

Abstract
Background:
The relationship between psychotic disorders and cannabis use is much debated. Shared underlying genetic risk is one potential explanation. Here, we investigate the genetic association between psychotic disorders [schizophrenia (SCZ) and bipolar disorder (BIP)] and cannabis phenotypes [lifetime cannabis use (LCU) and cannabis use disorder (CUD)].
Methods:
We estimated heritability, polygenicity, and discoverability of each phenotype. We additionally performed genome-wide and local genetic correlations (rg). Shared loci were identified and mapped to genes which were then tested for functional enrichment. Shared genetic liabilities to psychotic disorders and cannabis phenotypes were explored using causal analyses and polygenic scores.
Findings:
Psychotic disorders were more heritable than cannabis phenotypes and more polygenic than CUD. We observed positive genome-wide rgs across the psychotic-cannabis domains (range=0.22–0.35) with a mixture of positive and negative local rgs. A range of 3 to 27 shared loci were identified for psychotic-cannabis phenotype pairs. Enrichment of mapped genes implicated neuronal and olfactory cells as well as drug gene-targets for nicotine, alcohol, and duloxetine. Psychotic disorders exhibited a causal effect on cannabis phenotypes and a causal effect of LCU on BIP was observed. Polygenic scores for cannabis phenotypes predicted psychotic disorders independently and improved prediction beyond the psychotic disorder’s polygenic score.
Interpretation:
A subgroup of individuals may have a high genetic risk of developing SCZ, BIP, and using cannabis. This supports public health efforts to reduce cannabis use particularly in this patient group or high-risk individuals. Identified shared loci and their functional implications may facilitate development of novel treatments.
Introduction
Cannabis is among the most widely used substances globally. The prevalence of lifetime cannabis use (LCU) is estimated at 27.2% in the European Union.1 Among regular cannabis users, approximately 10% develop cannabis use disorder (CUD),2 defined as a problematic pattern of use resulting in clinically significant impairment.3 Cannabis use has been linked to disorders with psychotic symptoms, including schizophrenia (SCZ), with psychosis as a defining feature, and bipolar disorder (BIP), with an estimated prevalence of psychosis at 73.8%.4 Compared with the general population, persons who reported using cannabis suffer a higher risk and an earlier onset of psychotic disorders (i.e., SCZ and BIP), alongside more severe symptoms and longer hospitalizations.5–8 LCU is less strictly defined than CUD but is linked to adverse outcomes and is genetically associated with other substance use phenotypes and disorders.9 However, the nature of this connection between psychotic disorders and cannabis use has been the topic of much debate within the field of psychiatry and beyond.
While there are a variety of reasons for the observed relationship between psychotic disorders and cannabis phenotypes (i.e., LCU and CUD), including shared environmental risk, mutual genetic risk is plausible. SCZ, BIP, LCU, and CUD are partly heritable (heritability range is 0.50–0.80)10–12 and emerging evidence has suggested a shared genetic component that increases the likelihood of both developing psychotic disorders and using cannabis. For instance, a modest positive genome-wide genetic correlation (rg, ranging from 0.17 to 0.31) has been reported between psychotic disorders and cannabis phenotypes,9,13 indicating genetic overlap. Although, more detailed genetic and mechanistic insights remain elusive.
The bidirectional causal relationship between psychotic disorders and cannabis phenotypes is also often debated. A common hypothesis is that cannabis is a risk factor in the development of psychotic disorders,14 whereas a reverse causality hypothesis posits that psychotic disorders lead to cannabis use as a potential way to alleviate symptoms.14,15 Both causation and reverse causation are not mutually exclusive and have been assessed using mendelian randomization (MR), a statistical framework to test causal associations using genetic liability to the phenotypes of interest.16 For example, a bidirectional causal relationship has been suggested between LCU and SCZ.7,9,17 The accumulation of larger genome-wide association study (GWAS) datasets provides opportunities to improve assessment of causal relationships using MR.
Additional support for shared genetic liability comes from polygenic score (PGS) studies. A PGS is calculated as a weighted sum of phenotype-associated alleles and represents individual level genetic liability to a phenotype. Previous studies found the PGS for SCZ is positively associated with cannabis use18 and modulates the link between cannabis use and psychosis19 but one study found no link with CUD.20 Recent studies have shown that for a given phenotype, genetically correlated phenotypes may improve the prediction of the target phenotype by using their joint predictive power.21 Yet, to our knowledge, little is known about the potential to improve the prediction efficiency of psychotic disorders using joint genetic liability of psychotic disorders and cannabis phenotypes.
In the present study, we investigated the genetic foundations underlying the epidemiological associations between psychotic disorders and cannabis phenotypes, using statistical genetic approaches and the largest GWAS. We aimed to: (1) examine the genetic architecture of each phenotype, (2) estimate genetic overlap by investigating (a) genome-wide and local rgs, (b) specific shared genetic loci, and (c) putative biological mechanisms; (3) re-evaluate the causal and reverse causal hypotheses leveraging MR; and (4) improve the prediction of SCZ and BIP by integrating the genetic liability to psychotic disorders and cannabis phenotypes.
Methods
Genome-Wide Association Study (GWAS) Data
GWAS summary statistics on SCZ, BIP, LCU, and CUD were used in our discovery analyses.9,13,22,23 Details are provided in Supplementary Methods. Validation of SNP effect directions was conducted using summary statistics from independent samples for SCZ24 and BIP.25
Establishing Genetic Architecture using MiXeR
MiXeR v1.326 was used to estimate each phenotype’s heritability, polygenicity, and discoverability (Supplementary Methods). Briefly, MiXeR uses GWAS summary statistics to model additive genetic effects on a phenotype. Polygenicity is estimated as the number of trait-influencing variants expected to explain 90% of heritability. Discoverability is the average magnitude of additive genetic effects among trait-influencing variants. MiXeR estimates heritability as a function of the product of polygenicity and discoverability.
Genetic Correlations (rg)
To estimate a rg for each pair of phenotypes, we used linkage disequilibrium score regression (LDSR)27 and local analysis of covariant annotation (LAVA).28 LDSR is a method for estimating a rg at a genome-wide level. LAVA estimates rgs at a “local” level within 2,495 genomic regions. We used the default heritability thresholds for LAVA (p=0.05). The Benjamini-Hochberg correction (q<0.05) was applied.
Conjunctional False Discovery Rate (conjFDR)
To determine polygenic enrichment between pairs of phenotypes, we used conditional quantile-quantile plots (Supplementary Figure 1), which show the distribution of p-values for one phenotype conditioning on p-value cut-offs of another phenotype (p<0.1, p<0.01, p<0.001). Four complex LD regions (Supplementary Methods) were excluded from analysis to avoid potential inflation. Identification of shared loci between pairs of phenotypes was estimated using a conjunctional FDR (conjFDR) analysis.29 This method relies on two runs of a conditional FDR (condFDR) analysis. First, the association between variants and a secondary phenotype is used to re-rank the test statistic in the primary phenotype. The process is then repeated switching the roles of the primary and secondary phenotypes. The largest condFDR value between the two runs is then used as the conjFDR value. A SNP with a conjFDR<0.05 was considered as a shared SNP.30–32 Details for conjFDR, locus definition, lead SNP identification, and SNP sign tests are provided in Supplementary Methods.
Gene Mapping and Enrichment Analyses
All shared loci were then mapped to genes via FUMA (Supplementary Methods).33 For each psychotic disorder, the genes shared with LCU or CUD, located outside of the four complex LD regions, were combined for enrichment analyses. Enrichment analysis for Gene Ontology, KEGG pathways, cell types, and drug-gene interactions were performed (Supplementary Methods).
