amine (4 equiv of N,N-diisopropylethylamine) with graphite as the anode and cathode.
We next explored the substrate scope of our approach under the optimal reaction conditions. First, homocoupling of chlorosilanes was found to be efficient, providing a diverse range of disilanes in high yield (Scheme 3b, A1–A4). In particular, 1,1,2,2-tetramethyl-1,2-diphenyldisilane (A1), a commonly used silylating agent in organic synthesis, can be prepared on a 5 mmol scale. This reagent is prepared industrially via the Wurtz coupling using stoichiometric Na as the terminal reductant. Our electroreductive method thus offers a milder and potentially more practical alternative to prepare this and analogous disilanes. The strained silacyclophane A5 could be synthesized in 38% yield starting from a commercial bischlorosilane precursor, via the formation of two Si─Si bonds. This molecule and related cyclophane silanes have attracted interest for their luminescent properties, [29] although they have previously been synthesized in very low yields (<15%) using multi-step strategies (Pd-catalyzed cross coupling or Grignard addition). [30] Tetrasilane A6 was readily obtained from commercially available chloropentamethyldisilane without Si–Si cleavage. Chlorogermane was also found to be a suitable substrate, providing digermane A7 in 80% yield. Importantly, we successfully synthesized 4-fluorophenyl- and 2-thiophenyl-substituted disilanes (A8 and A9) from their respective chlorosilanes, thus introducing useful functional groups that could help tune the electronic and conductance properties of the silanes and provide handles for further functionalization.[21h], [31] We also attempted the homocoupling of ethoxychlorosilane (product A10), although only oxidized siloxane [(EtO)3Si]2O was isolated (see Supporting Information Section 10). From a mechanistic point of view, we note that the homocoupling of chlorosilanes could also be achieved via the dimerization of silyl radicals. However, this scenario is unlikely with chlorosilane substrates bearing a Ph, vinyl, or silyl substituent because the estimated reduction of the corresponding silyl radicals is substantially easier than that of the initial chlorosilane reduction (see Supporting Information Section 7 for computed potentials), implicating rapid conversion of the intermediate radicals to silyl anions.[32]
Scheme 3.

Scope of electroreductive coupling of monochlorosilanes. Isolated yields are shown unless noted otherwise. 1H NMR yields are provided in parentheses. [a]Prepared on 5 mmol scale passing 2 F/mol of charge. [b]The product was partially inseparable from oxidized disilane. [c]The oxidized siloxane [(EtO)3Si]2O was isolated as the only product. [d]Selectivity for cross coupling/homocoupling (determined using 1H NMR of the crude produce mixture) was >20:1 unless noted otherwise. [e]Selectivity (B7/A1) was 7:1 as determined by 1H NMR of the purified product mixture. [f]Homocoupling products of disilane and digermane were both detected as side products. [g]Selectivity (B12/A8) was 9:1 as determined by 1H NMR of the purified product mixture.
A diverse range of asymmetric disilanes were also synthesized using our electrochemical protocol in generally good yield and excellent selectivity (Scheme 3c). Cross coupling of Ph- and H-substituted chlorosilanes yielded the desired products (B1–B6) and no observed homocoupling products. Substitution of the hydrochlorosilane coupling partner with larger chlorotrimethylsilane yielded the heterodisilane (B7) in good yield (75%) with diminished, albeit good, selectivity (7:1 over homocoupling). Trisilane B8 and pentasilane B9 were likewise synthesized efficiently with excellent selectivity using the same protocol. Further, we also found that silylgermanes (B10–B11) could be prepared with good selectivity via Si–Ge cross coupling. Finally, heterodisilanes bearing functional groups such as arylfluoride, alkene, and thiophene could also be obtained using this method (B12–B13).
