

Abstract
Motivation
Minimal perfect hashing is the problem of mapping a static set of n distinct keys into the address space bijectively. It is well-known that bits are necessary to specify a minimal perfect hash function (MPHF) f, when no additional knowledge of the input keys is to be used. However, it is often the case in practice that the input keys have intrinsic relationships that we can exploit to lower the bit complexity of f. For example, consider a string and the set of all its distinct k-mers as input keys: since two consecutive k-mers share an overlap of symbols, it seems possible to beat the classic bits/key barrier in this case. Moreover, we would like f to map consecutive k-mers to consecutive addresses, as to also preserve as much as possible their relationship in the codomain. This is a useful feature in practice as it guarantees a certain degree of locality of reference for f, resulting in a better evaluation time when querying consecutive k-mers.
Results
Motivated by these premises, we initiate the study of a new type of locality-preserving MPHF designed for k-mers extracted consecutively from a collection of strings. We design a construction whose space usage decreases for growing k and discuss experiments with a practical implementation of the method: in practice, the functions built with our method can be several times smaller and even faster to query than the most efficient MPHFs in the literature.
1 Introduction
Given a universe set U, a function is a minimal perfect hash function (MPHF, henceforth) for a set with if for all , . In simpler words, f maps each key of S into a distinct integer in [n]. The function is allowed to return any value in [n] for a key . A classic result established that bits are essentially necessary to represent such functions for (Mehlhorn 1982). Minimal perfect hashing is a central problem in data structure design and has received considerable attention, both in theory and practice. In fact, many practical constructions have been proposed (see e.g. Pibiri and Trani 2021a and references therein). These algorithms find MPHFs that take space close to the theoretic-minimum, e.g. 2–3 bits/key, retain very fast lookup time, and scale well to very large sets. Applications of minimal perfect hashing range from computer networks (Lu et al. 2006) to databases (Chang and Lin 2005), as well as language models (Pibiri and Venturini 2019; Strimel et al. 2020), compilers, and operating systems. MPHFs have been also used recently in Bioinformatics to implement fast and compact dictionaries for fixed-length DNA strings (Almodaresi et al. 2018; Marchet et al. 2021; Pibiri 2022a,b).
In its simplicity and versatility, the minimal perfect hashing problem does not take into account specific types of inputs, nor the intrinsic relationships between the input keys. Each key is considered independently from any other key in the set and, as such, for any fixed . In practice, however, the input keys often present some regularities that we could exploit to let f act less randomly on S. This, in turn, would permit to achieve a lower space complexity for f.
We therefore consider in this article the following special setting of the minimal perfect hashing problem: the elements of S are all the distinct sub-strings of length k, for some , from a given collection of strings. The elements of S are called k-mers. The crucial point is that any two consecutive k-mers in a string of have indeed a strong intrinsic relationship in that they share an overlap of symbols. It seems profitable to exploit the overlap information to preserve (as much as possible) the local relationship between consecutive k-mers as to reduce the randomness of f, thus lowering its bit complexity and evaluation time.
In particular, we are interested in the design of a locality-preserving MPHF in the following sense. Given a query sequence Q, if for some k-mer , we would like f to hash to , to , and so on, where is the k-mer following x in Q (assuming and are in as well). This behavior of f is very desirable in practice, at least for two important reasons. First, it implies compression for satellite values associated to k-mers. Typical satellite values are abundance counts, reference identifiers (sometimes called “colors”), or contig identifiers (e.g. unitigs) in a de Bruijn graph. Consecutive k-mers tend to have very similar—if not identical—satellite values, hence hashing consecutive k-mers to consecutive identifiers induce a natural clustering of the associated satellite values which is amenable to effective compression. The second important reason is, clearly, faster evaluation time when querying for consecutive k-mers in a sequence. This streaming query modality is the query modality employed by k-mer-based applications (Almodaresi et al. 2018; Bingmann et al. 2019; Marchet et al. 2021; Robidou and Peterlongo 2021; Pibiri 2022b).
We formalize the notion of locality-preserving MPHF along with other preliminary definitions in Section 2. We show how to obtain a locality-preserving MPHF in very compact space in Section 3. To achieve this result, we make use of two algorithmic tools: random minimizers (Schleimer et al. 2003; Roberts et al. 2004) and a novel partitioning scheme for sub-sequences of consecutive k-mers sharing the same minimizers (super-k-mers) which allows a more parsimonious memory layout. The space of the proposed solution decreases for growing k and the data structure is built in linear time in the size of the input (number of distinct k-mers). In Section 4 we present experiments across a breadth of datasets to show that the construction is practical too: the functions can be several times smaller and even faster to query than the most efficient, albeit “general-purpose”, MPHFs. We conclude in Section 5 where we also sketch some promising future directions. Our C++ implementation of the method is publicly available at https://github.com/jermp/lphash.
2 Notation and definitions
Let be a set of strings over an alphabet . Throughout the article, we focus on the DNA alphabet to better highlight the connection with our concrete application but our algorithms can be generalized to work for arbitrary alphabets. A sub-string of length k of a string is called a k-mer of S.
Definition 1
(Spectrum). The k-mer spectrum of is the set of all distinct k-mers of the strings in . Formally:
Definition 2
(Spectrum-Preserving String Set). A spectrum-preserving string set (or SPSS) of is a set of strings such that (i) each string of has length at least k, and (ii) .
Since our goal is to build a MPHF for the k-mers in a SPSS, we are interested in a SPSS where each k-mer is seen only once, i.e. for each k-mer there is only one string of where x appears once. We assume that no k-mer appearing at the end of a string shares an overlap of symbols with the first k-mer of another string, otherwise we could reduce the number of strings in and obtain a smaller SPSS. In the following, we make use of this form of SPSS which is suitable for the minimal perfect hashing problem. We remark that efficient algorithms exist to compute such SPSSs (see e.g. Rahman and Medvedev 2020; Břinda et al. 2021; Khan and Patro 2021; Khan et al. 2022).