Mendelian Randomization (MR)
To estimate the potential causal relationship between psychotic disorders and cannabis phenotypes, we used MR (Supplementary Methods). We used the R package TwoSampleMR34 and reported results for three methods (i) inverse variance weighted [IVW],35 (ii) weighted median,36 and (iii) MR Egger.37 We also used MR Pleiotropy Residual Sum and Outlier (MR-PRESSO)38 and Causal Analysis Using Summary Effect estimates (CAUSE).39 Latent Causal Variable (LCV)40 analysis was also applied. The Benjamini-Hochberg correction (q<0.05) was applied across all MR analyses.
Polygenic Score (PGS) Calculation
Participants
The Norwegian Thematically Organized Psychosis (TOP) cohort was used for PGS analyses,41 including 2181 European participants (1,060 females, age: 33.1±11.8 years, nBIP=440, nSCZ=697, and ncontrols=1044). We also obtained information on recent cannabis use within 2 years prior to recruitment, and psychotic experience. Details are presented in the Supplementary Methods and Supplementary Table 1. All participants provided written informed consent and the study was approved by The Regional Committee for Medical and Health Research Ethics of South-East Norway.
Statistical Framework
LD-pred242 was used to calculate the PGS of SCZ, BIP, LCU, and CUD, separately, in TOP samples using the above GWAS datasets (Supplementary Methods). For each PGS, we examined the significance and extent (PGS.R2) of association with BIP and SCZ diagnosis using a generalized logistic regression model (‘single-PGS’ models) adjusting for sex, age, genetic batch ID, and the first 20 genetic principal components. The Benjamini-Hochberg correction (q<0.05) was performed.
Next, we established a ‘multi-PGS’ model43 for BIP and SCZ, separately, by combining the psychotic-specific PGS with LCU- and CUD- PGSs in a joint model, accounting for the same covariates. This multi-PGS model was compared with the single-PGS model for the psychotic-specific PGS to evaluate the difference in explained variance due to the addition of PGSs for cannabis phenotypes.
We utilized nonmelanoma skin cancer (NMSC) as a comparator, as NMSC does not appear to be associated with psychotic disorders.44 Meanwhile, we carried out sensitivity analyses leveraging 1031 participants without recent cannabis use in the past 2 years (Supplementary Methods).
Results
Genetic Architecture of Psychotic Disorders and Cannabis Phenotypes
Estimated heritability (range=7–38%) was greater among psychotic disorders than among cannabis phenotypes while, polygenicity was lowest for CUD (3.7k trait-influencing variants; Figure 1, Supplementary Table 2, and Supplementary Figure 2). Meanwhile, LCU was 75% genetically less discoverable than other phenotypes.
Figure 1. Genetic architecture of psychotic disorders and cannabis phenotypes.
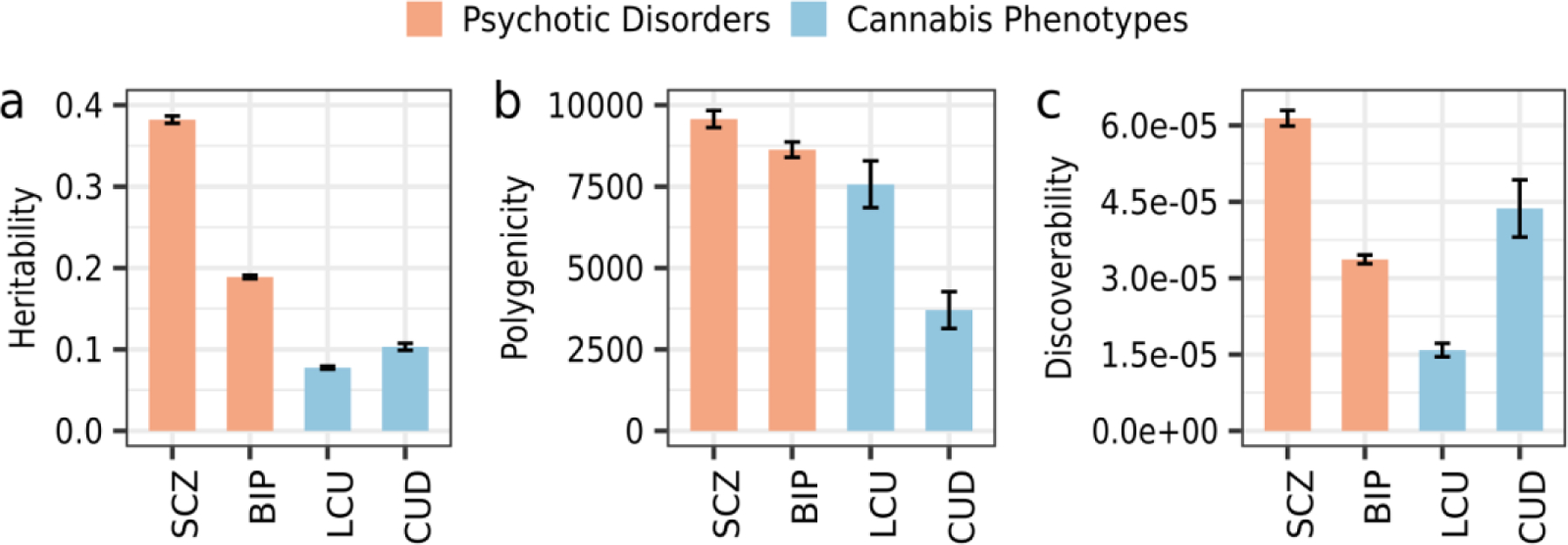
The MiXeR-estimated heritability, polygenicity, and discoverability for each phenotype. Error bars represent 1 standard deviation. SCZ: schizophrenia; BIP: bipolar disorder; LCU: lifetime cannabis use; CUD: cannabis use
Shared Genetic Architecture Between Psychotic Disorders and Cannabis Phenotypes
Genome-wide rgs between psychotic disorders and cannabis phenotypes range from 0.22, for BIP and CUD, to 0.35, for SCZ and CUD (Figure 2A). Local rgs, which give a more granular picture of genetic overlap in the presence of mixed effect directions, showed that on average only 65% of nominally significant local rgs were in the positive direction between each psychotic-cannabis phenotype pair (Supplementary Table 3). Therefore, in Figure 2B, a mixture of negative (i.e., blue points) and positive (i.e., red points) local rgs were observed for each psychotic-cannabis phenotype pair.
Figure 2. Genome-wide and Local Genetic Correlations.
