

Abstract
The B7/CD28 families of immune checkpoints play vital roles in negatively or positively regulating immune cells in homeostasis and various diseases. Recent basic and clinical studies have revealed novel biology of the B7/CD28 families and new therapeutics for cancer therapy. In this review, we discuss the newly discovered KIR3DL3/TMIGD2/HHLA2 pathways, PD-1/PD-L1 and B7-H3 as metabolic regulators, the glycobiology of PD-1/PD-L1, B7x (B7-H4) and B7-H3, and the recently characterized PD-L1/B7-1 cis-interaction. We also cover the tumor-intrinsic and -extrinsic resistance mechanisms to current anti-PD-1/PD-L1 and anti-CTLA-4 immunotherapies in clinical settings. Finally, we review new immunotherapies targeting B7-H3, B7x, PD-1/PD-L1, and CTLA-4 in current clinical trials.
Keywords: B7 family, immune checkpoints, metabolic regulators, glycobiology, therapy resistance
Subject terms: Tumour immunology, Tumour immunology
Introduction
The immune system can recognize and eliminate transformed and infected cells, with T cells playing an essential role in immunosurveillance. Thus, a hallmark of cancer is to evade these tumor-reactive T cells using immunosuppressive tumor-intrinsic and -extrinsic factors. Therefore, advancements in understanding cancer immunology are crucial in developing therapeutic strategies to eliminate the many mechanisms of tumor immune evasion, especially recent advances in understanding immune checkpoints and how they regulate the immune system. The relevance of immune checkpoints in disease and the therapeutic strategies targeting them using immune checkpoint inhibitors resulted in James Allison and Tasuku Honjo being awarded the Nobel Prize in Physiology or Medicine in 2018 [1, 2]. The B7/CD28 families of immune checkpoints are one of the most important families that regulates the immune system, but some of its members are not yet fully understood. The B7/CD28 families consists of three groups based on their phylogeny (Fig. 1): group I consists of CD28/CTLA-4/B7-1/B7-2 and ICOS/ICOS-L (B7h); group II consists of PD-1/PD-L1/PD-L2; and group III consists of TMIGD2/KIR3DL3/HHLA2, B7-H3, and B7x (B7-H4/B7S1/VTCN1) [3–6]. The receptors for B7x and B7-H3 have yet to be identified [7, 8]. Group I pathways are essential in regulating naïve T-cell activation and immune tolerance, while Groups II and III are important for regulating immunity in peripheral tissues. Understanding and targeting the cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1)/programmed death ligand-1 (PD-L1) pathways has led to significant therapeutic progress in cancer therapy and improved cancer patient outcomes. In this review, we focus on the recent advancements in the understanding and therapeutic targeting of the B7 family of immune checkpoints.
Fig. 1.
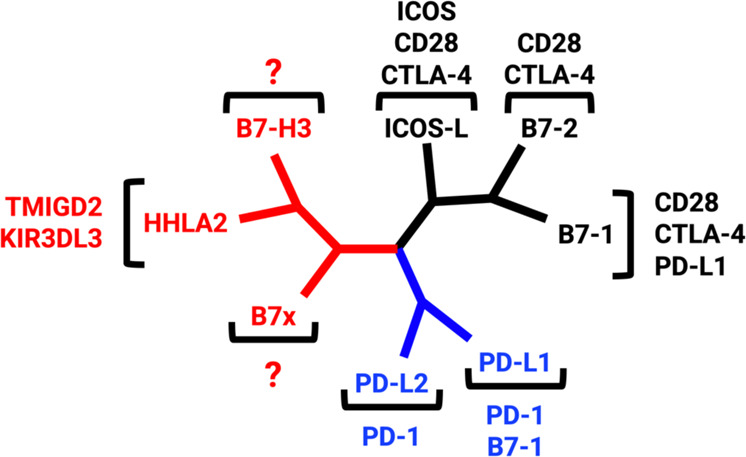
The phylogenetic tree of the B7 family and their respective receptors was generated by Phylogenetic Analysis Using Parsimony (PAUP). Group I (black) consists of CD28/CTLA-4/B7-1/B7-2 and ICOS/ICOS-L. Group II (blue) contains PD-1/PD-L1/PD-L2. Group III (red) includes TMIGD2/KIR3DL3/HHLA2, B7-H3, and B7x
HHLA2 and its two functionally-opposed receptors: TMIGD2 and KIR3DL3
The most recently described members of the B7 immune checkpoint family are HHLA2 (HERV-H LTR-associating 2) and its receptors TMIGD2 (transmembrane and immunoglobulin (Ig) domain containing 2) and KIR3DL3 (killer cell Ig-like receptor, three Ig domains, and long cytoplasmic tail) (Fig. 2). This pathway is phylogenetically related to the immune checkpoints B7x and B7-H3, which form the third arm of the B7/CD28 families (Fig. 1) [3, 9]. Unlike all other members of the B7/CD28 families, HHLA2, KIR3DL3, and TMIGD2 are found in various species but are absent from rodents such as mice and rats [6, 9–11]. Key differences between the TMIGD2/KIR3DL3/HHLA2 pathways and other B7/CD28 family members are most notable compared to the CD28/CTLA-4/B7-1/B7-2 pathways. Despite groups being composed of functionally opposed receptors that bind to common ligands, a major difference is the ability of HHLA2 to simultaneously bind its two receptors [6], while B7-1 and B7-2 cannot. Additionally, HHLA2 but not B7-1/B7-2 is highly expressed in various human cancers [6, 10, 12–18]. As new members of the B7/CD28 families, these immune checkpoints have become attractive targets for cancer immunotherapies [6, 19, 20].
Fig. 2.

Comparison of the newest HHLA2/TMIGD2/KIR3DL3 immunoregulatory pathway and the prototype B7-1/B7-2/CD28/CTLA-4 pathway reveals some similarities and important differences. Both pathways contain ligands with dual roles (B7-1/B7-2 or HHLA2) that bind to costimulatory (CD28 or TMIGD2) or coinhibitory (CTLA-4 or KIR3DL3) receptors on T and NK cells. While CD28 or CTLA-4 binding to B7-1/2 is mutually exclusive, KIR3DL3 and TMIGD2 can simultaneously bind to different sites on HHLA2. HHLA2 but not B7-1/B7-2, which is highly expressed in various human cancers. While the B7-1/B7-2/CD28/CTLA-4 pathways are expressed in humans and mice, the HHLA2/TMIGD2/KIR3DL3 pathways are found in humans but not in mice
HHLA2
HHLA2 (B7H7/B7y/B7-H5) is a type I transmembrane protein with an extracellular portion composed of tandem IgV1-IgC-IgV2 domains [3]. HHLA2 is expressed on human antigen-presenting cells (APCs) and can be induced on activated T and NK cells [6, 21], as well as exhausted PD-1+LAG-3+ T cells [22]. HHLA2 expression in normal tissue is limited to placental trophoblastic cells and the epithelium of the kidney, breast, gallbladder, and gut [10]. Despite its limited expression in normal human tissues, HHLA2 is highly expressed in various human cancers of the breast, lung, thyroid, skin, pancreas, ovary, liver, bladder, colon, prostate, kidney, and esophagus [10]. The expression of HHLA2 on APCs and cancer cells further differentiates it from B7-1 and B7-2, as they are mainly found on APCs (Fig. 2).
The associations between HHLA2 and cancer development and progression appear to be cancer specific. HHLA2 protein expression during cancer progression and recurrence is primarily associated with worse prognosis, clinical pathological features, and decreased overall survival. In clear cell renal cell carcinoma, HHLA2 expression correlates with pathological features such as tumor size, histological grade, and clinical stage, and is associated with poor overall survival [23]. Similarly, HHLA2 expression is associated with metastasis and decreased overall survival in lung cancer [24], increased metastasis and stage in triple-negative breast cancer (TNBC) [10], decreased overall survival in intrahepatic cholangiocarcinoma [25], and worse clinical features in prostate cancer [18]. Furthermore, HHLA2 expression in liver cancers, including intrahepatic cholangiocarcinoma (ICC) and hepatocellular carcinoma (HCC), is associated with worse prognosis [25, 26]. HHLA2 is a prognostic indicator of overall survival in ICC, correlates with cancer stage in HCC, and leads to a decrease in tumor-infiltrating T cells in both ICC and HCC [25, 26]. While most cancers with high HHLA2 expression show poorer outcomes, some studies have shown that higher HHLA2 expression is associated with better survival. A majority of pancreatic cancer, pancreatic ductal adenocarcinoma, and ampullary cancer samples express HHLA2, and HHLA2 expression in these cancers is associated with improved prognosis and postsurgical survival [15, 27]. In human non–small cell lung carcinoma (NSCLC), HHLA2 expression is positively associated with epidermal growth factor receptor (EGFR) mutational status [12].
Interestingly, a few studies have revealed that HHLA2 and PD-L1 have mutually exclusive expression in NSCLC [13], ICC [25], and clear cell renal cell carcinoma (ccRCC) [11] cells. In NSCLC, most (78%) PD-L1–negative cases express HHLA2, whereas only 3% express HHLA2 and PD-L1 [13]. In ccRCC, HHLA2 and PD-L1 coexpression occurs in only approximately 19% of samples but is significantly associated with TNM stage and acts as a prognostic factor of shorter progression-free survival and overall survival, while high HHLA2 expression alone occurs in >30% of samples with similar disease associations [18]. These results suggest that the molecular and cellular mechanisms regulating the expression of HHLA2 and PD-L1 are markedly different.
TMIGD2
TMIGD2 (CD28H/IGPR-1), which is the costimulatory receptor of HHLA2 [10, 21], is a transmembrane protein with a single IgV domain extracellular region and a cytoplasmic tail. TMIGD2 is an adhesion molecule on epithelial-derived tissues involved in angiogenesis [28]. Among immune cells, naïve T cells, memory T cells, tissue-resident T cells, NK cells, innate lymphoid cells, and plasmacytoid dendritic cells express TMIGD2 [6, 21, 29]. CD8 and CD4 T cells show similar patterns of TMIGD2 expression, and naïve T cells show high surface expression of TMIGD2, while memory T cells show lower expression of TMIGD2 in normal peripheral blood mononuclear cells, as indicated by an increase in CD31 and CD28 expression and a decrease in T-bet, interferon (IFN)-γ, and tumor necrosis factor (TNF)α expression. [29]. TMIGD2 is also expressed on immature NK cells, which are characterized as CD56bright CD16- KIR- NKG2A+ CD57- cells [30]. On NK cells, TMIGD2 functions as a strong activator that acts synergistically with NKp46 and 2B4 and can enhance activation through CD16 for antibody-dependent cellular cytotoxicity (ADCC) [30]. Despite the increased expression of TMIGD2 on CD56bright NK cells, degranulation, and cytotoxicity are only observed in CD56dim NK cells, further highlighting the specific role of TMIGD2 during activation [30]. TMIGD2 interacts with HHLA2 by binding to the IgV1 domain of HHLA2 [6], which promotes T-cell proliferation, differentiation, and cytokine production, as well as NK cell activation, cytokine production, and cytotoxicity [6]. The costimulatory signal is mediated by Tyr192 on the cytoplasmic tail of TMIGD2, which activates the PI3K/AKT pathway. In addition to a phosphotyrosine residue, the cytoplasmic tail of TMIGD2 contains a proline-rich region that associates with multiple Src homology 3 (SH3)-containing signaling molecules [28]. TMIGD2 stimulation alone is not sufficient for activation, however, as it synergizes with 2B4 and NKp46 to promote degranulation and cytokine secretion in NK cells [30]. Furthermore, upon stimulation with a CD3 agonist monoclonal antibody, the TMIGD2/HHLA2 interaction increases interleukin (IL)-2, IL-10, IFN-γ, and TNFα secretion and increases the proliferation of T cells in vitro [21]. Following T and NK cell activation, TMIGD2 expression decreases, indicating a specific role of TMIGD2 during activation [6]. The high levels of TMIGD2 mRNA in the thymus further support the role of TMIGD2/HHLA2 in the activation of naïve T cells [29].
KIR3DL3
KIR3DL3, which is a KIR family member, consists of extracellular tandem D0-D1-D2 domains and a cytoplasmic tail containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) [6, 9], indicating its role as the inhibitory receptor of HHLA2. KIR3DL3, which is a framework gene, is ubiquitous in humans and is inherited in all haplotypes but cannot be detected in most human tissues [31]. The domain responsible for KIR3DL3 binding to HHLA2 is the D0 domain, which alone is sufficient to bind to HHLA2 in the absence of D1 and D2 [6]. The D0 domain of KIR3DL3 is evolutionarily conserved between primate species, and loop 1 of D0 has complete amino acid sequence homology between primate species, indicating a critical role for KIR3DL3 in primates [31].
KIR3DL3 is predominantly expressed on CD56dim NK cells and terminally differentiated effector memory CD8 T cells (CD8 TEMRA) that are CCR7- CD45RA+ [6]. The limited expression of KIR3DL3 is due to the methylation of its promoter, and KIR3DL3- NK cells with abrogated methyltransferase activity exhibit spontaneous KIR3DL3 expression [32, 33]. Despite limited mRNA expression of KIR3DL3, its promoter has the strongest activity of all KIR genes [33]. KIR3DL3 mRNA is regulated by microRNAs [34], which act as posttranscriptional modulators to inhibit protein synthesis [35]. Antisense knockdown miRNAs targeting miR-26a-5p, miR-26b-5p, and miR-185-5p result in a significant decrease in KIR3DL3 mRNA expression, while knockdown of these miRNAs results in a significant increase in KIR3DL3 surface expression on K562 cells [35]. These data indicate two mechanisms controlling KIR3DL3 protein expression: methylation of its promoter to prevent transcription and inhibition of translation by miRNAs.
HHLA2 binding to KIR3DL3 is inhibitory in T and NK cells [6]. This inhibition is abrogated when HHLA2 or KIR3DL3 is blocked by monoclonal antibodies [6, 11]. Furthermore, in vitro coculture of HHLA2+ tumor cells with KIR3DL3+ CD8 T cells significantly decreased target cell lysis, T-cell degranulation, and IFN-γ and TNFα expression [6]. KIR3DL3+ CD8 T cells also secrete fewer cytokines in the presence of HHLA2+ tumor cells (IFN-γ, TNFα, CCL1, IL-13, GM-CSF, and IL-5) in vitro [6]. This interaction in NK cells leads to a significant decrease in cytokine production and target cell lysis, which is restored by KIR3DL3/HHLA2 blockade [6, 11].
HHLA2 engagement recruits KIR3DL3 to the immunological synapse and inhibits CD8 T and NK cell function and cytotoxicity [6]. The inhibitory signal following HHLA2 binding to KIR3DL3 depends on tyrosine 381 (Y381), which is phosphorylated to inhibit downstream signaling [6]. The residue Y381 is in the ITIM region of KIR3DL3 and is necessary for the inhibitory signal associated with HHLA2 engagement [6]. The wild-type KIR3DL3 ITIM recruits SHP-1/2, as shown by immunoprecipitation, indicating that the inhibitory potential of KIR3DL3 relies on SHP-1/2 recruitment [6]. Upon recruitment of SHP-1/2, there is a decrease in the phosphorylation of Vav1, ERK1/2, AKT, and NF-κB, which is restored by mutations in Y381 [6]. This reduction in downstream signaling leads to the inhibition of CD8 T-cell and NK cell function, as shown by decreased cytokine secretion and cytotoxicity, and mediates HHLA2+ tumor immune resistance [6].
Targeting KIR3DL3/HHLA2/TMIGD2 as novel immunotherapies
The immunosuppressive pathway associated with the KIR3DL3/HHLA2 interaction has recently become an attractive target for cancer immunotherapy [6]. Many cancer types highly express HHLA2 in primary samples. Importantly, KIR3DL3+ immune cells are often seen in HHLA2+ tumors, suggesting that the KIR3DL3/HHLA2 pathway is active within the tumor microenvironment [6]. A lack of the KIR3DL3/HHLA2 interaction by HHLA2 knockout or KIR3DL3/HHLA2 blockade increases lysis of NSCLC (HCC827) and HHLA2-expressing lymphoma by KIR3DL3+ NK cells in vitro [6]. Furthermore, the blockade of KIR3DL3/HHLA2 with an anti-KIR3DL3 mAb decreases the tumor burden in humanized mice in vivo [6], which provides the foundation for developing a novel cancer immunotherapy for use in clinical trials. In addition to targeting KIR3DL3, blockade of HHLA2/KIR3DL3 can maintain the TMIGD2/HHLA2 interaction. Interestingly, HHLA2 expression in melanoma patients predicts improved responsiveness to anti-PD-1/PD-L1 checkpoint blockade [36]. These findings highlight the potential for developing immunotherapies targeting KIR3DL3/HHLA2, which can be translated across multiple cancer types that express high levels of HHLA2, particularly PD-L1–negative tumors that are often HHLA2 positive [13], as well as the potential synergy of blocking KIR3DL3/HHLA2 and other therapies such as PD-1/PD-L1 blockade. Since HHLA2 has limited expression on normal cells but is highly expressed on various cancers, targeting HHLA2 is another attractive anticancer therapy. Since TMIGD2 functions as a costimulatory molecule for T and NK cells, a bispecific antibody targeting TMIGD2 and PD-L1 is being developed [37]. Further investigation of the roles of KIR3DL3/HHLA2 in cancer development and progression is needed to continue developing immunotherapies. Thus far, the focus on this pathway has been on the overexpression of HHLA2 on tumor cells, but it remains to be seen if this expression correlates with an increase in KIR3DL3 expression on T and NK cells in patient samples. These data would further support the use of anti-KIR3DL3 therapies for cancer treatment. We believe that new immunotherapies can be developed to target KIR3DL3, HHLA2, and TMIGD2 with different underlying mechanisms.
PD-1/PD-L1 and B7-H3 as metabolic regulators
The PD-1/PD-L1 and B7-H3 pathways are well-known regulators of immune cell function and exhaustion. However, recent studies have demonstrated that these immune checkpoint pathways are metabolic regulators of various immune cells, cancer cells, and adipocyte progenitor cells.
