

Abstract

In the context of the ever-growing interest in the cyclic diaryliodonium salts, this work presents synthetic design principles for a new family of structures with two hypervalent halogens in the ring. The smallest bis-phenylene derivative, [(C6H4)2I2]2+, was prepared through oxidative dimerization of a precursor bearing the ortho-disposed iodine and trifluoroborate groups. We also report, for the first time, the formation of cycles containing two different halogen atoms. These present two phenylenes linked by hetero-(I/Br) or -(I/Cl) halogen pairs. This approach was also extended to the cyclic bis-naphthylene derivative [(C10H6)2I2]2+. The structures of these bis-halogen(III) rings were further assessed through X-ray analysis. The simplest cyclic phenylene bis-iodine(III) derivative features the interplanar angle of ∼120°, while a smaller angle of ∼103° was found for the analogous naphthylene-based salt. All dications form dimeric pairs through a combination of π–π and C–H/π interactions. As the largest member of the family, a bis-I(III)-macrocycle was also assembled using the quasi-planar xanthene backbone. Its geometry enables the two iodine(III) centers to be bridged intramolecularly by two bidentate triflate anions. In a preliminary manner, the interaction of the phenylene- and naphthalene-based bis-iodine(III) dications with a new family of rigid bidentate bis-pyridine ligands was studied in solution and the solid state, with an X-ray structure showing the chelating donor bonding to just one of the two iodine centers.
Introduction
The term “diaryliodonium salt” refers to iodine(III) compounds in which the trivalent iodine center is bound to two aromatic rings and a third typically weakly coordinating anion (Figure 1). This structure class requires little introduction, given that the prototypical diaryliodonium motif (Figure 1, A)1,2 has been known for over 120 years,3 growing into an important class of aryl transfer agents. Diaryliodonium salts have been employed in a plethora of polar and radical processes, including metal-catalyzed and light-induced arylation reactions.4 In this context, the cyclic diaryliodonium derivatives containing mutually connected aryl groups have been of particular interest, with the iodine-containing ring structure imparting a series of new chemical and physical properties. For example, while the archetypal five-membered structure type B (Figure 1) is particularly stable toward conventional nucleophilic attack,5 this and the larger iodacycles (e.g., type C) undergo all sorts of ring-opening and ring-enlargement reactions under metal-catalyzed6,7 or single electron transfer (SET) conditions.8 Beyond this synthetic potential, cyclic diaryliodonium cations are also known to interfere with a variety of biological electron-transport systems. In fact, the parent diphenylene-iodonium (“DPI”) cation (archetype B) is widely used as a broad-spectrum go-to inhibitor of NADPH oxidases and other flavoenzymes,9 either as reporter/modulators of cellular activity9b or as therapeutic candidates.9c Recently, the ability of the Lewis acidic iodine(III) center to engage in highly directional intermolecular interactions, sometimes referred to as “halogen bonding”,10 has come under a spotlight in the fields of crystal engineering, molecular recognition, and Lewis acid organocatalysts.11,12 Another center of focus has been the revival (after decades of relatively little interest) of the hypervalent derivatives of the lighter two halogens: Br and Cl.13,14 Fresh examples include the bromonium-based cycloaddition reactions from the Wencel–Delord laboratory (Figure 1, D),14a−14c as well as chiral Br- and Cl-centered onium salts as enantioselective halogen-bonding catalysts by Yoshida et al. (structure E).14d
Figure 1.

A sampling of cyclic diaryl-halogen(III) structures with some applications. A: canonical diaryliodonium motif; B, C: most common cyclic diaryliodonium structure types; D, E: recent application of bromonium and chloronium salts; F, G: examples of molecules presenting multiple diaryliodonium groups.
In this context, our attention was drawn to the scarcely explored cyclic onium salts containing more than one iodine atom in a ring. A notable example of such compounds is the square-shaped macrocycle developed by Zhdankin and Stang, in which the near-90° C–I–C angles are used as corner pieces.15 Another example is the planar 3-fold symmetric iodine-doped sumanene derivative (Figure 1, F)16 obtained via an oxidation/EAS cyclization sequence. We note that routes such as this one are commonly used to prepare a variety of cyclic bis-diaryliodonium species,17,18 including the planar bis-diaryliodonium dication G (Figure 1). Interestingly, the latter exhibits exceptional Lewis acidity due to spatial convergence of the two iodonium C–I σ* vectors.19
With these precedents, and prompted by our own recent work on heteroatom-containing iodonium salts,20 we wondered about the properties of a hitherto unknown cyclic structure related to the archetype C (Figures 1 and 2), but having not one, but two iodine(III) bridges. Beyond the sheer synthetic challenge of making this cycle, this structure class appears to be an interesting platform for new “angle bar” molecular geometries (provided a near-90° C–I–C angle), and for exploring other classes of interhalogen synergistic effects. As part of this new program in our lab, we now report the synthesis and X-ray structures of not only this bis-λ3-iodane architecture (I–I), but also the analogous heterohalogen six-membered cycle with the I–Br and I–Cl bridges. The latter two represent, to the best of our knowledge, the first examples of a bona fide hetero-λ3-organo-halogen derivative. We also report the synthesis and structure of larger (macro)cyclic bis-λ3-iodane angle bars, as well as their interaction modes—including chelating and bridging—with bidentate donor structures.
Figure 2.

Principle bis-λ3-halonium angle bars developed in this study.