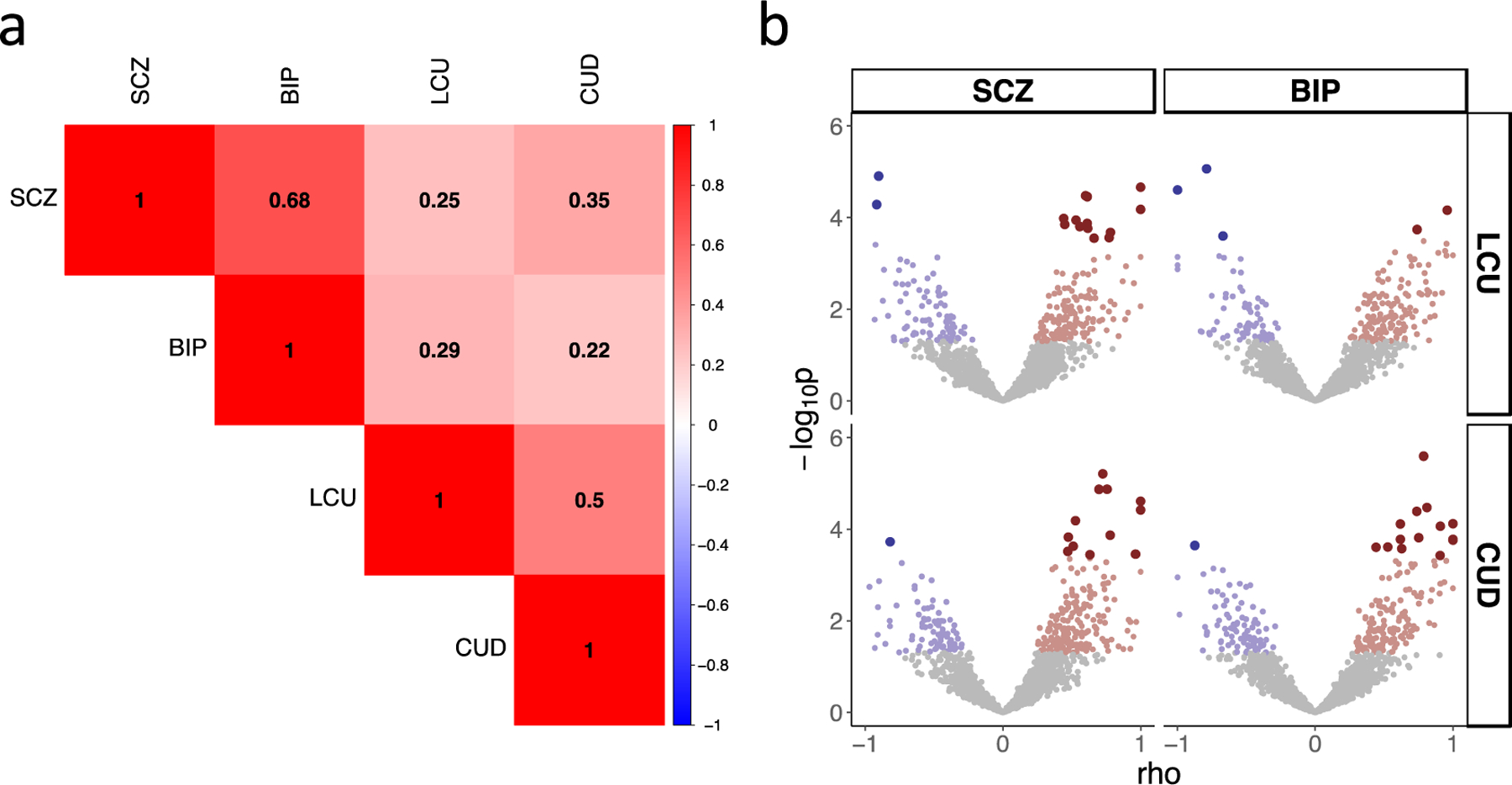
A) Results of genome-wide genetic correlations where numbers represent the correlation coefficient. All correlations were significant after correction for multiple comparisons. B) Results of local genetic correlations with positive (red) and negative (blue) correlation across regions of the genome (each represented by one point). Grey points are genetic correlations with a p>0.05. Correlations with p<0.05 are represented in red or blue depending on the direction of effect. Correlations surviving correction for multiple comparison are represented by larger points that are darker in color. SCZ: schizophrenia; BIP: bipolar disorder; LCU: lifetime cannabis use; CUD: cannabis use disorder.
Next, we identified shared loci for each psychotic-cannabis phenotype pair using the conjFDR approach. For SCZ and LCU, SCZ and CUD, BIP and LCU, and BIP and CUD, we identified 27, 21, 14, and 3 shared loci, respectively (Figure 3, Supplementary Table 4). Five loci were identified as shared by more than one phenotype pair (Supplementary Table 5). For example, three loci shared between LCU and SCZ were overlapped with loci shared between LCU and BIP. When investigating the direction of effects, the majority of shared lead SNPs for each pair exhibited concordant effects (ranging from 67% to 93%, Supplementary Table 4). Additionally, the SNP sign test replicated sign concordance in independent samples for SCZ and BIP at 67.5% and 73.3% of shared lead SNPs, respectively (Supplementary Tables 6–7).
Figure 3. Manhattan Plot of Shared Genetic Architecture.
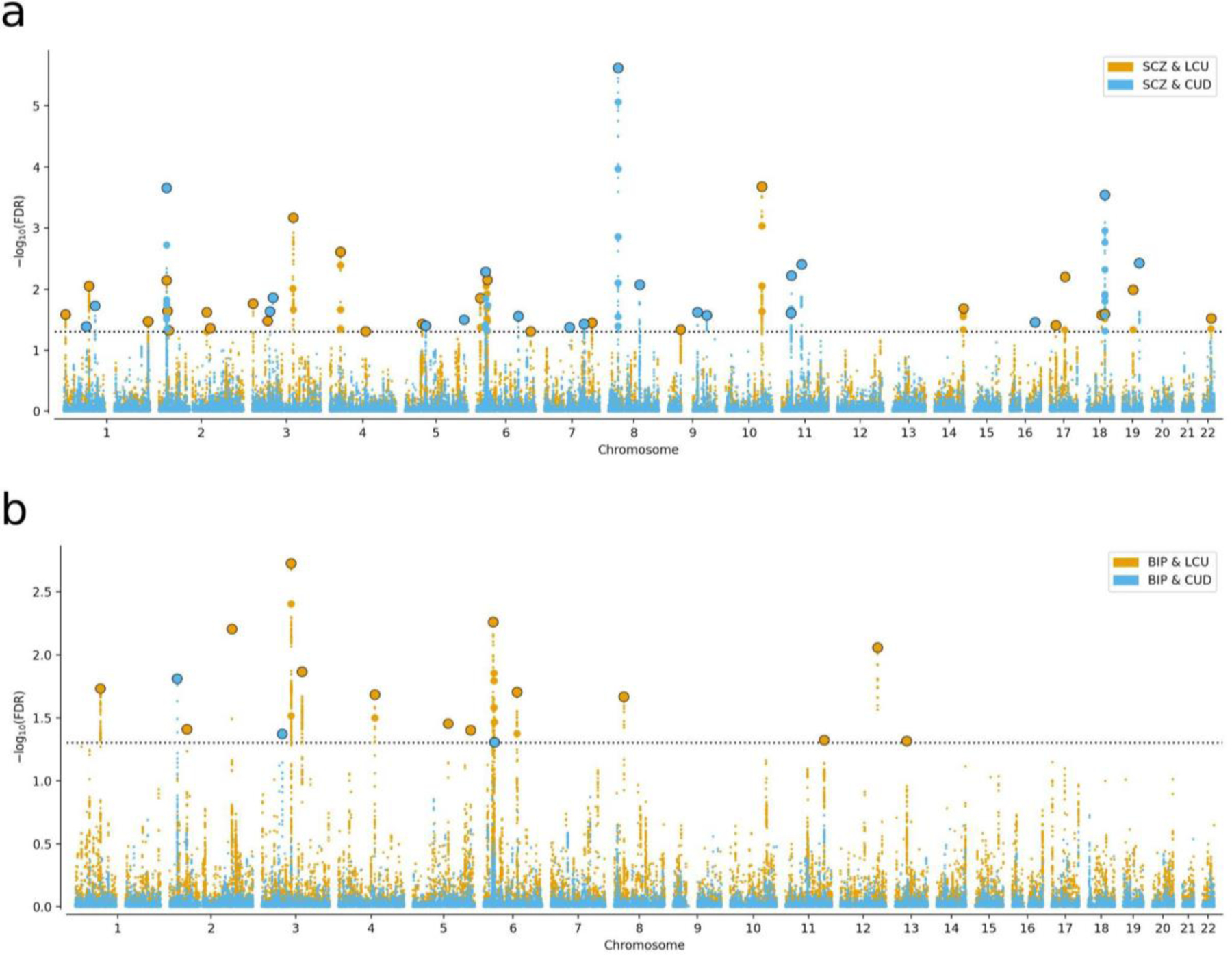
The conjunctional false discovery rate Manhattan plot for the shared genetic architecture between schizophrenia (SCZ) (A) and bipolar disorder (BIP) (B) with lifetime cannabis use (LCU) (orange) and cannabis use disorder (CUD) (blue). For each plot, lead variants are represented as larger dots with a black outline.