PD-1 as a metabolic regulator in immune cells
Most studies on PD-1/PD-L1-mediated regulation of metabolism have been conducted on T cells. The T-cell receptor (TCR), costimulation, and cytokine receptors target the Ras/MAPK and PI3K/Akt/mTOR pathways, which are two critical regulators of T-cell metabolism that promote glycolysis and T-cell activation (Fig. 3A) [38, 39]. However, TCR signaling also upregulates PD-1 cell-surface expression [40]. When PD-1 binds to its ligands PD-L1 or PD-L2, the ITIM and the immunoreceptor tyrosine switch motif (ITSM) of PD-1 are tyrosine phosphorylated by the Src family kinases Lck and Fyn [41–46]. The phosphorylation of the ITIM and ITSM of PD-1 recruits the phosphatase SHP-2, which downregulates TCR and CD28-costimulatory signaling and the Ras/MAPK and PI3K/Akt/mTOR metabolic pathways [47–49]. As a result, PD-1 decreases T-cell glycolysis, amino acid metabolism, and intracellular trafficking while upregulating T-cell lipid metabolism [50]. PD-1 signaling promotes lipolysis and fatty acid oxidation (FAO) of endogenous lipids by upregulating the expression of adipose triglyceride lipase (ATGL) and carnitine palmitoyltransferase 1 A (CPT1A) (Fig. 3A) [50]. Additionally, PD-1 upregulates the intracellular concentrations of polyunsaturated fatty acids (PUFAs) in T cells and their secretion in vitro [50]. PUFAs inhibit T-cell production of IL-2 production and proliferation in vitro and in vivo [51–53]. These PUFAs are often oxidized by reactive oxygen species (ROS) to form oxidized lipids. The scavenger receptor CD36 on T cells takes up these oxidized lipids and promotes lipid peroxidation, resulting in dysfunctional intratumoral CD8 T cells [54].
Fig. 3.

PD-1/PD-L1 and B7-H3 are metabolic regulators of effector and regulatory T cells (Tregs) and tumor cells. A In T effector cells, the TCR and CD28/B7-1/B7-2 costimulatory signals upregulate PD-1 via NFATc1 and activate RAS/MAPK and PI3K/AKT/mTOR, which stimulates glycolysis. PD-1/PD-L1 engagement inhibits the RAS/MAPK and PI3K/AKT/mTOR signaling pathways, thereby preventing the induction of glycolysis. It also inhibits the expression of the main glutamine transporters, SNAT1 and SNAT2, thus reducing amino acid uptake and metabolism. PD-1/PD-L1 promotes fatty acid β-oxidation (FAO) by increasing the expression of the mitochondrial FAO enzyme CPT1A and lipolysis, which generates fatty acids for FAO. B Tregs acquire lactate from the tumor microenvironment that is generated by tumor cell glycolysis, which is possibly regulated by PD-L1. The lactate transporter MCT1 transports lactate into Tregs, where lactate is converted into Ca2+, which induces NFATc1-mediated upregulation of PD-1. PD-1 promotes PI3K/AKT/mTOR signaling, thereby upregulating glycolysis. PD-1 inhibits the spare respiratory capacity and immunosuppressive activity of Tregs. C In intratumoral Tregs, PD-1 downstream signaling maintains the expression of FoxP3, inhibiting the expression of genes associated with glycolysis and promoting lipid metabolism, fatty acid β-oxidation (FAO), oxidation phosphorylation, peroxisome proliferator-activated receptor-β (PPAR-β) expression, proliferation, immunosuppression, and chemotaxis. These genes promote FAO, oxidation phosphorylation, and mitochondrial mass. PD-1 activates BATF to upregulate genes associated with the activation and function of Tregs. PD-1 maintains lipid uptake through unknown mechanisms. Tregs internalize lipids through CD36, activating the PPAR-β pathway to support mitochondrial biogenesis and fitness. PD-1 and CD36 may maintain the fitness, stability, and functions of intratumoral Tregs. D Tumor cells acquire glucose and lipids from the tumor microenvironment. PD-L1 promotes glucose metabolism by activating the AKT/GSK3β signaling pathway to upregulate the transcriptional repressor SNAI1 to inhibit SIRT3, which allows the expression of HK2 and LDHA. PD-L1 activates the AKT/mTOR pathway to upregulate the expression of PGK1, TPI, HK2, and LDHA. PD-L1 activates the PI3K/AKT and ERK pathways to induce the expression of HK2. Additionally, PD-L1 upregulates the expression of PFK-2/FBPase 3. Thus, PD-L1 promotes glucose metabolism by upregulating the expression of the glycolysis-related enzymes PGK1, TPI, HK2, LDHA, and PFK-2/FBPase 3. Lactate is produced from glycolysis and is exported out of the tumor cell, acidifying the tumor microenvironment. PD-L1 also promotes lipid uptake and metabolism by upregulating the expression of the fatty acid binding protein (FABP4) and FABP5, which are involved in lipid metabolism. E In tumor cells, B7-H3 inhibits the transcription factor NRF2, which governs the transcription of the antioxidant enzymes SOD1, SOD2, and PRX3; this leads to the accumulation of ROS, which stabilizes HIF-1α and increases the expression of LDHA and PDK1. LDHA participates in lactate production via glycolysis, and PDK1 inhibits pyruvate flux to the citric acid cycle. B7-H3 promotes the activation and phosphorylation of the STAT3 pathway, which upregulates the expression of HK2, which participates in glycolysis. B7-H3 promotes the transcription and translation of the transcription factor SCREBP-1, which regulates the mRNA and protein expression of fatty acid synthase (FASN), which is involved in lipogenesis
Recent studies have shown that PD-1 may also affect redox metabolism in T cells. PD-L1/PD-1 ligation decreases the amount of reduced glutathione (GSH) and increases the levels of cysteine-GSH disulfide, suggesting an intracellular oxidative environment within T cells [50]. In the mouse graft versus host disease (GVHD) model, alloreactive T cells have increased PD-1 expression and FAO-derived ROS, which increases T-cell sensitivity to F1F0-ATP synthase complex inhibitors [55]. Treatment with antioxidants or PD-1 blockade abrogates these effects, lowering ROS and desensitizing the cells to F1F0-ATP inhibition [55]. However, another study showed that PD-1 blockade increased intracellular ROS and mitochondrial mass, as well as tumoral CD8 T-cell activation and proliferation [56]. More studies are needed to clarify how PD-1 affects the redox metabolism of T cells.
PD-1 regulates the metabolism of various T-cell subsets and innate immune cells. A recent study showed that PD-1 signaling was necessary to regulate mTOR-dependent anabolic glycolysis and the FAO pathway to maintain the bioenergetic requirements of quiescent CD8 T cells [57, 58]. Thus, PD-1 signaling and its regulation of metabolism promote the development of protective long-lived memory CD8 T cells. In a model of chronic lymphocytic leukemia, PD-1 signaling impairs glycolysis, Bruton’s tyrosine kinase (BTK) signaling, and the phagocytosis of PD-1-expressing monocytes [59]. Myeloid-specific deletion of PD-1 results in PD-1-deficient myeloid progenitor cells with increased levels of metabolites involved in glycolysis, the pentose phosphate pathway, the citric acid cycle, and cholesterol in response to emergency myelopoiesis-related growth factors [60]. These results correlate with a reduction in tumor-infiltrating myeloid-derived suppressor cell (MDSC) accumulation and enhanced T effector memory cell functionality and antitumor protection in a myeloid-specific PD-1-deficient tumor mouse model. Once activated, NK cells upregulate PD-1 expression [61]. Their effector functions depend on the activation of the PI3K/Akt/mTOR pathway and glycolysis, similar to T-cell activation [62]. However, little is known about how the PD-1/PD-L1 pathway regulates NK cell metabolism. These studies highlight new mechanisms by which PD-1 governs the metabolism of various immune cells.
The function of PD-1 in regulating the metabolism and function of regulatory T cells (Tregs) remains controversial. Regarding metabolism, PD-1-deficient Tregs have reduced glycolysis due to decreased PI3K/Akt/mTOR signaling (Fig. 3B) [63]. In addition, these PD-1-deficient activated Tregs have enhanced mitochondrial spare respiration and maximal respiratory capacity [63]. Thus, these PD-1-deficient Tregs have reduced glycolysis but enhanced mitochondrial and immunosuppressive functions in mouse models of autoimmune encephalomyelitis and nonobese diabetes [63]. Consequently, these findings and others suggest that PD-1 expression on Tregs inhibits the immunosuppressive functions of Tregs [63–65]. However, other studies have reported that PD-1 on Tregs promotes Treg functionality and homeostasis in mouse models of chronic viral infection [66], autoimmune disease [67], and cancer [68]. PD-1 expression on intratumoral Tregs maintains Foxp3 expression in the tumor microenvironment and preserves the expression of genes associated with proliferation, suppression, and lipid metabolism, including target genes of the peroxisome proliferator-activated receptor-β (PPAR-β) signaling pathway (Fig. 3C) [68]. PD-1 expression on intratumoral Tregs also promotes lipid uptake and maintains mitochondrial mass. Thus, the clonal expansion and immunosuppressive functionality of intratumoral Tregs are promoted [68]. Interestingly, intratumoral Tregs highly express CD36 and PD-1 [69]. CD36 promotes lipid uptake and levels in intratumoral Tregs and upregulates the PPAR-β signaling pathway to promote mitochondrial fitness and biogenesis, thereby sustaining the survival and functionality of tumor-infiltrating Tregs (Fig. 3C) [69]. These studies are consistent with previous findings that intratumoral Tregs primarily use lipid metabolism [69–71] and suggest that PD-1 and CD36 modulate the same metabolic programs to maintain the function, metabolic fitness, and stability of intratumoral Tregs. Additionally, PD-1 regulates triglyceride metabolism in Tregs by upregulating the basic leucine zipper ATF-like transcription factor (BATF) to promote the immunosuppressive functions of Tregs in controlling airway allergic inflammation and IgE responses (Fig. 3C) [72]. Consistent with these findings, BATF epigenetically and transcriptionally regulates the expression of differentiation/activation-related genes in intratumoral Tregs, promoting the functionality of Tregs in the tumor microenvironment (Fig. 3C) [73]. These studies indicate that PD-1 is an important metabolic regulator in Tregs. However, more studies are needed to further clarify the functions and mechanisms of PD-1 in regulating Treg metabolism and functionality during homeostasis and in chronic infections, autoimmune diseases, and cancer.
PD-L1 regulates tumor-cell metabolism
Although PD-L1 is a ligand, recent studies have reported that PD-L1 may regulate the metabolism of cancer cells. In various mouse tumor cell lines, knockdown or blockade of PD-L1 reduces the expression of glycolysis-related enzymes, inhibits AKT and mTOR activity, and decreases the extracellular acidification rate (ECAR) in vitro, suggesting that PD-L1 may regulate tumor cell glucose metabolism via the ATK/mTOR pathway (Fig. 3D) [74]. In human cervical cancer cells, PD-L1 overexpression promotes glucose metabolism and epithelial-mesenchymal transition (EMT) by activating the AKT/GSK3β signaling pathway to upregulate the transcriptional repressor Snail Family Transcriptional Repressor 1 (SNAI1) and inhibit sirtuin-3 (SIRT3) promoter activity in vitro [75]. In human NSCLC cell lines, PD-L1 knockdown reduces the expression of hexokinase-2, an enzyme involved in glycolysis, inhibiting the PI3K/AKT and ERK pathways in vitro and thereby reducing glycolysis [76]. A recent study showed that PD-L1 regulates tumor cell glycolysis by regulating the expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFK-2/FBPase 3 or PFKFB3) [77]. In addition to glucose metabolism, PD-L1 promotes lipid uptake by upregulating the expression of fatty acid binding protein (Fabp) 4 and Fabp5 in gastric tumor cells in vitro [78]. In a tumor cell-T-cell coculture system, these gastric tumor cells outcompete tissue-resident memory T cells for lipids, leading to the death of tissue-resident memory T cells [78]. Taken together, these studies suggest that PD-L1 is a metabolic regulator of tumor cells and enables them to outcompete immune cells for metabolic resources, thereby evading antitumor immunity.
B7-H3 is a metabolic regulator of tumor cells and adipocyte progenitor cells
Recent studies have revealed that B7-H3 is a metabolic regulator in various cell types. B7-H3 promotes the Warburg effect by reprogramming tumor cell metabolism toward glycolysis and lactate production via ROS-dependent hypoxia inducible factor 1 subunit α (HIF1-α) stabilization, resulting in enhanced tumor growth in vitro and in breast cancer mouse models (Fig. 3E) [79]. Similarly, B7-H3 expression increases the glycolytic capacity of breast cancer cells and their survival, as well as their resistance to two anticancer mTOR inhibitors in vitro and in vivo [80]. Consistent with previous findings, knockdown or inhibition of B7-H3 with an mAb in metastatic melanoma cells in vitro reduces their glycolytic capacity, proliferative ability, and resistance to chemotherapy and various anticancer small-molecule inhibitors [81]. B7-H3 regulates aerobic glycolysis to promote colorectal cancer cell migration, invasion, and apoptotic resistance by promoting hexokinase 2 expression in vitro and in vivo (Fig. 3E) [82]. Furthermore, B7-H3 regulates lipid metabolism in tumor cells. B7-H3 regulates lipid synthesis in lung cancer cells by controlling the expression of sterol regulatory element-binding protein 1 (SREBP-1), which governs the expression of fatty acid synthase (FASN) in vitro (Fig. 3E) [83]. In colorectal cancer patients and human cancer cell lines, B7-H3 is positively correlated with isocitrate dehydrogenase 1 (IDH1), a metabolic enzyme in the tricarboxylic acid cycle, whose metabolites are associated with lipid synthesis [84].
In addition to tumor cells, B7-H3 is highly expressed in mouse and human adipose tissue during homeostasis and is positively correlated with obesity in mice and humans [85]. B7-H3 is preferentially expressed in adipocyte progenitor (AP) cells and is downregulated during AP cell differentiation into adipocytes. In AP cells, B7-H3 regulates many genes associated with glycolysis, fatty acid metabolism, and amino acid metabolic processes. Cell-intrinsic B7-H3 stimulates glycolysis and oxidative metabolism in AP cells. Adipocytes derived from B7-H3-deficient AP cells exhibit impaired oxidative metabolism, which correlates with an increase in fat storage, suggesting that B7-H3 establishes oxidative metabolic programming in AP cells that persists as these cells differentiate into adipocytes, thereby regulating lipid storage in adipocytes. Consistent with these findings, B7-H3-deficient mice develop spontaneous obesity when fed a regular chow diet, which is accompanied by the accumulation of white adipose tissue consisting of hypertrophic adipocytes. Additionally, these B7-H3-deficient mice exhibit dyslipidemia, hyperinsulinemia, hyperglycemia, and impaired white adipose tissue lipolysis and fatty acid oxidation. Obesity in B7-H3-deficient mice is also associated with inflammation in adipose tissue. These results demonstrate the physiological role of B7-H3 as a metabolic regulator.
Glycosylation biology of PD-1/PD-L1, B7x and B7-H3
Glyco-immuno-oncology is an emerging topic and a new discipline for studying tumor glycosylation consequences in the immune system. Studies have shown that normal and tumor cells have diverse glyco-coats [86, 87]. Protein glycosylation is involved in regulating protein localization, stability and mediating receptor‒ligand interactions [88]. Aberrant glycan patterns have also been shown to serve as non-invasive tumor biomarkers, such as carcinoma antigen 19–9 (CA19–9) in pancreatic cancer [89] and α-fetoprotein (AFP) in hepatocellular carcinoma [90, 91]. Two main glycoconjugate glycosylate proteins are N-linked glycoproteins attached to the nitrogen of asparagine side chains (Asn-X-Ser/Thr: NXT) [92] and O-linked glycoproteins attached to the hydroxyl oxygen of Ser/Thr [93]. Approximately 50% of eukaryotic proteins are glycosylated based on the different specificities of glycosyltransferases and glycosidase enzymes [94]. N-glycans are classified into three main units: high-mannose, hybrid, and complex glycans. In addition, fucosylation of the core N-acetylglucosamine (GlcNAc) residue following β-1,2-N-acetylglucosaminyltransferase I (GlcNAcT-I) modification in hybrid and complex N-linked synthesis is normal but is also a frequent cancer-associated change in N-glycosylation. The enzyme α-1,6-fucosyltransferase (FUT8) generates a-1,6-fucosylated structures on the core of N-glycans. Upregulated expression of FUT8 is involved in many cancers (breast, lung, prostate, etc.) and is associated with poor prognosis in patients in clinical settings [95, 96]. Likewise, protein glycosylation regulates protein structure, function, turnover rate, and intermolecular interactions [97]. This section discusses the regulation of immune checkpoint receptor/ligands (PD-1/PD-L1, B7x, and B7-H3) by glycosylation (Fig. 4).
Fig. 4.

