

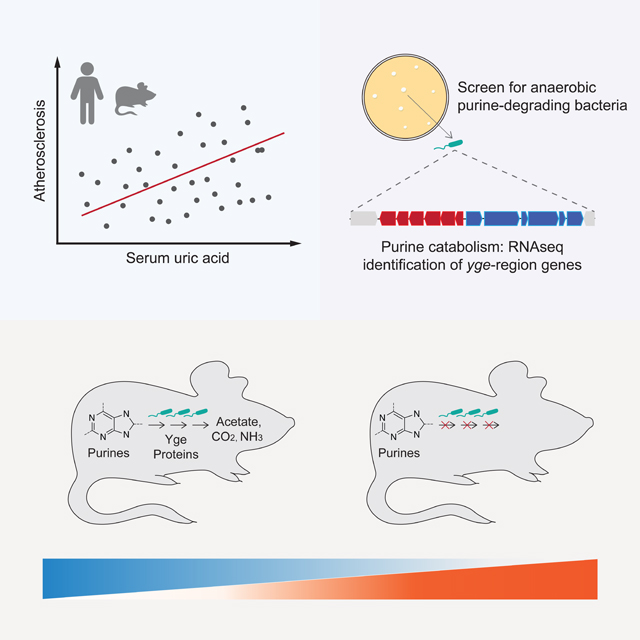
SUMMARY
The microbes and microbial pathways that influence host inflammatory disease progression remain largely undefined. Here we show that variation in atherosclerosis burden is partially driven by gut microbiota and is associated with circulating levels of uric acid in mice and humans. We identify gut bacterial taxa spanning multiple phyla, including Bacillota, Fusobacteriota and Pseudomonadota, that use multiple purines, including uric acid as carbon and energy sources anaerobically. We identify a gene cluster encoding key steps of anaerobic purine degradation that is widely distributed among gut-dwelling bacteria. Furthermore, we show that colonization of gnotobiotic mice with purine-degrading bacteria modulates levels of uric acid and other purines in the gut and systemically. Thus, gut microbes are important drivers of host global purine homeostasis and serum uric acid levels, and gut bacterial catabolism of purines may represent a mechanism by which gut bacteria influence health.
Graphical Abstract
INTRODUCTION
Metabolic disorders including obesity, type 2 diabetes, and atherosclerosis have historically been viewed as lipid conditions primarily driven by overindulgence of calorie-dense foods. However, it is now widely appreciated that chronic inflammation plays a central role in the development and progression of these disorders.1 Atherosclerosis, the leading cause of cardiovascular disease (CVD), is characterized by vascular inflammation and is influenced by multiple genetic and environmental factors.1–3 Large-scale genome-wide analyses in human populations have identified over 100 loci significantly associated with atherosclerosis itself4 and hundreds of additional loci for traits associated with atherosclerosis such as plasma lipids, obesity, and diabetes.5–7 Nevertheless, while genetics significantly influences atherosclerosis, the environment, especially diet also plays a major role in its progression. Furthermore, several recent studies have provided evidence suggesting that dietary contributions to disease progression are often mediated by the gut microbiome.8–10
The gut microbiome exerts profound influence on metabolism and inflammatory diseases.11,12 Diet and host-derived factors modulate the composition of the gut microbiome, which in turn transforms dietary components consumed by the host, generating bioactive molecules that interact with the immune system and virtually every host organ, including the vascular system. Changes over the last century in food production, dietary habits, antibiotic usage, and lifestyle have caused major changes in the microbiome and have affected human health in discordant ways:13–15 the prevalence of acute infectious diseases has decreased, while it increased for chronic inflammatory diseases. Furthermore, several diet-derived gut bacterially-produced metabolites have been uncovered as potential drivers of metabolic and cardiovascular ailments. These metabolites constitute a direct link between environmental exposures and host cellular function and encompass several uremic toxins (i.e., waste products that cannot be eliminated properly by subjects with impaired kidney function) including p-cresyl sulfate, indoxyl sulfate, trimethylamine-N-oxide (TMAO), and phenylacetylglutamine.16 For example, dietary choline, betaine and carnitine serve as substrates for TMAO production, which is generated in the liver from gut bacterially-produced trimethylamine (TMA).8 TMAO enhances inflammation and aortic thrombosis in mice and it is associated with CVD risks in humans.8,17,18 More recently it was found that phenylacetylglutamine, synthesized by the microbiota from dietary protein, enhances platelet activation and thrombosis via host G-protein coupled receptors.19 Together, this evidence supports the notion that gut bacteria metabolism contributes to CVD-related traits by modulating abundance of uremic toxins in circulation.
Several purines-derived metabolites, including xanthine, hypoxanthine and uric acid (UA) are also considered uremic toxins20 and contribute to several symptoms observed in subjects with chronic kidney disease.16,21 UA—the end product of the metabolic breakdown of purines in humans—is mostly studied for the complications it causes when its concentration reaches saturation levels, forming pro-inflammatory crystals that deposit in joints (e.g., gout). However, a recent study showed that concentrations of UA within the solubility range can promote atherosclerosis via induction of AMP-activated protein kinase (AMPK)-mediated inflammation.22 UA exacerbates inflammation, endothelial dysfunction, increases the renin-angiotensin-aldosterone system activity20 and it is increased in patients with hypertension and heart failure.21 Several studies have suggested that pharmacological interventions effective at reducing UA production or increasing its excretion in hyperuricemic patients improves cardiorenal outcomes,23,24 although these benefits are not consistently observed.25 While the kidneys play a major role in regulating levels of UA in circulation, a significant fraction of this metabolite is secreted into the intestine26 and a recent metagenomic study identified bacterial pathways associated with blood levels of UA.27 However, no causal relationships have been established between abundance of this uremic toxin and specific gut bacteria, and while the bacterial mechanisms of anaerobic purine metabolism have been studied biochemically their genetic underpinnings remains undefined.28,29
We sought to examine the role of the gut microbiome on atherosclerosis and identify potential microbial pathways that contribute to disease burden. First, we transplanted microbial communities derived from mouse strains with disparate atherosclerosis phenotypes into germ-free (GF) Apolipoprotein E knockout (ApoE KO) mice. We found that microbial-driven variation on atherosclerosis progression was associated with abundance of purine metabolites including UA. We also observed that this pro-inflammatory metabolite was associated with atherosclerosis burden and gut microbial features in a human cohort. We identified bacterial taxa able to degrade purines anaerobically, uncovered a gene cluster encoding key components needed for anaerobic purine degradation, demonstrated environmental factors affecting its activity, and showed that colonization with taxa containing this locus lowered multiple purines in the gut and UA systemically in mice. Altogether this work strengthens the connection between gut microbes and atherosclerosis and provides insights into how bacterial metabolism influences host biology.
RESULTS
Gut microbes modulate atherosclerosis progression and plasma metabolites associated with disease burden in mice.
Previous work revealed a large degree of variation in atherosclerosis burden among 100 inbred strains of mice from the Hybrid Mouse Diversity Panel (HMDP), which harbor distinct microbial communities.30,31 We hypothesized that the gut microbiome contributed to the variation observed in disease progression among HMDP strains. We transplanted cecal samples from four HMDP strains into GF ApoE KO recipient mice. We selected two strains that exhibited large atherosclerotic lesions (AXB10/PgnJ and BXD5/TyJ) and two strains that showed little signs of disease (BTBR T+tf/J and BXA8/PgnJ), hereinafter referred as “AXB10”, “BXD5”, “BTBR”, and “BXA8”, respectively. Transplanted mice were maintained on a chow diet supplemented with 0.2% cholesterol for 8 weeks. After this period, atherosclerotic lesions, gut microbiome composition, and disease biomarkers were evaluated (Fig 1A, Suppl. Fig 1). We found that mice colonized with cecal communities from HMDP donors prone to atherosclerosis development (i.e., AXB10 and BXD5) exhibited larger lesions compared to recipient mice colonized with samples from donors that showed little signs of atherosclerosis (i.e., BTBR and BXA8). These results support the notion that the gut microbiome contributes to the development of atherosclerosis and possibly to the variation in disease burden observed among the HMDP strains (Fig 1B–G). Neither traditional CVD risk factors such as body weight and cholesterol, nor previously identified gut microbiota-derived metabolites including lipopolysaccharides (LPS), TMAO, and short-chain fatty acids explained the differences in atherosclerosis burden observed among the transplanted mice (Suppl. Fig 1).
Figure 1. Plasma levels of purines are associated with atherosclerosis burden in transplanted gnotobiotic ApoE KO mice.

A) Experimental design. B-G) Representative sections and quantitative analysis of Oil Red O staining (B, C), MOMA-2 staining (D, E), and Masson’s trichrome staining (F, G) in the aortic sinus (n=11 for AXB10, n=12 for BXD5, n=12 for BTBR group, n=11 for BXA8). The data are expressed as box-and-whisker plots with individual data points, where the boxes indicate the median values and the interquartile ranges and the whiskers represent the minimum and maximum values. Significance was calculated by one-way ANOVA with the Tukey post-tests is indicated as follows: *, p-value <0.05; **, p-value of <0.01; ****, p-value of <0.0001. H) Principal component analysis of gut microbial functions from transplanted mice as determined by metagenomic analysis. I) Plasma metabolites positively or negatively associated with atherosclerotic lesion size, according to Spearman correlation analysis. J) Scatterplots showing associations between purines (relative mass spectrometry scaled intensities) and atherosclerosis lesion size (x104 μm2). GF; germ-free, ApoE; Apolipoprotein E, Chol; cholesterol, MOMA; monocytes and macrophages.
Shotgun metagenomic analyses of cecal contents from the transplanted mice identified 1649 functional features and 52 bacterial taxonomic features. Principal component analysis of functional features show distinct clustering by donor strain (p-value <0.001, PERMANOVA) suggesting unique gut bacterial function profiles for each of the four HMDP strains used (Fig. 1H). Pearson correlation analysis identified several bacterial functions associated with atherosclerosis lesion size among recipient mice (Suppl. Fig. 2A, B). These included several functions related to production and conversion of purines. We also observed that bacterial pathways including energy metabolism and amino acid metabolism were enriched among functions positively correlated with atherosclerosis lesion size, although these did not survive multiple hypothesis correction.
To further investigate whether microbiota transplants impacted circulating metabolites associated with disease, we performed metabolome analysis of plasma samples using ultra-High Performance Liquid Chromatography (uHPLC) - Tandem Mass Spectrometry (MS/MS). A total of 682 metabolites were measured (Suppl. Table 1). Pearson correlation analyses identified purine metabolites including xanthine, xanthosine, inosine, and UA, positively associated with atherosclerotic lesions size (Fig. 1I, J). Altogether, these results may suggest that gut microbes influence atherosclerosis progression and abundance of purines in the blood of transplanted mice.
Serum UA is correlated with gut microbial features and subclinical atherosclerosis in a human cohort.
UA is the end product of purine metabolism in humans, and it has been shown to cause inflammation,22,32,33 induce endothelial dysfunction34 and stimulate smooth muscle cell proliferation.35 We explored associations between atherosclerosis, gut bacteria, and UA in a human cohort previously characterized for gut microbiome and glucose homeostasis (n=998).36 Coronary artery calcium (CAC) score measurements were assessed for disease burden. Calcification of arteries is an accepted proxy for estimating overall plaque burden of atherosclerosis.37 We first classified individuals based on their CAC score status: CAC score = 0 (i.e., no detectable vascular calcification, n=492) vs. CAC >0 (n=497). Logistic regression analysis revealed that distribution of UA concentrations was significantly different between individuals from these two groups (Fig. 2A) with individuals with CAC > 0 showing higher mean and median UA levels. Furthermore, Spearman correlation analysis for individuals with CAC score >0 showed a significant positive association between UA and CAC score (rho coefficient = 0.14, p-value <0.001, Fig. 2B). We then applied extreme gradient boosting (XGBoost) regression to identify gut bacterial taxa correlated with UA levels. XGBoost is a decision-tree-based ensemble machine learning algorithm that uses the gradient boosting method. The top 10 features associated with UA levels are shown in Fig. 2C and Suppl. Table 2 after adjusted for covariates (CACS, BMI, gender, triglycerides and HbA1c) in a mixed linear regression model. Interestingly, we found multiple taxa within the Clostridia class that were negatively associated with levels of UA. Altogether these results suggest that the gut microbiome, particularly taxa within the Clostridia, may influence UA levels. This data is also consistent with gnotobiotic mouse work reported above and previous work connecting UA with CVD in humans.38–42
Figure 2. Plasma uric acid levels are positively associated with Coronary Artery Calcium (CAC) score in a human cohort.

A) Distribution of uric acid levels in serum from individuals with CAC score=0 and CAC score >0. B) Spearman correlation analysis between serum uric acid levels and CAC score. C) Top 10 taxa associated with serum uric acid levels. Blue and red bars show positive and negative associations, respectively.
Gut microbiome modulates purines in cecum and circulation.
We next investigated whether the gut microbiome modulated abundance of purines in the intestine and circulation. We quantified purine-related metabolites, including nucleotides, nucleosides and nucleobases by LC-MS/MS in cecal contents and plasma from GF mice and conventionally-raised (Conv) animals (Suppl. Table 3A, B). We found that most purines were decreased in the cecal contents from GF mice compared to Conv mice, with a few exceptions being increased in GF mice, especially UA and allantoin– both terminal purine metabolites in mice (Fig. 3A, B). Given that cecal/fecal purines arise from diet, bacterial and host turnover and metabolism, these analyses cannot establish the sources and fates of these compounds in the gut.
Figure 3. Gut microbiome modulates purines in cecum and circulation.