The number of genes mapped to shared loci (i.e., shared genes) ranged widely from 110 mapped to loci shared by SCZ and LCU to no genes mapped to loci shared by BIP and CUD (Supplementary Table 8). The shared genes between SCZ and cannabis phenotypes were enriched for mitochondrial, neuron projection cellular components (Supplementary Table 9), and targets of alcohol, nicotine, and pharmaceutical drugs for treating dementia, AIDS, and rheumatoid arthritis (Supplementary Tables 10–11). The shared genes for BIP and LCU exhibited enrichment for olfactory ensheathing glia cells (Supplementary Table 9), and the drug duloxetine, a serotonin-norepinephrine reuptake inhibitor (SNRI) (Supplementary Table 12).
Potential Causal Link Between Psychotic Disorders and Cannabis Phenotypes
For the MR analyses, we focus on more robust causal relationships supported by more than one MR method (Table 1). A putative causal link from LCU to BIP was observed. While the CUD GWAS lacked the power to estimate causal effects on psychotic disorders using genome-wide significant loci, a relaxed threshold revealed a putative causal link to SCZ (Supplementary Table 13). Strong evidence for reverse causal associations were observed where (i) the genetic liability to SCZ increased the odds of both LCU and CUD and (ii) the genetic liability to BIP increased the odds of LCU. LCV analyses did not support any causal association (Supplementary Table 14).
Table 1.
Bidirectional Mendelian Randomization Analysis
| Exposure | Outcome | Method | N SNPs | Estimate | OR | SE | P | PFDR |
|---|---|---|---|---|---|---|---|---|
| LCU | SCZ | Inverse variance weighted | 4 | β= 0.16 | 1.17 | 0.14 | 0.23 | 3.13e-1 |
| MR Egger | 4 | β= −0.96 | 0.38 | 0.88 | 0.39 | 4.74e-1 | ||
| Weighted Median | 4 | β= 0.23 | 1.26 | 0.11 | 0.03 | 5.37e-2 | ||
| MR-PRESSO (raw) | 4 | β= 0.16 | 1.17 | 0.14 | 0.32 | 4.18e-1 | ||
| CAUSE | 6236335 | γ= 0.05 | 1.05 | 0.05 | 0.61 | 6.48e-1 | ||
|
| ||||||||
| LCU | BIP | Inverse variance weighted | 4 | β= 0.37 | 1.45 | 0.13 | 5.37e-3 | 1.22e-2 |
| MR Egger | 4 | β= −0.63 | 0.53 | 0.90 | 0.55 | 6.23e-1 | ||
| Weighted Median | 4 | β= 0.40 | 1.49 | 0.11 | 1.78e-4 | 6.72e-4 | ||
| MR-PRESSO (raw) | 4 | β= 0.37 | 1.45 | 0.13 | 0.07 | 1.19e-1 | ||
| CAUSE | 6994919 | γ= 0.06 | 1.03 | 0.05 | 0.52 | 6.10e-1 | ||
|
| ||||||||
| CUD | SCZ | Inverse variance weighted | 2 | β= 0.45 | 1.57 | 0.13 | 6.40e-4 | 1.98e-3 |
| MR Egger | 2 | NA | NA | NA | NA | NA | ||
| Weighted Median | 2 | NA | NA | NA | NA | NA | ||
| MR-PRESSO (raw) | 2 | NA | NA | NA | NA | NA | ||
| CAUSE | 6006946 | γ= 0.05 | 1.05 | 0.05 | 0.60 | 6.48e-1 | ||
|
| ||||||||
| CUD | BIP | Inverse variance weighted | 2 | β= 0.03 | 1.03 | 0.10 | 0.79 | 8.14e-1 |
| MR Egger | 2 | NA | NA | NA | NA | NA | ||
| Weighted Median | 2 | NA | NA | NA | NA | NA | ||
| MR-PRESSO (raw) | 2 | NA | NA | NA | NA | NA | ||
| CAUSE | 6358021 | γ= 0.05 | 1.05 | 0.04 | 0.38 | 4.74e-1 | ||
|
| ||||||||
| SCZ | LCU | Inverse variance weighted | 128 | β= 0.09 | 1.09 | 0.02 | 7.45e-6 | 5.07e-5 |
| MR Egger | 128 | β= −0.02 | 0.98 | 0.09 | 8.45e-1 | 8.45e-1 | ||
| Weighted Median | 128 | β= 0.11 | 1.12 | 0.02 | 9.69e-6 | 5.49e-5 | ||
| MR-PRESSO (corrected) | 124 | β= 0.10 | 1.11 | 0.02 | 4.06e-7 | 3.45e-6 | ||
| CAUSE | 6236335 | γ= 0.05 | 1.05 | 0.01 | 9.90e-3 | 2.10e-2 | ||
|
| ||||||||
| SCZ | CUD | Inverse variance weighted | 129 | β= 0.21 | 1.23 | 0.03 | 3.50e-12 | 1.19e-10 |
| MR Egger | 129 | β= 0.36 | 1.43 | 0.12 | 4.02e-03 | 1.05e-2 | ||
| Weighted Median | 129 | β= 0.21 | 1.23 | 0.04 | 4.97e-08 | 5.63e-7 | ||
| MR-PRESSO (corrected) | 126 | β= 0.20 | 1.23 | 0.03 | 9.66e-12 | 1.64e-10 | ||
| CAUSE | 6006946 | γ= 0.09 | 1.09 | 0.02 | 0.02 | 3.78e-02 | ||
|
| ||||||||
| BIP | LCU | Inverse variance weighted | 36 | β= 0.11 | 1.12 | 0.03 | 1.21e-4 | 5.14e-4 |
| MR Egger | 36 | β= 0.69 | 1.99 | 0.15 | 7.90e-5 | 3.84e-4 | ||
| Weighted Median | 36 | β= 0.12 | 1.13 | 0.04 | 1.91e-3 | 5.41e-3 | ||
| MR-PRESSO (raw) | 36 | β= 0.11 | 1.12 | 0.03 | 4.89e-4 | 1.66e-3 | ||
| CAUSE | 6994919 | γ= 0.07 | 1.07 | 0.02 | 4.60e-3 | 1.12e-2 | ||
|
| ||||||||
| BIP | CUD | Inverse variance weighted | 38 | β= 0.06 | 1.06 | 0.05 | 0.18 | 2.66e-1 |
| MR Egger | 38 | β= 0.61 | 1.84 | 0.26 | 0.02 | 3.78e-2 | ||
| Weighted Median | 38 | β= 0.10 | 1.11 | 0.07 | 0.13 | 2.01e-1 | ||
| MR-PRESSO (raw) | 38 | β= 0.06 | 1.06 | 0.05 | 0.19 | 2.69e-1 | ||
| CAUSE | 6358021 | γ= 0.06 | 1.06 | 0.03 | 0.12 | 1.94e-1 | ||
Note: MR-PRESSO produces the same estimates as the inverse variance weighted method when no outlier SNP estimates are detected [i.e., MR-PRESSO (raw)]. When outliers are detected, those SNPs are removed, and the inverse variance weighted estimate is re-calculated [MR-PRESSO (corrected)]. NA (not applicable) is used when the particular MR method was unable to estimate the causal effect using so few SNPs. CAUSE uses the full set of overlapping SNPs between two genome-wide association studies to estimate the causal effect. The causal effect presented is the gamma (γ) estimate from the causal model and the p-value is from a test of whether the causal model is a better fit. LCU: Lifetime cannabis use; CUD: Cannabis use disorder; SCZ: Schizophrenia, BIP: Bipolar disorder; MR: Mendelian Randomization; N SNPs: number of single nucleotide polymorphisms (genetic variants) included in the analysis; OR: Odds Ratio: SE: Standard error; p: P-value; PFDR: P-value after the Benjamini-Hochberg correction.