Glycosylation of the immune checkpoints PD-1/PD-L1, B7x, and B7-H3. N-glycosylation occurs at the consensus sequence/sequon Asn-X-Ser/Thr (NXT) in the endoplasmic reticulum (ER) lumen, followed by complete synthesis/maturation in the Golgi and ending in the plasma membrane, where it is either secreted or embedded in the membrane. Nearly 90% of the protein undergoes this co-translational modification. Major types of N-glycans are high-mannose, hybrid, and complex N-glycans. PD-1 contains 15 kDa N-glycan moieties with four glycosylation NXT motifs. B3GNT2 and Fut8 induce the glycosylation of PD-1, whereas its ligand PD-L1 contains approximately 17 kDa of N-glycan moieties and four conserved NXT motifs. The glycosylation of PD-L1 is induced by B3GNT3, STT3, B4GALT1, MAN2A1 and GLT1D1. Monoclonal antibodies include STM418, BMS166, and MW11 h317; small-molecule inhibitors such as NG-1 and the sugar analog 2DG can inhibit the interaction between PD-1/PD-L1. B7x has a differential N-glycosylation pattern ranging from highly glycosylated (~50 kDa) to less glycosylated (~40 kDa) with five NXT motifs. B7x glycosylation is induced by STT3A and UGGG1. The small-molecule inhibitor NG-1 was shown to inhibit the addition of N-glycans to B7x. B7-H3 consists of a 40 kDa N-glycan moiety with 8 NXT motif sites in humans and 4 NXT motif sites in mice. B7-H3 glycosylation is induced by A4GALT and Fut8. The sugar analog 2F-Fuc inhibits the B7-H3 core fucosylation required for its N-glycosylation. B3GNT: Beta-1,3-N-acetylglucosaminyltransferase; STT3: STT3 oligosaccharyltransferase complex catalytic subunit; UGGG1: UDP-glucose glycoprotein glucosyltransferase 1; A4GALT: Alpha 1,4-Galactosyltransferase; MAN2A1: Mannosidase α class II member; GLT1D1: Glycosyltransferase 1 containing domain 1; 2-DG: 2-Deoxy-D-glucose; NG-1: Aminobenzamide-sulfonamide inhibitor; BMS166: Cathepsin L monoclonal antibody; 2F-Fuc: 2-Fluoro-L-fucose
PD-1/PD-L1 glycosylation
Heavy N-linked glycosylation composed of an extensive sugar moiety is important for the function of PD-1 and PD-L1 [98]. In TNBC, immunoblotting shows that PD-1 has a heterogeneous pattern showing two bands: 46 kDa and 32 kDa. In response to glycosidase (peptide-N-glycosidase F; PNGase F) and N-linked glycosylation inhibitor (tunicamycin, swainsonine, or castanospermine) treatment, an electrophoretic mobility shift of ~15 kDa was observed. No changes were observed after treatment with the O-linked glycosylation inhibitor (benzyl-GalNAc), which suggests that PD-1 is extensively N-glycosylated [99]. Subsequently, four conserved NXT motifs (N49, N58, N74, and N116) in the extracellular domain of PD-1 were identified. Of the identified sites, N58 is essential for PD-1/PD-L1 binding [100, 101]. Glycosylation of PD-1 is critical for maintaining PD-1 protein stability and membrane expression. Non-glycosylated PD-1 is more ubiquitylated than glycosylated PD-1 in response to treatment with the proteasome inhibitor MG132 [99]. The glycosyltransferases B3GNT2 and FUT8 may be responsible for regulating PD-1 glycosylation [102]. Inhibition of Fut8 by genetic ablation or pharmacologic inhibition reduces cell-surface expression of PD-1 and enhances T-cell activation, leading to more efficient tumor eradication [99, 103]. More ubiquitination is observed in Fut8−/− CD8 T cells than in wild-type cells, providing insight into core fucosylation-induced PD-1 stabilization [103]. Furthermore, the specific anti-PD-1 neutralizing mAb exhibited a higher affinity for PD-1 than nivolumab due to the heavy glycan moieties of PD-1 blocking its recognition by nivolumab, which does not recognize glycosylation. This mAb targets glycosylated PD-1 at N58, blocks the PD-1/PD-L1 interaction, and enhances the anti-tumor response in a humanized TNBC mouse model [99]. In addition, several small molecules, such as 2-DG (2-Deoxy-D-glucose), NG-1 (aminobenzamide-sulfonamide inhibitor), and BMS166 (Cathepsin L mAb), can disrupt the PD-1/PD-L1 interaction by modulating N-linked glycosylation [104, 105]. Consistent with previous findings, other studies have reported that PD-1 glycosylation is crucial for mediating the PD-1/PD-L1 interaction [98], its immunosuppressive function [106], and its repressive effects on chimeric antigen receptor T cells (CAR-T) [107].
PD-L1 is a heavily glycosylated B7 family protein found in various cancer cell types. A study revealed a range of bands for PD-L1 at ~ 50 kDa, whereas the nonglycosylated form of PD-L1 was detected at ~33 kDa. PD-L1 is primarily N-glycosylated, as demonstrated by treatment with PNGase F, and tunicamycin blocks N-linked but not O-linked glycosylation. A study revealed four Asn (N) residues of the consensus N-glycosylation motifs spanning the PD-L1 extracellular domain (N35, N192, N200, and N219). Furthermore, the glycosylation of N192, N200, and N219 but not N35 is crucial for preventing its degradation and enhancing its immunosuppressive properties. The turnover rate for glycosylated PD-L1 is slower than that for non-glycosylated PD-L1 in the presence of a protein synthesis inhibitor (CHX). Thus, glycosylation of PD-L1 stabilizes this protein [108]. The glycosyltransferases STT3, MAN2A1, and GLT1D1 are required to induce the glycosylation of PD-L1; likewise, B4GALT1 mediates PD-L1 galactosylation. Moreover, EGF/EGFR-induced N-linked glycosylation of PD-L1 via the glycosyltransferase B3GNT3 is necessary for its physical contact with PD-1 [109, 110]. Knocking out B3GNT3 in mouse breast cancer cells reduces PD-L1 expression and potentiates tumor rejection [111]. Non-glycosylated PD-L1 exhibits more ubiquitination, suggesting that the non-glycosylated PD-L1 form undergoes faster protein degradation. PD-L1 is also heavily glycosylated in T cells, and specific glycoforms are altered in response to T-cell activation, as shown by LC‒MS/MS analysis [110]. Notably, heavy glycosylation of PD-L1 hampers the identification of polypeptide antigenic regions by some Abs and reduces antibody binding affinity [112]. Glycosylated PD-L1 in melanoma is associated with poor PD-L1 detection by immunohistochemistry (IHC) [98]. Notably, deglycosylation methods to remove the N-glycan moiety enhance anti-PD-L1 antibody binding affinity and signal intensity, in addition to improving the false detection of PD-L1 levels in the clinic through detection methods such as IHC, ELISA, immunofluorescence microscopy, and immunoblotting [112]. Furthermore, a retrospective study showed that deglycosylation leads to a more accurate assessment of PD-L1 expression by reducing false-negative patient stratification [105, 113]. These findings indicate that deglycosylated PD-L1 could be a potential biomarker to predict anti-PD-1/PD-L1 immunotherapy by decreasing antigen heterogeneity and eliminating glycan structural hindrance.
Glycosylation of B7x
The immune checkpoint B7x remains an orphan ligand without a known receptor [7]. Its expression is limited in normal tissue; however, it is often overexpressed in many human cancers, such as skin, prostate, stomach, pancreas, brain, liver, lung, and TNBC, especially in immune-cold tumors. B7x regulates T cells and innate immune cells. The receptor for B7x is dynamic and occurs distinctly from the exhaustion mechanisms that induce PD-1 and Tim-3 expression [7]. B7x has different N-glycosylation forms, such as highly glycosylated (~50 kDa), less glycosylated (~40 kDa), and unglycosylated (~28 kDa). The turnover rate of the non-glycosylated form is faster than the glycosylated form of B7x, which enhances B7x stability. This turnover rate is further regulated by the ubiquitin‒proteasome pathway, indicating that inhibiting N-glycosylation improves B7x ubiquitination [114]. Glycosylation of B7x within the endoplasmic reticulum ultimately determines the cellular abundance of this protein [114]. A mass spectrometry study identified five asparagine residues that were N-linked glycosylation sites (N112, N140, N156, N160, and N255) and two ubiquitination sites at lysine (K) residues K146 and K138 in 293 T cells. However, the mutation of five asparagine sites (5NQ) partially reduces its glycosylation, perhaps because additional asparagine residues could compensate for the function of the mutated sites. The un-glycosylated B7x mutant (B7x-16NQ amino acids 47, 112, 119, 140, 142, 156, 160, 190, 196, 202, 205, 216, 220, 221, 229, and 255) completely blocks B7x glycosylation and preserves partial trafficking to the membrane. The glycosylation of B7x is catalyzed by specific glycosyltransferases (STT3A and UGGG1) to preclude protein degradation via ubiquitination by the E3 ligase autocrine motility factor receptor (AMFR). Additionally, the degradation of B7x is mediated by genetic or pharmacologic inhibition of STT3A [114]. The small-molecule oligosaccharide transferase inhibitor NGI-1, which inhibits the addition of N-glycans to B7x, can be used as an alternative therapeutic approach to cause its ubiquitylation and degradation [114, 115]. Inhibiting B7x glycosylation in combination with immunogenic chemotherapy and PD-L1 blockade could be an effective therapy to treat TNBC.
Glycosylation of B7-H3
B7-H3 overexpression is observed in various cancers, such as prostate cancer, medulloblastoma, NSCLC, pancreatic cancer, endometrial cancer, colorectal cancer, melanoma, glioma, breast cancer, renal cell carcinoma, ovarian carcinoma, and hepatocellular carcinoma, and is associated with poor prognosis [8]. Aberrant B7-H3 glycosylation predicts poor prognosis in patients with TNBC and oral cancer [116, 117]. B7-H3 in TNBC cells is ~110 kDa, which can be further reduced to ~70 kDa after PNGase F treatment. B7-H3 glycosylation is only partially reduced after the addition of recombinant glycosidase endoglycosidase H (Endo H) [117]. The glycosylation of endogenous B7-H3 is inhibited by the addition of an N-linked glycosylation inhibitor (tunicamycin) but not by O-glycosidase inhibitors (Thiamet G or PUGNAc). B7-H3 is a highly N-glycosylated protein with eight NXT motif sites in humans (N91, N104, N189, N215, N309, N322, N407, N433) and four NXT motif sites in mice (N91, N104, N189, N215). Additionally, the turnover rate of non-glycosylated B7-H3 was faster than that of glycosylated B7-H3 after treatment with a protein synthesis inhibitor (CHX), suggesting that nonglycosylated B7-H3 proteins are less stable and presumably more susceptible to degradation. The 26 S proteasome machinery revealed that the proteasome facilitated the degradation of ubiquitinated non-glycosylated B7-H3. Protein stability and cell surface expression of B7-H3 depend on the N-glycosylation of its NXT motif sites, which is also responsible for its immunosuppressive effects [117]. In the human genome, 13 different fucosyltransferases (FUTs) have been identified [96], but only FUT8 is responsible for positively regulating the glycosylation of B7-H3 and suppressing the immune response in TNBC. Core fucosylation is necessary for diverse protein functions regulated by FUT8 in the case of B7-H3. The sugar analog 2-Fluro L-fucose (2F-Fuc) inhibits B7-H3 core fucosylation and augments anti-PD-L1 immunotherapy in B7-H3-positive TNBC tumors in vivo. Different glycosyltransferases are involved in the glycosylation of B7-H3, but FUT8-mediated aberrant core fucosylation and degradation of B7-H3 by intervening with the core glycosylation modification of the B7-H3 protein (FUT8-B7H3 axis) would be a potential prognostic biomarker and important immunotherapeutic strategy for TNBC patients.
This crosstalk between glycosylation and immune checkpoints has special significance for improving immunotherapy and the immune response. It has been well established that N-glycosylation negatively impacts protein stabilization, signal intensity, and antibody binding affinity. The deglycosylation of proteins or biological samples could be a better biomarker of several pathological conditions. The engagement of glycan-binding immunoreceptors with the modified glycans facilitates immune evasion and tumor progression. Currently, advanced methods, tools, and techniques are being used to study the structure of complex glycans in cancer research, which will be further used to develop novel glyco-medicines to treat cancer in the future.
PD-L1 and B7-1 Cis-Interaction
The current dogma considers the PD-1/PD-L1 and CTLA-4/B7-1 pathways independent with no interactions. However, recent studies have demonstrated that the newly characterized PD-L1/B7-1 ligand‒ligand cis-interaction can bind to CD28 and CTLA-4 but not PD-1 (Fig. 5). These studies suggest that the PD-L1/B7-1 cis-interaction mediates significant crosstalk between the two immune checkpoint pathways [118].
Fig. 5.
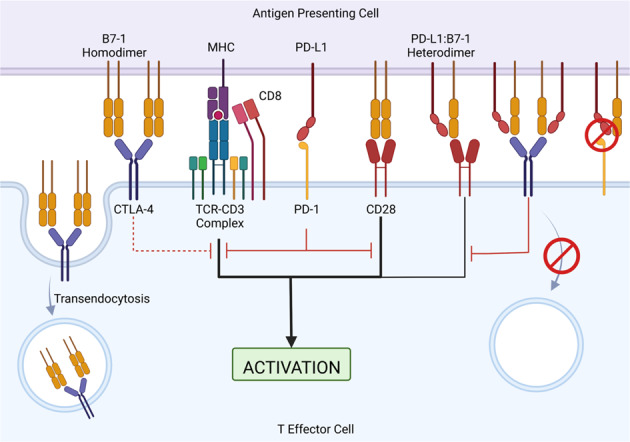
PD-L1/B7-1 cis-interaction in immune checkpoint pathways and T-cell activation. T-cell activation requires two signals: signal one is from the MHC-TCR complex and signal two is from the CD28/B7-1/B7-2 interaction. Inhibitory PD-1/PD-L1 signaling inhibits TCR and CD28 activation signals. CTLA-4 can bind to B7-1 to induce CTLA-4-mediated inhibition of the stimulatory signal. Additionally, CTLA-4 can sequester B7-1 from the cell surface of antigen-presenting cells by transendocytosis, thus removing available B7-1 from the system. The PD-L1/B7-1 heterodimer can bind to CD28 to induce a weak stimulatory signal and CTLA-4 to induce a weak inhibitory signal. CTLA-4 is unable to sequester the PD-L1/B7-1 heterodimer by transendocytosis, thereby allowing B7-1 to remain in the system. The PD-L1/B7-1 heterodimer is unable to bind to PD-1; thus, there is no PD-1-mediated inhibitory signal
PD-L1/B7-1 cis-interaction in vitro
Early studies using surface plasmon resonance and cell-protein adhesion assays reported that PD-L1/B7-1 interacts in trans [119, 120]. PD-L1/B7-1 trans-interaction decreased T-cell proliferation and the secretion of IL-2, IFN-γ, and TNFα in CD28/CTLA-4−/− cells, suggesting that the PD-L1/B7-1 trans-interaction plays an inhibitory role [119]. Consistent with these finding, B7-1-Ig could not inhibit the cells of CD28/CTLA-4/PD-L1−/− mice, thereby suggesting that the PD-L1/B7-1 interaction may be inhibitory [119]. A few mAbs targeting B7-1 or PD-L1 were shown to disrupt the PD-L1/B7-1 trans-interaction, PD-1/PD-L1, or both interactions [119, 120], suggesting that each mAb may have unique abilities to disrupt the PD-L1/B7-1 interaction, and they must be carefully characterized.
In contrast to earlier studies, recent work studying the PD-L1/B7-1 interaction showed that PD-L1 and B7-1 interact in cis [121–124]. ELISA and flow cytometry using purified proteins demonstrated that PD-L1 and B7-1 strongly interacted with each other only when PD-L1 was flexible, indicating that protein orientation is critical [121]. Further studies used multiple assays, such as protein-cell binding assays [121–124], fluorescence resonance energy transfer (FRET) assays [122], and proximity-based split luciferase assays [121, 124], and demonstrated that PD-L1 interacts with B7-1 in cis but not in trans (Fig. 5).
Interestingly, the PD-L1/B7-1 cis-heterodimer interferes with the PD-1/PD-L1 pathway. Recent studies showed that the PD-L1/B7-1 cis-interaction blocks the PD-1/PD-L1 interaction [122–125], decreases the formation of PD-1 microclusters [122], and inhibits PD-1-mediated immunosuppression [121–125]. Furthermore, the binding sites of B7-1 and PD-1 to PD-L1 partially overlap [119, 124]. Mutagenesis studies on the PD-L1 IgV domain identified specific amino acid mutations that may affect the PD-L1/B7-1 cis-interaction, PD-1/PD-L1, or both interactions [121–124]. In support of previous studies and these findings, a few anti-PD-L1 mAbs that can disrupt the PD-L1/B7-1 cis-interaction and PD-1/PD-L1 trans-interaction were identified [121, 125, 126]. These data suggest that the PD-L1/B7-1 cis-interaction interferes with PD-1/PD-L1 binding and can be targeted specifically by mAbs.
In addition to blocking PD-1/PD-L1 ligation, the PD-L1/B7-1 cis-heterodimer affects CTLA-4/B7-1 trans-interaction. The PD-L1/B7-1 binding sites overlap with the B7-1 homodimerization interface, which interferes with the formation of the B7-1 homodimer (Fig. 5) [124]. The binding sites of CTLA-4 and CD28 to B7-1 overlap [124]. The CTLA-4/B7-1 binding sites may partially overlap with the PD-L1/B7-1 interface [119] or be on the opposite side of the PD-L1/B7-1 interface [124]. Although the PD-L1/B7-1 cis-interaction may or may not directly hinder the CTLA-4/B7-1 trans-interaction, PD-L1/B7-1 heterodimerization reduces its interaction with the CTLA-4 homodimer and decreases CTLA-4-mediated transendocytosis of B7-1 (Fig. 5) [122]. Additionally, the PD-L1/B7-1 cis-interaction may reduce the CTLA-4/B7-1 interaction by weakening the avidity of the CTLA-4/B7-1 trans-interaction [122, 124]. Dimeric CTLA-4-Fc reduces the PD-L1/B7-1 cis-interaction, while monomeric CTLA-4 cannot [124]. Consistent with this finding, the PD-L1/B7-1 cis-interaction disrupts B7-1 homodimerization, preventing high-avidity CTLA-4/B7-1 lattice formation [122]. In contrast to these studies, one study suggested that the PD-L1/B7-1 cis-interaction did not alter high-avidity CTLA-4 pentamer formation and showed that CTLA-4 inhibited IL-2 release [123]. However, these data contradict another study showing that IL-2 secretion was reduced when atezolizumab, an anti-PD-L1 mAb that blocks the PD-L1/B7-1 cis-interaction, was added to CTLA-4+ Jurkat cells cocultured with B7-1+ PD-L1+ Raji cells compared with untreated controls [122]. Since the expression of immune checkpoints will likely determine the inhibitory or stimulatory effect [122, 123], further studies are needed for clarification.
Studies about the effects of the PD-L1/B7-1 cis-interaction on the CD28/B7-1 interaction have been conflicting. One study reported that the PD-L1/B7-1 cis-heterodimer moderately decreased T-cell activation compared to B7-1 alone using a T-cell activation reporter cell line [125]. Furthermore, the PD-L1 mAb that blocks PD-L1/B7-1 cis-heterodimer formation improved T-cell priming and increased the CD28-Fc and B7-1 interaction (Fig. 5) [125]. However, other studies reported that the PD-L1/B7-1 cis-interaction did not directly affect CD28/B7-1 binding [122, 123], the formation of TCR or CD28 microclusters [122], the enrichment of CD28 and B7-1 at the cell‒cell surface interface [122], the phosphorylation of CD28 [122], or the secretion of IL-2 [122, 123, 127]. The differences in the results of these studies may be due to the vastly different experimental technologies and conditions. Further studies are needed to clarify whether PD-L1/B7-1 affects the CD28/B7-1 interaction.