We next sought to synthesize more complicated oligosilanes via multi-component cross coupling involving a sequence of iterative electroreductive Si–Cl activation and Si–Si formation processes starting from polychlorosilanes (Scheme 4a). This strategy was first investigated in the synthesis of trisilanes (Scheme 4b, C1–C9). By using 4 equiv of the monochlorosilane coupling partner and passing 5 F/mol of charge, it was possible to convert a variety of dichlorosilanes to trisilanes (C1–C4) in good yields. Interestingly, an unexpected yet useful side product C2′ was isolated from the large-scale synthesis of compound C2, likely as a result of homocoupling of the chlorodisilane intermediate (Int4). Trisilanes C1–C3 feature terminal Si─H bonds that can be transformed into Si─Cl bonds in one step or engage in other dehydrogenative coupling and polymerization reactions;[21e], [33] the corresponding reagents from chlorination could be used to further extend the silicon chain into longer oligo- or polysilanes, or to construct cyclic silanes via cyclization (see below). Starting from a chlorodisilane coupling partner along with dichlorophenylmethylsilane, pentasilane C5 was obtained. Tetrasilanes with an aromatic spacer connecting two disilane moieties (C6, C9) were synthesized in a similar manner via dual Si─Si coupling. Lastly, phenyltrichlorosilane was also a compatible coupling partner, affording tetrasilanes C10 and C11 in synthetically useful yields.[34] However, attempts to react silicon tetrachloride with chlorotrimethylsilane proved unsuccessful, only yielding an inseparable mixture of Si-containing products. Of note, this electrochemically driven approach provides an efficient access to H-terminated oligosilanes with reduced step count, which would typically be synthesized from Ph-terminated silanes by dearylation and reduction.[26a]
Scheme 4.

Scope of electroreductive coupling of of polychlorosilanes towards polysilane synthesis. Isolated yields are shown for all products. [a]4 mmol scale passing 4.5 F/mol of charge. [b]Yield in parentheses was determined by 1H NMR using dibromomethane as internal standard. [c]6 equiv of the monochlorosilane coupling partner, a constant current of 10 mA, and while passing 8 F/mol of charge.
Cyclic oligosilanes have attracted increasing interest over the last few decades, owing to their potential use in molecular electronics[35] and as building blocks for poly(cyclosilane)s.[36] Nevertheless, their wider application has been limited by a relative dearth of methods for their controlled and selective synthesis. There exist seminal and elegant precedents for the preparation of cyclic oligosilane; however, these examples rely on different synthetic strategies and chemical transformations, and a unified approach for the synthesis of cyclosilanes with diverse structures and ring sizes remains elusive and desirable. For example, fully symmetric cyclohexasilane Si6Me12 was formed via the Wurtz coupling.[37] In contrast, site-selectively functionalized cyclosilanes are challenging to prepare by reductive coupling, as it was observed that in the reduction of a 1,6-dibromohexasilane, an unexpected silylene elimination took place to yield a Si5 instead of desired Si6 ring.[38] An alternative approach is to react silyl dianions with dichlorosilanes,[39] which has recently been employed for the synthesis of various cyclohexasilanes in excellent yields and with well-defined relative stereochemistry.[36], [40] Nonetheless, the empirical screening of additives is often necessary in these reactions to reduce undesired skeletal rearrangements.[40a]
We envisioned that by using our electroreductive coupling approach, it would be possible to couple dichlorooligosilanes of the same or different lengths to readily synthesize cyclic silanes of various sizes and substitution patterns, thereby dramatically expanding access to this desirable class of compounds without depending on the limited scope of achievable silyl dianions (Scheme 5). We first demonstrated this strategy in the synthesis of cyclopentasilanes D1 and D2 (Scheme 5), wherein using either (3+2) or (4+1) annulation, mono- or 1,3-diphenyl cyclopentasilanes were obtained. To our knowledge, our approach is the first to afford a general access to substituted cyclopentasilanes via the formation of Si─Si bonds.[38], [39b] By analogy, cyclohexasilanes could also be prepared via inter- or intramolecular Si─Si coupling (Scheme 5, D4–D6). In addition to dodecamethylcyclohexasilane (D5),[37a] 1,3- and 1,2-diphenyl substituted variants (D4, D6) could be obtained depending on the bond disconnection strategy. We could also synthesize dodecamethylcyclohexasilane (D5) using electrochemical (2+2+2) annulation from commercially available dichlorotetramethyldisilanes, albeit in poorer yield (15% versus 38% via (3+3) annulation). Lastly, hybrid carbosilacycle D3 was furnished via (4+1) annulation from the corresponding dichlorosilanes.[41] Of note, compounds D1, D2, D3, and D4 have no literature precedent, and the syntheses of structurally relevant cyclosilanes are provided in Scheme 5b for comparison. While D5 and D6 have been prepared previously, the reported method for the synthesis of D5 requires excess Li metal,[37a] and D6 needed to be synthesized from a more activated bromosilane.[40a] Most importantly, our strategy provides a unified approach to access these diverse cyclosilanes using the same transformation—the electroreductive coupling of readily available dichlorosilanes.[42] We anticipate that this modular synthesis can be further applied to the preparation of other structurally distinct cyclic Si-containing compounds.