Cannabis Use Polygenic Scores Improve Prediction of Psychotic Disorders
In single-PGS models, both LCU- and CUD- PGSs significantly predicted SCZ diagnosis (Figure 4A and Supplementary Table 15). A similar result was found for BIP where LCU- and CUD- PGSs predicted diagnosis (Figure 4B). As a comparator, the NMSC-PGS predicted neither SCZ nor BIP diagnoses. For SCZ and BIP, multi-PGS models including LCU- and CUD- PGSs showed a small yet significant improvement in explained variance beyond the psychotic-specific single-PGS models (Figure 4 and Supplementary Tables 16–17). Those improvements by LCU- and CUD- PGSs remained significant after including both psychotic disorders’ PGS in multi-PGS models (Supplementary Tables 16–17). Notably, adding NMSC-PGS did not show significant improvement.
Figure 4. Polygenic Risk Prediction.
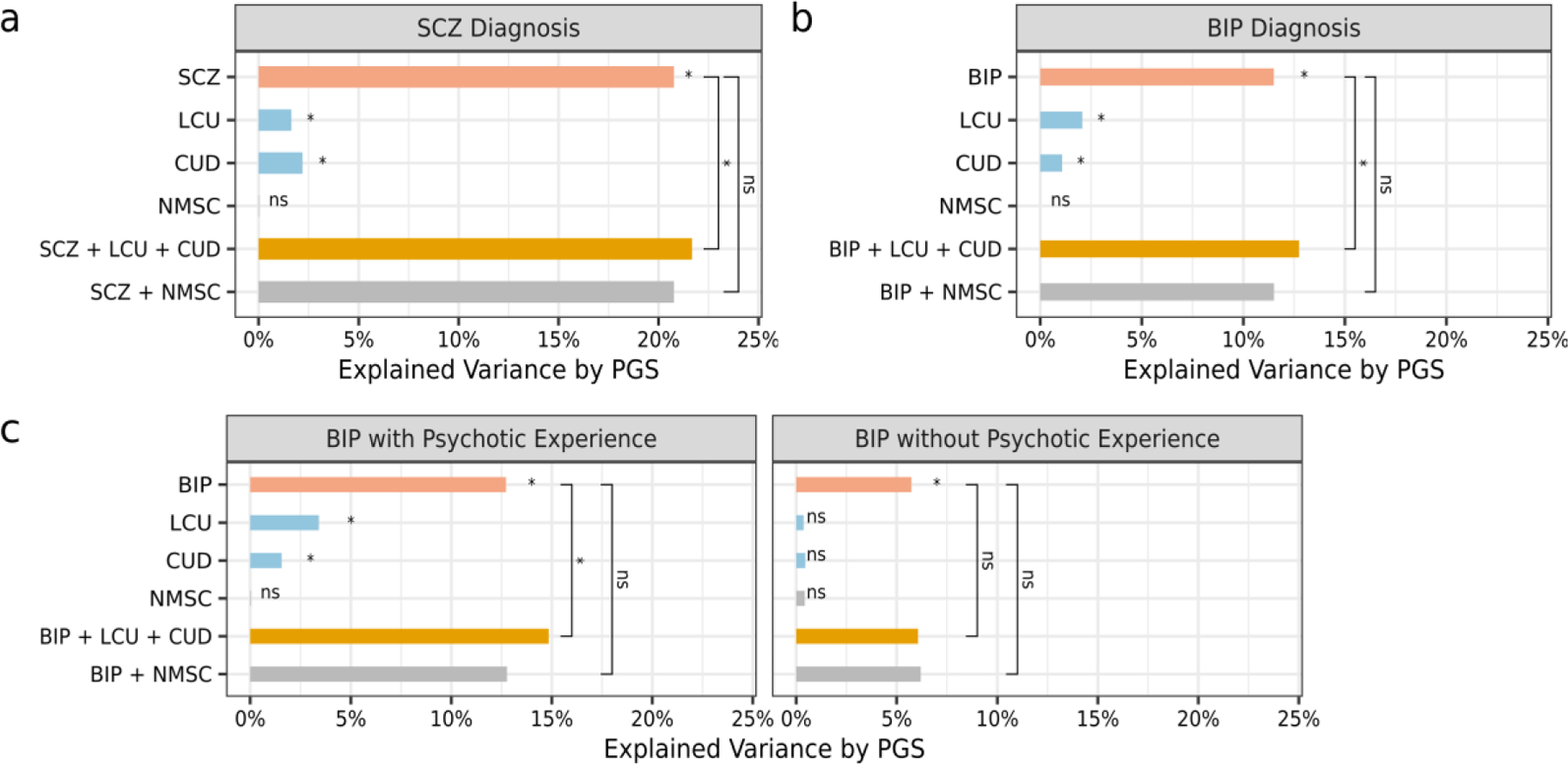
A comparison of variance explained by polygenic scores (PGS) in single- and multi-PGS models to predict patients from healthy controls, including schizophrenia (SCZ) (A), bipolar disorder (BIP) (B), and BIP with and without psychotic experience (C). Pink represents the single-PGS model with the psychotic-specific PGS and covariates only. Blue represents single-PGS models with covariates and PGS of lifetime cannabis use (LCU) or of cannabis use disorder (CUD). Orange represents the multi-PGS model with the psychotic-specific PGS, LCU- and CUD- PGSs, and covariates. Grey represents comparison models that include the PGS for nonmelanoma skin cancer (NMSC). Significance after Benjamini-Hochberg correction is indicated using * or ns for non-significant.
TOP participants exhibited much larger proportions of cannabis users with BIP and SCZ compared to controls (Supplementary Table 1). Therefore, we performed sensitivity analyses using participants without recent cannabis use, which showed the prediction efficiency of LCU- and CUD- PGSs remained significant in single-PGS models (Supplementary Table 15). Multi-PGS models demonstrated continued improvement in prediction for BIP (fold change R2=1.13, PFDR=0.04) but not for SCZ (fold change R2=1.03, PFDR=0.20; Supplementary Tables 16–17).
LCU-PGS was higher in BIP patients with psychotic experience than those without (P=0.02). We applied single- and multi- PGS analyses to predict BIP with psychotic experience, and BIP without psychotic experience from controls, separately. Single-PGS analyses revealed LCU- and CUD- PGSs predicted BIP with psychotic experience but not BIP without psychotic experience (Figure 4C). Multi-PGS models demonstrated significant improvement for BIP with psychotic experience (fold change R2=1.17, PFDR=7.72E-04) but not for those without. Details are provided in Supplementary Tables 18–21.
Discussion
The present study conducted a set of genetically informed analyses to investigate the nature of the association between psychotic disorders and cannabis phenotypes. We observed differences in the genetic architectures of SCZ, BIP, LCU and CUD. We found evidence of genetic overlap between each psychotic-cannabis phenotype pair at the genome-wide, regional, and locus levels. A group of shared loci, ranging from 3 to 27, with mixed effect directions was identified for each phenotype pair. Putative causal relationships were tested using MR, revealing evidence for some bidirectional causal associations. Additionally, combining the PGSs for cannabis phenotypes and psychotic disorders improved distinguishing SCZ and BIP patients from healthy participants. Overall, these findings suggest a shared genetic component underlying the phenotypic link between psychotic disorders and cannabis phenotypes with implications for guiding clinical practice and public policy.