The input for our problem is therefore a SPSS for with strings and distinct k-mers. Without loss of generality, we index k-mers based on their positions in , assuming an order of the strings of is fixed, and we indicate with the i-th k-mer in , for .
We want to build a MPHF for ; more precisely, for the n distinct k-mers in . We remark again that our objective is to exploit the overlap of symbols between consecutive k-mers from a string of to preserve their locality, and hence reduce the bit complexity of f as well as its evaluation time when querying k-mers in sequence.
We define a locality-preserving MPHF, or LP-MPHF, for as follows.
Definition 3
(LP-MPHF). Let be a MPHF forand A be the set . The function f is -locality-preserving for if .
Intuitively, the “best” LP-MPHF for is the one having the smallest , so we look for practical constructions with small . On the other hand, note that a “classic” MPHF corresponds to the case where the locality-preserving property is almost always not satisfied and, as a consequence, will be ∼1.
Two more considerations are in order. First, it should be clear that the way we define locality-preservation in Definition 3 is only pertinent to SPSSs where having consecutive hash codes for consecutive k-mers is a very desirable property as motivated in Section 1. A different definition of locality-preservation could instead be given if we were considering generic input keys. Second, we did not use the term “order-preserving” to stress the distinction from classic order-preserving functions in the literature (Fox et al. 1991) that make it possible to preserve any wanted order and, as such, incur in an avoidable -bit overhead per key. Here, we are interested in preserving only the input order of the k-mers which is the one that matters in practice.
Definition 4
(Fragmentation Factor). Given a SPSS with strings and distinct k-mers, we define the fragmentation factor of as .
The fragmentation factor of is a measure of how contiguous the k-mers in are. The minimum fragmentation is achieved for and, in this case, shares an overlap of symbols with for “all” . This ideal scenario is, however, unlikely to happen in practice. On the other hand, the worst-case scenario of maximum fragmentation is achieved when and k-mers do not share any overlap (of length ). This is also unlikely to happen given that k-mers are extracted consecutively from the strings of and, as a result, many overlaps are expected. A more realistic scenario happens, instead, when , resulting in . For the rest of the paper, we focus on this latter scenario to make our analysis meaningful.
From Definitions 3 and 4, it is easy to see that when , and when . In general, we have since there are at least indexes i for which . How small can actually be therefore depends on the input SPSS (and on the strategy used to implement f in practice, as we are going to illustrate in Section 3).
Lastly in this section, we define minimizers and super-k-mers that will be one of the main ingredients used in Section 3.
Definition 5
(Random Minimizer of a k-mer). Given a k-mer x and a random hash function h, the minimizer of x is any m-mer such that for any other m-mer y of x, for some .
In case the minimizer of x is not unique, we break ties by taking the leftmost m-mer in x. For convenience, we indicate with the number of m-mers in a k-mer. (Note that Definition 5 defines a minimizer as a specific m-mer inside a k-mer rather than a specific k-mer in a window of w consecutive k-mers, which is the more standard definition found in the literature.) Since h is a random hash function (with a wide range, e.g. ), each m-mer in a k-mer has probability of being the minimizer of the k-mer. We say that the triple defines a random minimizer scheme. The density of a minimizer scheme is the expected number of selected minimizers from the input.
Definition 6
(Super-k-mer). Given a string S, a super-k-mer g is a maximal sub-string of S where each k-mer has the same minimizer and appears only once in g.
3 Locality-preserving minimal perfect hashing of k-mers
In this section, we describe an algorithm to obtain locality-preserving MPHFs for a spectrum-preserving string set . The algorithm builds upon the following main insight.
Implicitly ranking k-mers through minimizers. Let g be a super-k-mer of some string and assume g is the only super-k-mer whose minimizer is . By definition of super-k-mer, all the k-mers in g contain the minimizer as a sub-string— being the i-th k-mer of g. If is the start position of in the first k-mer of g, then is the start position of in for . Figure 1 gives a practical example for a super-k-mer g of length 16 and .
Figure 1.
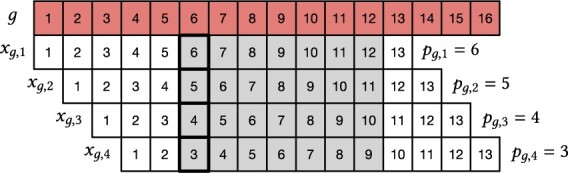
A super-k-mer g of length 16 with k-mers for and minimizer length . The shaded boxes highlight the minimizer whose start position is in k-mer . It is easy to see that for any .
| (1) |
The next property illustrates the relation between the size of the super-k-mer g and the position (we will come later on the implications of this property).
Property 1.
for any super-k-mer g.
Proof. Since is the start position of the minimizer in the first k-mer of g, there are at most k-mers that contain the minimizer as a sub-string, hence . However, g cannot contain more than w k-mers. □
Now, suppose we are given a query k-mer whose minimizer is . The k-mer must appear as a sub-string of g, i.e. it must be one among . We want to compute the rank of x among the k-mers of g, which we indicate by (assuming that it is clear from the context that is relative to g). Let p be the start position of in x. We can use this positional information p to compute as follows:
- if and , then
(2) otherwise ( or ), x cannot possibly be in g and, hence, indexed by f.
Our strategy is to compute as for any k-mer of g. Next, we show in Lemma 1 that this strategy maps the k-mers bijectively in and preserves their locality (i.e. their relative order in g).
Lemma 1.
The strategy in Equation (3) guaranteesfor any .
| (3) |
Proof. For Equation (3), . Therefore, . Since for Equation (1), then . □
To sum up, the position of the minimizer in the first k-mer of g, , defines an implicit ranking (i.e. achieved without explicit string comparison) of the k-mers inside a super-k-mer.