A) Heatmap of purines and related metabolites in cecal contents from Conv (n=9) and GF (n=8) mice. B) Values for adenine, xanthine, xanthosine, inosine, hypoxanthine, uric acid, and allantoin in cecal contents measured by LC/MS/MS and C) plasma uric acid levels analyzed by an enzymatic assay are shown using box-and-whisker plots with individual data points, where the boxes indicate the median values and the interquartile ranges and the whiskers represent the minimum and maximum values. Significance was calculated by unpaired two-tailed Student’s t-test and is designated as follows: **, p-value of <0.01; ****, p-value of <0.0001. Conv; Conventionally-raised, GF; germ-free.
Partial Least Squares-Discriminant Analysis (PLS-DA) analysis of cecal purines showed separation of the two groups by principal components 1 and 2. While the separation for plasma samples was less evident (Suppl. Fig. 3A, B), we found that GF mice had significantly increased UA levels in plasma compared to Conv mice (Suppl. Fig. 3A, C). This result was confirmed using an enzymatic assay to quantify UA (Fig. 3C). These results again suggested that the gut microbiome modulates abundance of purines both in the gut and systemically, and was the impetus for attempts to isolate anaerobic purine-degrading bacteria (PDB).
Human gut bacteria degrade purines anaerobically.
The intestine is a key organ for purine homeostasis. Dietary purines are absorbed in the gut, resident microbes produce and recycle purines needed for their anabolism and ~30% of the UA generated by the body is secreted into the intestine.43,44 We hypothesized that gut bacteria influence purine levels by metabolizing them to non-purine products. While this notion has been previously discussed, isolates from the human or mouse gut able to grow on these metabolites anaerobically have not been identified. We attempted anaerobic enrichments using fecal slurries (human samples) on media supplemented with UA as the primary source of carbon and energy. Culture medium included non-fermentable acetate (often beneficial for butyrate-producing Firmicutes) plus 0.1% yeast extract as the sole complex nutrient. Enrichments were plated and colonies isolated on bilayer agar plates bearing a top agar layer supplied with saturating amounts of UA or other purines as described in the Methods section. We obtained several isolates from the Bacillota (Firmicutes) and Pseudomonadota (Proteobacteria) phyla including species identified as Enterocloster bolteae and Escherichia coli by 16S rRNA gene sequencing. Additional confirmatory assays were used to verify the identity of the E. coli isolate (designated “I-11”, see Methods).
Using the same medium we verified growth and UA utilization by E. bolteae ATCC BAA-613—the species type strain, which has been previously sequenced. We observed that cultures of this strain supplemented with 12 mg UA degraded 49.6 μmoles of substrate in a 24-hour period, with accumulation of 106.9 μmoles acetate, a known fermentation product of this organism.45 HPLC and headspace Gas Chromatography (GC) analysis did not detect significant ethanol, formate, propionate, lactate and butyrate accumulation under this condition.
We then expanded our search for bacteria with this capability and by using methods described above screened a culture collection of 34 isolates encompassing gut-dwelling bacteria from six phyla (Suppl. Fig. 4; a representative subset of these species is shown in Fig. 4). The screen utilized monolayer plates containing no nitrogen (other than 0.1% yeast extract) or fermentable carbon or energy source, the same medium supplemented with soluble substrates (glucose or allantoin), or bilayer plates (briefly described above, see also Methods) containing insoluble purines (UA, adenine or hypoxanthine). These assays demonstrated the anaerobic allantoin and purine-dependent growth characteristics of these strains as evident by growth of the applied bacterial patch and a zone of disappearance of the insoluble purine substrates. Our screen showed evidence of purine utilization among the Bacillota (Firmicutes), Fusobacteriota and Pseudomonadota (Proteobacteria) phyla (Fig. 4, Suppl. Fig. 4), although this property was not universal among strains belonging to these phyla. Of particular note, these assays showed different purine-utilization capacities among UA-degrading strains: (i) E. coli MS 200–1 and lab isolate E. coli I-11 showed greater UA utilization compared to the commonly used E. coli K12 strain (Fig. 4); (ii) allantoin supported growth of E. bolteae and E. coli but not medically-relevant Clostridioides difficile or Edwardsiella tarda (Fig. 4); (iii) adenine supported the growth of several strains of Proteobacteria (E. coli MS200–1, E. coli I-11, E. tarda) and Firmicutes (E. bolteae, E. asparagifome), but not C. difficile, C. sporogenes or E. coli K12 (Fig. 4, Suppl. Fig. 4) (iv) consistent with previous work, growth of Proteobacteria strains on UA was enhanced by the addition of formate (Suppl. Fig. 4),46 while the addition of formate had more modest effects, if any, for strains from other phyla tested, and did not appear to enhance the utilization of the more reduced purines (adenine, hypoxanthine, Suppl. Fig. 5A) in any of the strains tested. In multiple organisms the ability to use UA was diminished or eliminated in media containing glucose or fructose, consistent with regulation by catabolite repression (Suppl. Figs. 4 and 5B). It is also important to note that none of the six Bacteroides strains tested showed any growth on UA, allantoin or adenine (Fig. 4, Suppl. Fig. 4).
Figure 4. Gut bacterial isolates use purines as carbon and energy sources.

Anaerobic growth of bacterial strains on plates containing soluble (glucose, allantoin), and insoluble (uric acid and adenine) substrates. As detailed in Methods, plates were inoculated with 4 μl of dense overnight cultures grown in rich medium then incubated for 2 days [no fermentable substrate, glucose, allantoin or uric acid conditions] or 7 days (adenine). Growth is indicated by the appearance of cell patches and a zone of clearing for the overlay plates. Details about strains are specified in the Key Resources Table, and a summary of all tested strains is presented in Supplemental Figure 4. Strains indicated in red were used for colonization of gnotobiotic mice.
Extensive biochemical analyses of environmental clostridial isolates has previously demonstrated molybdenum and selenium dependences for purine metabolism.47–49 While no attempt was made to limit trace minerals—i.e., the medium contained a high amount of phosphate buffer plus low levels of yeast extract and cysteine (a possible source of Se contamination)50 and the inocula were prepared in rich medium—a requirement for micromolar additions of Mo and Se was evident for both species of Proteobacteria tested on UA (Suppl. Fig. 5B). However, these effects were not observed with the two Firmicutes tested, C. difficile and E. bolteae (Suppl. Fig. 5B). Additional assays did not show an effect of Fe, Co, Mn, Ni, W, or Zn supplementation on UA utilization by any of the strains tested (E. bolteae, C. difficile, E. coli MS 200–1, E. tarda, again the media were not rigorously depleted of these metals). Altogether, the results presented above suggested that common gut bacteria can use purines for carbon and energy, and that availability of other carbon sources and metals could modulate this process.
Purine-degrading bacteria modulate abundance of purines in cecum and circulation.
To test the impact of PDB identified above in vivo, we created synthetic bacterial communities that varied in their capacity to degrade UA and used them to colonize GF mice. We colonized GF mice with a core community which included seven species spanning major phyla from the human gut and that did not degrade purines in vitro as determined by the assays described above (and as described below they lack genes encoding functions necessary for anaerobic purine metabolism). These included Bacteroides caccae, Bifidobacterium dentium, Blautia hansenii, Bacteroides thetaiotaomicron, Coprococcus comes, Mitsuokella multacida, and Ruminococcus torques. Half of the animals were also colonized with three PDB, including E. bolteae, Hungatella hathewayi, and E. coli isolate I-11 (Fig. 5A). In vitro tests of UA utilization for each of the strains used in these communities is shown in Fig. 4 (highlighted in red), and their combined activities measured using fecal innocula verified that colonization with PDB was required for UA degradation (Fig. 5A). A third group of animals remained germ-free throughout the experiment. The engrafted bacterial communities were analyzed by COPRO-Seq (community profiling by sequencing) analysis.51 All taxa included in these communities successfully colonized the gut of GF mice. E. bolteae was the most abundant among the three PDB (relative abundance: E. bolteae 19.5%, H. hathewayi 5.6% and E. coli 0.9%, Fig. 5B). We next performed targeted quantification of purines, pyrimidines and related metabolites in cecal contents and plasma (Suppl. Table 4A, B). Global analysis of the data showed distinct patterns for cecal purine-related metabolites between GF mice and mice with the “core” or the “core plus PDB”, where nucleosides were increased and nucleotides were decreased in the cecum of GF mice (Fig. 5C). Surprisingly, mice colonized with the “core plus PDB’’ community showed significantly higher levels of UA in the cecum compared to mice colonized with the “core” community, while cecal levels of other purines/nucleosides including hypoxanthine and allantoin were significantly reduced in mice co-colonized with the three PDB (Fig. 5D). It is important to note that these metabolites were detected at significantly higher concentrations in the gut of mice relative to UA (Fig. 5D and Suppl. Table 4A). PLS-DA analysis separated plasma samples from the core plus PDB from the ones from the other two groups (Suppl. Fig. 6A, B). Colonization with core plus PDB resulted in consistently lower levels of several purine metabolites in plasma, including UA (Suppl. Fig. 6A, C), recapitulating previous data (Fig. 3C). Plasma UA results were again confirmed by an enzymatic assay (Fig. 5E). Altogether these results suggested that purine-degrading bacteria impacted levels of several purines in the gut and specifically UA both locally and in circulation.
Figure 5. Purine-degrading bacteria (PDB) modulate abundance of purines in cecum and circulation.

A) Experimental design. Anaerobic uric acid degradation by fecal samples from different groups is indicated using uric acid overlay plates as detailed in Methods. B) Community profiling by sequencing (COPRO-Seq) analysis of fecal samples from gnotobiotic B6 mice colonized with the ‘core’ community (n=4) or the ‘core plus purine-degrading bacteria’ community (n=3). The bar charts show the abundance of each species in each community. C) Heatmap of purines and related metabolites in cecal contents from GF (n=5), ‘core’ (n=3) and ‘core plus PDB’ (n=5) mice analyzed by LC-MS/MS. D) Values for adenine, xanthine, xanthosine, inosine, hypoxanthine, uric acid, and allantoin in cecal contents of the three mouse cohorts analyzed by targeted metabolomics and E) plasma uric acid levels analyzed by enzymatic assay were expressed as box-and-whisker plots with individual data points, where the boxes indicate the median values and the interquartile ranges and the whiskers represent the minimum and maximum values. Significance was calculated by one-way ANOVA test with the Tukey post-tests and is designated as follows: ***, p-value of <0.001; ****, p-value of <0.0001.
Transcriptional analysis identifies bacterial genes required for anaerobic growth on multiple purines.
Having established the role of PDB on lowering levels of UA systemically and having identified gut isolates capable of anaerobic purine metabolism, we sought to identify genes encoding these functions. Cultures of E. bolteae were cultivated in medium supplemented with UA or xylose plus NH4Cl (henceforth, “xylose”). We selected xylose for comparison as the growth rate of E. bolteae on this substrate was similar to that of UA, with doubling times of 2.6 h for xylose and 4.6 h for UA. For both substrates log-phase cells were harvested and libraries subjected to sequencing. We obtained ~3.6 ×107 reads/sample, of which 99.2% mapped to the E. bolteae genome. Fig. 6A shows reads per million (RPM) normalized to gene size plotted against the relative expression level for growth on the two substrates, limited to the 3217 (of 5993) differentially expressed genes (FDR <0.01, Suppl. Table 5). As expected, genes encoding 30S and 50S RNA Polymerase (RNAP) subunits show a slight bias (1.6-fold, Suppl. Fig. 7) towards the xylose substrate side, consistent with the faster growth rate observed on this substrate and the rate-limiting nature of RNAP subunit expression.52 Growth on UA promoted higher expression of 51 genes relative to xylose (cut-off >25-fold; Fig. 6A), including several predicted to encode micronutrient transport functions, one glycine-cleavage system and a probable electron bifurcating hydrogenase (Suppl. Fig. 7).
Figure 6. Identification of a gene cluster necessary for anaerobic bacterial growth on purines.

A) Comparison of transcriptional profiles for Enterocloster bolteae showing differentially-expressed genes (FDR <0.01) and reads per million (RPM)/gene size (kb) for cultures grown on xylose + NH4Cl (“Xylose,” upregulated genes to the left) or uric acid (upregulated genes to the right), highlighting genes encoding two adjacent predicted operons encoding functions likely necessary for uric acid metabolism (blue and red circles). Descriptions of additional upregulated genes including those encoding micronutrient transport, one glycine cleavage system and a probable bifurcating hydrogenase system as well as genes upregulated during growth on xylose are described in Suppl. Fig. 7. B) Diagram of the adjacent upregulated predicted operons, including genes CGC65_RS20560-RS20625, color-matched with the filled circles in panel (A). C) Representative alignments of chromosomal regions from multiple organisms that anaerobically catabolize uric acid. The genetic regions from E. bolteae shown in panel B are compared to those from Clostridioides difficile CD196 (CD196_RS16070 – RS16115), Fusobacterium varium (C4N18_RS01955 – RS01995) and (C4N18_RS03270 – RS03290), Edwardsiella tarda (ETATCC_RS03320 – RS03390) and E. coli MS 200–1 (HMPREF9553_RS03160 - RS03225). Matched genes are color-coded, and the percent similarities of the encoded proteins are indicated. Although selected genes appear to be conserved, their organization differs in different organisms, and in the case of F. varium do not occur in a contiguous chromosomal region. D) Growth of E. coli MS 200–1 wild-type and deletion variants (FER039 [ΔallB::tetA-sacB], FER041 [Δ(ygeW-arcC)::tetA-sacB], and FER063 [ΔygeV::tetA-sacB]) on plates lacking a carbon source, supplemented with glucose or allantoin, or prepared with overlays containing saturating amounts of uric acid, adenine, or hypoxanthine. E) In vivo experimental design. Mice were colonized with the Core community as in Fig. 5 and with either E. coli MS 200–1 wild-type or the deletion variant FER041 [Δ(ygeW-arcC)::tetA-sacB]. Anaerobic uric acid degradation by fecal samples from different groups is indicated using uric acid overlay plates. F) The levels of fecal E. coli in the bacterial communities were assessed by qPCR and (G) plasma uric acid levels measured by HPLC were expressed as box-and-whisker plots with individual data points, where the boxes indicate the median values and the interquartile ranges and the whiskers represent the minimum and maximum values. Significance was calculated by one-way ANOVA test with the Tukey post-tests and is designated as follows: *, p-value of <0.05; ***, p-value of <0.001.