Both psychotic disorders exhibited greater heritability than cannabis phenotypes and were more polygenic than CUD. While the polygenicity findings for SCZ and BIP are in line with previous reports.45 To our knowledge, the polygenicity of LCU or CUD have not been previously estimated. Cannabis phenotypes exhibited distinctive genetic architectures from each other. CUD was more heritable and influenced by fewer genetic variants which maybe reflective of a more specific, clinically defined disorder, potentially more influenced by biological factors like an individuals’ physical response to the consumption of tetrahydrocannabinol46. LCU was less heritable and more polygenic likely reflecting a less specific, heterogeneous, behavioral phenotype more responsive to environmental factors. Moreover, the low discoverability of LCU suggests a large sample size is required to uncover its complete genetic architecture.
The current study adds support for the shared genetic hypothesis for psychotic disorders and cannabis phenotypes by confirming genome-wide rgs,9,13 identifying local rgs in smaller genomic regions, and discovering 57 distinct shared loci. Positive genome-wide rgs, positive shifts in local rgs, and concordant effects in the majority of lead shared variants for each psychotic-cannabis phenotype pair indicates that, in general, genetic liability to both cannabis use and psychotic disorders increase concurrently. This suggests, genetic factors underlie the robust positive phenotypic association linking both SCZ and BIP with cannabis phenotypes. PGS analyses revealed a link between genetic liability of cannabis phenotypes and psychotic experience in BIP. Although this adds supports to the established cannabis-psychosis connection,8,47 the associations of cannabis use and BIP with and without psychotic experience require validation. Further, shared genes showed significant enrichment in various biological processes. Some enriched gene ontology terms have been linked to cannabis use and psychotic disorders, such as neuron projection,48,49 while for others, such as glycosphingolipid biosynthesis50, the connection to cannabis phenotypes requires further investigations.
Part of the shared genetic component has opposite effects on psychotic and cannabis phenotypes, such as genomic regions with negative correlation coefficients and shared loci with discordant effect directions. These results may partly be explained by the fact that both SCZ and BIP are clinically and biologically heterogeneous disorders with a wide range of symptoms, that may exhibit mixed relationships with cannabis phenotypes. For instance, in a sample of SCZ patients, cannabis use was associated with severe positive symptoms but fewer negative symptoms.47 This mixed relationship may also be supported by the results of our enrichment analyses of drug gene-targets. Shared genes for SCZ and cannabis phenotypes showed significant enrichment for genes encoding targets of nicotine and alcohol. Use of nicotine or alcohol is prevalent in cannabis users, and co-users demonstrate a higher rate of psychotic disorder and symptom severity.51 Genes shared between BIP and cannabis phenotypes were enriched for drug targets of duloxetine, an antidepressant52 and reliever of chronic pain.53 Medicinal cannabis use has been linked to both lower self-reported depression54 and pain management.55 Although cannabis use/misuse is also associated with adverse effects, including higher risk of depression, suicidal behaviors,56 and worse analgesic outcomes.57 Further investigation is required to explore this potential biological mechanisms linked to cannabis, antidepressants, and analgesics. Taken together, the mixed effect directions and the gene-drug interactions help explain the mixed relationship between cannabis use and symptom dimensions in psychotic disorders.
The MR analyses provide putative evidence for bidirectional causal effects between psychotic disorders and the cannabis phenotypes. We observed robust evidence supporting the genetic liability to SCZ causally increases the risk of both cannabis phenotypes. This is in line with previous findings9,17,58. We present novel putative evidence that the genetic liability to LCU increases BIP risk. A previous bidirectional MR study only found the genetic liability to BIP increased the risk of LCU,59 which we also observed. Using the latest and largest BIP GWAS likely aided this discovery. However, the CAUSE method could not distinguish causality from effects due to a shared factor related to both LCU and BIP. Additionally, the lack of power in the LCU GWAS may affect the validity of this finding. We caution readers on concluding that psychotic disorders cause cannabis use, and that cannabis use does not cause psychotic disorders. It is important to consider the large difference in the number of genetic variants included in the analyses testing forward (cannabis-to-psychosis) and reverse (psychosis-to-cannabis) causal associations. Given current GWAS, the power to detect reverse causation is greater. As more genome-wide significant loci are discovered for cannabis phenotypes, the reliability of causal estimates will improve and may reveal more robust causal associations.
PGSs have become an important tool in understanding complex genetic phenotypes and for precision medicine. Consistent with prior reports,13 we found each cannabis phenotype PGS to be significantly associated with BIP and SCZ diagnosis. A multi-PGS approach provided a statistically significant improvement in prediction of BIP and SCZ by adding PGSs for cannabis phenotypes. These findings support the idea that incorporating additional PGSs alongside the psychotic-specific PGS improves prediction accuracy.21,43 However, the improvement of our multi-PGS models were small, which limits their clinical utility. Still, the potential of these models for risk stratification of patients is promising and may become useful with larger GWAS in the future.
There are several clinically relevant implications for the current findings. A bidirectional causal link between psychotic disorders and cannabis use suggests public efforts to reduce cannabis use, in individuals at high risk and patients, may prevent psychotic disorders and potentially reduce psychotic symptoms for a subset of the population. Moreover, the underlying genetic component that contributes to the co-occurrence of psychotic disorders and cannabis use suggests a subgroup of individuals are at high genetic risk for psychosis and cannabis use. Early identification of this subgroup is important for targeted interventions and our results suggest polygenic risk scores may help with this risk stratification and treatment in the future.
The present findings should be interpreted considering some limitations. The GWAS for BIP and SCZ may include cannabis users, which could bias the current findings. The power of CUD GWAS is limited, which further confines the shared locus discovery, MR analyses, and the prediction efficiency of CUD PGS. The exclusion of LD regions and the removal of the overlapping UK Biobank sample in BIP GWAS may affect power of the conjFDR analyses. Further, shared loci require validation in independent cohorts for cannabis phenotypes. Also, we only focused on the possibility for boosting prediction efficiency on psychotic disorders by integrating the PGSs of cannabis phenotypes. This decision was based on available data in the TOP sample, but the analyses also have potential for greater clinical utility than the prediction of cannabis phenotypes. We use “psychotic disorders” as a general term, but psychosis is not a defining feature of BIP. Therefore, most analyses relate cannabis use to SCZ and BIP, not psychosis. However, psychiatric disorders, such as depression, have been genetically associated with cannabis use and have a relevant links to psychosis. Thus, the current findings may extend beyond SCZ and BIP. Additionally, psychotic disorders and cannabis use share environmental factors, which may contribute to their covariation.60 Further work is required to disentangle shared genetics from environmental influences.
In summary, our study leveraged the largest genetic datasets and various genetic approaches to evaluate the relationship between cannabis phenotypes and psychotic disorders. The present findings support a shared genetic basis, with bidirectional causality, which helps explain the well-established co-occurrence of psychotic disorders and cannabis use. Also, a subgroup of individuals will exhibit a high genetic risk of both developing a psychotic disorder and using cannabis, supporting targeted public health efforts to reduce cannabis use particularly among these high-risk individuals. Identified shared genetic loci may also aid in treatment efforts. Ultimately, these results may help inform public health policies and aid in pursuits of customized care for patients.
Supplementary Material
Research in Context.