3.1 Basic data structure
From Equation (3) is evident that acts as a “global” component in the calculation of , which must be added to a “local” component represented by . We have already shown how to compute in Equation (2): Lemma 1 guarantees that this local rank computation bijectively maps the k-mers of g into . We are therefore left to show how to compute for each super-k-mer g. We proceed as follows.
Layout. Let be the set of all the distinct minimizers of . We build a MPHF for , . Assume, for ease of exposition, that each super-k-mer g is the only super-k-mer having minimizer . (We explain how to handle the case where more super-k-mers have the same minimizer in Section 3.3.) We allocate an array where and for every minimizer . We then take the prefix-sums of into another array L, that is, for all . We therefore have that indicates the number of k-mers before those in g (whose minimizer is ) in the order given by . The size of g can be recovered as . In conclusion, we compute as . The positions of each super-k-mer g are instead written in another array where .
It follows that the data structure is built in time, since a scan over the input suffices to compute all super-k-mers and can be built in expected time.
Lookup. With these three components—, and the two arrays L and P—it is easy to evaluate as shown in Algorithm 1. The complexity of the lookup algorithm is since this is the complexity of computing the minimizer (assuming each hash calculation to take constant time) and the overall evaluation of as well, since accessing the arrays L and P takes .
Algorithm 1.
Evaluation algorithm for f, given the k-mer x. The helper function computes the minimizer of x and the starting position p of in x.
1: function f(x):
2:
3:
4: return
Compression. The data structure for f itself is a compressed representation for , L, and P. To compute the space taken by the data structure we first need to know —the expected number of distinct minimizers seen in the input. Assuming again that there are no duplicate minimizers, if d indicates the density of a random minimizer scheme, then
, and
as a direct consequence of Lemma 1.
In particular, a result due to Zheng et al. (2020, Theorem 3) allows us to compute d for a random minimizer scheme as if for any . We will always operate under the condition that m is sufficiently large compared to k otherwise minimizers are meaningless.
Therefore, any random minimizer scheme gives us a -LP MPHF with (we omit lower order terms for simplicity) as illustrated in the following theorem (see the Supplementary Material for the proof).
Theorem 1.
Given a random minimizer scheme with for any and , there exists a -LP MPHF for a SPSS with which takeswhere and b is a constant larger than .
Note that the space bound in Theorem 1 decreases as w grows; for example, when m is fixed and k grows. Next we show how to improve this result using some structural properties of super-k-mers.
3.2 Partitioned data structure
Property 1 states that for any super-k-mer g. As an immediate implication we have that if then also (and, symmetrically, if then ). This suggests that, whenever a super-k-mer contains a maximal number of k-mers, then we can always implicitly derive that . We can thus save the space for the entries dedicated to such super-k-mers in the arrays L and P. Note that the converse is not true in general, i.e. if it could be that . Nonetheless, we can still save space for some entries of P in this case.
Depending on the starting position of the minimizer in the first and last k-mer of a super-k-mer, we distinguish between four “types” of super-k-mers (Definition 7).
Definition 7
(FL rule). Let g be a super-k-mer. The first/last (FL) rule is as follows:
if and , then g is left-right-max; else
if and , then g is left-max; else
if and , then g is right-max; else
if and , then g is non-max.
See Fig. 2 for a schematic illustration.
Figure 2.
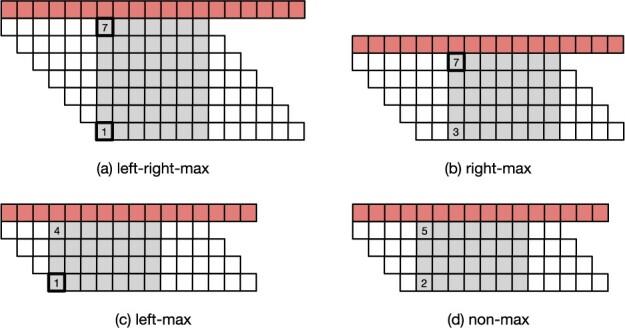
The four different types of super-k-mers. The example is for and minimizer length , so . The shaded boxes highlight the minimizer sub-string inside a k-mer. The start position of the minimizer is marked with a solid border when it is either max (7), or min (1).
Layout. Based on the FL-rule above, we derive a partitioned layout as follows. We store the type of each super-k-mer in an array , in the order given by . We can now exploit this labeling of super-k-mers to improve the space bound of Theorem 1 because:
for all left-right-max super-k-mers, we do not store L nor P;
for all left/right-max super-k-mers, we only store L—precisely, two arrays and for left- and right-max super-k-mers, respectively;
for all the other super-k-mers, i.e. non-max, we store both L and P as explained before—let us indicate them with and in the following.
Addressing the arrays , , and , can be achieved by answering queries on R: the result of this query is the number of super-k-mers that have type t in the prefix R[1.i]. If , then we read the type of the super-k-mer associated to as . Then we compute . Depending on the type t, we either do not perform any array access or access the j-th position of either , or , or and (see Algorithm 2).
Algorithm 2.
Evaluation algorithm for a partitioned representation of f. The quantities , , , and are, respectively, the number of left-right-max, left-max, right-max, and non-max super-k-mers of .
1: function f(x):
2:
3:
4:
5:
6: , ,
7: switch(t):
8: case left-right-max:
9: , ,
10: break
11: case left-max:
12: , ,
13: break
14: case right-max:
15: , ,
16: break
17: case non-max:
18: , ,
19: break
20: return
A succinct representation of R that also supports and queries is the wavelet tree (Grossi et al. 2003). In our case, we only have four possible types, hence a 2-bit integer is sufficient to encode a type. The wavelet tree therefore represents R in bits [The term is the redundancy needed to accelerate the binary rank queries. In practice, the term can be non-negligible, e.g. can be as high as bits using the Rank9 index (Vigna 2008, Section 3), but it is necessary for fast queries in practice (namely, time). Looking at Table 1a from Pibiri and Kanda (2021), we see that the redundancy is in between 3% and 25% of .] and supports both queries in time. The wavelet tree is also built in linear time, so the building time of the overall data structure remains . Refer to Fig. 3 for a pictorial representation of this partitioned layout.