Results and Discussion
Bis-Halogen-Linked Bis-Phenyelene Cores
Our initial approach to the cyclic bis-iodonium 12+ was an electrophilic ring closing of the ortho-iodo-diphenyliodonium precursor 2.21 However, attempts to cyclize 2 under oxidative conditions commonly used in the formation of cyclic iodonium salts were unsuccessful, likely due to the strongly electron-withdrawing effect exerted by the already-present iodine(III) group.22 Seeking to overcome this reluctance to C–H cyclization, we took notice of a recent report from the Legault laboratory on the oxidative dimerization of the ortho-iodo-phenyltrifluoroborate 4 to the interesting zwitterion 3.23 Since the latter contains both an iodine and an ortho trifluoroborate group, we hoped that 4 might be directly dimerized to 12+ via two consecutive oxidation/transmetalation events. Our initial attempts to accomplish this transformation using Selectfluor as oxidant in CH3CN inevitably stopped at the intermediate 3, mirroring the original report.23 In addition, attempts to use this solvent at higher temperatures led to significant amounts of the ortho-diiodobenzene side-product, ostensibly through the breakdown of 3 or of the putative cyclic target. Nevertheless, a breakthrough came thanks to the use of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as solvent. Hence, exposing 3 to excess Selectfluor at 40–50 °C in HFIP suppressed the formation of ortho-diiodobenzene, leading instead to a gradual appearance of a new species with only two 1H NMR resonances, found at 8.5 and 7.9 ppm (see Scheme 1, A and B), in a pattern consistent with the C2v-symmetric bis-diaryliodonium 12+. Further corroboration came from HR-MS(ESI+), which revealed a peak at m/z = 202.9358 for the target dication [(C6H4)2I2]2+ (calcd 202.9352 for z = 2). The product was initially formed as the tetrafluoroborate salt, i.e., 1-(BF4)2. Nevertheless, given the crucial influence often exerted by the counterions in the chemistry and applications of a diaryliodonium fragment, the family was expanded with additional counterions. Hence, the BF4 salt could be converted to the virtually insoluble derivatives, either ditosylate 1-(OTs)2 (71% based on 3) or the di-iodide 1-(I2). The −OTf, −BF4, and even the BArf24 derivatives [BArf24 = B(3,5-bis-trifluoromethyl-C6H3)4] were also synthesized through anion exchange with the corresponding silver salt. X-ray-quality crystals of 1-(OTf)2 were grown through vapor diffusion of Et2O into the CH3CN solution. The salt crystallized in space group P1 and the solid state structure featured the tricyclic dication 12+ having a local C2v-symmetry. The two phenyl-containing planes intersect (at the I···I line) at a 120° angle (Scheme 1, C); while the C–I–C angles were measured at ∼95°. The I···I distance of 3.52 Å is somewhat shorter than the sum of the iodine van der Waals radii (∼3.8–4.0 Å), suggesting a degree of steric crowding between the two halogens. The bis-iodonium fragments are arranged in pairs (see Scheme 1, C, right), which are held together through π-stacking on their inner (concave) sides. Not shown are the additional principal I···O interactions with neighboring triflate anions (see SI) completing the roughly square planar environment around each iodine(III) center.
Scheme 1. Synthesis and Characterization of the Cyclic Bis-diaryliodonium 12+ and the Hetero-Bis-diaryl-Halonium 8a2+ and 8b2+.
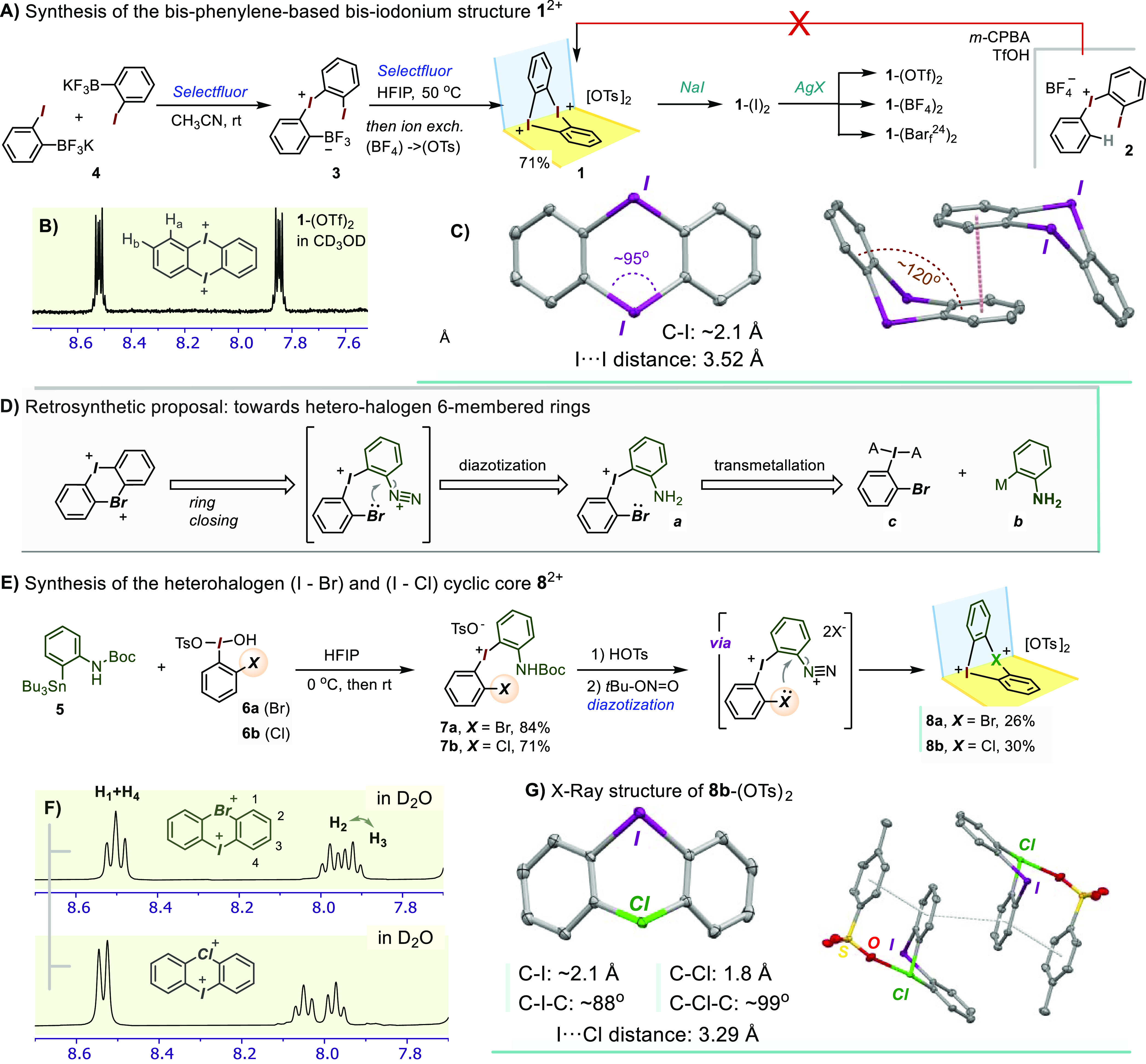
A and B: Synthesis of 12+, along with a portion of its 1H NMR spectrum. C: Solid state structure of 12+ along the molecular C2 axis (left) and a view of the mutually fitting dication pairs (right) (thermal ellipsoids shown with a 50% probability, H and OTf omitted for clarity). D: retrosynthetic approach to 82+. E: synthesis of 8a2+ and 8b2+. F: Aromatic 1H NMR regions of 8a (top) and 8b (bottom). G: Solid state structure of 8b2+; thermal ellipsoids shown with a 50% probability, H and OTs (fully or partially) omitted for clarity.