Evidence before this study
Cannabis use often co-occurs with disorders involving psychosis, psychotic symptoms, and mood dysregulation. Previous twin-based studies indicate that psychotic disorders and phenotypes associated with cannabis use are heritable. Yet, it remains unclear how genetics can inform our understanding of the connection between psychotic disorders and cannabis use. We searched PubMed and Google Scholar for genetic studies published in English before April 4th, 2022, investigating the relationship between two psychotic disorders [i.e., schizophrenia (SCZ) and bipolar disorder (BIP)] and cannabis use. The search terms included [“Genetic” OR “Genome wide association study” OR “GWAS” OR “Mendelian randomization” OR “Mendelian randomisation” OR “MR” OR “Genetic correlation” OR “Genetic overlap” OR “Polygenic score” OR “Polygenic risk score”] AND [“Schizophrenia” OR “Bipolar Disorder” OR “Bipolar”] AND [“Cannabis” OR “Marijuana”].
Previous studies have discovered modest genetic correlations (rg) between SCZ and BIP with cannabis use. However, deeper investigation into such shared genetics is lacking. Studies have shown some evidence for genetic liability to cannabis use being causally linked to increased risk of SCZ and BIP with additional evidence of a reverse causal association (from psychotic disorders to cannabis use). Recent large genome wide association studies (GWAS) for SCZ and BIP can improve our assessment of these causal associations. Meanwhile, many studies using polygenic scores (PGS) have reported positive associations between the genetic risk for psychotic disorders and cannabis use, although there is a lack of understanding on how genetic liability of cannabis use can be leveraged to improve prediction of psychotic disorders.
Added value of this study
The current study used a series of genetic analyses, leveraging data from the latest GWAS, to develop a more comprehensive understanding of the relationship between SCZ and BIP with two cannabis phenotypes: lifetime cannabis use (LCU) and cannabis use disorder (CUD). First, we observed that psychotic disorders are more heritable than cannabis phenotypes, and more polygenic than CUD, while each phenotype varies in their degree of genetic discoverability. Second, modest positive rgs at a genome-wide level were observed to be a result of a mixture of effect directions at the local level. That is, on average only 65% of nominally significant local rgs were in the positive direction between each psychotic-cannabis phenotype pair. Third, moving beyond rg, we identified a total of 57 distinct shared genetic loci for psychotic-cannabis phenotype pairs. Enrichment analyses of genes mapped to these loci reveal a potential neuronal and olfactory cell involvement and implicated genes encoding targets of drugs such as nicotine, alcohol, and duloxetine. Fourth, we provided a novel, putatively causal association between genetic liability to LCU and increased risk of BIP. Finally, we demonstrated that the genetic liability to LCU and CUD, captured by polygenic scores, significantly predict BIP and SCZ, and improved prediction of both psychotic disorders above and beyond polygenic scores specific to the psychotic disorder. Moreover, LCU and CUD predicted BIP in patients that experienced psychosis but not those without a psychotic experience.
Implications of all the available evidence
The accumulated evidence points to a genetic component that contributes to the co-occurrence of SCZ, BIP, and cannabis use. A subgroup of individuals will have a high risk for both disorders and cannabis use thus providing support for public health efforts to reduce cannabis use, particularly in this high-risk group. Moreover, identified genetic loci may inform targeted drug development.
Acknowledgments
Funding was provided by the National Institutes of Health (grants NS057198, EB00790 and 1R01MH124839), the Research Council of Norway (RCN grants 223273, 296030, 300309, 324252), the South-East Regional Health Authority (grant 2017-112, 2022-073), Stiftelsen Kristian Gerhard Jebsen (grants SKGJ-MED-008 and SKGJ-MED-021), EEA-RO-NO-2018-0535, European Union’s Horizon 2020 Research and Innovation Programme (Grant 847776; CoMorMent and 964874 REALMENT), the Marie Skłodowska-Curie Actions Grant 80113 (Scientia fellowship), and part of convergence environment (MultiModal Mental Models [4MENT]) at the University of Oslo (UiO) Life Science. The authors have also received internationalization support from UiO:Life Science.
This work was performed on resources provided by Sigma2 (the National Infrastructure for High Performance Computing and Data Storage in Norway) and the TSD (Tjeneste for Sensitive Data) facilities.
Funding:
Funding was provided by the NIH (grants NS057198, EB00790 and 1R01MH124839), the RCN (grants 223273, 296030, 300309, 324252), the South-East Regional Health Authority (grant 2017-112, 2022-073), SKGJ (grants SKGJ-MED-008 and SKGJ-MED-021), EEA-RO-NO-2018-0535, EU Horizon 2020 Research and Innovation Programme (Grant 847776; CoMorMent and 964874 REALMENT), the Marie Skłodowska-Curie Actions Grant 80113 (Scientia fellowship), part of convergence environment (MultiModal Mental Models [4MENT]) at the UiO Life Science, and internationalization support from UiO:Life Science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
All GWAS summary statistics included in this study are publicly available. TOP sample data can be made available upon request and with appropriate data transfer agreements.
Declaration of Interest
Dr. Andreassen reported personal fees from Lundbeck (speaker’s honorarium), Sunovion (speaker’s honorarium), Biogen (consultant) outside the submitted work and is a consultant to HealtLytix (stock options). Dr. Dale is a founder of and holds equity interest in CorTechs Labs and serves on its scientific advisory board. He is also a member of the Scientific Advisory Board of Healthlytix and receives research funding from General Electric Healthcare (GEHC).
References
- 1.Observatoire européen des drogues et des toxicomanies, editor. European drug report: trends and developments Luxembourg: Publications office of the European Union, 2021. [Google Scholar]
- 2.Off U. Drugs Crime (UNODC). 2019. World drug report 2019 2019.