The role of B7-2 in the new PD-L1/B7-1 cis-interaction paradigm remains to be clarified. CTLA-4 and CD28 bind to B7-2. PD-L1 does not bind to B7-2, as shown by CTLA-4/B7-2 depletion [122], protein-cell binding [119, 120, 123], and proximity-based assays [122, 124]. B7-2 can interact with CD28 in the presence of the PD-L1/B7-1 cis-heterodimer [122]. Treatment with the anti-PD-L1 mAb atezolizumab did not prevent B7-2 transendocytosis in a Treg-dendritic cell B7-1 and B7-2 transendocytosis assay, suggesting that PD-L1 does not affect CTLA-4/B7-2 transendocytosis [122]. More studies are needed to clarify how B7-2 is involved in T-cell activation in the context of the PD-L1/B7-1 cis-interaction.
PD-L1/B7-1 cis-interaction in vivo
In vivo studies using autoimmune disease and cancer mouse models are unclear regarding whether the PD-L1/B7-1 cis-interaction plays an inhibitory or stimulatory role. An early study using a nonobese diabetic (NOD) mouse model reported that the PD-L1/B7-1 interaction may be inhibitory [126]. The PD-1/PD-L1 interaction plays an inhibitory role in the development of diabetes in NOD mice by regulating pathogenic self-reactive effector T cells [128, 129]. However, the role of the PD-L1/B7-1 cis- or trans-interactions in the development of diabetes is unknown. Blocking mAbs targeting only the PD-L1/B7-1 interaction or both the PD-1/PD-L1 and PD-L1/B7-1 interactions accelerate diabetes development in NOD mice [126]. The blocking mAb targeting only the PD-L1/B7-1 interaction was more effective in inducing diabetes in old mice than in younger mice; the blocking mAb that dual-targeted the PD-1/PD-L1 and PD-L1/B7-1 interaction induced diabetes in both young and old mice [126]. In an adoptive-cell transfer mouse model of diabetes, the blocking mAb targeting only the PD-L1/B7-1 interaction accelerated diabetes development in recipients of T cells from established diabetic mice but not prediabetic mice [126]. The dual-targeting blocking mAb effectively induced diabetes in both settings [126]. These data suggest that the PD-L1/B7-1 interaction may be inhibitory.
A recent study used mutated PD-L1 and B7-1 in vitro and in vivo to further examine the role of the PD-L1/B7-1 cis-interaction [123]. Genetically engineered mice with PD-L1 Y56A or B7-1 L107E mutations that prevent the PD-L1/B7-1 cis-interaction in vivo reduced IFN-γ and IL-2 production by T cells in response to OVA protein and MHC-OVA peptide stimulation [123]. These genetically engineered mice have poorer antitumor responses than wild-type mice in an OVA-expressing E.G7 T lymphoma mouse model [123]. In the autoimmune disease experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis, PD-L1- and B7-1-mutated mice have fewer signs of EAE after myelin oligodendrocyte glycoprotein (MOG) peptide immunization than wild-type mice [123]. Splenocytes derived from PD-L1- and B7-1-mutated mice produce less IL-17 after MOG protein immunization [123]. These results suggest that the PD-L1/B7-1 cis-interaction may have a stimulatory role.
The newly characterized PD-L1/B7-1 cis-interaction should be of interest in examining clinical trials data. A recent study showed that PD-L1 and B7-1 are coexpressed in cancer patient-derived tumoral conventional dendritic cells, but these cells express 20-fold more PD-L1 than B7-1 [125]. RNA-sequencing data from NSCLC and renal cell cancer patients showed that those with a high dendritic cell signature who were treated with atezolizumab, an anti-PD-L1 mAb that blocks the PD-1/PD-L1 and PD-L1/B7-1 interactions, had higher overall survival [125]. Consistent with previous studies [130, 131], immune cell PD-L1 expression was associated with the response to atezolizumab [125]. More studies are needed to determine whether the PD-L1/B7-1 cis-interaction is involved in the response to immune checkpoint blockade therapy in the clinic.
Resistance mechanisms to anti-PD-1/PD-L1 and anti-CTLA-4 in clinical studies
Although immune checkpoint blockade (ICB) targeting PD-1/PD-L1 and CTLA-4 can lead to durable responses, most patients do not respond to ICB. This suggests that therapeutic resistance remains a critical limitation of the current ICB strategy. Patients can exhibit primary resistance to ICB, meaning their cancers possess inherent characteristics that make them unresponsive to treatment, or patients can experience acquired resistance, in which their cancer initially responds to therapy but later progresses despite continued treatment. Regardless of the type, resistance can be mediated through tumor cell-intrinsic and -extrinsic factors, depending on the treatment and cancer type. Many molecular and cellular resistance mechanisms to ICB have been proposed using preclinical models (reviewed in [132]). However, few of these mechanisms have been directly confirmed in clinical samples from patients treated with ICB, and their clinical relevance remains unclear. Here, we focused solely on resistance mechanisms that were uncovered using clinical samples from patients treated with ICB (Fig. 6).
Fig. 6.
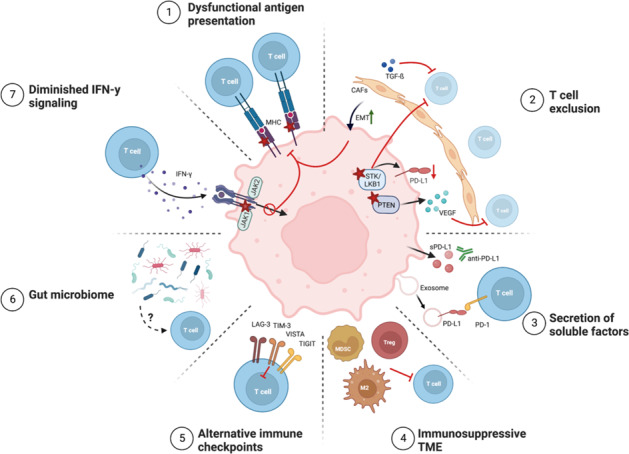
Mechanisms of resistance to immune checkpoint blockade. The decreased efficacy of ICB in cancer patients can occur through tumor cell-intrinsic and -extrinsic factors as follows: (1) defects in antigen presentation due to the loss or reduced expression of MHC molecules; (2) the exclusion of T cells mediated by tumor-intrinsic genetic changes, CAF TGF-β signaling, and EMT; (3) the secretion of soluble and exosomal PD-L1 that potentially competes with drug binding; (4) the presence of immunosuppressive cell types in the TME that inhibit T-cell functions; (5) T-cell exhaustion and the expression of alternative inhibitory immune checkpoints; (6) a lack of bacterial diversity or the enrichment of specific ‘bad’ microbes in the gut microbiome; and (7) insensitivity to IFN-γ-mediated prevention of cancer cell apoptosis and the expression of MHC molecules. Stars denote loss of function or expression. ICB, immune checkpoint blockade; MHC, major histocompatibility complex; CAF, cancer-associated fibroblasts; sPD-L1, soluble PD-L1; M2, M2 macrophage; Treg, regulatory T-cell; MDSC, myeloid-derived suppressor cell
Tumor-intrinsic mechanisms of resistance
Tumor-intrinsic resistance mechanisms can be divided into three groups: (1) defects in antigen processing and presentation; (2) insensitivity to IFN signaling; and (3) genetic T-cell exclusion (Fig. 6). The efficacy of ICB relies on the recognition of tumor-specific neoantigens in the context of major histocompatibility complex (MHC) class I or II molecules by cytotoxic CD8 and CD4 T cells, respectively. Consequently, a high tumor mutational burden (TMB) predicts the response to ICB, suggesting that tumor immunogenicity is a prerequisite for neoantigen recognition by cytotoxic T cells [133–139]. Thus, ICB resistance can be driven by tumor neoantigen presentation and processing defects, such as dysfunctional MHC variants that perturb antigen presentation. In melanoma, loss of heterozygosity (LOH) or deleterious mutations in β-2-microglobulin (β2M), a crucial component of the MHC I complex, are thought to cause primary and acquired resistance to PD-1 and CTLA-4 blockade [137, 140–142]. Other reports have shown that the loss of MHC class I protein expression correlates with primary resistance to CTLA-4 but not PD-1 blockade in melanomas. In contrast, reduced MHC class II expression is associated with PD-1 blockade resistance [143–145]. Similar to these observations, a NSCLC patient receiving combined PD-L1 and CTLA-4 blockade had homozygous β2M loss in the tumor at the time of acquired resistance [146]. The potential of B2M loss as a resistance mechanism was further validated using a mouse lung cancer model with β2m knockout [146]. Furthermore, the same study observed β2M downregulation, which resulted in the loss of MHC class I cell-surface expression in patient-derived xenografts from patients with acquired ICB resistance. Thus, the loss of neoantigen presentation machinery leads to poor immunogenicity and resistance to ICB.
IFN-γ is a pleiotropic cytokine produced by T and NK cells that is important for antitumor immune responses through mechanisms including direct induction of cancer cell apoptosis and the upregulation of MHC class I expression and antigen presentation [147]. Active IFN-γ signaling is a predictive marker of the response to PD-1 blockade [139, 148–151]. Thus, it is not surprising that the suppression of this pathway may be a tumor-intrinsic factor contributing to resistance. Truncating mutations in genes encoding kinases within the IFN-γ signaling pathway, such as Janus kinase 1 (JAK1) and JAK2, lead to IFN-γ insensitivity and T-cell escape, thereby conferring primary and acquired resistance to PD-1 and PD-L1 blockade in multiple cancer types [138, 140, 152]. Moreover, genetic defects or losses in multiple IFN-γ pathway genes and amplification of genes encoding suppressors of IFN-γ signaling are suggested to cause primary resistance to CTLA-4 blockade [153]. Taken together, these studies indicate that decreased IFN-γ signaling in tumor cells is a common mechanism of resistance to ICB.
Oncogenic signaling pathways can induce the exclusion of tumor-infiltrating lymphocytes (TILs). The absence of TILs, specifically CD8 T cells, in the tumor microenvironment (TME) is associated with resistance to ICB [139, 148, 151, 154, 155]. Mutations in the tumor suppressor STK11/LKB1 in NSCLC are associated with TIL exclusion from the TME and primary resistance to PD-1 blockade [138, 156]. Although the exact mechanism has yet to be determined, these mutations are accompanied by TIL exclusion, downregulation of PD-L1 expression, and increased neutrophil TME infiltration [138, 156]. Similarly, PTEN loss significantly correlates with TIL exclusion and confers primary and acquired resistance to PD-1 blockade in melanoma, and potentially led to acquired resistance in a single case of uterine leiomyosarcoma [157–159]. On a molecular level, PTEN loss may activate the PI3K/AKT signaling pathway, which induces the production of immunosuppressive cytokines, such as vascular endothelial growth factor (VEGF) [157]. VEGF promotes angiogenesis and modulates the TME by decreasing the trafficking and effector function of T cells and promoting suppressive immune cell populations, such as Tregs and MDSCs [160]. Increased expression of VEGF in early on-treatment tumor biopsies from melanoma patients is associated with PD-1 blockade resistance, and VEGF inhibition potentiates ICB by increasing the trafficking of CD8 T cells to the tumor [148, 161, 162].
In addition to VEGF, the release of other soluble factors is associated with ICB response (Fig. 6). Secreted variants of PD-L1 (sPD-L1), which originate from alternative splicing, are thought to confer resistance to ICB in multiple cancer types [163–165]. sPD-L1 is hypothesized to act as a decoy for PD-L1 blockade, reducing treatment efficacy through competitive binding of the drug [166, 167]. However, this does not explain why sPD-L1 is also associated with resistance to CTLA-4 and PD-1 blockade, suggesting that other mechanisms are at play. PD-L1 may contribute to resistance through other noncanonical mechanisms. Exosome release is another major route by which cancer cells can induce local and systemic changes in immune cells. Elevated plasma levels of exosomes carrying PD-L1 correlate with progressive disease in multiple cancer types treated with PD-1 blockade. However, it is unclear whether baseline levels or changes in PD-L1+ exosome levels during treatment predict responses [168–171].
Tumor cell-extrinsic mechanisms of resistance
Tumor cell-extrinsic mechanisms involve the composition and function of the other cells in the TME (Fig. 6). The TME is thought to play a prominent role in the efficacy of ICB. Processes involved in cell plasticity and dedifferentiation processes, including EMT, hypoxia, angiogenesis, and extracellular matrix remodeling, are related to resistance to ICB across different cancer types [151, 172–175]. By separating tumors from several cancer types into four distinct TME categories, cancer-associated fibroblasts (CAFs) in the TME lead to worse ICB outcomes [176]. Transforming growth factor β (TGF-β) signaling in CAFs is connected to CD8 T-cell exclusion from the TME and resistance to PD-L1 blockade in urothelial cancers and has been shown to induce an EMT phenotype and the downregulation of MHC class I expression in melanomas that are resistant to PD-1 blockade [175, 177]. Furthermore, increased TGF-β and EMT-related gene expression decreases the beneficial association between the presence of TILs and ICB outcomes in melanoma and urothelial cancer [174, 178].
Many other immunosuppressive cell types in the TME can contribute to immune escape. FoxP3+ Tregs, MDSCs, and immunosuppressive M2 macrophages can induce resistance to ICB in preclinical models [179–181]. However, very few clinical studies have examined the roles of these suppressive cells during ICB treatment in a clinical context. Surprisingly, the clinical activity of CTLA-4 blockade in melanoma is positively correlated with high FoxP3 levels in baseline tumor biopsies [182]. In contrast, one study showed an increase in Tregs in 56% of progression biopsies compared to baseline for melanoma patients with acquired PD-1- or combined PD-1 and CTLA-4 blockade resistance [159]. Other studies examining the percentages of Tregs before and during PD-1 blockade saw no differences in tumor biopsies from responding and nonresponding patients [183, 184]. An increase in peripheral Tregs early during ICB is associated with progression in melanoma patients [185, 186]. Moreover, higher Treg frequencies in baseline peripheral blood samples from melanoma patients are associated with unfavorable outcomes of CTLA-4 blockade [187]. The immunosuppressive function of PD-1+ Tregs is strengthened by PD-1 blockade and increased levels of this cell type before the start of treatment are considered a primary resistance mechanism in multiple cancer types and are associated with rapid disease progression in gastric cancer [64, 65]. Due to the inconsistent data, more studies are needed to pinpoint the role of Tregs in ICB resistance. Tregs are not the only suppressive cell type that can impact the ICB response. The frequency of peripheral MDSCs is increased in patients who do not respond to CTLA-4 blockade [187–190]. Regarding the presence of M2 macrophages in the TME, one study looking at pretreatment biopsies of lung cancer showed that low infiltration of PD-1+ CD8 T cells and high infiltration of M2 macrophages were predictive of worse responses to PD-1/PD-L1 blockade [191]. In sarcoma patients treated with PD-1 blockade, tumor infiltration of IDO+ M2 macrophages was observed in nonresponding patients [192]. However, it should be noted that these patients had PD-L1-negative tumors and low CD8 T-cell tumor infiltration, calling into question the exact mechanism of resistance in this study [192].
Mechanisms underlying acquired resistance can be linked to the functional impairment of chronically stimulated TILs (Fig. 6). In tumors, persistent neoantigen-induced TCR signaling drives T cells toward a state of dysfunction, which is also known as T-cell exhaustion, in which these cells exhibit decreased cytotoxic and effector functions [193]. The expression of multiple inhibitory immune checkpoint molecules further characterizes this T-cell subset. Ex vivo studies of TILs showed that high PD-1 expression and the coexpression of multiple inhibitory immune checkpoints indicated T cells with poor T-cell function restoration in response to ICB treatment [194, 195]. Patients with prostate cancer do not have a significant clinical response to CTLA-4 blockade. One possible explanation might be that these patients have increased expression levels of V-domain immunoglobulin suppressor of T-cell activation (VISTA) on TILs and M2 macrophages, indicating that VISTA is a potential mechanism of resistance [196]. In melanoma, an increased density of TILs expressing VISTA from baseline to progression was found in 67% of patients who were treated with PD-1 or combined PD-1 and CTLA-4 blockade [159]. Baseline expression of LAG-3, T-cell immunoglobulin and ITIM domain (TIGIT), and IDO in melanomas is significantly associated with nonresponse to PD-1 blockade [151], while the expression of LAG-3, but not PD-1 or T-cell immunoglobulin and mucin domain-containing protein 3 (Tim-3), on TILs is associated with shorter survival after PD-1 blockade in NSCLC [155, 197]. Tim-3 and CTLA-4 expression were upregulated in two lung cancer patients who progressed during PD-1 blockade [198], and neoantigen-specific TILs in nonresponding NSCLC tumors had increased expression of genes encoding proteins associated with T-cell dysfunction and inhibitory checkpoints, such as thymocyte selection-associated HMG BOX (TOX), CTLA-4, and Tim-3, during PD-1 blockade [199]. In contrast, tumors expressing PD-L1 and a second inhibitory checkpoint, such as LAG-3, Tim-3, or CTLA-4, showed a trend toward an increased response to PD-L1 blockade compared to the expression of PD-L1 alone across multiple cancer types [130]. Intriguingly, the only coexpressed immune checkpoint that reduced the response to PD-L1 blockade in this study was B7x. This existence of potentially nonredundant checkpoint receptors on dysfunctional T cells may serve as a resistance mechanism by preventing the targeting of a single checkpoint from restoring an efficient antitumor response. Alternatively, the coexpression of multiple checkpoint molecules may simply denote dysfunctional T cells that cannot be reinvigorated by ICB, thus serving as markers of terminally differentiated T cells rather than drivers of resistance.