Two adjacent and divergently-oriented putative operons, each encoding 6 genes, amounted to 12% of all RNA-Seq reads in E. bolteae grown on UA. These highly upregulated genes are indicated by the filled blue and red circles with their corresponding operons and putative gene products diagrammed (Fig. 6A, B). Notably, the encoded proteins are predicted to catalyze C-N cleavage and Se-dependent hydrolytic reactions. Also indicated are a purine permease, likely specific for UA uptake based on the conservation of residues S99 and S314 (T100 and S317 in E. coli UacT)53 and a knotted carbamoyl transferase/carbamate kinase, presumably required for ATP synthesis.54
Alignments of conserved chromosomal regions of purine-fermenting organisms illustrated conservation of five genes (E. coli nomenclature: dpaL, hydA, ssnA, ygeY, xdhD) among all taxa, although not with a conserved organization nor exclusively present in a contiguous genomic region across phyla (Fig. 6C). A variant of E. coli MS 200–1 bearing a deletion of the ygeW-dpaL-ygeY-hydA-arcC operon grew as the wild-type strain in medium supplied with glucose or allantoin but was unable to grow anaerobically using UA, adenine or hypoxanthine as the carbon and energy source. Conversely, a variant of E. coli MS 200–1 bearing a deletion of allB, encoding the enzyme catalyzing the first step of anaerobic allantoin metabolism,55 was unable to utilize allantoin but retained the ability to catabolize UA, adenine, and hypoxanthine, indicating distinct mechanisms of allantoin and purine metabolism in this organism (Fig. 6D), and in good agreement with low expression level of allB genes in UA-grown E. bolteae cells. Finally, a variant of E. coli MS 200–1 bearing a deletion of the ygeV gene (Fig. 6C) encoding a single-component sigma 54-type transcription factor grew normally on glucose and allantoin but failed to grow on all tested purines (Fig. 6D), consistent with previous data indicating transcription regulation of the adjacent ygeW operon by YgeV.46
To assess the role of this bacterial gene cluster involved in purine metabolism in vivo, we used the same approach as described for Fig. 5, where GF mice were colonized with the core community which lacks purine-degrading bacteria, the “core community plus E. coli MS 200–1 wild-type”, or the “core community plus the deletion variant FER041 [Δ(ygeW-arcC)::tetA-sacB] (Fig. 6E). Both the wild-type and the variant showed comparable levels of colonization in the gut under the conditions tested (Fig. 6F). Colonization with the wild-type strain resulted in lower levels of plasma UA compared with mice colonized with the core or the deletion variant FER041 (Fig. 6G). Collectively, these results suggest that the bacterial functions encoded by these genes contribute to uric acid homeostasis in vivo.
Detection of genes encoding purine degradation functions in bacterial genomes and transplanted mice.
Having identified genes required for anaerobic purine metabolism and uric acid homeostasis in mice we then sought to identify bacterial taxa containing these genes. We performed BLASTP of the NCBI RefSeq Genome Database (refseq_genomes) using parameters described in Methods. We detected 230 non-redundant bacterial taxa that had the five genes reliably detected among all experimentally confirmed purine-degrading taxa (dpaL, hydA, ssnA, ygeY, xdhD). These potential UA degraders included bacterial taxa belonging to Actinobacteria, Firmicutes, Proteobacteria, Fusobacteria and Spirochaetes (Suppl. Table 6).
Lastly we assessed the abundance of these genes in the cecum of ApoE gnotobiotic mice (Fig. 1, Fig. 7A) and correlated their abundance with levels of purine-related metabolites quantified in their cecum (Suppl. Table 7). We found that cecal levels of several purine-related metabolites including deoxyxanthosine, xanthosine and UA were negatively associated with the abundance of genes involved in anaerobic-purine degradation (Fig. 7B). Altogether these results highlight the potential of these genes as biomarkers for purine breakdown in the gut. Understanding how to manipulate the representation and function (as opposed to abundance of relevant genes) of purine-consuming species in the intestinal microbiota could potentially lead to means for preventing or treating hyperuricemia and associated conditions.
Figure 7. Correlation between cecal purines and gut bacterial genes encoding uric acid degradation in transplanted ApoE KO mice.

A) Abundance of genes encoding anaerobic purine degradation in gut metagenomes from gnotobiotic mice transplanted with cecal contents from strains with disparate atherosclerosis phenotypes (see Fig. 1). Differences between groups were evaluated using unpaired two-tailed Welch’s t-test. B) Correlation between abundance of nucleosides and their derivatives and bacterial functions was performed using Spearman correlation.
DISCUSSION
In this study we sought to identify microbially-regulated metabolites involved in atherosclerosis progression. Our initial gnotobiotic mouse transplant studies revealed that levels of several purines including UA were influenced by gut microbes and associated with atherosclerosis burden. These initial results motivated a deeper exploration into the role of the gut microbiome on purine homeostasis and led us to identify bacteria able to breakdown purines anaerobically, and to uncover a cluster of bacterial genes required for anaerobic catabolism of these substrates. We also demonstrated that taxa encoding these functions lowered circulating UA levels in mice. Altogether this work has implications beyond atherosclerosis: it provides insights into how gut bacterial metabolism may influence UA in the circulation and suggests that microbes able to catabolize purines anaerobically are important drivers of host purine homeostasis both locally (i.e., gut) and systemically. A recent preprint using complementary approaches, including hyperuricemic humans and mutagenesis in Clostridia species, identified the same bacterial gene cluster associated with uric acid degradation, and arrived to similar conclusions with regards to its role on host uric acid homeostasis.56
In humans, nearly two-thirds of purines are endogenously produced while the remainder comes from diet, which are primarily absorbed in the duodenum but absorption could occur in the large intestine as transporters of nucleosides are expressed in epithelial cells57–59 and their abundance increases when demand for purines increases, for example in colon cancer.60 Absorbed purines can be used by enterocytes or colonocytes or be degraded to UA. In most mammals, UA can be further metabolized to allantoin, but in hominoids the presence of an uricase gene bearing multiple mutations and premature stop codons results in the accumulation of UA.61 Since UA is relatively insoluble, humans are susceptible to diseases resulting from precipitation of UA including gout and kidney stones. In fact, the prevalence of hyperuricemia in the U.S. is ~20% among adults and has been steadily increasing in recent decades.62,63 This increase may be related to the prevalence of a high-purine diets, fructose beverages (known to increase UA levels), and alcohol consumption.64
The association between UA and CVD has been demonstrated for several conditions, such as hypertension,38 chronic kidney diseases,39 metabolic syndrome,40 atherosclerosis,41 and adverse cardiovascular outcomes,42 and even for UA levels in the normal to high range (from 5.2 to 6 mg/dL).65 Several mechanisms have been proposed to explain the role of UA in CVD, including endothelial dysfunction, systemic inflammation, and renin-angiotensin-aldosterone system activation. Our results in gnotobiotic ApoE KO mice (Fig. 1) and humans (Fig. 2) support this notion and suggest that levels of UA within the soluble range may contribute to disease progression. This is consistent with a recent mechanistic study suggesting that soluble UA activates the NLRP3 (NLR Family Pyrin Domain Containing 3) inflammasome.22 However, whether elevated serum UA levels are an independent risk factor for CVD remains controversial. There is conflicting evidence regarding the benefits of UA-reducing strategies for treating patients with CVD.24,25,66,67 Importantly, the beneficial effects of UA-lowering therapies on CVD are not yet established in large-scale randomized trials. Moreover, several Mendelian randomization studies showed mixed results for causal effects of serum uric acid on CVD outcomes.68,69 These inconsistent results are also observed in experimental animals. For example, in one study increasing the levels of UA with a high-purine diet did not change the development of atherosclerosis,70 while in a second study, lowering uric acid levels by administration of xanthine oxidase inhibitor or using uricase transgenic mice reduced the atherosclerosis development.22 Interestingly, the latter showed that UA promoted the production of the inflammatory cytokine IL-1b only in the presence of LPS, suggesting that the effects of UA on the cardiovascular system may be context-dependent. Collectively, more work is needed to clarify the contribution of hyperuricemia to atherosclerosis in humans: our study introduces an unappreciated factor, the microbial composition and nutritional milieu of the gut as discussed below.
The kidneys play a critical role in maintaining plasma UA levels through complex transport systems that mediate both reabsorption and secretion of UA.71 Intestinal secretion is a substantial contributor to extra-renal elimination of UA, accounting for about one-third of total elimination of UA.43,44 Our results showing that PDB lower the abundance of some purines in the intestine (Fig. 5) suggest that these organisms may lower circulating UA levels by decreasing the burden of purines bioavailable to the host. Alternatively, the increased levels of UA detected in the cecum of mice colonized with PDB (Fig. 5) may suggest that in the presence of these organisms, consumption of certain purines may trigger more secretion of UA into the intestine. More studies are needed to clarify how PDB influences gut and systemic levels of purines. Given the differential purine-metabolizing capabilities and environmental modulation of purine consumption among the examined PDB, this may vary widely depending on the specific PDB colonizing an individual.
Anaerobic purine utilization by bacteria was first described over 100 years ago, yet relatively few species have been identified and these isolates were obtained from environmental sources and are obligate purinolytic Firmicutes. The biochemistry of their purine catabolic pathway was delineated prior to the advent of genetic manipulations and only in the past decade have their genomes been sequenced, although the genetic elements encoding the process remain undefined.28,29 More nutritionally-diverse anaerobic purine-utilizing organisms, including Firmicutes and Proteobacteria isolated from termite intestines, metabolize purines and evidently recycle purine nitrogen in the nitrogen-limited host diet, but further biochemical and genetic analyses of these organisms have not been published.72 Operons encoding proposed pathways of anaerobic purine degradation have been identified through in silico analyses,73,74 through overexpression of individual genes54,75 or by using cultures and cell suspensions cultivated in complex media containing additional carbon and energy sources.46 While these efforts identified the appropriate genomic regions encoding functions necessary for anaerobic purine catabolism, the appropriate conditions allowing reproducible growth on multiple purines were not formulated and a full understanding of the encoded metabolism has remained elusive.
We asked to what extent UA could serve as a source of carbon and energy for gut bacteria, and to what extent the gut microbiota composition might affect host systemic purine concentrations. We tested 34 human gut isolates from six phyla cognizant of a) the demonstrated requirements of trace elements (Mo, Se) for metabolic function47 and b) the well-known phenomenon of catabolite repression reported for the yge operon of E. coli and likely present in other bacteria.76 We identified representatives for three different phyla, primarily belonging to the Firmicutes and Proteobacteria, that readily degraded UA. A limited subset of organisms was shown to utilize adenine and hypoxanthine and, independent of the ability to use UA or adenine, some organisms including strains of E. coli grew anaerobically in medium supplied with allantoin (Fig. 4, Suppl. Fig. 4, Suppl. Fig. 5A), in contrast to reports that this compound only serves as a source of nitrogen.55 However, these properties were not consistently present in any taxonomic group and even strain differences between species were identified. Moreover, the presence of an alternative carbon source (glucose and/or fructose) reduced or eliminated UA metabolism, confirming transcriptional regulation by catabolite repression, and indicating one nutritional parameter that could modulate purine utilization in the gut.
In two Proteobacteria the ability to utilize UA was influenced by the presence of Se and Mo (Suppl. Fig. 5B). However, the effects of metals were not evident in C. difficile and E. bolteae, likely reflecting the high expression of multiple metal uptake systems identified in the RNA-Seq data for E. bolteae (Suppl. Fig. 7) and the requirement for multiple metal-limited transfers to affect mineral limitation in other purinolytic Firmicutes.47,48,77 The notion that dietary Se might affect the metabolism of purines by human gut microbiota has been suggested49,74 but the in vitro results shown here indicate that any requirement could depend upon microbiota composition: necessary if purine utilizers mainly are members of the Proteobacteria but perhaps of less importance if Firmicutes predominate. Altogether, these results (i) suggest that phylogeny is a poor predictor of microbial purine utilization; (ii) indicate that the presence of the identified genes does not correlate with the breadth of purines utilized by an organism; (iii) demonstrate effects on purine metabolism of two nutritional parameters–i.e., carbon source and metals availability; and (iv) underscore the need for assessments beyond genomics when making predictions about purine metabolism by the gut microbiota.
Lastly, it is important to note that while the notion of using bacteria able to degrade purines might be an appealing strategy to lower pro-inflammatory UA, more work is needed to fully understand the consequences to the host. For example, the intestinal epithelium is the most vigorously self-renewing tissue of adult mammals, which imposes a high demand of nucleotides that are needed for proliferation and energy.78 Adenine–a purine consumed by several taxa in our study (Fig. 4, Suppl. Fig. 4)–is a precursor of nucleic acids in intestinal cells unable to synthesize purines de novo.79 Furthermore, recent work from the Colgan group showed that gut bacteria are a major source of purines that are used for nucleotide generation by the intestinal mucosa. Importantly, supplementation of purines directly through bacterial colonization improved intestinal epithelial cell wound healing and barrier restitution capabilities and suggested that purines play essential roles for colonic epithelial proliferation, energy balance, and mucin barrier integrity.80,81 Adenine also inhibits TNF (Tissue Necrosis Factor)-α signaling in intestinal epithelial cells and reduces mucosal inflammation in a dextran sodium sulfate-induced colitis mouse model.82 Consistent with these results, a recent study identified intestinal purine starvation associated with irritable bowel syndrome.83 Our results showing that PDB can metabolize a variety of purines and lower hypoxanthine levels in the intestine, and that the abundance of genes encoding for key proteins in anaerobic purine degradation is associated with lower purines in the gut suggests that the influence of these organisms on purine availability to intestinal epithelial cells and barrier function needs to be carefully examined, especially considering that many of the taxa identified as anaerobic purine degraders, including E. coli, E. bolteae, F. varium, and C. difficile, have been associated with disease.84,85 Thus, further studies are warranted to examine the contribution of purine degradation to the fitness of these taxa and host health.