Having succeeded in the formation of the six-membered bis-iodane 12+, we sought to expand our study to rings incorporating two different halogen atoms. Indeed, despite a renewed spotlight on the organo-chloro(III) and -bromo(III) derivatives,14,24 we could find no precedent of molecules having two distinct high-valent halogen atoms, let alone an example of a heterohalogen ring structure.25 Given that the synthetic route designed for 12+ was deemed unsuitable for hard-to-oxidize lighter halogens, an alternative retrosynthetic sequence was envisioned to access the six-membered bromine(III)–iodine(III) cycle. The route would rely on the ortho-NH2 diaryliodonium precursors a (Scheme 1, D), with ring closing achieved via diazotization followed by cyclizative N2 displacement. The precursor a, in turn, could arise via transmetalation using a suitably ortho-metalated aniline b. This route, however, presents a series of challenges. One is the question of whether the diazotization event is even possible on a diaryliodonium core. Another is the actual synthesis of the ortho-amino derivative a, a species with scarce precedent in the literature, having been described as difficult to prepare and highly unstable.26 Fortunately, albeit after considerable effort, a route was identified in which a reaction between the ortho-stannyl N-Boc aniline 5 and the Koser-type ortho-bromo λ3-iodane 6a took place in HFIP to give the diaryliodonium intermediate 7a in 84% yield (Scheme 1-E). A one-pot N-deprotection of 7a with HOTs, followed by the addition of the diazotizing reagent tBu-ON=O14a led to the precipitation of an off-white powder. To our delight, the 1H NMR spectrum of this product displayed an aromatic ABCD pattern (clustered into two groups of resonances), in line with the Cs-symmetric mixed iodine(III)–bromine(III) cyclic bis-halonium target 8a-(OTs)2 (Scheme 1, F, top). The structure was further corroborated via HR-MS(ESI+) analysis, which revealed the (8a-OTs)+ peak at m/z = 528.8974 (vs calcd 528.8964), including the expected Br isotope pattern (see SI).
Seeking to extend this approach to the analogous mixed iodine(III)-chlorine(III) derivative, the o-Cl Koser derivative 6b was transformed into the diaryliodinium salt 7b in 71% yield. As had been the case for 7a, this salt underwent diazotizative cyclization to give, in this case, the mixed I(III)–Cl(III) cyclic salt 8b-(OTs)2.27 The 1H NMR analysis confirmed, once again, a Cs-symmetric dicationic portion, albeit with only minimal chemical shift separation between the two ortho-halo positions H1 and H4 (Scheme 1, F, bottom). The HR-MS(ESI+) peak for (8b-OTs)+ was recorded at m/z = 484.9462, in line with the calculated value of 484.9470.
Although 8a and 8b were only sparingly soluble in water, their 1H NMR spectra in D2O showed them to be surprising stable in aqueous solutions, remaining unchanged for several weeks at room temperature (see SI, Figures S3 and S5, respectively). Paradoxically, after just a few hours in dmso-d6, both compounds were observed to undergo clean hydrolytic ring opening to the phenolic derivatives S4 and S6, as observed by NMR and confirmed by HR-MS (see SI).
This made it somewhat challenging to grow single crystals of 8a and 8b, with earlier attempts invariably leading to hydrolysis. Nevertheless, X-ray-quality crystals of both species were obtained by taking advantage of the low product solubility in the reaction mixture upon their synthesis. Hence, a solution containing precursor 7a (or 7b), HOTs, and tBu-ON=O in MeNO2 was briefly heated to 60 °C and then left undisturbed at room temperature. After several days, small colorless plate-like crystals of both 8a-(OTs)2 and 8b-(OTs)2 were observed. Their X-ray analysis revealed the expected cyclic heterohalogen structures which, as in the case of 1, form the π-stacked pairs of dications (Scheme 1, G, for 8b-(OTs)2; for 8a-(OTs)2 see SI and the CIF file); these pairs, in turn, are boxed in between two tosylate anions via additional π-stacking interaction with the counterion's tolyl group. For both 8a2+ and 8b2+ structures, the C–X–C angles for the lighter halogen bridges (93.5° for Br and 98.6° for Cl) are larger than those for the corresponding C–I–C fragments (90.0° and 87.9°, respectively). This trend is in line with the predominant halogen p-orbital contribution for iodine(III) bonding and a larger s-orbital component associated with lighter halogens, as discussed recently by Stuart and co-workers.28
Toward the Bis-diaryliodonium Angle Bar Structure
As part of our broader interest in the cyclic bis-halonium cyclic structures, we wondered whether the use of the 1,8-disubstituted naphthalene backbone, or even a wider-spaced anthracene scaffold, could afford rigid structures that approach a 90° “angle bar” geometry. We envisage that in addition to serving as a blueprint to other rigid halogen-based molecular architectures, such structures could allow for the study of new types of interplay between halogen(III) Lewis acidity (or halogen bonding) vectors. Hence, aiming to apply an oxidative head-to-tail dimerization process, as seen with 1, we began by synthesizing the peri-iodo-trifluoroborate precursor 10 from 1,8-diiodonaphthalene via a selective mono-magnesiation–borylation process (see SI). Gratifyingly, while the cyclization step in the synthesis of 1 required a stepwise approach and forcing conditions, simply treating 10 with excess Selectfluor in CH3CN at room temperature led, after 17 h, directly to the clean formation of the C2v-symmetric dication 92+ (Scheme 2, A).29 The resulting BF4 salt could be obtained in pure form in a 73% yield via reversed phase chromatography on C18 silica gel. Alternatively, treating the crude mixture with TsOH resulted in the precipitation of 9-(OTs)2 as a clean pale-yellow solid in an 86% yield. The OTf, PF6, and BArf24 derivatives were also obtained through a double anion exchange route via the insoluble 9-(I)2 form, followed by treatment with the silver salt of the corresponding counterion. Alternatively, 9-(BArf24)2 could also be obtained directly from the initially formed tetrafluoroborate via salt metathesis with NaBArf24. The C2v-symmetric dication 92+ presents three strongly deshielded 1H NMR resonances at ∼9.2, 8.5, and 7.9 ppm (see Scheme 2, B). The HR-MS(ESI+) analysis of the triflate salt (eluent spiked with formic acid) revealed a peak at m/z = 550.8991, in line with the value 550.8999 expected for [9]·(O2CH)+. Furthermore, the analysis of the BArf24 derivative led to m/z = 1368.9662, matching well the theoretical value of 1368.9672 for [9]·(BArf24)+ (Scheme 2, panel C). Crystals of 9-(OTf)2 suitable for X-ray structure determination grew in space group P1 through vapor diffusion of Et2O into a CH3CN solution. As expected, the peri C–I vectors in each naphthalene unit diverged at an angle of ∼22° due to the steric repulsion between iodine atoms, a situation further confirmed by the relatively short I···I contact distance of 3.38 Å. The structure showed a 103° angle between the two naphthalene planes, a value closer to an idealized 90° angle than the ∼120° angle observed in 1 (see panels D and E in Scheme 2). Once again, the bis-cationic fragments are arranged in tightly fitting mutually complementing pairs, which appear to be held together by a combination of π-stacking and CH−π interactions (panel E). Each iodine atom also presents two main I···O interactions with the neighboring triflate anions to form an approximate square plane geometry (see panel F), along with a number of secondary I····O interactions.
Scheme 2. Synthesis and Characterization of the Naphthalenyl-Based Bis-λ3-diaryliodonium Dication of 92+.