The gut microbiome has received increasing attention for its involvement in ICB resistance in multiple cancer types (Fig. 6). Decreased bacterial diversity in the microbiome is associated with resistance to ICB in some studies [200, 201], while others showed that the enrichment of specific microbes could predict outcomes [201, 202]. This finding was further confirmed by studies showing that using antibiotics in conjunction with ICB was associated with worse outcomes [200, 203]. The overlap in these studies is small, which might reflect the differences in geographical locations, patient cohorts, and technical differences. The precise underlying mechanism remains unknown, and the suggested mechanisms are mainly based on murine cancer models in which the mice were transplanted with fecal matter from human ICB responders and nonresponders [200, 201, 204]. These studies suggest that microbiome composition may affect ICB efficacy due to T-cell cross-reactivity between microbial antigens and tumor antigens or through microbial metabolites that modulate the immune system (reviewed in [205]).
Many recent studies have used comprehensive omics methods to identify signatures that encompass multiple genomic and transcriptomic features to predict response and nonresponse to ICB, highlighting that numerous pathways may need to be considered to comprehensively understand ICB resistance [139, 145, 148, 149, 172, 178, 206–212]. Although some common denominators have been found across studies, such as TMB, IFN-γ signaling, and the presence of TILs in the TME, these signatures have been challenging to validate across independent patient cohorts [145, 175, 212–214]. It is becoming increasingly clear that robust predictive signatures are complicated by the heterogeneity and adaptability of cancer, especially considering the selective pressure of immunotherapy. Accordingly, future studies need a more individualized approach to identify resistance mechanisms.
New immunotherapies targeting B7-H3, B7x, and PD-1/PD-L1 in clinical trials
Current approved immune checkpoint blockade therapies for the treatment of cancers include blocking mAbs targeting PD-1, PD-L1, or CTLA-4, combination therapies of anti-PD-1 plus anti-CTLA-4 or anti-LAG-3, and bispecific antibodies targeting PD-1 and CTLA-4. However, one of the biggest challenges is that the majority of cancer patients do not respond to these treatments. Thus, there is a pressing need to develop new and effective treatments. In this section, we discuss various new therapeutic strategies that have been developed and are currently in clinical trials.
New anti-B7-H3 immunotherapies in clinical trials
Currently, one humanized anti-B7-H3 IgG1 mAb is being evaluated in clinical trials (Table 1). This anti-B7-H3 mAb has five amino acid changes in its humanized Fc region, which resulted in enhanced ADCC and antitumor abilities, as demonstrated in bladder and renal cell xenograft tumor models [215]. This anti-B7-H3 mAb is being evaluated in the clinic as monotherapy or in combination with anti-CTLA-4, an anti-PD-1 mAb, or a PD-1/LAG-3 bispecific antibody to treat patients with multiple advanced solid tumors. In 2015, a phase I trial showed that this anti-B7-H3 mAb had an acceptable safety profile in cancer patients with B7-H3+ tumors and antitumor activities across different cancer types [216]. In 2022, a phase II trial showed that the anti-B7-H3 mAb was a neoadjuvant therapy before prostatectomy in patients with prostate cancer and was associated with a decrease in Gleason grading and increases in CD8 T cells, inflammation, and PD-1/PD-L1 expression, suggesting that it had promising antitumor abilities [217]. However, a 2022 phase I/II trial assessing the safety and efficacy of anti-B7-H3 and anti-PD-1 mAb combination therapy showed that the response to this combination treatment was limited in patients with head and neck squamous cell carcinoma (HNSCC), NSCLC, urothelial bladder cancer, and cutaneous melanoma [218]. Additionally, a phase II trial assessing combination treatment with an anti-B7-H3 mAb plus anti-PD-1 or a PD-1/LAG-3 bispecific antibody in patients with HNSCC was terminated due to seven hemorrhagic-associated fatalities. A planned phase II/III trial evaluating these combination treatments in patients with HNSCC was withdrawn.
Table 1.
New clinical therapeutic strategies targeting B7-H3
| Target | Platform | Tumor type | Phase |
|---|---|---|---|
| B7-H3 | Monoclonal antibody | Neuroblastoma, rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, Wilms tumors, desmoplastic small round cell, melanoma, NSCLC, head and neck cancer, HNSC, bladder cancer | I, II, II/III |
| B7-H3 |
Radiolabeled antibody. Antibody-drug conjugate |
Advanced and/or metastatic solid tumors, diffuse intrinsic pontine glioma, medulloblastoma, ependymoma, desmoplastic small round cell, peritoneal, leptomeningeal, Prostate, melanoma, pancreatic, hepatocellular, ovarian, HNSC, TNBC, NSCLC | I, I/II, II |
|
B7-H3 x CD3 |
Bispecific antibody | Advanced solid tumors, mesothelioma, bladder, melanoma, HNSC, NSCLC, clear cell renal cell, ovarian, thyroid, breast, pancreatic, prostate, colon, sarcoma | I |
| B7-H3 | CAR-T cells | Advanced solid tumors, glioblastoma, Osteosarcoma, neuroblastoma, gastric, lung, neuroblastoma, acute myeloid leukemia, melanoma, colorectal, germ cell, retinoblastoma, hepatoblastoma, Wilms tumors, teratoid/rhabdoid tumors, Ewing sarcoma, rhabdomyosarcoma, synovial sarcoma, clear cell sarcoma, peripheral nerve sheath, desmoplastic small round cell, sarcoma, glioma, ependymoma, medulloblastoma, neuroectodermal, choroid plexus, childhood pine blastoma, lung, TNBC, breast, brain, pancreatic, ovarian, hepatocellular, adrenocortical | I, I/II |
A few anti-B7-H3 radiolabeled mAbs and antibody‒drug conjugates (ADCs) are undergoing clinical trials (Table 1). Currently, there are three radiolabeled anti-B7-H3 mAbs in clinical trials. An anti-B7-H3 mAb radiolabeled with iodine 131 (131I) showed promising antitumor activity; 56% of patients with metastatic neuroblastoma were alive at the end of the study, and 45% of the patients survived beyond 36 months and 29% survived beyond 60 months. This is a significant improvement, since the same institution reported that their historical median overall survival time of patients with neuroblastoma was 6.6 months [219, 220]. A retrospective analysis of the same cohort showed that the anti-B7-H3 131I-antibody and conventional treatment did not increase the risk of radionecrosis in long-term survivors, supporting the safety of this treatment [221]. In a retrospective study, the anti-B7-H3 131I-antibody improved the survival of 23 recurrent rhabdomyosarcoma patients [222]. Additionally, the anti-B7-H3131I antibody was tolerable in patients with peritoneal tumors such as desmoplastic small round cell tumors [223]. The anti-B7-H3 mAb radiolabeled with 124I improved the median overall survival of patients with diffuse intrinsic pontine gliomas by 3–4 months compared to the historical data from other trials [224, 225]. Finally, an anti-B7-H3 mAb radiolabeled with 177Lutetium is in clinical trials to treat patients with advanced solid tumors and metastatic leptomeningeal tumors, but the results are not yet available.
Four anti-B7-H3 ADCs are in clinical trials to assess their safety, tolerability, and preliminary efficacy (Table 1). Some anti-B7-H3 ADCs are tolerable in patients with advanced cancers with evidence of antitumor effects [226]. However, one anti-B7-H3 ADC had an acceptable safety profile but with two dose-limiting toxicities: one grade 3 fatigue and one grade 4 neutropenia [227]. In a cohort expansion study, approximately 87.7% of cancer patients treated with this anti-B7-H3 ADC experienced at least one adverse event [228]. However, the same study observed tumor regression and a decrease in prostate-specific antigen (PSA) in the metastatic prostate cancer patient cohort [228], demonstrating that this anti-B7-H3 ADC has antitumor effects. The other two anti-B7-H3 ADCs are under evaluation in phase I trials to treat multiple solid cancers, but results have yet to be reported.
Bispecific antibodies (bsAbs) targeting B7-H3 and another target simultaneously are currently in development (Table 1). These bsAbs induce synergistic antitumor effects through various mechanisms, such as blocking inhibitory pathways or engaging immune cells. Multiple B7-H3-targeting bsAbs are being studied in the preclinical stage, such as B7-H3/4-1BB bsAbs [229], B7-H3/PD-1 bsAbs [230], a B7-H3/CD16 bsAb NK cell engager [231], and a B7-H3/CD3 bsAb T-cell engager [232]. These bsAbs exhibit antitumor effects in vitro, while B7-H3/4-1BB, B7-H3/CD16, and B7-H3/CD3 can suppress tumor growth in mouse xenograft models. Recently, a B7-H3/CD16/IL-15 tri-specific NK cell killer engager exerted antitumor effects in cancer models in vitro and in vivo [233]. Currently, a B7-H3/CD3 T-cell engager is the only anti-B7-H3 bsAb under clinical evaluation, alone or combined anti-PD-1 immunotherapy (NCT03406949).
Anti-B7-H3 CAR-T therapy has been effective in many preclinical cancer models and is currently being evaluated in clinical trials (Table 1). Case reports have demonstrated that B7-H3-targeting CAR-T cells are well tolerated and effective in reducing tumor growth in patients with relapsed basal cell carcinoma, recurrent anaplastic meningioma, and glioblastoma [234–236]. Early results of a phase I trial evaluating B7-H3 CAR-T-cell therapy to treat relapsed or refractory non-CNS tumors in children and young adults reported that no dose-limiting toxicity was observed after the first infusion, and the median persistence of circulating CAR-T cells was 28 days [237]. Three of the nine subjects infused showed disease stabilization. After the second infusion, one patient experienced significant CAR-T-cell expansion, transient grade 4 liver enzyme elevation, and partial metabolic response on FDG-PET on Day 28. The combination of B7-H3-specific CAR-T cells with chemotherapy (fludarabine, cyclophosphamide, MESNA, or temozolomide) is currently being investigated in clinical trials, but results have yet to be reported.
The new therapeutic strategy targeting B7x
Eight clinical trials assessing anti-B7x immunotherapy using various treatment modalities are ongoing (Table 2), and one clinical trial was recently completed. Currently, there are three phase I trials and one phase I/II trial evaluating anti-B7x ADCs carrying various cytotoxic payloads to treat many advanced solid cancer types; no results have been posted. Three different bsAbs targeting B7x/4-1BB or B7x/CD3 are currently being studied in phase I and phase I/II trials, but no results have been posted. One phase I/II clinical trial assessed an anti-B7x blocking mAb as a single agent to treat advanced or metastatic solid tumors. One completed phase I trial showed that the use of anti-B7x blocking mAbs as a single agent to treat advanced solid tumors had a favorable safety profile, but its antitumor effect has not been reported [238].
Table 2.
New clinical therapeutic strategies targeting B7x
| Target | Platform | Tumor type | Phase |
|---|---|---|---|
| B7x | Monoclonal antibody | Advanced and/or metastatic solid tumors, ovarian, NSCLC, breast, endometrial | I, I/II |
| B7x | Antibody-drug conjugate | Advanced solid tumors, TNBC, endometrial, ovarian, fallopian tube, peritoneal, cholangiocarcinoma, NSCLC, gallbladder, adenoid cystic | I, I/II |
|
B7x x CD3, 4-1BB |
Bispecific antibody | Advanced solid tumors, ovarian, endometrial, breast, uterine, squamous cell, NSCLC | I, I/II |
PD-L1-targeting bispecific antibodies
To improve upon anti-PD-L1 blocking mAbs, bsAbs simultaneously targeting PD-L1 and another target were developed and are currently being investigated in clinical trials (Table 3). A phase I trial showed that PD-L1/Tim-3 bsAbs were associated with unexpected immunogenicity from both arms of the bsAb; as a result, the study was terminated [239]. One PD-L1/TIGIT bsAb is being assessed for safety and efficacy in two phase I/II clinical trials, but the results have yet to be reported. There are three PD-L1/TGF-β bsAbs in phase I clinical trials. In 2021, a phase I trial showed that one PD-L1/TGF-β bsAb had an acceptable safety profile with evidence of antitumor activity in patients with advanced solid tumors, especially those who were previously treated with immune checkpoint inhibitors [240]. Combination treatment with this PD-L1/TGF-β bsAb and an anti-TIGIT mAb is currently in a phase I trial. In 2021, phase II and III trials showed that another PD-L1/TGF-β bsAb could not improve upon the standard-of-care treatment in patients with advanced NSCLC and biliary tract cancer. One PD-L1/OX40 bsAb is in a phase I trial, but the results have yet to be reported. Two PD-L1/LAG-3 bsAbs are in phase I trials. One PD-L1/LAG-3 bsAb is tolerable and has antitumor activity in patients with advanced tumors who are resistant to anti-PD-L1 immunotherapy [241]. A bsAb targeting PD-L1 and the tumor-specific antigen claudin 18.2 has been reported to be safe in patients with resistant/refractory advanced or metastatic solid tumors who previously failed standard-of-care therapies [242]. PD-L1/CD47 bsAbs were reported to have a manageable safety profile and antitumor activity in patients with advanced solid cancers [243]. BsAbs targeting PD-L1 and costimulatory molecules such as CD27, CD137, or 4-1BB are also in clinical trials. PD-L1/CD27 bsAbs and PD-L1/CD137 bsAbs were reported to be safe in patients with different cancer types in phase I trials [244, 245]. Four PD-L1/4-1BB bsAbs are in phase I or phase I/II trials. One of the four PD-L1/4-1BB bsAbs is tolerable and has antitumor effects in patients with advanced solid tumors, including those who are resistant to previous immune checkpoint immunotherapy [246].
Table 3.
New clinical therapeutic strategies targeting PD-1, PD-L1, or CTLA-4
| Target | Platform | Tumor type | Phase |
|---|---|---|---|
|
PD-L1 x LAG-3, TIGIT, Tim-3, 4-1BB, OX40, CD27, CD47, CD137, Claudin 18.2, TGF-β |
Bispecific antibody | Advanced and/or metastatic solid tumors or lymphomas, myeloid, thoracic, NSCLC, SCLC, breast, gastric, renal cell, ovarian, peritoneal, fallopian tube, cholangiocarcinoma, bladder, microsatellite instability-high colorectal, esophageal, hepatic, head and neck, HNSC, papillomavirus-related tumors, melanoma, gastric adenocarcinoma, esophageal adenocarcinoma, NSCLC, SCLC | I, I/II |
|
PD-1 x PD-L1, LAG-3, TIGIT, Tim-3, VEGF |
Bispecific antibody | Advanced solid tumors, NSCLC, lymphoma, nasal type, classical Hodgkin lymphoma, melanoma, SCLC, squamous cell esophageal, gastric, gastroesophageal-junction, ovarian | I, I/II |
|
CTLA-4 x PD-1, LAG-3, OX40, PD-L1 |
Bispecific antibody | Advanced and/or metastatic solid tumors, nasopharyngeal, melanoma, cervical, pancreatic, TNBC, hepatocellular, bladder, HNSC, clear cell renal cell, renal cell, NSCLC, SCLC, gastric adenocarcinoma, gastroesophageal-junction adenocarcinoma, prostate, ovarian, fallopian tube, cervical, peritoneal, cholangiocarcinoma, squamous cell esophageal, squamous cell anal, squamous cell penile, squamous cell vulvar, colorectal, endometrial, lymphoma, MSI-H/deficient mismatch repair (dMMR) gastric and colorectal, MSI-H/dMMR advanced solid tumors, anaplastic or oncocytic thyroid gland, thymoma and thymic, breast, mesothelioma, neuroendocrine, adnexal, non-squamous cell salivary gland, biliary tract, HER2 + solid tumors, B-lymphoblastic leukemia | I, I/II, II, III |
Bispecific antibodies targeting PD-1 and/or CTLA-4
BsAbs targeting PD-1 or CTLA-4 and other factors are also in clinical trials (Table 3). There are two PD-1/PD-L1 bsAbs in phase I and I/II trials. Preliminary results from a phase I trial evaluating one PD-1/PD-L1 bsAb in treating patients with advanced tumors showed an acceptable safety profile; the evaluation of its antitumor activity is ongoing [247]. A phase I trial evaluating other PD-1/PD-L1 bsAbs was recently completed, but the results have yet to be reported. BsAbs targeting PD-1 and TIGIT or Tim-3 are in ongoing phase I or I/II trials, but the results have yet to be reported. Currently, there is one PD-1/VEGF bsAb in clinical trials. A phase I trial evaluating PD-1/VEGF-A bsAb to treat patients with refractory/platinum-resistant ovarian cancer showed that it induced antitumor activity with a favorable safety profile [248]. Additionally, the combination of the PD-1/VEGF bsAb and chemotherapy has promising antitumor efficacy and safety in patients with advanced NSCLC compared to combination therapy with anti-VEGF and anti-PD-L1 mAbs [249]. PD-1/VEGF bsAbs and chemotherapy improved progression-free survival (PFS) and the overall response rate (ORR).
Four CTLA-4/PD-1 bsAbs are being assessed in clinical trials (Table 3). One CTLA-4/PD-1 bsAb was approved in China in 2022 to treat patients with relapsed or metastatic cervical cancer (r/m CC) who progressed after platinum-based chemotherapy. It is the first-in-class CTLA-4/PD-1 bsAb and the first dual-targeting immune checkpoint bsAb to be approved for clinical use worldwide. The CTLA-4/PD-1 bsAb showed promising results in a phase II trial treating patients with r/m CC [250]. Of the 100 evaluable patients, the ORR for this treatment was 33%, and the complete response (CR) rate was 12%. The response rates at 6 and 12 months were 77.6% and 52.9%, respectively. The median PFS was 3.75 months, and the overall survival was 17.5 months. Of the 64 patients with PD-L1+ tumors, the ORR was 43.8%, the median PFS was 6.34 months, and the median overall survival was not reached. Finally, this CTLA-4/PD-1 bsAb has a favorable safety profile, and 27% of the total 111 enrolled patients experienced grade ≥3 treatment-related adverse events. In another phase II trial, combination treatment with this CTLA-4/PD-1 bsAb plus platinum-based chemotherapy had an ORR of 79.3% regardless of PD-L1 expression and ORRs of 82.4% and 75% for patients with PD-L1+ and PD-L1- tumors, respectively [251]. Phase I and II trials evaluating this CTLA-4/PD-1 bsAb as a monotherapy showed that it had a favorable safety profile, was well tolerated among patients, and had evidence of antitumor activity in patients with mesothelioma [252] and metastatic nasopharyngeal carcinoma [253] who were resistant to prior therapies. Finally, a phase I trial reported that combination treatment with this CTLA-4/PD-1 bsAb, oxaliplatin, and capecitabine had a manageable safety profile and an ORR of 66.7%, including two complete and 14 partial responses out of 24 patients with advanced gastric or gastroesophageal junction cancer who received treatment [254]. Phase I trials also showed that the other CTLA-4/PD-1 bsAbs were safe, tolerable, and effective in treating patients with advanced solid tumors who were previously treated [255, 256] and those with advanced renal cell carcinoma [257]. The fourth CTLA-4/PD-1 bsAb is currently being studied as a monotherapy or combination with chemotherapy or an anti-B7-H3 ADC; the results have yet to be reported. Finally, combination treatment with CTLA-4/PD-1 bsAbs and anti-CD73 or anti-CD47 mAbs are currently being evaluated in ongoing phase I and I/II trials.