Scheme 5.

(a) Design principle for synthesis of cyclosilane via (n+m) annulation. (b) The scope of this work and the most relevant examples.
Conclusion
In summary, we have developed a general electrochemical method to synthesize a diverse array of disilanes and oligosilanes from chlorosilanes. Our strategy constitutes a milder alternative to the canonical Wurtz coupling, which utilizes harsh reducing metals, and allows for convenient access to linear and cyclic oligosilanes that have previously been difficult to prepare. We hope that our work will stimulate the broader use of electrochemistry in the synthesis of organosilanes and related materials.
Supplementary Material
Acknowledgements
Financial support was provided by NIGMS (R01GM134088; to S.L.) and Department of Energy (DOE), Office of Science, Basic Energy Sciences (DE-SC0020681; to R.S.K., chlorosilane building block synthesis). S.L. is grateful to Bristol Myers Squibb for an Unrestricted Grant in Synthetic Organic Chemistry and FMC Corporation for a New Investigator Award. A.F.G. thanks the National Science Foundation for a Graduate Research Fellowship. We thank Andrew Ressler for reproducing experiments and editing the manuscript.
References
- [1].(a) Miller RD, Michl J, Chem. Rev 1989, 89, 1359–1410; [Google Scholar]; (b) Marschner C, Oligosilanes. in Functional Molecular Silicon Compounds I, Scheschkewitz D, Ed.; Springer Verlag, 2014, p.163–228; [Google Scholar]; (c) Cheng C, Hartwig JF, Chem. Rev, 2015, 115, 8946–8975. [DOI] [PubMed] [Google Scholar]
- [2].(a) Xiao P, Gao L, Song Z, Chem. Eur. J, 2019, 25, 2407–2422; [DOI] [PubMed] [Google Scholar]; (b) Hiyama T, Oestreich M, Organosilicon Chemistry: Novel Approaches and Reactions. John Wiley & Sons, 2020 [Google Scholar]
- [3].(a) Yajima S, Hayashi J, Omori M, Chem. Lett 1975, 4, 931–934; [Google Scholar]; (b) Yajima S, Philos. Trans. R. Soc. Lond. A 1980, 294, 419–426. [Google Scholar]
- [4].Surampudi S, Yeh ML, Siegler MA, Hardigree JFM, Kasl TA, Katz HE, Klausen RS, Chem. Sci 2015, 6, 1905–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].(a) Mignani G, Krämer A, Puccetti G, Ledoux I, Soula G, Zyss J, Meyrueix R, Organometallics, 1990, 9, 2640–2643; [Google Scholar]; (b) Shimada M, Yamanoi Y, Matsushita T, Kondo T, Nishibori E, Hatakeyama A, Sugimoto K, Nishihara H, J. Am. Chem. Soc 2015, 137, 1024–1027. [DOI] [PubMed] [Google Scholar]
- [6].(a) Klausen RS, Widawsky J, Steigerwald ML, Venkataraman L, Nuckolls C, J. Am. Chem. Soc 2012, 134, 4541–4544; [DOI] [PubMed] [Google Scholar]; (b) Marro EA, Klausen RS, Chem. Mater 2019, 31, 2202–2211. [Google Scholar]
- [7].(a) West R, Carberry E, E. Science, 1975, 189, 179–186; [DOI] [PubMed] [Google Scholar]; (b) West R, Pure Appl. Chem, 1982, 54, 1041–1050; [Google Scholar]; (c) Hengge E, Janoschek R, Chem. Rev 1995, 95, 1495–1526. [Google Scholar]
- [8].(a) Fang F, Jiang Q and Klausen RS, J. Am. Chem. Soc, 2022, 144, 7834–7842; [DOI] [PubMed] [Google Scholar]; (b) Li H, Garner MH, Shangguan Z, Zheng Q, Su TA, Neupane M, Li P, Velian A, Steigerwald ML, Xiao S, Nuckolls C, Solomon G, Venkataraman L, Chem. Sci, 2016, 7, 5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].(a) Jenkner PK, Hengge E, Czaputa R, Kratky C, J. Organomet. Chem, 1993, 446, 83–90; [Google Scholar]; (b) Fischer J, Baumgartner J, Marschner C, Science, 2005, 310, 825-825; [DOI] [PubMed] [Google Scholar]; (c) Tsurusaki A, Koyama Y, Kyushin S, J. Am. Chem. Soc 2017, 139, 3982–3985. [DOI] [PubMed] [Google Scholar]