In summary, the work presented here shows that anaerobic purine utilization is widespread among gut-dwelling bacteria and suggests that microbial purine degraders are important modulators of host purine homeostasis in the gut and of UA levels in circulation. Studies are needed to dissect the contribution of aerobic vs. anaerobic purine consuming pathways to the purine economy, gut ecology and health conditions including atherosclerosis.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Federico Rey (ferey@wisc.edu).
Materials availability
E. coli strain constructs described in this manuscript are available upon request.
Data and code availability
The accession number for sequence data used in the study (Mouse Metagenomics data, Bacterial RNA-Seq data, Copro-Seq data, Human Metagenomics data) are PRJNA904303, PRJNA911264, PRJNA903666, and EGAS00001004480 (European Genome-Phenome Archive). Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code is available here: https://github.com/qijunz/Purine_paper
Any additional information required to reanalyze the data reported in this paper including the custom Python scripts employed in the processing of metagenomic data is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Bacterial culture conditions.
Cultures and plates were prepared in an anaerobic (ca. 75% N2/20% CO2/5% H2) chamber. Media formulations and preparations are detailed in Methods S1. Cultures were routinely grown at 37°C in anaerobic septum-stoppered “Hungate tubes” containing rich, well-buffered (pH 7) media, typically “Mega Medium”86 supplemented with maltose (0.9 g/l), cellobiose (0.86 g/l), fructose (0.46 g/l) and NaHCO3 (1.68 g/l) although most clostridial strains were reliably recovered from 20% glycerol freezer stocks using medium 11E (see Methods S1). Freshly-prepared cultures were combined at roughly equivalent levels (normalized to OD600) for the purpose of colonizing germ free mice. Growth on purines utilized medium 23B, which contains 0.1% yeast extract (0.05% for the RNA-Seq cultures) as the sole undefined component, with additions of carbohydrate and NH4Cl (25 and 10 mM, respectively) or purines (UA, 1 mg/ml; adenine, 1 mg/ml; allantoin, 45 mM) as carbon and nitrogen sources. These levels of UA and adenine did not dissolve fully, and growth in tubes was monitored by observing purine disappearance as well as culture OD600, measured using a Spectronic 20D+ (ca. 1.4 cm sample path length) after the saturating purine had settled (ca. 15 minutes). Commercially-available strains and isolates have been verified by 16S rRNA gene sequencing and are specified in the Key Resources Table. As described below, additional steps were employed to verify isolate Escherichia coli I-11.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Monocyte + Macrophage antibody [MOMA-2] | abcam | Cat#ab33451 |
| Anti-Actin, α-Smooth Muscle – FITC antibody, Mouse monoclonal | Sigma-Aldrich | Cat#3777 |
| Monoclonal Anti-FITC-Biotin antibody produced in mouse | Sigma-Aldrich | Cat#B0287 |
| Rabbit Anti-Rat IgG H&L (Biotin) | abcam | Cat#ab6733 |
| Bacterial and virus strains | ||
| Bifidobacterium dentium | ATCC | Cat#27678 |
| Collinsella aerofaciens | ATCC | Cat#25986 |
| Bacteroides caccae | ATCC | Cat#43185 |
| Bacteroides finegoldii | DSM | Cat#17565 |
| Bacteroides stercoris | ATCC | Cat#43183 |
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | Cat#29148 |
| Bacteroides xylanisolvens | DSM | Cat#18836 |
| Phocaeicola dorei | DSM | Cat#17855 |
| Blautia hansenii | ATCC | Cat#27752 |
| Blautia luti | DSM | Cat#14534 |
| Blautia producta | Lab isolate | N/A |
| Clostridioides difficile CD196 | Jan Peter van Pijkeren (University of Wisconsin-Madison) |
N/A |
| Clostridium scindens | Lab isolate | N/A |
| Clostridium sporogenes | ATCC | Cat#15579 |
| Clostridium symbiosum | ATCC | Cat#14940 |
| Coprococcus comes | ATCC | Cat#27758 |
| Dorea formicigenerans | ATCC | Cat#2755 |
| Dorea longicatena | DSM | Cat#13814 |
| Enterocloster asparagiformis | DSM | Cat#15981 |
| Enterocloster bolteae | ATCC | Cat#BAA-613 |
| Erysipelatoclostridium ramosum | ATCC | Cat#25582 |
| Hungatella hathewayi | DSM | Cat#13479 |
| Megamonas funiformis | DSM | Cat#19343 |
| Mitsuokella multacida | ATCC | Cat#27723 |
| Ruminococcus torques | ATCC | Cat#27756 |
| Fusobacterium nucleatum | ATCC | Cat#23726 |
| Fusobacterium varium | ATCC | Cat#8501 |
| Victivallis vadensis | ATCC | Cat#BAA-548 |
| Citrobacter youngae | ATCC | Cat#29220 |
| Edwardsiella tarda | ATCC | Cat#23685 |
| Escherichai coli I-11 | Lab isolate | N/A |
| Escherichia coli K12 | ATCC | Cat#29425 |
| Escherichia coli MS 200–1 | Emily Balskus (Harvard University) |
N/A |
| E. coli MS 200–1 with ΔallB::tetA-sacB | This manuscript | FER039 |
| E. coli MS 200–1 with Δ(ygeW-arcC)::tetA-sacB | This manuscript | FER041 |
| E. coli MS 200–1 with ΔygeV::tetA-sacB | This manuscript | FER063 |
| T-SACK | Thomason et al. 88 | N/A |
| Biological samples | ||
| Human peripheral plasma | Human study | N/A |
| Human stool samples | Human study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Adenine | Alfa Aesar | Cat#A14906 |
| Allantoin | Alfa Aesar | Cat#A15571.30 |
| Hypoxanthine | Thermo Fisher Scientific |
Cat#A11481.06 |
| Uric Acid | Sigma-Aldrich | Cat#U0881 |
| Oxonic acid potassium salt | Thermo Fisher Scientific |
Cat#J60400.14 |
| Phenol:Chloroform:Isoamyl alcohol | Invitrogen / Thermo Fisher Scientific | Cat#15593–049 |
| 0.1 mm zirconia/silic beads | BioSpec Products | Cat#11079101z |
| Bacterial media formulation, detailed preparation | This manuscript, Methods S1 | N/A |
| Oil Red O | Sigma-Aldrich | Cat#O0625 |
| Streptavidin/HRP | Agilent Technologies | Cat#P0397 |
| DAB, Liquid | Agilent Technologies | Cat#K3467 |
| Hematoxylin Solution, Mayer’s | Sigma-Aldrich | Cat#MHS16 |
| Tissue-Tek O. C.T. compound | Sakura Finetek, Tokyo, Japan | Cat# 4583 |
| D9-Choline | Cambridge Isomiddlee Laboratories | Cat#DLM-549–1 |
| D9-TMAO | Cambridge Isomiddlee Laboratories | Cat#DLM-4779-PK |
| Acetic acid-d4 | Sigma-Aldrich | Cat#233315 |
| Propionic acid-d6 | Sigma-Aldrich | Cat#490644 |
| Butyric acid-d7 | CDN Isomiddlees | Cat#D-0171 |
| N-tert-Butyldimethylsilyl-N-methyltrifluoroacetamide | Sigma-Aldrich | Cat#394882 |
| SsoAdvanced SYBR Green Supermix | BioRad Laboratories, Inc. | Cat#1725271 |
| Critical commercial assays | ||
| Monarch Total RNA Miniprep Kit | New England Biolabs Inc. |
Cat#T2010S |
| Broad Range Qubit | Invitrogen / Thermo Fisher Scientific | Cat#Q32853 |
| Nucleospin Gel and PCR Clean-up Kit | Macherey-Nagel GmbH & Co. | Cat#740609.250 |
| Trichrome Stain Kit | Abcam | Cat#ab150686 |
| Cholesterol E | FUJIFILM Wako | Cat#999–02601 |
| HDL-Cholesterol E | FUJIFILM Wako | Cat#997–01301 |
| Multi-Calibrator Lipids | FUJIFILM Wako | Cat#464–01601 |
| L-Type Triglyceride M Enzyme Color A | FUJIFILM Wako | Cat#461–08992 |
| L-Type Triglyceride M Enzyme Color B | FUJIFILM Wako | Cat#461–09092 |
| QCL-1000 Endpoint Chromogenic LAL Assay | Lonza | Cat#50–647U |
| Deposited data | ||
| Mouse Metagenomics data | This study | PRJNA904303 |
| Bacterial RNA-Seq data | This study | PRJNA911264 |
| COPRO-Seq data | This study | PRJNA903666 |
| Human Metagenomics data | Wu et al. 36 | EGAS00001004480 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| AXB10/PgnJ | The Jackson Laboratory | Cat#001681 |
| BXD5/TyJ | The Jackson Laboratory | Cat#000037 |
| BTBR T+tf/J | The Jackson Laboratory | Cat#002282 |
| BXA8/PgnJ | The Jackson Laboratory | Cat#001704 |
| Germ-free C57BL/6J | Germ-free mice raised in the gnotobiotic facility at the University of Wisconsin–Madison |
N/A |
| Germ-free ApoE KO | Germ-free mice raised in the gnotobiotic facility at the University of Wisconsin–Madison |
N/A |
| Oligonucleotides | ||
| qPCR primer 401F | Walker et al. 98 | N/A |
| qPCR primer 611R | Walker et al. 98 | N/A |
| 5’- ATATTGAACATTGAGTTAAAAACCAATCTGTATTTTA CAAGGAGTTTGTTTCCTAATTTTTGTTGACACTCTA- 3’ |
This manuscript | allBtetA-f |
| 5’- ACGGGCATTGCAGGGCCAGATTACTGCTGATGTTTA AGGATAAATTGACCATCAAAGGGAAAACTGTCCATA T-3’ |
This manuscript | allBsacB-r |
| 5’-CTTTATGGAACCGTTATCACG-3’ | This manuscript | allBseq-f |
| 5’-CAGAAAGTAGCGTAGGAGG-3’ | This manuscript | allBseq-r |
| 5’- AGTCACAGTGAAATAAACCACTTTGCCTGTCATTCC ACTACCGGGACTTTTCCTAATTTTTGTTGACACTCTA −3’ |
This manuscript | ygeWtetA-f |
| 5’- AGTGGCTTAAATGAAAATGGCCGGATGGTATTACCC ATCCGGCCTTTAAAATCAAAGGGAAAACTGTCCATA T-3’ |
This manuscript | arcCsacB-r |
| 5’-ACAAACAACCGGAAGCTGG-3’ | This manuscript | ygeWseq-f |
| 5’-GATATCGATCCTCGTGGTG-3’ | This manuscript | arcCseq-r |
| 5’- ACTTTTAAGTAAGTCTGGAAGGTAAAGCTATGGAGC TTGCGACTACGCAGTCCTAATTTTTGTTGACACTCTA −3’ |
This manuscript | ygeVtetA-f |
| 5’- CGGATTCTCAGAGGTTATGTGTTTAACAACTCATATT TCTTAATCTTGCGATCAAAGGGAAAACTGTCCATAT- 3’ |
This manuscript | ygeVsacB-r |
| 5’-GCAGGGTATCTCTCTTCAG-3’ | This manuscript | ygeV-seqF |
| 5’-GCGATGCTGGCAAAACCAC-3’ | This manuscript | ygeV-seqR |
| Recombinant DNA | ||
| Plasmid pSIM5 | Thomason et al. 88 | |
| Software and algorithms | ||
| R v3.6.3 or v4.0.3 | The R foundation | https://www.rproject.org/ |
| Python v3.7.4 | Python Software Foundation | https://www.python.org/ |
| FASTX | Cold Spring Harbor Laboratory | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Bowtie2 | Langmead et al. 100 | https://bowtiebio.sourceforge.net/bowtie2/ |
| BLAST | NCBI | https://blast.ncbi.nlm.nih.gov/ |
| RSEM | Li et al. 101 | https://github.com/deweylab/RSEM/ |
| Kraken2 | Wood et al. 104 | https://ccb.jhu.edu/software/kraken2/ |
| Xgboost | Chen et al. 105 | N/A |
| PLS-DA | Pang et al. 107 | https://www.metaboanalyst.ca/ |
| Prism | Graphpad | https://www.graphpad.com/ |
| COPRO-Seq pipeline | McNulty et al. 99 | https://github.com/nmcnulty/COPROSeq |
| caret | R package 106 | https://cran.rproject.org/web/packages/caret/ |
| ImageJ | National Institute of Health | Imagej.net |
| Syngo Calcium Scoring Software | Siemens | N/A |
| Other | ||
| Western diet | Research Diets | Cat#D10042101 |
| LabDiet 5021 (Autoclavable mouse breeder diet) | LabDiet | Cat#5021 |
| Standard chow supplemented with 0.2% cholesterol | Envigo | TD.07798 |
| Vistro DT60 II Analyzer | University of Massachusetts Medical School MMPC |
N/A |
| Shimadzu HPLC system (CBM-40 controller, LC-40D pumps, SIL-40C autosampler, CTO-40C column oven, SPD-M40 diode array detector) | Shimadzu Scientific Instruments, Columbia, MD |
N/A |
| Luna Omega 5 μm Polar C18 LC column (250 × 4.6 mm) | Phenomenex | Cat# 00G-4754-E0 |
| Dionex UltiMate 3000 uHPLC | Thermo Scientific | N/A |
| Q Exactive Orbitrap Mass Spectrometers | Thermo Scientific | N/A |
| Bio-Bond C4 3 μm column (150 × 2.1 mm) | Dikma | Cat#84413 |
| Agilent 7890B GC with 5977A MSD | University of Wisconsin Mass Spectrometry Facility |
N/A |
| Agilent DB-1 1 ms 0.25 mm x 60 m column | University of Wisconsin Mass Spectrometry Facility |
N/A |
| SH-Stabilwax column (30 m, 0.25 mm ID, 0.10 μm df) | Shimadzu Scientific Instruments, Columbia, MD |
Cat#227-36246-01 |
| Bacterial RNA sequencing | Microbial Genome Sequencing Center (MiGS) |
N/A |
| Untargeted UPLC Mass Spectrometry | Metabolon, Inc. Morrisville, NC | N/A |
| Targeted purine and pyrimidine Mass Spectrometry | TMIC, The Metabolomics Innovation Centre, UVic Genome BC Proteomics Centre, Victoria, Canada |
N/A |
| MiSeq | University of Wisconsin Biotechnology Center DNA Sequencing Facility |
N/A |
| HiSeq 2500 | University of Wisconsin Biotechnology Center DNA Sequencing Facility |
N/A |
| Roche Cobas analyzer | Sahlgrenska University University Hospital |
N/A |
| Dual-source CT scanner with a Stellar Detector (Siemens) | Sahlgrenska University University Hospital |
N/A |
| Nikon Eclipse TE300 Inverted Microscope | University of Wisconsin-Madison Experimental Animal Pathology Laboratory |
N/A |
| Mini-Bead Beater 96 | BioSpec Products, Barlesville, OK | Cat#1001 |
| 20 ml headspace vials | Restek, Bellefonte, PA | Cat#21163 |
| Amicon Ultra-0.5 3kDa ultrafiltration device | MilliporeSigma | Cat#UFC500324 |
Gnotobiotic husbandry.