Table 1.
Computed (compt.) probabilities with Theorem 2 versus measured (measr.) using the whole human genome for three representative configurations.
|
|
|
|
||||
|---|---|---|---|---|---|---|
| compt. | measr. | compt. | measr. | compt. | measr. | |
| 0.297 | 0.281 | 0.273 | 0.264 | 0.264 | 0.257 | |
| 0.248 | 0.261 | 0.249 | 0.256 | 0.250 | 0.254 | |
| 0.248 | 0.261 | 0.249 | 0.256 | 0.250 | 0.254 | |
| 0.207 | 0.197 | 0.228 | 0.224 | 0.236 | 0.235 | |
Figure 3.
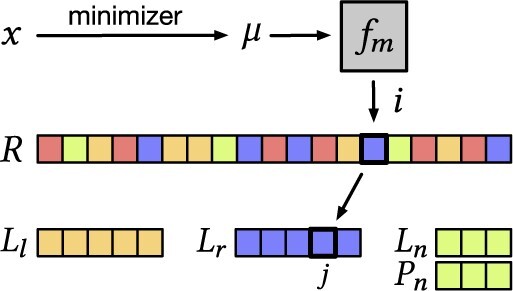
Partitioned data structure layout and the flow of Algorithm 2 for a query k-mer x, whose minimizer is , and with . Different colors in R are used to distinguish between the different super-k-mer types.
Lookup. Algorithm 2 gives the lookup algorithm for the partitioned representation of f. The complexity of the algorithm is still like that of the un-partitioned counterpart, Algorithm 1. The evaluation algorithm must now distinguish between the four different types of minimizer. On the one hand, this distinction involves an extra array access (to R) and a rank query as explained above but, on the other hand, it permits to save 2 array accesses in the left-right-max case or 1 in the left/right-max case compared to Algorithm 1 that always performs 2 array accesses (one access to L and one to P). Hence, the overall number of array accesses performed by Algorithm 2 is on average the same as that of Algorithm 1 assuming the four cases are equally likely (see next paragraph). For this reason we do not expect Algorithm 2 to incur in a penalty at query time compared to Algorithm 1 despite of its more complex evaluation. Compression. Intuitively, if the fraction of left-right-max super-k-mers and that of left/right-max super-k-mers is sufficiently high, we can save significant space compared to the data structure in Section 3.1 that stores both L and P for all minimizers. We therefore need to compute the proportions of the different types of super-k-mers as given by the FL rule. For ease of notation, let , , , , for any super-k-mer g.
Remark 1.
The FL rule is a partitioning rule, i.e. for any super-k-mer.
Our objective is to derive the expression for the probabilities , , , and , parametric in k (k-mer length) and m (minimizer length). To achieve this goal we propose a simple model based on a (discrete-time) Markov chain.
Let be a discrete random variable, modeling the starting position of the minimizer in a k-mer. The corresponding Markov chain is illustrated in Fig. 4. Each state of the chain is labeled with the corresponding value assumed by X, i.e. with each value in . Clearly, we have a left-right-max super-k-mer if, from state w we transition to state , then to , , down to state 1. Each state has a fallback probability to go to state w which corresponds to the event that the right-most m-mer (that coming next to the right) is the new minimizer. If the chain reaches state 1, instead, we know that we are always going to see a new minimizer next. If is the code assigned to the current minimizer by the coding function h used by , for some universe size u (e.g. if c is a 64-bit hash code, then ), the probability for any m-mer to become the new minimizer is equal to . Vice versa, the probability of keeping the same minimizer when sliding one position to the right, is . Whenever we change minimizer, we generate a new code c and, hence, the probability changes with every formed super-k-mer. Nonetheless, the following Theorem shows that the probabilities , , , and , do not depend on .
Figure 4.
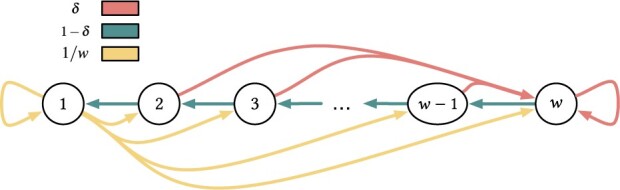
The chain is in state if the minimizer starts at position p in the k-mer. Different edge colors represent different probabilities.
Theorem 2.
For any random minimizer scheme we have
where and .
We give the following lemma to prove Theorem 2. (When we write “first”/“last” k-mer we are going to silently assume “of a super-k-mer”.)
Lemma 2.
and .
Proof. First note that for any . Then we have the following equivalences.
| (4) |
| (5) |
Now note that because the starting position of the minimizer of the first k-mer of any left-right-max and of any right-max super-k-mer is w. In a similar way, we have that because the starting position of the minimizer of the last k-mer of any left-right-max and of any left-max super-k-mer is 1. Now note that because:
| (6) |
| (7) |
From equation , we have which, using Equations (6) and (7), yields . The Lemma follows by using the latter equation into Equation (5). □
Now we prove Theorem 2.
Proof. Since the FL rule induces a partition:
| (8) |
Again exploiting the fact that , we also have
| (9) |
We have therefore to compute to also determine , , and .
| (10) |
Now letting and substituting [Equation (10)] into Equations (8) and (9), the Theorem follows. □
In Table 1, we report the probabilities , , , and computed using Theorem 2 for some representative combinations of k and m (these combinations are some of those used in the experiments of Section 4; see also Table 2). For comparison, we also report the probabilities measured over the whole human genome. We see that the probabilities computed with the formulas in Theorem 2 accurately model the empirical probabilities.