- [10].Jones RG, Holder SJ, Synthesis of polysilanes by the wurtz reductive-coupling reaction, Springer, Dordrecht, 2000, p. 353–373. [Google Scholar]
- [11].(a) Li Z, Iida K, Tomisaka Y, Yoshimura A, Hirao T, Nomoto A, Ogawa A, Organometallics, 2007, 26, 1212–1216; [Google Scholar]; (b) Yoshimura A, Tomisaka Y, Li Z, Nomoto A, Ogawa A, Heteroat. Chem 2014, 25, 684–689. [Google Scholar]
- [12].Corey JY, Adv. Organomet. Chem, 2014, 51, 1–52. [Google Scholar]
- [13].(a) Corey JY, Zhu XH, Bedard TC, Lange LD, Organometallics, 1991, 10, 924–930; [Google Scholar]; (b) Rosenberg L, Davis CW, Yao J, J. Am. Chem. Soc, 2001, 123, 5120–512. [DOI] [PubMed] [Google Scholar]
- [14].(a) Marschner C, Organometallics, 2006, 25, 2110–2125; [Google Scholar]; (b) Kayser C, Kickelbick G, Marschner C, Angew. Chem. Int. Ed, 2002, 41, 989–992. [DOI] [PubMed] [Google Scholar]
- [15].Shishido R, Uesugi M, Takahashi R, Mita T, Ishiyama T, Kubota K, Ito H, J. Am. Chem. Soc, 2020, 142, 14125–14133. [DOI] [PubMed] [Google Scholar]
- [16].(a) Hengge E, Litscher G, Angew. Chem. Int. Ed, 1976, 15, 370–370; [Google Scholar]; (b) Hengge E, Firgo H, J. Organomet. Chem, 1981, 212, 155–161. [Google Scholar]
- [17].(a) Shono T, Kashimura S, Ishifune M, Nishida R, J. Chem. Soc., Chem. commun, 1990, 17, 1160–1161; [Google Scholar]; (b) Kashimura S, Ishifune M, Yamashita N, Bu HB, Takebayashi M, Kitajima S, Yoshiwara D, Kataoka Y, Nishida R, Kawasaki S, Murase H, Shono T, J. Org. Chem 1999, 64, 6615–6621. [DOI] [PubMed] [Google Scholar]
- [18].(a) Kunai A, Kawakami T, Toyoda E, Ishikawa M, Organometallics, 1991, 10, 893–895; [Google Scholar]; (b) Ohshita J, Hino K, Iwawaki T, Kunai A, J. Electroanal. Chem, 2009, 625, 138–143. [Google Scholar]
- [19].Grogger C, Loidl B, Stueger H, Kammel T, Pachaly B, J. Organom. chem 2006, 691, 105–110. [Google Scholar]
- [20].(a) For further functionalization of vinylsilanes, see: Corriu RJP, Leclercq D, Mutin PH, Plane JM, Vioux A, Organometallics, 1993, 12, 454–462. [Google Scholar]; (b) Li Y, Kawakami Y, Macromolecules, 1998, 31, 5592–5597. [Google Scholar]; (c) Chen S, Zhu J, Ke J, Li Y, He C, Angew. Chem. Int. Ed, 2022, 61, e202117820. For further functionalization of Si-H, see: [DOI] [PubMed] [Google Scholar]; (d) Sanchez JC, Urbas SA, Toal SJ, Dipasquale AG, Rheingold AL, Trogler WC, Macromolecules, 2008, 41, 1237–1245 [Google Scholar]; (e) Lee PTKK, Skjel MK, Rosenberg L, Organometallics, 2013, 32, 1575–1578. [Google Scholar]; (f) Kanno K.-i., Awake Y, Kyushin S, Tetrahedron Lett, 2020, 61, 152274. [Google Scholar]; (g) Fan X, Zhang M, Gao Y, Zhou Q, Zhang Y, Yu J, Xu W, Yan J, Liu H, Lei Z, Ter YC, Chanmungkalakul S, Lum Y, Liu X, Cui G, Wu J, Nat. Chem, 2023, 1, 11. [DOI] [PubMed] [Google Scholar]; (h) For further functionalization of thienyl, see: Herrema JK, Wildeman J, Gill RE, Wieringa RH, van Hutten PF, Hadziioannou G, Macromolecules, 1995, 28, 8102–8116 [Google Scholar]
- [21].Ishifune M, Kashimura S, Kogai Y, Fukuhara Y, Kato T, Bu HB, Yamashita N, Murai Y, Murase H, Nishida RJ, J. Organomet. Chem 2000, 611, 26–31. [Google Scholar]