All GF C57BL/6J and ApoE KO mice were maintained in a controlled environment in either plastic flexible film gnotobiotic isolators or individually ventilated cages under a strict 12 h light cycle and received sterilized water and standard chow (LabDiet 5021) ad libitum unless otherwise stated. The age and sex of mice used in different experiments are specified below. Sterility of GF animals was assessed by routine PCR testing (16S rRNA gene) and by incubating freshly collected fecal samples under aerobic and anaerobic conditions using standard microbiology methods. The animal experiments were conducted according to relevant national and international guidelines and were approved by the UCLA Animal Research Committee, the UCLA IACUC, or the University of Wisconsin-Madison Animal Care and Use Committee, as appropriate.
Human studies.
The Impaired Glucose Tolerance (IGT) and Microbiota study is a prospective observational study of subjects aged between 50 and 64 years. In the IGT-Microbiota study, more than 5,000 men and women born in Sweden with a range of glucose tolerance based on their fasting glucose values and an oral glucose tolerance test (OGTT) were screened. Here we included the same sub-population (n=1011, 44% male) as in Wu et al 202036 where plasma urate levels were measured resulting in a cohort of 998 individuals after exclusion. Exclusion criteria were: known diabetes, other severe disease that may jeopardize interpretation of results, e.g. inflammatory bowel disease, rheumatic diseases, malignancy (unless no relapse during 5 years of follow-up), treatment with steroids or immune-modulating treatment, pharmacological treatment of infection during the last 3 months and major cognitive dysfunction. The ethics committee at Gothenburg University approved the study (Dnr 560–13) which was conducted in accordance with the Declaration of Helsinki. Participants gave written informed consent.
METHODS DETAILS
Plate assay of anaerobic purine utilization.
These assays employed saturating levels of UA, adenine, or hypoxanthine using bilayer plates similar to those described previously.87 Specifically, working in the glove bag a 25 ml base layer consisting of a 1:1 mixture of medium 26B plus 2.4% molten Bacto agar was poured into each 100 × 15 mm petri dish and allowed to solidify. The following day, the base layer was overlaid with a 7 ml top layer medium + agar plus (per 7 ml) 84 mg UA, 96 mg adenine, or 91 mg hypoxanthine (see Methods S1). As for the liquid cultures, these levels of purines were saturating and formed and opaque overlay. Where indicated, filter-sterilized additions of concentrated stock solutions (e.g., glucose, fructose, NH4Cl, formate) were added to indicated levels in both base and overlay medium layers. Plates were allowed to dry for two days, then spotted with 4 μl of an overnight culture grown in rich medium (CMM or 11E) and incubated anaerobically at 37°C for 2 (UA, soluble substrates), 3 (hypoxanthine) or 7 (adenine) days, unless otherwise specified. Cultures that utilized purines both grew on the medium and formed zones of clearing as the saturating purine was depleted. Attempts to prepare overlay plates containing xanthine or guanine at levels useful to support growth did not show clear zones of purine utilization. Similarly, monolayer plates containing UA resulted in relatively indistinct zones of clearing relative to bilayer plates, often with formation of a dense ammonium urate precipitate especially after storage at 4°C. Five cultures were spotted/plate--E. bolteae or E. coli MS 200–1 as a positive control plus 4 test strains.
Trace mineral requirements for growth on UA.
Tests utilized the standard phosphate-buffered basal medium formulation (“26B” with added 100 nM Mn, Ni, Zn, 50 nM Co, W) plus combinations of the following: 2.5 μM Fe, 5 μM Mo, 0.5 μM Se as indicated (specific mineral compounds are listed in Methods S1). No attempt was made to rigorously exclude the individual metal being tested, and it is likely that other medium components—particularly phosphate buffer, cysteine and yeast extract—supplied trace levels; therefore, evidence of mineral requirements indicates a substantial demand.
Inactivation of allB, ygeV, and ygeW-arcC.
The allB gene (Locus: NZ_GG773866; HMPREF9553_RS01540), the ygeV gene (HMPREF9553_RS03160), and the ygeW-arcC operon (HMPREF9553_RS03165 - RS03180) of E. coli MS 200–1 were deleted and replaced with the tetA-sacB cassette amplified from T-SACK in accord with standard recombineering methods, followed by elimination of the pSIM5 helper plasmid.88 The resulting constructs were verified by sequencing across the cassette-genome junctions. Primers used in these constructs are listed in the Key Resources Table. Unfortunately E. coli MS 200–1 proved to be resistant to phage P1 transduction and the constructs could not be transferred to naive recipients.
Isolation of E. coli strain I-11.
This strain was isolated from a de-identified human fecal sample collected in accord with University of Wisconsin Health Science Institutional Review Board. For the isolation, approximately 20 mg of aseptically-sampled material was injected into a 10-ml medium 23B anaerobic culture supplemented with 10 mg of UA, then incubated at 37°C. Upon observation of growth and disappearance of the UA precipitate, the culture was transferred (1:100 dilution) into the same medium, maintained under the same conditions, this was repeated then the enrichment was streaked to UA bilayer plates. Colonies demonstrating UA metabolism were purified and identified by sequencing of the 16S rRNA gene. Strain I-11 was a facultative rod-shaped organism, which fermented glucose, sucrose and lactose but not cellobiose, as expected for E. coli.89 A diagnostic PCR analysis confirmed the identification.90
RNA isolation and sequencing.
10 ml cultures were grown in triplicate in tubes containing medium 23B (with 0.05% yeast extract) plus a) 25 mM xylose + 10 mM NH4Cl or b) 12 mg UA and harvested at OD600 = 0.25 by plunging the tubes into an ice water slurry, then pelleting cells prior to storage at −80°C. RNA was isolated using the Monarch Total RNA Miniprep kit with yields of 2000 – 3000 ng RNA per sample, as assayed by dye binding (Qubit). Samples in were submitted to the Microbial Genome Sequencing Center (MiGS, Pittsburgh, PA) where Illumina Stranded RNA library preparation with RiboZero Plus rRNA depletion was performed, followed by Illumina sequencing [paired-end reads (2×50bp)]. The sequence data was processed by MiGS as follows: quality control and adapter trimming was performed with bcl2fastq [ver. 2.20.0.445]. Read mapping was performed with HISAT2 [ver. 2.2.0].91 Read quantification was performed using Subread’s featureCounts [ver. 2.0.1] functionality.92 Read counts were loaded into R [ver. 4.0.2, R Core Team (2020)] and normalized using edgeR’s Trimmed Mean of M values (TMM) algorithm (ver. 1.14.5).93 Subsequent values were then converted to counts per million (cpm).
Mouse experimental design.
We conducted four animal experiments as follows. i) At UCLA six-week-old female AXB10/PgnJ, BTBR T+tf/J, BXD5/TyJ, and BXA8/PgnJ conventionally-raised mice from the HMDP cohort were fed a Western diet (Research Diets D10042101) for 4 weeks, and cecal contents were collected. Frozen cecal samples were shipped to the University of Wisconsin-Madison for microbiota transplant. Ten-week-old GF female ApoE KO mice (C57BL/6J background) fed a standard chow (LabDiet 5021) were inoculated by oral gavage with 0.2 mL of resuspended cecal slurry from these HMDP strains. Mice were switched to a standard chow diet supplemented with 0.2% cholesterol (TD.07798, Envigo). Mice were then euthanized at 18 weeks of age after 4h fasting and tissues collected. ii) Cecal and plasma samples were collected from eighteen-week-old GF or conventionally-raised ApoE KO mice fed the 0.2% cholesterol-supplemented diet (TD.07798) for 8 weeks. iii) Three groups of adult gnotobiotic C57BL/6J mice on a standard chow were tested for purine metabolism: a) mice bearing a “core” community which included seven species that do not degrade purines in vitro, Bacteroides caccae, Bifidobacterium dentium, Blautia hansenii, Bacteroides thetaiotaomicron, Coprococcus comes, Mitsuokella multacida, and Ruminococcus torques; b) mice bearing the “core plus purine-degrading bacteria (PDB)” community that added three PDB to the “core” community mixture including E. bolteae, H. hathewayi, and E. coli; and c) one group remained germ-free. iv) Three groups of adult gnotobiotic C57BL/6J mice on a standard chow were tested for purine metabolism: a) the same “core” community as above; b) the “core plus E. coli MS 200–1 wild-type”; and c) the “core plus the E. coli MS 2001 deletion variant FER041”. In the latter two experiments, oxonic acid, a uricase inhibitor, was supplemented in the drinking water (1.5% w/v) as previously described94 and samples were collected 4 weeks after the colonization.
Atherosclerotic lesion assessments.
Atherosclerotic lesions were assessed as previously described.95 Briefly, mice were anesthetized and the aorta was perfused with PBS. To assess the atherosclerotic lesion size at the aortic sinus, the samples were cut in the ascending aorta, and the proximal samples containing the aortic sinus were embedded in Tissue-Tek OCT compounds. Five consecutive sections (10 μm thickness) taken at 100 μm intervals (i.e. 50, 150, 250, 350, and 450 μm from a bottom of the aortic sinus) were collected from each mouse and stained with Oil Red O. Plaque area and Oil Red O-positive area were measured using Image J software. The volume of atherosclerosis in the aortic sinus was expressed as mean size of the 5 sections for each mouse. Immunohistochemistry was performed on formalin-fixed cryosections of mouse aortic roots using antibodies to identify macrophages (1:50), followed by detection with biotinylated secondary antibodies (1:400) and streptavidin-horseradish peroxidase (1:500). Smooth muscle cells were identified by immunostaining with fluorescein isothiocyanate (FITC)-conjugated primary antibody against α- smooth muscle actin (1:100), followed by anti-FITC biotin-conjugated secondary antibody (1:400). Negative controls were prepared by substitution with an isotype control antibody. Staining with Masson’s trichrome was used to delineate the fibrous area according to the manufacturer’s instructions. Stained sections were digitally captured, and the percentage of the stained area (the stained area per total atherosclerotic lesion area) was calculated.
Plasma biochemical analysis.
Blood samples were collected by cardiac puncture into EDTA-rinsed syringes under anesthesia using isoflurane. Plasma was acquired by centrifugation and stored at −80°C until measurement. The levels of triglycerides, total cholesterol, and high-density lipoprotein cholesterol were measured with commercially available kits from Wako Chemicals. Plasma LPS levels were quantitated with the QCL-1000 Endpoint Chromogenic LAL Assay. Plasma UA levels were determined by the Vistro DT60 II Analyzer at the University of Massachusetts Medical School MMPC (National Mouse Metabolic Phenotyping Center). For the experiment shown in Fig. 6G, plasma UA was measured using the HPLC system described below, having first diluted plasma samples 8-fold in PBS and passing the diluted plasma through a 3kDa ultrafiltration device (Amicon Ultra-0.5).
DNA Extraction from cecal and fecal samples.
DNA was extracted from samples according to published bead-beating procedures.10,96,97 In short, fecal or cecal samples were resuspended in a solution containing 500 μl of 2× extraction buffer [200 mM Tris (pH 8.0), 200 mM NaCl, 20 mM EDTA], 210 μl of 20% SDS, 500 μl phenol:chloroform:isoamyl alcohol (pH 7.9, 25:24:1) and 500 μl of 0.1 mm diameter zirconia/silica beads. Cells were mechanically disrupted using a bead beater (BioSpec Products) for 3 min at room temperature. The aqueous layer was collected and DNA precipitated using 600 μl isopropanol and 60 μl 3M Na-acetate. Pellets were dried with ethanol and resuspended in TE buffer. A NucleoSpin Gel and PCR Clean-up Kit (Macherey-Nagel) was used to remove contaminants. Isolated DNA was stored at −80°C until downstream processing.
qPCR analysis.
The level of E. coli in the bacterial communities described in Fig. 6F were assessed by quantitative extraction of DNA from fecal samples by bead beating followed by qPCR analysis of 2 ng purified DNA samples using the E. coli-specific primers 401F and 611R and SsoAdvanced SYBR Green Supermix (BioRad) with 40 amplification cycles.98 The method generated a linear standard curve (0.02 – 20000 pg) using purified E. coli MS 200–1 DNA and a uniform melt curve for all standards and sample products.