Table 2.
Minimizer length m by varying k on the different datasets.
| 31 | 35 | 39 | 43 | 47 | 51 | 55 | 59 | 63 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Yeast | 15 | 15 | 16 | 16 | 16 | 16 | 18 | 18 | 18 | |
| Elegans | 16 | 18 | 18 | 20 | 20 | 20 | 20 | 20 | 20 | |
| Cod | 20 | 20 | 22 | 22 | 22 | 24 | 24 | 24 | 24 | |
| Kestrel | 20 | 20 | 22 | 22 | 22 | 24 | 24 | 24 | 24 | |
| Human | 21 | 21 | 23 | 23 | 26 | 26 | 28 | 28 | 28 |
The net result is that, for sufficiently large w, the probabilities in Theorem 2 are all approximately equal to 1/4, so that we have super-k-mers of each type. This also implies that the choice of 2-bit codes for the symbols of R is essentially optimal. Under this condition, we give the following theorem (see the Supplementary Material for the proof).
Theorem 3.
Given a random minimizer scheme with for any and , there exists a -LP MPHF for a SPSS with which takeswhere and b is a constant larger than .
3.3 Ambiguous minimizers
Let be the set of super-k-mers whose minimizer is . The rank computation in Equation (2) can be used as long as , i.e. whenever one single super-k-mer g has minimizer and, thus, the single unequivocally displace all the k-mers . When we say that the minimizer is ambiguous. It is a known fact that the number of such minimizers is very small for a sufficiently long minimizer length m (Chikhi et al. 2014; Jain et al. 2020; Pibiri 2022b), and the number decreases for growing m. For example, on the datasets used in Section 4, the fraction of ambiguous minimizers is in between 1% and 4%. However, they must be dealt with in some way.
Let be the fraction of k-mers whose minimizers are ambiguous. Our strategy is to build a fallback MPHF for these k-mers. This function adds bits/k-mer on top of the space of Theorem 1 and Theorem 3, where is the number of bits per key spent by a MPHF of choice. The fallback MPHF makes our functions -locality-preserving.
To detect ambiguous minimizers, one obvious option would be to explicitly use an extra 1-bit code per minimizer. This would however result in a waste of 1 bit per minimizer for most of them since we expect to have a small percentage of ambiguous minimizers. To avoid these problems, we use the following trick. Suppose is an ambiguous minimizer. We initially pretend that is not ambiguous. For the unpartitioned data structure from Section 3.1, we set . A super-k-mer of size 0 is clearly not possible, thus we use the value 0 to indicate that is actually ambiguous. We do the same for the partitioned data structure from Section 3.2: in this case, we set pretending the type of is right-max (but we could have also used the type left-max or non-max). To sum up, with just an extra check on the super-k-mer size we know if the query k-mer must be looked-up in the fallback MPHF or not.
We leave the exploration of alternative strategies to handle ambiguous minimizers to future work. For example, one can imagine a recursive data structure where, similarly to Shibuya et al. (2022), each level is an instance of the construction with different minimizer lengths: if level i has minimizer length , then level is built with length over the k-mers whose minimizers are ambiguous at level i.
4 Experiments
In this section, we report on the experiments conducted to assess the practical performance of the method presented in Section 3, which we refer to as LPHash in the following. Our implementation of the method is written in C++ and available at https://github.com/jermp/lphash.
Implementation details. We report here the major implementation details for LPHash. The arrays L and P are compressed with Elias-Fano (Fano 1971; Elias 1974) to exploit its constant-time random access (see also Pibiri and Venturini 2021, Section 3.4) for an explanation of such compressed encoding). Both the function and the fallback MPHF are implemented with PTHash using parameters , unless otherwise specified. Under this configuration, the space taken by a PTHash MPHF is 2.3–2.5 bits/key.
We do not compress the bit-vectors in the wavelet tree and we add constant-time support for rank queries using the Rank9 index (Jacobson 1989; Vigna 2008). The Rank9 index adds more space at each level of the wavelet tree, making the wavelet tree to take 2.5 bits per element in practice. Therefore, we estimate the little-Oh factor in Theorem 1 and Theorem 3 to be 0.5.
Competitors. We compare the space usage, query time, and building time of LPHash against PTHash (Pibiri and Trani a,b), the fastest MPHF in the literature, and the popular BBHash (Limasset et al. 2017). Both competitors are also written in C++. Following the recommendations of the respective authors, we tested two example configurations each:
PTHash-v1, with parameters ;
PTHash-v2, with parameters ;
BBHash-v1, with parameter ;
BBHash-v2, with parameter ;
We point the reader to the respective papers for an explanation of such parameters; we just report that they offer a trade-off between space, query efficiency, and building time as also apparent in the following experiments.
Testing machine. The experiments were executed on a machine equipped with a Intel i9-9900K CPU (clocked at 3.60 GHz), 64 GB of RAM, and running the Linux 5.13.0 operating system. The whole code (LPHash and competitors) was compiled with gcc 11.2.0, using the flags -O3 and -march=native.
Datasets. We use datasets of increasing size in terms of number of distinct k-mers; namely, the whole-genomes of: Saccharomyces cerevisiae (Yeast, 11.6 × 106k-mers), Caenorhabditis elegans (Elegans, 96.5 × 106k-mers), Gadus morhua (Cod, 0.56 × 109k-mers), Falco tinnunculus (Kestrel, 1.16 × 109k-mers), and Homo sapiens (Human, 2.77 × 109k-mers). For each dataset, we obtain the corresponding SPSS by first building the compacted de Bruijn graph using BCALM2 (Chikhi et al. 2016), then running the UST algorithm (Rahman and Medvedev 2020). At our code repository we provide detailed instructions on how to prepare the datasets for indexing. Also, all processed datasets are available at https://zenodo.org/record/7239205 already in processed form so that it is easy to reproduce our results.