- [22].Another paper from Hengge mentioned the synthesis of cyclobutasilanes but did not provide any experimental details or yield data, see: Hengge EF, J. Inorg. Organomet. Polym 1993, 3, 287–303. [Google Scholar]
- [23].(a) Zhang W, Lin S, J. Am. Chem. Soc 2020, 142, 20661–20670; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu L, Siu JC, Lai Y, Lin S, J. Am. Chem. Soc, 2020, 142, 21272–21278; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang W, Lu L, Zhang W, Wang Y, Ware S, Mondragon J, Rein J, Strotman N, Lehnherr D, See K, Lin S, Nature, 2022, 604, 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reduction potential of 1 was measured to be about −3 V vs Fc/Fc+ using cyclic voltammetry. For 2, the reduction potential is outside of the solvent window so it could not be measured by CV. The computed reduction potentials are overestimated because we only considered the electron transfer step (i.e., the formation of chlorosilane radical anions). If we take the subsequent irreversible chemical step (i.e., the loss of Cl−) into account, the reduction potentials will be much less negative (see Supporting Information Section 7 for details).
- [25].(a) For energy decomposition analysis, see Supporting Information Section 7: Su P, Jiang Z, Chen Z, Wu W, J. Phys. Chem. A, 2014, 118, 2531–2542. [DOI] [PubMed] [Google Scholar]; (b) Su P, Jiang Z, Chen Z, Wu W, Wiley Interdiscip. Rev.: Comput. Mol. Sci, 2020, 10, e1460. [Google Scholar]; (c) Tang Z, Song Y, Zhang S, Wang W, Xu Y, Wu D, Wu W, Su P, J. Comput. Chem, 2021, 42, 2341–2351. [DOI] [PubMed] [Google Scholar]; (d) Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA Jr. J. Comput. Chem, 1993, 14, 1347–1363. [Google Scholar]
- [26].(a) Nimoth JP, Müller T, Chem. Eur. J, 2022, 28, e202104318; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harrison DJ, Edwards DR, McDonald R, Rosenberg L, Dalton trans., 2008, 26, 3401–3411; [DOI] [PubMed] [Google Scholar]; (c) Lee PT, Rosenberg L, J. Organomet. Chem, 2016, 809, 86–93. [Google Scholar]
- [27].(a) Beil SB, Müller T, Sillart SB, Franzmann P, Bomm A, Holtkamp M, Karst U, Schade W, Waldvogel SR, Angew. Chem. Int. Ed, 2018, 57, 2450–2454. [DOI] [PubMed] [Google Scholar]; (b) Beil SB, Breiner M, Schulz L, Schüll A, Müller T, Schollmeyer D, Bomm A, Holtkamp M, Karst U, Schade W, Waldvogel SR, RSC Adv., 2020, 10, 14249–14253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].LeSuer RJ, Buttolph C, Geiger WE, Anal. Chem, 2004, 76, 6395–6401. [DOI] [PubMed] [Google Scholar]
- [29].Shimada M, Yamanoi Y, Ohto T, Pham ST, Yamada R, Tada H, Omoto K, Tashiro S, Shionoya M, Hattori M, Jimura K, Hayashi S, Koike H, Iwamura M, Nozaki K, Nishihara H, J. Am. Chem. Soc, 2017, 139, 11214–11221. [DOI] [PubMed] [Google Scholar]
- [30].A5 was previously made in 1.6% yield. See: Sakurai H, Hoshi S, Kamiya A, Hosomi A, Kabuto C, Chem. Lett, 1986, 15, 1781–1784. [Google Scholar]
- [31].Jiang Q, Gittens AF, Wong S, Siegler MA, Klausen RS, Chem. Sci, 2022, 13, 7587–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Silyl anion formation has been proposed under electrochemical conditions via trapping with proton donors, see:P Corriu RJ, Dabosi G, Martineau M, J. Organomet. Chem, 1980, 188, 63–72;Zeitouny J, Jouikov V, Phys. Chem. Chem. Phys, 2009, 11, 7161–7170.