COPRO-Seq analysis.
Bacteria communities resulting from inoculation of GF animals were analyzed using Illumina sequencing according to the COPRO-Seq (community profiling by sequencing) method.99 Feces were collected 4 weeks after the colonization. In short, DNA isolated from feces via bead beating was used to prepare libraries for shotgun Illumina sequencing. Five hundred nanograms of DNA from each sample was fragmented by sonication and subjected to enzymatic blunting and adenine tailing. Customized Illumina adapters containing maximally distant 8-bp barcodes were ligated to the poly (A)-tailed DNA. Gel-extracted DNA (size selection ~250 to 300bp) was amplified by PCR using primers and cycling conditions recommended by Illumina. Purified PCR products were submitted to the UW-Madison Biotechnology Center for a single end 50-bp Illumina MiSeq run. Results were processed using the software pipeline detailed by McNulty et al.99
Metagenomic shotgun DNA sequencing.
DNA was extracted from cecal contents of individual mice as described above. Following DNA extraction, Illumina paired end libraries were constructed using a previously described protocol,51 with a modification of gel selecting DNA fragments at ∼450 bp in length. Paired end (PE) reads (2 × 125) were generated using HiSeq 2500 platform.
Metagenomic reads processing.
Raw reads were preprocessed using Fastx Toolkit (ver. 0.0.13): (1) for demultiplexing raw samples, fastx_barcode_splitter.pl, with –partial 2, mismatch 2 was used; (2) when more than one forward and reverse read file existed for a single sample (due to being run on more than one lane, more than one platform, or at more than one time), read files were concatenated into one forward and one reverse read file; (3) barcodes were trimmed to form reads (fastx_trimmer -f 9 -Q 33); (4) and reads were trimmed to remove low quality sequences (fastq_quality_trimmer -t 20 -l 30 -Q33). Following trimming, unpaired reads were eliminated from the analysis using custom Python scripts. To identify and eliminate host sequences, reads were aligned against the mouse genome (mm10/GRCm38) using bowtie2 (ver. 2.3.4)100 with default settings and microbial DNA reads that did not align to the mouse genome were identified using samtools (ver. 1.3; samtools view -b -f 4 -f 8).
Microbiome trait quantification.
Quantification of microbial genes was done by aligning clean paired end reads from each sample to a previous published mouse gut microbiome non-redundant gene catalog using Bowtie2 (ver. 2.3.4) and default parameters. RSEM (ver. 1.3.1) was used to estimate microbial gene abundance.101 Relative abundance of microbial gene counts per million (CPM) were calculated using microbial gene expected counts divided by gene effective length then normalized by the total sum. To obtain abundance information for microbial functions, CPM of genes with the same KEGG Orthology (KO) annotation were summed together. In case there were multiple KO annotations for a single gene, we used all KO annotations. To obtain taxonomic abundance, CPM of genes with the same NCBI taxa annotation were summed together at phylum, order, class, family, and genus levels with a minimum of 10 genes in each taxon.
HPLC analysis.
Analyses of growth media and ultra-filtered serum samples (Fig. 6G) were performed using a Shimadzu system comprised of a CBM-40 controller, LC-40D pumps, SIL-40C autosampler, CTO-40C column oven, and SPD-M40 diode array detector. Samples were applied to a Phenomenx Luna Omega 5μm Polar C18 LC column maintained at 25°C with a 0.5 ml/min gradient composed of (A) 100 mM KxHxPO4 pH 2.4 and (B) A with 40% acetonitrile as follows (Time in minutes, %A, %B): 0, 99, 1 / 15, 96, 4 / 25, 25, 75 / 35, 25, 75 / 36, 99, 1 / 50, 99,1. Under these conditions formate, lactate and acetate eluted at 7.1, 10.1, and 10.6 minutes and 1 mM concentrations were readily detected at 205 nm. UA eluted at 17.4 minutes and 10 μM was detected at 284 nm. As some samples contained saturating substrate levels, for UA analyses all samples were vigorously mixed and immediately diluted 40-fold in phosphate buffered saline to allow full UA dissolution prior to analysis.
Headspace Gas Chromatography.
Analyses of short-chain fatty acids and ethanol in growth media were performed using a Shimadzu headspace GC/FID as previously described.102 Briefly, samples were added to chilled 20 ml headspace vials containing 2.0 g NaHSO4 and 1.0 ml of 60 μM 2-butanol (internal standard). Vials were crimp sealed immediately after sample addition and vortexed periodically to disperse and mix the contents. Headspace GC analyses were performed using a Shimadzu HS-20 headspace sampler connected to a Shimadzu GC-2010 Plus GC equipped with a 30 m SH-Stabilwax column linked to a FID. Samples were equilibrated with shaking to 80°C for 20 min and pressurized to 80 kPa for 3 min prior to column injection (2 ml injection loop, load time 0.2 min, sample and transfer line temperature 150°C, 1:15 split ratio, N2 column flow 1.2 ml/min), with the following column temperature program: 40°C/2 min, increased to 200°C (20°C/min), held 2 min, decreased to 120°C (20°C/min), decreased to 40°C (40°C/min), and stabilized 1 min prior to the subsequent injection. The GC cycle time was approximately 23 minutes. Standard mixtures were prepared and analyzed by the same method, and peak areas determined using Shimadzu Lab Solution software (version 5.92).
GC-MS measurement of short-chain fatty acids.
Sample preparation was based on a previously described procedure.10 Cecal contents were weighed into 4 ml polytetrafluoroethylene (PTFE) screw cap vials and 10 μl of a mixture of internal standards (20 mM of acetic acid-D4, propionic acid-D6, and butyric acid-D7) was subsequently added to each vial, followed by 20 μl of 33% HCl and 1 ml diethyl ether. For plasma samples, 50 μl of each sample, 1.25 μl of the internal standard mix, 5 μl of 33% HCl, and 0.75 ml of diethyl ether were mixed. The mixture was vortexed vigorously for 3 min and then centrifuged (4,000 × g, 10 min). The upper organic layer was transferred to another vial and a second diethyl ether extraction was performed. After combining the two ether extracts, a 60 μl aliquot was removed, combined with 2 μl N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) in a GC autosampler vial with a 200 μl glass insert, and incubated for 2 h at room temperature. Derivatized samples (1 μl) were injected onto an Agilent 7890B/5977A GC/MSD instrument with an Agilent DB1-ms 0.25 mm x 60 m column with 0.25 μm bonded phase. A discontinuous oven program was used starting at 40°C for 2.25 min, then ramping at 20°C/min to 200°C, then ramping at 100°C/min to 300°C and holding for 7 min. The total run time was 18.25 minutes. Linear column flow was maintained at 1.26 ml/min. The inlet temperature was set to 250°C with an injection split ratio of 15:1. Quantitation was performed using selected ion monitoring (SIM) acquisition mode and metabolites were compared to relevant labeled internal standards using Agilent Mass Hunter v. Acquisition B.07.02.1938. The m/z of monitored ions are as follows: 117 (acetic acid), 120 (acetic acid-D4), 131 (propionic acid), 136 (propionic acid-D6), 145 (butyric acid), and 151 (butyric acid-D7). Concentrations were normalized to mg of cecal contents.
uHPLC-MS/MS analysis of metabolites.
Plasma samples were prepared for analysis by precipitating proteins with 4 volumes of ice-cold methanol spiked with 2.5 μM deuterium-labeled choline and TMAO internal standards. Samples were centrifuged at 18,213 × g at 4°C for 3 min. The recovered supernatants were diluted 1:1 in uHPLC-grade water prior to screening. Identification and quantification of choline and TMAO was performed using a uHPLC (Dionex 3000) coupled to a high-resolution mass spectrometer (Thermo Scientific Q Exactive). Liquid chromatography separation was achieved on a Dikma Bio-Bond C4 column (150 mm by 2.1 mm; 3-μm particle size) using a 7-min isocratic gradient (50:50 methanol-water, 5 mM ammonium formate, and 0.1% formic acid). A heated electrospray ionization interface, working in positive mode, was used to direct column eluent to the mass spectrometer. Quantitation of TMAO and D9-TMAO was performed via targeted MS/MS using the following paired masses of parent ions and fragments: TMAO (76.0762 and 58.0659) and D9-TMAO (85.1318 and 68.1301). Quantitation of choline and d9-choline was performed in full-MS scan mode by monitoring their exact masses: 104.1075 and 113.1631, respectively.
UPLC-MS/MS for untargeted plasma metabolome.
Untargeted mass spectrometry data were collected at Metabolon Inc.. Plasma samples were prepared using the automated MicroLab STAR system (Hamilton Company). To remove protein, dissociate small molecules bound to protein or trapped in the precipitated protein matrix and to recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 min (Glen Mills GenoGrinder 2000) followed by centrifugation. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/UPLC-MS/MS methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by HILIC/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. The sample extracts were stored overnight under nitrogen before preparation for analysis.
All methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried then reconstituted in solvents compatible to each of the four methods. Each reconstitution solvent contained a series of standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion conditions, chromatographically optimized for more hydrophilic compounds. In this method, the extract was gradient eluted from a C18 column (Waters UPLC BEH C18–2.1×100 mm, 1.7 μm) using water and methanol containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). Another aliquot was also analyzed using acidic positive ion conditions, however it was chromatographically optimized for more hydrophobic compounds. In this method, the extract was gradient eluted from the aforementioned C18 column using methanol, acetonitrile, water, 0.05% PFPA and 0.01% FA and was operated at an overall higher organic content. Another aliquot was analyzed using basic negative ion optimized conditions using a separate dedicated C18 column. The basic extracts were gradient eluted from the column using methanol and water, amended with 6.5mM ammonium bicarbonate at pH 8. The fourth aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1 × 150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10mM ammonium formate, pH 10.8. Compounds were identified by comparison to library entries based upon retention time/index, mass to charge ratio (m/z) and chromatographic data, and peaks were quantified using area-under-the curve.
Purine and pyrimidine metabolite quantitation.
Targeted purine/pyrimidine mass spectrometry data were collected at The Metabolomics Innovation Centre (Victoria, Canada). An internal standard (IS) solution containing 13C- and/or 15N-labeled AMP, ATP, GMP, GTP, UMP, UTP, xanthine, guanine and adenine, hypothanxine, guanosine and adenosine was prepared in 80% methanol (for cecal samples) or 10% methanol (for plasma samples). Serially-diluted standard solutions containing standard substances of the targeted nucleotides, nucleosides and nucleobases were prepared in a concentration range of 0.00001 to 50 nmol/ml in 80% methanol (for cecal samples) or 0.00001 to 10 nmol/ml in 10% methanol (for plasma samples). The cecal samples were lyophilized to dryness. The powder of each sample was weighted into an Eppendorf tube and 80% methanol at 20 μl per mg of powder was added. The samples were homogenized on a mill mixer at 30 Hz for 2 min, three times, with the aid of 2 metal beads, followed by sonication in an ice-water bath for 5 min. The tubes were centrifuged at 21,000 × g and 5°C for 20 min. 100 μl of the clear supernatant of each sample or 100 μl of each standard solution was in turn mixed with an equal volume of the IS solution, 100 μl of water and 120 μl of dichloromethane. The mixtures were vortex-mixed at 3,000 rpm for 2 min and subsequently centrifuged at 21,000 × g for 5 min. 120 μl of the clear supernatant was collected and then dried at 30°C under a nitrogen gas flow. For plasma samples, 20 μl was mixed with 200 μl of the IS solution and 780 μl of methanol. After vortex mixing at 3000 rpm for 1 min and sonication in an ice-water bath for 2 min, the sample was centrifuged at 21,000 × g and 5°C for 15 min. 500 μl of the clear supernatant of each sample was transferred to another tube, 100 μl of water and 400 μl of dichloromethane then added. The mixture was vortex mixed at 3000 rpm for 1 min and then centrifuged. The upper aqueous phase of each sample was transferred to a LC microvial and dried under a nitrogen gas flow. For both cecal and plasma samples, the residues were reconstituted in 100 μl of 10% methanol. 10 μl aliquots of the sample solutions and the standard solutions were injected into a C18 LC column (2.1 × 100 mm, 1.8 μm) to run UPLC-MRM/MS on a Waters Acquity UPLC system coupled to a Sciex QTRAP 6500 Plus mass spectrometer operated in the negative-ion mode for detection of nucleotides. The mobile phase was a tributylamine acetate buffer and acetonitrile for binary gradient elution (5% to 35% acetonitrile over 25 min), at 0.25 ml/min and 40°C. For quantitation of nucleosides and nucleobases, 10 μl aliquots of the sample solutions and the standard solutions were injected into a polar C18 UPLC column (2.1 × 100 mm, 1.6 μm) to run UPLC-MRM/MS on the same LC-MS instrument operated in the positive-ion mode. The mobile phase was a 0.1% HFBA solution and acetonitrile for binary-solvent gradient elution (0% to 28% acetonitrile in 14 min), at 0.30 ml/min and 40°C.
Genes encoding for purine degradation.
The quantification of genes encoding purine-degradation functions in bacterial genomes were predicted by BLASTP of the NCBI RefSeq Genome Database (refseq_genomes) using five Enterocloster bolteae genes found in multiple purine-utilizing organisms as queries (ygeY, encoding a Se-dependent hydrolase; ygeX/dpaL, encoding a diamino- propionate ammonia-lyase; ssnA, encoding an amino- hydrolase; hydA/hyuA, encoding a dihydro- pyrimidinase; xdhD, encoding a Se-dependent Xanthine DH). Purine-degrading bacteria were defined as bearing the five “probe” genes listed above with identity > 25% to the E. bolteae genes. We removed redundancy if two bacteria genomes had identical proteins encoded by all five purine degradation genes. To quantify the abundance of these genes in gut microbiomes of transplanted mice, we mapped metagenomic reads to genes for purine utilization functions using RSEM (ver. 1.3.1).101 Relative abundance of microbial gene counts per million (CPM) were calculated using microbial gene expected counts divided by gene effective length then normalized by the total sum.