4.1 Space effectiveness
To build an instance of LPHash for a given k, we have to choose a suitable value of minimizer length (m). A suitable value of m should clearly be not too small (otherwise, most minimizers will appear many times), nor too large (otherwise, the space of will be too large as well). In general, a good value for m can be chosen around where N is the cumulative length of the strings in the input SPSS. Remember from our discussion in Section 3.3 that the fraction of ambiguous minimizers decreases for growing m. Therefore, testing LPHash for growing values of k allows us to progressively increase m, starting from , while keeping sufficiently large and reducing the fraction of ambiguous minimizers as well. Following this principle, for each combination of k and dataset, we choose m as reported in Table 2.
Figure 5 shows the space of LPHash in average bits/k-mer, by varying k from 31 to 63 with a step of 4, for both un-partitioned and partitioned data structures. We report the actual space usage achieved by the implementation against the space bounds computed using Theorem 1 (un-partitioned) and Theorem 3 (partitioned) for . The b parameter models the number of bits per key spent by a MPHF of choice for the representation of the minimizer MPHF and the fallback MPHF. (For all datasets, we use for the PTHash and fallback, except on the largest Human where we use to lower construction time at the expense of a larger space usage.)
Figure 5.

Space in average bits/k-mer for LPHash by varying k, for both unpartitioned and partitioned data structures. The flat solid line at bits/k-mer indicates the classic MPHF lower-bound. Lastly, the dashed lines corresponds to the space bounds computed using Theorem 1 and Theorem 3 with and including the space for the fallback MPHF.
We make the following observations.
The space bounds computed with Theorem 1 and Theorem 3 are very similar to the actual space usage of LPHash, thus confirming the correctness and accuracy of our analysis in Section 3.
As expected, the space of LPHash lowers for increasing k and the partitioned data structure is always considerably smaller than the un-partitioned counterpart.
We report the space taken by the tested competitive configurations in Table 3. Comparing the space values in Table 3 with those in Fig. 5, the net result is that the space of LPHash is much lower than that of the classic MPHFs traditionally used in the prior literature and in practice.
Table 3.
Space in average bits/k-mer for PTHash and BBHash.a As reference points, we also report the bits/k-mer for partitioned LPHash for three representative values of k (see also Fig. 5).
| Method | k | Yeast | Elegans | Cod | Kestrel | Human |
|---|---|---|---|---|---|---|
| LPHash | 31 | 1.18 | 1.47 | 1.55 | 1.43 | 1.74 |
| 47 | 0.72 | 0.85 | 1.01 | 0.82 | 1.14 | |
| 63 | 0.53 | 0.64 | 0.83 | 0.58 | 0.87 | |
|
| ||||||
| PTHash-v1 | 2.76 | 2.68 | 2.65 | 2.58 | 2.65 | |
| PTHash-v2 | 2.20 | 2.13 | 2.09 | 2.06 | 2.04 | |
| BBHash-v1 | 3.71 | 3.71 | 3.71 | 3.71 | 3.71 | |
| BBHash-v2 | 3.06 | 3.06 | 3.06 | 3.06 | 3.06 | |
The numbers reported in Table 3 were taken for k = 63 although the avg. bits/k-mer for PTHash and BBHash does not depend on k.
To make a concrete example, partitioned LPHash for achieves 0.53, 0.64, 0.83, 0.58, and 0.87 bits/k-mer on Yeast, Elegans, Cod, Kestrel, and Human respectively. These values are , , , , and smaller than the those achieved by PTHash-v1 (and even smaller when compared to BBHash). Even compared to the most succinct configuration, PTHash-v2 (around 2 bits/k-mer), LPHash still retains 2.3–3.7× better space.
We remark that, however, PTHash and BBHash are “general-purpose” MPHFs that can work with arbitrary keys, whereas the applicability of LPHash is restricted to spectrum-preserving string sets.
4.2 Query time
Table 4 reports the query time for LPHash in comparison to PTHash and BBHash. Timings were collected using a single core of the processor. We query all k-mers read from the Human chromosome 13, for a total of queries. First of all, we report that query timings for un-partitioned and partitioned LPHash are the same, so we do not distinguish between the two data structures in Table 4. This meets our expectation regarding the average number of array accesses that the two query algorithms perform as explained in Section 3.2.
Table 4.
Query time in average nanoseconds per k-mer.
| Method | k | Yeast |
Elegans |
Cod |
Kestrel |
Human |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| stream | random | stream | random | stream | random | stream | random | stream | random | ||
| LPHash | 31 | 29 | 110 | 40 | 118 | 79 | 144 | 84 | 145 | 107 | 162 |
| 35 | 28 | 125 | 35 | 124 | 65 | 147 | 69 | 149 | 90 | 166 | |
| 39 | 27 | 130 | 32 | 131 | 60 | 149 | 63 | 153 | 82 | 166 | |
| 43 | 25 | 137 | 30 | 135 | 52 | 152 | 54 | 155 | 73 | 169 | |
| 47 | 24 | 145 | 28 | 143 | 47 | 155 | 49 | 159 | 69 | 172 | |
| 51 | 24 | 152 | 28 | 150 | 45 | 159 | 46 | 162 | 63 | 174 | |
| 55 | 23 | 157 | 26 | 157 | 41 | 165 | 42 | 167 | 59 | 176 | |
| 59 | 23 | 165 | 25 | 165 | 39 | 171 | 39 | 173 | 57 | 182 | |
| 63 | 22 | 174 | 24 | 172 | 37 | 180 | 37 | 179 | 53 | 188 | |
|
| |||||||||||
| PTHash-v1 | 24 | 46 | 67 | 72 | 72 | ||||||
| PTHash-v2 | 38 | 64 | 130 | 155 | 175 | ||||||
| BBHash-v1 | 42 | 118 | 170 | 175 | 175 | ||||||
| BBHash-v2 | 42 | 125 | 180 | 190 | 190 | ||||||
We distinguish between streaming and random queries (lookups) for LPHash. Given a query string Q, we query for each k-mer read “consecutively” from Q, that is, for Q[1.k], , , etc. We refer to the this query modality as streaming; anything else different from streaming is a random lookup (i.e. “random” here means “without locality”). LPHash is optimized for streaming lookup queries, whereas PTHash and BBHash do not benefit from any specific query order. In fact, the locality-preserving nature of LPHash makes the calculation of hashes for consecutive k-mers very cheap, as consecutive k-mers are likely to be part of the same super-k-mer.