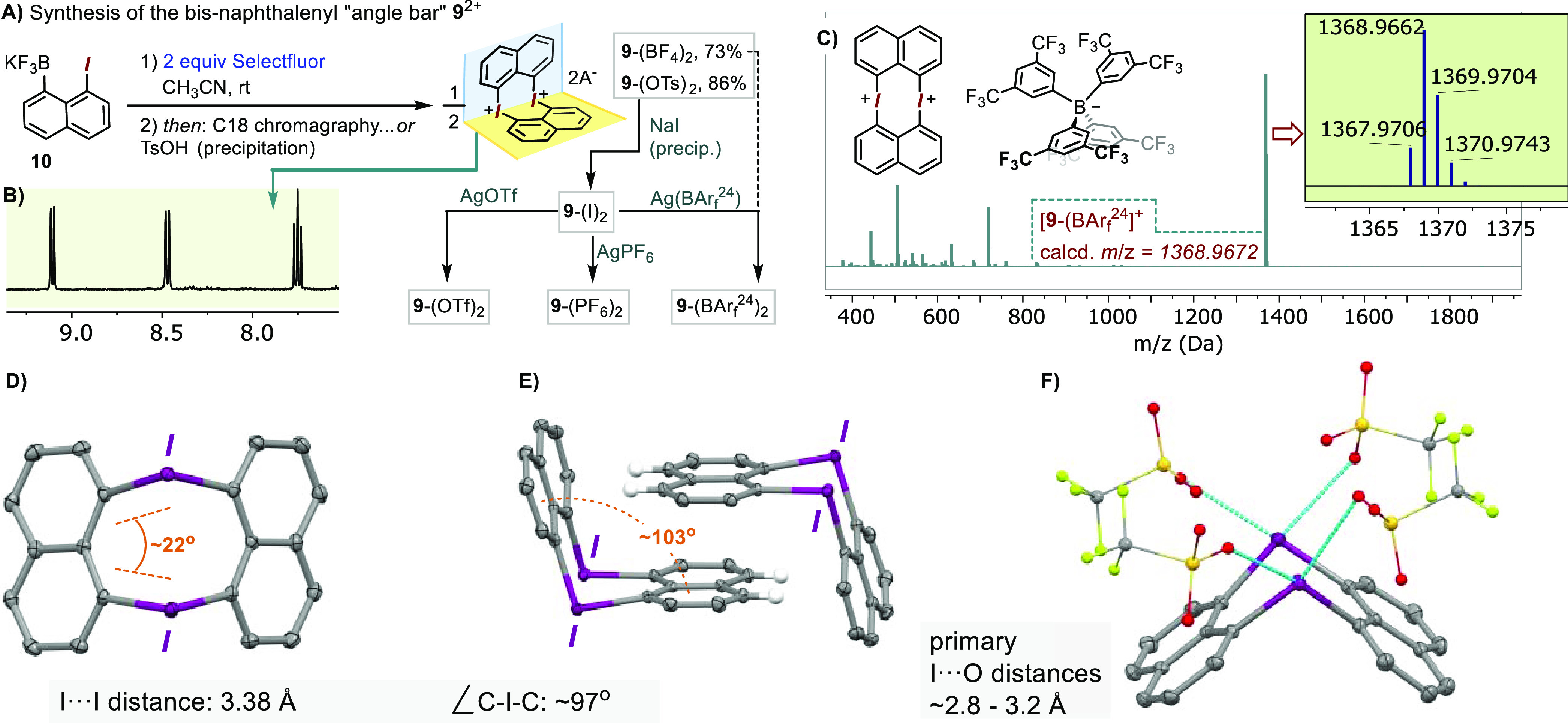
A and B: Synthetic route along with a portion of its 1H NMR spectrum. C: HR-MS(ESI+) analysis of 9-(BArf24)2. D, E, and F: partial X-ray ORTEP diagrams of 9-(OTf)2 at 50% thermal ellipsoids; most H omitted for clarity.
The newly obtained 9-(A)2 salts were found to degrade over time if not protected from light. Interestingly, perylene was identified as the main decomposition product. In fact, irradiation of 9-(PF6)2 in a DMF solution at 450 nm led to a rather clean formation of perylene in ∼70% yield, likely through a sequence of homolytic C–I cleavage and C–C coupling steps (Scheme 3).
Scheme 3. Photolytic Evolution of 9-(PF6)2 to Perylene.

To our initial disappointment, attempts to produce a wider-spaced anthracene analogue of 9 were unsuccessful, likely due to the poor solubility of the anthracene precursor and the facile oxidative degradation of the anthracene core. Instead, we turned our attention to the geometrically similar 9,9-dimethylxanthene backbone. Hence, as shown in Scheme 4, A, the bifunctional precursor 12 was prepared from the 4,5-disilyl-9,9-dimethylxanthene 11 by a selective exchange of one of the −SiMe3 groups for I using the I2/Selectfluor combination. Next, exposing 12 to Selectfluor led to an initial formation of a new compound, 13, tentatively identified by 1H and 19F NMR as bearing the hypervalent λ3-IF2 group.30 Heating this intermediate in the presence of BF3·Et2O helped induce the aryl transfer from silicon to iodine(III), leading to a gradual conversion of 13 to the more symmetric 12-membered macrocycle 142+. This result was supported by the observation of a simple three-resonance 1H NMR aromatic set for the new product (see SI). Furthermore, when measured in methanol-d4, the locked “angle bar” geometry leads to the splitting of the 9,9-di-Me signal into two Me singlets: one for the inner (a) and another for the outer (b) positions (Scheme 4, B). The 14-(BF4)2 salt was isolated in 40% yield via reversed-phase chromatography, while the corresponding triflate salt was obtained through double anion metathesis via the sparingly insoluble 14-(I)2 derivative. The HR-MS(ESI+) analysis produced a peak at m/z = 714.9847, consistent with 714.9837 calculated for [14]·(O2CH)+.
Scheme 4. Cyclic Bis-iodonium Structure 14 Based on 9,9-Dimethylxanthene: (A) Synthesis Starting with 9,9-Dimethylxanthene; (B) Upfield Portion of the 1H NMR Spectrum.

The structure of 14-(OTf)2 was further assessed through single-crystal X-ray analysis (Figure 3). As expected, the xanthene backbone imparts a relatively large I··I spacing of ∼4.2 Å, a value similar to that expected in a hypothetical anthracene-based structure. At the same time, the xanthene moiety was found to display certain conformational flexibility around the central pyran ring.31 Interestingly, while the solid state structures of 12+ and 92+ show pairs of divergent Lewis acidity vectors stemming from the neighboring iodine(III) centers, the corresponding vector pairs in the xanthene-based 142+ show a small degree of convergence (see Figure 3, C). Thanks to this, the two triflate ligands in the structure of 142+ are each capable of connecting adjacent iodine centers via crystallographically symmetric I··S–O–S··I bridges, leading to a geometry broadly reminiscent of the “paddle-wheel” bimetallic complexes (see Figure 3, B).32
Figure 3.