In addition to CTLA-4/PD-1 bsAbs, bsAbs targeting CTLA-4 and PD-L1, OX40, or LAG-3 are being assessed in clinical trials (Table 3). Combination therapy with a CTLA-4/PD-L1 bsAb and an anti-HER2 mAb had a favorable safety profile and evidence of antitumor activity against HER2+ solid tumors according to a phase I trial [258, 259]. The combination of this CTLA-4/PD-L1 bsAb and chemotherapy improved the PFS and ORR of patients with unresectable advanced or metastatic hepatocellular carcinoma in a phase II trial [260]. Finally, a CTLA-4/OX40 bsAb was safe and tolerable up to 200 mg in a phase I trial; dose escalation and antitumor studies are ongoing, but the results have yet to be reported [261].
Conclusion
The B7/CD28 families of immune checkpoints are essential in regulating immune homeostasis and play a significant role in diseases such as autoimmunity and cancer. Targeting CTLA-4 and the PD-1/PD-L1 pathway using immune checkpoint inhibitors directly improved patient clinical outcomes and highlighted the importance of immune checkpoints in cancer. Over the past decade, our understanding of the B7/CD28 families has exponentially grown and identified new avenues of research and targets to develop novel therapies. The studies described herein showcase recently developed topics and questions that remain. How do HHLA2, B7-H3, and B7x promote tumor immune evasion in various cancer types? What are the receptors for B7x and B7-H3? How do immune checkpoints regulate metabolism and the phenotype of different cell types? How does glycosylation affect the biology of the newest members of the B7 family? How do we overcome resistance mechanisms to current immune checkpoint inhibitors, including glycosylation? How do the structural interactions and crosstalk between CTLA-4 and the PD-1/PD-L1 pathway affect current and future immune checkpoint inhibitors? These studies lay the foundation for continued research to improve our understanding of immune checkpoints, improve our knowledge of the immune system, and develop successful therapeutics to treat autoimmunity and cancer.
Funding
Research in the Zang lab is supported by NIH R01CA175495 and R01CA262132, the Department of Defense (PC210331 and BC190403), and the Price Family Foundation. M.C.P. is supported by NIH 5TL1TR002557. A.T.M. is supported by Scandinavia/Borge.
Competing interests
XZ is the scientific co-founder of NextPoint Therapeutics. The other authors declare no competing interests.
References
- 1.Ledford H, Else H, Warren M. Cancer immunologists scoop medicine Nobel prize. Nature. 2018;562:20–1. doi: 10.1038/d41586-018-06751-0. [DOI] [PubMed] [Google Scholar]
- 2.Zang X. 2018 Nobel prize in medicine awarded to cancer immunotherapy: Immune checkpoint blockade - a personal account. Genes Dis. 2018;5:302–3. doi: 10.1016/j.gendis.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao R, Chinai JM, Buhl S, Scandiuzzi L, Ray A, Jeon H, et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc Natl Acad Sci USA. 2013;110:9879–84. doi: 10.1073/pnas.1303524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: A widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci USA. 2003;100:10388–92. doi: 10.1073/pnas.1434299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janakiram M, Shah UA, Liu W, Zhao A, Schoenberg MP, Zang X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol Rev. 2017;276:26–39. doi: 10.1111/imr.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei Y, Ren X, Galbo PM, Jr, Moerdler S, Wang H, Sica RA, et al. KIR3DL3-HHLA2 is a human immunosuppressive pathway and a therapeutic target. Sci Immunol. 2021;6:eabf9792. doi: 10.1126/sciimmunol.abf9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.John P, Wei Y, Liu W, Du M, Guan F, Zang X. The B7x immune checkpoint pathway: From discovery to clinical trial. Trends Pharm Sci. 2019;40:883–96. doi: 10.1016/j.tips.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. 2016;22:3425–31. doi: 10.1158/1078-0432.CCR-15-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zang X. New immune checkpoint pathways: HHLA2 and its receptors including TMIGD2. Cold Spring Harbor Asia Conference on Precision Cancer Biology: From Targeted to Immune Therapies, Suzhou, China 18 to 22 September. 2017.
- 10.Janakiram M, Chinai JM, Fineberg S, Fiser A, Montagna C, Medavarapu R, et al. Expression, clinical significance, and receptor identification of the newest B7 family member HHLA2 protein. Clin Cancer Res. 2015;21:2359–66. doi: 10.1158/1078-0432.CCR-14-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatt RS, Berjis A, Konge JC, Mahoney KM, Klee AN, Freeman SS, et al. KIR3DL3 Is an inhibitory receptor for HHLA2 that mediates an alternative immunoinhibitory pathway to PD-1. Cancer. Immunol Res. 2021;9:156–69. doi: 10.1158/2326-6066.CIR-20-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng H, Janakiram M, Borczuk A, Lin J, Qiu W, Liu H, et al. HHLA2, a new immune checkpoint member of the B7 Family, is widely expressed in human lung cancer and associated with EGFR mutational status. Clin Cancer Res. 2017;23:825–32. doi: 10.1158/1078-0432.CCR-15-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng H, Borczuk A, Janakiram M, Ren X, Lin J, Assal A, et al. Wide expression and significance of alternative immune checkpoint molecules, B7x and HHLA2, in PD-L1-negative human lung cancers. Clin Cancer Res. 2018;24:1954–64. doi: 10.1158/1078-0432.CCR-17-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koirala P, Roth ME, Gill J, Chinai JM, Ewart MR, Piperdi S, et al. HHLA2, a member of the B7 family, is expressed in human osteosarcoma and is associated with metastases and worse survival. Sci Rep. 2016;6:31154. doi: 10.1038/srep31154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boor PPC, Sideras K, Biermann K, Hosein Aziz M, Levink IJM, Mancham S, et al. HHLA2 is expressed in pancreatic and ampullary cancers and increased expression is associated with better post-surgical prognosis. Br J Cancer. 2020;122:1211–8. doi: 10.1038/s41416-020-0755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Liu J, Zhang C, Li F, Li L, Wang D, et al. Over-expression and prognostic significance of HHLA2, a new immune checkpoint molecule, in human clear cell renal cell carcinoma. Front Cell Dev Biol. 2020;8:280. doi: 10.3389/fcell.2020.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan Z, Gardiner JC, Maggi EC, Huang S, Adem A, Bagdasarov S, et al. B7 immune-checkpoints as targets for the treatment of neuroendocrine tumors. Endocr Relat Cancer. 2021;28:135–49. doi: 10.1530/ERC-20-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Q, Li K, Lai Y, Yao K, Wang Q, Zhan X, et al. B7 score and T cell infiltration stratify immune status in prostate cancer. J Immunother Cancer. 2021;9:e002455. doi: 10.1136/jitc-2021-002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janakiram M, Chinai JM, Zhao A, Sparano JA, Zang X. HHLA2 and TMIGD2: New immunotherapeutic targets of the B7 and CD28 families. Oncoimmunology. 2015;4:e1026534. doi: 10.1080/2162402X.2015.1026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamb M, Wei Y, Ren X, O’Connor R, Dulak A, Rausch M, et al. NPX267, a first-in-class monoclonal antibody targeting KIR3DL3, blocks HHLA2-mediated immunosuppression and potentiates T and NK cell-mediated antitumor immunity. J Immunother Cancer. 2022;10:A510. [Google Scholar]
- 21.Zhu Y, Yao S, Iliopoulou BP, Han X, Augustine MM, Xu H, et al. B7-H5 costimulates human T cells via CD28H. Nat Commun. 2013;4:2043. doi: 10.1038/ncomms3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luu K, Schwarz H, Lundqvist A. B7-H7 is inducible on T cells to regulate their immune response and serves as a marker for exhaustion. Front Immunol. 2021;12:682627. doi: 10.3389/fimmu.2021.682627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen D, Chen W, Xu Y, Zhu M, Xiao Y, Shen Y, et al. Upregulated immune checkpoint HHLA2 in clear cell renal cell carcinoma: A novel prognostic biomarker and potential therapeutic target. J Med Genet. 2019;56:43–9. doi: 10.1136/jmedgenet-2018-105454. [DOI] [PubMed] [Google Scholar]
- 24.Farrag M, Ibrahim E, El-Hadidy T, Akl M, Elsergany A, Abdelwahab H. Human endogenous retrovirus-H long terminal repeat-associating protein 2 (HHLA2) is a novel immune checkpoint protein in lung cancer which predicts survival. Asian Pac J Cancer Prev. 2021;22:1883–9. doi: 10.31557/APJCP.2021.22.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jing CY, Fu YP, Yi Y, Zhang MX, Zheng SS, Huang JL, et al. HHLA2 in intrahepatic cholangiocarcinoma: An immune checkpoint with prognostic significance and wider expression compared with PD-L1. J Immunother Cancer. 2019;7:77. doi: 10.1186/s40425-019-0554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo M, Lin Y, Liang R, Li Y, Ge L. Clinical significance of the HHLA2 protein in hepatocellular carcinoma and the tumor microenvironment. J Inflamm Res. 2021;14:4217–28. doi: 10.2147/JIR.S324336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan H, Qiu W, Koehne de Gonzalez AK, Wei JS, Tu M, Xi CH, et al. HHLA2 is a novel immune checkpoint protein in pancreatic ductal adenocarcinoma and predicts post-surgical survival. Cancer Lett. 2019;442:333–40. doi: 10.1016/j.canlet.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahimi N, Rezazadeh K, Mahoney JE, Hartsough E, Meyer RD. Identification of IGPR-1 as a novel adhesion molecule involved in angiogenesis. Mol Biol Cell. 2012;23:1646–56. doi: 10.1091/mbc.e11-11-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crespo J, Vatan L, Maj T, Liu R, Kryczek I, Zou W. Phenotype and tissue distribution of CD28H+ immune cell subsets. OncoImmunology. 2017;6:e1362529. doi: 10.1080/2162402X.2017.1362529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuang X, Long EO. CD28 homolog is a strong activator of natural killer cells for lysis of B7-H7+ tumor cells. Cancer Immunol Res. 2019;7:939–51. doi: 10.1158/2326-6066.CIR-18-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leaton LA, Shortt J, Kichula KM, Tao S, Nemat-Gorgani N, Mentzer AJ, et al. Conservation, extensive heterozygosity, and convergence of signaling potential all indicate a critical role for KIR3DL3 in higher primates. Front Immunol. 2019;10:24. doi: 10.3389/fimmu.2019.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trundley AE, Hiby SE, Chang C, Sharkey AM, Santourlidis S, Uhrberg M, et al. Molecular characterization of KIR3DL3. Immunogenetics. 2006;57:904–16. doi: 10.1007/s00251-005-0060-7. [DOI] [PubMed] [Google Scholar]
- 33.Trompeter H-I, Gómez-Lozano N, Santourlidis S, Eisermann B, Wernet P, Vilches C, et al. Three structurally and functionally divergent kinds of promoters regulate expression of clonally distributed killer cell Ig-like receptors (KIR), of KIR2DL4, and of KIR3DL3. J Immunol. 2005;174:4135–43. doi: 10.4049/jimmunol.174.7.4135. [DOI] [PubMed] [Google Scholar]
- 34.Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101. doi: 10.1186/s40425-017-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nutalai R, Gaudieri S, Jumnainsong A, Leelayuwat C. Regulation of KIR3DL3 expression via miRNA. Genes. 2019;10:603. doi: 10.3390/genes10080603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang FX, Wu JW, Cheng XQ, Wang JH, Wen XZ, Li JJ, et al. HHLA2 predicts improved prognosis of anti-PD-1/PD-L1 immunotherapy in patients with melanoma. Front Immunol. 2022;13:902167. doi: 10.3389/fimmu.2022.902167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramaswamy M, Kim T, Jones DC, Ghadially H, Mahmoud TI, Garcia A, et al. Immunomodulation of T- and NK-cell responses by a bispecific antibody targeting CD28 homolog and PD-L1. Cancer Immunol Res. 2022;10:200–14. doi: 10.1158/2326-6066.CIR-21-0218. [DOI] [PubMed] [Google Scholar]
- 38.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–38. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36:257–64. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181:4832–9. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 42.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 43.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–92. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8 T cells. EMBO Mol Med. 2011;3:581–92. doi: 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–17. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46. doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33:3091–8. doi: 10.1128/MCB.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–33. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kew S, Mesa MD, Tricon S, Buckley R, Minihane AM, Yaqoob P. Effects of oils rich in eicosapentaenoic and docosahexaenoic acids on immune cell composition and function in healthy humans. Am J Clin Nutr. 2004;79:674–81. doi: 10.1093/ajcn/79.4.674. [DOI] [PubMed] [Google Scholar]
- 52.Verlengia R, Gorjao R, Kanunfre CC, Bordin S, Martins De Lima T, Martins EF, et al. Comparative effects of eicosapentaenoic acid and docosahexaenoic acid on proliferation, cytokine production, and pleiotropic gene expression in Jurkat cells. J Nutr Biochem. 2004;15:657–65. doi: 10.1016/j.jnutbio.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 53.Jaudszus A, Gruen M, Watzl B, Ness C, Roth A, Lochner A, et al. Evaluation of suppressive and pro-resolving effects of EPA and DHA in human primary monocytes and T-helper cells. J Lipid Res. 2013;54:923–35. doi: 10.1194/jlr.P031260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8 T cells in tumors. Immunity. 2021;54:1561–77 e7. doi: 10.1016/j.immuni.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tkachev V, Goodell S, Opipari AW, Hao LY, Franchi L, Glick GD, et al. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J Immunol. 2015;194:5789–800. doi: 10.4049/jimmunol.1402180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci USA. 2017;114:E761–70. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalia V, Yuzefpolskiy Y, Vegaraju A, Xiao H, Baumann F, Jatav S, et al. Metabolic regulation by PD-1 signaling promotes long-lived quiescent CD8 T cell memory in mice. Sci Transl Med. 2021;13:eaba6006. doi: 10.1126/scitranslmed.aba6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnnidis JB, Muroyama Y, Ngiow SF, Chen Z, Manne S, Cai Z, et al. Inhibitory signaling sustains a distinct early memory CD8 T cell precursor that is resistant to DNA damage. Sci Immunol. 2021;6:eabe3702. doi: 10.1126/sciimmunol.abe3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qorraj M, Bruns H, Bottcher M, Weigand L, Saul D, Mackensen A, et al. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia. 2017;31:470–8. doi: 10.1038/leu.2016.214. [DOI] [PubMed] [Google Scholar]
- 60.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol. 2020;5:eaay1863. doi: 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu Y, Tsang JC, Wang C, Clare S, Wang J, Chen X, et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature. 2016;539:102–6. doi: 10.1038/nature20105. [DOI] [PubMed] [Google Scholar]
- 62.Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, et al. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J Immunol. 2014;193:4477–84. doi: 10.4049/jimmunol.1401558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med. 2021;218:e20182232. doi: 10.1084/jem.20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci USA. 2019;116:9999–10008. doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 66.Park HJ, Park JS, Jeong YH, Son J, Ban YH, Lee BH, et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8 T cell immune response via the interaction with PD-L1 expressed on CD8 T cells. J Immunol. 2015;194:5801–11. doi: 10.4049/jimmunol.1401936. [DOI] [PubMed] [Google Scholar]
- 67.Stathopoulou C, Gangaplara A, Mallett G, Flomerfelt FA, Liniany LP, Knight D, et al. PD-1 inhibitory receptor downregulates asparaginyl endopeptidase and maintains Foxp3 transcription factor stability in induced regulatory T cells. Immunity. 2018;49:247–63.e7. doi: 10.1016/j.immuni.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim MJ, Kim K, Park HJ, Kim GR, Hong KH, Oh JH, et al. Deletion of PD-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat Immunol. 2023;24:148–61. doi: 10.1038/s41590-022-01373-1. [DOI] [PubMed] [Google Scholar]
- 69.Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21:298–308. doi: 10.1038/s41590-019-0589-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, et al. Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol. 2019;20:1208–19. doi: 10.1038/s41590-019-0442-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lim SA, Wei J, Nguyen TM, Shi H, Su W, Palacios G, et al. Lipid signalling enforces functional specialization of Treg cells in tumours. Nature. 2021;591:306–11. doi: 10.1038/s41586-021-03235-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu C, Fu Y, Liu S, Trittipo J, Lu X, Qi R, et al. BATF regulates T regulatory cell functional specification and fitness of triglyceride metabolism in restraining allergic responses. J Immunol. 2021;206:2088–100. doi: 10.4049/jimmunol.2001184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Itahashi K, Irie T, Yuda J, Kumagai S, Tanegashima T, Lin Y-T, et al. BATF epigenetically and transcriptionally controls the activation program of regulatory T cells in human tumors. Sci Immunol. 2022;7:eabk0957. doi: 10.1126/sciimmunol.abk0957. [DOI] [PubMed] [Google Scholar]
- 74.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–41. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang S, Li J, Xie J, Liu F, Duan Y, Wu Y, et al. Programmed death ligand 1 promotes lymph node metastasis and glucose metabolism in cervical cancer by activating integrin beta4/SNAI1/SIRT3 signaling pathway. Oncogene. 2018;37:4164–80. doi: 10.1038/s41388-018-0252-x. [DOI] [PubMed] [Google Scholar]