Considering the result in Table 4, we see that LPHash’s streaming query time is in fact much smaller than random query time. Both timings are sensitive to the growth of k: while the streaming one slightly decreases for the better locality, the random one increases instead, for the more expensive hash calculations.
LPHash is as fast as PTHash-v1 (fastest configuration) for streaming queries on the smaller Yeast dataset, but actually up to 1.4–2× faster on the larger datasets Elegans, Cod, Kestrel, and Human. Instead, it is up to faster than PTHash-v2. We stress that this is a remarkable result given that PTHash is the fastest MPHF in the literature, being 2–6× faster than other methods. Compared to BBHash, LPHash is faster on Yeast and up to 4–5× faster on the larger datasets.
Random lookup time is, instead, slower for LPHash compared to PTHash: this is expected because the evaluation of LPHash is more complex (it involves computing the minimizer, accessing several arrays, and computing a rank using a wavelet tree). However, we do not regard this as a serious limitation since, as we already motivated, the streaming query modality is the one used in Bioinformatics tasks involving k-mers (Almodaresi et al. 2018; Bingmann et al. 2019; Marchet et al. 2021; Robidou and Peterlongo 2021; Pibiri 2022b). We also observe that the slowdown is more evident on the smaller datasets while it tends to diminish on the larger ones. Except for the smaller Yeast dataset, the random lookup time of LPHash is competitive with that of BBHash or better.
4.3 Building time
We now consider building time which is reported in Table 5. Both LPHash and PTHash were built limiting to 8GB the maximum amount of RAM to use before resorting to external memory. (There is no such capability in the BBHash implementation so BBHash took more RAM at building time than the other two constructions.)
Table 5.
Total building time, including the time to read the input and serialize the data structure on disk. All constructions were run with four processing threads.
| Method | Yeast | Elegans | Cod | Kestrel | Human |
|---|---|---|---|---|---|
| mm:ss | mm:ss | mm:ss | mm:ss | mm:ss | |
| LPHash | 00:01 | 00:15 | 05:30 | 03:50 | 07:25 |
| PTHash-v1 | 00:03 | 00:29 | 07:37 | 20:34 | 63:30 |
| PTHash-v2 | 00:03 | 00:46 | 14:15 | 40:00 | 124:00 |
| BBHash-v1 | 00:01 | 00:07 | 00:48 | 01:40 | 04:13 |
| BBHash-v2 | 00:01 | 00:08 | 01:05 | 02:22 | 07:50 |
The building time for un-partitioned and partitioned LPHash is the same. LPHash is competitive with the fastest BBHash and significantly faster than PTHash on the larger datasets. Specifically, it is faster than PTHash over the entire set of k-mers since it builds two smaller PTHash functions ( and fallback). The slowdown seen for Cod is due to the larger fallback MPHF, which is built with PTHash under a strict configuration () that privileges space effectiveness (and query efficiency) rather than building time. One could in principle use BBHash instead of PTHash for the fallback function, hence trading space for better building time. For example, recall that we use on Human for this reason.
5 Conclusion and future work
In this article, we initiate the study of locality-preserving MPHFs for k-mers. We propose a construction, named LPHash, that achieves very compact space by exploiting the fact that consecutive k-mers share overlaps of symbols. This allows LPHash to actually break the theoretical bit/key barrier for MPHFs.
We show that a concrete implementation of the method is practical as well. Before this paper, one used to build a BBHash function over the k-mers and spend (approximately) 3 bits/k-mer and 100–200 nanoseconds per lookup. This work shows that it is possible to do significantly better than this when the k-mers come from a spectrum-preserving string set: for example, less than 0.6–0.9 bits/k-mer and 30–60 nanoseconds per lookup. Our code is open-source.
As future work, we plan to further engineer the current implementation to accelerate construction and streaming queries. Other strategies for sampling the strings could be used other than random minimizers (Frith et al. 2022); for example, the Miniception (Zheng et al. 2020) achieving . Evaluating the impact of such different sampling schemes is a promising avenue for future research. Lastly, we also plan to investigate other strategies for handling the ambiguous minimizers. A better strategy is likely to lead to improved space effectiveness and faster construction.
Supplementary Material
Acknowledgments
The first author wishes to thank Piotr Beling for useful comments on an early draft of the article.
Contributor Information
Giulio Ermanno Pibiri, Ca’ Foscari University of Venice, Venice 30172, Italy; ISTI-CNR, Pisa 56124, Italy.
Yoshihiro Shibuya, University Gustave Eiffel, Marne-la-Vallée 77454, France.
Antoine Limasset, University of Lille, CRIStAL CNRS, UMR 9189 , Lille F-59000, France.
Supplementary data
Supplementary data are available at Bioinformatics online.
Conflict of interest
None declared.
Funding
Funding for this research has been provided by the European Union's Horizon Europe research and innovation programme (EFRA project, Grant Agreement Number 101093026) and by the French ANR AGATE (ANR-21-CE45-0012).