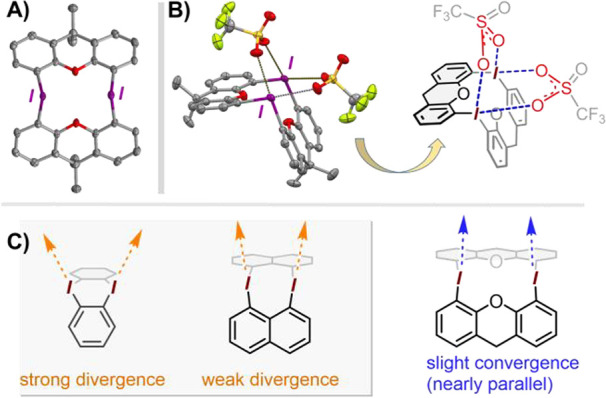
ORTEP-type diagram of the X-ray (single crystal) structure of 14-(OTf)2. Only one of the two independent molecules shown, with protons omitted for clarity. (A) Top view of 142+ showing the 12-membered macrocycle. (B) Side view of the full molecule illustrating the bridging (I–O–S–O-I) η2-triflate ligands and the overall “paddle-wheel” geometry. (C) Comparison of divergence in the hypothetical C–I σ* vectors in cyclic bis-iodonium dications.
Interaction with Rigid Bis-pyridine Ligands
Picking up on this last point, each halogen(III) center contributes with a pair of mutually perpendicular acidity directions. Therefore, all the bis-diarylhalonium dications shown thus far present four principal Lewis acidity vectors. In this scenario, a bidentate donor ligand could bind to such a dication in a number of modes. One possibility includes a ligand bridging between two adjacent iodine centers, as observed for the OTf ligands in 142+ (refer to Figure 3, B). Another is the chelating mode, i.e., two donor atoms binding to the same halogen site via its mutually perpendicular “vacant” sites.33 For the simple cyclic structure type B (see Figure 1), such scarcely precedented chelating mode was recently explored by the Huber laboratory employing bis-R(OR′)C=O: donor ligands based on a rigid bis-alkynyl-benzene backbone.34
Combining this idea with the recent advances in pyridine-stabilized I3+, I5+, and, especially, I1+ structures,35,36 a small family of rigid bis-pyridine donors was synthesized varying the N–N distances and chelation angles (Scheme 5, top panel). These were then used, along with pyridine itself, in binding studies with the bis-diaryliodonium Lewis acceptors. In fact, pyridine was previously used as model donor to measure and benchmark the Lewis acidities of various diaryliodonum cations,37 as well as of their bromine(III) and chlorine(III) analogues.28 Hence, in a very preliminary assay, a portionwise addition of pyridine to a CD2Cl2 solution of 1-(BArf24)2 caused a gradual upfield shift of the 12+ resonances, which were then used to extract the binding constant. Assuming a 1:1 binding model, a value of Ka ∼ 609 M–1 was obtained, which would be 27 times higher than for the noncyclic salt A (Figure 1) and ∼4.7 times higher than the value of 130 M–1 that had been previously measured for the simple cyclic diaryliodonium prototype B.28 Even for the naphthalene-based 9-(BArf24)2, this titration led to a Ka ∼ 308 M–1, which although not as high as in 12+ is still ∼2.4 times higher than for B. We are cognizant, however, that the assumption of the 1:1 binding stoichiometry in this model may break down, especially at higher pyridine concentration, where the potential rise of the 1:2 adduct (one pyridine per each iodine center) may affect the accuracy of these preliminary binding constants. Next, the NMR titration of the model dications 12+ and 92+ with this ligand set revealed good levels of binding, achieving binding constants as high as K ∼ 104 M–1 (see SI). We note, however, that in this preliminary module the exact binding modes could not be established unequivocally. Nevertheless, a combination of 1-(OTf)2 with ligand L3 did produce X-ray-quality single crystals with a one-to-one [1·L3]-(OTf)2 stoichiometry and the bis-pyridine chelating one of the two iodine atoms (Scheme 5). The adduct composition was further confirmed through elemental analysis of CHN, S, and I. Interestingly, the pyridine rings of the ligand appear to “push down” upon the phenylene groups, leading to an interplane angle of 108°, down from the 120° angle observed in the original 1-(OTf)2.
Scheme 5. Selected Rigid Bis-pyridine Ligand Set, along with the Formation of a 1:1 Adduct between 12+ and L3.

Last panel (bottom) shows an X-ray ORTEP diagram (50% probability plots) of [1·L3]-(OTf)2; H and OTf omitted for clarity.
Conclusions and Outlook
In conclusion, this work amplifies the structure space of diaryliodonium salts to a new family of cyclic diaryliodonium structures containing two halogens in a ring. Noteworthy, despite over half a century of the history of six-membered cyclic iodonium salts, this is the first report describing the synthesis and X-ray structure of even the simplest ring structure 12+ having two iodine atoms bridging between two phenylene rings. This delay reflects the need to solve a synthetically difficult ring-closing step, now possible through a formal head-to-tail dimerization approach. Thanks to this methodology, cyclic bis-iodonium salts based on wider-spaced naphthalene and xanthene scaffolds were also synthesized, showing geometric features resembling a right-angle “angle bar” structure. Using a complementary stepwise approach, the structure class was further expanded to heterohalogen iodine(III)–bromine(III) and iodine(III)–chlorine(III) analogues 82+, which showed remarkable stability in water. The new bis-iodonium structures allow for the study of new types of interplay between chelating ligands and pairs of iodine(III) Lewis acid vectors, which includes both chelating and bridging binding modes. We envisage that this chemistry and the structure archetypes presented herein will serve as a blueprint for the development of a wider range of cyclic multihalogen structures that would be of interest in the realms such as synthetic methodology, molecular recognition, materials, organocatalysis, and self-assembly, to name a few.
Acknowledgments
This work was supported through an MICINN grant (PID2020-113661GB-I00) and AGAUR (2021 SGR 00520, 2017 SGR 01051). IQS-URL is also acknowledged for the doctoral scholarship to W.W.C.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c02406.