Human study.
UA and coronary artery calcification (CAC) data were obtained from prediabetes Swedish cohort participating in a study examining the link between the gut microbiota and type 2 diabetes.36 The cohort comprised men and women aged 50–64 years from the Gothenburg area, Sweden, who were recruited at random from the census register (n=988). Exclusion criteria were: known diabetes; inflammatory diseases, such as Crohn’s disease, ulcerative colitis, and rheumatic diseases; treatments with steroids or immunomodulatory drugs; cancer (unless relapse-free for the preceding 5 years); cognitive dysfunction; and treatment for infectious diseases and with antibiotics in the past three months. UA was measured using a photometric technique on a Roche Cobas analyzer. Coronary artery calcifications (CAC) were assessed by computed tomography (CT) scanning using a dual-source CT scanner equipped with a Stellar Detector (Siemens). CAC images were obtained using electrocardiogram-gated non-contrast cardiac CT imaging at 120 kV. All non-contrast image sets were reconstructed (B35f HeartView medium CaScore) and CAC were identified and scored using the syngo.via calcium scoring software (Volume Wizard; Siemens) to obtain a CAC score according to Agatston. Previously reported microbiome data34 was mapped against Unified Human Gastrointestinal Genome (UHGG) v1.0 catalogue103 using Kraken2 v.2.1.2104 to examine association between UA levels and bacterial taxa. Xgboost105 and caret packages106 in R version 4.0.3 were used to select microbes associated with the serum urate levels based on regression analysis. These associations were further assessed for significance after adjustment for covariates (CACS, BMI, gender, triglycerides and HbA1c) in a mixed linear regression model.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis and statistical analysis.
Data integration and statistical analysis were performed in R (ver. 3.6.3) or Prism 9. The data were expressed as box-and-whisker plots with individual data points, where the boxes indicate the median values and the interquartile ranges and the whiskers represent the minimum and maximum values. Significance was calculated by unpaired two-tailed Student’s t-test or one-way ANOVA with the Tukey post-tests. Details of these analysis (n, p values) were presented in figure legends. The correlation analysis was performed using Spearman’s correlation by R function “cor.test()”. For multiple testing, Benjamini-Hochberg FDR procedure was used to adjust p-values. Heatmap plots were performed using R package pheatmap (ver. 1.0.12). Purine metabolites include ones from class “purine nucleotides”, “purine nucleosides”, “imidazopyrimidines”, and “azoles”. Pyrimidine metabolites include ones from class “pyrimidine nucleotides”, “pyrimidine nucleosides”, “diazines”, and “pyrazolopyrimidines”. A partial least squares discriminant analysis (PLS-DA) model was constructed using the function PLSR.Anal() in the MetanoAnalystR package and the nonlinear iterative partial least squares (NIPALS) algorithm to obtain the variable importance for the projection (VIP) of purine and pyrimidine metabolites.107 We performed the Leave-One-Out Cross-Validation (LOOCV) method for cross validation.
Supplementary Material
Methods S1. Media and purine overlay formulations used for growth and assessments of purine utilization, related to STAR Methods.
Supplementary Table 1. Metabolites detected in plasma samples from gnotobiotic ApoE KO mice, related to Figure 1.
Supplementary Table 2. Fecal bacterial taxa associated with uric acid levels in humans, related to Figure 2.
Supplementary Table 3. Abundance of purine related metabolites in germ-free and conventional mice, related to Figure 3. A) cecal contents, B) Plasma.
Supplementary Table 4. Abundance of purine related metabolites in gnotobiotic mice colonized with purine-degrading bacteria, related to Figure 5. A) cecal contents, B) Plasma.
Supplementary Table 5. Genes differentially expressed by E. bolteae grown on uric acid compared to xylose/NH4Cl, related to Figure 6.
Supplementary Table 6. Prediction of genes encoding proteins required for growth on uric acid, related to Figure 6.
Supplementary Table 7. Purine-related metabolites detected in the cecum of ApoE KO mice colonized with communities from mice with disparate atherosclerosis phenotypes, related to Figure 7.
ACKNOWLEDGMENTS
We thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing sequencing and support services, the University of Wisconsin Mass Spectrometry Facility for the SCFA measurement, the Metabolomics Innovation Centre (TMIC) for purine metabolomics analysis, and Metabolon for untargeted metabolomics analysis. This work was partly supported by grants from NIH HL144651 (F.E.R. and A.J.L.), NIH HL148577 (F.E.R. and A.J.L.) and NIH HL147883 (A.J.L.). This work was also supported by a grant from a Transatlantic Networks of Excellence Award from Foundation Leducq (17CVD01; to F.B. and F.E.R.), Knut and Alice Wallenberg Foundation (2017.0026; to F.B.), the Swedish Heart Lung Foundation (20210366; to F.B.), and AFA insurances (160337; to F.B.). F.B. is the Torsten Söderberg Professor in Medicine and a Wallenberg Scholar.
F.B. is co-founder and shareholder of Roxbiosens Inc and Implexion Pharma AB, receives research funding from Biogaia AB, and a member of the scientific advisory board of Bactolife A/S.
Footnotes
DECLARATION OD INTERESTS
The other authors declare no competing financial interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wolf D, and Ley K. (2019). Immunity and Inflammation in Atherosclerosis. Circ Res 124, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lusis AJ (2000). Atherosclerosis. Nature 407, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stylianou IM, Bauer RC, Reilly MP, and Rader DJ (2012). Genetic basis of atherosclerosis: insights from mice and humans. Circ Res 110, 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessler T, and Schunkert H. (2021). Coronary Artery Disease Genetics Enlightened by Genome-Wide Association Studies. JACC Basic Transl Sci 6, 610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selvaraj MS, Li X, Li Z, Pampana A, Zhang DY, Park J, Aslibekyan S, Bis JC, Brody JA, Cade BE, et al. (2022). Whole genome sequence analysis of blood lipid levels in >66,000 individuals. Nat Commun 13, 5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan J, Mägi R, et al. (2010). Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue A, Wu Y, Zhu Z, Zhang F, Kemper KE, Zheng Z, Yengo L, Lloyd-Jones LR, Sidorenko J, Wu Y, et al. (2018). Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun 9, 2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung Y-M, et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. (2013). Intestinal microbiota metabolism of l -carnitine, a nutrient in red meat, promotes atherosclerosis. Nature Medicine 19, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasahara K, Krautkramer KA, Org E, Romano KA, Kerby RL, Vivas EI, Mehrabian M, Denu JM, Bäckhed F, Lusis AJ, et al. (2018). Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol 3, 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tilg H, Zmora N, Adolph TE, and Elinav E. (2020). The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol 20, 40–54. [DOI] [PubMed] [Google Scholar]
- 12.Eelke Brandsma, Kloosterhuis Niels J., Koster Mirjam, Dekker Daphne C., Gijbels Marion J.J., van der Velden Saskia, Ríos-Morales Melany, van Faassen Martijn J.R., Loreti Marco G., de Bruin Alain, et al. (2019). A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circulation Research 124, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, and Lionetti P. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences 107, 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolte LA, Vila AV, Imhann F, Collij V, Gacesa R, Peters V, Wijmenga C, Kurilshikov A, Campmans-Kuijpers MJE, Fu J, et al. (2021). Long-term dietary patterns are associated with pro-inflammatory and anti-inflammatory features of the gut microbiome. Gut 70, 1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Modi SR, Collins JJ, and Relman DA (2014). Antibiotics and the gut microbiota. J Clin Invest 124, 4212–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beker BM, Colombo I, Gonzalez-Torres H, and Musso CG (2022). Decreasing microbiota-derived uremic toxins to improve CKD outcomes. Clin Kidney J 15, 2214–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, and Hazen SL (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med 368, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, et al. (2016). Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, Cajka T, Mohan ML, Li L, Wu Y, et al. (2020). A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 180, 862–877.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falconi CA, Junho C.V. da C., Fogaça-Ruiz F, Vernier ICS, da Cunha RS, Stinghen AEM, and Carneiro-Ramos MS (2021). Uremic Toxins: An Alarming Danger Concerning the Cardiovascular System. Front Physiol 12, 686249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valkenburg S, Glorieux G, and Vanholder R. (2021). Uremic Toxins and Cardiovascular System. Cardiol Clin 39, 307–318. [DOI] [PubMed] [Google Scholar]
- 22.Kimura Y, Yanagida T, Onda A, Tsukui D, Hosoyamada M, and Kono H. (2020). Soluble Uric Acid Promotes Atherosclerosis via AMPK (AMP-Activated Protein Kinase)-Mediated Inflammation. Arterioscler Thromb Vasc Biol 40, 570–582. [DOI] [PubMed] [Google Scholar]
- 23.Weisman A, Tomlinson GA, Lipscombe LL, Perkins BA, and Hawker GA (2019). Association between allopurinol and cardiovascular outcomes and all-cause mortality in diabetes: A retrospective, population-based cohort study. Diabetes Obes Metab 21, 1322–1329. [DOI] [PubMed] [Google Scholar]
- 24.Lai S-W, Lin C-L, and Liao K-F (2019). Case-control study examining the association between allopurinol use and ischemic cerebrovascular disease. J Investig Med 67, 48–51. [DOI] [PubMed] [Google Scholar]
- 25.Ju C, Lai RWC, Li KHC, Hung JKF, Lai JCL, Ho J, Liu Y, Tsoi MF, Liu T, Cheung BMY, et al. (2020). Comparative cardiovascular risk in users versus non-users of xanthine oxidase inhibitors and febuxostat versus allopurinol users. Rheumatology (Oxford) 59, 2340–2349. [DOI] [PubMed] [Google Scholar]
- 26.Méndez-Salazar EO, and Martínez-Nava GA (2022). Uric acid extrarenal excretion: the gut microbiome as an evident yet understated factor in gout development. Rheumatol Int 42, 403–412. [DOI] [PubMed] [Google Scholar]
- 27.Chu Y, Sun S, Huang Y, Gao Q, Xie X, Wang P, Li J, Liang L, He X, Jiang Y, et al. (2021). Metagenomic analysis revealed the potential role of gut microbiome in gout. npj Biofilms Microbiomes 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vogels GD, and Van der Drift C. (1976). Degradation of purines and pyrimidines by microorganisms. Bacteriol Rev 40, 403–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartwich K, Poehlein A, and Daniel R. (2012). The purine-utilizing bacterium Clostridium acidurici 9a: a genome-guided metabolic reconsideration. PLoS One 7, e51662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, Pan C, Packard RRS, Eskin E, Yan M, et al. (2015). Genetic Architecture of Atherosclerosis in Mice: A Systems Genetics Analysis of Common Inbred Strains. PLOS Genetics 11, e1005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Org E, Parks BW, Joo JWJ, Emert B, Schwartzman W, Kang EY, Mehrabian M, Pan C, Knight R, Gunsalus R, et al. (2015). Genetic and environmental control of host-gut microbiota interactions. Genome Res 25, 1558–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinon F, Pétrilli V, Mayor A, Tardivel A, and Tschopp J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- 33.Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, Castoldi A, Hiyane MI, Davanso MR, Latz E, et al. (2017). Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci Rep 7, 39884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, Krotova K, Block ER, Prabhakar S, and Johnson RJ (2005). Hyperuricemia induces endothelial dysfunction. Kidney International 67, 1739–1742. [DOI] [PubMed] [Google Scholar]
- 35.Rao GN, Corson MA, and Berk BC (1991). Uric acid stimulates vascular smooth muscle cell proliferation by increasing platelet-derived growth factor A-chain expression. J Biol Chem 266, 8604–8608. [PubMed] [Google Scholar]
- 36.Wu H, Tremaroli V, Schmidt C, Lundqvist A, Olsson LM, Krämer M, Gummesson A, Perkins R, Bergström G, and Bäckhed F. (2020). The Gut Microbiota in Prediabetes and Diabetes: A Population-Based Cross-Sectional Study. Cell Metab 32, 379–390.e3. [DOI] [PubMed] [Google Scholar]
- 37.Osawa K, Nakanishi R, and Budoff M. (2016). Coronary Artery Calcification. Glob Heart 11, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agarwal V, Hans N, and Messerli FH (2013). Effect of allopurinol on blood pressure: a systematic review and meta-analysis. J Clin Hypertens (Greenwich) 15, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohno I. (2011). Relationship between hyperuricemia and chronic kidney disease. Nucleosides Nucleotides Nucleic Acids 30, 1039–1044. [DOI] [PubMed] [Google Scholar]
- 40.Sun H-L, Pei D, Lue K-H, and Chen Y-L (2015). Uric Acid Levels Can Predict Metabolic Syndrome and Hypertension in Adolescents: A 10-Year Longitudinal Study. PLoS One 10, e0143786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drivelegka P, Forsblad-d’Elia H, Angerås O, Bergström G, Schmidt C, Jacobsson LTH, and Dehlin M. (2020). Association between serum level of urate and subclinical atherosclerosis: results from the SCAPIS Pilot. Arthritis Res Ther 22, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rahimi-Sakak F, Maroofi M, Rahmani J, Bellissimo N, and Hekmatdoost A. (2019). Serum uric acid and risk of cardiovascular mortality: a systematic review and dose-response meta-analysis of cohort studies of over a million participants. BMC Cardiovascular Disorders 19, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yun Y, Yin H, Gao Z, Li Y, Gao T, Duan J, Yang R, Dong X, Zhang L, and Duan W. (2017). Intestinal tract is an important organ for lowering serum uric acid in rats. PLoS One 12, e0190194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sorensen LB, and Levinson DJ (1975). Origin and extrarenal elimination of uric acid in man. Nephron 14, 7–20. [DOI] [PubMed] [Google Scholar]