- 3.Diagnostic and statistical manual of mental disorders : DSM-5 Arlington, VA: American Psychiatric Association, 2013. [Google Scholar]
- 4.Bergen AH van, Verkooijen S, Vreeker A, et al. The characteristics of psychotic features in bipolar disorder. Psychological Medicine 2019; 49: 2036–48. [DOI] [PubMed] [Google Scholar]
- 5.Hjorthøj C, Posselt CM, Nordentoft M. Development over time of the population-attributable risk fraction for cannabis use disorder in schizophrenia in Denmark. JAMA psychiatry 2021; 78: 1013–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto JV, Medeiros LS, da Rosa GS, et al. The prevalence and clinical correlates of cannabis use and cannabis use disorder among patients with bipolar disorder: A systematic review with meta-analysis and meta-regression. Neuroscience & Biobehavioral Reviews 2019; 101: 78–84. [DOI] [PubMed] [Google Scholar]
- 7.Vaucher J, Keating BJ, Lasserre AM, et al. Cannabis use and risk of schizophrenia: a Mendelian randomization study. Mol Psychiatry 2018; 23: 1287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Large M, Sharma S, Compton MT, Slade T, Nielssen O. Cannabis use and earlier onset of psychosis: a systematic meta-analysis. Archives of general psychiatry 2011; 68: 555–61. [DOI] [PubMed] [Google Scholar]
- 9.Pasman JA, Verweij KJH, Gerring Z, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal effect of schizophrenia liability. Nat Neurosci 2018; 21: 1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hilker R, Helenius D, Fagerlund B, et al. Heritability of schizophrenia and schizophrenia spectrum based on the nationwide Danish twin register. Biological psychiatry 2018; 83: 492–8. [DOI] [PubMed] [Google Scholar]
- 11.Verweij KJ, Zietsch BP, Lynskey MT, et al. Genetic and environmental influences on cannabis use initiation and problematic use: a meta‐analysis of twin studies. Addiction 2010; 105: 417–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pettersson E, Lichtenstein P, Larsson H, et al. Genetic influences on eight psychiatric disorders based on family data of 4 408 646 full and half-siblings, and genetic data of 333 748 cases and controls. Psychological medicine 2019; 49: 1166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson EC, Demontis D, Thorgeirsson TE, et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. The Lancet Psychiatry 2020; 7: 1032–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillespie NA, Kendler KS. Use of Genetically Informed Methods to Clarify the Nature of the Association Between Cannabis Use and Risk for Schizophrenia. JAMA Psychiatry 2021; 78: 467. [DOI] [PubMed] [Google Scholar]
- 15.Zammit S, Allebeck P, Andreasson S, Lundberg I, Lewis G. Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. Bmj 2002; 325: 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanderson E, Glymour MM, Holmes MV, et al. Mendelian randomization. Nat Rev Methods Primers 2022; 2: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson EC, Hatoum AS, Deak JD, et al. The relationship between cannabis and schizophrenia: a genetically informed perspective. Addiction; n/a. DOI: 10.1111/add.15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Power RA, Verweij KJ, Zuhair M, et al. Genetic predisposition to schizophrenia associated with increased use of cannabis. Molecular psychiatry 2014; 19: 1201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wainberg M, Jacobs GR, di Forti M, Tripathy SJ. Cannabis, schizophrenia genetic risk, and psychotic experiences: a cross-sectional study of 109,308 participants from the UK Biobank. Transl Psychiatry 2021; 11: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hjorthøj C, Uddin MJ, Wimberley T, et al. No evidence of associations between genetic liability for schizophrenia and development of cannabis use disorder. Psychological Medicine 2021; 51: 479–84. [DOI] [PubMed] [Google Scholar]
- 21.Krapohl E, Patel H, Newhouse S, et al. Multi-polygenic score approach to trait prediction. Mol Psychiatry 2018; 23: 1368–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullins N, Forstner AJ, O’Connell KS, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet 2021; 53: 817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trubetskoy V, Pardiñas AF, Qi T, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022; 604: 502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam M, Chen C-Y, Li Z, et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat Genet 2019; 51: 1670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurki MI, Karjalainen J, Palta P, et al. FinnGen: Unique genetic insights from combining isolated population and national health register data 2022; : 2022.03.03.22271360. [Google Scholar]
- 26.Frei O, Holland D, Smeland OB, et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun 2019; 10: 2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulik-Sullivan BK, Loh P-R, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47: 291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Werme J, Sluis S van der, Posthuma D, Leeuw CA de. LAVA: An integrated framework for local genetic correlation analysis 2021. [DOI] [PubMed] [Google Scholar]
- 29.Andreassen OA, Thompson WK, Schork AJ, et al. Improved Detection of Common Variants Associated with Schizophrenia and Bipolar Disorder Using Pleiotropy-Informed Conditional False Discovery Rate. PLOS Genetics 2013; 9: e1003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng W, van der Meer D, Parker N, et al. Shared genetic architecture between schizophrenia and subcortical brain volumes implicates early neurodevelopmental processes and brain development in childhood. Mol Psychiatry 2022; : 1–10. [DOI] [PubMed] [Google Scholar]
- 31.Smeland OB, Bahrami S, Frei O, et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol Psychiatry 2019; 25: 844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng W, Frei O, van der Meer D, et al. Genetic Association Between Schizophrenia and Cortical Brain Surface Area and Thickness. JAMA Psychiatry 2021; published online June 23. DOI: 10.1001/jamapsychiatry.2021.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017; 8: 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 2018; 7: e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013; 37: 658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol 2016; 40: 304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015; 44: 512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018; 50: 693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet 2020; 52: 740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 2018; 50: 1728–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steen NE, Aas M, Simonsen C, et al. Serum levels of second-generation antipsychotics are associated with cognitive function in psychotic disorders. World J Biol Psychiatry 2017; 18: 471–82. [DOI] [PubMed] [Google Scholar]
- 42.Privé F, Arbel J, Vilhjálmsson BJ. LDpred2: better, faster, stronger. Bioinformatics 2020; 36: 5424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoeler T, Choi SW, Dudbridge F, et al. Multi–Polygenic Score Approach to Identifying Individual Vulnerabilities Associated With the Risk of Exposure to Bullying. JAMA Psychiatry 2019; 76: 730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu D, Andersson TML, Fall K, et al. Clinical Diagnosis of Mental Disorders Immediately Before and After Cancer Diagnosis: A Nationwide Matched Cohort Study in Sweden. JAMA Oncology 2016; 2: 1188–96. [DOI] [PubMed] [Google Scholar]
- 45.Hindley G, Frei O, Shadrin AA, et al. Charting the Landscape of Genetic Overlap Between Mental Disorders and Related Traits Beyond Genetic Correlation. AJP 2022; 179: 833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferland J-MN, Hurd YL. Deconstructing the neurobiology of cannabis use disorder. Nat Neurosci 2020; 23: 600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quattrone D, Ferraro L, Tripoli G, et al. Daily use of high-potency cannabis is associated with more positive symptoms in first-episode psychosis patients: the EU-GEI case–control study. Psychological Medicine 2021; 51: 1329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danos P, Baumann B, Bernstein HG, et al. Schizophrenia and anteroventral thalamic nucleus: selective decrease of parvalbumin-immunoreactive thalamocortical projection neurons. Psychiatry Res 1998; 82: 1–10. [DOI] [PubMed] [Google Scholar]
- 49.Wu C-S, Zhu J, Wager-Miller J, et al. Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci 2010; 32: 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood PL, Filiou MD, Otte DM, Zimmer A, Turck CW. Lipidomics reveals dysfunctional glycosynapses in schizophrenia and the G72/G30 transgenic mouse. Schizophrenia Research 2014; 159: 365–9. [DOI] [PubMed] [Google Scholar]
- 51.Yurasek AM, Aston ER, Metrik J. Co-use of Alcohol and Cannabis: A Review. Curr Addict Rep 2017; 4: 184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de DIOS C, Ezquiaga E. Manic Switching in Patients Receiving Duloxetine. AJP 2007; 164: 1121–1121. [DOI] [PubMed] [Google Scholar]
- 53.Lunn MPT, Hughes RAC, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; : CD007115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vázquez M, Guevara N, Maldonado C, Guido PC, Schaiquevich P. Potential Pharmacokinetic Drug-Drug Interactions between Cannabinoids and Drugs Used for Chronic Pain. Biomed Res Int 2020; 2020: 3902740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bicket MC, Stone EM, McGinty EE. Use of Cannabis and Other Pain Treatments Among Adults With Chronic Pain in US States With Medical Cannabis Programs. JAMA Network Open 2023; 6: e2249797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gobbi G, Atkin T, Zytynski T, et al. Association of Cannabis Use in Adolescence and Risk of Depression, Anxiety, and Suicidality in Young Adulthood: A Systematic Review and Meta-analysis. JAMA Psychiatry 2019; 76: 426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boehnke KF, Scott JR, Litinas E, Sisley S, Williams DA, Clauw DJ. High frequency medical cannabis use is associated with worse pain among individuals with chronic pain. J Pain 2020; 21: 570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gage SH, Jones HJ, Burgess S, et al. Assessing causality in associations between cannabis use and schizophrenia risk: a two-sample Mendelian randomization study. Psychological Medicine 2017; 47: 971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jefsen OH, Speed M, Speed D, Østergaard SD. Bipolar disorder and cannabis use: A bidirectional two-sample Mendelian randomization study. Addiction Biology 2021; 26: e13030. [DOI] [PubMed] [Google Scholar]
- 60.Nesvåg R, Reichborn-Kjennerud T, Gillespie NA, et al. Genetic and Environmental Contributions to the Association Between Cannabis Use and Psychotic-Like Experiences in Young Adult Twins. Schizophr Bull 2017; 43: 644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.