Details on the synthetic procedures and product characterization, including NMR and MS data, and details on single-crystal X-ray structural characterization (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Yoshimura A.; Zhdankin V. V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]
- a Hypervalent Iodine Chemistry; Wirth T., Eds.; Springer International: Switzerland, 2016. [Google Scholar]; b Villo P.; Olofsson B., Arylations promoted by hypervalent iodine reagents. In PATAI’S Chemistry of Functional Groups; Olofsson B.; Marek I.; Rappoport Z., Eds.; Wiley, 2018; pp 1–42. [Google Scholar]
- Hartmann C.; Meyer V. Ueber eine neue Klasse jodhaltiger, stickstofffreier organischer Basen. Ber. Dtsch. Chem. Ges. 1894, 27, 426. 10.1002/cber.18940270183. [DOI] [Google Scholar]
- a Merritt E. A.; Olofsson B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]; b Olofsson B.Arylation with Diaryliodonium Salts. In Hypervalent Iodine Chemistry, Topics in Current Chemistry; Wirth T., Ed.; Springer International Publishing, 2016; Vol. 373, pp 135–166. [Google Scholar]
- Grushin V. V. Cyclic diaryliodonium ions: old mysteries solved and new applications envisaged. Chem. Soc. Rev. 2000, 29, 315–324. 10.1039/a909041j. [DOI] [Google Scholar]
- For a recent review, see:Chatterjee N.; Goswami A. Synthesis and Application of Cyclic Diaryliodonium Salts: A Platform for Bifunctionalization in a Single Step. Eur. J. Org. Chem. 2017, 2017, 3023–3032. 10.1002/ejoc.201601651. [DOI] [Google Scholar]
- For selected examples on metal-promoted ring opening, see (with Cu, atroposelective):; a Zhao K.; Duan L.; Xu S.; Jiang J.; Fu Y.; Gu Z. Enhanced Reactivity by Torsional Strain of Cyclic Diaryliodonium in Cu-Catalyzed Enantioselective Ring-Opening Reaction. Chem. 2018, 4, 599–612. 10.1016/j.chempr.2018.01.017. [DOI] [Google Scholar]; With Pd, enantioselective:; b Han J.; Xiao B.; Sun T.-Y.; Wang M.; Jin L.; Yu W.; Wang Y.; Fang D.-M.; Zhou Y.; Wu X.-F.; Wu Y.-D.; Liao J. Enantioselective Double Carbonylation Enabled by High-Valent Palladium Catalysis. J. Am. Chem. Soc. 2022, 144, 21800–21807. 10.1021/jacs.2c10559. [DOI] [PubMed] [Google Scholar]; With Pd, ring expansion:; c Mathew B. P.; Yang H. J.; Kim J.; Lee J. B.; Kim Y.-T.; Lee S.; Lee C. Y.; Choe W.; Myung K.; Park J.-U.; Hong S. Y. An Annulative Synthetic Strategy for Building Triphenylene Frameworks by Multiple C–H Bond Activations. Angew. Chem., Int. Ed. 2017, 56, 5007–5011. 10.1002/anie.201700405. [DOI] [PubMed] [Google Scholar]
- For an interesting recent example on TM-free radical-based ring expansions, see:Lee J. B.; Kim G. H.; Jeon J. H.; Jeong S. Y.; Lee S.; Park J.; Lee D.; Kwon Y.; Seo J. K.; Chun J.-H.; Kang S. J.; Choe W.; Rohde J.-U.; Hong S. Y. Rapid access to polycyclic N-heteroarenes from unactivated, simple azines via a base-promoted Minisci-type annulation. Nat. Commun. 2022, 13, 2421–2429. 10.1038/s41467-022-30086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a O’Donnell V. B.; Tew D. G.; Jones O. T. G.; England P. J. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem. J. 1993, 290, 41–49. 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Diaza J. M.; Plummer S.; Hansel C. M.; Andeer P. F.; Saito M. A.; McIlvin M. R. NADPH-dependent extracellular superoxide productionis vital to photophysiology in the marine diatom Thalassiosira oceanica,. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 16448–16453. 10.1073/pnas.1821233116. [DOI] [PMC free article] [PubMed] [Google Scholar]; For cyclic iodonium agents with low nanomolar activity against “problematic” bacteria, see:; c Nguyen N.; Wilson D. W.; Nagalingam G.; Triccas J. A.; Schneider E. K.; Li J.; Velkov T.; Baell J. Broad activity of diphenyleneiodonium analogues against Mycobacterium tuberculosis, malaria parasites and bacterial pathogens. Eur. J. Med. Chem. 2018, 148, 507–518. 10.1016/j.ejmech.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Mayer R. J.; Ofial A. R.; Mayr H.; Legault C. Y. Lewis Acidity Scale of Diaryliodonium Ions toward Oxygen, Nitrogen, and Halogen Lewis Bases. J. Am. Chem. Soc. 2020, 142, 5221–5233. 10.1021/jacs.9b12998. [DOI] [PubMed] [Google Scholar]
- For selected recent examples, see:; a Aliyarova I. S.; Ivanov D. M.; Soldatova N. S.; Novikov A. S.; Postnikov P. S.; Yusubov M. S.; Kukushkin V. Y. Bifurcated Halogen Bonding Involving Diaryliodonium Cations as Iodine(III)-Based Double-σ-Hole Donors. Cryst. Growth Des. 2021, 21, 1136–1147. 10.1021/acs.cgd.0c01463. [DOI] [Google Scholar]; Wolf J.; Huber F.; Erochok N.; Heinen F.; Guérin V.; Legault C. Y.; Kirsch S. F.; Huber S. M. Activation of a Metal-Halogen Bond by Halogen Bonding. Angew. Chem., Int. Ed. 2020, 59, 16496–16500. 10.1002/anie.202005214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Rahim A.; Peng W.; Jiang F.; Gu Z.; Wen S.. Recent Progress in Cyclic Aryliodonium Chemistry: Syntheses and Applications. Chem. Rev. 2023, 10.1021/acs.chemrev.2c00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For the earliest report on cyclic bromonium and chloronium salts, see:Sandin R. B.; Hay A. S. Stable Bromonium and Chloronium Salts. J. Am. Chem. Soc. 1952, 74, 274–275. 10.1021/ja01121a524. [DOI] [Google Scholar]; b Also, see:Egorova L. D.; Lisichkina I. N.; Tolstaya T. P.; Kostitsyna N. N. Synthesis of 10H-dibenzo[b,e]chlorinium tetrafluoroborate and deuterium exchange reaction in the methylene group of 10H-dibenzo[b,e]halogeninium cations. Izv. Akad. Nauk SSSR, Ser. Khim. 1985, 1347. [Google Scholar]