- 76.Kim S, Jang JY, Koh J, Kwon D, Kim YA, Paeng JC, et al. Programmed cell death ligand-1-mediated enhancement of hexokinase 2 expression is inversely related to T-cell effector gene expression in non-small-cell lung cancer. J Exp Clin Cancer Res. 2019;38:462. doi: 10.1186/s13046-019-1407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu Y, Liang Y, Li D, Wang L, Liang Z, Chen Y, et al. Glucose metabolism involved in PD-L1-mediated immune escape in the malignant kidney tumour microenvironment. Cell Death Disco. 2021;7:15. doi: 10.1038/s41420-021-00401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty acid oxidation controls CD8 tissue-resident memory T cell survival in gastric adenocarcinoma. Cancer Immunol Res. 2020;8:479–92. doi: 10.1158/2326-6066.CIR-19-0702. [DOI] [PubMed] [Google Scholar]
- 79.Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. Immunoregulatory protein B7-H3 reprograms glucose metabolism in cancer cells by ROS-mediated stabilization of HIF1-α. Cancer Res. 2016;76:2231–42. doi: 10.1158/0008-5472.CAN-15-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nunes-Xavier CE, Karlsen KF, Tekle C, Pedersen C, Øyjord T, Hongisto V, et al. Decreased expression of B7-H3 reduces the glycolytic capacity and sensitizes breast cancer cells to AKT/mTOR inhibitors. Oncotarget. 2016;7:6891–901. doi: 10.18632/oncotarget.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flem-Karlsen K, Tekle C, Andersson Y, Flatmark K, Fodstad Ø, Nunes-Xavier CE. Immunoregulatory protein B7-H3 promotes growth and decreases sensitivity to therapy in metastatic melanoma cells. Pigment Cell Melanoma Res. 2017;30:467–76. doi: 10.1111/pcmr.12599. [DOI] [PubMed] [Google Scholar]
- 82.Shi T, Ma Y, Cao L, Zhan S, Xu Y, Fu F, et al. B7-H3 promotes aerobic glycolysis and chemoresistance in colorectal cancer cells by regulating HK2. Cell Death Dis. 2019;10:308. doi: 10.1038/s41419-019-1549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Luo D, Xiao H, Dong J, Li Y, Feng G, Cui M, et al. B7-H3 regulates lipid metabolism of lung cancer through SREBP1-mediated expression of FASN. Biochem Biophys Res Commun. 2017;482:1246–51. doi: 10.1016/j.bbrc.2016.12.021. [DOI] [PubMed] [Google Scholar]
- 84.Wu J, Wang F, Liu X, Zhang T, Liu F, Ge X, et al. Correlation of IDH1 and B7-H3 expression with prognosis of CRC patients. Eur J Surg Oncol. 2018;44:1254–60. doi: 10.1016/j.ejso.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 85.Picarda E, Galbo PM, Jr., Zong H, Rajan MR, Wallenius V, Zheng D, et al. The immune checkpoint B7-H3 (CD276) regulates adipocyte progenitor metabolism and obesity development. Sci Adv. 2022;8:eabm7012. doi: 10.1126/sciadv.abm7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol. 2019;9:380. doi: 10.3389/fonc.2019.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bartish M, del Rincón SV, Rudd CE, Saragovi HU. Aiming for the sweet spot: Glyco-immune checkpoints and γδ T cells in targeted immunotherapy. Front Immunol. 2020;11:564499. doi: 10.3389/fimmu.2020.564499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Haas P, Hendriks WJAJ, Lefeber DJ, Cambi A. Biological and technical challenges in unraveling the role of N-glycans in immune receptor regulation. Front Chem. 2020;8:55. doi: 10.3389/fchem.2020.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Munkley J. The glycosylation landscape of pancreatic cancer. Oncol Lett. 2019;17:2569–75. doi: 10.3892/ol.2019.9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang C, Fang M, Feng H, Liu L, Li Y, Xu X, et al. N-glycan fingerprint predicts alpha-fetoprotein negative hepatocellular carcinoma: A large-scale multicenter study. Int J Cancer. 2021;149:717–27. doi: 10.1002/ijc.33564. [DOI] [PubMed] [Google Scholar]
- 91.Munkley J, Elliott DJ. Hallmarks of glycosylation in cancer. Oncotarget. 2016;7:35478–89. doi: 10.18632/oncotarget.8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346–66. doi: 10.1038/s41581-019-0129-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stanley P, Tanwar A. Regulation of myeloid and lymphoid cell development by O-glycans on Notch. Front Mol Biosci. 2022;9:979724. doi: 10.3389/fmolb.2022.979724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.An HJ, Froehlich JW, Lebrilla CB. Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr Opin Chem Biol. 2009;13:421–6. doi: 10.1016/j.cbpa.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ma M, Han G, Wang Y, Zhao Z, Guan F, Li X. Role of FUT8 expression in clinicopathology and patient survival for various malignant tumor types: A systematic review and meta-analysis. Aging. 2020;13:2212–30. doi: 10.18632/aging.202239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bastian K, Scott E, Elliott DJ, Munkley J. FUT8 Alpha-(1,6)-Fucosyltransferase in cancer. Int J Mol Sci. 2021;22:455. doi: 10.3390/ijms22010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Varki A. Biological roles of glycans. Glycobiology. 2017;27:3–49. doi: 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Y-N, Lee H-H, Hsu JL, Yu D, Hung M-C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J Biomed Sci. 2020;27:77. doi: 10.1186/s12929-020-00670-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun L, Li CW, Chung EM, Yang R, Kim YS, Park AH, et al. Targeting glycosylated PD-1 induces potent antitumor immunity. Cancer Res. 2020;80:2298–310. doi: 10.1158/0008-5472.CAN-19-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of core fucosylation reduces cell-surface expression of PD-1 and promotes anti-tumor immune responses of T cells. Cell Rep. 2017;20:1017–28. doi: 10.1016/j.celrep.2017.07.027. [DOI] [PubMed] [Google Scholar]
- 101.Tan S, Zhang H, Chai Y, Song H, Tong Z, Wang Q, et al. An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat Commun. 2017;8:14369. doi: 10.1038/ncomms14369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou S, Zhu J, Xu J, Gu B, Zhao Q, Luo C, et al. Anti‐tumour potential of PD‐L1/PD‐1 post‐translational modifications. Immunology. 2022;167:471–81. doi: 10.1111/imm.13573. [DOI] [PubMed] [Google Scholar]
- 103.Zhang N, Li M, Xu X, Zhang Y, Liu Y, Zhao M, et al. Loss of core fucosylation enhances the anticancer activity of cytotoxic T lymphocytes by increasing PD-1 degradation. Eur J Immunol. 2020;50:1820–33. doi: 10.1002/eji.202048543. [DOI] [PubMed] [Google Scholar]
- 104.Guzik K, Zak KM, Grudnik P, Magiera K, Musielak B, Törner R, et al. Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J Med Chem. 2017;60:5857–67. doi: 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- 105.Sun R, Kim AMJ, Lim SO. Glycosylation of immune receptors in cancer. Cells. 2021;10:1100. doi: 10.3390/cells10051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu K, Tan S, Jin W, Guan J, Wang Q, Sun H, et al. N-glycosylation of PD-1 promotes binding of camrelizumab. EMBO Rep. 2020;21:e51444. doi: 10.15252/embr.202051444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shi X, Zhang D, Li F, Zhang Z, Wang S, Xuan Y, et al. Targeting glycosylation of PD-1 to enhance CAR-T cell cytotoxicity. J Hematol Oncol. 2019;12:127. doi: 10.1186/s13045-019-0831-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li C-W, Lim S-O, Xia W, Lee H-H, Chan L-C, Kuo C-W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao L, Guan X, Xiang M, Wang Q, Long Q, Yue C, et al. B7 family protein glycosylation: promising novel targets in tumor treatment. Front Immunol. 2022;13:1088560. doi: 10.3389/fimmu.2022.1088560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201.e10. doi: 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71:606–20.e7. doi: 10.1016/j.molcel.2018.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. 2019;36:168–78.e4. doi: 10.1016/j.ccell.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ou-Yang F, Li CL, Chen CC, Shen YC, Moi SH, Luo CW, et al. De-glycosylated membrane PD-L1 in tumor tissues as a biomarker for responsiveness to atezolizumab (Tecentriq) in advanced breast cancer patients. Am J Cancer Res. 2022;12:123–37. [PMC free article] [PubMed] [Google Scholar]
- 114.Song X, Zhou Z, Li H, Xue Y, Lu X, Bahar I, et al. Pharmacologic suppression of B7-H4 glycosylation restores antitumor immunity in immune-cold breast cancers. Cancer Disco. 2020;10:1872–93. doi: 10.1158/2159-8290.CD-20-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rinis N, Golden JE, Marceau CD, Carette JE, Van Zandt MC, Gilmore R, et al. Editing N-glycan site occupancy with small-molecule oligosaccharyltransferase inhibitors. Cell Chem Biol. 2018;25:1231–41.e4. doi: 10.1016/j.chembiol.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen JT, Chen CH, Ku KL, Hsiao M, Chiang CP, Hsu TL, et al. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc Natl Acad Sci USA. 2015;112:13057–62. doi: 10.1073/pnas.1516991112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang Y, Zhang HL, Li ZL, Du T, Chen YH, Wang Y, et al. FUT8-mediated aberrant N-glycosylation of B7-H3 suppresses the immune response in triple-negative breast cancer. Nat Commun. 2021;12:2672. doi: 10.1038/s41467-021-22618-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nishimura CD, Pulanco MC, Cui W, Lu L, Zang X. PD-L1 and B7-1 cis-interaction: New mechanisms in immune checkpoints and immunotherapies. Trends Mol Med. 2021;27:207–19. doi: 10.1016/j.molmed.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Butte MJ, Pena-Cruz V, Kim MJ, Freeman GJ, Sharpe AH. Interaction of human PD-L1 and B7-1. Mol Immunol. 2008;45:3567–72. doi: 10.1016/j.molimm.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, Freeman GJ. PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol Res. 2018;6:921–9. doi: 10.1158/2326-6066.CIR-17-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 cis-Heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity. 2019;51:1059–73 e9. doi: 10.1016/j.immuni.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science. 2019;364:558–66. doi: 10.1126/science.aav7062. [DOI] [PubMed] [Google Scholar]
- 124.Garrett-Thomson SC, Massimi A, Fedorov EV, Bonanno JB, Scandiuzzi L, Hillerich B, et al. Mechanistic dissection of the PD-L1:B7-1 co-inhibitory immune complex. PLoS One. 2020;15:e0233578. doi: 10.1371/journal.pone.0233578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med. 2020;12:eaav7431. doi: 10.1126/scitranslmed.aav7431. [DOI] [PubMed] [Google Scholar]
- 126.Paterson AM, Brown KE, Keir ME, Vanguri VK, Riella LV, Chandraker A, et al. The programmed death-1 ligand 1:B7-1 pathway restrains diabetogenic effector T cells in vivo. J Immunol. 2011;187:1097–105. doi: 10.4049/jimmunol.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer. 2020;1:681–91. doi: 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 128.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA. 2005;102:11823–8. doi: 10.1073/pnas.0505497102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–62. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 132.Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. 2019;39:147–64. doi: 10.1200/EDBK_240837. [DOI] [PubMed] [Google Scholar]
- 133.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl J Med. 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2016;352:207–12. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl J Med. 2017;377:2500–1. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–7. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti–programmed cell death (PD)-1 and anti–programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–41. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gurjao C, Liu D, Hofree M, AlDubayan SH, Wakiro I, Su M-J, et al. Intrinsic resistance to immune checkpoint blockade in a mismatch repair–deficient colorectal cancer. Cancer Immunol Res. 2019;7:1230–6. doi: 10.1158/2326-6066.CIR-18-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Johnson DB, Estrada VM, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. 2018;10:eaar3342. doi: 10.1126/scitranslmed.aar3342. [DOI] [PubMed] [Google Scholar]
- 145.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD-1 blockade in patients with metastatic melanoma. Nat Med. 2019;25:1916–27. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Disco. 2017;7:1420–35. doi: 10.1158/2159-8290.CD-17-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in tumor progression and regression: a review. Biomark Res. 2020;8:1–16. doi: 10.1186/s40364-020-00228-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Disco. 2016;6:827–37. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Higgs BW, Morehouse CA, Streicher K, Brohawn PZ, Pilataxi F, Gupta A, et al. Interferon gamma messenger RNA-signature in tumor biopsies predicts outcomes in patients with non–small cell lung carcinoma or urothelial cancer treated with durvalumab. Clin Cancer Res. 2018;24:3857–66. doi: 10.1158/1078-0432.CCR-17-3451. [DOI] [PubMed] [Google Scholar]
- 151.Gide TN, Wilmott JS, Scolyer RA, Long VG. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res. 2018;24:1260–70. doi: 10.1158/1078-0432.CCR-17-2267. [DOI] [PubMed] [Google Scholar]
- 152.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Disco. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lopez de Rodas M, Nagineni V, Ravi A, Datar IJ, Mino-Kenudson M, Corredor G, et al. Role of tumor infiltrating lymphocytes and spatial immune heterogeneity in sensitivity to PD-1 axis blockers in non-small cell lung cancer. J Immunother Cancer. 2022;10:e004440. doi: 10.1136/jitc-2021-004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Disco. 2018;8:822–35. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Disco. 2016;6:202–16. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity. 2017;46:197–204. doi: 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol. 2017;30:1666–76. doi: 10.1038/modpathol.2017.89. [DOI] [PubMed] [Google Scholar]
- 160.Ott PA, Stephen Hodi F, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: An overview of rationale, preclinical evidence, and initial clinical data. Front Oncol. 2015;5:1–7. doi: 10.3389/fonc.2015.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–42. doi: 10.1158/2326-6066.CIR-14-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:1–8. doi: 10.1038/ncomms12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhou J, Mahoney KM, Giobbie-Hurder A, Zhao F, Lee S, Liao X, et al. Soluble PD-L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunol Res. 2017;5:480–92. doi: 10.1158/2326-6066.CIR-16-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Oh SY, Kim S, Keam B, Kim TM, Kim DW, Heo DS. Soluble PD-L1 is a predictive and prognostic biomarker in advanced cancer patients who receive immune checkpoint blockade treatment. Sci Rep. 2021;11:19712. doi: 10.1038/s41598-021-99311-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mahoney KM, Ross-Macdonald P, Yuan L, Song L, Veras E, Wind-Rotolo M, et al. Soluble PD-L1 as an early marker of progressive disease on nivolumab. J Immunother Cancer. 2022;10:e003527. doi: 10.1136/jitc-2021-003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non–small cell lung cancer. J Exp Med. 2019;216:982–1000. doi: 10.1084/jem.20180870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sagawa R, Sakata S, Gong B, Seto Y, Takemoto A, Takagi S, et al. Soluble PD-L1 works as a decoy in lung cancer immunotherapy via alternative polyadenylation. JCI Insight. 2022;7:e153323. doi: 10.1172/jci.insight.153323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Del ReM, Marconcini R, Pasquini G, Rofi E, Vivaldi C, Bloise F, et al. PD-L1 mRNA expression in plasma-derived exosomes is associated with response to anti-PD-1 antibodies in melanoma and NSCLC. Br J Cancer. 2018;118:820–4. doi: 10.1038/bjc.2018.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cordonnier M, Nardin C, Chanteloup G, Derangere V, Algros MP, Arnould L, et al. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J Extracell Vesicles. 2020;9:1–11. doi: 10.1080/20013078.2019.1710899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang C, Fan Y, Che X, Zhang M, Li Z, Li C, et al. Anti-PD-1 therapy response predicted by the combination of exosomal PD-L1 and CD28. Front Oncol. 2020;10:760. doi: 10.3389/fonc.2020.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, et al. MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res. 2017;77:4697–709. doi: 10.1158/0008-5472.CAN-17-0395. [DOI] [PubMed] [Google Scholar]
- 174.Wang L, Saci A, Szabo PM, Chasalow SD, Castillo-Martin M, Domingo-Domenech J, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun. 2018;9:3503. doi: 10.1038/s41467-018-05992-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, et al. Transcriptional downregulation of MHC class I and melanoma de-differentiation in resistance to PD-1 inhibition. Nat Commun. 2020;11:1897. doi: 10.1038/s41467-020-15726-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:845–65.e7. doi: 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 177.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGF-β attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD-1 efficacy. Sci Transl Med. 2014;6:237ra67. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.John P, Pulanco MC, Galbo PM, Jr, Wei Y, Ohaegbulam KC, Zheng D, et al. The immune checkpoint B7x expands tumor-infiltrating Tregs and promotes resistance to anti-CTLA-4 therapy. Nat Commun. 2022;13:2506. doi: 10.1038/s41467-022-30143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4:194–203. doi: 10.1158/2326-6066.CIR-15-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti–PD-1 therapy in human melanoma. J Clin Invest. 2016;126:3447–52. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–8. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63:675–83. doi: 10.1007/s00262-014-1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Martens A, Wistuba-Hamprecht K, Foppen MG, Yuan J, Postow MA, Wong P, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22:2908–18. doi: 10.1158/1078-0432.CCR-15-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63:247–57. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res. 2015;21:5453–9. doi: 10.1158/1078-0432.CCR-15-0676. [DOI] [PubMed] [Google Scholar]