Code availability
Data availability
References
- Almodaresi F, Sarkar H, Srivastava A. et al. A space and time-efficient index for the compacted colored de Bruijn graph. Bioinformatics 2018;34:i169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingmann T, Bradley P, Gauger F. et al. Cobs: a compact bit-sliced signature index. In: International Symposium on String Processing and Information Retrieval. 2019, 285–303.
- Břinda K, Baym M, Kucherov G.. Simplitigs as an efficient and scalable representation of de Bruijn graphs. Genome Biol 2021;22:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-C, Lin C-Y.. Perfect hashing schemes for mining association rules. Comput J 2005;48:168–79. [Google Scholar]
- Chikhi R, Limasset A, Jackman S. et al. On the representation of de Bruijn graphs. In: International Conference on Research in Computational Molecular Biology. 2014, 35–55. [DOI] [PubMed]
- Chikhi R, Limasset A, Medvedev P.. Compacting de Bruijn graphs from sequencing data quickly and in low memory. Bioinformatics 2016;32:i201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias P. Efficient storage and retrieval by content and address of static files. J ACM 1974;21:246–60. [Google Scholar]
- Fano RM. On the number of bits required to implement an associative memory. In: Memorandum 61, Computer Structures Group, MIT. 1971.
- Fox EA, Chen QF, Daoud AM. et al. Order-preserving minimal perfect hash functions and information retrieval. ACM Trans Inf Syst 1991;9:281–308. [Google Scholar]
- Frith MC, Shaw J, Spouge JL. How to optimally sample a sequence for rapid analysis. bioRxiv2022, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- Grossi R, Gupta A, Vitter JS. High-order entropy-compressed text indexes. In: Proceedings of the Fourteenth Annual ACM-SIAM Symposium on Discrete Algorithms. 2003, 841–50.
- Jacobson G. Space-efficient static trees and graphs. In: 30th Annual Symposium on Foundations of Computer Science. 1989, 549–54.
- Jain C, Rhie A, Zhang H. et al. Weighted minimizer sampling improves long read mapping. Bioinformatics 2020;36:i111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Patro R.. Cuttlefish: fast, parallel and low-memory compaction of de Bruijn graphs from large-scale genome collections. Bioinformatics 2021;37:i177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Kokot M, Deorowicz S. et al. Scalable, ultra-fast, and low-memory construction of compacted de bruijn graphs with cuttlefish 2. Genome Biol 2022;23:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limasset A, Rizk G, Chikhi R. et al. Fast and scalable minimal perfect hashing for massive key sets. In: 16th International Symposium on Experimental Algorithms, Vol. 11. 2017, 1–11. [Google Scholar]
- Lu Y, Prabhakar B, Bonomi F. Perfect hashing for network applications. In: 2006 IEEE International Symposium on Information Theory. 2006, 2774–8.
- Marchet C, Kerbiriou M, Limasset A.. Blight: efficient exact associative structure for k-mers. Bioinformatics 2021;37:2858–65. [DOI] [PubMed] [Google Scholar]
- Mehlhorn K. On the program size of perfect and universal hash functions. In: 23rd Annual Symposium on Foundations of Computer Science. 1982, 170–5.
- Pibiri GE. On weighted k-mer dictionaries. In: International Workshop on Algorithms in Bioinformatics (WABI). 2022a, 9:1–9:20. [Google Scholar]
- Pibiri GE. Sparse and skew hashing of k-mers. Bioinformatics 2022b;38:i185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pibiri GE, Kanda S.. Rank/select queries over mutable bitmaps. Inf Syst 2021;99:101756. [Google Scholar]
- Pibiri GE, Trani R. PTHash: revisiting FCH minimal perfect hashing. In: The 44th International ACM SIGIR Conference on Research and Development in Information Retrieval. 2021a, 1339–48.
- Pibiri GE, Trani R.. Parallel and external-memory construction of minimal perfect hash functions with PTHash. CoRR abs/2106.02350, 2021b. [Google Scholar]
- Pibiri GE, Venturini R.. Handling massive N-gram datasets efficiently. ACM Trans Inf Syst 2019;37:1–25:41. [Google Scholar]
- Pibiri GE, Venturini R.. Techniques for inverted index compression. ACM Comput Surv 2021;53:1–36. [Google Scholar]
- Rahman A, Medvedev P. Representation of k-mer sets using spectrum-preserving string sets. In: International Conference on Research in Computational Molecular Biology. 2020, 152–68. [DOI] [PMC free article] [PubMed]
- Roberts M, Hayes W, Hunt BR. et al. Reducing storage requirements for biological sequence comparison. Bioinformatics 2004;20:3363–9. [DOI] [PubMed] [Google Scholar]
- Robidou L, Peterlongo P. findere: fast and precise approximate membership query. In: International Symposium on String Processing and Information Retrieval. 2021, 151–63.
- Schleimer S, Wilkerson DS, Aiken A. Winnowing: local algorithms for document fingerprinting. In: Proceedings of the 2003 ACM SIGMOD International Conference on Management of Data. 2003, 76–85.
- Shibuya Y, Belazzougui D, Kucherov G.. Space-efficient representation of genomic k-mer count tables. Algorithms Mol Biol 2022;17:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimel GP, Rastrow A, Tiwari G. et al. Rescore in a Flash: compact, cache efficient hashing data structures for n-gram language models. In: Proceedings of the 21st Annual Conference of the International Speech Communication Association. 2020, 3386–90.
- Vigna S. Broadword implementation of rank/select queries. In: International Workshop on Experimental and Efficient Algorithms. 2008, 154–68.
- Zheng H, Kingsford C, Marçais G.. Improved design and analysis of practical minimizers. Bioinformatics 2020;36:i119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.