- a Lanzi M.; Dherbassy Q.; Wencel-Delord J. Cyclic Diaryl λ3-Bromanes as Original Aryne Precursors. Angew. Chem., Int. Ed. 2021, 60, 14852–14857. 10.1002/anie.202103625. [DOI] [PubMed] [Google Scholar]; b Lanzi M.; Ali Abdine R. A.; De Abreu M.; Wencel-Delord J. Cyclic Diaryl λ3-Bromanes: A Rapid Access to Molecular Complexity via Cycloaddition Reactions. Org. Lett. 2021, 23, 9047–9052. 10.1021/acs.orglett.1c03278. [DOI] [PubMed] [Google Scholar]; c Lanzi M.; Rogge T.; Truong T. S.; Houk K. N.; Wencel-Delord J. Cyclic Diaryl λ3-Chloranes: Reagents and Their C–C and C–O Couplings with Phenols via Aryne Intermediates. J. Am. Chem. Soc. 2023, 145, 345–358. 10.1021/jacs.2c10090. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yoshida Y.; Mino T.; Sakamoto M. Chiral Hypervalent Bromine(III) (Bromonium Salt): Hydrogen- and Halogen-Bonding Bifunctional Asymmetric Catalysis by Diaryl-λ3-bromanes. ACS Catal. 2021, 11, 13028–13033. 10.1021/acscatal.1c04070. [DOI] [Google Scholar]
- a Stang P. J.; Zhdankin V. V. Preparation and Characterization of a Macrocyclic Tetraaryltetraiodonium Compound, cyclo(Ar4I4)4+·4X–. A Unique, Charged, Cationic Molecular Box. J. Am. Chem. Soc. 1993, 115, 9808–9809. 10.1021/ja00074a061. [DOI] [Google Scholar]; b Radhakrishnan U.; Stang P. J. Synthesis and Characterization of Cationic Iodonium Macrocycles. J. Org. Chem. 2003, 68, 9209–9213. 10.1021/jo030246x. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhou D.; Zhang T.; Liu B.; Xu B. Iodine-doped sumanene and its application for the synthesis of chalcogenasumanenes and silasumanenes. Chem. Commun. 2017, 53, 10279–10282. 10.1039/C7CC05885C. [DOI] [PubMed] [Google Scholar]
- a Collette J.; McGreer D.; Crawford R.; Chubb F.; Sandin R. B. J. Am. Chem. Soc. 1956, 78, 3819–3820. 10.1021/ja01596a070. [DOI] [Google Scholar]; b Bielawski M.; Olofsson B. High-yielding one-pot synthesis of diaryliodonium triflates from arenes and iodine or aryl iodides. Chem. Commun. 2007, 2521–2523. 10.1039/b701864a. [DOI] [PubMed] [Google Scholar]; c Riedmüller S.; Nachtsheim B. J. Palladium-catalyzed synthesis of N-arylated carbazoles using anilines and cyclic diaryliodonium salts. Beilstein J. Org. Chem. 2013, 9, 1202–1209. 10.3762/bjoc.9.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For a systematic study, see:Postnikov P. S.; Guselnikova O. A.; Yusubov M. S.; Yoshimura A.; Nemykin V. N.; Zhdankin V. V. Preparation and X-ray Structural Study of Dibenziodolium Derivatives. J. Org. Chem. 2015, 80, 5783–5788. 10.1021/acs.joc.5b00741. [DOI] [PubMed] [Google Scholar]; b For synthesis and conformational analysis of six-membered iodonium salts, see:Caspers L. D.; Spils J.; Damrath M.; Lork E.; Nachtsheim B. J. One-Pot Synthesis and Conformational Analysis of Six-Membered Cyclic Iodonium Salts. J. Org. Chem. 2020, 85, 9161–9178. 10.1021/acs.joc.0c01125. [DOI] [PubMed] [Google Scholar]
- a Wu B.; Yoshikai N. Conversion of 2-Iodobiaryls into 2,2′-Diiodobiaryls via Oxidation-Iodination Sequences: A Versatile Route to Ladder-Type Heterofluorenes. Angew. Chem., Int. Ed. 2015, 54, 8736–8739. 10.1002/anie.201503134. [DOI] [PubMed] [Google Scholar]; b Heinen F.; Reinhard D. L.; Engelage E.; Huber S. M. A Bidentate Iodine(III)-Based Halogen-Bond Donor as a Powerful Organocatalyst. Angew. Chem., Int. Ed. 2021, 60, 5069–5073. 10.1002/anie.202013172. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Portela S.; Cabrera-Trujillo J. J.; Fernández I. Catalysis by Bidentate Iodine(III)-Based Halogen Donors: Surpassing the Activity of Strong Lewis Acids. J. Org. Chem. 2021, 86, 5317–5326. 10.1021/acs.joc.1c00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu Y.; Izquierdo S.; Vidossich P.; Lledós A.; Shafir A. NH-Heterocyclic Aryliodonium Salts and their Selective Conversion into N1-Aryl-5-iodoimidazoles. Angew. Chem., Int. Ed. 2016, 55, 7152–7156. 10.1002/anie.201602569. [DOI] [PubMed] [Google Scholar]; b Antonkin N. S.; Vlasenko Y. A.; Yoshimura A.; Smirnov V. I.; Borodina T. N.; Zhdankin V. V.; Yusubov M. S.; Shafir A.; Postnikov P. S. Preparation and Synthetic Applicability of Imidazole-Containing Cyclic Iodonium Salts. J. Org. Chem. 2021, 86, 7163–7178. 10.1021/acs.joc.1c00483. [DOI] [PubMed] [Google Scholar]
- Kervefors G.; Becker A.; Dey C.; Olofsson B. Metal-free formal synthesis of phenoxazine. Beilstein J. Org. Chem. 2018, 14, 1491–1497. 10.3762/bjoc.14.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NMR analysis seems to indicate the formation of I-oxidized noncyclized intermediates; see Figure S1 (SI). Similar reluctance to cyclize was recently documented during an attempted synthesis of an ortho-sulfone-bridged cyclic diaryliodonium salt:; Reinhard D. L.; Heinen F.; Stoesser J.; Engelage E.; Huber S. M. Tuning the Halogen Bonding Strength of Cyclic Diaryliodonium Salts. Helv. Chim. Acta 2021, 104, e2000221 10.1002/hlca.202000221. [DOI] [Google Scholar]; We don’t discard, however, that the cyclization of 2 to 1 may be possible under yet-to-be developed oxidative conditions.