- 190.Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical significance of circulating CD33+ CD11b+ HLA-DR- myeloid cells in patients with stage IV melanoma treated with ipilimumab. Clin Cancer Res. 2016;22:5661–72. doi: 10.1158/1078-0432.CCR-15-3104. [DOI] [PubMed] [Google Scholar]
- 191.Li L, Lu G, Liu Y, Gong L, Zheng X, Zheng H, et al. Low infiltration of CD8 PD-L1+ T cells and M2 macrophages predicts improved clinical outcomes after immune checkpoint inhibitor therapy in non-small cell lung carcinoma. Front Oncol. 2021;11:658690. doi: 10.3389/fonc.2021.658690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas a phase 2 clinical trial. JAMA Oncol. 2018;4:93–7. doi: 10.1001/jamaoncol.2017.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2015;37:457–95. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 194.Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res. 2015;3:1344–54. doi: 10.1158/2326-6066.CIR-15-0097. [DOI] [PubMed] [Google Scholar]
- 195.Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD-1 therapy by TIM-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. 2017;6:e1261779. doi: 10.1080/2162402X.2016.1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23:551–5. doi: 10.1038/nm.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non–small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res. 2019;25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:1–9. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EHC, et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. 2021;596:126–32. doi: 10.1038/s41586-021-03752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 201.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets VT, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28:1368–79. doi: 10.1093/annonc/mdx108. [DOI] [PubMed] [Google Scholar]
- 203.Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. 2018;29:1437–44. doi: 10.1093/annonc/mdy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–8. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Li X, Zhang S, Guo G, Han J, Yu J. Gut microbiome in modulating immune checkpoint inhibitors. EBioMedicine. 2022;82:104163. doi: 10.1016/j.ebiom.2022.104163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xiong D, Wang Y, You M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat Commun. 2020;11:1–12. doi: 10.1038/s41467-020-18546-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Du K, Wei S, Wei Z, Frederick DT, Miao B, Moll T, et al. Pathway signatures derived from on-treatment tumor specimens predict response to anti-PD-1 blockade in metastatic melanoma. Nat Commun. 2021;12:1–16. doi: 10.1038/s41467-021-26299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with Nivolumab. Cell. 2017;171:934–49.e15. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun. 2017;8:1–12. doi: 10.1038/s41467-017-01018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017;9:eaah3560. doi: 10.1126/scitranslmed.aah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984–97.e24. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24:1545–9. doi: 10.1038/s41591-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Carter JA, Gilbo P, Atwal GS. IMPRES does not reproducibly predict response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2019;25:1833–5. doi: 10.1038/s41591-019-0671-4. [DOI] [PubMed] [Google Scholar]
- 214.Xiao X, Xu C, Yang W, Yu R. Inconsistent prediction capability of ImmuneCells.Sig across different RNA-seq datasets. Nat Commun. 2021;12:4167. doi: 10.1038/s41467-021-24303-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti–B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. 2012;18:3834–45. doi: 10.1158/1078-0432.CCR-12-0715. [DOI] [PubMed] [Google Scholar]
- 216.Powderly J, Cote G, Flaherty K, Szmulewitz RZ, Ribas A, Weber J, et al. Interim results of an ongoing Phase I, dose escalation study of MGA271 (Fc-optimized humanized anti-B7-H3 monoclonal antibody) in patients with refractory B7-H3-expressing neoplasms or neoplasms whose vasculature expresses B7-H3. J Immunother Cancer. 2015;3:O8. doi: 10.1186/2051-1426-3-S2-O8. [DOI] [Google Scholar]
- 217.Shenderov E, Marzo AMD, Lotan TL, Wang H, Lim SJ, Allaf ME, et al. Targeting B7-H3 in prostate cancer: Phase 2 trial in localized prostate cancer using the anti-B7-H3 antibody enoblituzumab, with biomarker correlatives. J Clin Oncol. 2022;40:5015. doi: 10.1200/JCO.2022.40.16_suppl.5015. [DOI] [Google Scholar]
- 218.Aggarwal C, Prawira A, Antonia S, Rahma O, Tolcher A, Cohen RB, et al. Dual checkpoint targeting of B7-H3 and PD-1 with enoblituzumab and pembrolizumab in advanced solid tumors: Interim results from a multicenter phase I/II trial. J Immunother Cancer. 2022;10:e004424. doi: 10.1136/jitc-2021-004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Tomlinson U, Wolden SL, et al. A curative approach to central nervous system metastases of neuroblastoma. J Clin Oncol. 2017;35:10545. doi: 10.1200/JCO.2017.35.15_suppl.10545. [DOI] [Google Scholar]
- 220.Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, et al. Compartmental intrathecal radioimmunotherapy: Results for treatment for metastatic CNS neuroblastoma. J Neurooncol. 2010;97:409–18. doi: 10.1007/s11060-009-0038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kramer K, Pandit-Taskar N, Zanzonico P, Wolden SL, Humm JL, DeSelm C, et al. Low incidence of radionecrosis in children treated with conventional radiation therapy and intrathecal radioimmunotherapy. J Neurooncol. 2015;123:245–9. doi: 10.1007/s11060-015-1788-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.De B, Kinnaman MD, Wexler LH, Kramer K, Wolden SL. Central nervous system relapse of rhabdomyosarcoma. Pediatr Blood Cancer. 2018;65. 10.1002/pbc.26710. [DOI] [PMC free article] [PubMed]
- 223.Modak S, Zanzonico P, Grkovski M, Slotkin EK, Carrasquillo JA, Lyashchenko SK, et al. B7-H3-directed intraperitoneal radioimmunotherapy with radioiodinated omburtamab for desmoplastic small round cell tumor and other peritoneal tumors: Results of a phase I study. J Clin Oncol. 2020;38:4283–91. doi: 10.1200/JCO.20.01974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Souweidane MM, Kramer K, Pandit-Taskar N, Haque S, Zanzonico P, Carrasquillo JA, et al. Phase 1 dose-escalation trial using convection-enhanced delivery of radiolabeled monoclonal antibody for diffuse intrinsic pontine glioma following external radiation therapy. J Clin Oncol. 2021;39:2010. doi: 10.1200/JCO.2021.39.15_suppl.2010. [DOI] [Google Scholar]
- 225.Souweidane MM, Kramer K, Pandit-Taskar N, Zhou Z, Haque S, Zanzonico P, et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: A single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018;19:1040–50. doi: 10.1016/S1470-2045(18)30322-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Patel MR, Johnson ML, Falchook GS, Doi T, Friedman CF, Piha-Paul SA, et al. DS-7300 (B7-H3 DXd-ADC) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): A subgroup analysis of a phase 1/2 multicenter study. J Clin Oncol. 2022;40:87. doi: 10.1200/JCO.2022.40.6_suppl.087. [DOI] [Google Scholar]
- 227.Jang S, Powderly JD, Spira AI, Bakkacha O, Loo D, Bohac GC, et al. Phase 1 dose escalation study of MGC018, an anti-B7-H3 antibody-drug conjugate (ADC), in patients with advanced solid tumors. J Clin Oncol. 2021;39:2631. doi: 10.1200/JCO.2021.39.15_suppl.2631. [DOI] [Google Scholar]
- 228.Shenderov E, Mallesara GHG, Wysocki PJ, Xu W, Ramlau R, Weickhardt AJ, et al. MGC018, an anti-B7-H3 antibody-drug conjugate (ADC), in patients with advanced solid tumors: Preliminary results of phase I cohort expansion. Ann Oncol. 2021;32:S657–9. doi: 10.1016/j.annonc.2021.08.1133. [DOI] [Google Scholar]
- 229.You G, Lee Y, Kang Y-W, Park HW, Park K, Kim H, et al. B7-H3 × 4-1BB bispecific antibody augments antitumor immunity by enhancing terminally differentiated CD8 tumor-infiltrating lymphocytes. Sci Adv. 2021;7:eaax3160. doi: 10.1126/sciadv.aax3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Xu Y, Xiao Y, Luo C, Liu Q, Wei A, Yang Y, et al. Blocking PD-1/PD-L1 by an ADCC enhanced anti-B7-H3/PD-1 fusion protein engages immune activation and cytotoxicity. Int Immunopharmacol. 2020;84:106584. doi: 10.1016/j.intimp.2020.106584. [DOI] [PubMed] [Google Scholar]
- 231.Liu J, Yang S, Cao B, Zhou G, Zhang F, Wang Y, et al. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J Hematol Oncol. 2021;14:21. doi: 10.1186/s13045-020-01024-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Feng Y, Xie K, Yin Y, Li B, Pi C, Xu X, et al. A novel anti-B7-H3 × anti-CD3 bispecific antibody with potent antitumor activity. Life. 2022;12:157. doi: 10.3390/life12020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Vallera DA, Ferrone S, Kodal B, Hinderlie P, Bendzick L, Ettestad B, et al. NK cell-mediated targeting of various solid tumors using a B7-H3 tri-specific killer engager in vitro and in vivo. Cancers. 2020;12:2659. doi: 10.3390/cancers12092659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tang X, Liu F, Liu Z, Cao Y, Zhang Z, Wang Y, et al. Bioactivity and safety of B7-H3-targeted chimeric antigen receptor T cells against anaplastic meningioma. Clin Transl Immunol. 2020;9:e1137. doi: 10.1002/cti2.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tang X, Wang Y, Huang J, Zhang Z, Liu F, Xu J, et al. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct Target Ther. 2021;6:125. doi: 10.1038/s41392-021-00505-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hu G, Liang Y, Li G, Ding W. luo m. B7-H3 CAR-T therapy in relation to tumor growth in skin tumor. J Clin Oncol. 2022;40:e21502-e. doi: 10.1200/JCO.2022.40.16_suppl.e21502. [DOI] [Google Scholar]
- 237.Pinto NR, Albert CM, Taylor M, Wilson A, Rawlings-Rhea S, Huang W, et al. STRIVE-02: A first-in-human phase 1 trial of systemic B7-H3 CAR T cells for children and young adults with relapsed/refractory solid tumors. J Clin Oncol. 2022;40:10011. doi: 10.1200/JCO.2022.40.16_suppl.10011. [DOI] [Google Scholar]
- 238.Sachdev JC, Bauer TM, Chawla SP, Pant S, Patnaik A, Wainberg ZA, et al. Phase 1a/1b study of first-in-class B7-H4 antibody, FPA150, as monotherapy in patients with advanced solid tumors. J Clin Oncol. 2019;37:2529. doi: 10.1200/JCO.2019.37.15_suppl.2529. [DOI] [Google Scholar]
- 239.Hellmann MD, Bivi N, Calderon B, Shimizu T, Delafontaine B, Liu ZT, et al. Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clin Cancer Res. 2021;27:2773–81. doi: 10.1158/1078-0432.CCR-20-3716. [DOI] [PubMed] [Google Scholar]
- 240.Guo Y, Liu B, Lv D, Cheng Y, Zhou T, Zhong Y, et al. Phase I/IIa study of PM8001, a bifunctional fusion protein targeting PD-L1 and TGF-β, in patients with advanced tumors. J Clin Oncol. 2022;40:2512. doi: 10.1200/JCO.2022.40.16_suppl.2512. [DOI] [Google Scholar]
- 241.Yap T, Wong D, Hu-Lieskovan S, Papadopoulos K, Morrow M, Grabowska U, et al. A first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1, in patients with advanced cancer and resistance to PD-L1 therapy. J Immunother Cancer. 2020;8:A240–A. doi: 10.1158/1078-0432.CCR-22-1449. [DOI] [PubMed] [Google Scholar]
- 242.Gong J, Shen L, Hou J, Chen X, Yu Q, Zheng Y, et al. Safety results of Q-1802, a Claudin18.2/PD-L1 bsABs, in patients with relapsed or refractory solid tumors in a phase 1 study. J Clin Oncol. 2022;40:2568. doi: 10.1200/JCO.2022.40.16_suppl.2568. [DOI] [Google Scholar]
- 243.Wang J, Sun Y, Chu Q, Duan J, Wan R, Wang Z, et al. Phase I study of IBI322 (anti-CD47/PD-L1 bispecific antibody) monotherapy therapy in patients with advanced solid tumors in China. Cancer Res. 2022;82:CT513. doi: 10.1158/1538-7445.AM2022-CT513. [DOI] [Google Scholar]
- 244.Sanborn RE, Bordoni RE, Fleming GF, Khasraw M, Hawthorne T, Thomas LJ, et al. A phase 1 dose-escalation study of a PD-L1 x CD27 bispecific antibody CDX-527 in patients with advanced malignancies. J Clin Oncol. 2021;39:2585. doi: 10.1200/JCO.2021.39.15_suppl.2585. [DOI] [Google Scholar]
- 245.Prenen H, Kyi C, Van Lancker G, Patel SP, Mittag D, Weaver A, et al. Phase I dose escalation study of MCLA-145, a bispecific antibody targeting CD137 and PD-L1 in solid tumors. Ann Oncol. 2021;32:S1436. doi: 10.1016/j.annonc.2021.10.155. [DOI] [Google Scholar]
- 246.Garralda E, Geva R, Ben-Ami E, Maurice-Dror C, Calvo E, LoRusso P, et al. First-in-human phase I/IIa trial to evaluate the safety and initial clinical activity of DuoBody®-PD-L1×4–1BB (GEN1046) in patients with advanced solid tumors. J Immunother Cancer. 2020;8:A250–A1. [Google Scholar]
- 247.Xu R-H, Zhao H, Wei X-L, Zhang Y, Wang F, Wang Z, et al. Phase Ia dose escalation of IBI318, a first-in-class bispecific anti-PD-1/PD-L1, in patients with advanced tumors. J Clin Oncol. 2020;38:3062. doi: 10.1200/JCO.2020.38.15_suppl.3062. [DOI] [Google Scholar]
- 248.Coward J, Frentzas S, Mislang A, Gao B, Lemech C, Jin X, et al. Efficacy and safety of AK112, an anti-PD-1/VEGF-A bispecific antibody, in patients with platinum-resistant/refractory epithelial ovarian cancer in a Phase 1 study. J Immunother Cancer. 2021;9:A457–A. doi: 10.1136/jitc-2021-SITC2021.427. [DOI] [Google Scholar]
- 249.Zhao Y, Fang W, Yang Y, Chen J, Zhuang L, Du Y, et al. A phase II study of AK112 (PD-1/VEGF bispecific) in combination with chemotherapy in patients with advanced non-small cell lung cancer. J Clin Oncol. 2022;40:9019. doi: 10.1200/JCO.2022.40.16_suppl.9019. [DOI] [Google Scholar]
- 250.Wu X, Ji J, Lou H, Li Y, Feng M, Xu N, et al. Efficacy and safety of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, in previously treated recurrent or metastatic (R/M) cervical cancer: A multicenter, open-label, single-arm, phase II trial. Gynecologic Oncol. 2022;166:S47–S8. doi: 10.1016/S0090-8258(22)01293-8. [DOI] [Google Scholar]
- 251.Wang J, Lou H, Cai H-B, Huang X, Li G, Wang L, et al. A study of AK104 (an anti-PD-1 and anti-CTLA-4 bispecific antibody) combined with standard therapy for the first-line treatment of persistent, recurrent, or metastatic cervical cancer (R/M CC) J Clin Oncol. 2022;40:106. doi: 10.1200/JCO.2022.40.16_suppl.106. [DOI] [Google Scholar]
- 252.Millward M, Frentzas S, Gan HK, Prawira A, Tran B, Coward J, et al. Safety and antitumor activity of AK104, a bispecific antibody targeting PD-1 and CTLA-4, in patients with mesothelioma which is relapsed or refractory to standard therapies. Ann Oncol. 2020;31:S705–S6. doi: 10.1016/j.annonc.2020.08.1141. [DOI] [Google Scholar]
- 253.Mai H, Lin S, Chen D, Chen X, Qu S, Lin Q, et al. A phase II study of AK104, a bispecific antibody targeting PD-1 and CTLA-4, in patients with metastatic nasopharyngeal carcinoma (NPC) who had progressed after two or more lines of chemotherapy. J Immunother Cancer. 2021;9:A466–A. doi: 10.1136/jitc-2021-SITC2021.436. [DOI] [Google Scholar]
- 254.Ji J, Shen L, Li Z, Xu N, Liu T, Chen Y, et al. AK104 (PD-1/CTLA-4 bispecific) combined with chemotherapy as first-line therapy for advanced gastric (G) or gastroesophageal junction (GEJ) cancer: Updated results from a phase Ib study. J Clin Oncol. 2021;39:232. doi: 10.1200/JCO.2021.39.3_suppl.232. [DOI] [Google Scholar]
- 255.Shum E, Reilley M, Najjar Y, Daud A, Thompson J, Baranda J, et al. Preliminary clinical experience with XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in patients with advanced solid tumors. J Immunother Cancer. 2021;9:A553–A. doi: 10.1136/jitc-2021-SITC2021.523. [DOI] [Google Scholar]
- 256.Hickingbottom B, Clynes R, Desjarlais J, Li C, Ding Y. Preliminary safety and pharmacodynamic (PD) activity of XmAb20717, a PD-1 x CTLA-4 bispecific antibody, in a phase I dose escalation study of patients with selected advanced solid tumors. J Clin Oncol. 2020;38:e15001-e. doi: 10.1200/JCO.2020.38.15_suppl.e15001. [DOI] [Google Scholar]
- 257.Albiges L, Rodriguez LM, Kim S-W, Im S-A, Carcereny E, Rha SY, et al. Safety and clinical activity of MEDI5752, a PD-1/CTLA-4 bispecific checkpoint inhibitor, as monotherapy in patients (pts) with advanced renal cell carcinoma (RCC): Preliminary results from an FTIH trial. J Clin Oncol. 2022;40:107. doi: 10.1200/JCO.2022.40.16_suppl.107. [DOI] [Google Scholar]
- 258.Gong J, Shen L, Dong Z, Liu D, Xu J, Yang J, et al. Preliminary safety, tolerability and efficacy results of KN026 in combination with KN046 in patients with HER2 aberrated solid tumors. J Immunother Cancer. 2020;8:A485–A6. [Google Scholar]
- 259.Gong J, Dong Z, Liu D, Xu J, Yang J, Yang Y, et al. Preliminary safety, tolerability and efficacy results of KN026 (a HER2-targeted Bispecific Antibody) in combination with KN046 (an anti-PD-L1/CTLA-4 Bispecific Antibody) in patients (pts) with HER2 aberrated solid tumors. J Immunother Cancer. 2020;8:A207–A. [Google Scholar]
- 260.Xing B, Da X, Zhang Y, Ma Y. A phase II study combining KN046 (an anti-PD-L1/CTLA-4 bispecific antibody) and lenvatinib in the treatment for advanced unresectable or metastatic hepatocellular carcinoma (HCC): updated efficacy and safety results. J Clin Oncol. 2022;40:4115. doi: 10.1200/JCO.2022.40.16_suppl.4115. [DOI] [Google Scholar]
- 261.Yachnin J, Ullenhag GJ, Carneiro A, Nielsen D, Rohrberg KS, Kvarnhammar AM, et al. A first-in-human phase I study in patients with advanced and/or refractory solid malignancies to evaluate the safety of ATOR-1015, a CTLA-4 x OX40 bispecific antibody. J Clin Oncol. 2020;38:3061. doi: 10.1200/JCO.2020.38.15_suppl.3061. [DOI] [Google Scholar]