- 45.Song Y, Liu C, Molitoris DR, Tomzynski TJ, Lawson PA, Collins MD, and Finegold SM (2003). Clostridium bolteae sp. nov., isolated from human sources. Syst Appl Microbiol 26, 84–89. [DOI] [PubMed] [Google Scholar]
- 46.Iwadate Y, and Kato J-I (2019). Identification of a Formate-Dependent Uric Acid Degradation Pathway in Escherichia coli. J Bacteriol 201, e00573–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schiefer-Ullrich H, Wagner R, Dürre P, and Andreesen JR (1984). Comparative studies on physiology and taxonomy of obligately purinolytic clostridia. Arch Microbiol 138, 345–353. [DOI] [PubMed] [Google Scholar]
- 48.Dürre P, and Andreesen JR (1983). Purine and glycine metabolism by purinolytic clostridia. J Bacteriol 154, 192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dürre P, and Andreesen JR (1982). Anaerobic degradation of uric acid via pyrimidine derivatives by selenium-starved cells of Clostridium purinolyticum. Arch Microbiol 131, 255–260. [DOI] [PubMed] [Google Scholar]
- 50.Wilber CG (1980). Toxicology of selenium: a review. Clin Toxicol 17, 171–230. [DOI] [PubMed] [Google Scholar]
- 51.Faith JJ, McNulty NP, Rey FE, and Gordon JI (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaal T, Bartlett MS, Ross W, Turnbough CL, and Gourse RL (1997). Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science 278, 2092–2097. [DOI] [PubMed] [Google Scholar]
- 53.Papakostas K, Botou M, and Frillingos S. (2013). Functional identification of the hypoxanthine/guanine transporters YjcD and YgfQ and the adenine transporters PurP and YicO of Escherichia coli K-12. J Biol Chem 288, 36827–36840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Jin Z, Yu X, Allewell NM, Tuchman M, and Shi D. (2011). The ygeW encoded protein from Escherichia coli is a knotted ancestral catabolic transcarbamylase. Proteins 79, 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cusa E, Obradors N, Baldomà L, Badía J, and Aguilar J. (1999). Genetic analysis of a chromosomal region containing genes required for assimilation of allantoin nitrogen and linked glyoxylate metabolism in Escherichia coli. J Bacteriol 181, 7479–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Jarman JB, Low YS, Huang S, Chen H, DeFeo ME, Sekiba K, Hou B-H, Ganesan C, Pao AC, et al. (2022). A widely distributed gene cluster compensates for uricase loss in hominids. bioRxiv 2022.07.24.501321. [Google Scholar]
- 57.Errasti-Murugarren E, Pastor-Anglada M, and Casado FJ (2007). Role of CNT3 in the transepithelial flux of nucleosides and nucleoside-derived drugs. J Physiol 582, 1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernández-Calotti P, Casulleras O, Antolin M, Guarner F, and Pastor-Anglada M. (2016). Galectin-4 interacts with the drug transporter human concentrative nucleoside transporter 3 to regulate its function. FASEB J 30, 544–554. [DOI] [PubMed] [Google Scholar]
- 59.Pastor-Anglada M, and Pérez-Torras S. (2018). Emerging Roles of Nucleoside Transporters. Front Pharmacol 9, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Naes SM, Ab-Rahim S, Mazlan M, Hashim NAA, and Rahman AA (2022). Increased ENT2 expression and its association with altered purine metabolism in cell lines derived from different stages of colorectal cancer. Exp Ther Med 25, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, and Gaucher EA (2014). Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci U S A 111, 3763–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh G, Lingala B, and Mithal A. (2019). Gout and hyperuricaemia in the USA: prevalence and trends. Rheumatology (Oxford) 58, 2177–2180. [DOI] [PubMed] [Google Scholar]
- 63.Chen-Xu M, Yokose C, Rai SK, Pillinger MH, and Choi HK (2019). Contemporary Prevalence of Gout and Hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey, 2007–2016. Arthritis Rheumatol 71, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi HK (2010). A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol 22, 165–172. [DOI] [PubMed] [Google Scholar]
- 65.Feig DI, Kang D-H, and Johnson RJ (2008). Uric acid and cardiovascular risk. N Engl J Med 359, 1811–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh JA, Ramachandaran R, Yu S, and Curtis JR (2017). Allopurinol use and the risk of acute cardiovascular events in patients with gout and diabetes. BMC Cardiovasc Disord 17, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mackenzie IS, Hawkey CJ, Ford I, Greenlaw N, Pigazzani F, Rogers A, Struthers AD, Begg AG, Wei L, Avery AJ, et al. (2022). Allopurinol versus usual care in UK patients with ischaemic heart disease (ALL-HEART): a multicentre, prospective, randomised, open-label, blinded-endpoint trial. The Lancet 400, 1195–1205. [DOI] [PubMed] [Google Scholar]
- 68.Keerman M, Yang F, Hu H, Wang J, Wang F, Li Z, Yuan J, Yao P, Zhang X, Guo H, et al. (2020). Mendelian randomization study of serum uric acid levels and diabetes risk: evidence from the Dongfeng-Tongji cohort. BMJ Open Diabetes Research and Care 8, e000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu J, Zeng Y, Zhang H, Qu Y, Ying Z, Sun Y, Hu Y, Chen W, Yang H, Yang J, et al. (2022). The Association of Hyperuricemia and Gout With the Risk of Cardiovascular Diseases: A Cohort and Mendelian Randomization Study in UK Biobank. Front Med (Lausanne) 8, 817150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wakuda H, Uchida S, Ikeda M, Tabuchi M, Akahoshi Y, Shinozuka K, and Yamada S. (2014). Is hyperuricemia a risk factor for arteriosclerosis? Uric acid and arteriosclerosis in apolipoprotein e-deficient mice. Biol Pharm Bull 37, 1866–1871. [DOI] [PubMed] [Google Scholar]
- 71.Hediger MA, Johnson RJ, Miyazaki H, and Endou H. (2005). Molecular physiology of urate transport. Physiology (Bethesda) 20, 125–133. [DOI] [PubMed] [Google Scholar]
- 72.Thong-On A, Suzuki K, Noda S, Inoue J, Kajiwara S, and Ohkuma M. (2012). Isolation and characterization of anaerobic bacteria for symbiotic recycling of uric acid nitrogen in the gut of various termites. Microbes Environ 27, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barba M, Dutoit R, Legrain C, and Labedan B. (2013). Identifying reaction modules in metabolic pathways: bioinformatic deduction and experimental validation of a new putative route in purine catabolism. BMC Syst Biol 7, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haft DH, and Self WT (2008). Orphan SelD proteins and selenium-dependent molybdenum hydroxylases. Biol Direct 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Uo T, Yoshimura T, Nishiyama T, and Esaki N. (2002). Gene cloning, purification, and characterization of 2,3-diaminopropionate ammonia-lyase from Escherichia coli. Biosci Biotechnol Biochem 66, 2639–2644. [DOI] [PubMed] [Google Scholar]
- 76.Li Z, Pan Q, Xiao Y, Fang X, Shi R, Fu C, Danchin A, and You C. (2019). Deciphering global gene expression and regulation strategy in Escherichia coli during carbon limitation. Microb Biotechnol 12, 360–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schiefer-Ullrich H, and Andreesen JR (1985). Peptostreptococcus barnesae sp. nov., a Gram-positive, anaerobic, obligately purine utilizing coccus from chicken feces. Arch. Microbiol 143, 26–31. [Google Scholar]
- 78.Crosnier C, Stamataki D, and Lewis J. (2006). Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 7, 349–359. [DOI] [PubMed] [Google Scholar]
- 79.Savaiano DA, and Clifford AJ (1981). Adenine, the precursor of nucleic acids in intestinal cells unable to synthesize purines de novo. J Nutr 111, 1816–1822. [DOI] [PubMed] [Google Scholar]
- 80.Lee JS, Wang RX, Alexeev EE, Lanis JM, Battista KD, Glover LE, and Colgan SP (2018). Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J Biol Chem 293, 6039–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JS, Wang RX, Goldberg MS, Clifford GP, Kao DJ, and Colgan SP (2020). Microbiota-Sourced Purines Support Wound Healing and Mucous Barrier Function. iScience 23, 101226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fukuda T, Majumder K, Zhang H, Turner PV, Matsui T, and Mine Y. (2016). Adenine Inhibits TNF-α Signaling in Intestinal Epithelial Cells and Reduces Mucosal Inflammation in a Dextran Sodium Sulfate-Induced Colitis Mouse Model. J Agric Food Chem 64, 4227–4234. [DOI] [PubMed] [Google Scholar]
- 83.Mars RAT, Yang Y, Ward T, Houtti M, Priya S, Lekatz HR, Tang X, Sun Z, Kalari KR, Korem T, et al. (2020). Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome. Cell 182, 1460–1473.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chandra H, Sharma KK, Tuovinen OH, Sun X, and Shukla P. (2021). Pathobionts: mechanisms of survival, expansion, and interaction with host with a focus on Clostridioides difficile. Gut Microbes 13, 1979882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bartlett A, Padfield D, Lear L, Bendall R, and Vos M. (2022). A comprehensive list of bacterial pathogens infecting humans. Microbiology (Reading) 168. [DOI] [PubMed] [Google Scholar]
- 86.Romano KA, Vivas EI, Amador-Noguez D, and Rey FE (2015). Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio 6, e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barnes EM, and Impey CS (1974). The occurence and properties of uric acid decomposing anaerobic bacteria in the avian caecum. J Appl Bacteriol 37, 393–409. [DOI] [PubMed] [Google Scholar]
- 88.Thomason LC, Sawitzke JA, Li X, Costantino N, and Court DL (2014). Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 106, 1.16.1–39. [DOI] [PubMed] [Google Scholar]
- 89.Farmer JJ, Davis BR, Hickman-Brenner FW, McWhorter A, Huntley-Carter GP, Asbury MA, Riddle C, Wathen-Grady HG, Elias C, and Fanning GR (1985). Biochemical identification of new species and biogroups of Enterobacteriaceae isolated from clinical specimens. J Clin Microbiol 21, 46–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lindsey RL, Garcia-Toledo L, Fasulo D, Gladney LM, and Strockbine N. (2017). Multiplex polymerase chain reaction for identification of Escherichia coli, Escherichia albertii and Escherichia fergusonii. J Microbiol Methods 140, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 93.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dankers ACA, Mutsaers HAM, Dijkman HBPM, van den Heuvel LP, Hoenderop JG, Sweep FCGJ, Russel FGM, and Masereeuw R. (2013). Hyperuricemia influences tryptophan metabolism via inhibition of multidrug resistance protein 4 (MRP4) and breast cancer resistance protein (BCRP). Biochim Biophys Acta 1832, 1715–1722. [DOI] [PubMed] [Google Scholar]
- 95.Kasahara K, Tanoue T, Yamashita T, Yodoi K, Matsumoto T, Emoto T, Mizoguchi T, Hayashi T, Kitano N, Sasaki N, et al. (2017). Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J. Lipid Res. 58, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kreznar JH, Keller MP, Traeger LL, Rabaglia ME, Schueler KL, Stapleton DS, Zhao W, Vivas EI, Yandell BS, Broman AT, et al. (2017). Host genotype and gut microbiome modulate insulin secretion and diet-induced metabolic phenotypes. Cell Rep 18, 1739–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walker DI, McQuillan J, Taiwo M, Parks R, Stenton CA, Morgan H, Mowlem MC, and Lees DN (2017). A highly specific Escherichia coli qPCR and its comparison with existing methods for environmental waters. Water Res 126, 101–110. [DOI] [PubMed] [Google Scholar]
- 99.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. (2011). The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med 3, 106ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murga-Garrido SM, Hong Q, Cross T-WL, Hutchison ER, Han J, Thomas SP, Vivas EI, Denu J, Ceschin DG, Tang Z-Z, et al. (2021). Gut microbiome variation modulates the effects of dietary fiber on host metabolism. Microbiome 9, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, et al. (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol 39, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wood DE, Lu J, and Langmead B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen T, and Guestrin C. (2016). XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining KDD ‘16. (Association for Computing Machinery; ), pp. 785–794. [Google Scholar]
- 106.Kuhn M. (2008). Building Predictive Models in R Using the caret Package. Journal of Statistical Software 28, 1–26.27774042 [Google Scholar]
- 107.Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, and Xia J. (2022). Using MetaboAnalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc 17, 1735–1761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1. Media and purine overlay formulations used for growth and assessments of purine utilization, related to STAR Methods.
Supplementary Table 1. Metabolites detected in plasma samples from gnotobiotic ApoE KO mice, related to Figure 1.
Supplementary Table 2. Fecal bacterial taxa associated with uric acid levels in humans, related to Figure 2.
Supplementary Table 3. Abundance of purine related metabolites in germ-free and conventional mice, related to Figure 3. A) cecal contents, B) Plasma.
Supplementary Table 4. Abundance of purine related metabolites in gnotobiotic mice colonized with purine-degrading bacteria, related to Figure 5. A) cecal contents, B) Plasma.
Supplementary Table 5. Genes differentially expressed by E. bolteae grown on uric acid compared to xylose/NH4Cl, related to Figure 6.
Supplementary Table 6. Prediction of genes encoding proteins required for growth on uric acid, related to Figure 6.
Supplementary Table 7. Purine-related metabolites detected in the cecum of ApoE KO mice colonized with communities from mice with disparate atherosclerosis phenotypes, related to Figure 7.
Data Availability Statement
The accession number for sequence data used in the study (Mouse Metagenomics data, Bacterial RNA-Seq data, Copro-Seq data, Human Metagenomics data) are PRJNA904303, PRJNA911264, PRJNA903666, and EGAS00001004480 (European Genome-Phenome Archive). Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code is available here: https://github.com/qijunz/Purine_paper
Any additional information required to reanalyze the data reported in this paper including the custom Python scripts employed in the processing of metagenomic data is available from the lead contact upon request.