- a Robidas R.; Guérin V.; Provenćal L.; Echeverria M.; Legault C. L. Investigation of iodonium trifluoroborate zwitterions as bifunctional arene reagents. Org. Lett. 2017, 19, 6420–6423. 10.1021/acs.orglett.7b03307. [DOI] [PubMed] [Google Scholar]; b fFor a related report on the ortho-borylated diaryliodonium salts, see:Yoshimura A.; Fuchs J. M.; Middleton K. R.; Maskaev A. V.; Rohde G. T.; Saito A.; Postnikov P. S.; Yusubov M. S.; Nemykin V. N.; Zhdankin V. V. Pseudocyclic Arylbenziodoxaboroles: Efficient Benzyne Precursors Triggered by Water at Room Temperature. Chem.—Eur. J. 2017, 23, 16738–16742. 10.1002/chem.201704393. [DOI] [PubMed] [Google Scholar]
- a Nakajima M.; Miyamoto K.; Hirano K.; Uchiyama M. Diaryl-λ3-chloranes: versatile synthesis and unique reactivity as aryl cation equivalent. J. Am. Chem. Soc. 2019, 141, 6499–6503. 10.1021/jacs.9b02436. [DOI] [PubMed] [Google Scholar]; b For a recent perspective on hypervalent bromine reagents, see:Winterson B.; Patra T.; Wirth T. Hypervalent Bromine(III) Compounds: Synthesis, Applications, Prospects. Synthesis 2022, 54, 1261–1271. 10.1055/a-1675-8404. [DOI] [Google Scholar]
- We note, however, a mention by Olah and co-workers of in situ NMR observation of a species arising from 1-bromo-4-iodo-benzene upon accepting two Me+ cations:Olah G. A.; Mo Y. K.; Melby E. G.; Lin H. C. Onium ions. V. Di- and trihalonium ions. J. Org. Chem. 1973, 38, 367–372. 10.1021/jo00942a034. [DOI] [Google Scholar]
- a Beringer F. M.; Lillien I. Diaryliodonium Salts. XIII. Salts in which the Cations Bear Carboxyl, Hydroxyl, Alkoxyl or Amino Groups. J. Am. Chem. Soc. 1960, 82, 725–731. 10.1021/ja01488a056. [DOI] [Google Scholar]; b Linstad E. J.; Va̅vere A. L.; Hu B.; Kempinger J. J.; Snyder S. E.; DiMagno S. G. Thermolysis and radiofluorination of diaryliodonium salts derived from anilines. Org. Biomol. Chem. 2017, 15, 2246–2252. 10.1039/C7OB00253J. [DOI] [PubMed] [Google Scholar]
- The modest yields obtained for 8a2+ and 8b2+ reflect the challenge of the diazotiative cyclization in this case. Reaction byproducts (as per HPLC-MS) are mainly noncyclic iodonium species consistent with the entry of nucleophiles, e.g., OTs and CH3CN (Ritter-type), either to the diazonium intermediate or (via ring-opening) to the final cycle.
- Karandikar S. S.; Bhattacharjee A.; Metze B. E.; Javaly N.; Valente E. J.; McCormick T. M.; Stuart D. R. Orbital analysis of bonding in diarylhalonium salts and relevance to periodic trends in structure and reactivity. Chem. Sci. 2022, 13, 6532–6540. 10.1039/D2SC02332F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- At shorter reactions times (∼1 h), or with less Selectfluor, we do observe a certain amount of the noncyclized species consistent with the zwitterionic intermediate (see Figure S7 in SI), i.e., an analogue of 3.
- Ye C.; Twamley B.; Shreeve J. M. Straightforward Syntheses of Hypervalent Iodine(III) Reagents Mediated by Selectfluor. Org. Lett. 2005, 18, 3961–3964. 10.1021/ol051446t. [DOI] [PubMed] [Google Scholar]
- In fact, the asymmetric unit contained two crystallographically independent 142+ structures differing somewhat in their degree and sense of puckering around the central pyran ring.
- Cotton F. A.; Wilkinson G.. Advanced Inorganic Chemistry, 5th ed.; Wiley: New York, 1988. [Google Scholar]
- Of course, a third possibility is that of a bidentante ligand bridging between two discreet iodonium species, which is what we see for most X-ray structures measured in this work.
- a Heinen F.; Engelage E.; Cramer C. J.; Huber S. M. Hypervalent Iodine(III) Compounds as Biaxial Halogen Bond Donors. J. Am. Chem. Soc. 2020, 142, 8633–8640. 10.1021/jacs.9b13309. [DOI] [PMC free article] [PubMed] [Google Scholar]; b For an interesting recent approach to iodine(III) “noncovalent” chelation with metal-centered species acting as bis-O bidentate ligands, also see:Semenov A. V.; Baykov S. V.; Soldatova N. S.; Geyl K. K.; Ivanov D. M.; Frontera A.; Boyarskiy V. P.; Postnikov P. S.; Kukushkin V. Yu. Noncovalent Chelation by Halogen Bonding in the Design of Metal-Containing Arrays: Assembly of Double σ-Hole Donating Halolium with CuI-Containing O,O-Donors. Inorg. Chem. 2023, 62, 6128–6137. 10.1021/acs.inorgchem.3c00229. [DOI] [PubMed] [Google Scholar]
- a For selected examples of iodine(III) and (V) chemistry with pyridine donors, see:Zhdankin V. V.; Koposov A. Y.; Yashin N. V. Complexes of hypervalent iodine compounds with nitrogen ligands. Tetrahedron Lett. 2002, 43, 5735–5737. 10.1016/S0040-4039(02)01192-9. [DOI] [Google Scholar]; b Tierno A. F.; Walters J. C.; Vazquez-Lopez A.; Xiao X.; Wengryniuk S. E. Heterocyclic group transfer reactions with I(III) N-HVI reagents: access to N-alkyl(heteroaryl)onium salts via olefin aminolactonization. Chem. Sci. 2021, 12, 6385–6392. 10.1039/D1SC00187F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xiao X.; Roth J. M.; Greenwood N. S.; Velopolcek M. K.; Aguirre J.; Jalali M.; Ariafard A.; Wengryniuk S. E. Bidentate Nitrogen-Ligated I(V) Reagents, Bi(N)-HVIs: Preparation, Stability, Structure, and Reactivity. J. Org. Chem. 2021, 86, 6566–6576. 10.1021/acs.joc.1c00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For a wide-angle bis-pyridine donor adduct of monovalent iodine, see:Carlsson A.-C. C.; Gráfenstein J.; Budnjo A.; Laurila J. L.; Bergquist J.; Karim A.; Kleinmaier R.; Brath U.; Erdélyi M. Symmetric Halogen Bonding Is Preferred in Solution. J. Am. Chem. Soc. 2012, 134, 5706–5715. 10.1021/ja301341h. [DOI] [PubMed] [Google Scholar]; b Vanderkooy A.; Gupta A. K.; Földes T.; Lindblad S.; Orthaber A.; Pápai I.; Erdélyi M. Halogen Bonding Helicates Encompassing Iodonium Cations. Angew. Chem. Int.Ed. 2019, 58, 9012–9016. 10.1002/anie.201904817. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yu S.; Kalenius E.; Frontera A.; Rissanen K. Macrocyclic complexes based on [N···I···N]+ halogen bonds. Chem. Commun. 2021, 57, 12464–12467. 10.1039/D1CC05616F. [DOI] [PubMed] [Google Scholar]
- a Ochiai M.; Suefuji T.; Shiro M.; Yamaguchi K. Complexation of Diphenyl(Tetrafluoroborato)-λ3-Iodane with Pyridines. Heterocycles 2006, 67, 391. 10.3987/COM-05-S(T)8. [DOI] [Google Scholar]; b Mayer R. J.; Ofial A. R.; Mayr H.; Legault C. Y. Lewis Acidity Scale of Diaryliodonium Ions toward Oxygen, Nitrogen, and Halogen Lewis Bases. J. Am. Chem. Soc. 2020, 142, 5221–5233. 10.1021/jacs.9b12998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.