- [33].(a) Varaprath S, Stutts DH, J. Organomet. Chem, 2007, 692, 1892–1897; [Google Scholar]; (b) Matsuo T, Yamaguchi T, Hirohata T, Nakamoto M, Yamamoto Y, Maeda Y, Kawachi A, Eur. J. Inorg. Chem, 2021, 39, 4096–4102. [Google Scholar]; (c) Gittens AF, Jiang Q, Siegler MA, Klausen RS, Organometallics, 2022, 41, 3762–3769. [Google Scholar]
- [34].C10 was previously made by using excess Li as terminal reductant. See: von Hänisch C, Feierabend M, Z. Anorg. Allgem. Chem, 2013, 639, 788–793. [Google Scholar]
- [35].(a) Li H, Garner MH, Shangguan Z, Chen Y, Zheng Q, Su TA, Neupane M, Liu T, Steigerwald ML, Ng F, Nuckolls C, Xiao S, Solomon GC, Venkataraman L, J. Am. Chem. Soc 2018, 140, 15080–15088; [DOI] [PubMed] [Google Scholar]; (b) Garner MH, Li H, Chen Y, Su TA, Shangguan Z, Paley DW, Liu T, Ng F, Li H, Xiao S, Nuckolls C, Venkataraman L, Solomon CC, Nature, 2018, 558, 415–419. [DOI] [PubMed] [Google Scholar]
- [36].Press EM, Marro EA, Surampudi SK, Siegler MA, Tang JA, Klausen RS, Angew. Chem., Int. Ed, 2017, 56, 568–572. [DOI] [PubMed] [Google Scholar]
- [37].(a) Helmer BJ, West R, J. Organomet. Chem, 1982, 236, 21–32; [Google Scholar]; (b) Kumar K, Litt MH, Chadha RK, Drake JE, Can. J. Chem, 1987, 65, 437–440. [Google Scholar]
- [38].Ballestero-Martínez E, Ferguson JT, Siegler MA, Klausen RS, Eur. J. Org. Chem, 2021, 33, 4641–4646. [Google Scholar]
- [39].(a) Fischer R, Konopa T, Ully S, Baumgartner J, Marschner C, J. Organomet. Chem 2013, 685, 79–92; [Google Scholar]; (b) Purkait TK, Press EM, Marro EA, Siegler MA, Klausen RS, Organometallics, 2019, 38, 1688–1698. [Google Scholar]
- [40].(a) Marro EA, Press EM, Siegler MA, Klausen RS, J. Am. Chem. Soc, 2018, 140, 5976–5986; [DOI] [PubMed] [Google Scholar]; (b) Marro EA, Folster CP, Press EM, Hoyeon I, Ferguson JT, Siegler MA, Klausen RS, J. Am. Chem. Soc, 2019, 141, 17926–17936. [DOI] [PubMed] [Google Scholar]
- [41].Ishikawa M, Yamanaka T, Kumada M, J. Organomet. Chem, 1985, 292, 167–176. [Google Scholar]
- [42].It is difficult to directly compare known synthetic methods with ours in terms of reaction yield because these syntheses start with different substrates. We provide a more detailed comparison of known methods vs our approach in the Supporting Information Section 11. As stated in the main text, our method provided a unified bond disconnection strategy for oligosilane synthesis, which is lacking in the literature.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.